

Abstract
Long-term follow up of prospective studies has shown that continuous Bruton’s tyrosine kinase inhibitor (BTKi) therapy leads to durable remissions in previously untreated patients with TP53-altered chronic lymphocytic leukemia (CLL); however, it is unknown how variant allele frequency (VAF) of TP53 mutation (TP53-m) or percentage of cells with deletion of chromosome 17p [del(17p)] influences efficacy of firstline BTKi. We performed a retrospective analysis of 130 patients with CLL with baseline del(17p) and/or TP53-m treated with BTKi with or without the BCL2 inhibitor venetoclax (VEN) and with or without CD20 antibody in the firstline setting. A total of 104/131 (79%) patients had del(17p). TP53-m was noted in 89/110 (81%) patients tested; there were 101 unique TP53-m with an available VAF. The 4-year progression-free survival (PFS) and overall survival (OS) rates were 72.9% and 83.6%. No baseline characteristics including IGHV mutation status and number of TP53 alterations were associated with significant differences in PFS or OS, though a trend towards shorter PFS with increasing karyotypic complexity (hazard ratio 1.08, p=0.066) was observed. Del(17p) was identified in <25% of cells in 26/104 (25%) of patients, and 28/101 (28%) of TP53-m were low-burden with a VAF of <10%; outcomes of these patients were similar to those with high-burden lesions. This study suggests that low-burden TP53 alterations should not be ignored when assessing genomic risk in CLL in the era of targeted therapy.
Keywords: Bruton’s tyrosine kinase inhibitor, deletion 17p, TP53 mutation, treatment naïve CLL, allelic frequency, clonal burden, combination therapy
Graphical Abstract
INTRODUCTION
TP53 alterations, including TP53 gene mutation (TP53-m) and deletion of chromosome 17p [del(17p)], have historically been the strongest predictors of poor outcomes in patients with chronic lymphocytic leukemia (CLL) after first-line chemoimmunotherapy (CIT).1–4 The advent of targeted therapies such as Bruton’s tyrosine kinase inhibitors (BTKi) and the BCL2 inhibitor venetoclax (VEN) that circumvent the p53 pathway has transformed the therapeutic armamentarium for patients with TP53-altered CLL.5,6 The prevalence of baseline TP53 alteration ranges from 5–15% in patients with previously untreated CLL.1,3,7–9 Unlike with CIT and time-limited VEN with obinutuzumab,10 these high-risk patients can achieve durable remissions with continuous first-line BTKi therapy that potentially approximate those experienced by patients with wild-type disease.11 Single-arm prospective studies of 3412 and 2713 treatment-naïve patients treated with the first-generation BTKi ibrutinib with or without CD20 antibody (CD20 mAb) and a pooled analysis of 89 similarly-treated patients from several studies14 report 4-year progression-free survival (PFS) rates of close to 80%. Zanubrutinib, a second generation BTKi, led to an 18-month PFS of 88.6% for 109 patients with del(17p) in the firstline setting.15
The clinical impact of low-burden TP53 alterations in patients with CLL is an evolving area of research. Next generation sequencing (NGS) allows for detection of low-burden TP53 mutations with variant allele frequencies (VAF) as low as 0.1%, though the negative impact of “subclonal” mutations not detected by conventional Sanger sequencing is not clear, with heterogeneity in findings across studies.7,8,16–18 Conversely, increased percentage of cells harboring del(17p) does appear more consistently associated with shorter time to first treatment and overall survival (OS).9,19 The aforementioned studies were limited to patients who received CIT in the first-line setting and had received at times multiple lines of treatment. Brieghel and colleagues performed a detailed analysis of TP53 alterations in 51 ibrutinib-treated patients and found that a VAF cutoff of 1% was an appropriate threshold of TP53-m “positivity” that maintained the predictive value of TP53-m, and that patients with a single TP53 alteration [del(17p) or single TP53-m) enjoyed superior PFS and OS compared to those with multiple TP53 alterations [del(17p) and TP53-m, or multiple TP53-m). However, they did not stratify patients by size of the TP53-altered clone and included patients treated with ibrutinib in both firstline and relapsed settings.20 Notably, Tam and colleagues found that stratifying patients with del(17p) treated with firstline zanubrutinib by del(17p) positivity of 20% by FISH did not lead to differences in overall response or 18-month PFS.15 TP53-m VAF also does not appear to influence OS in patients treated with targeted therapies in the relapsed setting.18 As targeted therapies have supplanted CIT for treatment-naïve TP53-altered CLL, an updated analysis of the size of the TP53-altered clone and its influence on first-line treatment outcomes is needed.
Several ongoing phase II studies have reported on the outcomes of fixed-duration combination targeted therapy with BTKi + VEN +/− CD20 mAb in the firstline setting with high rates of undetectable measurable residual disease (U-MRD) and favorable PFS.21–26 Jain and colleagues report an 3-year PFS rate of 86% in 18 patients with TP53 alteration treated with firstline ibrutinib and VEN.22 The CAPTIVATE study included 32 patients with TP53 alteration treated with firstline ibrutinib and VEN and reported an overall 30-month disease free survival rate of approximately 95%.26 The CLL2-GIVe study treated 41 patients that uniformly harbored TP53 alteration with ibrutinib, VEN, and obinutuzumab, leading to a 2-year PFS rate of 95.1%.25 Though these findings are encouraging, without randomized data, it is not clear which treatment strategy (continuous BTKi versus fixed-duration combination BTKi + VEN +/− CD20 mAb) is better for TP53-altered CLL.
Here, we report on a single-institution retrospective analysis of the outcomes of patients with CLL with baseline TP53 alteration who received firstline treatment with BTKi-based therapy.
METHODS
Patients with a diagnosis of CLL or small lymphocytic lymphoma (SLL) with fluorescence in situ hybridization (FISH) and/or TP53 gene sequencing testing performed at The University of Texas MD Anderson Cancer Center (MDACC) between 1/1/2010 and 2/28/2021 demonstrating del(17p) and/or TP53-m were identified. Patients who met iwCLL indications for treatment27 who received a BTKi as part of their first CLL-directed treatment, either on or outside of a clinical trial, were included in this study. Patients could have received a CD20 mAb and/or VEN in combination with the BTKi. Nine patients not meeting criteria for treatment who were enrolled on an early intervention study with ibrutinib were excluded. Demographic information, pre-treatment disease characteristics, and survival outcomes were abstracted from the electronic medical record. Rai stage and beta 2-microglobulin (B2M) were recorded from time of diagnosis while all other characteristics were captured immediately pretreatment.
FISH for common abnormalities associated with CLL was performed on cultured bone marrow and/or peripheral blood cells using a multi-color probe panel designed to detect the deletions of 11q22.3 (ATM), 13q14.3 (D13S319), 13q34 (LAMP1) and 17p13.1 (TP53), and trisomy 12 (12p11.1-q11) according to the manufacturer’s instructions (Abbott Molecular, Abbott Park, IL). A total of 200 interphases were analyzed, and the cut off for detection del(17p) alone was 4.5% (independently validated). Conventional cytogenetics was performed on unstimulated and pokeweed mitogen/phorbol ester/CpG oligodeoxynucleotide stimulated cultures. Clonal cytogenetic abnormalities were enumerated by an expert cytogeneticist (GT); single-cell abnormalities were included if they were consistent with FISH results from the same sample or if consistent with a previously identified clone.
TP53 sequencing (of exons 2 and 4–11) was performed by amplicon-based NGS assay in the CLIA-certified Molecular Diagnostics Laboratory in 106 patients, and Sanger sequencing (exons 5–9) in the remaining 4 patients who were excluded from VAF analysis. Using NGS, the median coverage was at least 1500x with a limit of detection of 1%. A TP53 mutation was considered high-burden if the VAF was ≥10% and low-burden if <10%. TP53 variants of germline origin were excluded.
Patient characteristics were described using frequency (percentage) for categorical variables and median (range) for continuous variables. Fisher’s exact tests and Wilcoxon rank sum tests were performed to assess differences between groups. PFS was defined as the time interval between initiation of firstline BTKi-based therapy and progression of CLL meeting iwCLL 2018 criteria27 or death or censored at last follow-up date for non-progressors. OS was defined as time interval between initiation of BTKi and death (from any cause) or censored at last follow-up date for patients who were still alive. Patients who received VEN consolidation after initiation of BTKi therapy were censored with respect to time-to-event outcomes at time of VEN consolidation. The Kaplan-Meier method was used to estimate time-to-event outcomes. Cox proportional hazards regression models were fit to estimate the hazard ratio (HR) and confidence intervals (CI) for covariates and evaluate differences in time to event outcomes stratified by covariates. Forest plots were used to depict the association between each covariate and time-to-event outcome. Spearman rank correlation was used to assess the correlation between covariates. Statistical software used include SAS 9.4 (SAS, Cary, NC) and R 4.1.0 (R Core Team, Vienna, Austria).
RESULTS
A total of 130 patients with TP53 alteration who received first-line BTKi-based treatment for CLL/SLL were included. Pretreatment characteristics are summarized in Table 1. The median follow-up was 4.0 years (range, 0.2 to 9.3).
Table 1.
Pre-treatment Characteristics (N = 130)
| Characteristic | Number (%) or Median [range] |
|---|---|
| Age, years | 63 [36–88] |
| Age ≥ 65 years | 63 (48) |
| Male sex | 88 (68) |
| Rai stage at diagnosis | |
| 0 | 38 (29) |
| I-II | 78 (60) |
| III-IV | 14 (11) |
| B2M at diagnosis, mg/L (n=124) | 2.8 [0.1–14.3] |
| B2M >3.5 mg/L | 40 (32) |
| IGHV unmutated (n=119) | 95 (80) |
| TP53 mutated (n=110) | 89 (81) |
| Median TP53 mutation VAF, % | 42.6 [1–99.5] |
| Del(17p) present | 104 (80) |
| Median cells with del(17p), % | 58.5 [3.5–99] |
| Multi-hit TP53 alteration (n=110) | 68 (62) |
| Other FISH abnormalities | |
| Del(13q) | 76 (58) |
| Trisomy 12 | 29 (22) |
| Del(11q) | 18 (14) |
| Cytogenetic abnormalities (n=98) | 1 [0–22] |
| ≥ 3 | 41 (42) |
| ≥ 5 | 33 (34) |
| Additional mutations (n=75) | |
| NOTCH1 | 23 (31) |
| SF3B1 | 13 (17) |
| BIRC3 | 5 (7) |
Characteristics captured immediately pretreatment unless specified
B2M, beta 2-microglobulin; IGHV, immunoglobulin heavy chain variable region; VAF, variant allele frequency; del, deletion
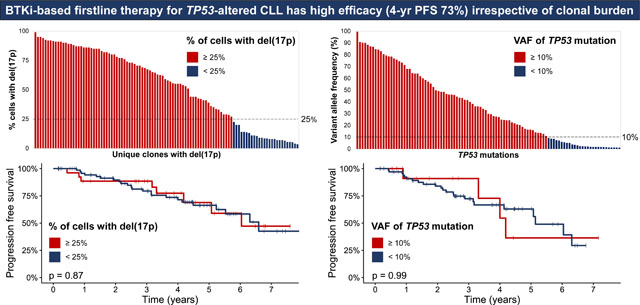
A total of 104/130 (80%) patients had pretreatment del(17p). The median % of cells with del(17p) was 58.5% (range, 3.5–99) (Fig 1A) and del(17p) was identified in <25% of cells in 26/104 (25%) of patients. TP53-m was noted in 89/110 (81%) patients tested. There were 106 unique TP53-m, of which 101 had an available VAF; the median VAF was 29.9% (range, 1–99.5), and 28/101 (28%) mutations were low-burden (Fig 1B–C, Table S1). Among the 110 patients tested for both del(17p) and TP53-m, 63 (57%) patients harbored both lesions, while 26 (24%) only had TP53-m, and 21 (19%) only had del(17p) (Fig 1D). A total of 42/110 (38%) patients had a single TP53 hit [del(17p) or single TP53-m] and 68 (62%) had multiple hits [both del(17p) and TP53-m (n=63) or multiple TP53-m (n=5)]. A depiction of the TP53-m VAF dynamics for patients with multiple timepoints captured is shown in Fig S1.
Figure 1.
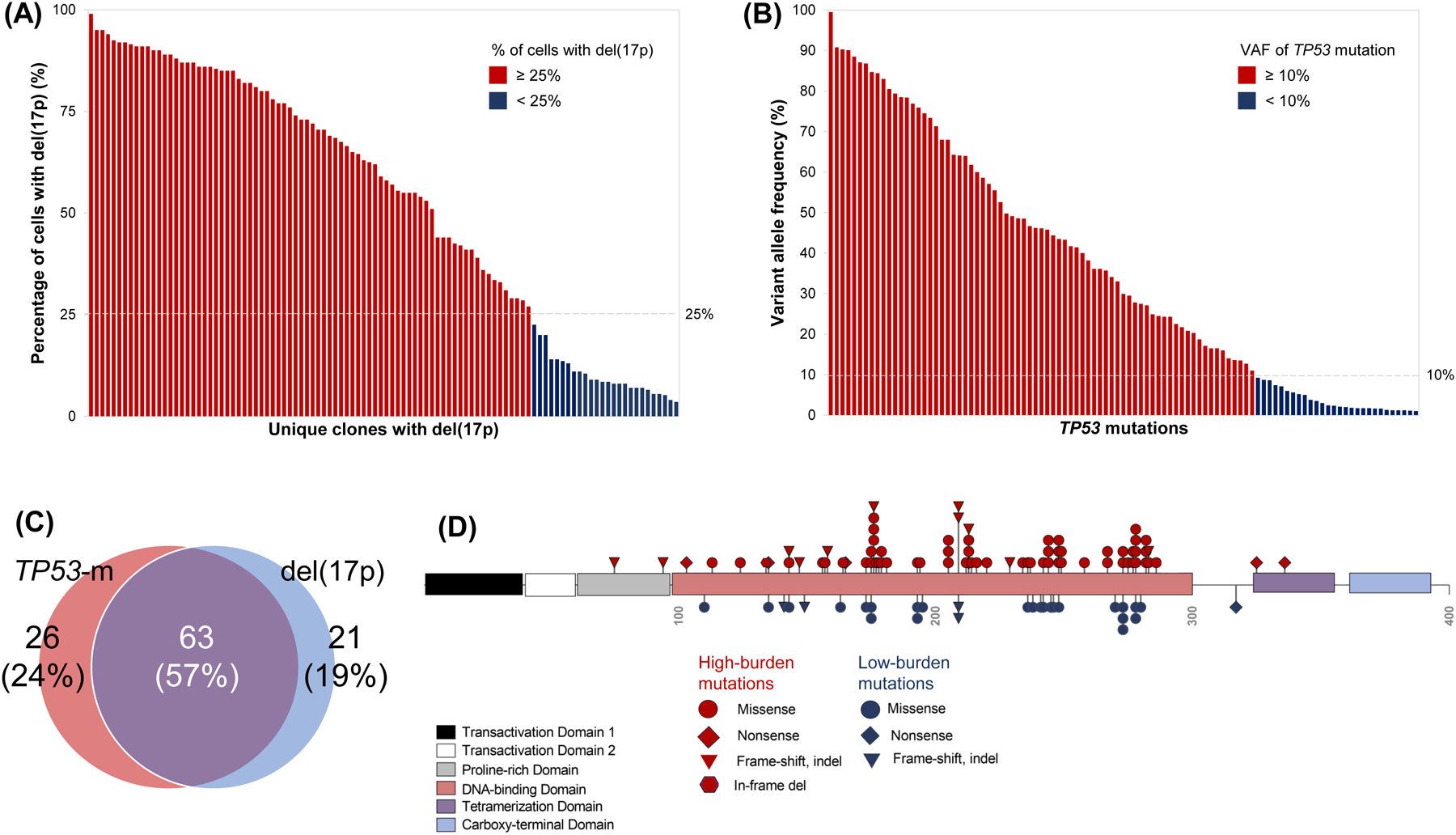
Molecular profiling of TP53 alterations. (A) Percentage of cells with del(17p) identified by fluorescence in situ hybridization for each patient with del(17p), ordered by decreasing percentage. Bars are colored red or blue by whether del(17p) was detected in at least or less than 25% of cells, respectively. (B) Variant allele frequency of 108 unique TP53 mutations identified by next generation sequencing, ordered by decreasing allele frequency. Bars are colored red or blue for high-burden (allele frequency at least 10%) or low-burden (allele frequency less than 10%) mutations, respectively. (C) Venn diagram of the 110 patients tested for both del(17p) and TP53 mutation depicting whether they harbored one or both alterations. (D) Diagram of the TP53 protein and position of 101 unique TP53 mutations along its functional domains. Shapes are colored red or blue for high or low-burden mutations, respectively. Missense mutations involving the DNA binding domain were most common regardless of clonal burden.
Overall, 37 (29%) patients experienced a progression event and 22 (17%) patients died. Six (5%) experienced Richter transformation (RT) as their first event. The 4-year PFS rate was 72.9% (95% CI 64.4–82.5) and the median PFS was 6.6 years (95% CI 5.23-NA) (Fig 2A). The 4-year OS rate was 83.6% (95% CI 76.3–91.5) and the median OS was not reached (Fig 2B). A depiction of the cumulative risk of CLL progression and RT over time for the entire cohort is depicted in Fig S2. By univariable analyses, no baseline characteristic reached statistical significance for their association with PFS or OS (Fig S3–4), though there was a trend towards shorter PFS for patients with TP53-m versus wild type TP53 (p=0.08, HR 3.61 95% CI 0.86–15.18, Fig S5A) and with multiple TP53 hits versus a single TP53 hit (p=0.091, HR 2.07 95% CI 0.89–4.84, Fig S5B). There was no significant difference in PFS when comparing patients with both del(17p) and TP53-m, del(17p) only, and TP53-m only (p=0.15, Fig S5C). Patients with unmutated IGHV had similar PFS versus those with mutated IGHV (p=0.41, HR 0.73 95% CI 0.34–1.56).
Figure 2.
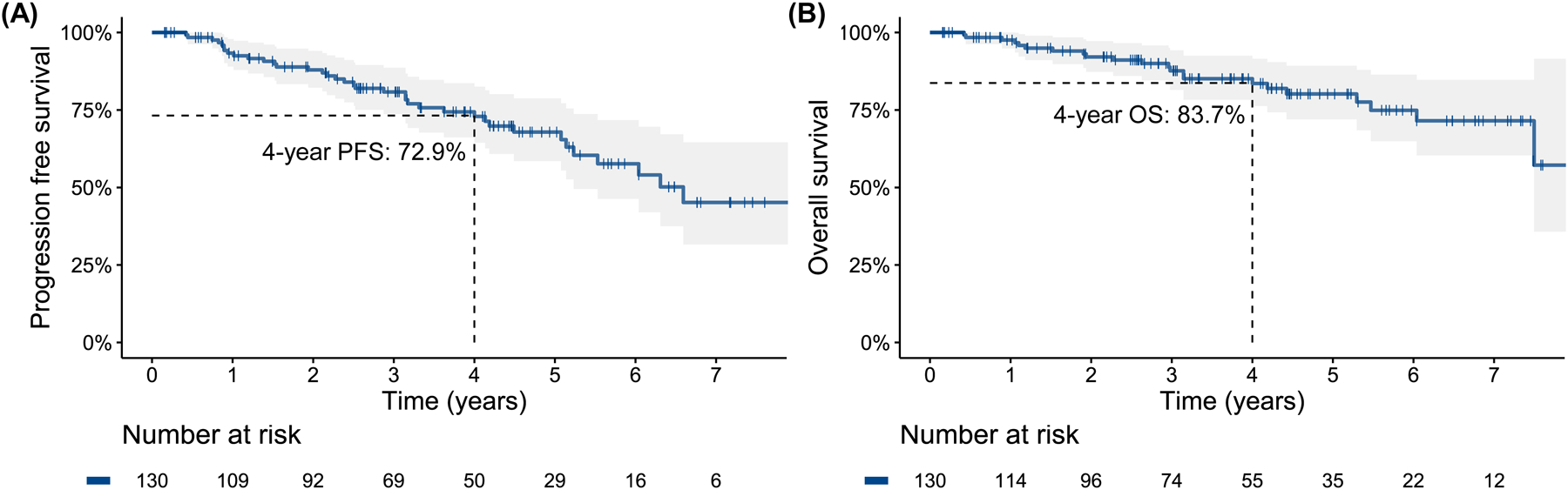
Progression free survival (A) and overall survival (B) for the entire TP53-altered cohort
Patients with high-burden TP53-m (VAF ≥10%) had a higher percentage of cells with del(17p) compared to those with only low-burden TP53-m (median 71% versus 34%, p=0.018, Table S2). Patients with del(17p) affecting ≥25% of cells compared to those with del(17p) affecting <25% of cells were more likely to have TP53-m (85% versus 48%, p=0.001) and harbored more cytogenetic abnormalities (median 3.5 versus 0.5, p=0.018) (Table S3). Patients with multiple TP53 hits compared to those with a single TP53 hit harbored more cytogenetic abnormalities (median 3 versus 0, p=0.0004). Increasing number of cytogenetic abnormalities was correlated with increasing percentage of cells with del(17p) (Spearman’s rank ρ=0.41, p<0.0001, Figure S6A) but not increasing VAF of TP53-m (ρ=0.19, p=0.079, Figure S6B).
PFS was not significantly different for TP53-m patients stratified by a VAF threshold of 10% (p=0.99, HR 1.0 95% CI 0.34–2.88, Fig 3A) and patients with del(17p) based on a threshold of 25% of cells affected (p=0.87, HR 1.07 95% CI 0.48–2.40, Fig 3B). Though PFS was not shorter for patients with at least 3 (p=0.57, HR 1.25 95% CI 0.59–2.65, Fig S7A) or 5 (p=0.13, HR 1.81 95% CI 0.84–3.90, Fig S7B) cytogenetic abnormalities, karyotypic complexity as a continuous variable trended towards associated with PFS. For each additional cytogenetic abnormality, the risk of a PFS event increased by 8% (HR 1.08, 95% CI 1.00–1.18, p=0.066).
Figure 3.
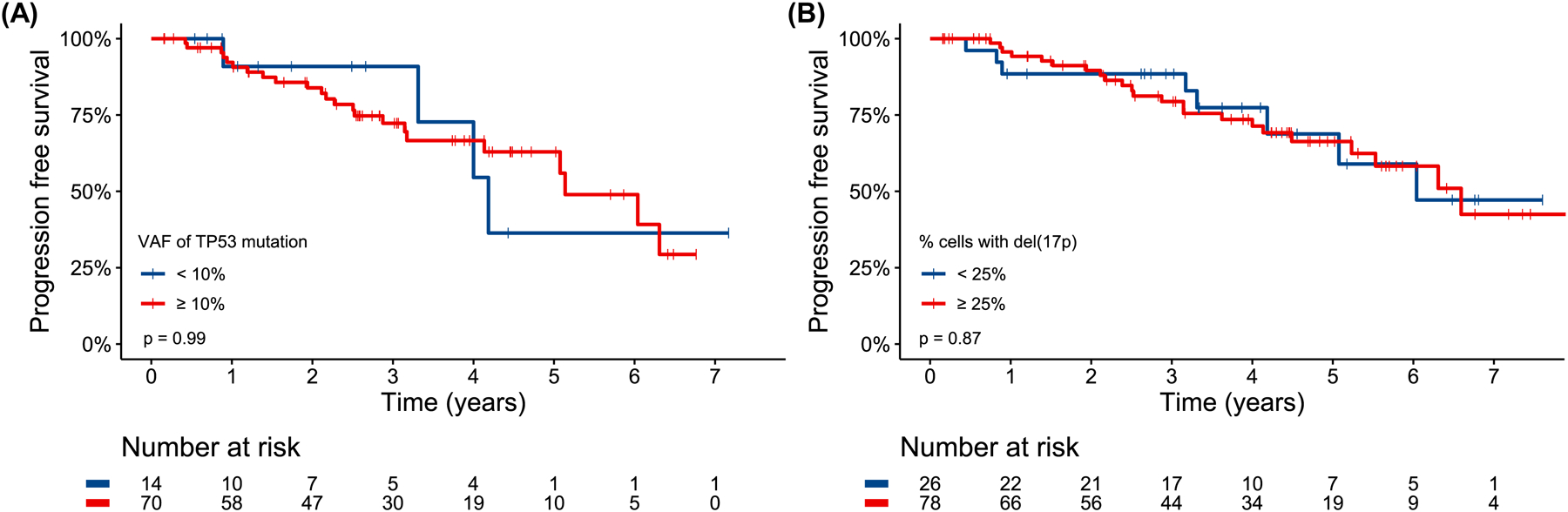
Progression free survival for TP53-mutated patients stratified by a variant allele frequency threshold of 10% (A) and patients with del(17p) with a “cells affected” threshold of 25% (B). In the case of a patient harboring multiple TP53 mutations, the mutation with the highest variant allele frequency was used to stratify patients.
A total of 38/130 (29%) patients were treated on a BTKi and VEN combination protocol (with CD20 mAb, n=4; no CD20 mAb, n=34. A total of 92 (71%) patients received a BTKi (with CD20 mAb, n=24; no CD20 mAb, n=68) without VEN; 13 of these 92 patients received VEN as a consolidation therapy after durable responses to ibrutinib and were censored at the time of VEN consolidation. BTKi was ibrutinib for 114 (88%) and acalabrutinib for 16 (12%) patients. A total of 26 (20%) patients discontinued BTKi because of toxicities. There was no significant association between the addition of a CD20 mAb (p=0.83, HR 0.92 95% CI 0.42–2.02, Fig S8) with PFS. Subsequent therapy choices after CLL progression or RT for 26 patients are described in Table S4 and causes of death without disease progression for the remaining 11 patients are described in Table S5. A total of 6/26 (23%) patients who progressed went on to receive an allogeneic stem cell transplant and 2 patients received chimeric antigen receptor T-cell therapy.
PFS was significantly longer for patients who received BTKi + VEN (±CD20 mAb) versus those who received BTKi without VEN (±CD20 mAb) (p=0.036, HR 0.28 95% CI 0.08–0.92, Fig 4A) with a 4-year PFS rate of 90.5% (95% CI 80.8–100) versus 66.8% (95% CI 56.5–79.0) and median PFS not reached versus 6.0 years (95% CI 5.1-NA). OS was numerically longer for BTKi + VEN (±CD20 mAb) treated patients versus BTKi (±CD20 mAb) treated patients (p=0.15, HR 0.34 95% CI 0.08–1.46, Fig 4B) with a 4-year OS rate of 93.7% (95% CI 8–100) versus 80.0% (95% CI 71.0–90.2), respectively. Median follow-up was 3.3 years (range 0.2–4.8) for BTKi + VEN treated patients and 4.9 years (range 0.2–9.3) for BTKi treated patients. Of the 39 patients who received BTKi + VEN (±CD20 mAb), 23 successfully completed their planned combination therapy (1–2 years depending on the protocol); 13/23 stopped both BTKi and VEN and have been followed for a median of 1.8 years (range 0.3–2.4) after discontinuation without experiencing disease progression.
Figure 4.
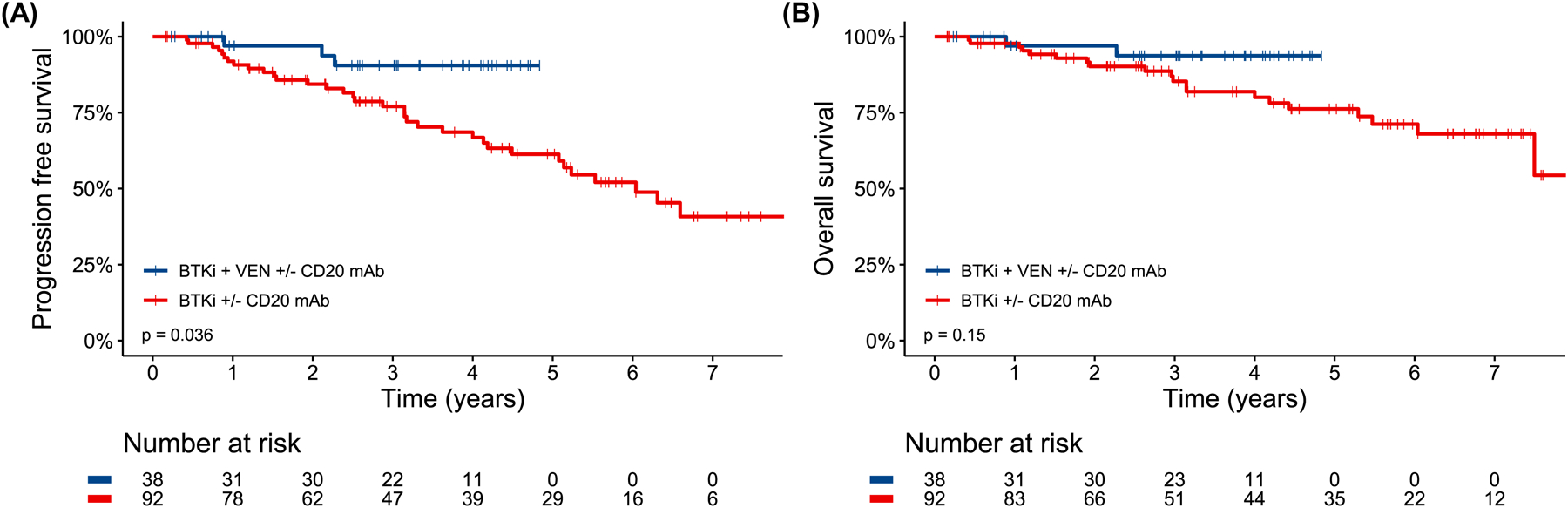
Progression free survival (A) and overall survival (B) by receipt of Bruton’s tyrosine kinase inhibitor and venetoclax (with or without CD20 antibody) combination therapy versus Bruton’s tyrosine kinase inhibitor (with or without CD20 antibody) alone.
Patients who received BTKi + VEN (±CD20 mAb) were less likely to harbor TP53-m (68% vs. 88%, p=0.022) or multi-hit TP53 (45% vs. 71%, p=0.013) compared to those who received BTKi (±CD20 mAb), but no other characteristics were significantly different (Table S6). Multivariable Cox regression incorporating TP53-m or multi-hit TP53 with number of cytogenetic abnormalities and VEN combination found that in both models, only number of cytogenetic abnormalities and the addition of VEN were associated with differences in PFS (Figure S9A–B). No baseline characteristics were associated with PFS by univariable analysis when analyzing only the 92 patients who received BTKi without VEN (Figure S10).
DISCUSSION
Here, we describe a large single-institution retrospective analysis of patients with baseline TP53 alteration treated with firstline BTKi-based therapy for CLL, including patients who received combination therapy with VEN. We report favorable 4-year PFS and OS rates of 72.9% and 83.6%, comparable to reported outcomes of smaller prospective studies.12–14 The clone size either by percentage of cells with del(17p) or by TP53-m VAF was not associated with differences in PFS, though patients with high versus low-burden lesions were more likely to harbor other markers of chromosomal instability. No baseline disease characteristics including multi-hit TP53 and IGHV mutation were statistically associated with difference in PFS by univariate analysis.
The lack of association between clonal burden of TP53 alteration and PFS may reflect the relatively high efficacy of BTKi in the studied patient population or conversely may indicate that even low burden TP53 alterations confer a negative impact on outcomes. We are unable to distinguish between these two possibilities with this study without a comparator group of patients with wild-type TP53. The A041202 trial did not demonstrate a difference in outcomes between patients with TP53-wild-type and TP53-altered CLL treated with firstline ibrutinib, but it was not powered to make this comparison.11 Mato and colleagues performed a large retrospective analysis of patients with or without del(17p) treated with firstline ibrutinib and found that the former cohort experienced significantly shorter time to next treatment and OS as well as more common discontinuation due to progression.28 These results suggest that BTKi efficacy is affected by TP53-alteration in real-world-treated patients. Testing and documentation of TP53 alterations regardless of clonal burden should still be performed for patients receiving first-line BTKi for CLL. We did not find a significant association between multi-hit TP53 and PFS which differs from the findings of other studies20,29 which did report worse outcomes for patients with multi-hit TP53. This difference may be explained by the fact those studies analyzed patients uniformly treated with ibrutinib monotherapy and Brieghel and colleagues20 analyzed a significant number of patients treated in the relapsed setting.
We did note that increasing karyotypic complexity may have contributed to shorter PFS, particularly by multivariate analysis when incorporating VEN combination as a treatment variable. The contribution was much less significant compared to as reported by Kittai and colleagues who found that increasing number of cytogenetic abnormalities was strongly associated with both inferior PFS and OS in ibrutinib-treated patients.30 This may suggest that TP53-aberration remains the primary driver of progression in CLL when complex karyotype is present, but nonetheless, conventional cytogenetics analysis must still be performed in TP53-aberrant CLL to fully characterize patients.
We observed a potential lower risk of progression for first-line combination therapy of BTKi + VEN (±CD20 mAb) compared to BTKi (±CD20 mAb) without VEN for patients with TP53-altered CLL. The BTKi + VEN and BTKi without VEN cohorts were relatively well balanced for baseline characteristics besides prevalence of TP53-m and multi-hit TP53, but those covariates were not associated with outcomes. While PFS appeared improved with combination therapy, there was no significant difference in OS, so the question of whether combination novel therapy with BTKi + VEN is preferable to sequential therapy with each novel agent alone remains unanswered. Some patients were however able to enjoy time off all CLL-directed therapy with the combination approach despite their high-risk disease and maintain their durable remissions.
Some limitations of this study stem from its retrospective single-institution nature and from the inclusion of patients treated on and off clinical trials over a decade-long time period. Patients who received BTKi + VEN (±CD20 mAb) were all enrolled on more recent clinical trials (multiple protocols with differing study designs) and had shorter follow-up while BTKi (±CD20 mAb) patients were treated on and off protocol. The inclusion of “real-world” patients who received BTKi without VEN could have contributed to a relatively inferior 4-year PFS rate of 67% and likely introduced differences between cohorts not captured by baseline characteristic comparisons such as co-morbidities and differences in clinical management or monitoring. Heterogeneity in patient follow-up and response assessment schedules precluded us from describing response rates, MRD assessments, or adverse events. The lower limit of detection of the NGS assays used at our institution is a VAF of 1%, higher than that of other studies,7,17,18,20 which likely led to a difference of distribution between high-burden and low-burden TP53 mutations and an underestimation of low-burden mutations. VAF values were missing for the several patients with TP53-m detected by Sanger sequencing in this study. Brieghel and colleagues20 did find that excluding TP53-m with a VAF < 1% did not affect predictive performance for patients treated with ibrutinib. We captured VAF dynamics of TP53-m in a minority of patients only, though other studies suggest that clonal expansion of TP53-m during treatment with targeted therapy (unlike CIT) is uncommon and not associated with inferior outcomes.18,31
Studies demonstrating the efficacy of the combination BTKi + VEN in treatment naïve CLL continue to be reported, but whether a fixed duration of combination therapy (that may be associated with increased toxicity) has advantages over sequencing novel agents or over continuous BTKi is still unknown. This is particularly pertinent to patients with high-risk genomics. Though multiple large phase III randomized studies comparing combination novel therapy with single novel therapy are planned and/or enrolling, no studies are specifically enrolling patients with TP53 alteration. Studies dedicated to enrolling such high-risk patients are needed to clarify optimal treatment and should not exclude patients with low-burden alterations.
Supplementary Material
Acknowledgments
This research was funded by the MD Anderson NIH/NCI Cancer Support Grant under award number P30 CA016672 and the MD Anderson Chronic Lymphocytic Leukemia Moon Shot program.
Conflicts of interest
JB serves on the speaker bureau for Beigene, TG Therapeutics, Novartis, Gilead, Pharmacyclics LLC, Janssen, reports research funding from Beigene, TG Therapeutics, Gilead, Pharmacyclics LLC, and consultancy for AstraZeneca, Gilead, Pharmacyclics LLC, and Janssen. PT reports research funding from Genentech, Amgen, Pharmacyclics, Adaptive Biotechnologies, AbbVie, consultancy for Genentech, Janssen, Pharmacyclics, Adaptive Biotechnologies, Gilead, and AbbVie, and has received honoraria from Genentech, Amgen, Janssen, Pharmacyclics, Adaptive Biotechnologies, Gilead, and AbbVie. AF reports research funding from AstraZeneca and BeiGene and serves on the advisory board for Janssen, AstraZeneca, and BeiGene. KS serves on the advisory board for Daiichi-Sankyo, Novartis, and Pfizer, and reports research funding from Novartis and honoraria from Otsuka Pharmaceuticals. DS reports serving on the advisory board for and receiving personal fees/stock options from Newave. HK reports research funding from AbbVie, Amgen, Ascentage, BMS, Daiichi-Sankyo, Immunogen, Jazz, Novartis, and Pfizer and honoraria from AbbVie, Amgen, Aptitude Health, Ascentage, Astellas Health, Astra Zeneca, Ipsen, Pharmaceuticals, KAHR Medical Ltd, NOVA Research, Novartis, Pfizer, Precision Biosciences, and Taiho Pharmaceutical Canada. WW reports research funding from Juno Therapeutics, KITE Pharma, Loxo Oncology, Inc., Miragen, Cyclacel, Sunesis, Acerta Pharma Inc., Pharmacyclics LLC, Oncternal Thererapeutics, Inc., Karyopharm, Gilead Sciences, Xencor, Janssen, Genentech, AstraZeneca, GSK/Novartis, AbbVie, and consultancy for Genzyme Corporation. NJ has received research funding from Pharmacyclics, AbbVie, Genentech, AstraZeneca, BMS, Pfizer, Servier, ADC Therapeutics, Cellectis, Adaptive Biotechnologies, Incyte, Precision Biosciences, Aprea Therapeutics, Fate Therapeutics, Mingsight, Takeda, Medisix, Loxo Oncology, Novalgen, and has received honoraria / served on advisory board for Pharmacyclics, Janssen, AbbVie, Genentech, AstraZeneca, BMS, Adaptive Biotechnologies, Servier, Precision Biosciences, Beigene, Cellectis, TG Therapeutics, ADC Therapeutics, MEI Pharma, Ipsen, CareDX
Authors not listed declare no potential conflicts of interest.
Footnotes
Ethics Committee Approval
This study was approved by the Institutional Review Board of MD Anderson Cancer Center under protocol 2021-0441.
References
- 1.Zenz T, Eichhorst B, Busch R, et al. TP53 mutation and survival in chronic lymphocytic leukemia. Journal of Clinical Oncology. 2010;28(29):4473–4479. doi: 10.1200/JCO.2009.27.8762 [DOI] [PubMed] [Google Scholar]
- 2.Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123(21):3247–3254. doi: 10.1182/BLOOD-2014-01-546150 [DOI] [PubMed] [Google Scholar]
- 3.An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. The Lancet Oncology. 2016;17(6):779–790. doi: 10.1016/S1470-2045(16)30029-8 [DOI] [PubMed] [Google Scholar]
- 4.Strati P, Keating MJ, O’Brien SM, et al. Outcomes of first-line treatment for chronic lymphocytic leukemia with 17p deletion. Haematologica. 2014;99(8):1350. doi: 10.3324/HAEMATOL.2014.104661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burger JA. Treatment of Chronic Lymphocytic Leukemia. New England Journal of Medicine. 2020;383(5):460–473. doi: 10.1056/NEJMra1908213 [DOI] [PubMed] [Google Scholar]
- 6.Cherng HJJ, Jain N. First-Line Therapy for Chronic Lymphocytic Leukemia: Bruton Tyrosine Kinase or BCL2 or Both? Hematol Oncol Clin North Am. 2021;35(4):725–738. doi: 10.1016/J.HOC.2021.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123(14):2139–2147. doi: 10.1182/blood-2013-11-539726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nadeu F, Delgado J, Royo C, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127(17):2122–2130. doi: 10.1182/blood-2015-07-659144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dyke DL Van, Werner L, Rassenti LZ, et al. The Dohner fluorescence in situ hybridization prognostic classification of chronic lymphocytic leukaemia (CLL): the CLL Research Consortium experience. British Journal of Haematology. 2016;173(1):105. doi: 10.1111/BJH.13933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Sawaf O, Zhang C, Tandon M, et al. Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (CLL14): follow-up results from a multicentre, open-label, randomised, phase 3 trial. The Lancet Oncology. 2020;21(9):1188–1200. doi: 10.1016/S1470-2045(20)30443-5 [DOI] [PubMed] [Google Scholar]
- 11.Woyach JA, Ruppert AS, Heerema NA, et al. Long-Term Results of Alliance A041202 Show Continued Advantage of Ibrutinib-Based Regimens Compared with Bendamustine Plus Rituximab (BR) Chemoimmunotherapy. Blood. 2021;138(Supplement 1):639–639. doi: 10.1182/BLOOD-2021-153146 [DOI] [Google Scholar]
- 12.Ahn IE, Tian X, Wiestner A. Ibrutinib for Chronic Lymphocytic Leukemia with TP53 Alterations. N Engl J Med. 2020;383(5):498–500. doi: 10.1056/NEJMC2005943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sivina M, Kim E, Wierda WG, et al. Ibrutinib induces durable remissions in treatment-naïve patients with CLL and 17p deletion/TP53 mutations. Blood. Published online September 14, 2021. doi: 10.1182/BLOOD.2021012315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allan JN, Shanafelt T, Wiestner A, et al. Long-term efficacy of first-line ibrutinib treatment for chronic lymphocytic leukaemia in patients with TP53 aberrations: a pooled analysis from four clinical trials. British Journal of Haematology. Published online December 5, 2021. doi: 10.1111/BJH.17984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tam CS, Robak T, Ghia P, et al. Zanubrutinib monotherapy for patients with treatment naïve chronic lymphocytic leukemia and 17p deletion. Haematologica. 2020;106(9):2354–2363. doi: 10.3324/HAEMATOL.2020.259432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu L, Kim HT, Kasar SN, et al. Survival of Del17p CLL depends on genomic complexity and somatic mutation. Clinical Cancer Research. 2017;23(3):735–745. doi: 10.1158/1078-0432.CCR-16-0594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brieghel C, Kinalis S, Yde CW, et al. Deep targeted sequencing of TP53 in chronic lymphocytic leukemia: Clinical impact at diagnosis and at time of treatment. Haematologica. 2019;104(4):789–796. doi: 10.3324/haematol.2018.195818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malcikova J, Pavlova S, Kunt Vonkova B, et al. Low-burden TP53 mutations in CLL: Clinical impact and clonal evolution within the context of different treatment options. Blood. 2021;138(25):2670–2685. doi: 10.1182/BLOOD.2020009530/1806970/BLOOD.2020009530.PDF [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tam CS, Shanafelt TD, Wierda WG, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009;114(5):957–964. doi: 10.1182/BLOOD-2009-03-210591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brieghel C, Aarup K, Torp MH, et al. Clinical Outcomes in Patients with Multi-Hit TP53 Chronic Lymphocytic Leukemia Treated with Ibrutinib. Clinical Cancer Research. 2021;27(16):4531–4538. doi: 10.1158/1078-0432.CCR-20-4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain N, Keating M, Thompson P, et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. New England Journal of Medicine. 2019;380(22):2095–2103. doi: 10.1056/nejmoa1900574 [DOI] [PubMed] [Google Scholar]
- 22.Jain N, Keating M, Thompson P, et al. Ibrutinib Plus Venetoclax for First-line Treatment of Chronic Lymphocytic Leukemia: A Nonrandomized Phase 2 Trial. JAMA Oncology. 2021;7(8):1213–1219. doi: 10.1001/JAMAONCOL.2021.1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davids MS, Lampson BL, Tyekucheva S, et al. Acalabrutinib, venetoclax, and obinutuzumab as frontline treatment for chronic lymphocytic leukaemia: a single-arm, open-label, phase 2 study. The Lancet Oncology. 2021;22(10):1391–1402. doi: 10.1016/S1470-2045(21)00455-1 [DOI] [PubMed] [Google Scholar]
- 24.Soumerai JD, Hochberg E, Barnes JA, et al. Zanubrutinib, obinutuzumab, and venetoclax with minimal residual disease-driven discontinuation in previously untreated patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: a multicentre, single-arm, phase 2 trial. The Lancet Haematology. 2021;8(12):e879–e890. doi: 10.1016/S2352-3026(21)00307-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huber H, Edenhofer S, von Tresckow J, et al. Phase 2 study of obinutuzumab (GA-101), ibrutinib and venetoclax (CLL2-GIVe) in patients with untreated high-risk chronic lymphocytic leukemia. Blood. Published online December 15, 2021. doi: 10.1182/BLOOD.2021013208 [DOI] [PubMed] [Google Scholar]
- 26.Wierda WG, Allan JN, Siddiqi T, et al. Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results From the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J Clin Oncol. 2021;39(34):3853–3865. doi: 10.1200/JCO.21.00807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760. doi: 10.1182/blood-2017-09-806398 [DOI] [PubMed] [Google Scholar]
- 28.Mato AR, Tang B, Azmi S, et al. A clinical practice comparison of patients with chronic lymphocytic leukemia with and without deletion 17p receiving first-line treatment with ibrutinib. Haematologica. Published online April 21, 2020. doi: 10.3324/HAEMATOL.2021.280376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Visentin A, Mauro FR, Cibien F, et al. Continuous treatment with Ibrutinib in 100 untreated patients with TP53 disrupted chronic lymphocytic leukemia: A real-life campus CLL study. American Journal of Hematology. 2022;97(3):E95–E99. doi: 10.1002/AJH.26437 [DOI] [PubMed] [Google Scholar]
- 30.Kittai AS, Miller CR, Goldstein D, et al. The impact of increasing karyotypic complexity and evolution on survival in patients with CLL treated with ibrutinib. Blood. 2021;138(23):2372–2382. doi: 10.1182/BLOOD.2020010536 [DOI] [PubMed] [Google Scholar]
- 31.Cafforio L, Raponi S, Cappelli LV, et al. Treatment with ibrutinib does not induce a TP53 clonal evolution in chronic lymphocytic leukemia. Haematologica. Published online October 14, 2020. doi: 10.3324/HAEMATOL.2020.263715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.