

Abstract
Background
Gorlin-Goltz syndrome (GS) is an inherited disease characterized by predisposition to basal cell carcinomas (BCCs) and various developmental defects, whose numerous disease-causing PTCH1 mutations have been identified in the hedgehog (Hh) signaling pathway.
Methods
In this study, whole exome sequencing was used to screen for both somatic and germline deleterious mutations in three sisters with a lethal GS. The mutations we found were confirmed by subcloning and Sanger sequencing of the genomic DNA. RNA-seq was performed to profile gene expression in paired BCCs samples and the expression levels for selected genes were validated by quantitative PCR.
Results
The clinical and histopathologic features were analyzed for the proband in the three-generation GS family. We identified the insertion mutation PTCH1 c.1341dupA (p. L448Tfs*49), which segregated with BCC phenotype and contributed to the death of two in four patients from a Chinese family with GS. Compared with adjacent non-cancerous tissues (ANCT), four second-hit mutations were found in four of the six pairs of BCC from three patients. Of note, somatic genomic alterations in all six BCC samples were mainly clustered into non-clock-like Signature 7 (ultraviolet mutagenesis) and 11 (related to certain alkylating agents). Both RNA-seq and quantitative RT-PCR confirmed that the mRNA levels of PTCH1 and its effector GLI1 were markedly upregulated in six pairs of BCC samples versus ANCT.
Conclusions
The distinct non-clock-like signatures of BCCs indicated that GS was not a life-threatening illness. The main reasons for untimely death of GS patients were PTCH1 mutation, exposure to intense ultraviolet radiationand the poor economic conditions.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-022-01324-7.
Keywords: Gorlin-Goltz syndrome, PTCH1, Mutational signatures
Background
Gorlin-Goltz syndrome (GS, OMIM #109400), also called nevoid basal cell carcinoma syndrome (NBCCS), is a rare autosomal dominant disorder that predispose to early onset tumours, such as multiple BCCs and medulloblastoma [1]. It is generally recognized that the life expectancy is not significantly affected by GS [2]. Here, we collected one three-generation family with GS from Hainan province of China. Unfortunately, two patients died of cachexia resulting from aggressive BCCs at the age of 54 and 39 years, respectively. To investigate the genetic cause of the untimely death, we performed WES to detect the causative germline and somatic mutations. Moreover, RNA-Seq based transcriptomics, combined with quantitative RT-PCR, were used to determine preferential expressions of the genes related to GS. It is of note that GS is mostly caused by mutations in the patched 1 (PTCH1) gene in hedgehog (Hh) signaling pathway [3]. More recently, patched 2 (PTCH2) is no longer considered a highly susceptible gene for GS [4]. Although the germline mutation in PTCH1 c.1341dupA (p. L448Tfs*49) we found in this family has been listed in the Human Gene Mutation Database (HGMD®), no clinical data are available from those patients. This study provided the clinical information and PTCH1 mutations identified in three sisters with a lethal GS.
Methods
Sample collection
A three-generation GS family was identified from Hainan Province of China (Fig. 1a). All family members and non-familial cases were carefully examined by at least 2 dermatologists, and the diagnosis was confirmed by histological examination of skin biopsy specimens on the proband (II-6). With patents’ written informed consent, 13 blood samples were collected from the family members. Moreover, six pairwise BCCs and ANCT were dissected from 3 affected females (II-2, II-4, II-6). All procedures followed the guidelines of the Helsinki Declaration and were approved by the Scientific Ethics Committee of Affiliated Haikou People’s Hospital, Xiangya School of Medicine.
Fig. 1.

a The pedigree chart of three-generation familial GS. Squares and circles denote males and females, respectively. Affected and unaffected individuals are represented by black and open symbols, respectively. Slash lines and dot marks indicate death and disease-carrying genotype, respectively. The initial proband was indicated by an arrow. b Sanger sequencing chromatograms of proband (II-6, affected) and normal control (II-7, unaffected) at the c.1341dupA mutation site indicated by arrow
Whole exome sequencing (WES) and data analyses
Genomic DNA (gDNA) was extracted from peripheral blood and the frozen tissues with DNeasy Blood & Tissue Kit (Qiagen, Germany) according to the manufacturer’s protocol. The purity and concentration of the gDNA met the sequencing requirements. WES was performed using gDNA with a SureSelectXT Reagent kit (Agilent, USA), SureSelectXT Human All Exon V6 (Agilent, USA), TruSeq PE Cluster Kit v3-cBot-HS (Illumina, USA), and HiSeq SBS Kit V4 (Illumina, USA). Quantification was performed with an Agilent Bioanalyzer (Agilent Technologies, USA), and multiplexed sequencing was done on HiSeq 2500 sequencers with 2 × 150 paired-end modules (Illumina, USA). Total sequencing depth was performed at 100× coverage.
Before variant calling, the raw sequence reads were mapped to human genome reference (hg19). Then, PCR duplicates were marked to mitigate the possible biases and the base quality scores were recalibrated by using the Genome Analysis Toolkit (GATK) [5]. For germline single nucleotide variants (SNVs) and insertion/deletion variants (indels), the GATK4 HaplotypeCaller was used to produce a genomic variant call format (GVCF) file. Next, the GVCFs from multiple samples were consolidated into a GenomicsDB datastore, followed by joint genotyping and variant quality score recalibration (VQSR) filtering to produce the final callsets with the balance between sensitivity and precision [5]. Exome-wide sequence variants were filtered with the sequencing depth < 5×. For somatic short variants (SNVs and Indels), the GATK4 MuTect2 was used to analyze somatic mutations in BCCs and ANCT, and the confident somatic calls were filtered by GATK4 FilterMutectCalls and FilterByOrientationBias tools [5].
The Ensembl variant effect predictor (VEP) and vcf2maf tools were applied to generate the somatic mutation annotation format (MAF) files [6]. Moreover, ANNOVAR was also used to annotate population frequencies of variations [7]. The variants were identified as low-frequent functional mutation with < 0.01 frequency in ExAC03 database [8] and 1000 genome database [9], < 0.05 frequency in GeneskyDB database. According to the results, the variants were extracted as one of these functional annotations: “Frame_Shift_Del”, “Frame_Shift_Ins”, “In_Frame_Del”, “In_Frame_Ins”, “Missense_Mutation”, “Nonsense_Mutation”, “Nonstop_Mutation”, “Splice_Site” or “Translation_Start_Site”.
Subcloning and Sanger re-sequencing of the genomic DNA
Genomic DNA was extracted from each pair of BCCs and ANCT using a TIANamp Genomic DNA Kit (TransGen Biotech, Beijing, China). PCR amplification products were subcloned into the pEASY®-Blunt Cloning Vector (TransGen Biotech, Beijing, China) followed by Escherichia coli transformation and blue-white screening. The colonies were analyzed by colony PCR, moreover, capillary electrophoresis was performed on an ABI Prism 3130xl Genetic Analyzer (Applied Biosystems, USA) to screen the somatic mutations through the diverse length of colony PCR products. Fragment analysis was performed using the Peak Scanner Software v1.0 (Applied Biosystems, USA).
RNA-seq
RNA quality was accessed by an Agilent Bioanalyzer and those samples with RNA integrity number (RIN) higher than 7 were used to construct RNA-seq libraries by adapter ligation. RNA-seq libraries were prepared from 1 μg of total RNA using VAHTS mRNA-seq V3 Library Prep Kit (Vazyme Biotech) according to manufacturer’s instruction. mRNA was purified with oligo-dT magnetic beads, followed by fragmentation, end repair, adapter ligation and PCR amplification. The analysis of final RNA quality and integrity was performed with Agilent BioAnalyzer and Qubit Fluorometer (Invitrogen, USA). All libraries were sequenced on Illumina NovaSeq platform to generate 150 bp paired-end reads. Raw sequencing data (.fastq files) was processed using STAR (spliced transcripts alignment to a reference) to generate read alignments with hg19. Effective read counts for genes were obtained with featureCounts under default settings, standardized and analyzed using DEseq2.
Real-time quantitative PCR (qPCR)
The mRNA expression levels of PTCH1 (NM_000264.3) and GLI1 (NM_005269.3) were evaluated by real-time qPCR, using total RNA from BCC and ANCT from three sisters with GS. The total RNA of each sample was reverse transcribed to cDNA using the PrimeScript RT Reagent Kit (Takra, Japan). qPCR reactions were conducted in triplicate using the TB Green Premix Ex Taq II (Takara, Japan) and the ABI 7300 system (Applied Biosystems, USA). The primers were designed with the Primer 3 software (http://bioinfo.ut.ee/primer3-0.4.0). qRT-PCR data were analyzed by two-tailed paired-sample t-tests and statistical comparisons were considered significantly different at P < 0.01.
Results
Clinical features
In this family, the earliest onset of the disease was at 16 years old, and the average onset age was 19 years. The proband (II-6) complained of multiple hyperpigmented nodules on her body (Fig. 2a). She had ocular hypertelorism, but no obvious palmar and/or plantar pits. Cranial CT and upper-limb X-ray showed ectopic calcifications of the falx cerebri and abnormalities in bone density, respectively (Fig. 2b and c). Histopathologic examination of biopsy specimen from her right face revealed compact basaloid cell nests with peripheral palisading that extend into the dermis (Fig. 3a). Except for CK10 (Fig. 3b), Bcl-2 and BerEp4 immunoreactivity were observed (Fig. 3c and d). There were three other family members affected by this disease. Hence, the diagnosis of GS was made based on three major and two minor criteria, established in 2011 [10]. Among them, the patient (II-2) died of cachexia resulting from aggressive BCCs on her neck four months after the diagnosis. Similarly, it was told that the patient (I-1) died of giant BCCs on his nose in 2010.
Fig. 2.
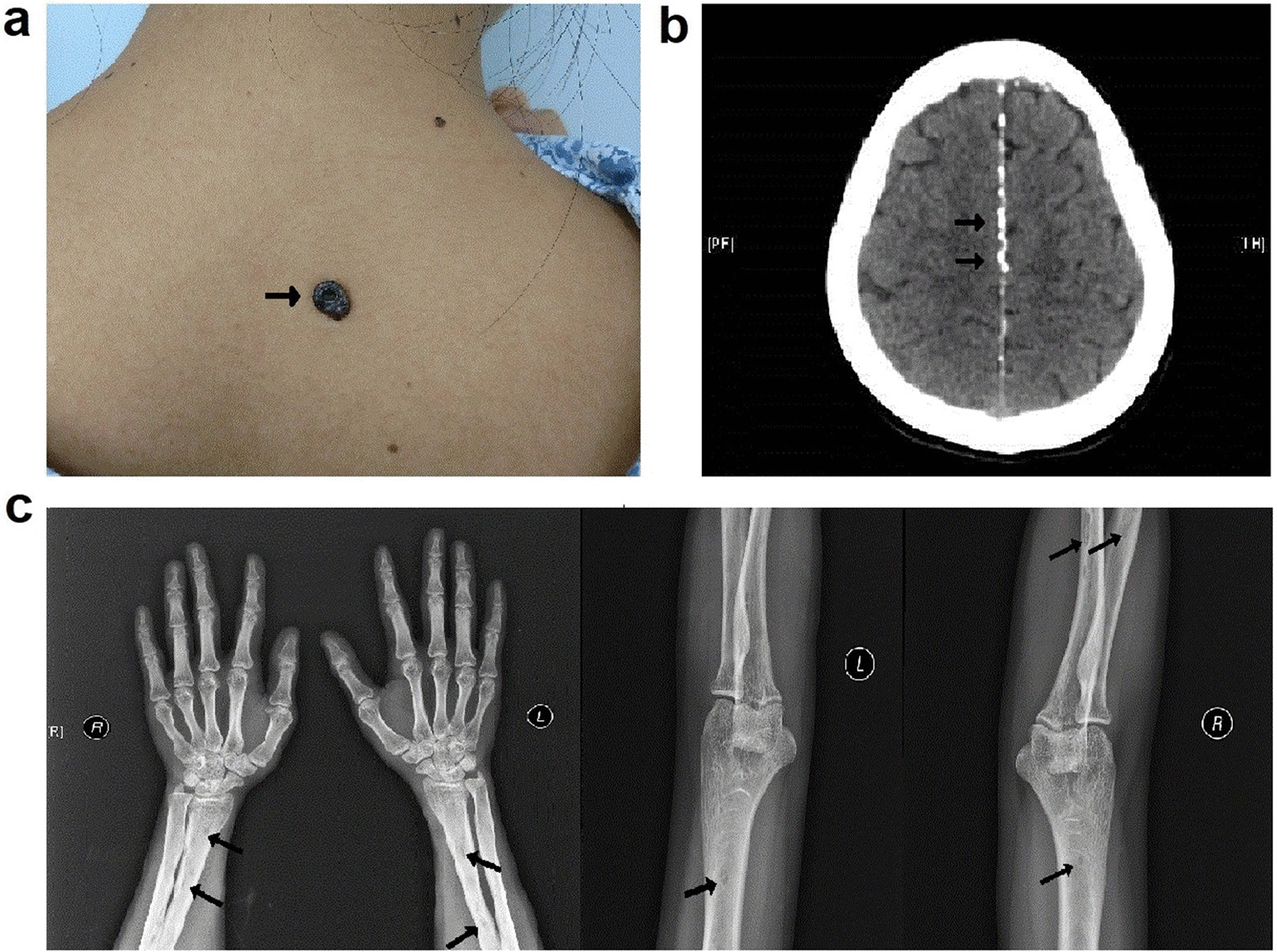
BCC lesions on the back (a), falx cerebri calcification on cranial CT b and low-density shadow on upper-limb X-ray c of the proband, which is indicated by arrows
Fig. 3.
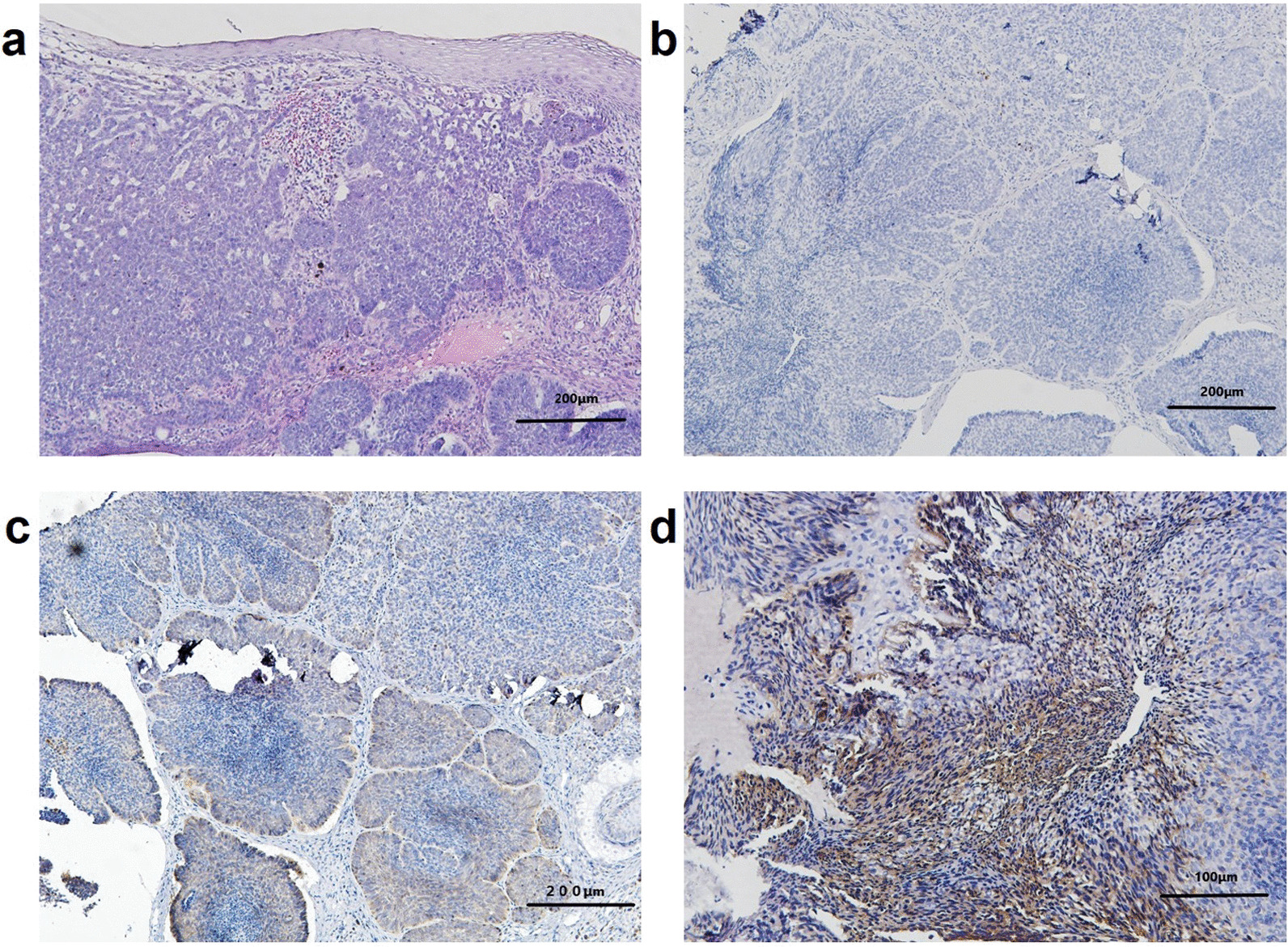
Histopathologic examination of biopsy specimen showed compact basaloid cell nests with peripheral palisading that extend into the dermis (a, HE × 100). Except for CK10 (b, SP × 100), Bcl-2 and BerEp4 immunoreactivity were observed (c and d, SP × 100 and 200, respectively)
Germline and somatic mutations in the PTCH1 tumor suppressor gene
In this study, the paternal germline mutation c.1341dupA (p.L448Tfs*49) in PTCH1 cosegregated with multiple BCCs (Fig. 1b), which was identified by WES and confirmed by Sanger sequencing. To our knowledge, this insertion mutation has been listed on HGMD® and molecular genetics of Kitasato University School of Medicine website. But no clinical data are available from those patients. Moreover, we analyzed WES data from 6 paired BCC/ANCT and their matched blood samples. As Table 1 indicated, different somatic PTCH1 mutations were found in four of the six paired BCC samples. Those somatic mutations were present on the wild-type maternal allele, which was confirmed by subcloning of genomic DNA and Sanger sequencing. Moreover, additional driver mutations in ERBB2 and PPP6C were found in two of the six BCCs (Additional file 1). Loss of heterozygosity of PPP6C and PTCH1 were observed in two and three of the six BCCs, respectively (Additional file 2). These results showed the two-hit mutational inactivation of PTCH1 and the genomic heterogeneity in BCCs. These results revealed the genomic heterogeneity in BCCs, as supported by previous findings [11].
Table 1.
Somatic PTCH1 mutations found in four of the six paired BCC samples
| No. in the pedigree chart | Sex | Age (years) | Location of skin biopsies | Paired skin Samples | No. of somatic mutation | TMB (mutations/MB) | Somatic mutations of PTCH1 in BCC | Reads in BCC, no. | Reads in ANCT, no. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | Non-ref. | Ref. | Non-ref. | ||||||||
| II. 2 | Female | 39 | Left side of the neck | II.2-1_BCC/ ANCT | 346 | 10.81 | c.391G > T (p.Glu131*) | 33 | 10 | 66 | 0 |
| Abdomen | II.2-2_BCC/ANCT | 39 | 1.22 | c.3271G > C (p.Gly1091Arg) | 196 | 13 | 148 | 0 | |||
| II. 4 | Female | 36 | Behind the right ear | II.4-1_BCC/ANCT | 386 | 12.06 | Negative | ||||
| Back of the neck | II.4-2_BCC/ANCT | 200 | 6.25 | c.1396C > T (p.Gln466*) | 139 | 28 | 222 | 0 | |||
| II. 6 | Female | 29 | Right side of the nose | II.6-1_BCC/ANCT | 567 | 17.71 | Negative | ||||
| Left side of the nose | II.6-2_BCC/ANCT | 989 | 30.9 | c.1130dupA (p.Tyr379Valfs*58) | 142 | 11 | 159 | 0 | |||
non-ref. non-reference; ref. reference; TMB tumor mutation burden; MB mega base
Distinct mutational signatures of the BCCs from three sisters
The mutational 0signature analysis was performed on the exomes of six BCC-ANCT pairs, inferring DNA damage and repair processes during the evolution of cancer. As indicated in Fig. 4, Signature 7 and 11 were distinctly detected in BCCs with either one or two PTCH1 mutations using the deconstructSigs R package. Of note, Signature 7 in a brown color predominated over Signature 11 in a yellow color. It is known that Signature 7 represents ultraviolet (UV) light related patterns of mutations, which show C > T transitions at dipyrimidines and CC > TT double nucleotide substitutions [12]. Whereas Signature 11 is statistically associated with the C > T substitutions pattern of agents treatment [13]. Both Signature 7 and 11 are non-clock-like mutational signature patterns, which are associated with better prognosis [14]. It might explain why GS has little effect on the life expectancy of patients.
Fig. 4.

Two distinct mutational signatures were present in six pairs BCC and ANCT, which are Signature7 (brown) and Signature11 (yellow)
Loss-of-function and over-expression of PTCH1 leading to activation of the Hh pathway in BCCs
Transcriptional analysis by RNA-seq showed up-regulation of the Hh pathway genes in BCCs (Fig. 5a). In particular, our finding showed that PTCH1 and PTCH2 shared similar expression patterns with GLI1 and GLI2. We quantified the expression of PTCH1 and its target GLI1 genes in six pairwise samples to confirm the results obtained by RNA-seq. The results indicated that the transcript levels of PTCH1 (Fig. 5b) and GLI1 (Fig. 5c) were relatively higher in BCCs compared with ANCT. The presence of inactivating mutations in PTCH1 promote autocrine activation of Hh signaling and genesis of BCCs. It is reported that the binding of Hh ligands to up-regulated PTCH1 receptors can activate smoothened (SMO) and lead to the overexpression of down-stream transcription factors. Moreover, transcription factor GLI2 directly activates GLI1 and forms a positive feedback loop to promote BCCs [15, 16].
Fig. 5.

a Hierarchical clustering and heatmap representation of 10 differentially expressed Hedgehog signaling pathway genes. Clustering was performed on six pairs of BCC and ANCT from three sisters with GS. Expression levels are colored with a red-to-green gradient, where red and green represented up-regulation and down-regulation, respectively. The expression levels of PTCH1 b and GLI1 c were significantly up-regulated in six pairs of BCC versus ANCT, confirmed by real-time qPCR. **p value < 0.01 (two-tailed paired-sample t-tests; n = 12)
Discussion
It is known that GS is not a life-threatening illness and has a near-normal life expectancy [2]. Except that complication was given as the underlying cause of death, around 10% (3/32) of deaths has been reported due to invasive lesions and intracranial involvement [17, 18]. However, in this three-generation GS family, one male (I-1) and one female (II-2) died as a direct result of aggressive BCCs, whose ages were 54 and 39 years respectively. There were three main reasons for their untimely death, including driver gene mutations, exposure to intense UV radiation and the poor economic conditions.
To our knowledge, the activation of Hh pathway represents the initial tumorigenic event in BCCs. In this study, the PTCH1 c.1341dupA (p.L448Tfs*49) mutation was identified in this GS family. Besides this germline mutation, different somatic PTCH1 mutations were found in four of the six paired BCC samples. These findings provided the evidence of two-hit mutational inactivation of PTCH1 and the genomic heterogeneity of BCCs, which supported the Knudson’s two-hit tumor progression hypothesis [19]. It is generally accepted that PTCH1 is one of the most common driver genes in BCCs. The concept of “driver” and “passenger” genes in cancer was proposed by Stratton et al. [20]. In contrast to the passenger gene, the driver gene is critical for oncogenesis. The driver and passenger gene mutations might be regarded as “mountain” and “hill” [21], respectively. The driver mutations are defined as making important contributions to cancer development, as compared to the passenger mutations. Previous studies have suggested the driver pivotal role of PTCH1, TP53, and SMO in BCCs development [22]. Moreover, PPP6C and ERBB2 are reported as additional driver genes in BCCs [11], which were also found in two of the six BCC samples from three sisters. The major limitation of this study was that we didn’t perform additional confirmation of these driver genes due to a limited budget and time.
The three-generation family with GS lives in a rural area of Hainan province, located in the southernmost part of China. This is the reason that they are exposed to intense UV radiation for a long time. It is believed that UV radiation plays a crucial role in the onset of skin carcinogenesis through DNA damage and immune suppression. Of note, UVB is directly absorbed by DNA and induce UV-signature DNA damages [23]. In this study, the results of WES showed that Signature 7 and 11 were mainly present with similarities in six pairs BCC versus ANCT, making it possible to decipher their mutational patterns. It is discovered that Signature 7 shows a higher prevalence of C > T mutations [13], attributed to pyrimidine dimers caused by UV exposure. In addition, Signature 11 exhibits a mutational pattern resembling that of alkylating agent treatment, associated with mutations occurred on guanine [24]. However, it is said that this family never underwent chemical treatment, and possible reasons need to be further explored. As mentioned above, both Signature 7 and 11 belong to non-clock-like mutational patterns, which means those mutational processes may occurred in an episodic manner, generating sudden mutations in a short time, rather than at a steady rate [14]. By comparison, clock-like signature is associated with worse prognosis, tumor progression and immune resistance to immune checkpoint inhibitor therapy [25]. Moreover, those non-clock-like mutational signatures might be consistent with the near-normal life expectancy of GS patients.
Notably, poor economic conditions of this family were the primary reason for the delay in seeking medical treatment. In recent years, our government provided special funding to support medical examinations in rural areas. The two patients who died might have survived longer if they went to hospital and received surgical treatment in time.
Conclusions
Taken together, this study provided clinical information and mutation analyses of PTCH1 in a three-generation family with GS, which has been listed on HGMD®. The main reasons for untimely deaths of GS patients were driver gene mutations, exposure to intense UV radiation and the poor economic conditions. Although the genetic background can’t be changed, it has been well established that restricting exposure to UV light is mandatory for GS patients.
Supplementary Information
Additional file 1: Somatic single nucleotide variant (SNV) sites in whole exome sequencing of matched BCC and ANCT.
Additional file 2: Somatic loss of heterozygosity (LOH) in whole exome sequencing of matched BCC and ANCT.
Acknowledgements
We thank all patients and family members who participated in this study.
Abbreviations
- GS
Gorlin-Goltz syndrome
- BCCs
Basal cell carcinomas
- Hh
Hedgehog
- WES
Whole exome sequencing
- ANCT
Adjacent non-cancerous tissues
- UV
Ultraviolet
- NBCCS
Nevoid basal cell carcinoma syndrome
- PTCH1
Patched 1
- PTCH2
Patched 2
- SUFU
Suppressor of fused
- GATK
Genome Analysis Toolkit
- SNVs
Single nucleotide variants
- GVCF
Genomic variant call format
- VQSR
Variant quality score recalibration
- VEP
Variant effect predictor
- MAF
Mutation annotation format
Author contributions
Conception and design: ZZ, OF, QZ; Acquisition of data (acquired and managed patients, etc.): LY, LW, ZT, XF, QC, JC, JL, ZZ, QZ; Analysis and interpretation of data (statistical analysis, etc.): OF, CL, ZZ; Writing, review, and/or revision of the manuscript: ZZ, KP, LY, LW, OF; Study supervision: ZZ, QZ. All authors read and approved the final manuscript.
Funding
This work was supported by the grants from National Natural Science Foundation of China (No. 81673073, 81974471), Hainan Provincial Natural Science Foundation of China (No. 821MS155) and Shanghai Municipal Key Clinical Specialty (No. shslczdzk01002).
Availability of data and materials
The datasets generated during the current study are available in the [Mendeley Data] repository, Peng, Kexin; Zhang, Zhenghua (2022), “Distinct Non-Clock-Like Signatures of the Basal Cell Carcinomas from Three Sisters with a Lethal Gorlin-Goltz Syndrome”, Mendeley Data, V3, https://doi.org/10.17632/5s4pk4rj32.3.
Declarations
Ethics approval and consent to participate
All methods were performed in line with the principles of the Declaration of Helsinki. Study approval was granted by the Scientific Ethics Committee of Haikou People’s Hospital, Xiangya Medical College (Approval no: 2020-201, Jul 14, 2020). Informed consent was obtained from all individual participants included in the study.
Consent for publication
All individual participants gave written informed consents that their clinical details and images could be used for scientific publication.
Competing interests
The authors have no competing financial or non-financial interests to disclose.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Lihua Ye, Li Wang and Kexin Peng contributed equally to this work
Contributor Information
Zhenghua Zhang, Email: verzhang@foxmail.com.
Qiaoan Zhang, Email: zqa1976@163.com.
References
- 1.Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6(6):530–539. doi: 10.1097/01.GIM.0000144188.15902.C4. [DOI] [PubMed] [Google Scholar]
- 2.Wilding A, et al. Life expectancy in hereditary cancer predisposing diseases: an observational study. J Med Genet. 2012;49(4):264–269. doi: 10.1136/jmedgenet-2011-100562. [DOI] [PubMed] [Google Scholar]
- 3.Onodera S, Nakamura Y, Azuma T. Gorlin syndrome: recent advances in genetic testing and molecular and cellular biological research. Int J Mol Sci. 2020;21(20):7559. doi: 10.3390/ijms21207559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith MJ, Evans DG. PTCH2 is not a strong candidate gene for Gorlin syndrome predisposition. Fam Cancer. 2022;21(3):343–346. doi: 10.1007/s10689-021-00269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLaren W, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.1000 Genomes Project Consortium. Auton A, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bree AF, Shah MR, BCNS Colloloquium Group Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS) Am J Med Genet A. 2011;155A(9):2091–2097. doi: 10.1002/ajmg.a.34128. [DOI] [PubMed] [Google Scholar]
- 11.Bonilla X, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. 2016;48(4):398–406. doi: 10.1038/ng.3525. [DOI] [PubMed] [Google Scholar]
- 12.Alexandrov LB. Understanding the origins of human cancer. Science. 2015;350(6265):1175. doi: 10.1126/science.aad7363. [DOI] [PubMed] [Google Scholar]
- 13.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov LB, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47(12):1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regl G, et al. Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene. 2002;21(36):5529–5539. doi: 10.1038/sj.onc.1205748. [DOI] [PubMed] [Google Scholar]
- 16.Ikram MS, et al. GLI2 is expressed in normal human epidermis and BCC and induces GLI1 expression by binding to its promoter. J Invest Dermatol. 2004;122(6):1503–1509. doi: 10.1111/j.0022-202X.2004.22612.x. [DOI] [PubMed] [Google Scholar]
- 17.Evans DG, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64(5):959–961. doi: 10.1038/bjc.1991.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andreani A, Prato W, Vittone L. La sindrome del nevo basocellulare [Basal cell nevus syndrome] Minerva Med. 1986;77(34–35):1533–1538. [PubMed] [Google Scholar]
- 19.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De S, Ganesan S. Looking beyond drivers and passengers in cancer genome sequencing data. Ann Oncol. 2017;28(5):938–945. doi: 10.1093/annonc/mdw677. [DOI] [PubMed] [Google Scholar]
- 22.Pellegrini C, et al. Understanding the molecular genetics of Basal Cell Carcinoma. Int J Mol Sci. 2017;18(11):2485. doi: 10.3390/ijms18112485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu-Smith F, Jia J, Zheng Y. UV-induced molecular signaling differences in melanoma and non-melanoma skin cancer. Adv Exp Med Biol. 2017;996:27–40. doi: 10.1007/978-3-319-56017-5_3. [DOI] [PubMed] [Google Scholar]
- 24.Alexandrov LB, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chong W, et al. Association of clock-like mutational signature with immune checkpoint inhibitor outcome in patients with melanoma and NSCLC. Mol Ther Nucleic Acids. 2020;23:89–100. doi: 10.1016/j.omtn.2020.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Somatic single nucleotide variant (SNV) sites in whole exome sequencing of matched BCC and ANCT.
Additional file 2: Somatic loss of heterozygosity (LOH) in whole exome sequencing of matched BCC and ANCT.
Data Availability Statement
The datasets generated during the current study are available in the [Mendeley Data] repository, Peng, Kexin; Zhang, Zhenghua (2022), “Distinct Non-Clock-Like Signatures of the Basal Cell Carcinomas from Three Sisters with a Lethal Gorlin-Goltz Syndrome”, Mendeley Data, V3, https://doi.org/10.17632/5s4pk4rj32.3.