

Abstract
Background/Aim: The role of nuclear respiratory factor 1 (NRF1) on the prostate cancer progression is controversial. We aimed to investigate the effect of NRF1 overexpression on the metastasis potential of PC3 prostate cancer cells and the associated molecular mechanisms.
Materials and Methods: Cell survival, migration capacity, mitochondrial biogenesis, the expression of TGF-β signaling and EMT markers were examined after overexpression and silencing of NRF1 in PC3 cells.
Results: We found that NRF1-overexpressing cells exhibited a decreased cell viability and proliferation ability as well as a reduced migration capacity compared to control cells. Moreover, ectopic expression of NRF1 increased the mitochondrial biogenesis and inhibited the EMT characteristics, including a decrease in the mesenchymal marker, α-SMA and an increase in the epithelial cell marker, E-cadherin. We also demonstrated that overexpression of NRF1 suppressed the expression of TGF-β signaling in PC3 cells. As expected, silencing of NRF1 reversed the abovementioned effects.
Conclusion: This study demonstrated that upregulation of NRF1 holds the potential to inhibit the metastasis of prostate cancer, possibly through an elevation of mitochondrial biogenesis and the subsequent repression of TGF-β-associated EMT. Therapeutic avenues that increase NRF1 expression may serve as an adjunct to conventional treatments of prostate cancer.
Keywords: Nuclear respiratory factor 1, mitochondrial biogenesis, prostate cancer, TGF-β signaling, epithelial mesenchymal transition
Over the past decades, prostate cancer has remained one of the frequently diagnosed cancers among males worldwide (1,2). Although the mortality rate has greatly decreased due to the improved treatment and/or early detection, prostate cancer still accounts for more than 1 in 5 newly diagnosed cancers in men (1,2). Moreover, metastatic prostate cancer has been shown to carry a dismal survival rate. Aside from epithelial-to-mesenchymal transition (EMT), the dysfunction of mitochondrial biogenesis has also been implicated in prostate cancer metastasis (3). It has been known that several factors mediated the regulation of mitochondrial biogenesis, such as peroxisome proliferator-activated receptor gamma co-activator (PGC-1α) or nuclear respiratory factors (NRF1 and NRF2). The role of PGC-1α in the development of prostate cancer has been addressed in several studies. For instance, PGC-1α was highly expressed in prostate cancer PC3 and DU145 cells, and PGC-1α could promote tumor growth of the androgen-dependent prostatic cancer cells through activation of the androgen receptor (4). On the other hand, the studies concerning the effect of NRFs on the cancer progression have been limited.
It has been shown that NRF1 protected breast cancer MCF-7 cells from tamoxifen-induced apoptosis and increased cancer cell survival (5). NRF1 has also been found to increase the cell cycle genes in E2-treated MCF-7 (6). Nevertheless, NRF1 seems to play both oncogenic and tumor suppressor roles in prostate cancer. It has been demonstrated that NRF1 collaborates with Oct4 to acquire chemo-resistance in the absence of androgen receptor (AR), while both NRF1 and Oct4 are elevated in castration-resistant prostate cancer tissues (7). Contrarily, another study revealed that an upregulation of NRF1 by sulforaphane treatment resulted in a decrease of cell viability in PC3 cells (8). Hence, we sought to evaluate the functional role of NRF1 in the prostate cancer cells with a higher metastatic potential and elucidate the changes of the downstream factors.
In the current study, we examined the effect of overexpressing NRF1 on the cell morphology, cell viability, migration ability and mitochondrial biogenesis of PC3 prostate cancer cells. Moreover, the expression of EMT markers and transforming growth factor beta (TGF-β) cascades were also assessed in the NRF1-overexpressing cells in order to unravel the implication of NRF1 in the metastatic capacity of prostate cancer. These findings provide a novel insight into the significance of NRF1 in the aggressiveness of prostate cancer and the associated mechanism.
Materials and Methods
Cell culture. The metastasis-derived PC3 prostate cancer cell line was obtained from the American Tissue Culture Collection (Rockville, MD, USA). Cells were cultivated and maintained in MEM medium (Sigma Chemical Co., St. Louis, MO, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) at 37˚C in a humidified atmosphere containing 5% CO2 and passaged twice a week.
Plasmid construction and transfection procedure. The plasmid DNA containing NRF-1 was a generous gift from Dr. Chang and NRF-1 full-length cDNA was listed in the previous study (9). The cDNA of NRF1 was cloned into the pcDNA3.1 plasmid (Clontech Inc., Palo Alto, CA, USA) following EcoRI/ BamHI digestion. The recombinant plasmids were then transduced into Escherichia coli DH5α cells (Tiangen, Beijing, PR China) and extracted using a plasmid DNA extraction kit (Takara Biotechnology Co., Ltd.). Serum-free MEM mixed with Fugene HD Transfection Reagent (Roche Molecular Biochemicals, Indianapolis, IN, USA) was prepared and then incubated at room temperature for 5 min. The PC3 cells were transfected with 2 μg/ml pcDNA.3.1-NRF1 using Fugene HD Transfection reagent (Roche Molecular Biochemicals; catalog no. E2311) and incubated for 37˚C and 5% CO2 for 24 h according to the manufacturer’s protocol. Transfection efficiency was measured by western blot. As for the knockdown experiment, the shRNA constructs shLacZ (TRCN0000072226) and shNRF-1 (TRCN0000016904) were obtained from the National RNAi Core Facility. Plasmid transfections were performed using FuGENE HD Transfection Reagent (Roche Diagnostics, Inc.) and incubated at 37˚C and 5% CO2 for 24 h according to the manufacturer’s protocol. After transfection, the cells were incubated for 24 h and harvested for protein extraction.
Lactate dehydrogenase (LDH) assay. Cells were seeded in a 96-well plate at a density of 2×104 cells/well and incubated at 37˚C in 5% CO2 overnight. LDH activity in medium collected from PC3 cells expressing empty vector or pcDNA NRF1 was measured using an LDH cytotoxicity detection kit (Clontech, Mountain View, CA, USA), according to the manufacturer’s protocol. 1% Triton X-100 was used as a positive control for maximum LDH release.
MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Cells were plated in a 96-well plate at a density of 1×104 cells/dl and incubated for 24 h, followed by treatment with culture medium containing pcDNA-NRF1 plasmids. After the incubation, 10 μl of MTT dye were added to each well for another 6 h. Subsequently, 100 μl of acidic isopropanol was used as a solubilizing agent to dissolve the formazan. Spectrometric absorbance at 595 nm was measured with the absorbance at 655 nm for reference.
Cell viability examination. To discriminate the viable and dead cells, trypan blue was added to the medium and the cell viability was determined by counting the number of cells that excluded the dye. Percentage of cell viability was calculated using the following formula:
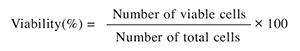
Wound healing assay. Cells were seeded overnight at a concentration of 1.75×104/100 μl/well in each individual compartment of the Ibidi culture insert. The culture plate was then filled with medium before the Ibidi culture inserts were removed. A live cell imaging light microscope (Leica AF 6000 LX; Leica Microsystems, GmbH; magnification, ×200) was used to monitor and capture images of the cells at 0 h and after 24 h of incubation at 37˚C. For each image, areas between one side of the gap and the other were measured using Quantity One software (version 4.6.6; Bio-Rad Laboratories, Inc.). Migration rate was quantified by dividing the change in wound area by the time spent in migration and was expressed as a percentage. To quantify the effects of NRF1 overexpression on migration, the percentage of gap closure after 24 h was analyzed using an inverted phase-contrast microscope at 0 and 24 h.
Examination of mitochondrial biogenesis. Mitochondrial biogenesis was analyzed using a MitoBiogenesis™ InCell ELISA Kit (Abcam, Cambridge, UK) according to the manufacturer’s instructions. Briefly, 2×104 cells/well were seeded in a 96-well plate and allowed to adhere overnight, followed by fixation with 4% paraformaldehyde. After washing with PBS, 0.5% acetic acid was added to block the endogenous alkaline phosphatase activity and 0.1% Triton X-100 was used to permeabilize the cells. Subsequent to blocking with 2X blocking solution, cells were incubated with primary antibodies against mitochondrial DNA-encoded cyclooxygenase (COX-I) and nuclear DNA-encoded succinate dehydrogenase complex flavoprotein subunit A (SDH-A) (ab110217, Abcam). Secondary antibodies containing an HRP-labeled antibody for COX-I and an AP-labeled antibody for SDH-A were utilized. A 15-min kinetic reaction with 1 min interval was recorded using a microplate reader. AP detection of SDH-A and HRP detection of COX-I were analyzed at 405 nm and 650 nm, respectively. The ratio of COX-I/SDHA was calculated to determine mitochondrial biogenesis.
Western blot. Cell proteins were extracted using the lysis buffer containing PhosphoSafe Extraction Reagent (Novagen Inc., Madison, WI, USA) supplemented with 1% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). BCA Protein Assay Kit (Sigma-Aldrich) was used for protein determination. Separation of proteins was conducted by SDS-PAGE and then transferred onto nitrocellulose membranes. Following incubation of primary antibodies against NRF1 (1:2,000; cat. #AB34682), E-cadherin (1:2,000; cat. #AB53033), α-SMA (1:2,000; cat. #AB5694), TGFβRI (1:2,000; cat. #sc-9048), TGFβRII (1:2,000; cat. #sc-1700), Smad2/3 (1:2,000; cat. #sc-8332), Smad4 (1:200; cat. #sc-7154), Smad7 (1:2,000; cat. #sc-11392), phospho-Smad2/3 (1:2,000; cat. #sc-11769) and β-actin (1:4,000; cat. #A5441), the membranes were incubated with the corresponding secondary antibodies (1:4,000; goat anti-rabbit IgG HRP and goat anti-mouse IgG HRP; Cell Signaling Technology). The Pierce™ ECL Western Blotting Substrate enhanced chemiluminescence system (cat. #32209; Thermo Fisher Scientific, Inc.) was used for visualization and detection using a Multi function Gel Image System (cat. #MQIS 21 C2; Tangshan Top Bio Technology, Co., Ltd.)
Statistical analysis. Each experiment was performed at least three times, and representative results are shown. Values in bar graphs are presented as mean±standard deviation (SD). Statistically significant differences were determined by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test or two-way ANOVA of repeated measures followed by Bonferroni post hoc test. A p-Value less than 0.05 was considered to indicate a statistically significant difference.
Results
Overexpression of NRF1 reduces the cell survival and migration capacity of PC3 prostate cancer cells. In an attempt to investigate the effect of NRF1 on PC3 cells, we transfected cells with pcDNA.3.1-NRF1 and showed that the cell count was reduced after 24 h (Figure 1). Results from the LDH assay demonstrated that overexpression of NRF1 increased the level of plasma membrane damage in PC3 cells (Figure 1A). This finding was consistent with the MTT assay showing a decreased cell metabolic activity in NRF1-overexpressing cells (Figure 1B). Likewise, the trypan blue exclusion test showed that there was a reduced percentage of viable cells after overexpression of NRF1 (Figure 1C). Next, we examined the wound healing ability using the scratch assay and showed the impairment of cell migration when NRF1 levels were increased (Figure 1D and E). These results demonstrate that NRF1 possesses a tumor-suppressive potential in PC3 prostate cancer cells.
Figure 1. Overexpression of NRF1 reduces the cell survival and migration capacity of PC3 prostate cancer cells. (A) LDH release assay was carried out to show that overexpression of NRF1 increased the cytotoxicity of PC3 cells. Triton X-100 served as a positive control; (B) MTT and (C) trypan blue exclusion assays were conducted to examine the cell proliferation and viability of PC3 cells after transfection of pcDNA-NRF1. (D) Representative migration capacity image and (E) quantitative analysis of wound healing assay in PC3 cells transfected with pcDNA NRF1 or pcDNA™3.1(-) vector. Magnification scale was 200×. The data is presented as mean±SD from three independent experiments. *p<0.05 compared to the control group; #p<0.05 compared to the pcDNA™3.1(-) vector group.

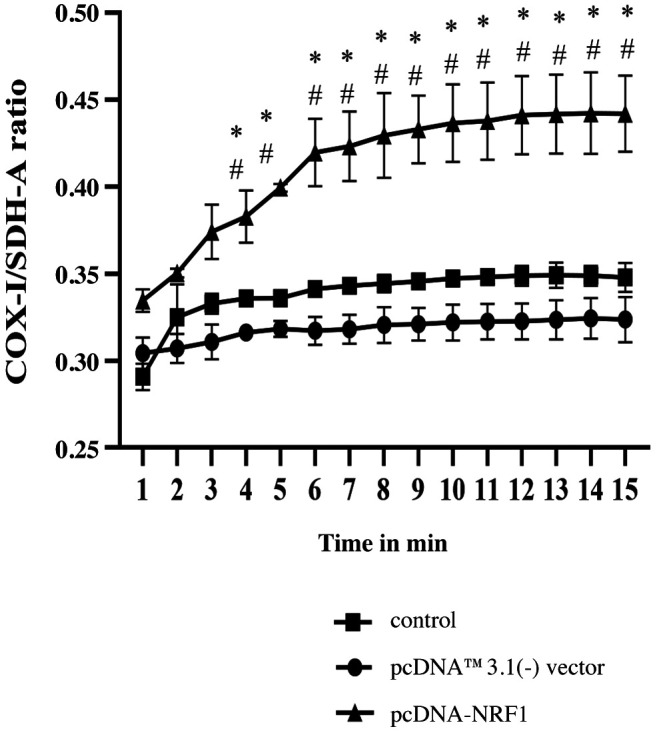
Mitochondrial biogenesis is enhanced after overexpression of NRF1. In light of the fact that NRF1 was a crucial regulator of mitochondrial DNA transcription and replication, we measured the mitochondrial biogenesis using the ratio of mitochondrial DNA encoded cytochrome C oxidase I (COX-I) and nuclear DNA encoded flavoprotein subunit A of succinate dehydrogenase (SDH-A) levels. Our results showed that ectopic expression of NRF1 markedly increased the COX-1/ SDH-A ratio, suggesting that mitochondrial biogenesis was elevated in NRF1-overexpressing cells (Figure 2).
Figure 2. Mitochondrial biogenesis is enhanced after overexpression of NRF1. The COX1/SDH-A ratio was used to show mitochondrial biogenesis over the course of 15 min using the MitoBiogenesis™ InCell ELISA Kit. The data is presented as mean±SD from three independent experiments. *p<0.05 compared to the control group. #p<0.05 compared to the pcDNA™3.1(-) vector group.
Overexpression of NRF1 downregulates the expression of EMT markers. Given that alteration of mitochondrial biogenesis may be associated with the EMT phenotypes (3), we sought to examine whether NRF1 affected the expression level of EMT markers in PC3 prostate cancer cells. As shown in Figure 3A and B, the protein expression of the epithelial cell marker, E-cadherin, was increased, while the mesenchymal marker, α-SMA, was reduced in cells transfected with pcDNA-NRF1. Immunofluorescent images of E-cadherin and α-SMA also showed similar results, i.e., the fluorescence intensity of E-cadherin was increased in NRF1-overexpressing cells, whereas the intensity of α-SMA was downregulated (Figure 3C).
Figure 3. Overexpression of NRF1 downregulates the expression of EMT markers. (A) Representative western blots and (B) densitometric analysis of E-cadherin, α-SMA and NRF1; β-actin was used as an internal control; (C) Immunofluorescence staining of E-cadherin and α-SMA in cells transfected with pcDNA NRF1 or pcDNA control (pcDNA™3.1(-) vector). Magnification scale was 200×. 4’,6-diamidino-2-phenylindole (DAPI) was used to detect nuclei. The data is presented as mean±SD from three independent experiments. *p<0.05 compared to the control group; #p<0.05 compared to the pcDNA™3.1(-) vector group.

Ectopic expression of NRF1 suppresses the TGF-β/Smad pathway. Subsequently, we assessed the expression of TGF-β signaling to ascertain if NRF1 modulated EMT through the upstream regulators. We showed that the protein expression of TGF-β RI, TGF-β RII, Smad2/3 and phosphorylated Smad2/3 were all downregulated, whereas the expression of Smad7 was upregulated (Figure 4A and B). Immunofluorescent images of the inhibitory Smad7 also showed that NRF1-overexpressing cells exhibited a higher expression of Smad7 (Figure 4C).
Figure 4. Ectopic expression of NRF1 suppresses the TGF-β/Smads pathway. (A) Representative western blots and (B) densitometric analysis of TGF-β RI, RII, Smad2/3, p-Smad2/3, Smad4, and Smad7; β-actin served as an internal control; (C) Immunofluorescence staining of Smad7 and NRF1 in cells 24 h after transfection of pcDNA NRF1 or pcDNA control [pcDNA™3.1(-) vector]. Magnification scale was 200X. DAPI was used to detect nuclei. The data is presented as mean±SD from three independent experiments. *p<0.05 compared to the control group; #p<0.05 compared to the pcDNA™3.1(-) vector group.


NRF1 may regulate the progression of prostate cancer through modulation of mitochondrial biogenesis and TGF-β-related EMT. To confirm the functional role of NRF1 in the modulation of mitochondrial biogenesis, TGFβ/Smad pathway, and EMT-associated markers, we conducted a short hairpin RNA mediated-knockdown experiment with shRNA-NRF1 in PC3 cells. As expected, silencing of NRF1 downregulated the mitochondrial biogenesis in PC3 cells, as evident from the decreased COX-1/ SDH-A ratio (Figure 5). Moreover, the protein expression of TGF-β RI, TGF-β RII, Smad2/3 and phosphorylated Smad2/3 were all increased, while the expression of Smad7 was decreased (Figure 6A and B). The expression of E-cadherin was downregulated, whereas the protein level of α-SMA was upregulated in NRF1-suppressing cells (Figure 6C and D).
Figure 5. Silencing NRF1 downregulated mitochondrial biogenesis. The COX1/SDH-A ratio was used to show mitochondrial biogenesis over the course of 15 min using the MitoBiogenesis™ In-Cell ELISA Kit. The data is presented as mean±SD from three independent experiments. *p<0.05 compared to the control group. #p<0.05 compared to the pcDNA™3.1(-) vector group.
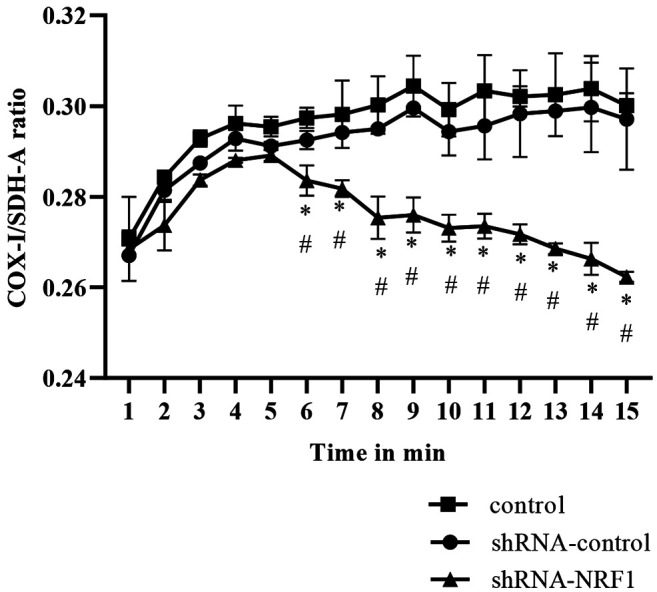
Figure 6. Knockdown of NRF1 increased the expression of TGFβ/Smad signaling and EMT markers. (A) Representative western blots of TGF-β RI, RII, Smad2/3, p-Smad2/3, and Smad7; (B) Densitometric analysis of TGF-β RI, RII, Smad2/3, p-Smad2/3 and Smad7. (C) Representative western blots of EMT marker, E-cadherin and α-SMA; (D) Densitometric analysis of E-cadherin and α-SMA in cells 24 h after transfection of shRNAcontrol (scrambled shRNA) or shRNA-NRF1; β-actin served as an internal control. Magnification scale was 200X. DAPI was used to detect nuclei. The data is presented as mean±SD from three independent experiments. *p<0.05 compared to the control group; #p<0.05 compared to the shRNAcontrol group.

Taken together, these results demonstrate that alteration of NRF1 affected the expression of Smad7, which may antagonize the TGF-β signaling and the subsequent EMT program (Figure 7). Our findings demonstrate the significance of NRF1 in the regulation of cancer progression on AR-negative prostate cancer cells (PC-3 cells) in terms of cell proliferation and migration capacities.
Figure 7. Schematic diagram showing the possible mechanism underlying the regulatory effect of NRF1 on TGF-β signaling and EMT markers in PC3 prostate cancer cells.

Discussion
The role of NRF1 in tumorigenesis of various cancers is controversial. As an oncogenic factor, NRF1 was found to interact with the metastasis suppressor KISS1 in the human melanoma cells (10). It has been shown that NRF1 upregulates various cell cycle genes following Akt activation in E2-treated breast cancer MCF-7 cells (6). NRF1 has also been reported to enhance tumor formation and metastasis in vivo, which was associated with its stimulation of mitochondrial OXPHOS, cell migration, invasion, and mesenchymal transition (11). Another study showed that NRF1 promotes cancer stemness features in estrogen-induced breast cancer through regulating CXCR4 signaling (12). Das et al. showed that NRF1 contributed to the acquisition of invasive mesenchymal phenotypes of breast cancer stem cells via EMT (12). Moreover, NRF1 has been found to form a complex with Oct4 to gain chemoresistance in AR-negative prostate cancer cells (7).
On the contrary, NRF1 seems to exert a tumor-suppressive capacity in prostate cancer. A recent study has revealed that the decreased translocation of NRF1 to nucleus in the African-American (AA) prostate cancer cells is implicated in the cytochrome c deficiency, which is associated with the higher therapeutic resistance in the AA prostate cancer patients (13). Results from Negrette-Guzmán et al. demonstrated that upregulation of NRF1 by sulforaphane treatment diminished the cell viability of PC3 cells (8). Moreover, their data suggested that the corresponding elevation in mitochondrial mass and markers of mitochondrial biogenesis induced PC3 prostate cancer cell death (8). In line with these findings, we showed that overexpression of NRF1 using pcDNA3.1(+) vector downregulated the cell viability of PC3 cells and markedly elevated mitochondrial biogenesis. Additionally, the wound healing assay demonstrated that the 2D cell migration and proliferation capacities of PC3 cells were also suppressed by overexpression of NRF1. Our results suggest that this effect may be related to the modification of TGF-β-mediated EMT following upregulation of mitochondrial biogenesis.
Emerging evidence has indicated that mitochondrial dysfunction may serve as a novel driver of EMT in various cancers (14). It has been demonstrated that PGC-1α facilitates mitochondria biogenesis and enhances the invasive potential of 4T1 breast cancer cells both in vitro and in vivo (15). Knockdown of mitochondrial transcriptional factor A (TFAM) resulted in the decreased expression of several EMT markers, including Snail, N-cadherin and vimentin in SW620 colorectal cancer cells (16). Herein, we showed that the increased mitochondrial biogenesis by ectopic expression of NRF1 downregulated the expression of mesenchymal marker, α-SMA and upregulated the epithelial cell marker, E-cadherin, along with impaired migration capacity in PC3 cells. Our data supports that alteration of mitochondrial biogenesis affected the EMT program, and we also demonstrated that this phenomenon may be associated with the repression of TGF-β signaling after NRF1 overexpression. TGF-β is a well-known inducer of EMT (17,18), and mitochondrial dysfunction has been shown to mediate the TGF-β-elicited EMT in A549 non-small cell lung cancer cells (19). In PC3 prostate cancer cells, we observed that the enhanced mitochondrial biogenesis by NRF1 may lead to suppression of TGF-β signaling and subsequent EMT. This finding is consistent with a recent study showing that NRF1 served as a Smad4 binding protein to inhibit TGF-β signaling in tumorigenesis (20). On the other hand, the present study has several limitations. For example, verification of these results using other prostate cancer cell lines and in vivo models is of great importance and will be our future direction in order to understand the role of NRF1 during the development of prostate cancer. Furthermore, assessment of prostate cancer treatment efficacy using NRF1 as a biomarker is worthy of investigation.
In summary, we demonstrated that overexpression of NRF1 inhibits the proliferation and migration potential of prostate cancer cells. Our results show that manipulation of NRF1 affects mitochondrial biogenesis and expression of several factors associated with TGF-β signaling and EMT. These findings indicate that NRF1 may function as a tumor suppressor in prostate cancer, possibly through its capacity to mitigate the TGF-β-mediated EMT. Our results suggest that approaches to upregulate NRF1 (such as dietary supplements) may help in preventing the progression and reducing metastasis of prostate cancer.
Conflicts of Interest
The Authors declare no conflicts of interest.
Authors’ Contributions
Methodology, Chun-Hsien Wu and Pei-Fang Hsieh; Validation, Chih-Hsin Hung, Ming-Lin Hsieh and See-Tong Pang; Investigation and data curation, Yen-Hsi Lee, Wade Wei-Ting Kuo, Richard Chen-Yu Wu and Yung-Yao Lin; Resources, See-Tong Pang, Yu-Lin Yang and Victor C. Lin; Writing-original draft preparation, Pei-Fang Hsieh; Writing-review and editing, Chun-Hsien Wu and See-Tong Pang; Supervision and project administration, Yu-Lin Yang and Victor C. Lin; Funding acquisition, Yu-Lin Yang and Victor C. Lin. All Authors have read and agreed to the published version of the manuscript.
Acknowledgements
The authors would like to thank Dr. Wen-Teng Chang (Department of pharmaceutical science and technology, Chung Hwa University of Medical Technology, Taiwan), for providing the Plasmid construct of pcDNA-NRF1/shRNA-NRF1. This research was funded by E-Da Hospital Research Grants (EDAHP106006), E-Da Hospital Research Grants (EDPJ106038), E-Da Hospital Research Grants (EDPJ107032), National Science Council Grants, Taiwan (NSC 108-2314-B-273 -001-) and National Science Council Grants, Taiwan (MOST 109-2314-B-650-013-MY2).
References
- 1.Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev. 2016;25(1):16–27. doi: 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Denisenko TV, Gorbunova AS, Zhivotovsky B. Mitochondrial involvement in migration, invasion and metastasis. Front Cell Dev Biol. 2019;7:355. doi: 10.3389/fcell.2019.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shiota M, Yokomizo A, Tada Y, Inokuchi J, Tatsugami K, Kuroiwa K, Uchiumi T, Fujimoto N, Seki N, Naito S. Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol Endocrinol. 2010;24(1):114–127. doi: 10.1210/me.2009-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivanova MM, Luken KH, Zimmer AS, Lenzo FL, Smith RJ, Arteel MW, Kollenberg TJ, Mattingly KA, Klinge CM. Tamoxifen increases nuclear respiratory factor 1 transcription by activating estrogen receptor beta and AP-1 recruitment to adjacent promoter binding sites. FASEB J. 2011;25(4):1402–1416. doi: 10.1096/fj.10-169029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okoh VO, Garba NA, Penney RB, Das J, Deoraj A, Singh KP, Sarkar S, Felty Q, Yoo C, Jackson RM, Roy D. Redox signalling to nuclear regulatory proteins by reactive oxygen species contributes to oestrogen-induced growth of breast cancer cells. Br J Cancer. 2015;112(10):1687–1702. doi: 10.1038/bjc.2014.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takayama KI, Kosaka T, Suzuki T, Hongo H, Oya M, Fujimura T, Suzuki Y, Inoue S. Subtype-specific collaborative transcription factor networks are promoted by OCT4 in the progression of prostate cancer. Nat Commun. 2021;12(1):3766. doi: 10.1038/s41467-021-23974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Negrette-Guzmán M, Huerta-Yepez S, Vega MI, León-Contreras JC, Hernández-Pando R, Medina-Campos ON, Rodríguez E, Tapia E, Pedraza-Chaverri J. Sulforaphane induces differential modulation of mitochondrial biogenesis and dynamics in normal cells and tumor cells. Food Chem Toxicol. 2017;100:90–102. doi: 10.1016/j.fct.2016.12.020. [DOI] [PubMed] [Google Scholar]
- 9.Chang WT, Huang AM. Alpha-Pal/NRF-1 regulates the promoter of the human integrin-associated protein/CD47 gene. J Biol Chem. 2004;279(15):14542–14550. doi: 10.1074/jbc.M309825200. [DOI] [PubMed] [Google Scholar]
- 10.Liu W, Beck BH, Vaidya KS, Nash KT, Feeley KP, Ballinger SW, Pounds KM, Denning WL, Diers AR, Landar A, Dhar A, Iwakuma T, Welch DR. Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Res. 2014;74(3):954–963. doi: 10.1158/0008-5472.CAN-13-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y, Xu Z, Quan D, Zhang F, Zhang H, Xiao T, Hou S, Qiao H, Harismendy O, Wang JYJ, Suo G. Nuclear respiratory factor 1 promotes spheroid survival and mesenchymal transition in mammary epithelial cells. Oncogene. 2018;37(47):6152–6165. doi: 10.1038/s41388-018-0349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das JK, Felty Q, Poppiti R, Jackson RM, Roy D. Nuclear respiratory factor 1 acting as an oncoprotein drives estrogen-induced breast carcinogenesis. Cells. 2018;7(12):234. doi: 10.3390/cells7120234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar R, Bhat TA, Walsh EM, Chaudhary AK, O’Malley J, Rhim JS, Wang J, Morrison CD, Attwood K, Bshara W, Mohler JL, Yadav N, Chandra D. Cytochrome c deficiency confers apoptosome and mitochondrial dysfunction in African-American men with prostate cancer. Cancer Res. 2019;79(7):1353–1368. doi: 10.1158/0008-5472.CAN-18-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerra F, Guaragnella N, Arbini AA, Bucci C, Giannattasio S, Moro L. Mitochondrial dysfunction: a novel potential driver of epithelial-to-mesenchymal transition in cancer. Front Oncol. 2017;7:295. doi: 10.3389/fonc.2017.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16(10):992–1003, 1-15. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CS, Liu LT, Ou LH, Pan SC, Lin CI, Wei YH. Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol Rep. 2018;39(1):316–330. doi: 10.3892/or.2017.6087. [DOI] [PubMed] [Google Scholar]
- 17.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suliman HB, Healy Z, Zobi F, Kraft BD, Welty-Wolf K, Smith J, Barkauskas C, Piantadosi CA. Nuclear respiratory factor-1 negatively regulates TGF-β1 and attenuates pulmonary fibrosis. iScience. 2021;25(1):103535. doi: 10.1016/j.isci.2021.103535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Zhang W, Zhang T, Zhou Q, Liu J, Liu Y, Kong D, Yu W, Liu R, Hai C. TGF-β1 induces epithelial-to-mesenchymal transition via inhibiting mitochondrial functions in A549 cells. Free Radic Res. 2018;52(11-12):1432–1444. doi: 10.1080/10715762.2018.1500020. [DOI] [PubMed] [Google Scholar]
- 20.Rajasekaran N, Song K, Lee JH, Wei Y, Erkin ÖC, Lee H, Shin YK. Nuclear respiratory Factor-1, a novel SMAD4 binding protein, represses TGF-β/SMAD4 signaling by functioning as a transcriptional cofactor. Int J Mol Sci. 2021;22(11):5595. doi: 10.3390/ijms22115595. [DOI] [PMC free article] [PubMed] [Google Scholar]