

SUMMARY
Metabolomics describes a high-throughput approach for measuring a repertoire of metabolites and small molecules in biological samples. One utility of untargeted metabolomics, unbiased global analysis of the metabolome, is to detect key metabolites as contributors to, or readouts of, human health and disease. In this perspective, we discuss how artificial intelligence (AI) and machine learning (ML) have promoted major advances in untargeted metabolomics workflows and facilitated pivotal findings in the areas of disease screening and diagnosis. We contextualize applications of AI and ML to the emerging field of high-resolution mass spectrometry (HRMS) exposomics, which unbiasedly detects endogenous metabolites and exogenous chemicals in human tissue to characterize exposure linked with disease outcomes. We discuss the state of the science and suggest potential opportunities for using AI and ML to improve data quality, rigor, detection, and chemical identification in untargeted metabolomics and exposomics studies.
INTRODUCTION
Chemical reactions in the body produce the myriad metabolites essential for human life, a process known as metabolism. Metabolism itself falls in two main types: catabolism, or the breakdown of molecules to obtain energy, and anabolism, or the synthesis of compounds required by cells. Metabolism also encompasses deactivation, detoxification, and elimination of foreign or unwanted substances. Insight into these processes is crucial for understanding human physiology in health and disease. There are multiple ways to study these processes individually or collectively, but one comprehensive, high-throughput approach is metabolomics, which relies on measurement of small molecules (<2,000 Da) in a biological sample, typically blood, urine, or saliva (Figure 1, bottom). The metabolomics framework can capture endogenous metabolites and signal molecules that participate in regulation of gene expression, protein function, and enzyme activity. Its high-throughput nature is particularly valuable, given that the scale of small molecule-enzyme interactions varies by organism from around 500 to a few thousand reactions and metabolite intermediates.1
Figure 1. Metabolomics and exposomics facilitate discovery.
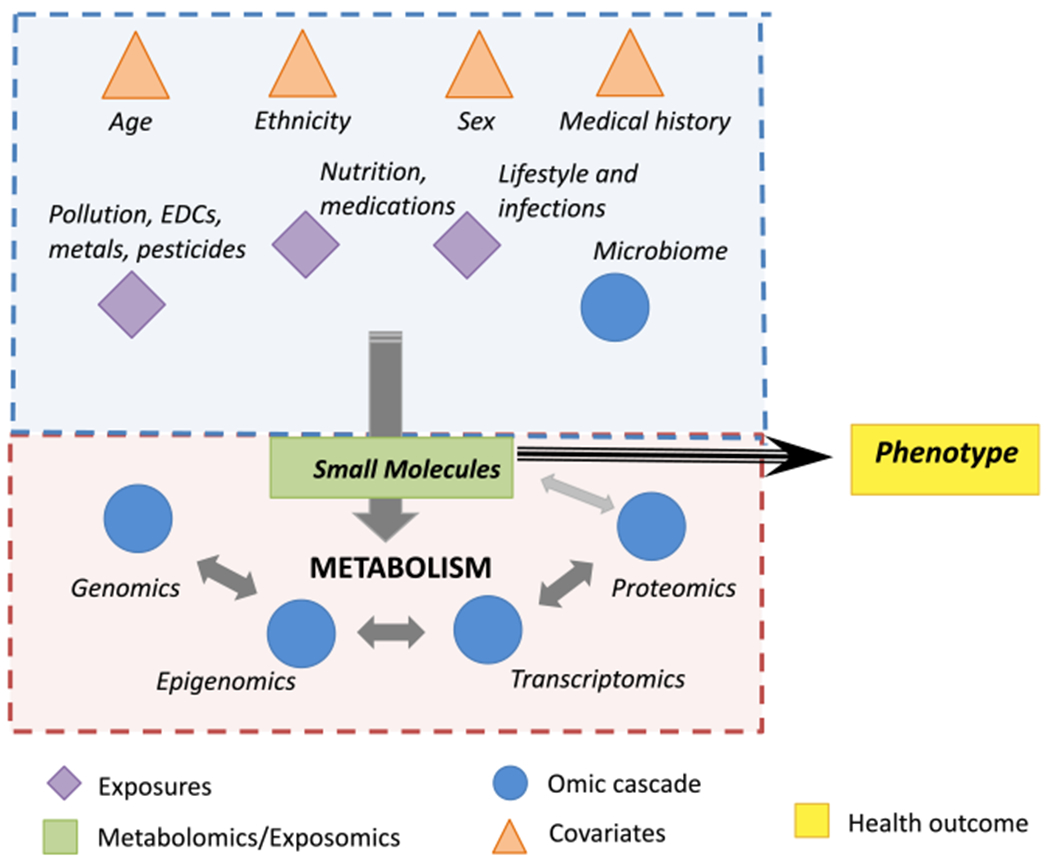
Metabolomics (pink shading, bottom) focuses on measures of endogenous small molecules as outputs of metabolic pathways. Perturbations in the ‘omics’ layers can lead to changes in metabolite profiles linked with phenotypes. Exposomics (blue shading, top) expands on this to include measurements of exogenous small molecules as well as the influence of exogenous and non-genetic factors on the ‘omics’ cascade that can lead to changes in metabolite profiles linked with phenotype. The timing of sample collection relative to phenotype characterization helps determine whether the metabolite biomarker or pathway is linked to etiology, diagnosis, or progression of disease.
Within the metabolomics framework, different approaches enable different kinds of insights into these metabolic processes. One approach, targeted metabolomics, typically measures concentrations of tens to approximately 100 endogenous metabolites determined a priori. This quantitative approach enables comparisons across studies and populations as well as development of thresholds describing average or expected ranges to aid medical diagnosis and intervention. A specific type of targeted metabolomics is metabolic flux analysis, which monitors the fate of stable isotope tracers (e.g., 13C-glucose, 15N-glutamine), allowing research into the flow of metabolites.1–5 Together, flux and concentration provide a fuller understanding of metabolism.
Complementing these quantitative approaches, untargeted metabolomics is an unbiased and semi-quantitative measure of thousands of small molecules simultaneously. This approach circumvents the logistical and economical challenges that restrict how many chemicals can be measured in a quantitative assay. Here, high-resolution mass spectrometry (HRMS) typically pairs with liquid chromatography (LC) or gas chromatography (GC) to easily separate and detect thousands of chemical peaks—unitless measurements of semi-quantitative features applicable for downstream analysis. With particular study designs, untargeted metabolomics can generate new hypotheses on altered pathways and individual metabolites that can be linked to disease initiation, diagnosis, progression, or prognosis (Figure 1).6,7
Untargeted metabolomics offers a particularly valuable approach when considering that beyond the core set of metabolites studied in targeted and flux analysis lies the vast unknown metabolome described holistically as the “exposome.”The exposome stems from a hypothesis that most diseases and disorders are heterogeneous and that non-genetic influence or “exposure” from environmental chemicals, diet, lifestyle, psychosocial factors, and disease history throughout life may play pivotal roles in health. Indeed, exogenous chemicals from food (genistein, vitamin E), lifestyle (nicotine, caffeine), drugs (cefuroxime, acetaminophen), and pollution (phthalates, perfluoroalkyl substances) enter the body and circulate in the blood to cells and organs. Biological fluids and tissues contain chemical readouts of these exposures, such as cortisol from stress, di(2-ethylhexyl)phthalate (DEHP) from plasticizers, caffeine from coffee, cholesterol from high-fat diets, and antibiotics used before surgery. Over the last decade, untargeted metabolomics strategies have expanded to include detection and measurement of exposure chemicals. “Exposomics” analysis leverages HRMS-based strategies to capture, in the same analytical assay, the endogenous metabolites typically measured in untargeted metabolomics analysis and exogenous chemicals resulting from various exposures (Figure 1, top).
Despite extensive LC-MS studies collecting exposome data, most of the human exposome remains unknown, with only a small fraction identified and incorporated into in-house libraries or databases. Even though only about 5,000 chemicals likely have wide enough dispersal in the environment to pose a global threat to the human population, many thousands more are expected to affect individuals.8 Because such chemicals can be converted to metabolites or environmental transformation products, first-order reaction products could number in the millions.9 Unlike typical endogenous metabolites and nutritional chemicals detected in high concentrations in most study participants, environmental chemicals often have concentrations orders of magnitude lower; they can be transient and then rapidly disappear, or they can fall below detection limits. Current hypotheses in environmental epidemiology purport that individuals commonly experience chemical exposure in mixtures rather than individually and that mixture effects underlie phenotypic changes in health or during disease.10,11
These phenomena are important when considering which suitable analytic approach to take. Although untargeted assays with HRMS can detect tens of thousands of peaks from each sample, the complexity of these data and inherent analytical variability in HRMS bring forth computational challenges. Advanced artificial intelligence (AI) and machine learning (ML) algorithms can assist with alignment of the data; feature selection to pinpoint important exposures, metabolites, and biomarkers as mixtures; and annotation of unknown metabolites. Thus, developing and optimizing such applications is necessary to advance exposomics discovery and further research.
Here we highlight some of the most recent AI/ML tools applied to the field of untargeted metabolomics data processing. Although AI/ML is now being applied to new technologies, including MS imaging and single-cell MS metabolomics,12–15 we focus our discussion on the most widely used LC- or GC-HRMS techniques and critical gaps required to overcome to advance exposomics research. We discuss these as key steps required for successful application of untargeted metabolomics within an exposomics framework.
Typical untargeted workflow
For biological matrices like serum, plasma, or urine, LC or GC column chromatography is used to first separate the complex mixture before detection and measurement by HRMS. The LC/GC-HRMS workflow typically follows a series of steps: sample preparation; data acquisition; data pre- and post-processing; data analysis, including feature selection; and identification/annotation of chemicals (Figure 2).16–18 Metabolites and chemicals are extracted from the biological sample using a high percentage of organic solvent that also removes proteins. In LC analysis, the extracts are usually analyzed using hydrophilic interaction LC (HILIC), which uses a column that retains polar chemicals, and reverse-phase (RP) chromatography, which uses a column that retains neutral and non-polar compounds. Together, these complementary methods maximize the total number of measurable small molecules. For GC analysis, a clean-up step using solid-phase extraction can precede protein precipitation; then the extracts are derivatized to make them more volatile so they can be analyzed on a capillary column.19 Physicochemical properties of the small molecules dictate their extraction efficiency as well as their retention on the LC or GC column, and ionization/detection on the MS, which provides an opportunity for ML approaches to support chemical space prediction for selecting chromatographic columns and buffer or temperature gradients.20 The untargeted approach uses minimal sample processing steps to maximize the breadth of chemicals measured because multi-step extractions trade this breadth of measurement for one of maximizing the signal of particular chemical classes. Most of the MS features measured in an untargeted analysis are unknown, precluding a method to optimize conditions a priori based on properties of all intended targets or to determine all of the chemicals that are missing or lost from the analysis because of the selected conditions.
Figure 2. Untargeted analysis workflow for biomedical applications.
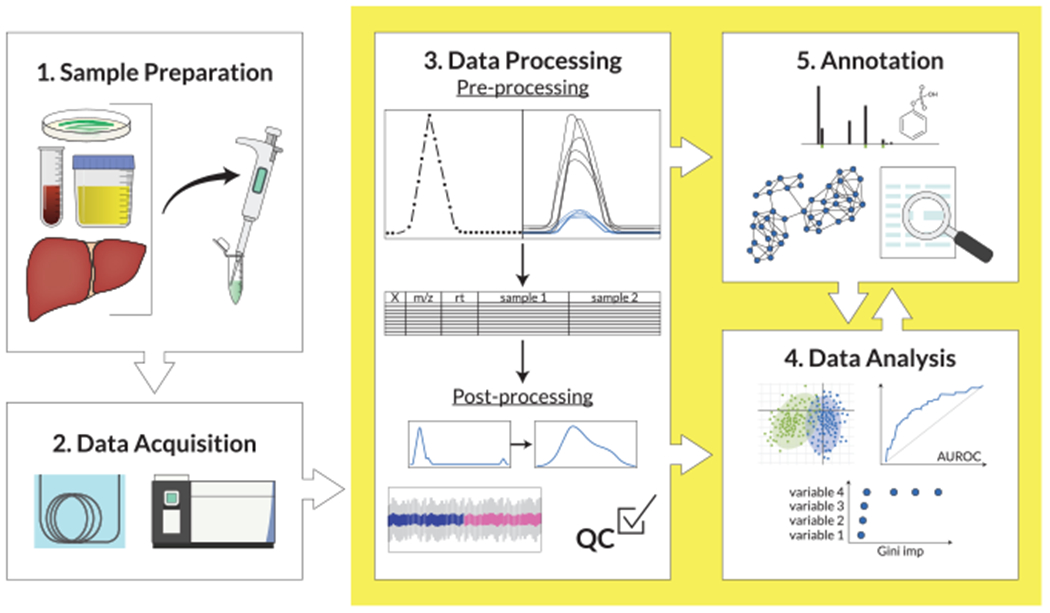
Step 1: biological samples, including biofluids, tissues, and/or cells, are collected from the study groups or phenotypes of interest (e.g., exposed or not exposed, cases or controls). Metabolites and chemicals are isolated through sample preparation. Step 2: the metabolites and chemicals are analyzed by liquid or gas chromatography-high resolution mass spectrometry (LC- or GC-HRMS, respectively), which separates the molecules in the complex mixture and detects and measures them. Step 3: data undergo processing to convert the 3D data collected on each sample (m/z, retention time, and abundance) to a 2D table for analysis. In pre-processing, the peaks are found and aligned across all samples, and the area under the peak curve is calculated. In post-processing, the data are normalized and scaled, batch correction is performed when needed, as well as filtering for QC. Step 4: data are analyzed using AI/ML and classic statistical approaches to identify individual metabolites and chemicals or pathways that are predictive of the phenotype of interest. Step 5: metabolites and chemicals of interest are annotated by matching to spectral libraries, spectral prediction, and spectral/chemical similarities.
Data acquisition can occur in one stage of mass spectrometry (MS1) or two stages with tandem mass spectrometry (MS/MS), in which ions from MS1 are selectively fragmented to generate a molecular fingerprint of the molecule to aid identification (Figure 3). Most studies use combinations of collecting MS1 and MS2 data; MS1 is usually used for semi-quantification of the feature, and MS/MS fragmentation data are usually used for annotation of the feature.
Figure 3. MS and MS/MS data acquisition.
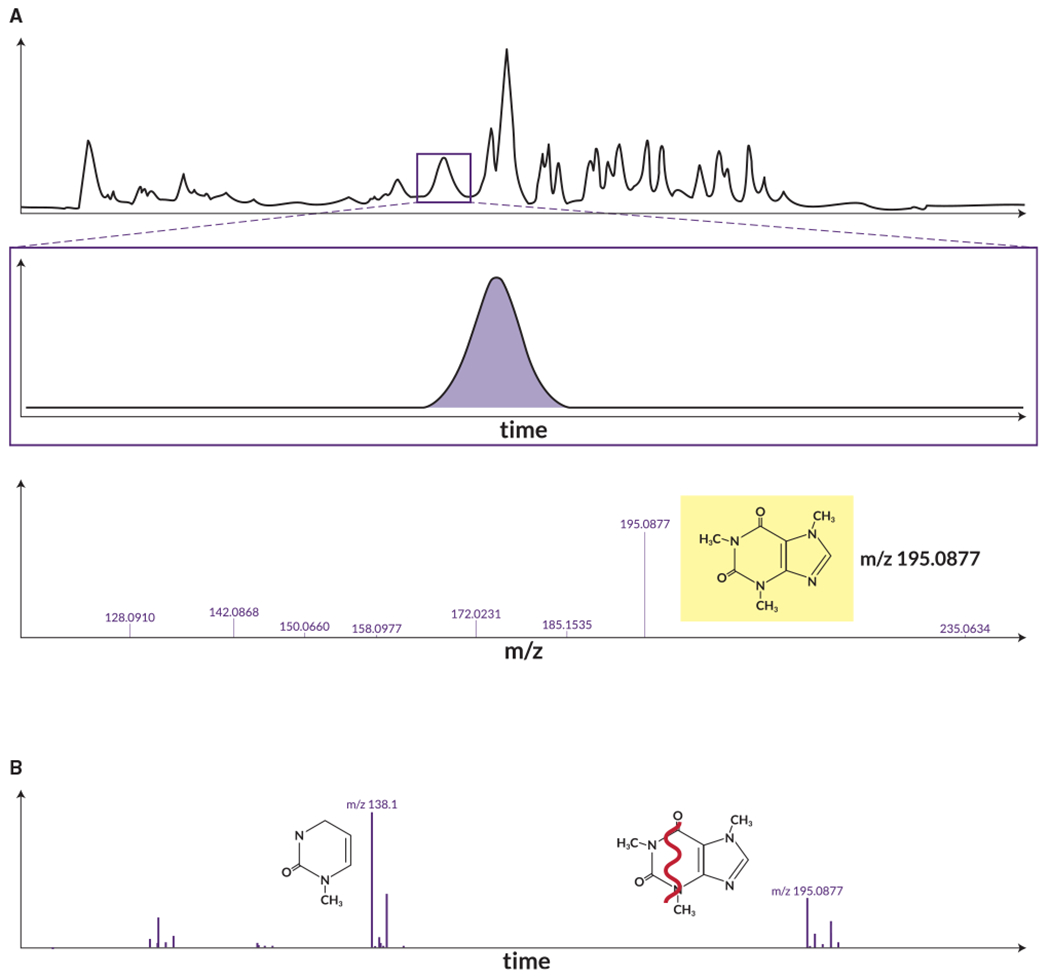
(A) A sample after injection into a chromatography column enters the mass spectrometer, where eluting chemicals are ionized, accelerated, and analyzed by mass spectrometry (MS1). Each chemical elutes at a characteristic time and m/z.
(B) Tandem mass spectrometry (MS/MS) can then be performed, where the ions of interest (e.g., m/z 195.0877) can be selectively fragmented to generate fragment ions (e.g., m/z 138.1). These fragment ions are characteristic of the molecule. Therefore, MS/MS spectra can be used to aid chemical identification.
Data are acquired for each analytical mode (HILIC-MS, RP-MS, or GC-MS) in three dimensions: mass-to-charge ratio (m/z), retention time (rt), and abundance. AI/ML play major roles in subsequent steps of metabolomics workflows. MS1 data are pre- and post-processed using a variety of algorithms to transform the large amount of raw spectral data into a much smaller, statistically manageable set of peaks or features (Figure 2). Software processes include selecting the peaks and aligning the peaks across the samples. The output is a peak table for each analytical mode that has peak intensity values (abundance) for every metabolite feature for every sample. When the data are adjusted to remove unwanted technical variation, feature selection takes place, often using statistical approaches or ML to focus on the small molecules associated with health outcomes or exposure. Finally, these features are identified for biological interpretation, with many ML and AI tools developed to facilitate metabolite annotation using MS/MS data.
Innovation in the first two steps of the workflow (Figure 2, steps 1 and 2) stems largely from instrumentation and automation, which enable more reproducible sample preparation, better detection in smaller sample volumes, and a broader range of measurable metabolites. In contrast, innovations in the last three steps (Figure 2, steps 3–5) stem from computational advances. Most efforts in AI/ML to date have focused on the latter part of the data processing workflow—feature selection and metabolite identification—because of an urgent need for computational tools to draw biologically interpretably connections between complex MS metabolomics data and health and exposure outcomes. However, recent efforts demonstrate a shift toward developing and applying advanced ML methods to enhance quality control and cleaning—data processing—of untargeted MS data before downstream analysis. This shift reflects that most readily used peak-picking algorithms can successfully measure high-concentration, Gaussian-shaped peaks typical of endogenous metabolites21 but are less successful with low-abundance signals. This measurement is a fundamental challenge for HRMS exposomics, which seeks to capture a handful of needles in a large and variable haystack. Therefore, advancing the field of HRMS exposomics requires robust peak picking of low-abundance features and development or optimization of novel computational tools for data processing.
Data processing
Metabolomics raw data are inherently complex because of multiple linear and nonlinear interactions among the metabolites as well as challenges with mass spectrometry data structure.22 These challenges include features (e.g., peaks) that can massively outnumber the samples, high levels of noise, batch and run order effects during measurements, and missing values during peak detection. The data pre-processing step of this workflow is crucial for accurate translation of the 3D data obtained from LC-MS (m/z, rt, abundance) into a 2D aligned peaktable (aligned peaks [of specific m/z and rt] and their respective abundances in every sample) that is required for downstream data analyses.23,24 This is crucial because the peak area correlates with chemical concentration in a sample, which are the data ultimately analyzed to draw statistical and biological inferences. Even though data pre-processing is easily performed using automated software, it is challenging to precisely, accurately, and robustly synthesize the data across the full range of metabolite features, concentrations, and sample acquisition times into a manageable dataset.16,25
Algorithms for pre-processing include open-source XCMS,23 MZmine3,24 MS-Dial,26 and MetAlign27 as well as several types of proprietary software. XCMS and MZmine are the most widely used, but no algorithm has been accepted as the benchmark in the fields of metabolomics or exposomics. Consequently, concordance across methods can be less than 50%.28 Recent evidence demonstrates that false positives and poorly integrated peaks (low quality) are retained in the data at large numbers for public and private software platforms,29,30 which can propagate errors into downstream analyses.31 These findings have stoked increased interest in development of different quality control (QC) measures to improve the quality and reliability of data reporting for high-throughput untargeted analysis.32–34
After peak picking, application of subsequent filtering strategies based on predetermined thresholds, such as mean/median value across samples, variability across biological samples, and levels of missing values, is routine to remove noisy peaks.35 The most commonly used is a pooled QC that is generated by thoroughly combining a small volume from all samples or from a representative subset of the samples and realiquoting this into multiple samples. These replicates can be evenly distributed throughout the analytical batch for acquisition. Because the pooled QC sample is a representative sample comprising the metabolite compositions of the study samples, features present in all pooled QC samples with a low quantitative coefficient of variation across the analytical batches are retained. However, with complex samples, contaminants can be greater than 50% in some experiments,36 which may be difficult to discriminate from true positives of low abundance. Although QCs allow removal of many false positive features (noise or baseline recognized as a peak), correct features will also be discarded. A recent study illustrated this phenomenon by re-mining untargeted metabolomics data using minimal thresholds to reveal additional metabolites and pathways associated with the outcome that were not identified in the primary publication.37 Therefore, to retain important but low-abundance features for downstream analysis, there is a need for new approaches that comprehensively retain all high-quality peaks.38 Here, new peak-picking algorithms and ML-based filtering have entered the arena.
The comprehensive peak characterization (CPC) algorithm with user-based peak criterion filtering removes 35% of the peak-picked XCMS features and demonstrates, for a subset of retained peaks, a 90% true positive rate and 87% true negative rate.39 Similarly, Finnee introduces algorithms to correct baseline drift and background noise and uses a clustering and targeted analysis approach to reduce false-positives.40 These resulted in more biomarkers than XCMS or MS-DIAL, on a limited demonstration of five controls, five with an asthma medical diagnosis and five with a chronic obstructive pulmonary disease medical diagnosis, suggesting that algorithm development and optimization may be needed to enable detection and measurement of new chemicals in datasets for statistical analysis.41
Recent work introduced applications of ML classification approaches that use peak quality to train models. The first tool, WiPP, introduced in 2019, uses support vector machine (SVM) to classify high-concentration peaks from GC-MS data but performs worse for low-concentration peaks.42 MetaClean assesses 24 different classifiers—combinations of eight algorithms and three sets of peak quality metrics—at filtering peaks based on quality of peak boundaries, revealing that the adaBoost algorithm and a set of 11 peak quality metrics perform best.43 However, the distributions of low-and high-abundance peaks between the true positive and true negative rates were not assessed. Peakonly44 and NeatMS45 utilize deep learning (DL) neural networks for LC-MS peak classification. Peakonly has a true positive detection rate of 97% but deliberately does not consider narrow peaks and uncertain peaks with noisy shape, achieving confidence to detect only true positive peaks. On the other hand, NeatMS demonstrates a greater ability to retain high-quality peaks even at lower concentrations. In the tool NPFimg, raw GC-MS data are flattened into a 2D image for processing using a neural networks model. NPFimg performs better than or to the same as XCMS, with true positive and true negative rates of more than 97% with a limited demonstration of application to human breath samples from a single participant.46 Finally, the software EVA uses convolutional neural networks (CNN) to classify good and bad peak shapes; applying this to 22 publicly available LC-MS-based metabolomics datasets yielded a classification accuracy greater than 90%.47
Although these tools show promise based on their initial demonstrations, their utility must be tested through full evaluations on large datasets. It is critical to determine whether exposure data, and not just endogenous metabolites, are retained in the analysis, especially after data processing,48–51 to determine where along the workflow critical improvements are needed. Anticipated future advancements and developments in algorithms for untargeted metabolomics data mining52 will contribute to robust data analysis and, ultimately, discovery of new biomarkers for health and disease.
Feature selection
Discovery of molecular biomarkers and metabolomics signatures requires analyzing the complex untargeted data in biological samples. Analyses using traditional univariate and multivariate linear models perform multiple hypothesis tests (one hypothesis per feature) and apply a correction method to adjust for multiple hypothesis testing (false discovery rate or Bonferroni). Borrowing from the concept of genome-wide association studies (GWASs), environment-wide association studies (EWASs) analytically validate associations between metabolite features and a phenotype. Such studies are comprehensive in that each measured metabolite is assessed for possible association with the target phenotype.53 These methods enable successful biomarker discovery in metabolomics data across disease contexts.54–57 However, these methods cannot consider the highly correlated structure of metabolomics data a priori and do not address interactions between molecules,58 thus increasing the probability of obtaining false positives and false negatives. In contrast, AI and ML approaches use the data to build models and then test those models with the data. For both epidemiological and clinical studies, AI and ML of metabolomics data can unveil important relationships between phenotypes and exposures or phenotypes and disease groups. AI and ML can identify variation between phenotypes through dimension reduction, metabolites and chemicals that can predict disease status or phenotypes, and biological pathways that are different between phenotypes, demonstrating the power of these approaches to answer a range of important clinical, environmental health, and precision health questions.59
The most widely used AI/ML tools in metabolomics include absolute shrinkage and selection operator (LASSO), principal-component analysis (PCA), hierarchical clustering analysis (HCA), self-organizing maps (SOMs), partial least square-discriminant analysis (PLS-DA), and random forest (RF).60,61 Recent studies also applied hidden-layer artificial neural networks (ANNs) and DL (CNN and deepNN [DNN]).62 These multivariate methods are advantageous in that they can consider all features simultaneously and, consequently, deal with correlation among the metabolites.63,64 As a result, these techniques have helped uncover significant biomarkers and metabolite signatures.
There are several examples of ML algorithm use in metabolite feature selection across disease contexts. In a recent metabolomics study, feature selection with RF identified 17 metabolites that, in combination, accurately detected cirrhosis resulting from non-alcoholic fatty liver disease (NAFLD), and whose levels were sufficient to discriminate NAFLD cirrhosis from control probands in a PCA. These findings yielded a potential non-invasive stool signature for disease prediction of NAFLD cirrhosis.65 In another study, application of RF identified metabolites predictive of coronavirus disease 2019 (COVID-19) severity.66 Similarly, RF and SVM uncovered a targeted metabolomics signature of Alzheimer’s disease (AD) in the brain.67 When tested in blood samples, this panel identified distinct metabolites belonging to the sphingolipid and glycerophospholipid classes that are related to the severity of AD pathology in the brain, and their concentrations in blood are associated with preclinical disease progression. In another example, application of LASSO defined metabolites as a metabolic clock of gestational age in maternal plasma during pregnancy.68 Finally, an HSIC LASSO-based prediction model showed better predictive power than LASSO, SVM, PLS, RF, and neural network for predicting depression symptoms in a study of more than 800 Japanese adults.69 These examples highlight the number and breadth of applications of ML algorithms—alone or in combination—to discover metabolomic biomarkers to support prediction of disease incidence or severity, demonstrating the versatility of models for use in the field.
To date, AI/ML feature selection applications have resulted primarily in identification of significant endogenous metabolites and pathways and not exogenous chemicals. Although use of classic statistical techniques, correlation analysis, and meet-in-the-middle approaches identified links between environmental and dietary exposure and disease outcomes,70–72 success using AI/ML feature selection tools to identify non-endogenous metabolites remains limited. It is possible that these tools have been less successful in selecting robust peaks; however, to our knowledge, the necessary comprehensive comparison of tools with an exposomics focus has yet to be undertaken.
The complexity of environmental toxicity relationships (e.g., U-shaped toxicity, nonlinear associations, and unknown interactions) may require more advanced AI or deep ML algorithms. Emerging applications of CNN and DNN to metabolomics showed successful feature selection of predictors of estrogen receptor (ER) status in breast cancer73 or of predictors of Alzheimer’s disease.74 Indeed, a DL framework yielded the highest area-under-the-curve point estimate for classifying individuals with breast cancer by ER+/ER−status based on metabolomics data compared with that of six other ML algorithms. Biological interpretation of the first hidden layer identified by the DL framework revealed enrichment of eight cancer-relevant metabolic pathways that were not identified through conventional ML algorithms. Although DL methods do not always outperform traditional ML methods,75 these results suggest that further development and applications of tools in ML, and especially DL, for feature selection may help uncover novel exposure risk factors. Several existing projects aim to leverage such opportunities, using HRMS exposomics data integrated with other exposures and measures with planned AI and ML strategies; for example, in the areas of women’s health76 and chronic gut inflammation.77
Metabolite identification
Metabolite identification is a critically important aspect in the biomarker discovery pipeline. Accordingly, many researchers devote efforts in software and tool development to support this process.78 After feature selection, important peaks or features, minimally defined by a specific m/z and rt, must be annotated to determine biological plausibility for eventual translation into intervention and prevention strategies or clinical practice. This step often relies on use of metabolite databases and spectral libraries containing experimental and in silico spectra, including GNPS,79 Metlin,80 the Human Metabolome Database,81 MassBank,82 and others.83 Users match to databases on m/z alone for low-confidence annotations or include additional orthogonal data (e.g., presence of isotopes and their ratios, MS/MS fragmentation data, neutral losses, and characteristic fragments) to increase the confidence of the annotations.84,85 For an annotated peak, the chemical or metabolite is confirmed by analyzing a commercially available or synthesized standard under the same experimental conditions and matching across all available parameters (m/z, retention time, MS/MS, etc.).
However, the list of chemicals available in databases is small compared with the more than 68 million known available chemicals,86 and spectral matching rates for specialized chemicals remain low. Thus, there is a need for additional tools to help generate annotations of unknown chemicals identified in an untargeted chemical assay. One approach is cognitive metabolomics computing using ML and natural language processing (NLP), which extracts information from the scientific literature and understands its semantic context.87 Although promising for annotation and biological interpretation of exposomics data,88 applications remain limited thus far, likely because of entry barriers such as required subscriptions to the databases and the need for expert user knowledge to successfully execute this type of analysis. Recent efforts to overcome these challenges produced a protocol with suggestions for free and open-source tools for NLP,89 showing promise for further expansion of use in metabolomics and exposomics.
Easier-to-use in silico tools provide a widely adopted alternative for annotation. CSI:FingerID uses SVM to predict MS/MS spectra, then suggests candidate compounds to match those spectra.90 Other tools, like LipidBlast,26 MetFrag,91 MIDAS,92 and CFM-ID,93 take molecular structures as inputs and predict the spectra. In CFM-ID, a pre-trained neural network model is mixed with rule-based fragmentation.93 The addition of rules, compared with ML alone, improves prediction of classes of metabolites found in food and endogenous metabolite databases but not exposomic chemicals, possibly because the approach lacks rules specific to industrial chemicals.
To match the spectral pairs between a user’s spectra and those of a database, there is a ranking system or score available to help the user understand the robustness of a match. One common metric for matching MS/MS spectral data is the cosine similarity score, which ranks the overlap between MS/MS spectral data but performs poorly at matching chemical analogs with several structural modifications.94,95 Recent improvements to this approach added ML algorithms focused on molecular structural similarity. Spec2Vec uses an unsupervised ML method to learn from co-occurrence of ion fragments across large datasets.96 This method is computationally faster than cosine similarity, and its results correlate better to structural similarity than cosine-based scores, suggesting better matching. Similarly, MS2Deep uses neural networks to predict structural similarity scores of MS/MS data without requiring a known molecular formula.97 Finally, SteroidXtract uses CNN on a training set of manually curated steroid MS/MS spectra to predict other steroid-like chemicals in an untargeted dataset.98
Further expansion of spectral libraries will facilitate confident metabolite and chemical identification, but this step must be supplemented with new annotation tools. In addition to the 1,500 new chemicals that are being produced annually by the United States,99 new sample types that require atypical sample processing are likely to generate new spectral adducts and unidentified chemicals. For example, using MS/MS spectral matching to Metlin, GNPS, and an in-house library resulted in just 4% of chemicals being annotated from a study on the tooth exposome.100 This example, with all 267 metabolites discriminating prenatal and postnatal tooth fractions unannotated, highlights that the large percentage of unannotated chemicals in a study poses a major challenge for biomarker discovery. Development of additional tools for annotation and identification of unknowns can be facilitated by the publicly available spectral databases that are now large enough to provide substantial training, validation, and testing data. Whether using network maps to expand annotations of unknowns through chemical similarity to those in databases101 or using biological information to drive development of chemical class prediction, ML and DL hold promise for robust MS/MS data interpretation and endogenous and exogenous chemical elucidation.102
Challenges for the future
Significant advances in untargeted chemical analysis instrumentation enable large numbers of measurements through several orders of magnitude including at trace-level concentrations. Similar to other fields, such as DNA sequencing, the decrease in cost of these technologies now facilitates cost-effective measurement of thousands of samples for epidemiological and clinical studies. Critically, over the last decade, AI/ML tools have been developed to support extraction and formatting of data, mining the data, and annotating the data generated from these massive datasets,103 already playing an important role in accelerating discovery.
Many applications of ML in metabolomics have focused on the forward-facing step of the untargeted analysis pipeline—the feature selection process. In this step, aided by ML algorithms, thousands of features are siphoned down to the tens of features that are predictive of a health outcome or phenotype. Although current applications are limited largely to an individual “omics” dataset, recent advances include using ML to combine data across different types of “omics” levels in a system’s biology approach.104–106 This development will lead to identification of additional and combined biomarkers to attain higher specificity or to assist with unraveling the cascade of factors associated with disease initiation and progression. However, these achievements require that metabolites are sufficiently annotated.
When features are selected, researchers hit the ultimate bottleneck of untargeted chemical analysis—annotation of the unknown metabolites. Without this critical step, selected metabolites cannot be biologically interpreted or further validated. This issue prompts burgeoning efforts to develop experimental databases of spectra along with AI/ML-based in silico prediction models, retention time predictors, and chemical similarity algorithms to facilitate annotation of metabolites at different levels of confidence from molecular formulas, to chemical classes, to absolute identification of a metabolite or chemical with the potential to drastically improve the breadth of annotations needed for exposomics.
Much work remains to be done. Evidence suggests that, when provided with information that an exogenous or non-endogenous chemical compound exists within an untargeted dataset, by screening the data for a chemical in an in-house library or a priori hypothesis, we can uncover associations between those chemicals and health outcomes. However, examples of successfully extracting these chemicals directly from the data using untargeted data analysis workflows (e.g., pre-processing, feature selection) are limited beyond food and microbiome metabolites.54,107 Recent work has shown that using typical filtering criteria of untargeted data can miss up to 80% of significant peaks,37 suggesting that pre-processing may play a pivotal role in this challenge. However, only in the last several years has there been advancement in addressing this peak quality aspect via ML classifiers and AI algorithm development.16
The ability to maximize features while decreasing false positives is a critical challenge to overcome in the field of untargeted metabolomics. This issue is exacerbated in exposomics, in which exposure biomarkers may be difficult to discriminate from noise. Although important strides are being made through development of ML classifiers to improve retention of high-quality peaks, these classifiers remain largely untested on the diverse range of concentrations seen in complex biological samples, and the little data available suggest that current algorithms and classifiers are insufficient to robustly capture low-concentration chemicals. Therefore, there is an important gap to fill, highlighting a critical need for ML algorithms with a focus on retaining quality peaks across the full dynamic range of measured chemicals in a biological sample.
The “functional exposomics” concept suggests that the complexity of the exposome can be reduced by focusing on a biology-driven approach.108 Such an approach is meant to complement measurement-based approaches; for example, using SteroidXtract,98 ML might predict and identify spectral patterns of exogenous chemicals by chemical class, focusing on those with similar biological activity, such as endocrine-disrupting chemicals or those for which analytical standards are not readily available, such as conjugated phthalates. This principle—that exogenous metabolites with potential to alter biology are likely to consist of several building blocks that mimic biochemical machinery—drives development of tools for determining structures of natural products.109 In this case, using a biological approach rather than a statistical approach to focus on feature candidates within the HRMS data may reveal previously unknown combinations of chemicals working synergistically to affect health. Many of these high-confidence annotations require collection of MS/MS data, which may not be possible for low-concentration chemicals. However, this problem might be surmountable by utilizing MS1 data collected on every sample. Existing HRMS exposomics tools that focus on reaction-level chemical changes110,111 and “molecular gatekeepers” that focus on determining active molecular networks can be expanded with information from in-source fragments, retention time prediction, and cognitive computing.112,113
AI/ML advances in data processing have triggered significant discoveries in metabolomics and are poised to do the same in the field of exposomics. The success of DL algorithms for unstructured data and use of new AI/ML approaches not readily implemented in metabolomics and exposomics, combined with available datasets or samples with known chemicals of low and high concentrations114,115 for training and validating peak picking algorithms and the QC step, are key starting points for catalyzing this new era of discovery toward environmental and precision health.
ACKNOWLEDGMENTS
L.P. is supported by National Institute of Environmental Health Sciences grants U2CES026561, U2CES030859, P30ES023515, R21ES030882, and R01ES031117 and National Cancer Institute grant UH2CA248974.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Jang C, Chen L, and Rabinowitz JD (2018). Metabolomics and isotope tracing. Cell 173, 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahu A, Blätke MA, Szymański JJ, and Töpfer N (2021). Advances in flux balance analysis by integrating machine learning and mechanism-based models. Comput. Struct. Biotechnol. J 19, 4626–4640. 10.1016/j.csbj.2021.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martínez-Reyes I, and Chandel NS (2021). Cancer metabolism: looking forward. Nat. Rev. Cancer 21, 669–680. [DOI] [PubMed] [Google Scholar]
- 4.Antoniewicz MR (2018). A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med 50, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weitzel M, Nöh K, Dalman T, Niedenführ S, Stute B, and Wiechert W (2013). 13CFLUX2—high-performance software suite for 13C-metabolic flux analysis. Bioinformatics 29, 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monteiro MS, Carvalho M, Bastos ML, and Guedes de Pinho P (2013). Metabolomics analysis for biomarker discovery: advances and challenges. Curr. Med. Chem 20, 257–271. [DOI] [PubMed] [Google Scholar]
- 7.Zhang A, Sun H, Yan G, Wang P, and Wang X (2015). Metabolomics for biomarker discovery: moving to the clinic. Biomed. Res. Int 2015, 354671. 10.1155/2015/354671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landrigan PJ, Fuller R, Acosta NJR, Adeyi O, Arnold R, Basu NN, Baldé AB, Bertollini R, Bose-O’Reilly S, Boufford JI, et al. (2018). The Lancet Commission on pollution and health. Lancet 391, 462–512. 10.1016/s0140-6736(17)32345-0. [DOI] [PubMed] [Google Scholar]
- 9.Vermeulen R, Schymanski EL, Barabási AL, and Miller GW (2020). The exposome and health: where chemistry meets biology. Science 367, 392–396. 10.1126/science.aay3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlin DJ, Rider CV, Woychik R, and Birnbaum LS (2013). Unraveling the health effects of environmental mixtures: an NIEHS priority. Environ. Health Perspect 121,A6–A8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joubert BR, Kioumourtzoglou MA, Chamberlain T, Chen HY, Gennings C, Turyk ME, Miranda ML, Webster TF, Ensor KB, Dunson DB, and Coull BA (2022). Powering research through innovative methods for mixtures in epidemiology (PRIME) program: novel and expanded statistical methods. Int. J. Environ. Res. Public Health 19, 1378. 10.3390/ijerph19031378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdelmoula WM, Lopez BGC, Randall EC, Kapur T, Sarkaria JN, White FM, Agar JN, Wells WM, and Agar NYR (2021). Peak learning of mass spectrometry imaging data using artificial neural networks. Nat. Commun 12, 5544. 10.1038/s41467-021-25744-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Behrmann J, Etmann C, Boskamp T, Casadonte R, Kriegsmann J, and Maaß P (2018). Deep learning for tumor classification in imaging mass spectrometry. Bioinformatics 34, 1215–1223. [DOI] [PubMed] [Google Scholar]
- 14.Xie YR, Castro DC, Bell SE, Rubakhin SS, and Sweedler JV (2020). Single-cell classification using mass spectrometry through interpretable machine learning. Anal. Chem 92, 9338–9347. 10.1021/acs.analchem.0c01660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu R, Zhang G, and Yang Z (2019). Towards rapid prediction of drug-resistant cancer cell phenotypes: single cell mass spectrometry combined with machine learning. Chem. Commun 55, 616–619. 10.1039/c8cc08296k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rampler E, Abiead YE, Schoeny H, Rusz M, Hildebrand F, Fitz V, and Koellensperger G (2021). Recurrent topics in mass spectrometry-based metabolomics and lipidomics—standardization, coverage, and throughput. Anal. Chem 93, 519–545. 10.1021/acs.analchem.0c04698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Shea K, and Misra BB (2020). Software tools, databases and resources in metabolomics: updates from 2018 to 2019. Metabolomics 16, 36. 10.1007/s11306-020-01657-3. [DOI] [PubMed] [Google Scholar]
- 18.Spicer R, Salek RM, Moreno P, Cañueto D, and Steinbeck C (2017). Navigating freely-available software tools for metabolomics analysis. Metabolomics 13, 106. 10.1007/s11306-017-1242-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musharraf SG, Mazhar S, Siddiqui AJ, Choudhary MI, and Atta-ur-Rahman. (2013). Metabolite profiling of human plasma by different extraction methods through gas chromatography–mass spectrometry—an objective comparison. Anal. Chim. Acta 804, 180–189. 10.1016/j.aca.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 20.Matyushin DD, Sholokhova A.Yu, and Buryak AK (2021). Deep learning based prediction of gas chromatographic retention indices for a wide variety of polar and mid-polar liquid stationary phases. Int. J. Mol. Sci 22, 9194. 10.3390/ijms22179194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji H, Zeng F, Xu Y, Lu H, and Zhang Z (2017). KPIC2: an effective framework for mass spectrometry-based metabolomics using pure ion chromatograms. Anal. Chem 89, 7631–7640. 10.1021/acs.analchem.7b01547. [DOI] [PubMed] [Google Scholar]
- 22.Yu H, and Huan T (2022). Comprehensive assessment of the diminished statistical power caused by nonlinear electrospray ionization responses in mass spectrometry-based metabolomics. Anal. Chim. Acta 1200, 339614. 10.1016/j.aca.2022.339614. [DOI] [PubMed] [Google Scholar]
- 23.Smith CA, Want EJ, O’Maille G, Abagyan R, and Siuzdak G (2006). XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem 78, 779–787. 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 24.Pluskal T, Castillo S, Villar-Briones A, and Orešič M (2010). MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sindelar M, and Patti GJ (2020). Chemical discovery in the era of metabolomics. J. Am. Chem. Soc 142, 9097–9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsugawa H, Cajka T, Kind T, Ma Y, Higgins B, Ikeda K, Kanazawa M, VanderGheynst J, Fiehn O, and Arita M (2015). MS-DIAL: data independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lommen A (2009). MetAlign: interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Anal. Chem 81, 3079–3086. 10.1021/ac900036d. [DOI] [PubMed] [Google Scholar]
- 28.Rafiei A, and Sleno L (2015). Comparison of peak-picking workflows for untargeted liquid chromatography/high-resolution mass spectrometry metabolomics data analysis. Rapid Commun. Mass Spectrom 29, 119–127. [DOI] [PubMed] [Google Scholar]
- 29.Myers OD, Sumner SJ, Li S, Barnes S, and Du X (2017). Detailed investigation and comparison of the XCMS and MZmine 2 chromatogram construction and chromatographic peak detection methods for preprocessing mass spectrometry metabolomics data. Anal. Chem 89, 8689–8695. 10.1021/acs.analchem.7b01069. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Lu Y, Guo Y, Cao H, Wang Q, and Shui W (2018). Comprehensive evaluation of untargeted metabolomics data processing software in feature detection, quantification and discriminating marker selection. Anal. Chim. Acta 1029, 50–57. 10.1016/j.aca.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Myers OD, Sumner SJ, Li S, Barnes S, and Du X (2017). One step forward for reducing false positive and false negative compound identifications from mass spectrometry metabolomics data: new algorithms for constructing extracted ion chromatograms and detecting chromatographic peaks. Anal. Chem 89, 8696–8703. 10.1021/acs.analchem.7b00947. [DOI] [PubMed] [Google Scholar]
- 32.Broadhurst D, Goodacre R, Reinke SN, Kuligowski J, Wilson ID, Lewis MR, and Dunn WB (2018). Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 14, 72. [Internet][cited 2020 Dec 22]. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5960010/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quintás G, Sánchez-Illana Á, Piñeiro-Ramos JD, and Kuligowski J (2018). Chapter six - data quality assessment in untargeted LC-MS metabolomics. In Comprehensive Analytical Chemistry, Jaumot J, Bedia C, and Tauler R, eds. (Elsevier; ), pp. 137–164. https://www.sciencedirect.com/science/article/pii/S0166526X18300564. [Google Scholar]
- 34.Beger RD, Dunn WB, Bandukwala A, Bethan B, Broadhurst D, Clish CB, Dasari S, Derr L, Evans A, Fischer S, et al. (2019). Towards quality assurance and quality control in untargeted metabolomics studies. Metabolomics 15, 4. 10.1007/s11306-018-1460-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, and Xia J (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahieu NG, and Patti GJ (2017). Systems-level annotation of a metabolomics data set reduces 25 000 features to fewer than 1000 unique metabolites. Anal. Chem 89, 10397–10406. 10.1021/acs.analchem.7b02380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barupal DK, Baygi SF, Wright RO, and Arora M (2021). Data processing thresholds for abundance and sparsity and missed biological insights in an untargeted chemical analysis of blood specimens for exposomics. Front. Public Health 9, 653599. 10.3389/fpubh.2021.653599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baygi SF, Kumar Y, and Barupal DK (2022). IDSL.IPA Characterizes the Organic Chemical Space in Untargeted LC/HRMS Data Sets. J. Proteome Res 21, 1485–1494. Published online 2022 May 17. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pirttilä K, Balgoma D, Rainer J, Pettersson C, Hedeland M, and Brunius C (2022). Comprehensive peak characterization (CPC) in untargeted LC-MS analysis. Metabolites 12, 137. 10.3390/metabo12020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erny GL, Acunha T, Simó C, Cifuentes A, and Alves A (2016). Finnee — a Matlab toolbox for separation techniques hyphenated high resolution mass spectrometry dataset. Chemometr. Intell. Lab. Syst 155, 138–144. 10.1016/j.chemolab.2016.04.013. [DOI] [Google Scholar]
- 41.Erny GL, Gomes RA, Santos MSF, Santos L, Neuparth N, Carreiro-Martins P, Marques JG, Guerreiro ACL, and Gomes-Alves P (2020). Mining for peaks in LC-HRMS datasets using finnee – a case study with exhaled breath condensates from healthy, asthmatic, and COPD patients. ACS Omega 5, 16089–16098. 10.1021/acsomega.0c01610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borgsmüller N, Gloaguen Y, Opialla T, Blanc E, Sicard E, Royer AL, Bizec BL, Durand S, Migné C, and Pétéra M (2019. Sep). WiPP: workflow for improved peak picking for gas chromatography-mass spectrometry (GC-MS) data. Metabolites 9, 171. 10.3390/metabo9090171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chetnik K, Petrick L, and Pandey G (2020). MetaClean: a machine learning-based classifier for reduced false positive peak detection in untargeted LC–MS metabolomics data. Metabolomics 16, 117. [Internet]. [cited 2020 Oct 24]. 10.1007/s11306-020-01738-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melnikov AD, Tsentalovich YP, and Yanshole VV (2020). Deep learning for the precise peak detection in high-resolution LC–MS data. Anal. Chem 92, 588–592. 10.1021/acs.analchem.9b04811. [DOI] [PubMed] [Google Scholar]
- 45.Gloaguen Y, Kirwan J, and Beule D (2022). Deep learning assisted peak curation for large scale LC-MS metabolomics. Anal. Chem 94, 4930–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jirayupat C, Nagashima K, Hosomi T, Takahashi T, Tanaka W, Samransuksamer B, Zhang G, Liu J, Kanai M, and Yanagida T (2021). Image processing and machine learning for automated identification of chemo-/biomarkers in chromatography-mass spectrometry. Anal. Chem 93, 14708–14715. 10.1021/acs.analchem.1c03163. [DOI] [PubMed] [Google Scholar]
- 47.Guo J, Shen S, Xing S, Chen Y, Chen F, Porter EM, Yu H, and Huan T (2021). EVA: evaluation of metabolic feature fidelity using a deep learning model trained with over 25000 extracted ion chromatograms. Anal. Chem 93, 12181–12186. 10.1021/acs.analchem.1c01309. [DOI] [PubMed] [Google Scholar]
- 48.Deng K, Zhao F, Rong Z, Cao L, Zhang L, Li K, Hou Y, and Zhu ZJ (2021). WaveICA 2.0: a novel batch effect removal method for untargeted metabolomics data without using batch information. Metabolomics 17, 87. 10.1007/s11306-021-01839-7. [DOI] [PubMed] [Google Scholar]
- 49.Brunius C, Shi L, and Landberg R (2016). Large-scale untargeted LC-MS metabolomics data correction using between-batch feature alignment and cluster-based within-batch signal intensity drift correction. Metabolomics 12, 173. 10.1007/s11306-016-1124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuligowski J, Sánchez-Illana Á, Sanjuán-Herráez D, Vento M, and Quintás G (2015). Intra-batch effect correction in liquid chromatography-mass spectrometry using quality control samples and support vector regression (QC-SvRc). Analyst 140, 7810–7817. 10.1039/c5an01638j. [DOI] [PubMed] [Google Scholar]
- 51.Tokareva AO, Chagovets VV, Kononikhin AS, Starodubtseva NL, Nikolaev EN, and Frankevich VE (2021). Normalization methods for reducing interbatch effect without quality control samples in liquid chromatography-mass spectrometry-based studies. Anal. Bioanal. Chem 413, 3479–3486. [DOI] [PubMed] [Google Scholar]
- 52.Pomyen Y, Wanichthanarak K, Poungsombat P, Fahrmann J, Grapov D, and Khoomrung S (2020). Deep metabolome: applications of deep learning in metabolomics. Comput. Struct. Biotechnol. J 18, 2818–2825. 10.1016/j.csbj.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel CJ (2017). Analytic complexity and challenges in identifying mixtures of exposures associated with phenotypes in the exposome era. Curr Epidemiol Rep. 4, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nemet I, Saha PP, Gupta N, Zhu W, Romano KA, Skye SM, Cajka T, Mohan ML, Li L, Wu Y, et al. (2020). A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell 180, 862–877.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zacharias HU, Hertel J, Johar H, Pietzner M, Lukaschek K, Atasoy S, Kunze S, Vӧlzke H, Nauck M, Friedrich N, et al. (2021). A metabolome-wide association study in the general population reveals decreased levels of serum laurylcarnitine in people with depression. Mol Psychiatry 26, 7372–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson O, Keski-Rahkonen P, Chatzi L, Kogevinas M, Nawrot T, Pizzi C, Plusquin M, Richiardi L, Robinot N, Sunyer J, et al. (2018). Cord blood metabolic signatures of birth weight: a population-based study. J. Proteome Res 17, 1235–1247. 10.1021/acs.jproteome.7b00846. [DOI] [PubMed] [Google Scholar]
- 57.Gumpenberger T, Brezina S, Keski-Rahkonen P, Baierl A, Robinot N, Leeb G, Habermann N, Kok D, Scalbert A, Ueland PM, et al. (2021). Untargeted metabolomics reveals major differences in the plasma metabolome between colorectal cancer and colorectal adenomas. Metabolites 11, 119. 10.3390/metabo11020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antonelli J, Claggett BL, Henglin M, Kim A, Ovsak G, Kim N, Deng K, Rao K, Tyagi O, Watrous JD, et al. (2019). Statistical workflow for feature selection in human metabolomics data. Metabolites 9, 143. 10.3390/metabo9070143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mazzella M, Sumner SJ, Gao S, Su L, Diao N, Mostofa G, Qamruzzaman Q, Pathmasiri W, Christiani DC, Fennell T, and Gennings C (2020). Quantitative methods for metabolomic analyses evaluated in the children’s health exposure analysis resource (CHEAR). J. Expo. Sci. Environ. Epidemiol 30, 16–27. 10.1038/s41370-019-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liebal UW, Phan ANT, Sudhakar M, Raman K, and Blank LM (2020). Machine learning applications for mass spectrometry-based metabolomics. Metabolites 10, 243. 10.3390/metabo10060243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mendez KM, Reinke SN, and Broadhurst DI (2019). A comparative evaluation of the generalised predictive ability of eight machine learning algorithms across ten clinical metabolomics data sets for binary classification. Metabolomics 15, 150. 10.1007/s11306-019-1612-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sen P, Lamichhane S, Mathema VB, McGlinchey A, Dickens AM, Khoomrung S, and Orešič M (2021). Deep learning meets metabolomics: a methodological perspective. Brief. Bioinform 22, 1531–1542. [DOI] [PubMed] [Google Scholar]
- 63.Sharma A, Lysenko A, Boroevich KA, Vans E, and Tsunoda T (2021). DeepFeature: feature selection in nonimage data using convolutional neural network. Brief. Bioinform 22, bbab297. 10.1093/bib/bbab297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deep learning (2022). [Internet]. [cited 2022 Mar 1]. https://www.deeplearningbook.org/.
- 65.Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S, Singh S, Madamba EV, Bettencourt R, Richards L, et al. (2020). A universal gut-microbiome-derived signature predicts cirrhosis. Cell Metabol. 32, 901. 10.1016/j.cmet.2020.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, Quan S, Zhang F, Sun R, Qian L, et al. (2020). Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182, 59–72.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varma VR, Oommen AM, Varma S, Casanova R, An Y, Andrews RM, O’Brien R, Pletnikova O, Troncoso JC, Toledo J, et al. (2018). Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: a targeted metabolomics study. PLoS Med. 15, e1002482. 10.1371/journal.pmed.1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liang L, Rasmussen MLH, Piening B, Shen X, Chen S, Rӧst H, Snyder JK, Tibshirani R, Skotte L, Lee NC, et al. (2020). Metabolic dynamics and prediction of gestational age and time to delivery in pregnant women. Cell 181, 1680–1692.e15. 10.1016/j.cell.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takahashi Y, Ueki M, Yamada M, Tamiya G, Motoike IN, Saigusa D, Sakurai M, Nagami F, Ogishima S, Koshiba S, et al. (2020). Improved metabolomic data-based prediction of depressive symptoms using nonlinear machine learning with feature selection. Transl. Psychiatry 10, 157. 10.1038/s41398-020-0831-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gaskins AJ, Tang Z, Hood RB, Ford J, Schwartz JD, Jones DP, Laden F, and Liang D (2021). Periconception air pollution, metabolomic biomarkers, and fertility among women undergoing assisted reproduction. Environ. Int 155, 106666. 10.1016/j.envint.2021.106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeong A, Fiorito G, Keski-Rahkonen P, Imboden M, Kiss A, Robinot N, Gmuender H, Vlaanderen J, Vermeulen R, Kyrtopoulos S, et al. (2018). Perturbation of metabolic pathways mediates the association of air pollutants with asthma and cardiovascular diseases. Environ. Int 119, 334–345. 10.1016/j.envint.2018.06.025. [DOI] [PubMed] [Google Scholar]
- 72.Niedzwiecki MM, Walker DI, Howell JC, Watts KD, Jones DP, Miller GW, and Hu WT (2019). High-resolution metabolomic profiling of Alzheimer’s disease in plasma. Ann. Clin. Transl. Neurol 7, 36–45. 10.1002/acn3.50956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alakwaa FM, Chaudhary K, and Garmire LX (2018). Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. J. Proteome Res 17, 337–347. 10.1021/acs.jproteome.7b00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stamate D, Kim M, Proitsi P, Westwood S, Baird A, Nevado-Holgado A, Hye A, Bos I, Vos SJ, Vandenberghe R, et al. (2019). A metabolite-based machine learning approach to diagnose Alzheimer-type dementia in blood: results from the European Medical Information Framework for Alzheimer disease biomarker discovery cohort. Alzheimers Dement. 5, 933–938. 10.1016/j.trci.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trainor PJ, DeFilippis AP, and Rai SN (2017). Evaluation of classifier performance for multiclass phenotype discrimination in untargeted metabolomics. Metabolites 7, E30. 10.3390/metabo7020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Merino Martinez R, Müller H, Negru S, Ormenisan A, Arroyo Mühr LS, Zhang X, Trier Møller F, Clements MS, Kozlakidis Z, Pimenoff VN, et al. (2021). Human exposome assessment platform. Environ Epidemiol 5, e182. 10.1097/ee9.0000000000000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pero-Gascon R, Hemeryck LY, Poma G, Falony G, Nawrot TS, Raes J, Vanhaecke L, De Boevre M, Covaci A, and De Saeger S (2022). FLEXiGUT: rationale for exposomics associations with chronic low-grade gut inflammation. Environ. Int 158, 106906. 10.1016/j.envint.2021.106906. [DOI] [PubMed] [Google Scholar]
- 78.Blaženović I, Kind T, Ji J, and Fiehn O (2018). Software tools and approaches for compound identification of LC-MS/MS data in metabolomics. Metabolites 8, E31. 10.3390/metabo8020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aron AT, Gentry EC, McPhail KL, Nothias LF, Nothias-Esposito M, Bouslimani A, Petras D, Gauglitz JM, Sikora N, Vargas F, et al. (2020). Reproducible molecular networking of untargeted mass spectrometry data using GNPS. Nat. Protoc 15, 1954–1991. [DOI] [PubMed] [Google Scholar]
- 80.Xue J, Guijas C, Benton HP, Warth B, and Siuzdak G (2020). METLIN MS 2 molecular standards database: a broad chemical and biological resource. Nat. Methods 17, 953–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, et al. (2007). HMDB: the human metabolome database. Nucleic Acids Res. 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, et al. (2010). MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom 45, 703–714. [DOI] [PubMed] [Google Scholar]
- 83.Vinaixa M, Schymanski EL, Neumann S, Navarro M, Salek RM, and Yanes O (2016). Mass spectral databases for LC/MS- and GC/MS-based metabolomics: state of the field and future prospects. TrAC Trends Anal. Chem 78, 23–35. 10.1016/j.trac.2015.09.005. [DOI] [Google Scholar]
- 84.Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, and Hollender J (2014). Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol 48, 2097–2098. 10.1021/es5002105. [DOI] [PubMed] [Google Scholar]
- 85.Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TWM, Fiehn O, Goodacre R, Griffin JL, et al. (2007). Proposed minimum reporting standards for chemical analysis chemical analysis working group (CAWG) metabolomics standards initiative (MSI). Metabolomics 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, et al. (2019). PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Y, Elenee Argentinis J, and Weber G (2016). IBM Watson: how cognitive computing can Be applied to big data challenges in life sciences research. Clin. Therapeut 38, 688–701. 10.1016/j.clinthera.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 88.Warth B, Spangler S, Fang M, Johnson CH, Forsberg EM, Granados A, Martin RL, Domingo-Almenara X, Huan T, Rinehart D, et al. (2017). Exposome-scale investigations guided by global metabolomics, pathway analysis, and cognitive computing. Anal. Chem 89, 11505–11513. 10.1021/acs.analchem.7b02759. [DOI] [PubMed] [Google Scholar]
- 89.Majumder ELW, Billings EM, Benton HP, Martin RL, Palermo A, Guijas C, Rinschen MM, Domingo-Almenara X, Montenegro-Burke JR, Tagtow BA, et al. (2021). Cognitive analysis of metabolomics data for systems biology. Nat. Protoc 16, 1376–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dührkop K, Shen H, Meusel M, Rousu J, and Bӧcker S (2015). Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 112, 12580–12585. 10.1073/pnas.1509788112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ruttkies C, Schymanski EL, Wolf S, Hollender J, and Neumann S (2016). MetFrag relaunched: incorporating strategies beyond in silico fragmentation. J. Cheminform 8, 3. 10.1186/s13321-016-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y, Kora G, Bowen BP, and Pan C (2014). MIDAS: a database-searching algorithm for metabolite identification in metabolomics. Anal. Chem 86, 9496–9503. 10.1021/ac5014783. [DOI] [PubMed] [Google Scholar]
- 93.Wang F, Liigand J, Tian S, Arndt D, Greiner R, and Wishart DS (2021). CFM-ID 4.0: more accurate ESI-MS/MS spectral prediction and compound identification. Anal. Chem 93, 11692–11700. 10.1021/acs.analchem.1c01465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bittremieux W, Schmid R, Huber F, van der Hooft JJ, Wang M, and Dorrestein PC (2022). Comparison of cosine, modified cosine, and neutral loss based spectral alignment for discovery of structurally related molecules. Preprint at bioRxiv. [Internet][cited 2022 Jun 12]. https://doi.org/10.1101/2022.06.01.494370. https://biorxiv.org/lookup/doi/10.1101/2022.06.01.494370. [DOI] [PubMed]
- 95.Schollée JE, Schymanski EL, Stravs MA, Gulde R, Thomaidis NS, and Hollender J (2017). Similarity of high-resolution tandem mass spectrometry spectra of structurally related micropollutants and transformation products. J. Am. Soc. Mass Spectrom 28, 2692–2704. 10.1007/s13361-017-1797-6. [DOI] [PubMed] [Google Scholar]
- 96.Huber F, Ridder L, Verhoeven S, Spaaks JH, Diblen F, Rogers S, and van der Hooft JJJ (2021). Spec2Vec: improved mass spectral similarity scoring through learning of structural relationships. PLoS Comput. Biol 17, e1008724. 10.1371/journal.pcbi.1008724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huber F, van der Burg S, van der Hooft JJJ, and Ridder L (2021). MS2DeepScore: a novel deep learning similarity measure to compare tandem mass spectra. J. Cheminform 13, 84. 10.1186/s13321-021-00558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xing S, Jiao Y, Salehzadeh M, Soma KK, and Huan T (2021). SteroidXtract: deep learning-based pattern recognition enables comprehensive and rapid extraction of steroid-like metabolic features for automated biology-driven metabolomics. Anal. Chem 93, 5735–5743. 10.1021/acs.analchem.0c04834. [DOI] [PubMed] [Google Scholar]
- 99.United States Government Accountability Office. (2019). HIGH-RISK SERIES substantial efforts needed to achieve greater progress on high-risk areas. [Internet]. https://www.gao.gov/assets/gao-19-157sp.pdf.
- 100.Yu M, Tu P, Dolios G, Dassanayake PS, Volk H, Newschaffer C, Fallin MD, Croen L , Lyall K, Schmidt R, et al. (2021). Tooth biomarkers to characterize the temporal dynamics of the fetal and early-life exposome. Environ. Int 157, 106849. 10.1016/j.envint.2021.106849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fox Ramos AE, Evanno L, Poupon E, Champy P, and Beniddir MA (2019). Natural products targeting strategies involving molecular networking: different manners, one goal. Nat. Prod. Rep 36, 960–980. [DOI] [PubMed] [Google Scholar]
- 102.Liu Y, De Vijlder T, Bittremieux W, Laukens K, and Heyndrickx W (2021). Current and future deep learning algorithms for tandem mass spectrometry (MS/MS)-based small molecule structure elucidation. Rapid Commun. Mass Spectrom e9120. 10.1002/rcm.9120. [DOI] [PubMed] [Google Scholar]
- 103.Dekermanjian J, Labeikovsky W, Ghosh D , and Kechris K (2021). MSCAT: a machine learning assisted catalog of metabolomics software tools. Metabolites 11, 678. 10.3390/metabo11l00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meng C, Kuster B, Culhane AC, and Gholami AM (2014). Amultivariate approach to the integration of multi-omics datasets. BMC Bioinformatics 15, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Picard M, Scott-Boyer MP, Bodein A, Périn O, and Droit A (2021). Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J 19, 3735–3746. 10.1016/j.csbj.2021.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reel PS, Reel S, Pearson E,Trucco E, and Jefferson E (2021). Using machine learning approaches for multi-omics data analysis: a review. Biotechnol. Adv 49, 107739. 10.1016/j.biotechadv.2021.107739. [DOI] [PubMed] [Google Scholar]
- 107.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chung MK, Rappaport SM, Wheelock CE, Nguyen VK, van der Meer TP, Miller GW, Vermeulen R, and Patel CJ (2021). Utilizing a biology-driven approach to map the exposome in health and disease: an essential investment to drive the next generation of environmental discovery. Environ. Health Perspect 129, 085001. 10.1289/ehp8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.van der Hooft JJJ, Mohimani H, Bauermeister A, Dorrestein PC, Duncan KR, and Medema MH (2020). Linking genomics and metabolomics to chart specialized metabolic diversity. Chem. Soc. Rev 49, 3297–3314. 10.1039/d0cs00162g. [DOI] [PubMed] [Google Scholar]
- 110.Yu M, and Petrick L (2020). Untargeted high-resolution paired mass distance data mining for retrieving general chemical relationships. Commun. Chem 3, 157. 10.1038/s42004-020-00403-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu M, Teitelbaum SL, Dolios G, Dang LHT, Tu P, Wolff MS, and Petrick LM (2022). Molecular gatekeeper discovery: workflow for linking multiple exposure biomarkers to metabolomics. Environ. Sci. Technol 56, 6162–6171. [Internet]. 2022 Feb 7 [cited 2022 Mar 15]. 10.1021/acs.est.1c04039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bonini P, Kind T, Tsugawa H, Barupal DK, and Fiehn O (2020). Retip: retention time prediction for compound annotation in untargeted metabolomics. Anal. Chem 92, 7515–7522. 10.1021/acs.analchem.9b05765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Witting M, and Bӧcker S (2020). Current status of retentiontime prediction in metabolite identification. J. Separ. Sci 43, 1746–1754. [DOI] [PubMed] [Google Scholar]
- 114.Human health exposure analysis resource (HHEAR) data center (2022). [Internet]. [cited 2022 Apr 3]. https://hheardatacenter.mssm.edu/.
- 115.Sud M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Nair KS, et al. (2016). Metabolomics Workbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 44, D463–D470. 10.1093/nar/gkv1042. [DOI] [PMC free article] [PubMed] [Google Scholar]