

Abstract

In the context of cross-coupling chemistry, the competition between the cross-coupling path itself and the oxidative homocoupling of the nucleophile is a classic issue. In that case, the electrophilic partner acts as a sacrificial oxidant. We investigate in this report the factors governing the cross- versus homocoupling distribution using aryl nucleophiles ArMgBr and (hetero)aryl electrophiles Ar′Cl in the presence of an iron catalyst. When electron-deficient electrophiles are used, a key transient heteroleptic [Ar2Ar′FeII]− complex is formed. DFT calculations show that an asynchronous two-electron reductive elimination follows, which governs the selective evolution of the system toward either a cross- or homocoupling product. Proficiency of the cross-coupling reductive elimination strongly depends on both π-accepting and σ-donating effects of the FeII-ligated Ar′ ring. The reactivity trends discussed in this article rely on two-electron elementary steps, which are in contrast with the usually described tendencies in iron-mediated oxidative homocouplings which involve single-electron transfers. The results are probed by paramagnetic 1H NMR spectroscopy, experimental kinetics data, and DFT calculations.
Keywords: iron catalysis, cross-coupling, homocoupling, two-electron processes, kinetics, mechanisms
Introduction
In the field of transition-metal-catalyzed transformations, Fe-mediated cross-couplings have been intensely developed in the last few decades, thanks to the pioneer work of Kochi,1,2 Cahiez,3 Fürstner,4,5 Nakamura,6,7 and Bedford.8−10 Thanks to its abundance and its good eco-compatibility, this cheap metal led to a significant breakthrough in transition-metal catalysis.11−14 Moreover, its rich redox chemistry (with a formal oxidation state panel ranking from Fe–II to Fe+VI) opens the way to a huge variety of reactivity patterns, involving both one- and two-electron redox chemistry.15 From a synthetic standpoint, the classic procedures of Fe-mediated couplings between Grignard reagents and organic halides developed by Cahiez and later on by Fürstner are particularly appealing because they allow the obtention of high yields using simple ferrous or ferric salts as catalysts in the absence of additional ligands, with THF/NMP solvent mixtures [FeXn, Fe(acac)n (X = Br, Cl; n = 2, 3)].3,4
However, the use of simple iron salts as catalysts also leads in several cases to a broad distribution of byproducts. When aryl nucleophiles are used as coupling partners, notable quantities of homocoupled bisaryls can also be formed (Scheme 1a).8 Formation of this side product thus hampers a full conversion of the reactant, limiting the possible extension of those methods to industrial processes. A fine understanding of the redox events undergone by the iron catalyst during the catalytic process is thus highly desirable to finally more efficiently control the factors governing the formation of homocoupling byproducts.
Scheme 1. (a) Distribution of Cross- and Homocoupled Products in an Aryl–Alkyl System Developed by Bedford; (b) General Scheme of the Cross-Coupling (i) and Homocoupling (ii) Catalytic Cycles Relying on a One-Electron Process Involving the FeII/FeIII Couple; (c) Off-Cycle Homocoupling Process in an Aryl–Alkyl Coupling System Mediated by the Fe0/FeII Couple (Chp = Cycloheptyl; (P,P) = SciOPP = 1,2-C6H4((3,5-C6H3tBu2)2P)2); and (d) Representative Example of a Cross- versus Homocoupling Competition Depending on the Nature of the Electrophilic Partner.
From a mechanistic standpoint, the active iron oxidation state in a cross-coupling cycle strongly depends on the nature of the coupling partners. For example, organoiron(II) intermediates proved to be often highly reactive toward alkyl halides, initiating the coupling cycle by a single electron-transfer (SET) step. The catalytic cycle thus features a FeII/FeIII redox couple (Scheme 1b).14 In that case, the oxidation of an on-cycle organoiron(II) intermediate by the electrophilic partner R′–X affords a transient organoiron(III) species along with radical R′•. A radical rebound of those species allows one to close the cross-coupling cycle (Scheme 1b, path i). On the other hand, homocoupling of the nucleophile can also occur from the organoiron(III) intermediate in a second catalytic process, in which the organic halide acts as a sacrificial oxidant (path ii). Alternatively, multiple transmetallations of the nucleophile onto on-cycle FeII species can also lead to the sacrificial homocoupling of the former along with a Fe0 complex (Scheme 1c). In that case, the organic electrophile acts as a sacrificial oxidant allowing the regeneration of the FeII precatalyst from the reduced Fe0 species. This mechanism has been probed by Neidig in the aryl–alkyl cross-coupling using a diphosphino-ligated (P,P)FeIICl2 as resting state, a system in which a bis-aryliron(II) intermediate (P,P)FeII(Ph)2 undergoes a two-electron reductive elimination leading to the formation of a biphenyle molecule ligated to a Fe0 center. The latter species then reaffords the precatalyst (P,P)FeIICl2 after oxidation by the electrophilic partner (herein Chp-Br, bromocycloheptane), this step mostly affording the β-elimination product (cycloheptene).16 It must also be stated that in the absence of a well-defined exogenous ligand, several multinuclear species formed by reduction of Fe(acac)3 with PhMgBr were structurally characterized, such as the ferrous dinuclear complex [Mg(acac)(THF)4]2[FePh2(μ-Ph)]2·4THF as well as the tetranuclear species Fe4(μ-Ph)6(THF)4. The latter cluster is a rare example of a reduced iron species (with an average oxidation state lower than +II) displaying a catalytic activity in a cross-coupling involving aliphatic halides.17
In terms of harsh competitions between the cross-coupling path and the off-cycle homocoupling process, the example of the aryl–aryl cross-coupling series is particularly illustrative. Selective formation of aryl–aryl cross-coupling products has remained a challenge for a long time, the bisaryl originating from the homocoupling of the nucleophilic partner being often obtained as the major compound. This point was described by Kharasch during the 1940s in a series of reports, showing that aryl halides with a significantly high oxidative power acted as sacrificial oxidants in transition-metal-promoted Grignard oxidative homocoupling. Simple halide salts such as FeCl3, CoCl2 or NiCl2 were used as catalysts for those transformations.18 This point was later on confirmed by Fürstner, who reported efficient aryl–heteroaryl couplings involving heteroaryl chlorides as coupling partners, procedures which are difficultly applicable to more easily reduced substrates such as electron-poor aryl chlorides, for example, methyl 4-chlorobenzoate (in the latter case, the oxidative homocoupling of the Grignard reagent is the preferred path).4 This representative example is detailed in Scheme 1d. Among the scarce successful reported iron-mediated cross aryl–aryl bond formations, Knochel reported that the use of arylcopper nucleophiles, weaker than their Mg-based analogues, and aryl iodides substituted by electron-withdrawing groups associated with a Fe(acac)3 catalyst mostly led to the expected cross-coupling product.19 Nakamura reported a more general procedure allowing the suppression of the Grignard bisaryl in aryl–aryl cross-coupling systems using FeF3 as a catalyst combined with a N-heterocyclic Carbene (NHC) ligand.20 Similarly, Duong described an association of NHC ligands and alkoxide salts leading to efficient Fe-catalyzed aryl–aryl cross-couplings.21
The competition between cross-coupling and Grignard oxidative homocoupling paths promoted by transition-metal catalysts in the presence of strongly oxidant aryl halides has been well documented since the work of Kharasch. Under reducing conditions, it has been demonstrated in several cases that low-valent iron complexes acted as one-electron reductants of Ar–X bonds (X = Cl, Br, I) in single electron-transfer (SET) steps, leading to the formation of the corresponding C–C-coupled products by a formal radical dimerization. Bis(2-halophenyl)methylamines (in the chloro and bromo series) were thus converted into the corresponding carbazole using strongly reductive iron ate complexes generated by action of MeLi onto iron precursors in the presence of Mg metal.22 However, much less is known regarding the reactivity trends of similar systems involving less reducing iron intermediates, which difficultly promote single electron transfers. In a recent work, we demonstrated that (hetero)aryl halides Ar′Cl such as C6F5Cl and 2-chloropyridine (2-PyCl) could be activated by transient Fe0 complexes in a two-electron oxidative addition process in the presence of aryl Grignard reagents ArMgBr to afford well-defined heteroleptic species [Ar2Ar′FeII]−.23 In that case, the reactive Fe0 intermediate is generated by reduction of iron precursors using an excess of PhMgBr. This bielectronic activation is particularly interesting in the case of C6F5Cl, whose first reduction potential (Ered = −2.05 V vs SCE)24 is close to that of other aryl chlorides (e.g., ethyl 4-chlorobenzoate, Ered = −2.02 V vs SCE),25 which traditionally act as monoelectronic sacrificial oxidants.1−10,22 Owing to its more negative reduction potential (Ered = −2.37 V vs SCE),25 2-PyCl displays a less pronounced tendency to undergo one-electron reduction events.
This prompted us to investigate the mechanistic aspects of classic aryl–aryl bond formation systems following this two-electron reactivity pattern, with a particular focus on the factors governing the competition between the formation of the cross-coupling (cc) and the homocoupling (hc) products. We demonstrate in this work that the reactivity of both C6F5Cl and 2-PyCl in the presence of aryl nucleophiles and of an iron catalyst is driven by two-electron processes, regardless of the preferred path (selective oxidative homocoupling of the aryl nucleophile in the presence of C6F5Cl or possible formation of a cross-coupling product using 2-PyCl). The selectivity displayed by the system for one path or the other is on the other hand strongly influenced by the nature of the electronic effects governing the reactivity of the Ar/Ar′ couple. Those results are sustained by experimental kinetics experiments as well as by DFT calculations.
Results and Discussion
Oxidative Homocoupling in the Presence of an Aryl Halide as a Sacrificial Oxidant
Iron-catalyzed oxidative homocoupling of aromatic Grignard reagents can be performed in excellent yields via several procedures, which require sacrificial oxidants. Some of us already described in the past an iron-catalyzed oxidative homocoupling reaction using atmospheric oxygen (Scheme 2).26
Scheme 2. Oxidative Homocoupling of an Aryl Grignard Reagent Mediated by FeCl3 in the Presence of O2 as a Sacrificial Oxidant.

The methodology described in Scheme 2 is thus a very convenient way to prepare symmetric bisaryls from the corresponding Grignard reagents (ArMgBr) using a cheap and abundant sacrificial oxidant. From a mechanistic standpoint, first, a one-electron reduction of the FeIII precursor by ArMgBr usually occurs leading to the formation of the FeII oxidation state.27 The final bisaryl is then obtained after a two-electron reductive elimination of a transient in situ-generated organoiron(II) species with a transmetallation degree Ar: Fe > 1, such as a ate [Ar3FeII]− complex.28 The reduced iron species obtained after this step is then reoxidized at the FeII stage by the sacrificial oxidant (O2 herein), which initiates a new catalytic cycle.
In a cross-coupling context though, the classic methodology consists in promoting a C–C bond formation between an organometallic nucleophile (e.g., ArMgBr in the aromatic series) and an organic electrophile (usually a halide or a pseudohalide). As outlined in the introduction of this article, bisaryls Ar–Ar are commonly obtained as side products in a parallel catalytic homocoupling process, the organic halide playing likely the role of a monoelectronic sacrificial oxidant. The reactivity of C6F5Cl and 2-PyCl toward a scope of aryl Grignard reagents in the presence of an iron catalyst [FeCl3 or Fe(acac)3] was then examined and a particular focus was put on the analysis of the cross-coupling versus homocoupling ratio. As outlined in the introduction, the choice of those substrates was motivated by their ability to undergo two-electron activation processes. FeCl3 and Fe(acac)3 were chosen for this benchmark work because they are among the most used ferric precatalysts in Fe-mediated coupling chemistry. Both were moreover used successfully by Fürstner or Cahiez in several coupling systems.3,4 Very poor yields are obtained in cross-coupling attempts using C6F5Cl (Table 1), and the oxidative homocoupling of the nucleophile is observed, almost quantitatively in some cases (2a, entry 2). On the other hand, the use of 2-PyCl as a coupling partner resulted in a more productive cross-coupling pathway because up to 22% of cross-coupling product 6b is obtained when p-Me2N–C6H4MgBr is used as a nucleophile (Table 1, Entry 6). The stark contrast between the reactivity of C6F5Cl and that of 2-PyCl can hardly be solely explained on the basis of the difference between their reduction potentials, which are quite close (vide supra). In other words, one cannot expect that C6F5Cl behaves exclusively as a classic sacrificial oxidant while 2-PyCl would act as a more efficient coupling partner.
Table 1. Iron-Catalyzed Coupling of Aryl Grignard Reagents with C6F5Cl or 2-PyCl [Isolated Yields; Non-Isolated Products Were Detected as Minor Peaks by GC–MS Analysis (<5%)].

| Ar–Ar | Ar–Ar′ | Ar–Ar | Ar–Ar′ | ||
|---|---|---|---|---|---|
| entry | Ar | Ar′ = C6F5; conditions i: C6F5Cl, ArMgBr 1 M (1.2 equiv), FeCl3 3 mol %, THF, 20 °C, 4 h | Ar′ = 2-Py; conditions ii: 2-PyCl, ArMgBr 1 M (1.2 equiv), Fe(acac)3 5 mol %, THF, 0 °C, 3 h | ||
| 1 | Ph | 87 (1a) | traces | 26 (1a) | 8 (1b) |
| 85a (1a) | traces | n.d. | 9b (1b) | ||
| 2 | p-Me-C6H4 | 94 (2a) | traces | 29 (2a) | 13 (2b) |
| 3 | m-Me-C6H4 | 28 (3a) | 11 (3b) | ||
| 4 | p-MeO-C6H4 | 88 (4a) | traces | 32 (4a) | 16 (4b) |
| 5 | o-MeO-C6H4 | 75 (5a) | traces | ||
| 6 | p-Me2N-C6H4 | 85 (6a) | traces | 22 (6a) | 22 (6b) |
| 7 | p-CF3-C6H4 | 48 (7a) | traces | ||
| 8 | p-F-C6H4 | 22 (8a) | 3 (8b) | ||
| 9 | 2-mesityl | 20 (9a) | traces | ||
| 10 | 1-naphthyl | 70 (10a) | traces | ||
| 11 | 2-naphthyl | 90 (11a) | traces | ||
Conditions ii were used.
In the presence of 5 equiv TEMPO per mole of iron.
In contrast with its reduction potential, which should allow an activation by single electron transfer (Ered = −2.05 V vs SCE), C6F5Cl did not lead to the formation of detectable quantities of homocoupling product C6F5–C6F5 arising from the recombination of C6F5• radicals. This behavior is in stark contrast with what was reported for an important number of cross-couplings involving other aryl halides, which proved to undergo a SET step to afford the corresponding radical R′• followed by dimerization of the latter.22 Noteworthy, activation of organic halides by a SET (followed by homodimerization of the corresponding radical) promoted by iron complexes under reducing conditions is also widely described when aliphatic electrophiles are used. Alkyl radicals are indeed more easily formed than their sp2 analogues, due to the weaker bond dissociation energy of the former.29 It was, for example, demonstrated that 1,2-dichloroisobutane (DCIB) acted as a one-electron sacrificial oxidant in Nakamura’s Fe-mediated C–H functionalization systems within a FeII/FeIII/FeI two-step redox sequence. In that case, the key formation of a cross-coupled product is allowed by monoelectronic oxidation of a transient bis-hydrocarbyliron(II) intermediate to the FeIII stage, a fast reductive elimination occurring in the latter. A FeI species is finally obtained, again oxidized by DCIB in a one-electron process.30,31 A similar FeII/FeIII/FeI sequence has been reported by Deng for a bis-phenyl FeII complex stabilized by N-heterocyclic carbenes (NHCs), (IPr2Me2)2FeII(Ph)2, which generates the corresponding bisaryl Ph–Ph upon single-electron oxidation by a ferrocenium salt followed by a two-electron reductive elimination. In that case, the corresponding FeI complex is trapped in situ by PMe3 to afford the 15-electron adduct [(IPr2Me2)2FeI(PMe3)2]+.32
In the system discussed herein though, no monoelectronic oxidation of ate [Ar3FeII]− intermediates by C6F5Cl occurs to induce formation of Ar–Ar by a subsequent FeIII-to-FeI reductive elimination, as attested by our previous studies (Ar = Mes).23 From a mechanistic standpoint, it is known that FeII or FeIII precursors such as FeCln (n = 2, 3) or Fe(acac)3 quickly afford ate ferrous species such as [Ar3FeII]− in the presence of an excess of ArMgBr (Ar = Ph,28,33 Mes10). Those complexes then evolve in the absence of a stabilizing co-ligand to lower Fe0 and FeI oxidation states, the zerovalent Fe0 species being predominant (ca. 85% of the iron distribution),27 and transiently stabilized by arene ligation with suitable species (toluene co-solvent, or biphenyl formed by oxidation of PhMgBr). This arene-stabilized complex (η4-arene)2Fe0 finally evolves to non-reactive aggregates. Among this distribution of FeII, FeI, and Fe0 intermediates obtained in the reaction medium, we recently demonstrated that the more reduced one was the only active species toward the C6F5Cl or 2-PyCl electrophiles (Scheme 3).
Scheme 3. Evolution of Transient ate-FeII Complexes toward Fe0 and FeI Intermediates; the Fe0 Promotes the Two-Electron Activation of Electron-Poor Electrophiles (C6F5Cl or 2-PyCl).

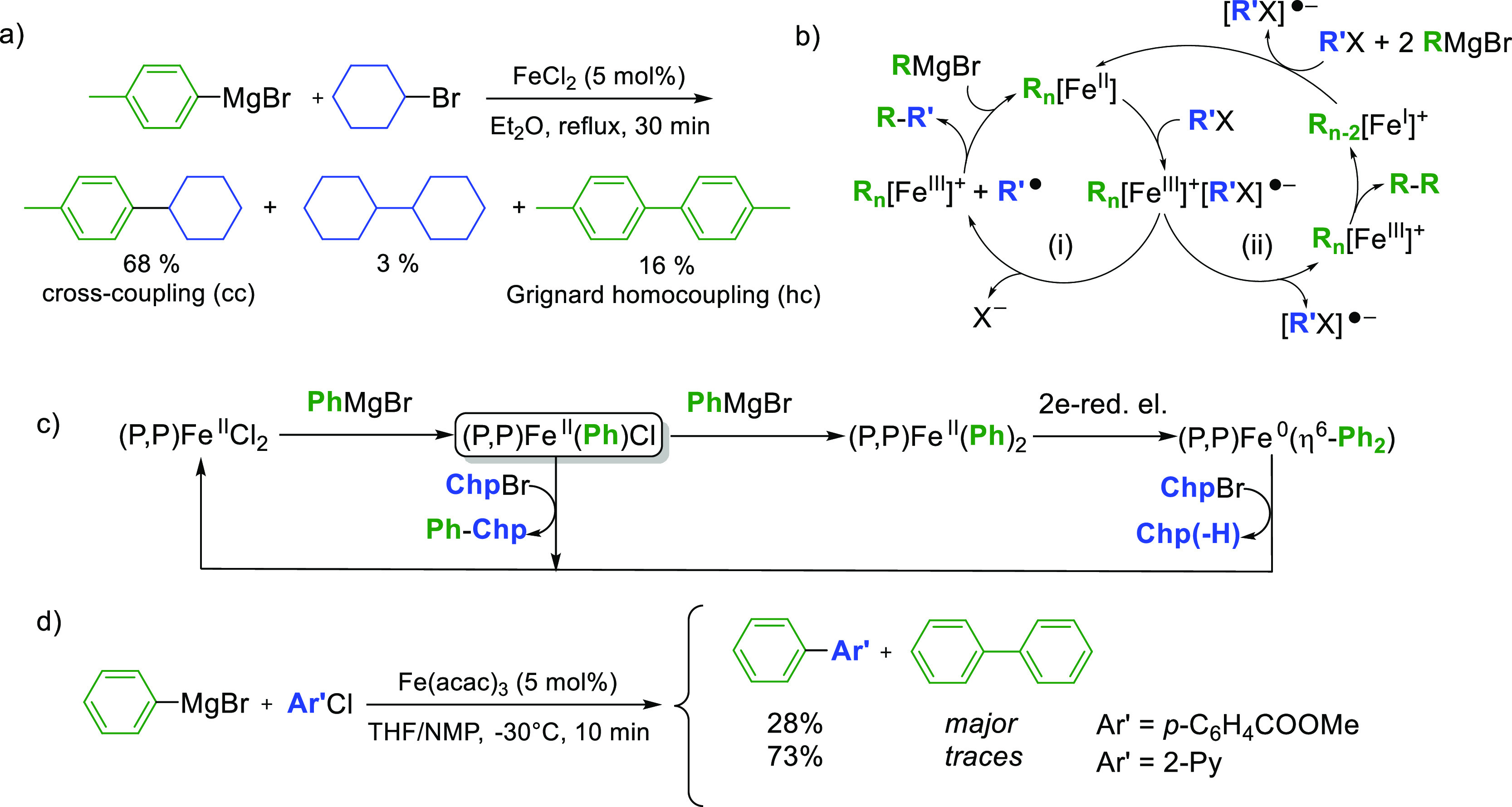
This led to the observation of heteroleptic adducts [Ar2Ar′FeII]− (Ar = Mes, Ar′ = C6F5 or 2-Py) by 1H and 19F paramagnetic NMR, those complexes being formed by a two-electron oxidative addition between Fe0 and the electrophilic partner Ar′X.23 In line with the occurrence of this bielectronic activation, the yield of the cross-coupling between PhMgBr and 2-PyCl is not affected by the presence of a radical scavenger (TEMPO herein, see Table 1, entry 1), suggesting that the coupling mechanism does not rely on monoelectronic steps. Therefore, the formation of the Fe0 oxidation state is crucial to the proficiency of the two-electron oxidative addition allowing the activation of Ar′X. Thus, we first confirmed that in situ reduction of classic iron precursors (FeCl3 and FeCl2) by a variety of aromatic Grignard reagents occurred efficiently. As shown below (Table 2), the addition of an excess (10 equiv) of various aryl magnesium bromides [substituted by either electron-donating (entries 2 and 3) or electron-withdrawing groups (entry 4)] to a solution of FeCl3 in THF leads to 1.4–1.6 equiv of biaryl with a short 5 min reaction time. This is consistent with the average reduction of FeIII to the Fe0 stage, with an overall transfer of three electrons per mole of starting iron precursor. In a similar way, FeCl2 (associated or not to LiCl to circumvent solubility issues) is reduced to the Fe0 oxidation state, showing that the formation of the latter can be achieved in those conditions regardless of the electronic properties of the aryl Grignard reagent. Some of us demonstrated that reduction of FeCl3 or Fe(acac)3 by an excess of PhMgBr also afforded a minor ate-FeI complex (ca. 10–15% of the overall iron quantity), [(η6-arene)FeI(Ph)2]− (arene = toluene when used as a co-solvent,27 or Ph–Ph formed by oxidation of the nucleophile34). Analysis of the reaction medium by X-band EPR spectroscopy also revealed that the reduction of Fe(acac)3 by several aryl Grignard reagents (ArMgBr) in conditions of Table 2 also afforded similar low-spin FeI intermediates at low concentrations (S = 1/2, see Figure 1a for Ar = Ph (g = 2.206; 2.021; 1.999) and Supporting Information for Ar = p-MeO-C6H4 and p-F-C6H4). FeI oxidation state indeed represents ca. 5.4% of the overall iron quantity after the reduction of Fe(acac)3 by PhMgBr, and 9.6% when p-F-C6H4MgBr is used. Traces of FeI species (0.2% of the iron quantity) are detected upon the reduction by p-MeO-C6H4MgBr. In all cases, formation of the homocoupling bisaryl product Ar–Ar as well as of the Fe0 oxidation state as a major product upon reduction of the iron precursor by the aryl Grignard reagent ensures a first catalyst turnover in the homocoupling process.
Table 2. Reduction of Iron Salts FeCln (n = 2 or 3) by Several Aryl Grignard Reagents Leading to the Corresponding Bisaryls; Reactions Performed on a 13 mmol Scale.

| ArAr vs Fe |
||||
|---|---|---|---|---|
| entry | ArMgBr | FeCl3 | FeCl2 | % [FeI] (vs total [Fe])b |
| 1 | PhMgBr | 1.4 equiv (1a) | 1.1 equiv (1a) | 5.4 |
| 2 | p-Me-C6H4MgBr | 1.5 equiv (2a) | 1 equiva (2a) | n.d. |
| 3 | p-MeO-C6H4MgBr | 1.4 equiv (4a) | 0.2 | |
| 4 | p-F-C6H4MgBr | 1.6 equiv (8a) | 9.6 | |
The same result was obtained from FeCl2·2LiCl.
Speciation of low-spin EPR-active FeI formed upon reduction of Fe(acac)3 by several Grignard reagents (15 equiv vs Fe).
Figure 1.
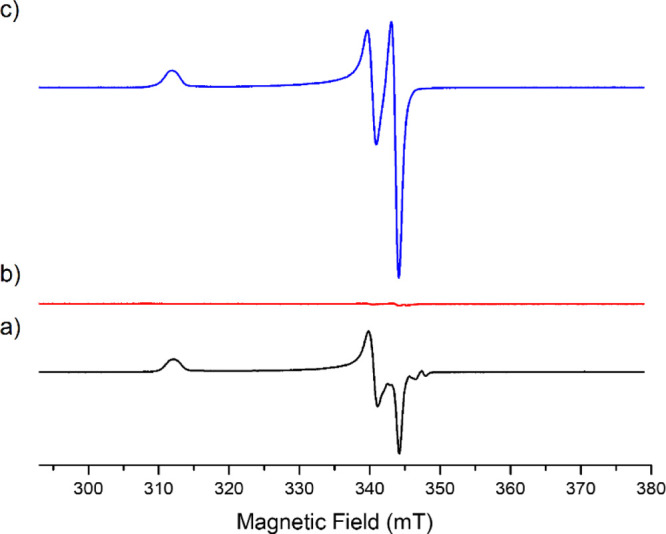
X-band EPR analysis (T = 90 K) of a solution of (a) Fe(acac)3 (9 mM in a 98:2 THF/2-MeTHF mixture) treated by 15 equiv PhMgBr and (b,c): the same, after addition of resp. C6F5Cl or 2-PyCl (10 equiv vs Fe); samples frozen after a 10 min reaction time at room temperature.
Reactivity of the minor FeI oxidation state toward C6F5Cl and 2-PyCl has also been monitored by EPR spectroscopy (Figure 1b,c). As discussed above, reduction of a solution of Fe(acac)3 by 15 equiv PhMgBr affords 5.4% of [(η6-PhPh)FeI(Ph)2]− (vs total iron concentration). The addition of 10 equiv C6F5Cl on this solution led to the disappearance of the FeI signal (Figure 1b), suggesting a possible electron transfer between those two species. It is however difficult to delineate the nature of the organic products formed by the reaction of [(η6-PhPh)FeI(Ph)2]− with C6F5Cl because the former complex only represents a small quantity of the overall iron distribution. However, the genuine reactivity of [(η6-PhPh)FeI(Ph)2]− toward a variety of organic halides has already been described by Hu34 who reported that it mostly led to the homocoupling Ph–Ph product along with small amounts of cross-coupling. On the other hand, the FeI oxidation state is not affected by the addition of 10 equiv 2-PyCl, attesting to the absence of electron transfer between those two species. This result is in line with the respective reduction potentials of C6F5Cl and 2-PyCl, the latter being less easily reduced. A slight alteration of the symmetry of the FeI signal is observed (Figure 1c), which might be due to the formation of a new species involving 2-PyCl as a ligand to the FeI ion. Similar trends are observed regarding the reactivity of the FeI species generated by the reduction of Fe(acac)3 with p-F-C6H4MgBr toward C6F5Cl and 2-PyCl (see Figure S3). The evolution of the distribution of FeI species formed by reduction of Fe(acac)3 with p-MeO-C6H4MgBr after the reaction with 2-PyCl is more unclear (Figure S4), but this oxidation state does not represent more than 1.5% of the overall iron quantity. In the next section, the productive pathways followed in the cross- and homocoupling reactions involving the major Fe0 species obtained by the reduction of the iron precursor are discussed.
Fate of the C6F5 and 2-Py Groups in the Catalytic Process
Because only traces of cross-coupling products Ar–C6F5 are detected (Table 1) in spite of the evidence of the formation of C6F5-[FeII] species upon activation of the C6F5–Cl bond, the fate of the C6F5 group in the overall process was then investigated. When p-MeO-C6H4MgBr and C6F5Cl (in a 1:0.55 ratio) were used as coupling reagents, an electrophilic quench of the medium by octanoic anhydride shows a very good 88% formation yield of the expected (4,4′)-bisanisyl homocoupling product 4a, along with 55% of ketone n-C7H15C(O)C6F5 (12, Scheme 4). In other words, the C6F5 group is catalytically released in the reaction medium as its two-electron reduced anion C6F5–—the presence of MgII cations moreover probably triggers the transfer of the C6F5– anion from the iron center to afford the corresponding Grignard, C6F5MgBr. The latter is afterward quantitatively trapped by electrophilic quenching using octanoic anhydride.35
Scheme 4. Electrophilic Quench of the C6F5– Anion after Its Release in the Reaction Medium.

The quantitative electrophilic quenching of the C6F5– anion at the end of the attempt of cross-coupling between p-MeO-C6H4MgBr and C6F5Cl suggests that the FeII-ligated C6F5– anion formed by the oxidative addition of Fe0 onto C6F5Cl is unreactive in the coupling catalytic process. The intrinsic stability of the C6F5-[FeII] bond has also been investigated by transmetallation of C6F5MgBr with an FeII precursor. Reaction of 2 equiv C6F5MgBr with FeCl2 led to the formation of a new organoiron(II) intermediate, characterized by a paramagnetic resonance at 283 ppm in 19F NMR (Figure 2a), attesting to the transmetallation of a C6F5– anion with a paramagnetic center. Further addition of an excess of MesMgBr (20 equiv vs Fe) after 3 h at 20 °C led to the sole observation of [Mes3FeII]− species in 1H NMR (characterized by two sharp downfielded peaks at 112 and 127 ppm in a 3:2 ratio, Figure 2b10) and to a 19F NMR silent spectrum. This shows that [Mes3FeII]− was formed by the substitution of the FeII-ligated C6F5– anions by their mesityl analogues. More importantly, this demonstrates that the addition of C6F5MgBr onto a FeII salt did not lead to the reduction of the ferrous ion, suggesting that the C6F5-[FeII] bond displays to a certain extent an appreciable thermal stability.
Figure 2.

(a) 19F NMR spectrum (377 MHz, THF d8) of a solution of FeCl2 treated by 2 equiv C6F5MgBr and (b) 1H NMR spectrum (400 MHz) recorded 3 h after the addition of 20 equiv MesMgBr at 20 °C.
The intermediate C6F5-[FeII] adduct generated by transmetallation of C6F5MgBr onto FeCl2 also proved to be stable at −10 °C up to 15 h because 88% of C6F5I is obtained by iodometric titration under those conditions. This stability is in stark contrast with other well-known Ar-[FeII] species (such as [Ph3FeII]−), which readily undergo decomposition toward lower Fe0 or FeI oxidation states along with either arenes Ar–H or bisaryls Ar–Ar by reductive elimination.28 Evolution of the C6F5-[FeII] adduct at 30 °C has further been monitored by iodolysis (Table 3). It is interesting to note that the decomposition of the latter occurs slowly at this temperature and mainly gives C6F5H as a reduction byproduct, rather than the bisaryl C6F5–C6F5.
Table 3. GC Monitoring (Internal Standard: Undecane) of the Decomposition Product Formed after Transmetallation of C6F5MgBr (2 equiv) onto FeCl2 in THF at 30 °C, after Iodolysis Quench.

| time (h) | C6F5H (%) | C6F5I (%) | C6F5–C6F5 (%) |
|---|---|---|---|
| 0.25 | 38 | 47 | 7 |
| 3 | 42 | 38 | 10 |
| 24 | 52 | 21 | 14 |
| 90 | 59 | 3 | 19 |
Those results overall demonstrate a reactivity of the C6F5-[FeII] bond significantly lower than that of a classic Ar-[FeII] compound, which usually follows much faster reductive pathways. A weak nucleophilicity is also observed for those C6F5-[FeII] adducts. Indeed, Mg-to-Fe transmetallation of C6F5MgCl onto FeCl2 (2 equiv per mole of iron) also results in the absence of detection of ketone C6F5C(O)(n-C7H15) (12) upon electrophilic quenching of the reaction medium by ester n-C7H15C(O)Et, whereas treatment of C6F5MgCl by this ester quantitatively affords the expected ketone 12 along with the tertiary alcohol (C6F5)2C(OH)(n-C7H15) in a global 92% yield. The C6F5-[FeII] bond thus shows a decreased nucleophilic reactivity compared to C6F5MgCl. In line with its greater reactivity as a cross-coupling partner, 2-PyCl only affords traces (<5%) of 2-PyI upon iodolysis quench of a coupling medium (see conditions in Table 1, entry 1). In other words, a much smaller quantity of the 2-Py– anion is released in the reaction medium compared to that of C6F5– anion. In order to rationalize the mechanistic facets of the cross versus homocoupling competition in those two-electron processes as well as the striking differences displayed by C6F5Cl and 2-PyCl, the evolution of the [Ar2Ar′FeII]− intermediate (Ar′ = C6F5 or 2-Py) in the catalytic medium was then investigated more precisely.
Key Two-Electron Reductive Elimination in Cross- and Homocoupling Pathways
Heteroleptic species [Ar2Ar′FeII]− is formed by the oxidative addition of a Fe0 intermediate onto Ar′Cl (Ar′ = C6F5 or 2-Py) in the presence of ArMgBr at early stages of the catalytic process (Scheme 3). This complex is likely involved as a key intermediate in both cross-coupling and homocoupling pathways through two-electron reductive elimination steps, as detailed in Scheme 5. Tris-coordinated (t) [Ar2Ar′FeII]− can indeed evolve toward the formation of either the cross-coupling Ar–Ar′ (path i) or homocoupling Ar–Ar (path ii) products. Alternatively, [Ar2Ar′FeII]− can undergo an additional transmetallation with an equivalent of ArMgBr28 leading to quaternized ate-FeII species (q) such as [Ar3Ar′FeIIMg(THF)]). Formation of quaternized adducts involving an ipso coordination of a Li+ cation were also reported earlier by Fürstner and structurally characterized.36 Such quaternized intermediates can also provide both cross-coupling or homocoupling products (following respectively paths iii and iv). [Ar3Ar′FeIIMg(THF)] can also release 1 equiv of Ar′MgBr, leading to the homoleptic species [Ar3FeII]−, which is solely able to afford the homocoupling product Ar–Ar by reductive elimination (path v).
Scheme 5. Formation of Cross-Coupling (cc) and Homocoupling (hc) Products by Two-Electron Reductive Elimination Occurring in Heteroleptic [Ar2Ar′FeII]− and [Ar3Ar′FeIIMg(THF)] and in Homoleptic [Ar3FeII]− Complexes; Mg = Mg(THF)2+.

Intermediates [Ar2Ar′FeII]−, [Ar3Ar′FeIIMg(THF)], and [Ar3FeII]− being accessible in usual coupling conditions,23,28 the feasibility of the reaction paths i–v was then investigated by DFT calculations (Ar = Ph and Ar′ = C6F5 or 2-Py). The corresponding computed activation energies are reported in Table 4. OPBE functional was used, associated with the following basis sets: 6-31+G* (C, H, O, N, Mg, F, and Br), SDD and pseudo-potential (Fe). The solvent (THF herein) was described using the PCM model. Similar trends were obtained using the more computationally costly Ahlrich’s basis set and pseudo-potential def2TZVPP for Fe (see Table S1). In those calculations, the tris-coordinated ate species [Ar2Ar′FeII]− and [Ar3FeII]− have been computed using a high-spin (S = 2) ground multiplicity, whereas the other structures (including the reductive elimination transition states for paths i–v) have been computed on the triplet spin surface (S = 1). Those choices have been motivated by previous reported results showing that those were the ground spin multiplicities of the analogous paths involving [Ph3FeII]− and [Ph4FeIIMgBr(THF)]− species (see the Supporting Information for the computational details and for full calculated surfaces of paths i and ii with Ar′ = 2-Py).28
Table 4. Thermal Activation Energies of the Cross- and Homocoupling Paths Discussed in Scheme 5; Energies Given in kcal·mol–1 with Respect to [Ar2Ar′FeII]− (Paths i and ii), [Ar3Ar′FeIIMg(THF)] (Paths iii and iv), or [Ar3FeII]− (path v).
| path i | path ii | path iii | path iv | path v | |
|---|---|---|---|---|---|
| ΔEcc,t‡ | ΔEhc,t‡ | ΔEcc,q‡ | ΔEhc,q‡ | ΔEhc,t′‡ | |
| Ar = Ph, Ar′ = C6F5 | 28.5 | 16.7 | 18.3 | 11.9 | 17.0 |
| Ar = Ph, Ar′ = 2-Py | 12.5 | 16.9 | 10.6 | 4.6 |
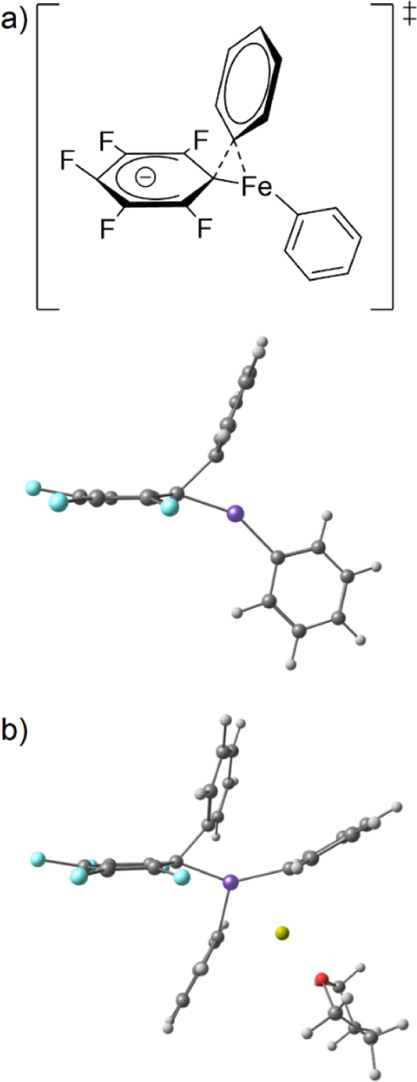
In all cases, the formation of the homocoupling product (PhPh) is favored when C6F5Cl is used as an electrophile. Regardless of the nature of the heteroleptic intermediate ([Ph2(C6F5)FeII]−, or [Ph3(C6F5)FeIIMg(THF)]), the activation energy of the cross-coupling (paths i or iii) is indeed much higher than that of the homocoupling (paths ii and iv). This is in line with the experimental results reported in Table 1, showing that only traces of cross-coupling products Ar–C6F5 are formed in such conditions. It is interesting to notice that the transition states computed on the cross-coupling surfaces for the formation of Ph-C6F5 by reductive elimination (paths i and iii) differ from the classic 3-center synchronous structure. Those transition states are more accurately described by the iron-to-carbon migration of the electron-rich phenyl anion onto the electron poor FeII-ligated C6F5 group. This mechanism is much similar to the first step of a SNAr process, involving the formation of a dearomatized Meisenheimer intermediate (Scheme 6a, top). This is attested in the computed structure of those transition states by a strong pyramidalization of the C atom in the para position of the C6F5 ring, with computed CipsoCparaF angles of resp. 166° and 167° in the tris- (path i) and tetra-coordinated (path iii) structures (Schemes 6a, bottom, and 6b). Accordingly, a negative charge develops at the Cpara atom within this migration: the computed Mulliken charge, for example, decreases from 0.1 |e| in [Ph2(C6F5)FeII]− to −0.3 |e| in the corresponding transition state. A similar evolution is observed in the tetracoordinated series (evolution from a 0.0 |e| charge on the Cpara atom of the C6F5 ring in [Ph3(C6F5)FeIIMg(THF)] to a more negative −0.2 |e| charge in the transition state).
Scheme 6. DFT-Computed Reductive Elimination Transition States of Ph-C6F5 from (a) [Ph2(C6F5)FeII]− and (b) [Ph3(C6F5)FeIIMg(THF)] on the Triplet (S = 1) Surface.
The situation is quite different in the coupling series involving 2-PyCl. [Ph2(2-Py)FeII]− indeed preferentially follows the cross-coupling path, whose activation energy is lower by 4.4 kcal·mol–1 with respect to that of the homocoupling (resp. path i and ii). Quaternized adduct [Ph3(2-Py)FeIIMg(THF)] evolves, on the other hand, more easily along the homocoupling path, akin to its C6F5-ligated analogue (comparison of paths iii and iv). The remarkably low activation barrier for the homocoupling path involving the quaternized species [Ph3(2-Py)FeIIMg(THF)] (4.6 kcal·mol–1) is due to a N-ligation of the iron center in the transition state, which does not occur in the cross-coupling reductive elimination (path iii: 10.6 kcal·mol–1, Table 4). Because the sole intermediate allowing the formation of the cross-coupling product requires a low Ph/Fe transmetallation degree (Ph/Fe = 2 in [Ph2(2-Py)FeII]−, whereas Ph/Fe = 3 in [Ph3(2-Py)FeIIMg(THF)]), this also explains why a slow addition of the Grignard reagent is crucial in those coupling systems. Indeed, a fast addition of this reagent would lead to an increased concentration of the quaternized species, which favors the formation of the homocoupled bisaryl [either directly (path iv) or after formation of the homoleptic species [Ph3FeII]− (path v)].
The reductive elimination of Ph–Ph from a tris-coordinated species moreover does not seem to be affected by the nature of the third ligand because the computed activation energy is quite the same (ca. 17 kcal·mol–1, paths ii and v, Table 4) starting from either [Ph2(C6F5)FeII]−, [Ph2(2-Py)FeII]−, or [Ph3FeII]−. Conversely, reductive elimination of the Ph-Ar′ cross-coupling product is strongly affected by the electronic properties of the Ar′ ring because it evolves from 12.5 kcal·mol–1 (Ar′ = 2-Py) to 28.5 kcal·mol–1 (Ar′ = C6F5) (Table 4). Similar observations can be made for the quaternized species [Ph3(Ar′)FeIIMg(THF)] (Ar′ = C6F5, 2-Py). When the coupling mechanism is dominated by two-electron processes, the reductive elimination leading to the cross-coupling product starting from heteroleptic species [Ar2Ar′FeII]− involves a migration of the electron-rich Ar group onto the electron-poor Ar′ moiety (the former being derived from the nucleophile ArMgBr, the later from the electrophile Ar′Cl), as shown in Scheme 6a. Existence of an energetically accessible antibonding π* system borne by the FeII-ligated Ar′− ligand is thus a first prerequisite to ensure the initiation of the reductive elimination on the cross-coupling path. Owing to their electron-poor character, both C6F5 and 2-Py rings fulfill this condition in the present study. However, completion of the reductive elimination process also requests in a second time an efficient transfer of the two electrons located onto the Ar′ ring in the transition state onto the FeII ion, allowing its reduction to the Fe0 stage. This requires a sufficient reductive power of the Ar′–[FeII] σ bond. Therefore, although the electron-poor character of the Ar′ group enables the transfer of the Ar ring thanks to an accessible π* system, it also translates into a decreased reducing power of the Ar′–[FeII] σ bond. The balance between these two opposite electronic requirements thus governs the strong discrepancy observed between C6F5Cl and 2-PyCl. The former is far too electron-poor to afford a reductive C6F5-[FeII] intermediate: no cross-coupling product is observed, and only byproducts derived from anion C6F5– are detected (C6F5H, C6F5–C6F5). On the other hand, 2-PyCl reaches a good compromise between its π-accepting effects and σ-donating properties of the 2-Py–[FeII] bond, overall allowing its use as a cross-coupling partner.
In the last section, the role of the electronic properties of the Ar ligand in the cross versus homocoupling competition has been investigated more closely.
Electronic Effects at Play in the Cross- versus Homocoupling Competition
Analysis of the cross-coupling (cc) versus homocoupling (hc) ratio depending on the electronic effects of the substituents borne by the Grignard reagent ArMgBr was performed using 2-PyCl as an electrophile (see Table 1). The cross-coupling ratio rcc (rcc = [cc]/([cc] + [hc])) was found to decrease with the value of the σ Hammett parameter of the nucleophilic partner, as shown in Figure 3a. This demonstrates that the cross-coupling pathway is more easily followed when electron-rich nucleophiles are used, rcc being higher for negative σ parameters (a maximum value of rcc = 0.5 is obtained when p-Me2N–C6H4MgBr is used).
Figure 3.

(a) Experimental evolution of the cross-coupling (cc) vs homocoupling (hc) ratio rcc for ArMgBr/2-PyCl coupling systems reported in Table 1 and (b) DFT-computed evolution of the activation free energy gap between the cross- and homocoupling paths undergone by [Ar2(2-Py)FeII]− intermediates.
This tendency has also been reproduced in silico (Figure 3b). DFT calculations indeed show that the gap between the computed free energy activations of the homo- and the cross-coupling paths involving ate [Ar2Ar′FeII]― species (paths i and ii in Scheme 5) increases when the Ar– anion is substituted with electron-donating substituents. Those results are consistent with the two-electron reductive elimination mechanism discussed above for Ar–C6F5 coupling products, with a key migration of the electron-rich Ar anion onto the electron-poor Ar′ ring: the electron-richer the Ar anion, the more efficient the migration. Overall, this also explains why the best match leading to a good cross-coupling versus homocoupling ratio in the aryl-heteroaryl series is obtained, for a given electrophile, using electron-rich nucleophiles.
In addition to the role played by the electronic effect of the substituents borne by the nucleophile on the kinetics trends of the reductive elimination, the nature of the nucleophile may also affect the preferred pathway (cross-coupling or homocoupling) at an earlier stage of the catalysis. Previous DFT calculations indeed suggested that the stable resting state of the Fe0 species generated in situ by reduction of the iron precursor (Scheme 7) may involve an aryl anion σ-bonded to the Fe0, by transmetallation of ArMgBr with the complex (η4-arene)2Fe0.23
Scheme 7. Formation of a Fe0 Resting State σ-Coordinated by an Ar– Anion Leading to [Ar2(2-Py)FeII]− after Oxidative Addition with 2-PyCl; Mg = MgBr(THF)+ (Model Structures Investigated by DFT Calculations).

Therefore, when electron-rich Grignard reagents ArMgBr are used as nucleophilic partners, the Fe0 resting state (η6-ArAr)Fe0(ArMgBr(THF)) displays an enhanced reducing power. Due to the electron richness of the iron center, a faster oxidative addition of intermediate Ar-[Fe0] onto 2-PyCl should occur, then leading to a higher dynamic concentration of the [Ar2(2-Py)FeII]− species and thus to a lower ArMgBr/[Ar2(2-Py)FeII]− ratio. Consequently, this also would slightly inhibit the quaternization process and therefore decrease the formation of the bisaryl Ar–Ar, given that the quaternized species selectively affords the homocoupling product (see paths iii and iv, Scheme 5, and Table 4).
Conclusions
Mechanistic patterns involved in iron-mediated cross-couplings are highly complex because the nature of the active species and of the elementary steps are strongly dependent on the coupling partners’ physical properties (hybridization, oxidoreduction potentials, ...). Therefore, generalities are drawn with difficultly from mechanistic studies carried out on specific substrates because no universal mechanism can describe this variety of transformations. When two-electron processes dominate the catalytic activity in aryl-(hetero)aryl couplings between ArMgBr nucleophiles and electron-poor Ar′Cl electrophiles, an heteroleptic complex [Ar2Ar′FeII]− can be formed by the oxidative addition of an Fe0 species onto the Ar′–Cl bond. This ate-complex is a key intermediate, which can evolve by two-electron reductive elimination along both cross- and homocoupling paths (directly or with the involvement of quaternized species such as [Ar3(Ar′)FeIIMg(THF)]). Such a bielectronic pattern is observed, for example, with Ar′–X substrates bearing an electron-poor Ar′ ring and a difficultly reduced Ar′–X bond (typically with X = Cl). A combination of those two electronic effects allows the two-electron mechanism to overcome the more usual monoelectronic reduction of the electrophilic partner in classic Kharasch-type Grignard oxidative homocouplings. Owing to an asynchronous reductive elimination mechanism involving a migration of the Ar– anion onto the Ar′ ring, the cross-coupling path is more favored compared to the homocoupling when electron-rich Ar nucleophiles are used. Those results also demonstrate that the competition between aryl–aryl cross-coupling and nucleophile oxidative homocoupling cannot be rationalized solely on the basis of the reduction potential of the electrophilic partner when the reactivity of the system is driven by two-electron elementary steps.
Acknowledgments
This work was financially supported by the joint program PheroChem (CNRS/M2i Company). The M2i Company is thanked for CIFRE PhD grants to E.Z. and P.C.; we also thank the “Fondation de la Maison de la Chimie” for a post-doctoral grant to N.L.; G.L. thanks the ERC (Project DoReMI StG, 852640) and the CNRS (Project IrMaCAR) for their financial support. The PSL Research University is thanked in frame of the NIMCOS project. The NMR shared facilities of Chimie ParisTech (Dr. M.-N. Rager) are thanked for the technical support. G.L. dedicates this article to the memory of G.C., who initiated this collaborative work in 2018.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.2c00002.
General procedures; 1H, 13C, and 19F NMR spectra; theoretical methods; and Cartesian coordinates of the computed structures (PDF)
Author Contributions
E.Z. and P.C. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Tamura M.; Kochi J. K. Vinylation of Grignard reagents. Catalysis by iron. J. Am. Chem. Soc. 1971, 93, 1487–1489. 10.1021/ja00735a030. [DOI] [Google Scholar]
- Neumann S. M.; Kochi J. K. Synthesis of olefins. Cross-coupling of alkenyl halides and Grignard reagents catalyzed by iron complexes. J. Org. Chem. 1975, 40, 599–606. 10.1021/jo00893a013. [DOI] [Google Scholar]
- Cahiez G.; Avedissian H. Highly Stereo- and Chemoselective Iron-Catalyzed Alkenylation of Organomagnesium Compounds. Synthesis 1998, 1998, 1199–1205. 10.1055/s-1998-2135. [DOI] [Google Scholar]
- Fürstner A.; Leitner A.; Méndez M.; Krause H. Iron-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc. 2002, 124, 13856–13863. 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]
- Martin R.; Fürstner A. Cross-Coupling of Alkyl Halides with Aryl Grignard Reagents Catalyzed by a Low-Valent Iron Complex. Angew. Chem., Int. Ed. 2004, 43, 3955–3957. 10.1002/anie.200460504. [DOI] [PubMed] [Google Scholar]
- Nakamura M.; Matsuo K.; Ito S.; Nakamura E. Iron-Catalyzed Cross-Coupling of Primary and Secondary Alkyl Halides with Aryl Grignard Reagents. J. Am. Chem. Soc. 2004, 126, 3686–3687. 10.1021/ja049744t. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Hashimoto T.; Kondo Y.; Fujiwara Y.; Seike H.; Takaya H.; Tamada Y.; Ono T.; Nakamura M. Iron-Catalyzed Suzuki–Miyaura Coupling of Alkyl Halides. J. Am. Chem. Soc. 2010, 132, 10674–10676. 10.1021/ja103973a. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Betham M.; Bruce D. W.; Danopoulos A. A.; Frost R. M.; Hird M. Iron–Phosphine, −Phosphite, −Arsine, and −Carbene Catalysts for the Coupling of Primary and Secondary Alkyl Halides with Aryl Grignard Reagents. J. Org. Chem. 2006, 71, 1104–1110. 10.1021/jo052250+. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Huwe M.; Wilkinson M. C. Iron-catalysed Negishi coupling of benzylhalides and phosphates. Chem. Commun. 2009, 600–602. 10.1039/b818961g. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Brenner P. B.; Carter E.; Cogswell P. M.; Haddow M. F.; Harvey J. N.; Murphy D. M.; Nunn J.; Woodall C. H. TMEDA in Iron-Catalyzed Kumada Coupling: Amine Adduct versus Homoleptic “ate” Complex Formation. Angew. Chem., Int. Ed. 2014, 53, 1804–1808. 10.1002/anie.201308395. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Brenner P. B.. The development of iron catalysts for cross-coupling reactions. Iron Catalysis II; Bauer E., Ed.; Springer Intl., 2015. [Google Scholar]; For complete reviews on Fe-catalyzed cross-couplings, see refs (12) and (13) and for a recent review on one-electron processes mediated by iron catalysts, see ref (14).
- Bauer I.; Knölker H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. Section 2.4.1 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]
- Rana S.; Biswas J. P.; Paul S.; Paik A.; Maiti D. Organic synthesis with the most abundant transition metal-iron: from rust to multitasking catalysts. Chem. Soc. Rev. 2021, 50, 243–472. section 6 10.1039/d0cs00688b. [DOI] [PubMed] [Google Scholar]
- Kyne S. H.; Lefèvre G.; Ollivier C.; Petit M.; Ramis Cladera V.-A.; Fensterbank L. Iron and cobalt catalysis: new perspectives in synthetic radical chemistry. Chem. Soc. Rev. 2020, 49, 8501–8542. 10.1039/d0cs00969e. [DOI] [PubMed] [Google Scholar]
- Fürstner A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. 10.1021/acscentsci.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daifuku S. L.; Kneebone J. L.; Snyder B. E. R.; Neidig M. L. Iron(II) Active Species in Iron-Bisphosphine Catalyzed Kumada and Suzuki-Miyaura Cross-Couplings of Phenyl Nucleophiles and Secondary Alkyl Halides. J. Am. Chem. Soc. 2015, 137, 11432–11444. 10.1021/jacs.5b06648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S. H.; Baker T. M.; Muñoz S. B. III; Brennessel W. W.; Neidig M. L. Multinuclear iron-phenyl species in reactions of simple iron salts with PhMgBr: identification of Fe4(μ-Ph)6(THF)4 as a key reactive species for cross-coupling catalysis. Chem. Sci. 2018, 9, 7931–7939. 10.1039/c8sc02915f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharasch M. S.; Fields E. K. Factors Determining the Course and Mechanisms of Grignard Reactions. IV. The Effect of Metallic Halides on the Reaction of Aryl Grignard Reagents and Organic Halides1. J. Am. Chem. Soc. 1941, 63, 2316–2320. 10.1021/ja01854a006. [DOI] [Google Scholar]
- Sapountzis I.; Lin W.; Kofink C. C.; Despotopoulou C.; Knochel P. Iron-Catalyzed Aryl-Aryl Cross-Couplings with Magnesium-Derived Copper Reagents. Angew. Chem., Int. Ed. 2005, 44, 1654–1658. 10.1002/anie.200462013. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Nakamura M. Iron-Catalyzed Selective Biaryl Coupling: Remarkable Suppression of Homocoupling by the Fluoride Anion. J. Am. Chem. Soc. 2007, 129, 9844–9845. 10.1021/ja073084l. [DOI] [PubMed] [Google Scholar]
- Chua Y.-Y.; Duong H. A. Selective Kumada biaryl cross-coupling reaction enabled by an iron(iii) alkoxide-N-heterocyclic carbene catalyst system. Chem. Commun. 2014, 50, 8424–8427. 10.1039/c4cc02930e. [DOI] [PubMed] [Google Scholar]
- Uchiyama M.; Matsumoto Y.; Nakamura S.; Ohwada T.; Kobayashi N.; Yamashita N.; Matsumiya A.; Sakamoto T. Development of a Catalytic Electron Transfer System Mediated by Transition Metal Ate Complexes: Applicability and Tunability of Electron-Releasing Potential for Organic Transformations. J. Am. Chem. Soc. 2004, 126, 8755–8759. 10.1021/ja039674a. [DOI] [PubMed] [Google Scholar]
- Wowk V.; Rousseau L.; Lefèvre G. Importance of Two-Electron Processes in Fe-Catalyzed Aryl-(hetero)aryl Cross-Couplings: Evidence of Fe0/FeII Couple Implication. Organometallics 2021, 40, 3253–3266. 10.1021/acs.organomet.1c00338. [DOI] [Google Scholar]
- Muthukrishnan A.; Sangaranarayanan M. V. Mechanistic Analysis of the Reductive Cleavage of Carbon–Halogen Bonds in Halopentafluorobenzenes. J. Electrochem. Soc. 2009, 156, F23–F28. 10.1149/1.3025830. [DOI] [Google Scholar]
- Enemærke R. J.; Christensen T. B.; Jensen H.; Daasbjerg K. Application of a New Kinetic Method in the Investigation of Cleavage Reactions of Haloaromatic Radical Anions. J. Chem. Soc., Perkin Trans. 2 2001, 1620–1630. 10.1039/b102835a. [DOI] [Google Scholar]
- Cahiez G.; Moyeux A.; Buendia J.; Duplais C. Manganese- or Iron-Catalyzed Homocoupling of Grignard Reagents Using Atmospheric Oxygen as an Oxidant. J. Am. Chem. Soc. 2007, 129, 13788–13789. 10.1021/ja075417k. [DOI] [PubMed] [Google Scholar]
- Clémancey M.; Cantat T.; Blondin G.; Latour J.-M.; Dorlet P.; Lefèvre G. Structural Insights into the Nature of Fe0 and FeI Low-Valent Species Obtained upon the Reduction of Iron Salts by Aryl Grignard Reagents. Inorg. Chem. 2017, 56, 3834–3848. 10.1021/acs.inorgchem.6b02616. [DOI] [PubMed] [Google Scholar]
- Rousseau L.; Herrero C.; Clémancey M.; Imberdis A.; Blondin G.; Lefèvre G. Evolution of Ate- Organoiron(II) Species towards Lower Oxidation States: Role of the Steric and Electronic Factors. Chem.—Eur. J. 2020, 26, 2417–2428. 10.1002/chem.201904228. [DOI] [PubMed] [Google Scholar]
- This translates into lower bond dissociation energies of the C–X bonds in the aliphatic series. For instance, the C–I (resp. C–Br) bond at 298 K increases from 55.6 kcal·mol–1 in tBu–I to 67 kcal·mol–1 in Ph–I (resp. from 72.6 kcal·mol–1 in tBu–Br to 84 kcal·mol–1 in Ph–Br), seeBlanksby S. J.; Ellison G. B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255. 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Tang H.; Chen K.; Hu L.; Yao J.; Shaik S.; Chen H. Two-State Reactivity in Low-Valent Iron-Mediated C-H Activation and the Implications for Other First-Row Transition Metals. J. Am. Chem. Soc. 2016, 138, 3715–3730. 10.1021/jacs.5b12150. [DOI] [PubMed] [Google Scholar]
- Boddie T. E.; Carpenter S. H.; Baker T. M.; DeMuth J. C.; Cera G.; Brennessel W. W.; Ackermann L.; Neidig M. L. Identification and Reactivity of Cyclometalated Iron(II) Intermediates in Triazole-Directed Iron-Catalyzed C-H Activation. J. Am. Chem. Soc. 2019, 141, 12338–12345. 10.1021/jacs.9b05269. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Xiao J.; Wang L.; Song Y.; Deng L. Carbon-Carbon Bond Formation Reactivity of a Four-Coordinate NHC-Supported Iron(II) Phenyl Compound. Organometallics 2015, 34, 599–605. 10.1021/om501061b. [DOI] [Google Scholar]
- Rousseau L.; Desaintjean A.; Knochel P.; Lefèvre G. Iron-Catalyzed Cross-Coupling of Bis-(aryl)manganese Nucleophiles with Alkenyl Halides: Optimization and Mechanistic Investigations. Molecules 2020, 25, 723–734. 10.3390/molecules25030723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurkin F. E.; Wodrich M. D.; Hu X. A Monometallic Iron(I) Organoferrate. Organometallics 2017, 36, 499–501. 10.1021/acs.organomet.6b00841. [DOI] [Google Scholar]
- Control experiments confirmed that pentafluorophenylmagnesium halide C6F5MgCl was not formed by direct magnesium-chloride exchange from ArMgBr and C6F5Cl. In the absence of catalyst, the exchange takes place with a low 3% yield.
- Fürstner A.; Martin R.; Krause H.; Seidel G.; Goddard R.; Lehmann C. W. Preparation, Structure, and Reactivity of Nonstabilized Organoiron Compounds. Implications for Iron-Catalyzed Cross Coupling Reactions. J. Am. Chem. Soc. 2008, 130, 8773–8787. 10.1021/ja801466t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.