

Abstract
The combination of histone deacetylase inhibitor and BRAF inhibitor (BRAFi) has been shown to enhance the antineoplastic effect and reduce the progress of BRAFi resistance. In this study, a series of (thiazol-5-yl)pyrimidin-2-yl)amino)-N-hydroxyalkanamide derivatives were designed and synthesized as novel dual inhibitors of BRAF and HDACs using a pharmacophore hybrid strategy. In particular, compound 14b possessed potent activities against BRAF, HDAC1, and HDAC6 enzymes. It potently suppressed the proliferation of HT-29 cells harboring BRAFV600E mutation as well as HCT116 cells with wild-type BRAF. The dual inhibition against BRAF and HDAC downstream proteins was validated in both cells. Collectively, the results support 14b as a promising lead molecule for further development and a useful tool for studying the effects of BRAF/HDAC dual inhibitors.
Keywords: BRAF, histone deacetylase, dual inhibitor, colorectal cancer, HDAC
1 Introduction
RAF kinases are key components of the mitogen-activated protein kinase (MAPK) signaling cascade (Wellbrock et al., 2004). Three isoforms of the RAF kinases (i.e., ARAF, BRAF, and CRAF) have been identified, among which BRAF is the most-defined proto-oncogene. BRAF mutations are reported in 6%–8% of all kinds of human cancers, including 50–80% of melanoma, ∼100% of hairy cell leukemia, 45% of papillary thyroid carcinoma, and 11% of colorectal cancer (Davies et al., 2002; Fransén et al., 2004; Ahmadzadeh et al., 2014). More than 90% of the mutations occur on a valine to glutamine at position 600 (BRAFV600E), leading to constitutively active BRAF and sustained MAPK activation independent of upstream signaling activity (Chang et al., 2003). Inhibition of mutated BRAF kinase is a well-validated approach for cancer therapy and three selective small molecular BRAF inhibitors, that is, vemurafenib (1, Figure 1), encorafenib (2), and dabrafenib (3) have achieved significant clinical benefits in patients of melanoma and colon cancer harboring BRAF mutations (Bollag et al., 2010; Rheault et al., 2013; Holderfield et al., 2014; Karoulia et al., 2017; Koelblinger et al., 2018). However, almost all patients displayed intrinsical resistance or secondary resistance to these drugs although they also harbor BRAFV600E mutation (Mauri et al., 2021). Dual inhibitors simultaneously targeting BRAF and other critical proliferative pathways were reported to overcome drug resistance or improve efficacy (Kim, 2016; Ma et al., 2018; Palušová et al., 2020; Pinzi et al., 2021).
FIGURE 1.

Chemical structures of BRAF inhibitors and HDAC inhibitors and the pharmacophore hybrid strategy for BRAF/HDAC inhibitors.
Histone deacetylases (HDACs) are critical enzymes that regulate the lysine acetylation balance of histones and other proteins (Grunstein, 1997). Dysregulation of HDACs involved in cancer initiation and HDAC inhibition has been proven as an effective therapeutic approach for human malignancies (Marks and Breslow, 2007; Yao and Seto, 2011). To date, five small molecule HDAC inhibitors have been approved for clinical management of hematologic cancers, including the hydroxamic acid–containing belinostat (4), panobinostat (5), and vorinostat (6), macrocyclic romidepsin and benzamide-containing chidamide, while many others are in clinical development (Luan et al., 2019; Ho et al., 2020). However, the clinical indication of HDAC inhibitors is limited in hematologic malignancies, including cutaneous T-cell lymphoma, peripheral T-cell lymphoma (PTCL), and multiple myeloma. HDAC inhibitors alone were not as effective in solid tumors, which severely limits their clinical application (Chen et al., 2020). To overcome these flaws, multitarget or hybrid HDAC inhibitors with better efficacy or ability to overcome drug resistance were thus developed.
Increasing evidence has been reported for the synergistic and additive effects of the joint use of HDAC inhibitors and BRAF inhibitors in colon cancer and melanoma (Lai et al., 2013; Carson et al., 2015; Fu et al., 2019). The combination of PLX4032 and HDAC inhibitors have been displayed complete elimination of cancer cells, and to reduce the progress of resistance to BRAF inhibitors (Madorsky Rowdo et al., 2020). Recently, Emmons et al. revealed that HDAC8 inhibitors could enhance the durability of BRAF inhibitor therapy (Mer et al., 2019). Hence, development of dual inhibitors of BRAF and HDACs may be a valuable strategy to overcome BARFi resistance.
Until now, only one class of phenoxybenzamide compounds has been reported to be BRAF/HDAC1 inhibitors. The IC50 values of the optimized compound were 0.073 μM against BRAF V600E and 1.17 μM against HDAC1, respectively (Geng et al., 2019). Previously, we have developed N-(3-ethynyl-2, 4-difluorophenyl) sulfonamide derivatives as selective BRAF inhibitors with potent activities against BRAF-mutant CRC cells, and also reported BRAF/epidermal growth factor receptor (EGFR) dual inhibitors for the treatment of drug-resistant CRCs (Cheng et al., 2014; Li et al., 2015; Li et al., 2016). In a continuing effort to identify BRAFi-based therapeutics for CRC, herein, we designed and synthesized a novel series of hydroxamic acid–containing compounds such as BRAF and HDAC dual-targeted inhibitors. The enzymatic inhibitory activities against BRAFV600E, HDAC1/6 and the structure-activity relationship (SAR) study were reported herein.
2 Results and Discussion
The pharmacophore hybrid strategy was used for the design of BRAF/HDAC inhibitors by analyzing the canonical pharmacophore of the HDAC inhibitors and the key features of dabrafenib binds to BRAF (Figure 1). As illustrated by vorinostat, the hydroxamic acid–containing HDAC inhibitors share three common structural components: a zinc-binding warhead group, a linker that fits the hydrophobic tunnel, and a cap group that sits outside and interacts with the surface residues of HDAC (Zhang et al., 2018). Importantly, a wide variety of cap groups could be accommodated for HDAC inhibition which allows for the replacement of the cap with another target pharmacophore to design dual-targeted inhibitors. On the other hand, the crystal structure of dabrafenib bound to BRAFV600E revealed its aminopyrimidine moiety pointed toward the solvent-exposed entrance of the pocket, which could accommodate a wide variety of modifications (Rheault et al., 2013; Waizenegger et al., 2016). It was postulated that incorporating the HDAC pharmacophore into this amine group will retain BRAF activity while giving rise to potentially HDAC inhibition activity. Thus, hydroxamic acid and aminopyrimidyl-pharmacophores were tethered via appropriate alkyl linkers, resulting in potential hybridized BRAF/HDAC inhibitors. The alkyl-linker, R 1 substitution on thiazole, and R 2 substitutions on sulfonamide were modified to study the SAR.
The synthesis of compounds began with the esterification of 2-fluoro-3-nitrobenzoic acid (7) and is detailed in Scheme 1 and Scheme 2. Subsequent nitroreduction of 8 and then replacement of NH2 with 2,6-difluorobenzenesulfonyl chloride obtained the sulfonamide compound 10. Intermediate 10 was then condensed with the lithium anion of 2-chloro-4-methylpyrimidine to generate ketone 11. Bromination of 11 with N-bromosuccinimide (NBS) followed by cyclization with thioamides afforded the desired thiazole cores 12a–12g, which were then tethered with different amines to give rise to compounds 13a–13j. The ethyl ester was reacted with hydroxylamine in the presence of potassium hydroxide (KOH) in MeOH to obtain the desired compounds 14a–14j with good yields (Liang et al., 2019). An alternative synthesis route was used for the convenient replacement of the R 2 group. Initially, intermediate 9 was protected by the trifluoroacetyl group. Then, a two-step synthesis of condensation and cyclization afforded the desired thiazole cores 17a and 17b. Following the nucleophilic substitution, intermediates 18a and 18b were obtained. After deprotection of the trifluoroacetyl under the conditions of HCl in MeOH, the corresponding amines 19a and 19b were further reacted with different sulfonyl chlorides to yield 20a–20f. The hydroxamic acid warhead group was introduced at the last step to afford the targeted compounds 21a–21f.
SCHEME 1.

Synthesis of compounds 14a–14j. a Reagents and conditions: (A) SOCl2, MeOH, reflux, 1 h, 87%; (B) 10% Pd/C, H2, rt, 100%, (C) 2,6-difluorobenzenesulfonyl chloride, pyridine, DCM, rt, overnight, 98%; (D) 2-chloro-4-methylpyrimidine, LiHMDS, 0°C to rt, 1 h, 92%; (E) NBS, 2,2,2-trimethylthioacetamide, DMA, rt to 60°C, 1 h, 30%–44%; (F) amines, CsCO3, NMP, 60°C, 17 h, 42%–60%; (G) NH2OH/KOH in MeOH, 0°C, 0.25 h, 50%–74%.
SCHEME 2.

Synthesis of compounds 21d–21f.a Reagents and conditions: (A) rifluoroacetic anhydride, DCM, rt, overnight, 1 h, 88%; (B) 2-chloro-4-methylpyrimidine, LiHMDS, 0°C to rt, 1 h, 90%; (C) NBS, 2,2,2-trimethylthioacetamide, DMA, rt to 60°C, 1 h, 40%; (D) 7-amino-heptanoic acid ethyl ester hydrochloride, CsCO3, NMP, 60°C, 17 h, 42%; (E) HCl in EtOH, 60°C, 2 h, 94%; (F) sulfonyl chlorides, pyridine, DCM, rt, overnight, 74–91%; (G) NH2OH/KOH in MeOH, 0°C, 0.25 h, 61%–84%.
The BRAFV600E, HDAC1, and HDAC6 enzymatic inhibitory activities of novel compounds are shown in Table 1. Vorinostat and dabrafenib were used as references. Initially, we synthesized compounds with dabrafenib as cap and different n-alkyl linkers ranging from five carbons (C5, 14a) to eight carbons (C8, 14d). Unsurprisingly, all these analogs exhibited potent BRAF inhibition with IC50 values ranging from 1.71 nM to 3.82 nM. On the other hand, different linker groups resulted in clear SAR in HDAC1/6 activities. A shorter linker, as in 14a (C5), brought the cap group and hydroxamate acid too close together and led to significantly reduced potency against HDAC1 and HDAC6. Compound 14b with a C6 linker had the most potent HDAC6 inhibition with an IC50 of 49.20 nM. Further linker extension to C7 (14c) or C8 (14d) led to a decrease in HDAC6 activity (IC50 = 127.92 nM and IC50 = 321.80 nM, respectively). HDAC1 inhibitory activities of 14a–14d showed a similar trend. However, C7 compound 14c (IC50 = 312.14 nM) was slightly more potent than C6 compound (14b, IC50 = 493.95 nM). We then fixed the linker to be C6 and sought to perform structural modifications on R 1 and R 2 groups.
TABLE 1.
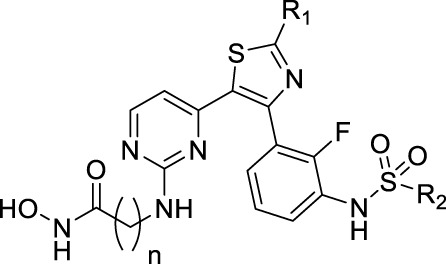
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | Cn | R1 | R2 | Enzymatic IC50 (nM) | Cellular IC50 (μM) | |||
| BRAFV600E | HDAC1 | HDAC6 | HCT116 | HT-29 | ||||
| Vorinostat | NA | 50.30 | 24.09 | 0.68 | 3.10 | |||
| Dabrafenib | 1.11 | NA | NA | >10 | 0.21 | |||
| 14a | 5 | t-Butyl | 2,6-Difluorophenyl | 3.25 | 678.50 | 1211.07 | 3.00 | 1.00 |
| 14b | 6 | t-Butyl | 2,6-Difluorophenyl | 1.71 | 493.95 | 49.20 | 1.68 | 0.31 |
| 14c | 7 | t-Butyl | 2,6-Difluorophenyl | 2.27 | 312.14 | 127.92 | 4.47 | 0.88 |
| 14d | 8 | t-Butyl | 2,6-Difluorophenyl | 3.82 | 765.39 | 321.80 | 3.66 | 0.48 |
| 14e | 6 | Methyl | 2,6-Difluorophenyl | 3.50 | 42.25 | 14.60 | 7.53 | 6.39 |
| 14f | 6 | Ethyl | 2,6-Difluorophenyl | 1.66 | 61.30 | 26.50 | 4.27 | 1.27 |
| 14g | 6 | i-Propyl | 2,6-Difluorophenyl | 1.51 | 129.8 | 30.01 | 2.68 | 0.99 |
| 14h | 6 | Cyclopropyl | 2,6-Difluorophenyl | 1.31 | 49.84 | 27.10 | 4.71 | 2.21 |
| 14i | 6 | n-Propyl | 2,6-Difluorophenyl | 1.07 | 104.50 | 64.60 | 3.08 | 0.97 |
| 14j | 6 | Phenyl | 2,6-Difluorophenyl | 5.14 | 15.59 | 140.20 | 1.08 | 0.95 |
| 21a | 6 | t-Butyl | Methyl | 1.03 | 112.55 | 22.17 | 8.52 | 5.02 |
| 21b | 6 | t-Butyl | n-Propyl | 1.71 | 432.07 | 72.07 | 5.44 | 0.78 |
| 21c | 6 | t-Butyl | Phenyl | 1.26 | 474.40 | 100.70 | 2.06 | 0.11 |
| 21d | 6 | Methyl | Methyl | 258.00 | 71.04 | 21.81 | >10 | >10 |
| 21e | 6 | Methyl | Ethyl | 107.49 | 52.25 | 18.82 | 9.15 | >10 |
| 21f | 6 | Methyl | Phenyl | 3.92 | 49.43 | 18.68 | 4.41 | 4.96 |
Kinase activity assays were performed by a FRET-based Z′-Lyte assay.
Antiproliferative activities were evaluated using a CCK-8, assay. Data are means of three independent experiments.
Aliphatic (methyl (14e), ethyl (14f), i-propyl (14g), cyclopropyl (14h), n-propyl (14i)), or phenyl (14j) substitutions on the 2-thiozole (R 1 ) had no dramatic influence on BRAFV600E inhibition. With reference to their HDAC inhibitory activities, these compounds were generally more potent than 14b against HDAC1. The SAR indicated aromatic and aliphatic R 1 with small steric hindrance was beneficial. The sulfonamide substitution (R 2 ) was shown to be very important for BRAF inhibition (Li et al., 2015). With compounds 21a–21f, when the R 1 group was t-butyl, aliphatic or phenyl substitution of sulfonamide exhibited comparable potency to dabrafenib and 14b. On the other hand, methyl, the methyl (21d) or ethyl (21e) sulfonamide group led to a significant decrease in BRAF inhibition. Compounds with the R 1 group of methyl (14h and 21d–21f) displayed overall increased activities against HDACs1/6 than compounds with the R 1 group of t-butyl (14b and 21a–21c). Next, we evaluated the efficacy of dabrafenib, 14b, 14j, and 21c for their inhibition of wild-type BRAF (Table 2). It was found dabrafenib, 14b, 14j, and 21c exerted nanomolar range IC50 against wild-type BRAF, with IC50 of 2.44 nM, 4.07 nM, 3.86 nM, and 2.86 nM, respectively. Dabrafenib, 14b, and 21c displayed week (about 2-fold) selectivity of BRAFV600E over wild-type BRAF, while 14j with R 1 of phenyl was slightly more potent against wild-type BARF.
TABLE 2.
Enzymatic activity of dabrafenib, 14b, 14j and 21c against wild-type BRAF kinase.
| Compound | BRAFwt enzymatic IC50 (nM) |
|---|---|
| Dabrafenib | 2.44 |
| 14b | 4.07 |
| 14j | 3.86 |
| 21c | 2.86 |
Given the potent BRAFV600E and HDAC1/6 inhibitory activities, the antiproliferation effects of novel inhibitors were investigated using two human colorectal adenocarcinoma cell lines (HT-29 cells harboring BRAFV600E and HCT116 cells with wild-type BRAF) by a Cell Counting Kit-8 (CCK-8) assay (Li et al., 2015). The results revealed that dabrafenib showed better antiproliferative activity against HT-29 cells than that of HCT116 cells. HDAC inhibitor vorinostat inhibited the proliferation of both HCT116 and HT-29 cells, with IC50 of 0.68 μM and 3.1 μM, respectively. Most of the novel dual inhibitors exhibited cell growth inhibitory activity except for compounds 21d and 21e. The overall results indicated that the BRAF/HDAC inhibitors were broadly efficacious compared to the selective BRAF inhibitor alone. Although 14f–14h and 21a displayed improved BRAF and HDAC1/6 activities and some other compounds displayed better efficacy against HDAC1/6 enzyme, this did not lead to improved cellular activities compared to 14b or 21c. Among the novel inhibitors, 14b displayed good antiproliferative activities with IC50 of 1.68 μM against HCT116 cells and 0.31 μM against HT-29 cells, respectively. Noticeably, it is well-documented that the electron-withdrawing fluorine substitution of phenyl ring reduced the liability to oxidative metabolism (Puszkiel et al., 2019). Compared to 21c (R 2 = phenyl) which also exerts good cellular activity, 14b with the R 2 of 2,6-difluorophenyl is superior in the metabolic setting. Therefore, 14b was selected for further studies.
To elucidate the efficacy and selectivity against other enzymes of HDAC family, compound 14b was tested against 8 HDAC isoforms (Table 3) together with vorinostat as a positive control (de Ruijter et al., 2003). Compound 14b showed IC50 against class I isoforms HDAC1, HDAC2, HDAC3, and HDAC8 of 0.493 μM, 0.747 μM, 0.913 μM, and 3.004 μM, respectively. HDAC8 is a promising therapeutic anticancer target that modifies non-histone proteins such as p53 (Spreafico et al., 2020). Compounds 14j and 21c exerted IC50 of 0.876 μM and 0.623 μM against HDAC8 respectively, which was 3.4- and 4.8-fold more potent than 14b (Supplementary Table S1), indicating that the subtle structural difference may change the selectivity between isomers of HDAC. The inhibition of 14b against HDAC6 (class IIB) was > 10-fold more potent than class I isoforms and > 800-fold more potent than class IIA HDACs (HDAC4, HDAC5, and HDAC7). Overall, 14b was a pan-HDAC inhibitor similar to vorinostat, displaying weak selectivity for HDAC6. HDAC6 is the key regulator in oncogenic cell transformation by targeting several non-histone cytoplasmic proteins including Hsp90, α-tubulin, cortactin, HSF1 (Li et al., 2018). The inhibition of class I HDACs and HDAC6 were reported to decrease cell motility and induce apoptosis in cancerous cells, partially contributing to the broadly antitumor effect of 14b (Miyake et al., 2016; Pulya et al., 2021).
TABLE 3.
Selectivity of 14b and vorinostat in HDAC enzyme.
| Enzyme inhibition (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Class I | Class IIA | Class IIB | |||||
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC4 | HDAC5 | HDAC7 | HDAC6 | |
| 14b | 0.493 | 0.747 | 0.913 | 3.004 | >100 | 52.850 | 43.290 | 0.049 |
| Vorinostat | 0.030 | 0.091 | 0.056 | 1.380 | >100 | >100 | 89.200 | 0.014 |
In an attempt to understand the interaction between 14b and its targets, it was docked into the BRAFV600E, HDAC1, and HDAC6 (Butler et al., 2010; Millard et al., 2013; Rheault et al., 2013). As shown in Figure 2A, the hydroxamic acid and linker were pointed to the entrance of the ATP pocket of BRAFV600E. The aminopyrimidine core of 14b formed two hydrogen bonds with the backbone amide of C532. Two more hydrogen bonds were formed between the ligand and the residues D594 and F595. In addition, two water-bridged hydrogen-bonding networks were found between the residues C532, G534; the residues K483, D594; and the ligand, respectively. The multiple hydrogen bonds indicated a crucial and strong binding between the inhibitor and BRAFV600E. On the docking mode of 14b to HDAC1 (Figure 2B), the dabrafenib moiety of 14b projected outside toward the solvent, loosely contacting the hydrophobic residues on the protein surface. The C6 linker was passed through the long tubular channel and stacked between F205 and F150. Hydroxamic acid moiety entered the active site by chelating the essential catalytic zinc ion and formed a hydrogen bond with H140. The binding of 14b to HDAC6 showed a similar mode to that of HDAC1 except that one more hydrogen bond between the ligand and Y745, and π–π interactions between the thiazole and aromatic rings of the ligand and the residues P464 and H463, respectively, were formed in HDAC6 (Figure 2C). (Miyake et al., 2016) As a result, though with the metal coordination bond present, the absence of the extra hydrogen bond and the π–π interactions between the ligand and HDAC1 pointed to a critical cause of its reduced activity. Given the satisfied inhibition enzymatically and cellularly, it was selected for further biological studies.
FIGURE 2.

Predicted binding mode of 14b into BRAFV600E [(A), PDB code: 4xv2], HDAC1 [(B), PDB code: 4bkx] and HDAC6 [(C), PDB code: 5eei]. The targeted proteins are shown in the turquoise picture with selected residues in stick and carbon atoms in green. Water molecules and metal zinc are shown as red and gray spheres, respectively.
Having established compound 14b as the desirable BRAF/HDAC inhibitor in both BRAF inhibitor sensitive cells and resistant cells, we then proceeded to determine whether or not the compound inhibit dual pathways simultaneously using Western blot analysis. In HT-29 and HCT116 cells, acetylated histone 3 (Ac-histone H3), acetylated α-tubulin (Ac-α-tubulin), and phosphorylated extracellular signal–regulated kinase (p-ERK) were analyzed as effectors of HDAC1, HDAC6, and BRAF signal, respectively (Haggarty et al., 2003). In both cells, the acetylation of α-tubulin and histone H3 was upregulated upon 14b treatment, indicating the effective inhibition of HDAC6 and HDAC1 signal by 14b (Figure 3). Phosphorylation of ERK, the downstream effector of BRAF, was significantly inhibited at the lowest concentration of 0.12 μM in HT-29 cells and almost completed inhibition of p-ERK was observed at higher concentrations. It was reported that BRAF inhibitors, including vemurafenib and dabrafenib, induced paradoxical MAPK pathway activation and are contraindicated for the treatment of cancers with RAS mutation (Poulikakos et al., 2010; Adelmann et al., 2016). HCT116 express the mutated active K-RAS (G13D) protein and we found that dabrafenib causes paradoxical activation of ERK in these cells (Supplementary Figure S1). The paradoxical hyperactivation of ERK signaling by BRAFi in BRAF wild-type cells is associated with the emergence of squamous cell carcinoma, a common side-effect of BRAFi (Li et al., 2015; Peng et al., 2015). We were delighted to find that 14b significantly inhibited phosphorylation of ERK in HCT116 cells, with an effective concentration close to the IC50 of HCT116 proliferation (Figure 3A). Similarly, p-ERK inhibition was observed in the presence of 14b but neither dabrafenib in mouse melanoma B16 cells harbored wild-type BRAF (Supplementary Figure S2). In addition, 14b did not affect the expression of BRAF itself in both cells. By comparison, vorinostat only upregulated acetylated α-tubulin and acetylated histone H3 in HT-29 cells and HCT116 cells, but had little impact on the p-ERK. On the other hand, dabrafenib reduced the phosphorylation of ERK, but had little impact on the protein acetylation status in HT-29 cells. These results supported that 14b not only successfully achieved dual inhibition of BRAF and HDAC pathways, but also broke the paradoxical ERK activation in cells with wild-type BRAF.
FIGURE 3.

Characterization of dual inhibition of HDAC and RAF pathway. Western blot analysis of 14b in HCT116 cells (A) and in HT-29 cells (B). Cells were treated with 0 μM, 0.12 μM, 0.37 μM, 1.1 μM, 3.3 μM, and 10 μM of drugs and incubated for 6 h. The cell lysis was subjected to Western blot analysis.
3 Conclusion
An epigenetic malfunction is a common event in most cancer, and HDAC inhibitors were shown to synergize with BRAF/MEK/ERK inhibition (Fu et al., 2019; Maertens et al., 2019). Selective BRAF inhibitors are associated with rapid adaptation and drug resistance as well as cause a high occurrence of secondary skin carcinoma. Developing dual inhibitors of BRAF and HDACs is a valuable strategy to both overcome resistance and enhance effects for BARFi. In summary, we designed a series of hydroxamate acid and 2-aminopyridinyl-containing BRAF/HDAC inhibitors with anti-CRC activity using the pharmacophore hybrid strategy. The compounds potently inhibited BRAFV600E, HDAC1, and HDAC6 enzymes and suppressed the proliferation of colorectal cancer cells harboring both wide-type BRAF and V600E mutated BRAF. Furthermore, the representative compound 14b potently inhibited the activation of the MAPK pathway and upregulated Ac-histone-H3 and Ac-a-tubulin in both cells. Selective BRAF inhibitors vemurafenib and dabrafenib were reported to induce paradoxically upregulation of ERK in HCT116 cells harboring wild-type BRAF. More importantly, the effectiveness of ERK inhibition and cytotoxicity of 14b in HCT116 cells, suggests its potential as a “paradox breaker” to reduce the side effect related to selective BRAFi. Collectively, our studies provided valuable tool compounds for dual pathway inhibition with a single molecule. BRAF mutations are linked to more advanced and aggressive colorectal cancer, lung cancer, and thyroid carcinoma. So it is worthwhile to explore the efficacy of BRAF/HDAC inhibitors against these types of tumors harboring mutated BRAF. In addition, further studies to elucidate the crosstalk of BRAF/HDAC pathways using tool compounds will be highly worthwhile to explore the clinical potential of the dual inhibitors.
4 Materials and Methods
4.1 Chemistry
4.1.1 General Conditions
All reagents and solvents were used directly as purchased from commercial sources. Flash chromatography was performed using silica gel (200–300 mesh). All reactions were monitored by thin-layer chromatography (TLC), using silica gel plates with fluorescence F254 and UV light visualization. 1H NMR spectra were recorded on a Bruker AV-400 spectrometer at 400 MHz. Coupling constants (J) are expressed in hertz (Hz). Chemical shifts (δ) of NMR are reported in parts per million (ppm) units relative to an internal control (TMS). Low-resolution ESI–MS was recorded on an Agilent 1200 HPLC-MSD mass spectrometer and high-resolution ESI–MS on an Applied Biosystems Q-STAR Elite ESI–LC–MS/MS mass spectrometer. HPLC instrument, Dionex Summit HPLC (column: Diamonsil C18, 5.0 μM, 4.6 × 250 mm (Dikma Technologies); detector, PDA-100 photodiode array; injector, ASI-100 autoinjector; pump, p-680A). A flow rate of 0.5 ml/min was used with a mobile phase of ACN in H2O with a 0.1% modifier (TFA, v/v).
4.1.2 Synthetic Procedures for 14a–14j
4.1.2.1 Methyl 2-Fluoro-3-Nitrobenzoate (8)
Into a 500 ml two-neck round bottom flask was placed 50 g (270 mmol) of 2-fluoro-3-nitrobenzoic acid (7) and 270 ml of anhydrous methanol (MeOH). A measure of 29 ml thionyl chloride was added dropwise in an ice bath. Then, the reaction was transferred to an oil bath and heated to reflux for 1 h (hour). After cooling to room temperature (rt), 1-fold of ice-cold water was added to the flask and the mixture was filtered through a Buchner funnel. The filter residue was washed three times with water and dried under reduced pressure to give 47 g (yield: 87%) of the yellowish compound 8. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.28–8.10 (m, 2H), 7.30–7.37 (m, 1H), and 3.99 (s, 3H). MS (ESI), m/z: 200 (M+ + H+).
4.1.2.2 Methyl 3-Amino-2-Fluorobenzoate (9)
In a 500 ml round bottom flask was placed 20 g methyl 2-fluoro-3-nitrobenzoate (8) and 200 ml MeOH. The air in the flask was exchanged for argon and then 1 g wet palladium on carbon (Pd/C) was added to the flask. Then, the reaction vessel was filled with hydrogen gas using a balloon and the reaction was stirred at rt. After 6 h, the completion of the reaction was monitored using TLC. The mixture was filtered through celite and washed with extra EtOAc. The filtrate was concentrated under reduced pressure and dried to get 17 g compound 9 as an orange oil (yield: 100%). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 6.92–7.01 (m, 3 H), 5.37 (s, 2 H), and 3.81 (s, 3 H). MS (ESI), m/z: 170 (M+ + H+).
4.1.2.3 Methyl 3-[(2,6-Difluorophenyl)Sulfonamido]-2-Fluorobenzoate (10)
In o a 500 ml flask was placed methyl 3-amino-2-fluorobenzoate (6.5 g, 35.8 mmol) and DCM (150 ml), and pyridine (3.5 ml, 35.8 mmol). Then, 2,6-difluorobenzenesulfonyl chloride (9 g, 42.3 mmol) in DCM (70 ml) was added dropwise via a funnel and the reaction mixture was allowed to stir at room temperature overnight. The reaction mixture was filtered through celite and washed twice with EtOAc. The filtrate was concentrated and purified by column chromatography [PE (petroleum ether): EtOAc = 5:1] to give 12.2 g (98%) of compound 10. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.98 (s, 1 H), 7.64–7.82 (m, 3H), 7.46–7.61 (m, 1 H), 7.29 (t, J = 8.8 Hz, 2 H), and 3.81 (s, 3 H). MS (ESI), m/z: 346 (M+ + H+).
4.1.2.4 N-{3-[2-(2-Chloropyrimidin-4-yl)Acetyl]-2-Fluorophenyl}-2,6-Difluorobenzene-Sulfonamide (11)
In a 250 ml flask was placed methyl 3-[(2,6-difluorophenyl)sulfonamido]-2-fluorobenzoate (10, 12.2 g, 35.3 mmol) and THF (70 ml). The flask was placed in an ice–water bath and 80 ml of 1M lithium bis(trimethylsilyl)amide (LiHMDS) was added dropwise via an addition funnel and then 2-chloro-4-methylpyrimidine (4.5 g, 35.0 mmol) was added dropwise via syringe. Then, the reaction was allowed to warm to rt and stirred for 4 h. The completion of the reaction was monitored by TLC. The solvent volume was reduced to half under reduced pressure and then treated with 6 N HCl to neutralize the mixture. EtOAc was added and the organic layers were separated. The aqueous layer was extracted twice with EtOAc and the combined organic layer was washed once with brine, dried over Na2SO4, and concentrated onto silica gel. The residue was purified by column chromatography (PE: EtOAc = 3:1) to give 14 g (92%) of compound 11. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 13.40 (s, 1 H), 10.82–11.12 (m, 2 H), 8.48–8.87 (m, 2 H), 7.67–7.80 (m, 3 H), 7.61–7.69 (m, 1 H), 7.52–7.61 (m, 2 H), 7.50 (d, J = 5.31 Hz, 1 H), 7.43 (td, J = 7.60, 1.28 Hz, 1 H), 7.21–7.38 (m, 6 H), 6.13 (s, 1 H), and 4.49 (s, 2 H). MS (ESI), m/z: 442 (M+ + H+).
4.1.2.5 N-{3-[2-(Tert-Butyl)-5-(2-Chloropyrimidin-4-yl)Thiazol-4-yl]-2-Fluorophenyl}-2,6-Difluorobenzenesulfonamide (12a)
In a solution of N-{3-[2-(2-chloropyrimidin-4-yl)acetyl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (11, 14 g, 32 mmol) in 50 ml N,N-dimethylacetamide (DMA), 6.42 g of N-bromosuccinimide (NBS, 32 mmol) was added via small potions and the solution was allowed to stir for 2 h at rt. Then, 2,2-dimethylpropanethioamide (4.2 g, 32 mmol) was then added. The reaction was heated to 60°C for 2 h. After completion of the reaction, the mixture was diluted with water and extracted two times with EtOAc. The combined EtOAc layers were washed three times with water to remove DMA, dried over Na2SO4, filtered, and concentrated onto silica gel. The residue was purified by column chromatography (PE: EtOAc = 3:1) to give 7.35 g (yield: 34%) of compound 12a. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.30 (d, J = 5.2Hz, 1H), 7.72 (t, J = 8.0Hz, 1H), 7.48–7.55 (m, 1H), 7.31–7.36 (m, 2H), 7.22–7.26 (m, 1H), 6.99 (t, J = 8.8Hz, 1H), 6.74 (d,J = 5.2Hz, 1H), and 1.48 (s, 9H). MS (ESI): 539.1 (M+ + H+).
4.1.2.6 Ethyl 7-{[4-(2-(Tert-Butyl)-4-(3-((2,6-Difluorophenyl)Sulfonamido)-2-Fluorophenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}Heptanoate (13b)
In o a 250 ml flask was placed 1.11g of N-{3-[2-(tert-butyl)-5-(2-chloropyrimidin-4-yl)thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (12a, 2 mmol), 1.3g of CsCO3 (4 mmol), 0.63 mg of 7-amino-heptanoic acid ethyl ester hydrochloride (3 mmol), and 5 ml of N-methylpyrrolidone (NMP). The reaction mixture was heated to 60°C for 17 h. After cooling to rt, water was added and the mixture was neutralized with dilute hydrochloric acid. The mixture was extracted three times with EtOAc. The combined EtOAc washings were washed two times with water, dried over Na2SO4, filtered, and concentrated onto silica gel. The residue was purified by column chromatography (PE: EtOAc = 2:1) to give 0.56 g (41.5%) of compound 13b. 1H NMR (400 MHz, DMSO-d6): δ ppm 7.90 (s, 1H), 7.71 (t, J = 8.0Hz, 1H), 7.50 (td, J = 7.2, 2.4Hz, 1H), 7.32 (t, J = 6.4Hz, 1H), 7.20 (t, J = 8.0Hz, 1H), 6.98 (t, J = 8.4Hz, 1H), 6.08 (s, 1H), 4.13 (q, J = 7.2 Hz, 2H), 3.17–3.27 (m, 2H), 2.30 (t, J = 7.2Hz, 2H), 1.55–1.66 (m, 4H), 1.47 (s, 9H), 1.35–1.40 (m, 4H), and 1.25 (t, J = 7.2Hz, 3H). MS (ESI), m/z: 675 (M+ + H+).
4.1.2.7 7-{[4-(2-(Tert-Butyl)-4-(3-((2,6-Difluorophenyl)Sulfonamido)-2-Fluorophenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (14b)
Compound 14b was synthesized using the following procedures as previously described.[38] a: preparation of NH2OH/KOH in MeOH solution: 9.34 g of hydroxylamine hydrochloride in 48 ml of MeOH was added dropwise to a solution of 11.2 g potassium hydroxide (KOH) in 28 ml MeOH in an ice–water bath. After that, the mixture was stirred for 0.5 h and then filtered. The filtrate was sealed and stored at −20°C for further usage. b: in a 50 ml flask was placed 0.675 g of ethyl 7-{[4-(2-(tert-butyl)-4-(3-((2,6-difluorophenyl)sulfonamido)-2-fluorophenyl)thiazol-5-yl)pyrimidin-2-yl]amino}heptanoate (13b, 1 mmol) and 5 ml of MeOH. A measure of 5 ml of NH2OH/KOH MeOH solution was added and stirred for 15 min in an ice–water bath. The completion of the reaction was monitored by TLC. Then, 20 ml of water was added and the pH was adjusted to 6 using 1N HCl. The precipitation was filtered and washed three times with water and dried under reduced pressure to obtain compound 500 mg 14b as light-yellow solid (yield: 74%). 1H NMR (500 MHz, DMSO-d6): δ (ppm) 10.86 (s, 1H), 10.32 (s, 1H), 8.64 (s, 1H), 8.02 (d, J = 5.0Hz, 1H), 7.65–7.67 (m, 1H), 7.42 (t, J = 7.0Hz, 1H), 7.30–7.35 (m, 1H), 7.20–7.28 (m, 4H), 5.80–5.95 (m, 1H), 3.05–3.20 (m, 2H), 1.94 (t, J = 7.0Hz, 1H), 1.45–1.52 (m, 4H), 1.40 (s, 9H), and 1.22–1.37 (m, 4H). HRMS (ESI) calcd for C30H33F3N6O4S2 (M-H)−: 661.1884; found 661.1879. HPLC purity = 95%, Rt 27.6 min. Melting point: 95.3°C.
4.1.2.8 6-{[4-(2-(Tert-butyl)-5-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)Thiazol-4-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyhexanamide (14a)
Yield: 50%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.84 (s, 1H), 10.31 (s, 1H), 8.02 (s, 1H), 7.64–7.71 (m, 1H), 7.42 (t, J = 7.0Hz, 1H), 7.33–7.35 (m, 1H), 7.20–7.30 (m, 4H), 5.78–6.00 (brs, 1H), 3.10–3.20 (m, 2H), 1.95 (t, J = 7.2Hz, 2H), 1.49–1.53 (m, 4H), and 1.24–1.28 (m, 2H). HRMS (ESI) calcd for C29H31F3N6O4S2 (M-H)−: 647.1727; found 647.1774.
4.1.2.9 8-{[4-(2-(Tert-Butyl)-4-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyoctanamide (14c)
Yield: 62%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.86 (s, 1H), 10.31 (s, 1H), 8.64 (s, 1H), 8.02 (d, J = 4.0Hz, 1H), 7.66–7.68 (m, 1H), 7.41–7.44 (m, 1H), 7.34–7.40 (m, 1H), 7.21–7.28 (m, 4H), 5.76–5.91 (brs, 1H), 3.13–3.17 (m, 2H), 1.93 (t, J = 6.0Hz, 2H), 1.47–1.53 (m, 4H), 1.40 (s, 9H), and 1.20–1.27 (m, 6H). HRMS (ESI) calcd for C3 1H35F3N6O4S2 (M-H)+: 677.2186; found 677.2177.
4.1.2.10 9-{[4-(2-(Tert-Butyl)-4-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxynonanamide (14d)
Yield: 71%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.86 (s, 1H), 10.31 (s, 1H), 8.6 3 (s, 1H), 8.02 (d, J = 4Hz, 1H), 7.60–7.69 (m, 1H), 7.42 (t, J = 5.6Hz, 1H), 7.20–7.31 (m, 5H), 5.78–5.88 (brs, 1H), 3.10–3.20 (m, 2H), 1.92 (t, J = 6.0Hz, 2H), 1.47–1.50 (m, 4H), 1.40 (s, 9H), and 1.20–1.30 (m, 8H). HRMS (ESI) calcd for C32H37F3N6O4S2 (M-H)−: 689.2197; found 689.2190.
4.1.2.11 7-{[4-(4-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)-2-Methylthiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (14e)
Yield: 71%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.88 (s, 1H), 10.32 (s, 1H), 8.65 (s, 1H), 8.02 (d, J = 4.4Hz, 1H), 7.63–7.72 (m, 1H), 7.38–7.45 (m, 1H), 7.20–7.36 (m, 5H), 5.80–5.92 (brs, 1H), 3.08–3.20 (m, 2H), 2.68 (s, 3H), 1.94 (t, J = 7.0Hz, 2H), 1.43–1.53 (m, 4H), and 1.22–1.30 (m, 4H). HRMS (ESI) calcd for C27H27F3N6O4S2 (M-H)−: 619.1414; found 619.1409.
4.1.2.12 7-{[4-(4-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)-2-Ethylthiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (14f)
Yield: 65%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.86 (s, 1H), 10.31 (s, 1H), 8.02 (d, J = 4.8Hz, 1H), 7.64–7.71 (m, 1H), 7.42 (t, J = 7.5Hz, 1H), 7.35 (t, J = 6.7Hz, 1H), 7.21–7.30 (m, 4H), 5.80–5.95 (brs, 1H), 3.10–3.20 (m, 2H), 3.02 (q, J = 7.6Hz, 2H), 1.94 (t, J = 7.2Hz, 2H), 1.43–1.55 (m, 4H), 1.32 (t, J = 7.6Hz, 3H), and 1.27 (m, 4H). HRMS (ESI) calcd for C28H29F3N6O4S2 (M-H)−:633.1571; found 633.1571.
4.1.2.13 7-{[4-(4-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)-2-Isopropylthiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (14g)
Yield: 70%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.87 (s, 1H), 10.32 (s, 1H), 8.65 (s, 1H), 8.02 (d, J = 5.5Hz, 1H), 7.65–7.70 (m, 1H), 7.43 (t, J = 7.5Hz, 1H), 7.35 (t, J = 6.7Hz, 1H), 7.22–7.29 (m, 4H), 5.74–5.94 (m, 1H), 3.27–3.30 (m, 1H), 3.06–3.20 (m, 2H), 1.94 (t, J = 7.5Hz, 2H), 1.47–1.52 (m, 4H), 1.35 (d, J = 7.0Hz, 6H), and 1.25–1.30 (m, 4H). HRMS (ESI) calcd for C29H31F3N6O4S2 (M-H)−: 647.1727; found 647.1774.
4.1.2.14 7-{[4-(2-Cyclopropyl-4-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (14h)
Yield: 54%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.88 (s, 1H), 10.33 (s, 1H), 8.04 (d, J = 4.4Hz, 1H), 7.66–7.70 (m, 1H), 7.34–7.45 (m, 3H), 7.20–7.30 (m, 4H), 5.80–6.00 (brs, 1H), 3.00 (m, 2H), 1.94 (m, 2H), 1.49 (m, 4H), and 1.27–1.34 (m, 9H). HRMS (ESI) calcd for C29H29F3N6O4S2 (M-H)−: 645.1571; found 645.1514.
4.1.2.15 7-{[4-(4-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)-2-Propylthiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (14i)
Yield: 67%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.86 (s, 1H), 10.32 (s, 1H), 8.64 (s, 1H), 8.02 (d, J = 4.8Hz, 1H), 7.63–7.71 (m, 1H), 7.43 (t, J = 7.2Hz, 1H), 7.34 (t, J = 6.0Hz, 1H), 7.20–7.29 (m, 4H), 5.80–5.60 (m, 1H), 3.10–3.25 (m, 2H), 2.95 (t, J = 7.6Hz, 2H), 1.94 (t, J = 7.2Hz, 2H), 1.70–1.80 (m, 2H), 1.40–1.52 (m, 4H), 1.22–1.30 (m, 4H), and 0.97 (t, J = 7.6Hz, 3H). HRMS (ESI) calcd for (M-H)−: 647.1728; found 647.1730.
4.1.2.16 7-{[4-(4-(3-(2,6-Difluorophenylsulfonamido)-2-Fluorophenyl)-2-Phenylthiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (14j)
Yield: 59%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.93 (s, 1H), 10.33 (s, 1H), 8.65 (s, 1H), 8.07 (d, J = 5.2Hz, 1H), 7.88–8.02 (m, 2H), 7.60–7.70 (m, 1H), 7.53–7.58 (m, 3H), 7.47 (t, J = 7.2Hz, 1H), 7.38–7.42 (m, 1H), 7.25–7.33 (m, 2H), 7.23 (t, J = 9.0Hz, 2H), 5.85–6.00 (brs, 1H), 3.15–3.25 (m, 2H), 1.95 (t, J = 7.2Hz, 2H), 1.47–1.57 (m, 4H), and 1.25–1.35 (m, 4H). HRMS (ESI) calcd for C32H29F3N6O4S2 (M-H)−: 681.1571; found 681.1558.
4.1.3 Synthetic Procedures for 21a–21f
4.1.3.1 Methyl 2-Fluoro-3-(2,2,2-Trifluoroacetamido)Benzoate (15)
In a 250 ml round bottom flask was added 10 g of methyl 3-amino-2-fluorobenzoate (9, 60 mmol) in 180 ml dry tetrahydrofuran (HTF). Then, 5.8 ml of trifluoroacetic anhydride (72 mmol), 12.5 ml of triethylamine (Et3N, 90 mmol), and 73 mg of 4-dimethylaminipyridine (DMAP) were successively added and the reaction was stirred for 3 h. The mixture was concentrated under reduced pressure. The residue was washed with DCM/PE (1:1) and filtered. The filtrate was further concentrated on silica gel and purified by column chromatography (PE: EtOAc = 7:1). Combined filtered solid and chromatographic elution obtained 11.7 g (90%) of compound 15. 1H NMR (400 MHz, DMSO-d6): δ (ppm) .8.32–8.17 (m, 1 H), 7.76–7.70 (m, 1H), 7.30 (t, J = 7.6Hz, 1H), and 3.91 (s, 3 H). MS (ESI), m/z: 266 (M+ + H+).
4.1.3.2 N-{3-[2-(2-Chloropyrimidin-4-yl)Acetyl]-2-Fluorophenyl}-2,2,2-Trifluoroacetamide (16)
In a three-neck 250 ml round bottom flask was placed 11.7 g of methyl 2-fluoro-3-(2,2,2-trifluoroacetamido)benzoate (15, 53 mmol) and 100 ml of THF (70 ml). The flask was placed in an ice–water bath and 132 ml 1M LiHMDS was added dropwise via an addition funnel and then 6.8 g 2-chloro-4-methylpyrimidine (53 mmol) via syringe. After the addition was complete, the reaction was allowed to warm to rt and stirred for 4 h. The completion of the reaction was monitored by TLC. The solvent volume was reduced to half under reduced pressure and then treated with 6 N HCl to neutralize the mixture. The solution was extracted three times with EtOAc and the combined organic layer was washed once with brine, dried over Na2SO4, and concentrated onto silica gel. The residue was purified by column chromatography (PE: EtOAc = 3:1) to give 18.5 g (yield: 98%) of compound 16. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 13.80 (s, 1H), 8.46 (d, J = 5.2Hz, 1H), 8.32 (t, J = 7.6Hz, 1H), 8.14–8.21 (m, 1H), 7.76 (t, J = 7.6Hz, 1H), 7.32 (t, J = 7.6Hz, 1H), 6.95 (d, J = 5.2Hz, 1H), and 6.17 (s, 1H). MS (ESI), m/z: 362 (M+ + H+).
4.1.3.3 N-{3-[2-(Tert-Butyl)-5-(2-Chloropyrimidin-4-yl)Thiazol-4-yl]-2-Fluorophenyl}-2,2,2-Trifluoroacetamide (17a)
In a solution of 5 g of N-(3-(2-(2-chloropyrimidin-4-yl)acetyl)-2-fluorophenyl)-2,2,2-trifluoroacetamide (16, 14 mmol) in 50 ml of DMA, 2.5 g NBS (14 mmol) was added and the solution and was allowed to stir for 2 h at rt. Then, 1.05 g of 2,2-dimethylpropanethioamide (14 mmol) was added at rt. The reaction was heated to 60°C for 2 h. After completion of the reaction, the mixture was diluted with water and extracted two times with EtOAc. The combined EtOAc washings were washed three times with water to remove DMA, dried over Na2SO4, filtered, and concentrated onto silica gel. The residue was purified by column chromatography (PE: EtOAc = 3:1) to give 4 g (yield: 68%) of compound 17a. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.41 (d, J = 5.6Hz, 1H), 8.36 (td, J = 7.6, 1.2Hz, 1H), 8.09 (s, 1H), 7.43 (td, J = 7.2, 1.2Hz, 1H), 7.35 (t, J = 8.0Hz, 1H), 6.92 (d, J = 5.5Hz, 1H), and 1.50 (s, 9H). MS (ESI), m/z: 457 (M+ + H+).
4.1.3.4 Ethyl 7-{[4-(2-(Tert-Butyl)-4-(2-Fluoro-3-(2,2,2-Trifluoroacetamido)Phenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}Heptanoate (18a)
In a 250 ml flask was placed 4 g of N-{3-[2-(tert-butyl)-5-(2-chloropyrimidin-4-yl)thiazol-4-yl]-2-fluorophenyl}-2,2,2-trifluoroacetamide (17, 10 mmol), 9.8 g of CsCO3 (30 mmol), 3 g of 7-amino-heptanoic acid ethyl ester hydrochloride (15 mmol), and 40 ml of NMP. The reaction mixture was heated to 60°C for 17 h. After cooling to rt, water was added and dilute hydrochloric acid was then added to neutralize the mixture. The mixture was extracted three times with EtOAc. The combined EtOAc washings were washed two times with water, dried over Na2SO4, filtered, and concentrated onto silica gel. The residue was purified by column chromatography (PE: EtOAc = 3:1) to give 3.6 g (65%) of compound 18a. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.95–8.03 (m, 1H), 7.67 (t, J = 7.6Hz, 1H), 7.36 (t, J = 6.8Hz, 1H), 7.23 (t, J = 8.0Hz, 1H), 6.21 (d, J = 5.2Hz, 1H), 4.12 (q, J = 7.2Hz, 2H), 3.23–3.45 (m, 2H), 2.76 (m, 1H), 2.29 (t, J = 7.6Hz, 2H), 1.53–1.67 (m, 4H), 1.45–1.52 (m, 4H), 1.41 (s, 9H), and 1.24 (t, J = 7.2Hz, 2H). MS (ESI), m/z: 596 (M+ + H+).
4.1.3.5 Ethyl 7-{[4-(4-(3-Amino-2-Fluorophenyl)-2-(Tert-Butyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}Heptanoate (19a)
A measure of 3.6 g ethyl of 7-{[4-(2-(tert-butyl)-4-(2-fluoro-3-(2,2,2-trifluoroacetamido)phenyl)thiazol-5-yl)pyrimidin-2-yl]amino}heptanoate (18a) was dissolved in 60 ml HCl/EtOH solution and heated to 60°C for 2 h. The completion of the reaction was monitored by TLC. The solvent was removed under reduced pressure. The product was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6): δ(ppm) 7.95–8.03 (m, 1H), 7.67 (t, J = 7.6Hz, 1H), 7.36 (t, J = 6.8Hz, 1H), 7.23 (t, J = 8.0Hz, 1H), 6.21 (d, J = 5.2Hz, 1H), 4.12 (q, J = 7.2Hz, 2H), 3.23–3.45 (m, 2H), 2.76 (m, 1H), 2.29 (t, J = 7.6Hz, 2H), 1.53–1.67 (m, 4H), 1.45–1.52 (m, 4H), 1.41 (s, 9H), and 1.24 (t, J = 7.2Hz, 2H). MS(ESI), m/z: 458 (M+ + H+).
4.1.3.6 7-{[4-(2-(Tert-Butyl)-4-(2-Fluoro-3-(Methylsulfonamido)Phenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (21a)
Yield: 61%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.32 (s, 1H), 9.65–9.75 (brs, 1H), 8.65 (s, 1H), 8.10 (d, J = 5.2Hz, 1H), 7.51 (t, J = 7.5Hz, 1H), 7.30–7.36 (m, 1H), 7.28–7.31 (m, 1H), 7.23–7.26 (m, 1H), 6.00–6.20 (m, 1H), 3.07–3.20 (m, 2H), 2.98 (s, 3H), 1.93 (t, J = 6.0Hz, 1H), 1.55–1.63 (m, 4H), 1.43 (s, 9H), and 1.20–1.30 (m, 4H). HRMS (ESI) calcd C25H33FN6O4S2 for (M-H)−:563.1916; found 563.1910.
4.1.3.7 7-{[4-(2-(Tert-Butyl)-4-(2-Fluoro-3-(Propylsulfonamido)Phenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (21b)
Yield: 70%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.34 (s, 1H), 9.73 (s, 1H), 8.67 (s, 1H), 8.11 (d, J = 5.0Hz, 1H), 7.52 (t, J = 6.0Hz, 1H), 7.24–7.31 (m, 3H), 6.10–6.17 (m, 1H), 3.05–3.25 (m, 2H), 3.02 (t, J = 7.5Hz, 2H), 1.95 (t, J = 7.0Hz, 2H), 1.65–1.70 (m, 2H), 1.46–1.50 (m, 4H), 1.44 (s, 9H), 1.24–1.28 (m, 4H), and 0.91 (t, J = 7.5Hz, 3H). HRMS (ESI) calcd for C27H37FN6O4S2 (M-H)−: 591.2229; found 591.2228.
4.1.3.8 7-{[4-(2-(Tert-Butyl)-4-(2-Fluoro-3-(Phenylsulfonamido)Phenyl)Thiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (21c)
Yield: 75%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.30–10.34 (brs, 2H), 8.64 (s, 1H), 8.03 (d, J = 4.8Hz, 1H), 7.74 (d, J = 4.2Hz, 1H), 7.54–7.58 (m, 1H), 7.48–7.54 (m, 2H), 7.35–7.37 (m, 1H), 7.23 (t, J = 5.2Hz, 1H), 7.13–7.18 (m, 2H), 5.80–5.95 (brs, 1H), 3.08–3.20 (m, 2H), 1.94 (t, J = 7.2Hz, 2H), 1.44–1.52 (m, 4H), 1.40 (s, 9H), and 1.23–1.30 (m, 4H). HRMS (ESI) calcd for C30H35FN6O4S2 (M-H)−: 625.2072; found 625.2065.
4.1.3.9 7-{[4-(4-(2-Fluoro-3-(Methylsulfonamido)Phenyl)-2-Methylthiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (21d)
Yield: 72%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.32 (s, 1H), 9.71 (s, 1H), 8.65 (s, 1H), 8.09 (d, J = 4.8Hz, 1H), 7.51 (t, J = 7.6Hz, 1H), 7.35–7.39 (m, 1H), 7.28–7.33 (m, 1H), 7.26 (t, J = 5.2Hz, 1H), 6.00-.620 (brs, 1H), 3.08–3.25 (m, 2H), 3.00 (s, 3H), 2.71 (s, 3H), 1.94 (t, J = 7.2Hz, 2H), 1.42–1.53 (m, 4H), and 1.20–1.33 (m, 4H). HRMS (ESI) calcd for C22H27FN6O4S2 (M-H)−: 521.1446; found 521.1440.
4.1.3.10 7-{[4-(4-(3-(Ethylsulfonamido)-2-Fluorophenyl)-2-Methylthiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (21e)
Yield: 84%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 10.33 (s, 1H), 9.74 (s, 1H), 8.65 (s, 1H), 8.09 (d, J = 4.8Hz, 1H), 7.52 (t, J = 7.6Hz, 1H), 7.37 (t, J = 6.8Hz, 1H), 7.26–7.32 (m, 2H), 6.00–6.11 (brs, 1H), 3.10–3.20 (m, 2H), 3.05 (q, J = 7.2Hz, 2H), 2.71 (s, 1H), 1.94 (t, J = 7.2Hz, 2H), 1.48 (m, 4H), 1.26 (m, 4H), and 1.19 (t, J = 7.2Hz, 3H). HRMS (ESI) calcd for C23H29FN6O4S2 (M+H)+: 689.1748; found 537.1749.
4.1.3.11 7-{[4-(4-(2-Fluoro-3-(Phenylsulfonamido)Phenyl)-2-Methylthiazol-5-yl)Pyrimidin-2-yl]Amino}-N-Hydroxyheptanamide (21g)
Yield: 80%. 1H NMR (500 MHz, DMSO-d6): δ (ppm) 10.26–10.35 (m, 2H), 8.65 (s, 1H), 8.02 (d, J =5.0 Hz, 1H), 7.75 (d, J = 5.0Hz, 2H), 7.61 (d, J = 7.5Hz, 1H), 7.51–7.55 (m, 2H), 7.39 (t, J = 7.5Hz, 1H), 7.20–7.27 (m, 3H), 5.75–5.90 (m, 1H), 3.05–3.20 (m, 2H), 2.67 (s, 1H), 1.94 (t, J = 7.5Hz, 2H), 1.42–1.52 (m, 4H), and 1.20–1.30 (m, 4H). HRMS (ESI) calcd for C27H29FN6O4S2 (M-H)−: 583.1603; found 583.1599.
4.2 Biological Study
4.2.1 Cells and Agents
HCT116 and HT-29 cells were obtained from the Shanghai Cell Bank (Type Culture Collection (TCC), Chinese Academy of Sciences) and cultured in DMEM (10–013-CVR, corning) supplemented with 10% fetal bovine serum (04-001-1A, BI), 1% penicillin/streptomycin (03-031-1B, BI). Primary antibodies against ERK1/2 (4695s), phosphor-ERK1/2 (4370s), AKT (4685s), phosphor-AKT (13038, 4060), α-tubulin (2125S), acetyl-α-tubulin (5335), BRAF (9434), phosphor-BRAF (2696S), GAPDH 2118), and anti-rabbit or anti-mouse IgG horseradish peroxidase (HRP)–linked secondary antibodies were purchased from Cell Signaling Technology (Boston, MA, United States).
4.2.2 Antiproliferation Cell Assay
The cells in the logarithmic phase were placed in 96-well plates (∼3000 cells/well) in a complete medium. After incubation overnight, the cells were exposed to the corresponding compounds or vehicle control at the indicated concentration for a further 72 h. Cell proliferation was evaluated using Cell Counting Kit 8 (CCK8, CK04, Dojindo Laboratories, Kumamoto, Japan). OD450 and OD650 were determined using a microplate reader. Absorbance rate (A) for each well was calculated as OD450 −OD650. The cell viability rate for each well was calculated as V% = (As − Ac)/(Ab − Ac) × 100%, and IC50 values were further calculated by concentration response curve fitting using GraphPad Prism 5.0 software. Each IC50 value is expressed as mean ± SD. As is the absorbance rate of the test compound well, Ac is the absorbance rate of the well without either the cell or test compound, and Ab is the absorbance rate of the well with the cell and vehicle control.
4.2.3 In Vitro BRAF Enzymatic Activity Assay
BRAFV600E (as BRAFV599E in supplier’s catalog), BRAF (wild-type) and the Z′-Lyte Kinase Assay Kit were purchased from Invitrogen. The experiments were performed according to the instructions of the manufacturer. The final 10 μl reaction consists of 0.002 ng of BRAF, 10 ng of inactive MAP2K1 (MEK1), 100 ng of inactive MAPK1 (ERK2), 2 μM Ser/Thr3 peptide in 1×kinase buffer. For each assay, 10 μl kinase reactions were added to 384-well plate, mixed thoroughly, and incubated for 1 hour at room temperature. Then, a 5 μl development solution was added to each well and the plate was incubated for another 1 h at room temperature. Then, a 5 μl stop reagent was loaded to stop the reaction. For the control setting, a 5 μl phospho-peptide solution instead of kinase/peptide mixture was used as 100% phosphorylation control. Then, 2.5 μl 1.33×kinase buffer instead of ATP solution was used as 100% inhibition control, and 2.5 μl of 4% DMSO instead of compound solution was used as the 0% inhibitor control. The plate was measured on an EnVision Multilabel Reader (Perkin-Elmer). Curve fitting and data presentations were performed using GraphPad Prism, version 5.0. Every experiment was repeated at least two times.
4.2.4 In Vitro HDAC Enzymatic Activity Assay
The purified recombinant Human HDACs and their corresponding substrates were purchased from BPS Bioscience (BPS Bioscience Inc., United States). The assays were carried out in a 384-well format using the BPS fluorescent–based HDAC activity assay according to the manufacturer’s protocol. In brief, 10 ml of the HDAC reaction mixture was composed of HDAC assay buffer, 100 mg BSA, serial diluted test compounds, appropriate concentration of HDACs, and 20 mM fluorogenic substrate, the mixture was incubated at 37°C for 60 min, and then stopped by the addition of developer containing trypsin and TSA. After 20 min of incubation, the fluorescence was detected at the excitation wavelength of 360 nm and the emission wavelength of 460 nm using the EnVision Multilabel Reader (PerkinElmer Inc., United States). The analytical software, GraphPad Prism 5.0 (GraphPad Software, Inc., United States) was used to generate IC50 value via non-linear regression analysis.
4.2.5 Western Blot Analysis
The cells were treated with various concentrations of 14b for 6 h. Then, the cells were lysed using 1×SDS sample lysis buffer (CST recommended) with protease and phosphatase inhibitors. Cell lysates were loaded and electrophoresed onto 8–12% SDS-PAGE gel, and then the separated proteins were transferred to a PVDF film. The film was blocked with 5% BSA (Sigma-Aldrich, St. Louis, MO, United States) in a TBS solution containing 0.5% Tween-20 for 4 h at room temperature, then incubated with the corresponding primary antibody (1:1000–1:200) overnight at 4°C. After washing with TBST, HRP-conjugated secondary antibody was incubated for 2 h. The protein signals were visualized by the ECL Western Blotting Detection Kit (Thermo Scientific, Grand Island, NY, United States), and detected with Amersham Imager 600 system (GE, Boston, MA, United States).
4.2.6 In Silico Docking Study
The crystal structures of HDACs and BRAF V600E complexes (PDB IDs: 4BKX, 5EEI, and 4XV2) were used as receptors to predict the binding modes of 14b using Autodock (Morris et al., 2009). Before docking, those protein structures were processed by removing water and adding hydrogens using Ambertools18 (Case et al., 2005). A single chain of all the protein structures was selected for docking. The protonation states of all amino acids and 14b were determined using PROPKA3 (Olsson et al., 2011), followed by 10000 steps of energy minimization. The structure of 14b was optimized at the B3LYP level of theory with the 6–31+g(d) basis set using Gaussian 16 (Frisch et al., 2016). Partial charges of the receptors and ligands for the docking study were calculated using AutodockTools following the Gasteiger’s method (Gasteiger and Marsili, 1980). The center of the native ligand was set as the grid box center with the grid spacing of 0.375 Å. The number of energy evaluations was set to 2500000. The Autodock4Zn zinc force field (Santos-Martins et al., 2014) was used in HDAC studies in addition to the Autodock standard force field to properly handle the metal zinc coordination.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
YL and ZZ initiated and designed the project; YL, HC, LP, and YY performed the syntheses of tested compounds; YH, FX, ZT, and ZZ performed the biology activity experiments; BD and RQ carried out the in silico docking study. All authors reviewed and approved the manuscript.
Funding
The project was supported by the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2020A1515110361), National Natural Science Foundation of China (Grant No. 82150206, 81973158), Ministry of Science and Technology of the People’s Republic of China (SQ2019YFE010401), Natural Science Foundation of Guangdong Province (2019A1515011235), and Guangzhou Municipal Science and Technology Bureau (202002030414).
Conflict of Interest
Author HC is employed by XtalPi Inc (Shenzhen Jingtai Technology Co., Ltd).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.910353/full#supplementary-material
Reference
- Adelmann C. H., Ching G., Du L., Saporito R. C., Bansal V., Pence L. J., et al. (2016). Comparative Profiles of BRAF Inhibitors: the Paradox Index as a Predictor of Clinical Toxicity. Oncotarget 7 (21), 30453–30460. 10.18632/oncotarget.8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadzadeh A., Shahrabi S., Jaseb K., Norozi F., Shahjahani M., Vosoughi T., et al. (2014). BRAF Mutation in Hairy Cell Leukemia. Oncol. Rev. 8 (2), 253. 10.4081/oncol.2014.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G., Hirth P., Tsai J., Zhang J., Ibrahim P. N., Cho H., et al. (2010). Clinical Efficacy of a RAF Inhibitor Needs Broad Target Blockade in BRAF-Mutant Melanoma. Nature 467 (7315), 596–599. 10.1038/nature09454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K. V., Kalin J., Brochier C., Vistoli G., Langley B., Kozikowski A. P. (2010). Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. J. Am. Chem. Soc. 132 (31), 10842–10846. 10.1021/ja102758v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson R., Celtikci B., Fenning C., Javadi A., Crawford N., Perez-Carbonell L., et al. (2015). HDAC Inhibition Overcomes Acute Resistance to MEK Inhibition in BRAF-Mutant Colorectal Cancer by Downregulation of C-FLIPL. Clin. Cancer Res. 21 (14), 3230–3240. 10.1158/1078-0432.ccr-14-2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A., Cheatham T. E., III, Darden T., Gohlke H., Luo R., Merz K. M., Jr, et al. (2005). The Amber Biomolecular Simulation Programs. J. Comput. Chem. 26 (16), 1668–1688. 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang F., Steelman L. S., Lee J. T., Shelton J. G., Navolanic P. M., Blalock W. L., et al. (2003). Signal Transduction Mediated by the Ras/Raf/MEK/ERK Pathway from Cytokine Receptors to Transcription Factors: Potential Targeting for Therapeutic Intervention. Leukemia 17 (7), 1263–1293. 10.1038/sj.leu.2402945 [DOI] [PubMed] [Google Scholar]
- Chen I. C., Sethy B., Liou J. P. (2020). Recent Update of HDAC Inhibitors in Lymphoma. Front. Cell Dev. Biol. 8, 576391. 10.3389/fcell.2020.576391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H., Chang Y., Zhang L., Luo J., Tu Z., Lu X., et al. (2014). Identification and Optimization of New Dual Inhibitors of B-Raf and Epidermal Growth Factor Receptor Kinases for Overcoming Resistance against Vemurafenib. J. Med. Chem. 57 (6), 2692–2703. 10.1021/jm500007h [DOI] [PubMed] [Google Scholar]
- Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., et al. (2002). Mutations of the BRAF Gene in Human Cancer. Nature 417 (6892), 949–954. 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- de Ruijter A. J., van Gennip A. H., Caron H. N., Kemp S., van Kuilenburg A. B. (2003). Histone Deacetylases (HDACs): Characterization of the Classical HDAC Family. Biochem. J. 370 (Pt 3), 737–749. 10.1042/BJ20021321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransén K., Klintenäs M., Osterström A., Dimberg J., Monstein H. J., Söderkvist P. (2004). Mutation Analysis of the BRAF, ARAF and RAF-1 Genes in Human Colorectal Adenocarcinomas. Carcinogenesis 25 (4), 527–533. 10.1093/carcin/bgh049 [DOI] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., et al. (2016). Gaussian 16 Rev. C.01. Wallingford, CT: Gaussian, Inc. [Google Scholar]
- Fu H., Cheng L., Jin Y., Cheng L., Liu M., Chen L. (2019). MAPK Inhibitors Enhance HDAC Inhibitor-Induced Redifferentiation in Papillary Thyroid Cancer Cells Harboring BRAF (V600E): An In Vitro Study. Mol. Ther. - Oncol. 12, 235–245. 10.1016/j.omto.2019.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger J., Marsili M. (1980). Iterative Partial Equalization of Orbital Electronegativity—A Rapid Access to Atomic Charges. Tetrahedron 36 (22), 3219–3228. 10.1016/0040-4020(80)80168-2 [DOI] [Google Scholar]
- Geng A., Cui H., Zhang L., Chen X., Li H., Lu T., et al. (2019). Discovery of Novel Phenoxybenzamide Analogues as Raf/HDAC Dual Inhibitors. Bioorg. Med. Chem. Lett. 29 (13), 1605–1608. 10.1016/j.bmcl.2019.04.047 [DOI] [PubMed] [Google Scholar]
- Grunstein M. (1997). Histone Acetylation in Chromatin Structure and Transcription. Nature 389 (6649), 349–352. 10.1038/38664 [DOI] [PubMed] [Google Scholar]
- Haggarty S. J., Koeller K. M., Wong J. C., Grozinger C. M., Schreiber S. L. (2003). Domain-selective Small-Molecule Inhibitor of Histone Deacetylase 6 (HDAC6)-Mediated Tubulin Deacetylation. Proc. Natl. Acad. Sci. U.S.A. 100 (8), 4389–4394. 10.1073/pnas.0430973100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T. C. S., Chan A. H. Y., Ganesan A. (2020). Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 63 (21), 12460–12484. 10.1021/acs.jmedchem.0c00830 [DOI] [PubMed] [Google Scholar]
- Holderfield M., Deuker M. M., McCormick F., McMahon M. (2014). Targeting RAF Kinases for Cancer Therapy: BRAF-Mutated Melanoma and beyond. Nat. Rev. Cancer 14 (7), 455–467. 10.1038/nrc3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoulia Z., Gavathiotis E., Poulikakos P. I. (2017). New Perspectives for Targeting RAF Kinase in Human Cancer. Nat. Rev. Cancer 17 (11), 676–691. 10.1038/nrc.2017.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. H. (2016). Dual Inhibition of BRAF and MEK in BRAF-Mutated Metastatic Non-small Cell Lung Cancer. J. Thorac. Dis. 8 (9), 2369–2371. 10.21037/jtd.2016.09.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelblinger P., Thuerigen O., Dummer R. (2018). Development of Encorafenib for BRAF-Mutated Advanced Melanoma. Curr. Opin. Oncol. 30 (2), 125–133. 10.1097/cco.0000000000000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F., Guo S. T., Jin L., Jiang C. C., Wang C. Y., Croft A., et al. (2013). Cotargeting Histone Deacetylases and Oncogenic BRAF Synergistically Kills Human Melanoma Cells by Necrosis Independently of RIPK1 and RIPK3. Cell Death Dis. 4 (6), e655. 10.1038/cddis.2013.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Cheng H., Zhang Z., Zhuang X., Luo J., Long H., et al. (2015). N-(3-Ethynyl-2,4-difluorophenyl)sulfonamide Derivatives as Selective Raf Inhibitors. ACS Med. Chem. Lett. 6 (5), 543–547. 10.1021/acsmedchemlett.5b00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Shen J., Tan L., Zhang Z., Gao D., Luo J., et al. (2016). Design and Synthesis of N-(4-aminopyridin-2-yl)amides as B-Raf(V600E) Inhibitors. Bioorg. Med. Chem. Lett. 26 (12), 2760–2763. 10.1016/j.bmcl.2016.04.076 [DOI] [PubMed] [Google Scholar]
- Li T., Zhang C., Hassan S., Liu X., Song F., Chen K., et al. (2018). Histone Deacetylase 6 in Cancer. J. Hematol. Oncol. 11 (1), 111. 10.1186/s13045-018-0654-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X., Zang J., Li X., Tang S., Huang M., Geng M., et al. (2019). Discovery of Novel Janus Kinase (JAK) and Histone Deacetylase (HDAC) Dual Inhibitors for the Treatment of Hematological Malignancies. J. Med. Chem. 62 (8), 3898–3923. 10.1021/acs.jmedchem.8b01597 [DOI] [PubMed] [Google Scholar]
- Luan Y., Li J., Bernatchez J. A., Li R. (2019). Kinase and Histone Deacetylase Hybrid Inhibitors for Cancer Therapy. J. Med. Chem. 62 (7), 3171–3183. 10.1021/acs.jmedchem.8b00189 [DOI] [PubMed] [Google Scholar]
- Ma R., Xu L., Qu X., Che X., Zhang Y., Fan Y., et al. (2018). AZ304, a Novel Dual BRAF Inhibitor, Exerts Anti-tumour Effects in Colorectal Cancer Independently of BRAF Genetic Status. Br. J. Cancer 118 (11), 1453–1463. 10.1038/s41416-018-0086-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madorsky Rowdo F. P., Barón A., Gallagher S. J., Hersey P., Emran A. A., Von Euw E. M., et al. (2020). Epigenetic Inhibitors Eliminate Senescent Melanoma BRAFV600E Cells that Survive Long-Term BRAF Inhibition. Int. J. Oncol. 56 (6), 1429–1441. 10.3892/ijo.2020.5031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O., Kuzmickas R., Manchester H. E., Emerson C. E., Gavin A. G., Guild C. J., et al. (2019). MAPK Pathway Suppression Unmasks Latent DNA Repair Defects and Confers a Chemical Synthetic Vulnerability in BRAF-, NRAS-, and NF1-Mutant Melanomas. Cancer Discov. 9 (4), 526–545. 10.1158/2159-8290.cd-18-0879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P. A., Breslow R. (2007). Dimethyl Sulfoxide to Vorinostat: Development of This Histone Deacetylase Inhibitor as an Anticancer Drug. Nat. Biotechnol. 25 (1), 84–90. 10.1038/nbt1272 [DOI] [PubMed] [Google Scholar]
- Mauri G., Bonazzina E., Amatu A., Tosi F., Bencardino K., Gori V., et al. (2021). The Evolutionary Landscape of Treatment for BRAF(V600E) Mutant Metastatic Colorectal Cancer. Cancers 13 (1), 137. 10.3390/cancers13010137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mer A. S., Ba-Alawi W., Smirnov P., Wang Y. X., Brew B., Ortmann J., et al. (2019). Integrative Pharmacogenomics Analysis of Patient-Derived Xenografts. Cancer Res. 79 (17), 4539–4550. 10.1158/0008-5472.can-19-0349 [DOI] [PubMed] [Google Scholar]
- Millard C. J., Watson P. J., Celardo I., Gordiyenko Y., Cowley S. M., Robinson C. V., et al. (2013). Class I HDACs Share a Common Mechanism of Regulation by Inositol Phosphates. Mol. Cell 51 (1), 57–67. 10.1016/j.molcel.2013.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y., Keusch J. J., Wang L., Saito M., Hess D., Wang X., et al. (2016). Structural Insights into HDAC6 Tubulin Deacetylation and its Selective Inhibition. Nat. Chem. Biol. 12 (9), 748–754. 10.1038/nchembio.2140 [DOI] [PubMed] [Google Scholar]
- Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., et al. (2009). AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 30 (16), 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M. H. M., Søndergaard C. R., Rostkowski M., Jensen J. H. (2011). PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 7 (2), 525–537. 10.1021/ct100578z [DOI] [PubMed] [Google Scholar]
- Palušová V., Renzová T., Verlande A., Vaclová T., Medková M., Cetlová L., et al. (2020). Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds. Cancers 12 (6), 1516. 10.3390/cancers12061516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S.-B., Henry J. R., Kaufman M. D., Lu W.-P., Smith B. D., Vogeti S., et al. (2015). Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 28 (3), 384–398. 10.1016/j.ccell.2015.08.002 [DOI] [PubMed] [Google Scholar]
- Pinzi L., Foschi F., Christodoulou M. S., Passarella D., Rastelli G. (2021). Design and Synthesis of Hsp90 Inhibitors with B-Raf and PDHK1 Multi-Target Activity. ChemistryOpen 10 (12), 1177–1185. 10.1002/open.202100131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos P. I., Zhang C., Bollag G., Shokat K. M., Rosen N. (2010). RAF Inhibitors Transactivate RAF Dimers and ERK Signalling in Cells with Wild-type BRAF. Nature 464 (7287), 427–430. 10.1038/nature08902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulya S., Amin S. A., Adhikari N., Biswas S., Jha T., Ghosh B. (2021). HDAC6 as Privileged Target in Drug Discovery: A Perspective. Pharmacol. Res. 163, 105274. 10.1016/j.phrs.2020.105274 [DOI] [PubMed] [Google Scholar]
- Puszkiel A., Noé G., Bellesoeur A., Kramkimel N., Paludetto M.-N., Thomas-Schoemann A., et al. (2019). Clinical Pharmacokinetics and Pharmacodynamics of Dabrafenib. Clin. Pharmacokinet. 58 (4), 451–467. 10.1007/s40262-018-0703-0 [DOI] [PubMed] [Google Scholar]
- Rheault T. R., Stellwagen J. C., Adjabeng G. M., Hornberger K. R., Petrov K. G., Waterson A. G., et al. (2013). Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med. Chem. Lett. 4 (3), 358–362. 10.1021/ml4000063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Martins D., Forli S., Ramos M. J., Olson A. J. (2014). AutoDock4Zn: An Improved AutoDock Force Field for Small-Molecule Docking to Zinc Metalloproteins. J. Chem. Inf. Model. 54 (8), 2371–2379. 10.1021/ci500209e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreafico M., Gruszka A. M., Valli D., Mazzola M., Deflorian G., Quintè A., et al. (2020). HDAC8: A Promising Therapeutic Target for Acute Myeloid Leukemia. Front. Cell Dev. Biol. 8, 844. 10.3389/fcell.2020.00844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger I. C., Baum A., Steurer S., Stadtmüller H., Bader G., Schaaf O., et al. (2016). A Novel RAF Kinase Inhibitor with DFG-Out–Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol. Cancer Ther. 15 (3), 354–365. 10.1158/1535-7163.mct-15-0617 [DOI] [PubMed] [Google Scholar]
- Wellbrock C., Karasarides M., Marais R. (2004). The RAF Proteins Take Centre Stage. Nat. Rev. Mol. Cell Biol. 5 (11), 875–885. 10.1038/nrm1498 [DOI] [PubMed] [Google Scholar]
- Yao T.-P., Seto E. (2011). Histone Deacetylases: The Biology and Clinical Implication, 206. Berlin, Heidelberg: Springer Science & Business Media. 10.1007/978-3-642-21631-2 [DOI] [Google Scholar]
- Zhang L., Zhang J., Jiang Q., Zhang L., Song W. (2018). Zinc Binding Groups for Histone Deacetylase Inhibitors. J. Enzyme Inhib. Med. Chem. 33 (1), 714–721. 10.1080/14756366.2017.1417274 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.