

Abstract
Humans and our ancestors have evolved since the most ancient times with a commensal microbiota. The conservation of indicator species in a niche-specific manner across all of the studied human population groups suggests that the microbiota confer conserved benefits on humans. Nevertheless, certain of these organisms have pathogenic properties and, through medical practices and lifestyle changes, their prevalence in human populations is changing, often to an extreme degree. In this Essay, we propose that the disappearance of these ancestral indigenous organisms, which are intimately involved in human physiology, is not entirely beneficial and has consequences that might include post-modern conditions such as obesity and asthma.
“We have met the enemy, and he is us.”
Walt Kelly, 1953.
“The enemy of my enemy is my friend.”
Arabic Proverb.
This is an era of great change. We are exploring the outer reaches of the universe and can travel across continents in hours, but we are also warming the Earth and depleting its oceans. These major changes in our macroecology represent the price for the past century’s technological progress. We have also begun to explore the microscopic universe within us. Is this smaller-scale biosphere affected by similar changes? In particular, has the progress that has lowered infant mortality and prolonged lifespan also caused unanticipated alterations in our microecology and, consequently, our health?
In this Essay, we focus on our intimate interface with microorganisms: residents and transients, and symbionts and pathogens. We will explore whether and how our microecology is changing, in analogy to our altered macroecology.
What are our microbiota?
We live in a microbial world1. When animals emerged, microorganisms had long (>3 billion years) been part of a vast community of life that included complex endosymbiotic relationships, and it has been calculated that animals have carried resident microorganisms since at least the emergence of sponges2,3. Humans have become host to a myriad of microorganisms that assemble into complex, largely beneficial communities that outnumber human cells by tenfold. The dominant forms of human–microorganism interactions are those in which microorganisms benefit the host without causing harm (commensal relationships) and those in which both host and microorganism benefit (symbiotic or mutualistic relationships). Co-evolution, co-adaptation and codependency are all features of our relationships with our indigenous microbiota4,5.
The vertebrate microbiota can be characterized as: ancient, with deep ancestries; conserved in their host species; often present for defined life cycle events or persisting for life; and host niche specific. These properties imply that the microbiota have been selected, but it is becoming increasingly clear that although they are bounded by these rules they are also highly individual.
The human microbiome is the subject of intensive studies, including the international Human Microbiome Project6 (BOX 1). Although possibly germ-free (gnotobiotic) before birth, humans develop a resident microbiota shortly after birth. In the neonatal period, the community assembly process is dynamic and is influenced by early environmental (in particular, maternal) exposure and stochastic effects7. The composition of the indigenous microbiota evolves in a generally orderly manner in response to diet and other environmental factors and is also influenced by diverse human genetic backgrounds.
Box 1 |. The Human Microbiome Project.
In recent years, there has been a growing appreciation of the extent of the commensal microbial populations in humans.
Collectively, these populations have been termed ‘the human microbiome’, originally by Joshua Lederberg107, and also the ‘human microbiota’. Owing to advances in DNA sequencing technologies and improvements in bioinformatics, it has become possible to characterize the great diversity in the human microbiota. In 2007, the US National Institutes of Health (NIH) launched the Human Microbiome Project (see Further Information) as one of its major roadmap initiatives, earmarking ~US$140 million for its completion108. This major scientific endeavour has the following aims:
to determine whether individuals share a core human microbiome;
to understand whether changes in the human microbiome can be correlated with changes in human health;
to develop the technological tools to support these goals;
to address the ethical, legal and social implications of human microbiome research.
One value of the Human Microbiome Project is that it can help us to ascertain how much the microbiota have been changing. Work is now ongoing, and the European Union and other countries are committed to similar projects, all working under the umbrella of the International Human Microbiome Consortium (see Further Information).
The bacterial diversity in the human body is striking in its richness of distinct species and strains, but it is noteworthy that a limited number of phyla are commonly found in indigenous microbial communities. Only 4 of the more than 50 bacterial phyla that have been identified in the environment (Firmicutes, bacteroidetes, Actinobacteria and Proteobacteria) dominate human mucosal and cutaneous habitats, which suggests that strong selective forces have limited diversity over at least hundreds of thousands of years of co-evolution8–16. despite this stereotypical assembly process, each individual in a single mammalian species, including Homo sapiens, has a virtually unique microbiota10,11. The composition of the microbiota and the phenotypes that are expressed affect, as well as reflect, the overall biological diversity of humans.
Most research has focused on defining the core elements of the microbiota that are shared by all, or at least most, humans8. To date, the rules that govern life on and in humans are poorly understood, and we are still in the earliest stages of discerning the functions of the microbiota in health and disease4. The human microbiota facilitate the extraction of energy from food, provide accessory growth factors, promote post-natal terminal differentiation of mucosal structure and function, stimulate both the innate and adaptive immune systems, and provide ‘colonization resistance’ against pathogen invasion17–20. However, studies of gnotobiotic animals have shown that microorganisms (or at least bacteria) are not necessary for life or for the completion of an animal life cycle19–21. Paradoxically, therefore, there seems to be no absolute requirement for a functional resident microbiota, but nor does gnotobiosis occur in nature.
Although the relationship between animals and their resident microbiota is thought to be largely beneficial, it can also become detrimental19,22. In humans, commensal α-haemolytic streptococci colonize the mouth and intestinal tract and, in part, protect us against incursions by pathogenic streptococci17,23. However, these commensal organisms are often transiently present in the bloodstream after we chew food, brush our teeth or defecate24,25. If there is damaged tissue present, such as an ageing heart valve, these long-term residents can cause a fatal infection. Thus, our generally beneficial microbiota have the capacity to cause our demise. They thin the herd, particularly if the normal defences that hold microorganisms in check are compromised in some manner.
It is clear that we share a complex biological relationship with our microscopic companions. Are these organisms symbionts or parasites? (BOX 2). The ecologist Theodor rosebury created the term ‘amphibiosis’ to describe relationships that can be symbiotic or parasitic, depending on the biological context18. This concept is the guiding principle for the remainder of this Essay.
Box 2 |. What is a pathogen?
Microbial pathogens can range from bacteria to metazoa to viruses or prions. All pathogens are excellent cell biologists, taking advantage of chinks in the armour of their hosts to propagate. There is no intrinsic difference between a pathogen and a symbiont, only context. However there are fingerprints that are sufficiently characteristic of pathogens or symbionts to be used to help distinguish between them:
Pathogens induce host responses. They usually cannot do their deeds without a trace.
Infection with a pathogen can sometimes cause disease, which is a function of the interaction between the pathogen and the specific host, as determined by a matrix of host, microbial and environment circumstances.
The gene pool of the host includes the genes of its resident microorganisms, which may be why many members of the microbiota are selected for by the host: to prevent colonization by pathogens instead.
When influenza sweeps across the globe, infecting millions and causing disease and sometimes death, we agree that the virus is a pathogen, as is Yersinia pestis, the cause of bubonic plague. However, the distinction between a commensal resident microorganism and a pathogen can be blurred at times, in part because some commensals cause disease, albeit often in immunocompromised hosts, whereas some of the most feared pathogens can persist in humans for a life-time without causing any symptoms26. For example, several members of the nasopharyngeal microbiota, including Streptococcus pneumoniae and Neisseria meningitidis, regularly cause disease. such ‘commensal pathogens’ colonize a substantial proportion of the human population, most of whom are asymptomatic carriers27. Are these organisms pathogens, or should they be considered to be members of the indigenous microbiota that have evolved to live in perilous locations28, such as in respiratory tract-associated lymphatic tissue? From this viewpoint, the specific indigenous microbiota in such niches would regularly come into contact with elements of an immune system that hold them at bay most of the time22 but that occasionally fail to do so, resulting in disease. Immunization against these microorganisms not only protects against disease but also prevents host colonization, in an antigen-specific manner (reviewed in REF. 29). serotype replacement is an acknowledged deleterious effect of such immunization programmes, but may there be other deleterious effects that we have yet to uncover? It is crucial to improve our understanding of the relationships between humans and our microbiota, so that we can assess the risks, in addition to the potential benefits, of modifying the composition of the microbiota deliberately or inadvertently. The use of molecular Koch’s postulates30 may assist in this risk assessment.
How are the microbiota stabilized?
Is there any underlying biological regulation that stabilizes the diverse microbial populations that we carry, even in the face of host immunity, competing organisms and the daily continuous flux of newly introduced microorganisms? Cooperation between competing life forms is a challenging concept and, although much of the literature on microorganisms focuses on the evolution of virulence31,32, there is an increasing focus on the rules that govern the evolution of cooperation33,34.
In an earlier report, a hypothesis was presented that Nash equilibria enable pathogens such as Mycobacterium tuberculosis and Salmonella enterica subsp. enterica serovar Typhi to persist35. such a view has also been applied to Helicobacter pylori, the dominant member of the gastric microbiota16. In game theory, a nash equilibrium represents a particular circumstance in which any player who deviates from the rules of the game is in an inferior position compared to those who have played by the rules36,37. such conditions can exist in a limited number of situations in nature, and biological co-evolution is a prime example.
For any equilibrium to function, there must be boundary conditions or limits and strong penalties against transgressors that, in net, remove any advantage from ‘cheating’38. This is characteristic of a nash equilibrium, but the complex evolved biology of metazoans such as humans requires solutions that operate on multiple scales. A series of nested dynamic equilibria was envisioned, in which each stratum contributes to creating the boundary conditions for the others35. This is made possible, in part, by the fractal nature of biological communities, in which ecosystems nest within one another, a structure that provides both stability and resilience in a world of opportunists, invaders and cheaters. In steady state, the equilibria form a continuum, analogous to a Mobius strip, with no beginning or end. This population structure permits the co-evolution of competing organisms (for example, host and microorganism) that would otherwise lead to an ‘arms race’39 and the destruction of one organism or the other. However, even in such a derived evolutionarily stable strategy40, all is not fixed. The equilibrium has sufficient elasticity for invasions (by pathogens or ‘cheaters’) to be contained under normal conditions of flux. However, even with such capacitance, there must be boundaries beyond which the systems lose equilibria and descend towards the extinction of one partner or the other.
In the human biosphere, the boundaries for flux are unknown. Even a catastrophe like the AIds epidemic has so far been contained within the overall population, in part because of the inefficiencies of HIv transmission41. However, scenarios involving more transmissible agents in a ‘smaller world’ in which population connectivity selects for virulence and the ecosystem has been perturbed are worrisome42. This is directly germane to considerations of the stability of the microbiota. On the basis of humanity’s long association with our indigenous microorganisms and their conservation4,10, the equilibria that maintain the composition of our microbiota should be considered to be major pillars of our biological stability. seen from this view, pathogens that arise are ‘cheaters’ that can break the equilibria. In principle, ‘cheaters’ can cause the extinction of the microorganism, the host or even both, although in reality variants usually emerge and equilibria ultimately rule. For example, if the host-mediated penalties against ‘cheating’ (such as host immunity to the microorganism or the microorganism’s loss of a niche in the host) are sufficiently high, then less virulent variants that can evade biological surveillance can become successful.
The ‘disappearing microbiota’ hypothesis
In general, it seems that the human microbiota and their hosts evolved their equilibria together, in an orderly way10, as diet, geography and occasional ecological disturbances had their effects on distinct, albeit diverse, human genetic backgrounds43. Our complex protective commensal microbiota facilitate nutrient and vitamin acquisition, promote tissue development and integrity, and stimulate multiple aspects of immunity 18–22. However, in the face of modern global ecological changes, has there been stability for the descendents of the microbiota that colonized humans during the ~2 million years that they lived in small groups as hunter–gatherers?
As human health and longevity have improved in developed countries, new diseases have arisen without obvious explanation. The ‘disappearing microbiota’ hypothesis44,45 has been advanced to explain the rise and fall of several common diseases in developed countries. beginning in the nineteenth century and accelerating in the twentieth century, there have been dramatic changes in human ecology (TABLE 1), including cleaner water, smaller families, an increase in the number of Caesarian sections, increased use of pre-term antibiotics, lower rates of breastfeeding and more than 60 years of widespread antibiotic use, particularly in young children. How have these changes affected the transmission and maintenance of the indigenous microbiota44?
Table 1 |.
Changes in human ecology that might affect microbiota composition
| change | consequence |
|---|---|
| clean water | reduced faecal transmission |
| Increase in caesarean sections | reduced vaginal transmission |
| Increased use of pre-term antibiotics | reduced vaginal transmission |
| reduced breastfeeding | reduced cutaneous transmission and a changed immunological environment |
| smaller family size | reduced early life transmission |
| Widespread antibiotic use | selection for a changing composition |
| Increased bathing, showering and use of antibacterial soaps | selection for a changing composition |
| Increased use of mercury-amalgam dental fillings | selection for a changing composition |
We postulate that the important factor in modern allergic and metabolic diseases might not be our decreased sampling of the microorganisms in food, air, water or soil, as has been postulated by the ‘hygiene hypothesis’ (REF. 46), but instead could reflect the loss of our ancestral microorganisms. As the representation of particular species diminishes in one generation, the potential for vertical transmission to the next generation47 can decrease in a stepwise manner (FIG. 1). diminished horizontal transmission resulting from changes in human ecology (TABLE 1) makes it more difficult to overcome losses in vertical transmission, and this then manifests as a birth cohort phenomenon. We believe that alterations in human macroecology have progressively affected the composition of our indigenous microbiota, which in turn has affected human physiology and, ultimately, disease risk. The increases that have occurred in recent years in the prevalence of conditions such as obesity and asthma, as well as oesophageal disorders that are a consequence of reflux, have been so rapid that an environmental cause must be present48–50. Is one of these causes the loss of one or more constituents of the indigenous microbiota?
Figure 1 |. The effect of maternal status on the resident microbiota of the next generation.

We propose that, since the earliest days of the evolution of mammals, there has been major maternal transmission of microbiota to their offspring (vertical transmission). However, loss of the conserved microbiota in one generation leads to its loss in the next. For humans, until recently, horizontal microbial transmission also occurred and could compensate for the loss of vertical transmission. Members of the microbiota were horizontally transmitted through faecally contaminated drinking and bathing water, and high physical contact as a result of social crowding and large families; in many modern societies, these routes have diminished. The progressive loss of vertically transmitted microorganisms without horizontal replacement represents a cumulative birth cohort phenomenon.
H. pylori, an ancient member of the human microbiota51, generally dominates the gastric niche (BOX 3). Accordingly, its presence can be used as a means to assess the status of our microbiota. surprisingly, H. pylori has been progressively disappearing52–54 from individuals in developed countries (FIG. 2) during the twentieth century, with secondary alterations in gastric secretory, hormonal and immune physiology 55,56. These alterations have been associated with a progressively declining incidence of important illnesses with long latent periods, such as gastric cancer. We now have multiple tools to detect H. pylori57. but if a lesser known ancestral indigenous microorganism disappeared in the colon, mouth, skin or vagina, could we identify that change and, crucially, could such a change (whether it was a loss or a replacement and subsequent ‘overgrowth’ by a different indigenous component) contribute to some of the diseases that are becoming more prevalent? The Human Microbiome Project6 should begin to address such questions (BOX 1).
Box 3 |. The gastric microbiome.
The human body is colonized by a highly complex microbiota, but the stomach is an exceptional niche. When Helicobacter pylori can be detected by conventional means, including culture or biochemical assays, tissue histology and host serological responses, it is the numerically dominant organism, representing >50% of all of the bacterial cells in the niche16,103. This phenomenon is unlike other human niches that house a large range of bacterial species but no single predominant organism (in the vagina, a single Lactobacillus species often predominates, but its identity differs in different women113). The dominance of the gastric niche by H. pylori is consistent with its adaptations that enable gastric persistence55. In persons in whom H. pylori cannot be detected by conventional means, the organism may still be detected by PCR-based techniques, usually as more minor populations16. These observations suggest that there may be a ‘stealth’ phenomenon in which H. pylori exists below the radar of histological, biochemical and immunological fingerprints, but its characteristics are not yet understood.
In addition to H. pylori, numerous other bacterial species may be present, but whether they are resident in the stomach or transient from the upper gastrointestinal tract has not been determined16. The phyla that are observed (chiefly Firmicutes, Actinobacteria, Proteobacteria and Bacteroidetes) seem to be similar in H. pylori-positive and H. pylori-negative hosts16.
Figure 2 |. Helicobacter pylori prevalence in the United States by age and year of birth.
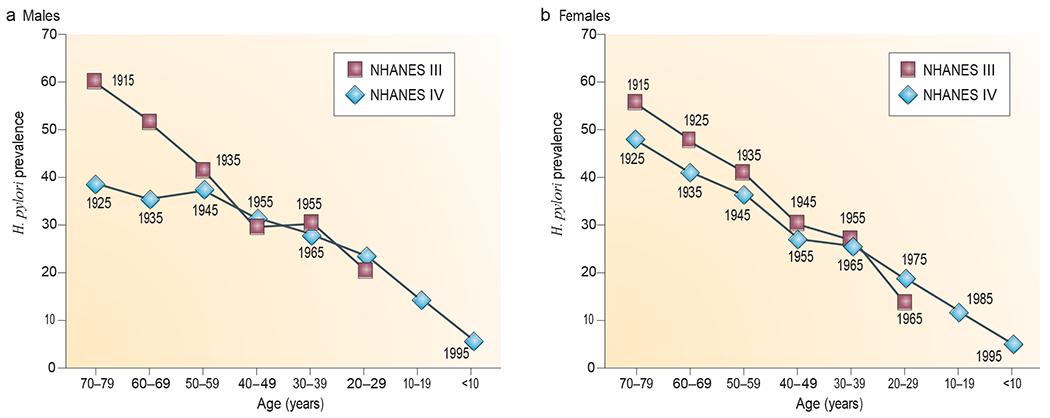
Helicobacter pylori prevalence in males (a) and females (b) was determined in two large national studies (the National Health and Nutrition survey (NHANEs) III and NHANEs Iv), which were conducted in 1988–1991 and 1999–2000, respectively. These data, involving 15,000 subjects, are consistent with a now well-recognized birth cohort effect52–54 in which H. pylori acquisition, and thus prevalence, has been declining in industrialized countries for >100 years. The numbers next to each point represent the midpoint year of birth for each group of survey subjects (with each group covering one decade of births). Image is modified, with permission, from REF. 56 © (2008) University of chicago Press.
The consequences of loss
Modelling of the specific host–microorganism relationships suggests that there are several potential physiological relationships that can occur (FIG. 3). several specific examples illustrate the concept that the loss of an indigenous microorganism will have consequences for the host.
Figure 3 |. interactions between host and microbiota.

a | For luminal microbiota that do not interact with host cells, the host-specific regulatory networks are essentially unaffected by transient microbiota or by luminal microbiota that are distant from the epithelium. This could represent a large class of organisms in the microbiota. b | For microbiota that interact with epithelial cells, there is an equilibrium relationship involving signalling between the microbial populations and the host35. c | For co-evolved microbiota that interact with multiple cell types, the cross-signalling in the most fully co-evolved states involves multiple host cell types, including epithelial, immunological and neuroendocrine cells. The interaction of Helicobacter pylori with the gastric mucosa is representative of this model. The introduction of drugs, such as the commonly used proton pump inhibitors that reduce gastric acidity, affects equilibrium values and selects for a differing microbiota59,114. d | In the absence of a microbiota, the indigenous host regulatory mechanisms predominate and reach different homeostasis values than are reached when interacting microbiota are present. In the situation of a disappearing microbiome, a and d predominate.
Los of. H. pylori.
H. pylori colonization and the response that it induces in the host affects the regulation of gastric hormones, including gastrin and somatostatin58. As such, H. pylori status affects gastric pH and its regulation55. Over the decades of H. pylori colonization, the mass of gastric acid-secreting glands in the host progressively decreases, owing to the long-term effects of inflammation58,59. Although the rates of glandular decrease reflect both host-specific and population-specific factors, overall there is a progressive decrease in acid production60 that is greater than the decrease observed in H. pylori-negative hosts60,61. The increasing gastric atrophy and hypochlorhydria that are associated with the presence of H. pylori60–62 contribute to the risk of gastric cancer63,64, but the sustained acidity in H. pylori-negative hosts increases the risk of gastroesophageal reflux disease (GErd) and its consequences, including oesophageal and gastric cardia adenocarcinomas64,65.
As H. pylori is disappearing from human populations, reflecting both diminishing transmission and increasing antibiotic treatment (TABLE 1), both ‘idiopathic’ peptic ulcer disease and gastric cancer rates are diminishing 66,67, which is clearly salutary. However, oesophageal reflux, barrett’s oesophagus and adenocarcinoma are increasing, which is clearly deleterious50,66. Are these reciprocal phenomena? The cag pathogenicity island55 (or perhaps a better term would be the cag ecological fitness island68) status of the H. pylori population has a strong influence on the development of barrett’s oesophagus and adenocarcinoma, as predicted by the strong interaction between cag-positive strains and host cells60,63,69,70. The stomach also produces the hormones ghrelin and leptin, both of which have multiple roles in energy homeostasis55. Patients in whom antibiotic treatment eliminates H. pylori have increased circulating ghrelin levels71. Investigations are ongoing, but it is already clear that in recent generations of children growing up in developed countries there has been little gastric H. pylori-mediated56 regulation of these adipokines at the developmental stage, when long-term adiposity is being programmed. It is possible that the disappearance of H. pylori might be contributing to the current epidemics of early-life obesity, type 2 diabetes and related metabolic syndromes.
Gastric T cell and b cell populations also differ in H. pylori-positive and H. pylori-negative hosts72–74. This might be expected, as the H. pylori-negative stomach has a diminished presence of the cellular elements of the immune system and reduced cytokine traffic, whereas the H. pylori-positive stomach has a rich population of immune cells (FIG. 3). The enhanced T cell populations in H. pylori-positive hosts include greater proportions of particular T cell subsets, including cells expressing forkhead box protein P3 (FOXP3), which regulate immune functions73,74. The stomachs of H. pylori-negative hosts have much lower numbers of these cells, which have systemic, as well as local, activities. several epidemiological studies (reviewed in REF. 75) show that H. pylori-positive individuals (especially individuals carrying cag-positive strains) have lower risks of childhood asthma, allergic rhinitis and skin allergies than those without H. pylori56,76,77. The rise in childhood asthma and related disorders has occurred while H. pylori has been disappearing; the loss of gastric T cell populations and their systemic effects could provide a mechanism for these allergic diseases78,79.
Subtherapeutic antibiotic treatment.
In the early 1950s, not long after the discovery of antibiotics, scientists found that feeding low doses of antibiotics (termed subtherapeutic antibiotic treatment (sTAT)) to farm animals increased their rate of growth and ability to convert food into body mass (otherwise known as their ‘feed efficiency’; reviewed in REF. 80). The younger the animals are when sTAT is started, the stronger the effect80. The fact that several antibiotics can produce this effect in poultry, cattle and swine80–82 has led to the widespread usage of antibiotics as animal feed supplements in the united states and other developed countries (this practice has now been banned by the European union because of the spread of antibiotic-resistant bacteria).
Why does giving sTAT change energy homeostasis in this broad group of vertebrates? As growth promotion can be induced by many different antibacterial (but not antifungal) agents, this activity is not a side effect but a consequence of the antibacterial activities of these agents, which presumably affect the microbiota of the exposed animals82. However, the affected microbial populations and metabolic pathways are unknown. Given that this manipulation of the microbiota has such effects on early life energy homeostasis and body mass development in farm animals, what might the effects be of the widespread exposure of children to antibiotics early in life? Instead of the continuous, low-dose antibiotics that are administered on the farm, we are giving our children short, high-dose pulses. How is this affecting their microbiota, energy homeostasis and ‘feed efficiency’? Even single courses of widely used antibiotics can change the stable structure of microbial communities, with the presence of selected organisms continuing for years after the antibiotic exposure has ceased83,84. We speculate that the widespread treatment of young children with antibiotics has caused alterations in the compositions of their intestinal microbiota, and the lumenal signals to the host (FIG. 3), that are directly contributing to the epidemic of obesity in developed countries.
The pneumococcus and Staphylococcus aureus.
S. pneumoniae (known as the pneumococcus) is an important human pathogen, causing pneumococcal pneumonia, infections of the upper respiratory tract and its appendages, and occasionally lethal diseases such as meningitis and endocarditis85. Consequently, the development of a pneumococcal vaccine has been a priority for >100 years. However, pneumococci are carried by healthy persons in the nasopharynx, often for months, and are part of the consortia of microorganisms inhabiting this niche. Pneumococci are naturally transformable and vary extensively, with a major source of variation being the presence and type of polysaccharide capsule86. Most human disease is caused by encapsulated pneumococci, and particular serotypes predominate87,88. Consequently, preventative strategies have focused on the most virulent capsular subtypes, and polyvalent vaccines have been developed89. These vaccines are effective and have reduced the incidence of serious pneumococcal infections in high-risk populations90. Immunization not only protects against disease but also prevents colonization by those pneumococci with the capsule types that are present in the vaccine89. One of the earliest concerns of researchers was that increasing the immunity to certain capsular types would lead to serotype replacement88–90, which might result in new pneumococcal infections, but presumably at a lower rate than rates of infection by the more virulent pneumococci that are targeted by the vaccine. Although serotype replacement and the subsequent pneumococcal disease would lower the net ‘effectiveness’ of vaccination, there would still be residual utility.
All of these predicted consequences have come to pass89–92. However, in addition to replacement with non-vaccine serotypes of S. pneumoniae, replacement with an unanticipated organism, Staphylococcus aureus, has occured92–95. A growing body of evidence indicates that the encapsulated pneumococci and S. aureus are ecological competitors93,94, and that the loss of the former is leading to the expansion of the latter95. This can take place through direct competition or through microbial manipulation of host immunity96–98. This is occurring just as we are witnessing an unprecedented epidemic of methicillin-resistant S. aureus (MrsA) infections in the community among persons who have not been recently hospitalized or received antibiotics. In the absence of obvious exposure or selection pressure, these community-acquired MrsA (CA-MrsA) strains are being widely transmitted from person to person and with occasional, but often serious, clinical phenotypes99,100. The ecology of S. aureus has clearly changed34. Whether or not this reflects the consequences of pneumococcal vaccination, antibiotic pressure selecting for particularly virulent clones, ecosystem degradation due to effects on the nasopharyngeal and cutaneous microbiota of widespread antibiotic use, or a combination of these factors remains to be determined.
Recolonizing a vacated niche
If the indigenous microbiota are disappearing, have there been replacements? Are there model systems in which to explore this question? As discussed above, H. pylori, the ancient and dominant microbial inhabitant of the human stomach, is disappearing with remarkable speed. In two or three generations, human societies have moved from near-ubiquitous (>80%) H. pylori prevalence rates to rates in single digits among contemporary native-born children in the united states (FIG. 2) and western Europe56,101,102. This is an unprecedented change in human microecology.
What about the gastric niche itself: with H. pylori disappearing, will there be new tenants and, if so, how will they interact with the host? The stomach is bounded proximally by the oropharynx and oesophagus and distally by the intestine, all of which possess resident microbiota8–11. The gastrointestinal tract is also a portal to the outside world through ingested food and drink. Although H. pylori is the dominant gastric microorganism, there is dnA evidence for the presence of other transient or residential microorganisms16,103 and indications that a more diverse microbial population is present in the H. pylori-vacated stomach103.
For occupancy of a vacated niche to occur, the transmission potential to other hosts is crucial104. several possible scenarios should be considered. The first possibility is that each individual will become colonized by their own host-specific microorganisms that can change over time but that are essentially unsuccessful at transmitting to new hosts. This would lead to billions of ongoing long-term experiments (one per human birth) in microbial transmission fitness. The second possibility is that the overall level of resident microorganisms will remain low, as other microorganisms cannot use the resources of the H. pylori-free stomach as efficiently as H. pylori. The third possibility is that, without H. pylori competition and inhibition, other microorganisms will better use the niche’s resources. such organisms could be a microbial consortium involving a few or many species, or a single species with a varied and ultimately robust pan-genome. Alternatively, they may be exogenous organisms that are acquired during ongoing environmental exposure. The transmission potential will determine whether the replacement organisms will be host-specific populations or emergent, highly selected organisms that are common between hosts. A potential consequence of the third scenario is the emergence of a highly transmissible microorganism that is well-adapted for the now sparsely populated niche. because in this situation selection is based on the ability to transmit to new hosts, a range of virulence is possible, with the potential for positive selection for virulence104. This could be dangerous, because the next main inhabitant of the gastric niche might not be as benign as H. pylori, which has co-evolved with humans at a moderate level of virulence. A newly acquired organism that evolved to colonize and efficiently use the resources would probably be more virulent105 and could selectively sweep through the population.
These possibilities are not gastric specific but represent the general consequences that might accompany the extinction of species from the microbiota of any niche. The more dominant the species that are lost, the larger the void and the greater the potential risk of replacement with a highly virulent organism.
Future therapeutics?
Will there be cases in which human health is improved by the replacement of some of the lost microbiota? Certainly, a trip through the dairy section of any super-market in the Western world would lead one to believe so. nonetheless, translating this marketing concept into scientifically based beneficial activities is limited by several outstanding questions. These include: which particular microorganisms should be replaced; what will their sources be; and what will the timing of their replacement be? We should begin to think about these options and their expected outcomes. replacement will necessarily lead to the development of new classes of probiotics; we speculate that one day we will see microbial additives that are specific for particular hosts, for specific durations of exposure and eradication. This is one vision for the future, but it will require a much deeper understanding of the metabolic, hormonal and immunological interactions with our microbiota (FIG. 3).
One emerging concept in microbial pathogenesis is the notion of the ‘community as a pathogen’, in which a conserved broad swathe of the microbial community, rather than any one specific member, contributes to disease. This concept might be relevant to a range of inflammatory processes of the skin and mucosa, including inflammatory bowel disease and chronic periodontitis. It suggests that studies of pathogenesis should consider the tgeneral properties of microbial communities, such as resilience, or conserved functional interactions, such as syntrophic interactions and the importance of gene transfer, rather than the role of single microorganisms, especially for the development of new approaches for maintaining or restoring health. Thus, under some circumstances, restoration of communities might be more appropriate than replacing single microorganisms.
Conclusions
It is predictable that social and medical progress that affects the composition of the microbiota will also have consequences for our physiology and health. However, the specific outcomes will only be learned empirically, as the human ecosystem is too complex for anything more than the most superficial predictions. Improving our prognostication is an important technical challenge.
Is the selective disappearance of the microbiota contributing to oesophageal dis-eases, obesity and its consequences, asthma and related disorders, and the epidemic spread of high-grade pathogens? Further investigation will clarify these points, but the theoretical basis exists: ecological changes involving our ancient microbiota have the power to affect physiology and, ultimately, health. Individuals who are ’normal’ in modern societies might be representative of the population at large but not of our historical heritage106. studies of persons who are indigenous to those developing countries that have had little impact from modern health practices (including exposure to antibiotics) could be ideal to define our historic ‘norms’.
Moving forward, we must learn to better distinguish between pathogens and amphibionts and to better assess in whom to eliminate, leave alone or restore the microorganism (or metabolic pathway) in question. Or is it even wise to contemplate any action until we understand more about our most intimate residents? Public health deals with trends in populations, but medical care inevitably involves individuals. A greater understanding of the characteristics of a host’s genome and microbiota, and their interactions, will lead to individualized approaches to the prevention and treatment of specific diseases. We are at a scientific frontier.
Acknowledgements
M. Blaser gratefully acknowledges funding from the US National Institutes of Health (NIH; grants RO1GM62370 and UH2AR057506) and from the Diane Belfer Program for Human Microbial Ecology. S. Falkow gratefully acknowledges funding from NIH (grants R01AI026195 and R01AI038459).
glossary
- Commensal relationship
An intimate, although generally benign, relationship between a resident microorganism and its host. Probably the product of a long evolutionary interplay between the microorganism and the host. The relationship need not be symbiotic.
- Gene transfer
The transfer of individual genes or their components, islands of genes, entire organisms, or communities of organisms from parent to offspring (vertical transfer) or between individuals not in a direct lineage (horizontal transfer).
- Microbiome
The collective genomes of the microorganisms that reside in an environmental niche.
- Nash equilibrium
In game theory, a solution concept in which players in a game are aware of the strategies of the other players but do not deviate from their own, because they do not have anything to gain; it will be disadvantageous to deviate (to ‘cheat’).
- Pan-genome
The set of all of the genes that are found in members of a single species.
- Parasite
An organism that diminishes the reproductive fitness of another organism or benefits from another organism without reciprocity.
- Pathogen
In medicine, any organism that causes disease. In biological terms, a pathogen is a microorganism that has the inherent capacity to cross anatomical barriers and resist host defences that ordinarily restrict most other microorganisms.
- Symbiont
An organism that has a biological relationship with one or more organisms that leads to mutual benefit.
- Syntrophic interactions
Interactions in which organisms do more together than alone.
- Virulence
A quantitative estimate of the ability of one organism to harm another.
Contributor Information
Martin J. Blaser, Department of Medicine and the Department of Microbiology, New York University Langone Medical Center, New York, New York 10017, USA
Stanley Falkow, Department of Microbiology and Immunology and the Department of Medicine, Stanford University School of Medicine, Stanford, California 94307, USA.
References
- 1.Whitman WB, Coleman DC & Wiebe WJ Prokaryotes: the unseen majority. Proc. Natl Acad. Sci. USA 95, 6578–6583 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iyer LM, Aravind L, Coon SL, Klein DC & Koonin EV Evolution of cell-cell signaling in animals: did late horizontal gene transfer from bacteria have a role? Trends Genet 20, 292–299 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Hoffmeister M & Martin W Interspecific evolution: microbial symbiosis, endosymbiosis and gene transfer. Environ. Microbiol 5, 641–649 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Dethlefsen L, McFall-Ngai M & Relman DA An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449, 811–818 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hickman CS in Influence of Cooperative Bacteria on Animal Host Biology(eds McFall-Ngai MJ, Henderson B & Ruby EG) 3–34 (Cambridge Univ. Press, Cambridge, UK, 2005). [Google Scholar]
- 6.Tumbaugh PJ et al. The human microbiome project. Nature 449, 804–810 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer C, Bik EM, Digiulio DB, Relman DA & Brown PO Development of the human infant intestinal microbiota. PLoS Biol.5, e177 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eckburg PB et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gill SR et al. Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ley RE, Lozupone CA, Hamady M, Knight R & Gordon JI Worlds within worlds: evolution of the vertebrate gut microbiota. Nature Rev. Microbiol 6, 776–788 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pei Z et al. Bacterial biota in the human distal esophagus. Proc. Natl Acad Sci. USA 101, 4250–4255 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lepp PW et al. Methanogenic Archaea and human periodontal disease. Proc. Natl Acad. Sci. USA 101, 6176–6181 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aas JA, Paster BJ, Stokes LN, Olsen I & Dewhirst FE Defining the normal bacterial flora of the oral cavity J. Clin. Microbiol 43, 5721–5732 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao Z, Tseng CH, Pei Z & Blaser MJ Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl Acad. Sci. USA 104, 2927–2932 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fierer N, Hamady M, Lauber CL & Knight R The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Natl Acad. Sci. USA 105, 17994–17999 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bik EM et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl Acad. Sci. USA 103, 732–737 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crowe CC, Sanders WE Jr & Longley S Bacterial interference. II. Role of the normal throat flora in prevention of colonization by group A Streptococcus. J. Infect. Dis 128, 527–532 (1973). [DOI] [PubMed] [Google Scholar]
- 18.Rosebury T Microorganisms Indigenous to Man (McGraw Hill, New York, 1962). [Google Scholar]
- 19.Mackowiak PA The normal microbial flora. N. Engl. J. Med 307, 83–93 (1982). [DOI] [PubMed] [Google Scholar]
- 20.Smith K, McCoy KD & Macpherson AJ Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol 19, 59–69 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Gordon HA & Pesti L The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol. Rev 35, 390–429 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pamer EG Immune responses to commensal and environmental microbes. Nature Immunol. 8, 1173–1178 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Roos K, Holm SE, Grahn-Hakansson E & Lagergren L Recolonization with selected α-streptococci for prophylaxis of recurrent streptococcal pharyngotonsillitis— a randomized placebo-controlled multicentre study. Scand J. Infect Dis 28, 459–462 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Durack DT Prevention of infective endocarditis. N. Engl. J. Med 332, 38–44 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Roberts GJ Dentists are innocent! “Everyday” bacteremia is the real culprit: a review and assessment of the evidence that dental surgical procedures are a principal cause of bacterial endocarditis in children. Pediatr. Cardiol 20, 317–325 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Falkow S Is persistent bacterial infection good for your health? Cell 124, 699–702 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Murphy TV, Pastor P, Medley F, Osterholm MT & Granoff DM, Decreased Haemophilus colonization in children vaccinated with Haemophilus influenzae type b conjugate vaccine. J. Pediatr 122, 517–523 (1993). [DOI] [PubMed] [Google Scholar]
- 28.Mazmanian SK, Round JL & Kasper DL A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Segal S & Pollard AJ Vaccines against bacterial meningitis. Br. Med. Bull 72, 65–81 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Falkow S Molecular Koch’s postulates applied to bacterial pathogenicity — a personal recollection 15 years later. Nature Rev. Microbiol 2, 67–72 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Maynard Smith J & Szathmary E The Major Transitions in Evolution (Oxford Univ. Press, 1995). [Google Scholar]
- 32.Messenger SL, Molineux IJ & Bull JJ Virulence evolution in a virus obeys a trade-off. Proc. R. Soc. Lond. B 266, 1480–1482 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michod RE Evolution of the individual. Am. Nat 150, S5–S21 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Sachs JL & Bull JJ Experimental evolution of conflict mediation between genomes. Proc. Natl Acad. Sci. USA 102, 390–395 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blaser MJ & Kirschner D The equilibria that allow bacterial persistence in human hosts. Nature 449, 843–849 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Nash JF Equilibrium points in n-person games. Proc. Natl Acad. Sci. USA 36, 48–49 (1950). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nash J Non-cooperative games. Ann. Math 54, 286–295 (1951). [Google Scholar]
- 38.Ma C-T, Moore J & Turnbull S Cheating and equilibrium. Stopping agents from ‘cheating.’ J. Econ. Theory 46, 355–372 (1988). [Google Scholar]
- 39.Obbard DJ, Jiggins FM, Halligan DL & Little TJ Natural selection drives extremely rapid evolution in antiviral RNAi genes. Curr. Biol 16, 580–585 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Smith JM in Evolution and the Theory of Games 10–27 (Cambridge Univ. Press, Cambridge, UK: 1982). [Google Scholar]
- 41.Gray RH et al. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet 357, 1149–1153 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Galvani AP Epidemiology meets evolutionary ecology. Trends Ecol. Evol 18, 132–139 (2003). [Google Scholar]
- 43.Turnbaugh PJ et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blaser MJ Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 7, 956–960 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blaser MJ Disappearing microbiota: Helicobacter pylori protection against esophageal adenocarcinoma. Cancer Prev. Res. (Phila. Pa) 1, 308–311 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Strachan DP Hayfever, hygiene, and household size. BMJ 299, 1259–1260 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nahar S et al. Evidence of intra-familial transmission of Helicobacter pylori by PCR-based RAPD fingerprinting in Bangladesh. Eur. J. Clin. Microbiol. Infect. Dis 28, 767–773 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Flegal KM, Graubard BI, Williamson DF & Gail MH Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA 298, 2028–2037 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Eder W, Ege MJ & von Mutius E The asthma epidemic. N. Engl. J. Med 355, 2226–2235 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Pohl H & Welch HG The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J. Natl Cancer Inst 97, 142–146 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Linz B et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 445, 915–918 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banatvala N et al. The cohort effect and Helicobacter pylori. J. Infect. Dis 168, 219–221 (1993). [DOI] [PubMed] [Google Scholar]
- 53.Roosendaal R et al. Helicobacter pylori and the birth cohort effect. Evidence of a continuous decrease of infection rates in childhood. Am. J. Gastroenterol 92, 1480–1482 (1997). [PubMed] [Google Scholar]
- 54.Harvey RF et al. Relationship between the birth cohort pattern of Helicobacter pylori infection and the epidemiology of duodenal ulcer. QJM 95, 519–525 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Blaser MJ & Atherton JC Helicobacter pylori persistence: biology and disease. J. Clin. Invest 113, 321–333 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y & Blaser MJ Helicobacter pylori colonization is inversely associated with childhood asthma. J. Infect. Dis 198, 553–560 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suerbaum S & Michetti P Helicobacter pylori infection. N. Engl. J. Med 347, 1175–1186 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Odum L, Petersen HD, Andersen IB, Hansen BF & Rehfeld JF Gastrin and somatostatin in Helicobacter pylori infected antral mucosa. Gut 35, 615–618 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kanno T et al. Gastric acid reduction leads to an alteration in lower intestinal microflora. Biochem. Biophys. Res. Commun 381, 666–670 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Kuipers EJ et al. Long-term sequelae of Helicobacter pylori gastritis. Lancet 345, 1525–1528 (1995). [DOI] [PubMed] [Google Scholar]
- 61.Kuipers EJ, Perez-Perez GI, Meuwissen SG & Blaser MJ Helicobacter pylori and atrophic gastritis: importance of the cagA status. J. Natl Cancer Inst 87, 1777–1780 (1995). [DOI] [PubMed] [Google Scholar]
- 62.Argent RH et al. Toxigenic Helicobacter pylori infection precedes gastric hypochlorhydria in cancer relatives, and H. pylori virulence evolves in these families. Clin. Cancer Res 14, 2227–2235 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Peek RM Jr. & Blaser MJ Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nature Rev. Cancer 2, 28–37 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Kamangar F et al. Opposing risks of gastric cardia and noncardia gastric adenocarcinomas associated with Helicobacter pylori seropositivity. J. Natl Cancer Inst 98, 1445–1452 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Islami F & Kamangar F Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev. Res. (Phila. Pa) 1, 329–338 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.el-Serag HB & Sonnenberg A Opposing time trends of peptic ulcer and reflux disease. Gut 43, 327–333 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Howson CP, Hiyama T & Wynder EL The decline in gastric cancer: epidemiology of an unplanned triumph. Epidemiol. Rev 8, 1–27 (1986). [DOI] [PubMed] [Google Scholar]
- 68.Hacker J & Carniel E Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2, 376–381 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Backert S & Selbach M Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 10, 1573–1581 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Viala J et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nature Immunol. 5, 1166–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Nwokolo CU, Freshwater DA, O’Hare P & Randeva HS Plasma ghrelin following cure of Helicobacter pylori. Gut 52, 637–640 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mattsson A, Lonroth H, Quiding-Jarbrink M & Svennerholm AM Induction of B cell responses in the stomach of Helicobacter pylori-infected subjects after oral cholera vaccination. J. Clin. Invest 102, 51–56 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson K et al. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut 57, 1375–1385 (2008). [DOI] [PubMed] [Google Scholar]
- 74.Lundgren A et al. Mucosal FOXP3-expressing CD4+CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect. Immun 73, 523–531 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blaser MJ, Chen Y & Reibman J Does Helicobacter pylori protect against asthma and allergy? Gut 57, 561–567 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Y & Blaser MJ Inverse associations of Helicobacter pylori with asthma and allergies. Arch. Intern. Med 167, 821–827 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Reibman J et al. Asthma is inversely associated with Helicobacter pylori status in an urban population. PLoS ONE 3, e4060 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Codolo G et al. The neutrophil-activating protein of Helicobacter pylori down-modulates Th2 inflammation in ovalbumin-induced allergic asthma. Cell Microbiol. 10, 2355–2363 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Del Prete G et al. Immunosuppression of T 2 responses in Trichinella spiralis infection by Helicobacter pylori neutrophil-activating protein. J. Allergy Clin. Immunol 122, 908–913.e5 (2008). [DOI] [PubMed] [Google Scholar]
- 80.Jukes T Antibiotics in animal feeds and animal production. Bioscience 22, 526–534 (1972). [Google Scholar]
- 81.Butaye P, Devriese LA & Haesebrouck F Antimicrobial growth promoters used in animal feed: effects of less well known antibiotics on Gram-positive bacteria. Clin. Microbiol. Rev 16, 175–188 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gaskins HR, Collier CT & Anderson DB Antibiotics as growth promotants: mode of action. Anim. Biotechnol 13, 29–42 (2002). [DOI] [PubMed] [Google Scholar]
- 83.Sjölund M et al. Long-term persistence of resistant Enterococcus species after antibiotics to eradicate Helicobacter pylori. Ann. Intern. Med 139, 483–487 (2003). [DOI] [PubMed] [Google Scholar]
- 84.Sjölund M et al. Persistence of resistant Staphylococcus epidermidis after single course of clarithromycin. Emerg. Infect. Dis 11, 1389–1393 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Musher DM et al. Bacteremic and nonbacteremic pneumococcal pneumonia. A prospective study. Medicine (Baltimore) 79, 210–221 (2000). [DOI] [PubMed] [Google Scholar]
- 86.MacLeod LC & Kraus MR Relation of virulence of pneumococcal strains for mice to the quantity of capsular polysaccharide formed in vitro. J. Exp. Med 92, 1–9 (1950). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Briles DE, Novak L, Hotomi M, van Ginkel FW & King J Nasal colonization with Streptococcus pneumoniae includes subpopulations of surface and invasive pneumococci. Infect. Immun 73, 6945–6951 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weinberger DM et al. Epidemiologic evidence for serotype-specific acquired immunity to pneumococcal carriage. J. Infect. Dis 197, 1511–1518 (2008). [DOI] [PubMed] [Google Scholar]
- 89.Ghaffar F et al. Effect of the 7-valent pneumococcal conjugate vaccine on nasopharyngeal colonization by Streptococcus pneumoniae in the first 2 years of life. Clin. Infect. Dis 39, 930–938 (2004). [DOI] [PubMed] [Google Scholar]
- 90.Albrich WC, Baughman W, Schmotzer B & Farley MM Changing characteristics of invasive pneumococcal disease in Metropolitan Atlanta, Georgia, after introduction of a 7-valent pneumococcal conjugate vaccine. Clin. Infect. Dis 44, 1569–1576 (2007). [DOI] [PubMed] [Google Scholar]
- 91.Veenhoven R et al. Effect of conjugate pneumococcal vaccine followed by polysaccharide pneumococcal vaccine on recurrent acute otitis media: a randomised study. Lancet 361, 2189–2195 (2003). [DOI] [PubMed] [Google Scholar]
- 92.Madhi SA et al. Long-term effect of pneumococcal conjugate vaccine on nasopharyngeal colonization by Streptococcus pneumoniae and associated interactions with Staphylococcus aureus and Haemophilus influenzae colonization in HIV-infected and HIV-uninfected children. J. Infect. Dis 196, 1662–1666 (2007). [DOI] [PubMed] [Google Scholar]
- 93.Bogaert D et al. Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363, 1871–1872 (2004). [DOI] [PubMed] [Google Scholar]
- 94.Regev-Yochay G et al. Association between carriage of Streptococcus pneumoniae and Staphylococcus aureus in children. JAMA 292, 716–720 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Regev-Yochay G et al. Does pneumococcal conjugate vaccine influence Staphylococcus aureus carriage in children? Clin. Infect. Dis 47, 289–291 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Selva L et al. Killing niche competitors by remote-control phage induction: the case of Streptococcus pneumoniae-Staphylococcus aureus interference. Proc. Natl Acad Sci. USA 106, 1234–1238 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lysenko ES, Ratner AJ, Nelson AL & Weiser JN The role of innate immune responses in the outcome of interspecies competition for colonization of mucosal surfaces. PLoS Pathog. 1, e1 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Regev-Yochay G et al. The pneumococcal pilus predicts the absence of Staphylococcus aureus co-colonization in pneumococcal carriers. Clin. Infect Dis 48, 760–763 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Herold BC et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 279, 593–598 (1998). [DOI] [PubMed] [Google Scholar]
- 100.Dufour P et al. Community-acquired methicillin-resistant Staphylococcus aureus infections in France: emergence of a single clone that produces Panton-Valentine leukocidin. Clin. Infect. Dis 35, 819–824 (2002). [DOI] [PubMed] [Google Scholar]
- 101.Tindberg Y et al. Helicobacter pylori infection in Swedish school children: lack of evidence of child-to-child transmission outside the family. Gastroenterology 121, 310–316 (2001). [DOI] [PubMed] [Google Scholar]
- 102.Rothenbacher D, Bode G & Brenner H History of breastfeeding and Helicobacter pylori infection in pre-school children: results of a population-based study from Germany. Int. J. Epidemiol 31, 632–637 (2002). [DOI] [PubMed] [Google Scholar]
- 103.Andersson AF et al. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3, e2836 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brown NF, Wickham ME, Coombes BK & Finlay BB Crossing the line: selection and evolution of virulence traits. PLoS Pathog. 2, e42(2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lenski RE & May RM The evolution of virulence in parasites and pathogens: reconciliation between two competing hypotheses. J. Theor. Biol 169, 253–265 (1994). [DOI] [PubMed] [Google Scholar]
- 106.Marini E et al. Helicobacter pylori and intestinal parasites are not detrimental to the nutritional status of Amerindians. Am. J. Trop. Med. Hyg 76, 534–540 (2007). [PubMed] [Google Scholar]
- 107.Lederberg J & McCray AT ‘Ome sweet’ omics. A genealogical treasury of words. The Scientist 15, 8 (2001). [Google Scholar]
- 108.Peterson J The NIH Human Microbiome Project. Genome Res. 9 October 2009. (doi: 10.1101/gr.096651.109). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tibayrenc M & Ayala FJ The clonal theory of parasitic protozoa: 12 years on. Trends Parasitol. 19, 405–410 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Rich SM, Sawyer SA & Barbour AG Antigen polymorphism in Borrelia hermsii, a clonal pathogenic bacterium. Proc. Natl Acad. Sci. USA 98, 15038–15043 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sarkar SF & Guttman DS Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl. Environ. Microbiol 70, 1992–2012 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kennedy AD et al. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc. Natl Acad. Sci. USA 105, 1327–1332 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhou X et al. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 150, 2565–2573 (2004). [DOI] [PubMed] [Google Scholar]
- 114.Vesper BJ et al. The effect of proton pump inhibitors on the human microbiota. Curr. Drug Metab 10, 84–89 (2009). [DOI] [PubMed] [Google Scholar]