

Abstract
Adenosine receptor (AR) radiotracers for positron emission tomography (PET) have provided knowledge on the in vivo biodistribution of ARs in the central nervous system (CNS), which is of therapeutic interest for various neuropsychiatric disorders. Additionally, radioligands that can image changes in endogenous adenosine levels in different physiological and pathological conditions are still lacking. The binding of known antagonist adenosine A1 receptor (A1R) radiotracer, [11C]MDPX, failed to be inhibited by elevated endogenous adenosine in a rodent PET study. Since most of the known AR PET radiotracers were antagonists, we propose that an A1R agonist radioligand may possess higher sensitivity to measure changes in endogenous adenosine concentration. Herein, we report our latest findings toward the development of a full agonist adenosine A1 radioligand for PET. Based on a 3,5-dicyanopyridine template, 16 new derivatives were designed and synthesized to optimize both binding affinity and functional activity, resulting in two full agonists (compounds 27 and 29) with single-digit nanomolar affinities and good subtype selectivity (A1/A2A selectivity of ~1000-fold for compound 27 and 29-fold for compound 29). Rapid O-[11C]methylation provided [11C]27 and [11C] 29 in high radiochemical yields and radiochemical purity. However, subsequent brain PET imaging in rodents showed poor brain permeability for both radioligands. An in vivo PET study using knockout mice for MDR 1a/a, BCRP, and MRP1 indicated that these compounds might be substrates for brain efflux pumps. In addition, in silico evaluation using multiparameter optimization identified high molecular weight and high polar surface area as the main molecular descriptors responsible for low brain penetration. These results will provide further insight toward development of full agonist adenosine A1 radioligands and also highly potent CNS A1AR drugs.
Keywords: Adenosine receptors, central nervous system, blood—brain barrier, PET imaging, multiparameter optimization, A1 antagonists
Graphical Abstract
INTRODUCTION
Adenosine is an important neuromodulator in the central nervous system (CNS) implicated in many physiological processes and neurological disorders.1–4 For the last two decades, various adenosine A1 and A2A receptors (A1R and A2AR) radiotracers for positron emission tomography (PET) have been developed to investigate the in vivo CNS adenosine system in both animals and humans. In general, antagonist radiotracers, such as [11C]preladenant (1),5 [11C]MDPX (2),6 [18F]CPFPX (3),7 (Figure 1) allow to measure adenosine receptor density, but as of now have been unable to measure endogenous adenosine,8,9 which would be valuable to investigate neuropathology.1,10,11 For [11C]MDPX, the studies to assess sensitivity in competition with endogenous adenosine showed negative results.8,9 In general, PET agonist radiotracers are more sensitive to changes in endogenous agonist concentrations.12 In the case of AR radiotracers, the only reported agonist [18F]FNECA (4) derived from endogenous adenosine did not cross the blood—brain barrier (BBB),13,14 presumably due to its high polar surface area resulting from the hydrophilic ribose substituents.
Figure 1.

Representative A1R PET ligands.
To overcome this problem, we aimed to develop agonist radiotracers based on a known 3,5-dicyanopyridine scaffold.15 Our previous study demonstrated that the 3,5-dicyanopyridine template could offer a wide range of chemical and functional properties, which could be fine-tuned en route to develop subtype selective agonist radioligands.15 For example, [11C]MMPD (5) is a potent (Ki = 0.49 nM), selective A1R partial agonist with high BBB permeability.14 These properties also make them a great starting point for developing selective partial agonist drug candidates without adverse cardiovascular side effects, such as high-grade atrioventricular block, extensive bradycardia, atrial fibrillation, and vasodilation.16
Herein, we report our recent progress toward the second generation of adenosine A1 agonist radioligands based on a 3,5-dicyanopyridine pharmacophore. Previously, through the systematic comparison of two similar templates, namely 3,5-dicyanopyridine and 5-cyanopyrimidine, we concluded that the 3,5-dicyanopyridine pharmacophore is superior in terms of binding affinity toward A1R and A2AR. Therefore, we focused solely on the 3,5-dicyanopyridine core and synthesized a library of molecules, studying their structure–activity relationship (SAR) for binding and functional activity, along with PET imaging in rodents and in silico estimation of BBB permeability with multiparameter optimization (MPO).
RESULTS AND DISCUSSION
Design, Synthesis, and SAR Study.
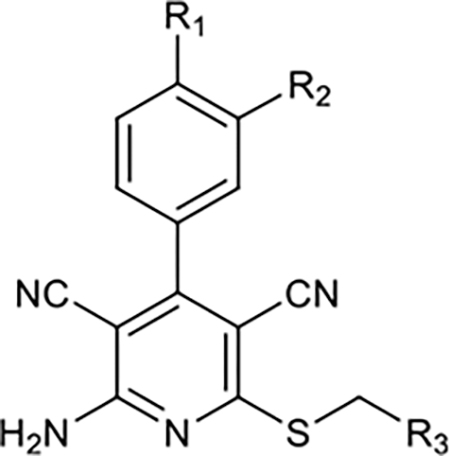
Taking into consideration physicochemical guidelines for CNS PET radiotracers,17,18 we explored the SAR of 3,5-dicyanopyridine derivatives to optimize both functional activity and binding affinity, as shown in Scheme 1. The A-ring was modified with an acetamido substituent in the para-position and hydroxy or alkoxy group in the meta-position, since they were shown to influence both binding and functional activities.15 A strategy of introducing heterocycles in the B-ring led to the synthesis of compounds 10–30, where most compounds were designed for facile radiolabeling with [11C] or [18F]. We screened the binding affinity of compounds 10–30 through a detailed SAR study for A1R, A2AR, and A3R subtypes and investigated their selectivity for the A1R subtype to further refine our prediction of target molecules as PET imaging probes (Table 1). It is noteworthy that compounds 10, 12,19 14, 15,20 and 2415 have been synthesized elsewhere, while compounds 11, 13, and 16–29 were newly synthesized and reported herein. Moreover, compound 30 in this series is a 5-cyanopyrimidine congener of the 3,5-dicyanopyridine 27.
Scheme 1.

Parallel Synthesis of 3,5-Dicyanopyridines for a Potential Adenosine Agonistsa
aReagents and conditions: (i) Malononitrile, piperidine, EtOH, reflux, 2 h; (ii) malononitrile, PhSH, Et3N, EtOH, reflux, 5 h; (iii) Na2S, DMF, 80 °C; (iv) HCl (aq); (v) Et3N, MeCN, 50 °C, overnight.
Table 1.
SAR of Compounds 10–30 at Adenosine Receptors
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Affinity, Ki, nMa (or %inhib.)b |
A1 functional efficacy (Emax %)c | ||
| A1 | A2A | A3 | |||||
|
| |||||||
| 10 |
|
-H |
|
53±18 | 2.8±0.1 | 22±6.7 | 122±5.2% |
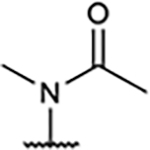
| 11 |
|
-H |
|
67±23 | 38±7.3 | 35±11% | −5.2±1.9% |
| 12 |
|
-H |
|
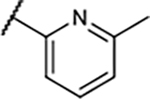
217±44 | 10±2.0 | 8.2±1.1 | 98±2.5% |
| 13 |
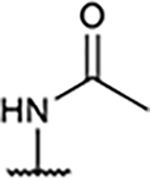
|
-H |
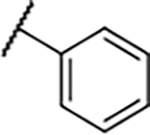
|
109±30 | 27±6.6 | 156±45 | 141±13% |
| 14 | -H | -OH |
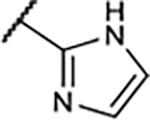
|
9.6±4.0 | 32±0.4 | 30±15 | 102±2.0 |
| 15 | -H | -OMe |
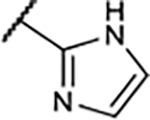
|
3.1±0.4 | 38±5.5 | 71±13 | 23±3.9% |
| 16 | -H | -OH |
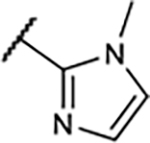
|
41±19 | 81±15 | 55±14% | 13±0.1% |
| 17 | -H | -OMe |
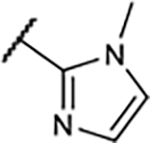
|
4.2±0.4 | 155±36 | 45±10% | −3.0±2.4% |
| 18 | -H | -F |
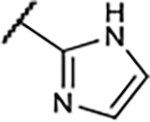
|
13±2.6 | 43±5.9 | 261±82 | 32±5.4% |
| 19 | -H | -OMe |
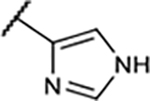
|
13±1.9 | 44±5.8% | 44±8.4% | 14±7.5% |
| 20 | -H | -OMe |
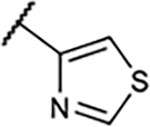
|
2.7±1.0 | 74±3.3 | 52±9.3% | 2.1±3.1% |
| 21 | -H | -OMe |
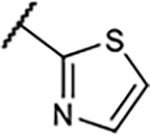
|
2.7±0.3 | 173±24 | 38±16% | −1.2±1.7% |
| 22 | -H | -OMe |
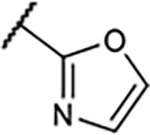
|
6.2±0.1 | 318±72 | 42±12% | 2.6±2.9% |
| 23 | -H | -OMe |
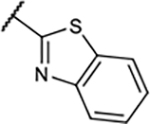
|
39±5.9 | 34±14% | 32±11 % | 3.2±2.7% |
| 24 | -H | -OH |
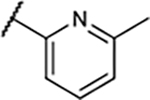
|
3.5±1.3 | 15±3.1 | 74±3.4 | 109±7.8% |
| 25 | -H |
|
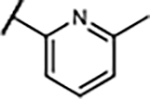
|
18±4.8 | 709±198 | 378±81 | −1.0±3.6% |
| 26 | -H | -F |
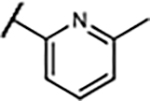
|
16±4.6 | 72±24 | 50±11% | 20±3.5% |
| 27 | -OCH2O- |
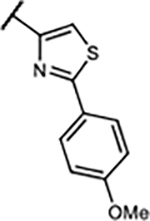
|
1.6±0.4 | 54±8.5% | 47±16% | 104±5.9% | |
| 28 | -OCH2O- |
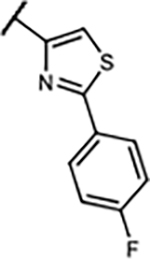
|
2.3±0.7 | 47±12% | 26±13% | 92±2.9% | |
| 29 | -H | -H |
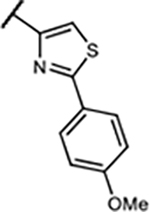
|
6.1±1.1 | 175±31 | 507±35 | 97±18% |
| 30 |
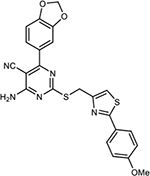
|
29±4.2 | 13±6.8% | 25±12% | 54±27% | ||
Competition radioligand binding assays were conducted with membranes prepared from HEK-293 cells expressing recombinant A1, A2A; or A3Rs (human). The incubation was performed for 1 h at 25 °C. The radioligands used were: for A1R, [3H]8-cyclopentyl-1,3-dipropylxanthine (0.5 nM); for A2AR, [3H]ZM241385 (0.8 nM); and for A3R, [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide ([125I]I-AB-MECA, 0.1 nM). Nonspecific binding was determined using 10 μM 8-[4-[[[[(2-aminoethyl)amino]carbonyl]-methyl]oxy]phenyl]-1,3-dipropylxanthine, 10 μM for A1 and A2A and 100 μM for A3. Values are expressed as the mean ± SEM from 2–4 independent experiments.
Percent inhibition at 1 μM.
cAMP accumulation assay (N6-cyclopentyladenosine expressed as 100%).
Previously, compound 10 was reported as a potent A2AR agonist and a weak A1R agonist.15 When the pyridyl ring was replaced with an imidazolyl or phenyl group (12 and 13), binding affinity was significantly reduced for both A1R and A2AR, while their full A1R agonism was maintained. This is presumably due to the presence of the acetamido group in para-position of the A-ring. Since we had previously reported15 that 3,5-dicyanopyridine compounds bearing an acetamido group showed inadequate BBB permeability due to a high polar surface area (PSA), we also synthesized N-methyl acetamido derivative (11) to reduce the PSA, considering the possible radiolabeling route as well as BBB penetration. Interestingly, agonistic activity was completely abolished, indicating the sensitive interaction of this position with A1R.
A similar trend on affinity was also observed at the meta-position of the A-ring in this compound series with an 2-imidazolyl group in the B-ring (14–17). For instance, the A1R binding affinity was improved by replacement of phenol with m-anisole as the A-ring, but the agonistic activity decreased significantly, which was consistent with our previously published results.15 In compounds 16 and 17, methylation on a nitrogen of the imidazole ring also lowered binding affinity, particularly for A1R and A3R subtypes, along with lower A1R agonistic activity.
It became clear that a hydroxy group and a methoxy group on the A-ring were critical for functional and binding activities, respectively. We therefore investigated replacing those groups in compound 18 with fluoro, which is a bioisosteric replacement for hydroxy and methoxy groups and is also a site for the introduction of a radioisotope. Unfortunately, the A1 binding affinity was not improved compared with the parent compounds 14 and 15, and functional activity was only slightly improved, compared with the methoxy substituent.
In the next step, the imidazolyl group in B-ring was replaced with various heteroaryl groups (19–23) to examine their influence on functional activity at the A1R, while the OMe group was retained to maintain the higher binding potency. In all these cases, A1R functional activity was not improved and the A1R binding affinity worsened with the large benzothiazole group in compound 23.
Since the methoxy group in MMPD (5) decreased functional activity despite its increased binding potency,15 we also replaced this substituent with the bioisosteric F and fluoroethoxy groups to give compounds 25 and 26. Although the A1R agonism was slightly improved, the binding affinities were significantly reduced.
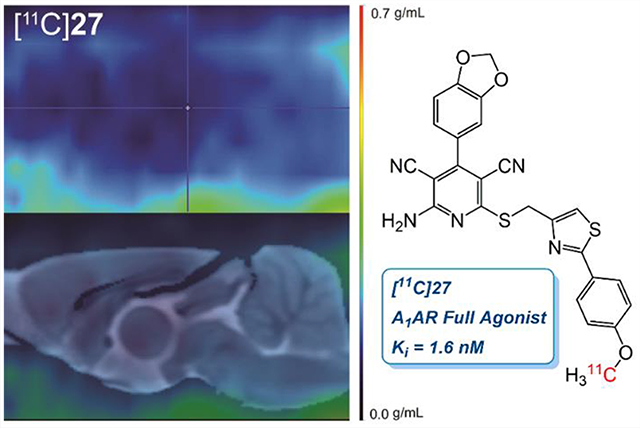
Lastly, inspired by a previously reported A1R full agonist,21 30, it was reasoned that a phenylthiazole group in the B-ring might improve functional activity. Surprisingly, while 30 turned out a weak A1R partial agonist (Ki = 29 nM, A1R Emax = 54%) in our assay setup, all three new compounds (27–29) showed full agonism. Furthermore, the binding affinity was also significantly improved, reflecting that the 3,5-dicyanopyridine platform was superior to its 5-cyanopyrimidine congener. We chose to pursue PET experiments of 27 since they demonstrated full agonism with a single digit nanomolar affinity (Ki = 1.6 nM, A1R Emax = 104%) along with compound 29, a smaller variant of 27 with low molecular weight and yet full A1R agonist functional activity. Based on known procedures,22,23 both A1R full agonists turned out to be highly subtype selective not only against A2A and A3 but also against the A2B subtype (27, Ki = 436 ± 115 nM; 29, Ki = 272 ± 99 nM).
It is also worth mentioning that compound 12 was serendipitously discovered to possess high binding affinity toward the A3R subtype (8.2 nM) with partial agonism (A3R Emax = 60%). Since most known A3R agonists are nucleoside derivatives,4 compound 12 could be a structural basis for BBB penetrable molecular probes as well as for the development of pain medications targeting A3R.4
Radiochemistry.
Two full agonists (27 and 29) were chosen for radiolabeling with [11C] for further in vivo imaging studies. The precursors (34 and 36) were synthesized by coupling of the corresponding free thiols (33 and 35) with 4-(chloromethyl)-2-(4-hydroxyphenyl)thiazole (32). Compound 32 was synthesized through a demethylation reaction with excess boron tribromide from the commercially available 4-(chloromethyl)-2-(4-methoxyphenyl)thiazole (31), as shown in Scheme 2.
Scheme 2.

Synthetic Reagents and Conditions for Synthesis of [11C]27 and [11C]29a
aReagents and conditions: (i) Anhydrous DCM, 10 equiv BBr3, 0 °C to rt, overnight; (ii) Et3N, MeCN, 50 °C, overnight; (iii) [11C]MeOTf, MeCN, TBAOH, 80 °C, 3 min; (iv) Et3N, MeCN, 50 °C, overnight; (v) [11C]MeOTf, MeCN, TBAOH, 80 °C, 3 min.
Both [11C]27 and [11C]29 were successfully radiolabeled via O-methylation using [11C]methyl triflate24 in moderate RCY ([11C]27, 22.2 ± 5.8% nondecay corrected, n = 6; and [11C] 29, 24.3 ± 6.7%, nondecay corrected, n = 5) and high molar activities ([11C]27, 832 ± 411 GBq/μmol @ EOB; and [11C] 29, 1195 ± 492 GBq/μmol @ EOB). Radiochemical purity was also high ([11C]27, 99%<, n = 6; and [11C]29, 99%<, n = 5), and the averaged total synthesis time was 32 min. It is worth noting that O-[11C]methylation using [11C]methyl iodide also led to the formation of [11C]27 and [11C]29, albeit in significantly lower RCYs of 3–7% (n = 2).
PET Imaging Studies in Rodents.
Initially, preclinical PET scans were performed on male Wistar rats with [11C]27 (Figure 2A). Compared with [11C]MMPD (5),15 the averaged whole brain uptake of [11C]27 was very low (standard uptake value (SUV) = 0.28 ± 0.03 g/mL, Figure 3A) with homogeneous distribution, which was reasoned to be due to the high polar surface area of 27 (tPSA, 127.1). We then tested compound 29 (Figure 2B) as its PSA is lower than that of 27 and is similar to that of MMPD (108.6). However, [11C]29 also a showed lack of BBB permeability, though it was slightly improved from that of [11C]27 (Figure 3A).
Figure 2.

In vivo PET imaging using [11C]27 and [11C]29. Averaged PET images (0–70 min) were normalized with injection dose and body weight into a single representative SUV image (upper panels) and coregistered to a Wistar rat MRI brain template (lower panels).
Figure 3.

Time–activity curves of (A) [11C]27 and [11C]29 baseline. (B) [11C]29 baseline and pretreatment with DPCPX (2 mg/kg) in the same subject. No significant reduction of brain uptake with DPCPX pretreatment was observed, indicating little binding specificity of [11C]29.
To investigate lack of BBB permeability, ex vivo studies of [11C]27 were performed using triple knockout (tKO) mice for the three most abundant brain efflux pumps, MDR 1a/a, BCRP, MRP1, along with control mice. As shown in Figure 4, the averaged brain uptake of [11C]27 of tKO mice was four times higher than that of control mice, reflecting that [11C]27 is a substrate of these cell membrane proteins. In short, [11C] 27 is likely to be a substrate for either MDR 1a/a or MRP1, not BCRP (refer to SI).
Figure 4.

Comparison of whole brain SUV of [11C]27 in control mice (n = 3) and tKO mice (n = 3) for the three most abundant brain efflux pumps MDR 1a/a, BCRP, and MRP1. The difference between average tKO mice SUV (0.165 ± 0.026) and control mice SUV (0.037 ± 0.006) was statistically significant using a two-sample t test of unequal variance with one tail.
In Silico Calculation: BBB Permeability.
MPO analysis was also used to uncover molecular properties which may be responsible for their lack of BBB permeability. According to Wager’s method,25 CNS scores were generated and compared for all the compounds in this report including previously published radioligands by our group (Figure 5A).15 The CNS MPO score was calculated using the StarDrop software package by Optibrium Inc.26,27 as the sum of the six physicochemical parameters, namely molecular weight (Mw), pKa of the most basic center, calculated logP (clogP), calculated logD at pH 7.4 (clogD), topological polar surface area (tPSA), number of hydrogen-bond donors (HBD).
Figure 5.

(A) MPO CNS scores for selected adenosine A1R (radio)ligands. [11C] Compounds shown as white circles, and all other compounds shown as dark circles. [11C]A = N-(4-(2-amino-3,5-dicyano-6-((methyl-11C)thio)pyridin-4-yl)phenyl)acetamide, [11C]B = N-(4-(6-amino-5-cyano-2-((methyl-11C)thio)pyrimidin-4-yl)phenyl)acetamide, [11C]C = 2-amino-4-(4-methoxyphenyl)-6-((methyl-11C)thio)pyridine-3,5-dicarbonitrile, [11C]D = 4-amino-6-(4-methoxyphenyl)-2-((methyl-11C)thio)pyrimidine-5-carbonitrile, and [11C]5 = 2-amino-4-(3-(methoxy-11C)phenyl)-6-(((6-methylpyridin-2-yl)methyl)thio)pyridine-3,5-dicarbonitrile. (B) Correlation of whole brain uptake area under the curve (AUC) vs MPO CNS score of [11C] labeled 3,5-dicyanopyridine derivatives synthesized in our laboratory. AUC was calculated from time–activity curves presented as the SUV using the trapezoidal sum from 0 to 30 min. Linear regression calculated using sum of least-squares residuals (R2 = 0.911).
While most of the previously reported brain penetrable radiolabeled compounds based on the 3,5-dicyanopyridine template ([11C]C, [11C]D, [11C]5)15 showed high MPO CNS scores (>5), both [11C]27 and [11C]29 showed relatively low (<4) scores (Figure 5A). Overall, the brain uptake was highly correlated with the CNS score (R2 = 0.91) when compared with AUCs for 30 min of each radiolabeled compound (Figure 5B). Initial brain uptake (t < 2.5 min) was also highly correlated with the CNS score (R2 = 0.78, SI). Therefore, we concluded that the main molecular property that limits the BBB permeability of [11C]27 and [11C]29 is their molecular weights despite their favorable calculated lipophilicity (clogP).
CONCLUSION
In summary, we developed A1R full agonist ligands with nanomolar affinity based on a 3,5-dicyanopyridine pharmacophore through comprehensive SAR studies and optimized both binding affinity and functional activity. Compounds 27 and 29 were chosen for labeling with [11C]MeOTf for preclinical brain imaging. Our rodent PET imaging studies indicated a lack of BBB permeability of these full agonists, consistent with low scores in predictions of physicochemical properties for CNS radioligand candidates. Although no successful CNS adenosine full agonist radioligand was discovered herein, this report provides valuable SAR information that might be broadly applicable in further studies of AR PET ligands including peripheral adenosine imaging as well as in potential pharmaceutical development.
METHODS
Detailed synthesis and characterization of all the new compounds are presented in the SI.
Radiosynthesis of Compound [11C]27 and [11C]29.
A solution of precursor (0.8–1.0 mg) was suspended in 200 μL of anhydrous MeCN, and 2.5 μL of tetrabutylammonium hydroxide (1 M in methanol) was added through the reaction flask wall. The mixture was then vortexed for 1 min which led to formation of a clear yellow solution. The mixture was allowed to react with [11C]CH3I in a stream of helium at 80 °C for 3 min before being injected into a semipreparative HPLC column (Phenomenex Onyx Monolithic C18 LC Column 100 × 10 mm). The mixture was eluted at 5 mL/min with an isocratic mixture of 60% solvent A (90% 0.01 M phosphate buffer, 10% EtOH pH = 7.2–7.4) and 40% solvent B (100% EtOH) and monitored for absorbance at 280 nm and radioactivity using the flow count detector (NaI(Tl)) built into the FX-M. The product [11C]27 was collected in 10.0 min ([11C]27), while the product [11C] 29 was collected in 11.9 min (Figure S2), and radioactivity was measured by a dose calibrator (Capintec, CRC 712M) to determine RCY. RCY for [11C]27 was calculated to be 22.2 ± 5.8% (n = 6) and molar activity (832 ± 411 GBq/μmol @ EOB, n = 4). The collected product solution was formulated with 2.5 mL of sterile water for rodent PET studies (final ethanol content ≤10%). RCY for [11C]29 24.3 ± 6.7%, n = 5) and molar activity (1195 ± 492 GBq/μmol @ EOB, n = 5). The collected product (1 mL) solution was formulated with 3.0 mL of sterile water (final ethanol content ≤10%) for rodent PET studies. Radiochemical synthesis data obtained from the Tracerlab FXM for compounds [11C]27 and [11C]29 are shown in Figures S1 and S2.
PET Imaging.
For PET studies, rats were anesthetized under isoflurane (Forane, 99.9%; 5.0% induction for 5 min, 1.0–2.5% maintenance) prior to catheter placement and for the duration of scanning. A catheter was inserted into the penile vein for radiotracer injection. Subjects were placed prone position side-by-side into a Siemens microPET Focus 220 scanner. Vitals (heart rate, respiratory rate, spO2, body temperature) were monitored using a PhysioSuite (Kent Scientific no. PS-04). Temperature was maintained close to 36 °C with a homeothermic blanket with negative feedback control (Harvard Apparatus no. 507222F). A 10 min transmission scan with a Co-57 point source was collected for attenuation correction prior to 90 min emission scans. Radiotracer was injected as a bolus over 1 min using a syringe pump (Harvard Apparatus no. HA1100WD) and immediately flushed with heparinized saline (250 μL). The injected dose was 16.6 ± 3.2 MBq. For the [11C]29 blocking study, DPCPX (2.0 mg/kg, 550 μL) was injected intraperitoneally 10 min prior to radiotracer injection. PET data were collected in list mode and reconstructed into 23 frames (6 × 20 s, 5 × 60 s, 4 × 120 s, 3 × 300 s, 3 × 600 s, and 2 × 1200 s), and sinogram reconstruction was performed using 2D Filtered Back Projection. Time–activity curves were generated in PMOD (version 3.807) and normalized to subject weight and injected activity and represented as SUV. Averaged image (0–70 min) was generated to evaluate BBB permeability for each compound.
Ex Vivo Studies.
For ex vivo studies, all mice were placed under anesthesia as described above. Catheters were constructed using 20 cm BPTE-10 polyethlyene tubing (Instech Las), sharp tip needles (27 GA, Becton Dickinson), and blunt tip needles (30 GA, Component Supply). Catheters were flushed with heparinized saline (0.6% heparin, 0.9% HCl saline) and placed in the tail vein. [11C]27 (2.63 ± 1.54 MBq, 100 μL) was administered intravenously and immediately flushed with heparinized saline (100 μL). Animals were euthanized 15 min postinjection via decapitation. Whole brain tissue and ventricular whole blood were extracted, stored in preweighed glass vials, and placed in an automatic well-type gamma counter (Wallac Wizard 3″; PerkinElmer) to measure radioactivity. Vials were postweighed to obtain tissue mass, and the SUV was generated using total injected activity and subject weight.
Supplementary Material
ACKNOWLEDGMENTS
This research is supported by the National Institutes of Health Intramural Research Program (ZIAAA000550 and Y1AA 3009, N.D.V.; ZIADK031117, K.A.J.). The authors are grateful to the NIH PET department team (Mr. Kris Kim, Dr. Peter Herscovitch, and Dr. Michael Channing) for cyclotron operation. We also thank the NIMH PET imaging team (Dr. Robert Innis, Dr. Jeih-San Liow, and Dr. Victor Pike) for PET operation and scientific input.
ABBREVIATIONS
- AR
adenosine receptors
- PET
positron emission tomography
- CNS
central nervous system
- BBB
blood—brain barrier
- [11C]MDPX
8-dicyclopropylmethyl-1-11C-methyl-3-propyl-xanthine
- MDR
multidrug resistance
- BCRP
breast cancer resistance protein
- MRC1
multidrug resistance-associated protein 1
- [18F]CPFPX
8-cyclopentyl-3-(3-(fluoro-18F)-propyl)-1-propyl-1,2,3,7-tetrahydro-6H-purin-6-one
- FNECA
(2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-N-(2-(fluoro-18F)-ethyl)-3,4-dihydroxytetrahydrofuran-2-carboxamide
- [11C]MMPD
2-amino-4-(3-(methoxy-11C)phenyl)-6-(((6-methylpyridin-2-yl)methyl)thio)pyridine-3,5-dicarbonitrile
- SAR
structure–activity relationship
- SUV
standard uptake value
- AUC
area under the curve
- RCY
radiochemical yield
- MPO
multiparameter optimization
- PSA
polar surface area
- tPSA
topological polar surface area
- clogP
calculated logP
- clogD
calculated logD at pH 7.4
- HBD
number of hydrogen-bond donors
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.1c00397.
Synthesis and characterization of newly synthesized compounds, radiochemistry, and small animal PET studies (PDF)
Contributor Information
Min Guo, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States.
Abolghasem Bakhoda, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States.
Zhan-Guo Gao, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892-0810, United States.
Joseph M. Ramsey, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States
Yang Li, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States.
Kelly A. O’Conor, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States
Andrew C. Kelleher, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States
Seth M. Eisenberg, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States
Yeona Kang, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States; Department of Mathematics, Howard University, Washington, D.C. 20059, United States.
Xuefeng Yan, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States.
Cameron Javdan, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States.
Joanna S. Fowler, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States
Kenner C. Rice, Drug Design and Synthesis Section, National Institute on Drug Abuse, National Institutes of Health, Rockville, Maryland 20850, United States
Jacob M. Hooker, Athinoula A. Martinos Center for Biomedical Imaging, Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Kenneth A. Jacobson, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892-0810, United States.
Sung Won Kim, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland 20892-1013, United States;.
Nora D. Volkow, Laboratory of Neuroimaging, National Institute on Alcohol Abuse and Alcoholism and National Institute on Drug Abuse, National Institutes of Health, Bethesda, Maryland 20892-1013, United States;
REFERENCES
- (1).Boison D Adenosine as a neuromodulator in neurological diseases. Curr. Opin. Pharmacol. 2008, 8, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Jacobson KA; Gao ZG Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discovery 2006, 5, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chen JF; Eltzschig HK; Fredholm BB Adenosine receptors as drug targets–what are the challenges? Nat. Rev. Drug Discovery 2013, 12, 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jacobson KA; Tosh DK; Jain S; Gao Z-G Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell. Neurosci. 2019, 13, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhou X; Boellaard R; Ishiwata K; Sakata M; Dierckx RAJO; de Jong JR; Nishiyama S; Ohba H; Tsukada H; de Vries EFJ; Elsinga PH In Vivo Evaluation of 11C-Preladenant for PET Imaging of Adenosine A2A Receptors in the Conscious Monkey. J. Nucl. Med. 2017, 58, 762–767. [DOI] [PubMed] [Google Scholar]
- (6).Noguchi J; Ishiwata K; Furuta R; Simada J-I; Kiyosawa M; Ishii S-I; Endo K; Suzuki F; Senda M Evaluation of Carbon-11 Labeled KF15372 and Its Ethyl and Methyl Derivatives as a Potential CNS Adenosine A1 Receptor Ligand. Nucl. Med. Biol. 1997, 24, 53–59. [DOI] [PubMed] [Google Scholar]
- (7).Bauer A; Holschbach MH; Meyer PT; Boy C; Herzog H; Olsson RA; Coenen HH; Zilles K In vivo Imaging of Adenosine A1 Receptors in the Human Brain with [18F]CPFPX and Positron Emission Tomography. NeuroImage 2003, 19, 1760–1769. [DOI] [PubMed] [Google Scholar]
- (8).Paul S; Khanapur S; Sijbesma JW; Ishiwata K; Elsinga PH; Meerlo P; Dierckx RA; van Waarde A Use of 11C-MPDX and PET to Study Adenosine A1 Receptor Occupancy by Nonradioactive Agonists and Antagonists. J. Nucl. Med. 2014, 55, 315–320. [DOI] [PubMed] [Google Scholar]
- (9).Paul S; Khanapur S; Rybczynska AA; Kwizera C; Sijbesma JWA; Ishiwata K; Willemsen ATM; Elsinga PH; Dierckx RAJO; van Waarde A Small-Animal PET Study of Adenosine A1 Receptors in Rat Brain: Blocking Receptors and Raising Extracellular Adenosine. J. Nucl. Med. 2011, 52, 1293–1300. [DOI] [PubMed] [Google Scholar]
- (10).Diógenes MJ; Neves-Tomé R; Fucile S; Martinello K; Scianni M; Theofilas P; Lopatář J; Ribeiro JA; Maggi L; Frenguelli BG; Limatola C; Boison D; Sebastião AM Homeostatic Control of Synaptic Activity by Endogenous Adenosine is Mediated by Adenosine Kinase. Cereb. Cortex 2014, 24, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Haskó G; Pacher P; Sylvester Vizi E; Illes P Adenosine receptor signaling in the brain immune system. Trends Pharmacol. Sci. 2005, 26, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shalgunov V; van Waarde A; Booij J; Michel MC; Dierckx RAJO; Elsinga PH Hunting for the high-affinity state of G-protein-coupled receptors with agonist tracers: Theoretical and practical considerations for positron emission tomography imaging. Med. Res. Rev. 2019, 39, 1014–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lehel S; Horváth G; Boros I; Mikecz P; Márián T; Szentmiklósi AJ; Trón L Synthesis of 5′-N-(2-[18F]Fluoroethyl)-Carboxamidoadenosine: A Promising Tracer for Investigation of Adenosine Receptor System by PET Technique. J. Labelled Compd. Radiopharm. 2000, 43, 807–815. [Google Scholar]
- (14).Márián T; Lehel S; Lengyel Z; Balkay L; Horváth G; Mikecz P; Miklovicz T; Fekete I; Szentmiklósi AJ The [18F]-FNECA Serves as a Suitable Radioligand for PET Investigation of Purinergic Receptor Expression. Orv. Hetil. 2002, 143, 1319–1322. [PubMed] [Google Scholar]
- (15).Guo M; Gao Z-G; Tyler R; Stodden T; Li Y; Ramsey J; Zhao W-J; Wang G-J; Wiers CE; Fowler JS; Rice KC; Jacobson KA; Kim SW; Volkow ND Preclinical Evaluation of the First Adenosine A1 Receptor Partial Agonist Radioligand for Positron Emission Tomography Imaging. J. Med. Chem. 2018, 61, 9966–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wu L; Belardinelli L; Zablocki JA; Palle V; Shryock JC A partial agonist of the A1-adenosine receptor selectively slows AV conduction in guinea pig hearts. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H334–H343. [DOI] [PubMed] [Google Scholar]
- (17).Van de Bittner GC; Ricq EL; Hooker JM A Philosophy for CNS Radiotracer Design. Acc. Chem. Res. 2014, 47, 3127–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pike VW Considerations in the Development of Reversibly Binding PET Radioligands for Brain Imaging. Curr. Med. Chem. 2016, 23, 1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Catarzi D; Varano F; Varani K; Vincenzi F; Pasquini S; Dal Ben D; Volpini R; Colotta V Amino-3,5-Dicyanopyridines Targeting the Adenosine Receptors. Pharmaceuticals 2019, 12, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Beukers MW; Chang LCW; von Frijtag Drabbe Kunzel JK; Mulder-Krieger T; Spanjersberg RF; Brussee J; IJzerman AP New, Non-Adenosine, High-Potency Agonists for the Human Adenosine A2B Receptor with an Improved Selectivity Profile Compared to the Reference Agonist N-Ethylcarboxamidoadenosine. J. Med. Chem. 2004, 47, 3707–3709. [DOI] [PubMed] [Google Scholar]
- (21).Louvel J; Guo D; Agliardi M; Mocking TA; Kars R; Pham TP; Xia L; de Vries H; Brussee J; Heitman LH; Ijzerman AP Agonists for the Adenosine A1 Receptor with Tunable Residence Time. A Case for Nonribose 4-Amino-6-Aryl-5-Cyano-2-Thiopyrimidines. J. Med. Chem. 2014, 57, 3213–3222. [DOI] [PubMed] [Google Scholar]
- (22).Corset V; Nguyen-Ba-Charvet KT; Forcet C; Moyse E; Chédotal A; Mehlen P Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2B receptor. Nature 2000, 407, 747–750. [DOI] [PubMed] [Google Scholar]
- (23).Herrera C; Casadó V; Ciruela F; Schofield P; Mallol J; Lluis C; Franco R Adenosine A2B Receptors Behave as an Alternative Anchoring Protein for Cell Surface Adenosine Deaminase in Lymphocytes and Cultured Cells. Mol. Pharmacol. 2001, 59, 127–134. [PubMed] [Google Scholar]
- (24).Jewett DG A simple synthesis of [11C]methyl triflate. Appl. Radiat. Isot. 1992, 43, 1383–1385. [DOI] [PubMed] [Google Scholar]
- (25).Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach to Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Segall MD Multi-parameter optimization: identifying high quality compounds with a balance of properties. Curr. Drug Metab. 2012, 18, 1292–1310. [DOI] [PubMed] [Google Scholar]
- (27).StarDrop; Optibrium: Cambridge, UK https://www.optibrium.com/downloads/MPO_Explorer_Profile_Builder.pdf (accessed July 2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.