

Abstract
Medicinal chemistry continues to be impacted by new synthetic methods. Particularly sought after, especially at the drug discovery stage, is the ability to enact the desired chemical transformations in a concise and chemospecific fashion. To this end, the field of organic synthesis has become captivated by the idea of ‘molecular editing’—to rapidly build onto, change or prune molecules one atom at a time using transformations that are mild and selective enough to be employed at the late stages of a synthetic sequence. In this Review, the definition and categorization of a particularly promising subclass of molecular editing reactions, termed ‘single-atom skeletal editing’, are proposed. Although skeletal editing applies to both cyclic and acyclic compounds, this Review focuses on heterocycles, both for their centrality in medicinal chemistry and for the definitional clarity afforded by a focus on ring systems. A classification system is presented by highlighting methods (both historically important examples and recent advances) that achieve such transformations, with the goal to spark interest and inspire further development in this growing field.
Modern approaches to drug discovery are increasingly impacted by the development of novel synthetic methods, with motivations that arise from the ability to enact the desired chemical transformations in a concise and chemospecific fashion1–3. Such innovations provide the ability to circumvent traditional hurdles in drug development, especially those associated with the synthesis of derivatives of a promising lead at late stages. To this end, the field of organic synthesis has become captivated by the idea of ‘molecular editing’, which has metaphorical roots in macroscopic, digital technologies; for example, can we make synthesis as easy as ChemDraw?
Although a clear definition of molecular editing remains elusive, tied up in this pursuit is the idea of being able to rapidly build and/or subsequently tolerate complexity—privileging those transformations that are mild and selective enough to be employed at late stages of a synthetic sequence or in the context of the total synthesis of complex natural products. As often occurs in burgeoning fields, the nomenclature has become muddled, with a veritable zoo of terms that claim to describe the ways in which our collective works can be organized. For molecules we have skeletons, frameworks, scaffolds, structural motifs and cores. For reactions we have editing, surgery, modification, remodelling, distortion, deconstruction, scaffold hopping and diversification. Despite the chemical precision that many of the pioneering contributions offer, precision of language has been slow to develop alongside these efforts. Indeed, the term ‘molecular editing’ itself is inherently vague. Strictly speaking, any chemical transformation is a molecular edit of one flavour or another, which leaves room for the criticism that this blossoming of nomenclature is unnecessary, or worse, intentionally obfuscatory.
Although we cannot single-handedly resolve the definitional issues of the field, our laboratories are intimately involved in such efforts, and therefore we aim here to instead propose the definition and categorization of a subclass of molecular editing reactions that we suggest are particularly fertile territory for the discovery of new, powerful chemical transformations: single-atom skeletal editing. In this Review, we demonstrate our classification system specifically for cyclic systems by highlighting methods (both historically important examples and recent advances) that achieve such transformations, with the hope to spark interest and inspire further development in this growing field.
Definitions
Any discussion of molecular or skeletal editing immediately raises several important questions, the answers of which define the boundaries of the task at hand. We propose several definitions here.
What constitutes a molecular skeleton?
Depending on the context, molecular editing either subsumes or stands in contrast to C–H functionalization. Most chemists probably agree that C–H bonds are not part of the molecular skeleton, but instead occupy peripheral sites of the molecule. As soon as one moves beyond hydrogen atoms, however, the situation becomes murkier. For example, is a primary amine peripheral? What about a secondary methylamine?
Rings offer a natural dividing line, wherein a skeleton is defined by a molecule’s cyclic system(s). In this dichotomy, we propose that a change in the component atoms that comprise a ring system constitutes skeletal editing, whereas the analogous modification to an exocyclic atom is better construed as peripheral editing (Fig. 1a). We favour such a definition because there is no unambiguous way to distinguish the skeleton of an acyclic molecule from its periphery, although a strategy akin to IUPAC nomenclature, wherein the longest chain of heavy atoms is privileged, may be used when necessary for such systems. Although modification of the heavy-atom chains in acyclic systems no doubt represents a similarly important challenge, in this Review we have chosen to focus on heterocyclic compounds not only for the definitional line they enable, but also for their prevalence in both naturally occurring and anthropogenic bioactive small molecules4–6. As such, methods capable of directly modifying late-stage ring systems have the propensity to substantially impact lead optimization and analogue synthesis in medicinal chemistry2.
Fig. 1 |. Definition and classification of single-atom molecular editing.
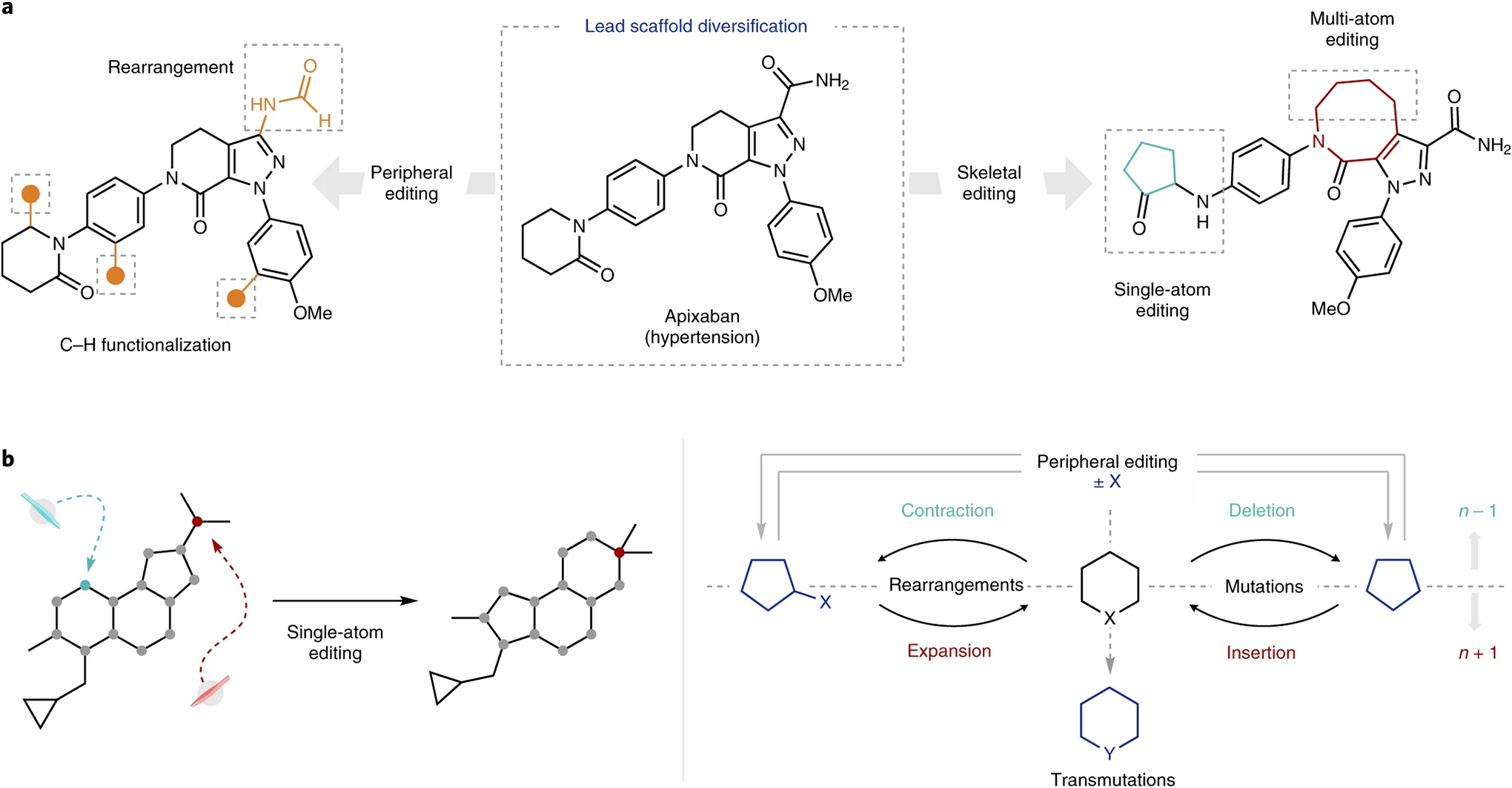
a, The anatomy of molecular editing. Examples of peripheral editing (left) and skeletal editing (right) conceptually applied towards the diversification of apixaban. b, Classification of single-atom editing. A depiction of single-atom editing applied to a representative molecule (left) and the related classification scheme (right).
What is a single-atom skeletal edit?
For ring systems, the scale by which the ring is modified offers further classification categories. As the carbon and nitrogen atoms themselves bear exocyclic (peripheral) substituents, a focus on the constituent atoms of the ring system again serves as a natural demarcation. Regardless of the number or size of the exocyclic substituents, the essence of a single-atom edit is that the ring system being modified is changed by one atom (an n-membered ring is converted to the n + 1 or n − 1 analogue; Fig. 1b). As we discuss in greater detail in the Outlook section, single-atom editing can also include ‘transmutations’ wherein one of the constituent atoms is exchanged for another. This focus on the synthetic logic enabled by such single-atom changes is deliberate, for two major reasons:
‘Editing’ evokes precision. Single-atom changes represent the most elementary of possible changes to a molecular skeleton, with more complex changes possible, in principle, as the combination of multiple single-atom edits.
The retrosynthetic simplicity of single-atom logic is enabling. Rearrangements of molecular skeletons often suffer from being difficult to see in a retrosynthetic sense, which limits their adoption. Indeed, the most used reactions in medicinal chemistry (cross-coupling, amide bond formation, nucleophilic aromatic substitution and reductive amination) have a common retrosynthetic simplicity that, in addition to their broad scope, has propelled their implementation.
We note that the focus in this Review does not represent a value statement, but instead a constraint on the ensuing discussion—many molecular editing strategies not covered in this Review are of immense potential interest. For example, advances in multi-atom editing, C–H functionalization, metathesis, homologation of acyclic chains, ring cleavage, transannular bond formation or annulation are simply beyond the scope of this Review7–14.
A classification scheme
Within single-atom skeletal editing, two major dividing lines can be drawn, which result in four general reaction classes. The first is straightforward as it depends on whether the ring system in question is expanded (n → n + 1) or contracted (n → n − 1). Within this classification, there remains a question of whether the atom being edited begins (or is retained, respectively) as an exocyclic substituent of the ring system. We refer to such transformations as rearrangements, subdivided into contraction and expansion. Juxtaposed, deletions and insertions (collectively called mutations) denote skeletal edits in which the edited atom is not directly attached to the ring system in question15. Fig. 1b (right) provides an overview of this classification scheme for a simple (hetero)cyclic ring system.
Carbonyl chemistry has historically dominated the development of these kinds of transformations, with many named reactions that enable the manipulation of cyclic ketones in a precise fashion. Modern approaches have sought to expand the mechanistic and conceptual template of these pioneering examples to aliphatic and aromatic heterocycles. Accordingly, we seek below to highlight classical, carbonyl-based approaches as context for the selected recent advances in single-atom skeletal editing of heterocycles, as well as specific transformations that exemplify this emerging area.
As a number of the transformations described herein require multiple steps to achieve the overall single-atom skeletal edit, there are several instances in which the classification of a particular transformation could vary based on the defined starting and ending points. We note these cases where they arise in our discussion. Indeed, the ability to toggle between rearrangements and mutations is itself a problem of peripheral editing, which highlights the synergy between the two pursuits. Ultimately, our selected transformations should serve as an inspiration for the future development of analogous, one-step methods that could serve as powerful, ideal chemical transformations to enable molecular editing with single-atom precision.
n − 1 single-atom editing
Contractions.
For carbocyclic systems, anionic, carbene and cationic intermediates have all been classically employed to realize ring contractions of cyclic ketones16. A widely exploited reaction for carbocyclic ring contraction is the Favorskii rearrangement. In this reaction, treatment of α-haloketones with a base results in a skeletal rearrangement, which occurs through a cyclopropanone intermediate 2 (although this can be bypassed in some systems, for example, a quasi-Favorskii rearrangement17). Ultimately, the carbonyl motif undergoes an endo-to-exocyclic transposition, which gives rise to acyl n − 1 cycloalkane products (Fig. 2a, top). The utility of this transformation has been demonstrated in several natural product syntheses. For example, starting from a (+)-pulegone derivative, a Favorskii ring contraction forms the corresponding methyl cyclopentane carboxylate 418. This versatile intermediate has been further elaborated in the total syntheses of several terpenoid natural products, such as (+)-epoxydictymene by Schreiber and co-workers19 and (−)-iridomyrmecin by Wolf and co-workers20.
Fig. 2 |. Ring contractions that leverage classical carbonyl chemistry.
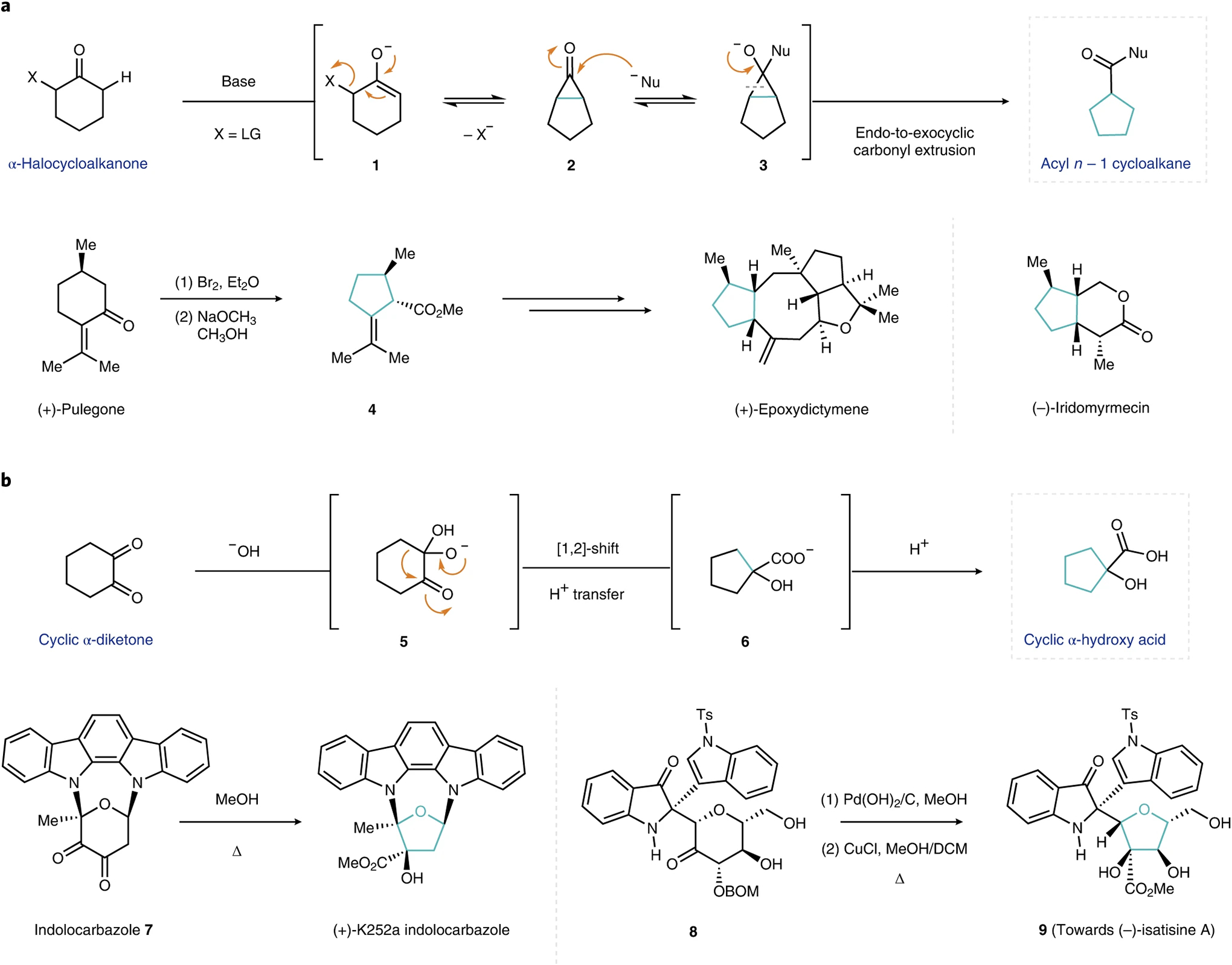
a, General reaction mechanism for the Favorskii rearrangement reaction (top) with synthetic applications demonstrated in the syntheses of (+)-epoxydictymene19 and (−)-iridomyrmecin20 (bottom). b, General reaction mechanism for the benzilic acid rearrangement reaction (top) and applications towards (+)-K252a21 and (−)-isatisine A22 (bottom). Contracted rings are highlighted in blue for clarity. LG, leaving group; Nu, nucleophile; DCM, dichloromethane; BOM, benzyloxymethyl acetal group; Ts, para-toluenesulfonyl group.
The benzilic acid rearrangement is another powerful method to achieve ring contractions (Fig. 2b). Treatment of cyclic α-diketones with a base triggers a rearrangement sequence initiated by the addition of the nucleophile across the C=O bond. Collapse of the resulting tetrahedral intermediate 5 and subsequent alkyl migration furnishes the corresponding ring-contracted cyclic α-hydroxy acid. This transformation has been applied toward the total syntheses of numerous natural products. In their 1996 synthesis of (+)-K252a, Stoltz and Wood used a stereoselective benzilic acid rearrangement to convert a precursor pyranoindolocarbazole 7 into the desired furanoindolocarbazole core (Fig. 2b, bottom left)21. More recently, Xie and co-workers demonstrated the synthetic applicability of this transformation in the total synthesis of (−)-isatisine A (Fig. 2b, bottom right)22. Here a biomimetic benzilic acid rearrangement converts indole glucoside 8 into the densely functionalized indole furanoside 9 upon contraction.
Recent advances have enabled similar ring-contraction rearrangements in non-carbonyl-containing ring systems. Saturated azacyclic systems, for example, can be induced via bicyclic quaternary ammonium intermediates to undergo a formal ring contraction following nucleophilic attack on an endocyclic electrophile23,24. Building on previous accounts by Suárez and co-workers25,26, Sarpong and co-workers recently reported a photomediated ring contraction of acyl azacyclic scaffolds, wherein the nitrogen atom undergoes an endocyclic to exocyclic migration (Fig. 3a)27. Under a radical-polar-crossover manifold, irradiation of α-acylated 10 results in excitation and intersystem crossing to give 11 in the triplet state. A Norrish type II 1,5-hydrogen atom abstraction then affords the corresponding 1,4-diradical 12, which undergoes a C–N bond fragmentation to generate tethered imine-enol 13. The desired cyclopentane product is then formed following an intramolecular Mannich reaction. The versatility of this transformation was demonstrated by its application towards the late-stage remodelling of several biologically active small-molecule derivatives (Fig. 3a, 14–16). This rearrangement was also rendered enantioselective with the use of chiral phosphoric acid derivatives—a combination of hydrogen-bonding and ion-pairing interactions leads to an organized transition state, and the chiral phosphoric acid backbone favours the attack on one enantio-face, which gives rise to (+)-18 in up to a 90% e.e.
Fig. 3 |. Select developments in single-atom ring contractions of heterocycles.
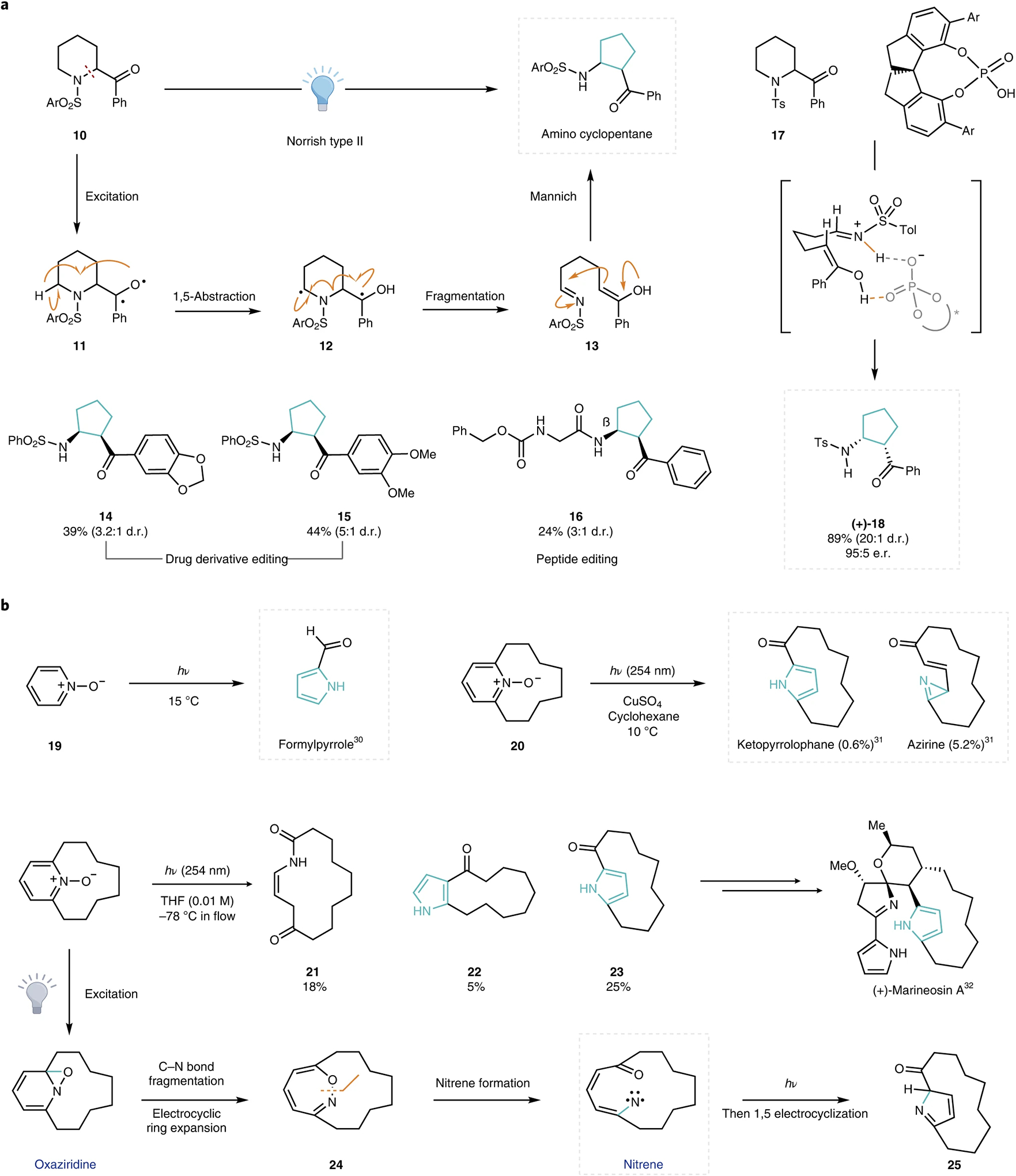
a, Photomediated ring contractions of acyl piperidine scaffolds27. b, Photochemical ring contractions of pyridine N-oxides30–33. Contracted rings are highlighted in blue for clarity. d.r., diastereomeric ratio; e.r., enantiomeric ratio.
Photochemical transformations have also been successfully employed for the skeletal editing of aromatic heterocycles28,29. In particular, pyridine N-oxides undergo unique rearrangements on photoirradiation; Streith et al. reported a ring contraction rearrangement that affords 2-formyl pyrrole products (Fig. 3b, top left)30. Building on this precedent, Weber and Rohn demonstrated pyridinophane ring contractions to afford ketopyrrolophane and macrocyclic azirene products, albeit in low yields31. After reaction optimization, Harran and co-workers recently demonstrated the utility of this skeletal modification in their synthesis of the prodigi-nine alkaloid, (+)-marineosin A32. Mechanistically, excitation of the N-oxide generates the corresponding oxaziridine33. An ensuing electrocyclic ring expansion generates 1,2-oxazepine 24. Under the photoirradiation conditions, excitation of 24 leads to N–O bond lengthening and subsequent nitrene formation, which then undergoes a 1,5-electrocyclization and tautomerization to provide ketopyrrolophane 23. A competitive 1,3-electrocyclization process forms the corresponding azirene, which was converted into lactam 21 on silica gel chromatography. Under the reaction conditions, the in situ generated azirine was also reported to be unstable, and was converted into bicyclic pyrrole 22 after rearrangement through a nitrile ylide intermediate, which could then cyclize to give the observed product34,35. The unique photoreactivity of such heteroaromatic N-oxides was also applied in photo-Dimroth-like rearrangements of pyridazines (not shown), which further highlights the versatility of these photoinduced ring-opening processes to achieve dramatic skeletal editing through formally contractive rearrangements36.
Deletions.
The formal deletion of a single, selected atom from a cyclic skeleton to afford ring-contracted products can conceptually (and chemically) be related to ring-contraction rearrangements by removal of the extruded exocyclic substituent. For carbonyls, in an extension of the Favorskii rearrangement discussed above, decarboxylation of the rearranged substrates affords ring-contracted products in which the carbonyl carbon of the starting material is excised from the skeleton (Fig. 4a). This sequence to achieve ring contraction by formal atom deletion (specifically, contraction followed by removal of the resulting peripheral substituent), was elegantly applied in the first synthesis of cubane by Eaton and Cole in 1964. In that case, treatment of α-bromoketone 27 with potassium hydroxide effected the Favorskii rearrangement to afford cuban-ecarboxylic acid 28. Conversion into the related t-butyl perester 29 facilitated a subsequent thermal decarboxylation, and ultimately generated cubane on overall deletion of the highlighted carbon (Fig. 4a)from the skeleton37,38.
Fig. 4 |. Single-atom deletions that leverage carbonyl chemistry.
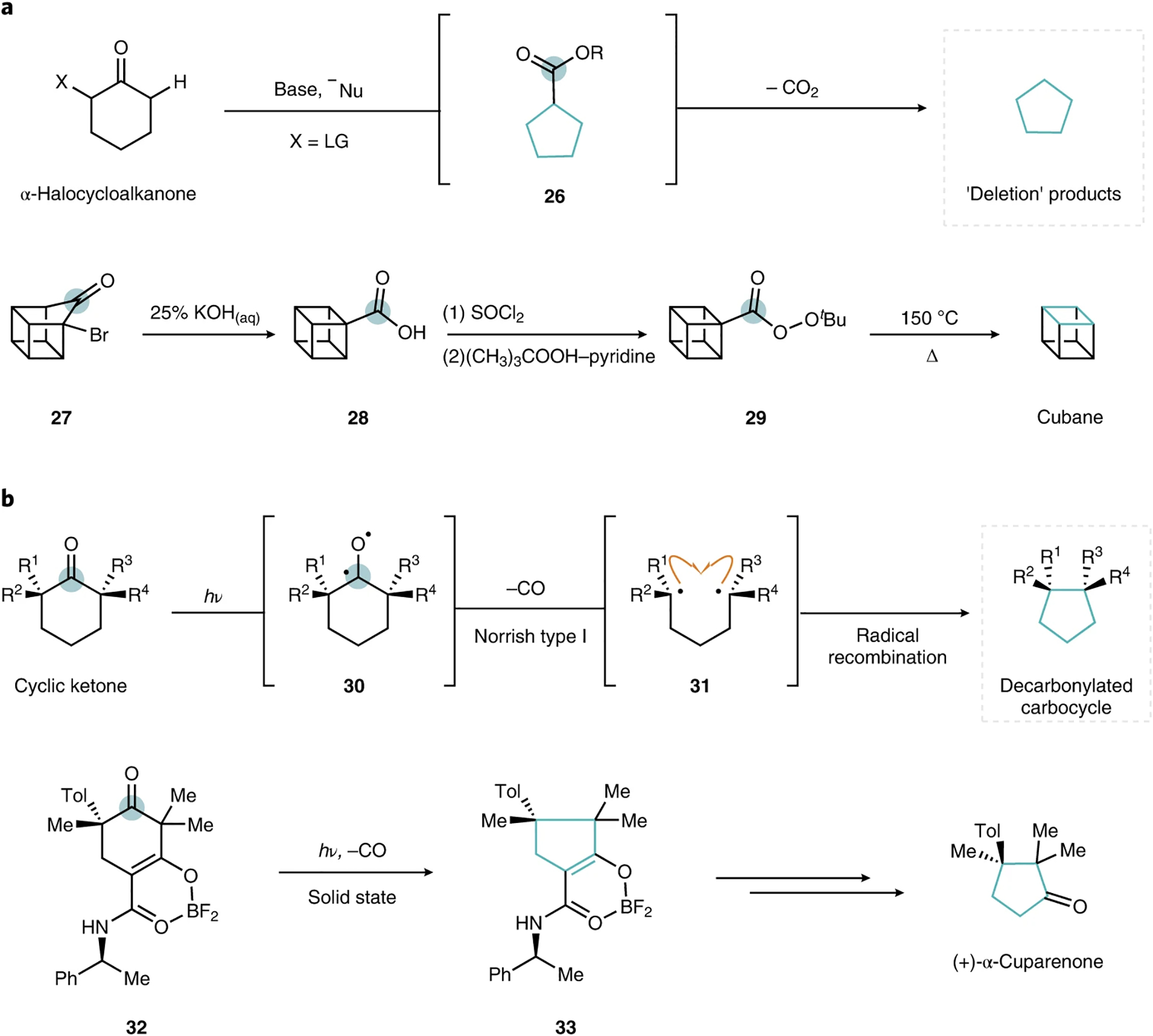
a, General reaction mechanism for the Favorskii rearrangement reaction and decarboxylation (top) with the application demonstrated in the synthesis of cubane37,38 (bottom). b, General reaction mechanism for photodecarbonylation (top) and the application towards (+)-α-cuparenone42 (bottom). Deleted carbon atoms are circled, and contracted rings are highlighted in blue for clarity. Tol, tolyl group.
Alternatively, carbonyl carbon deletion can also be achieved directly (Fig. 4b). Photodecarbonylation of carbocycles that bear photoreactive carbonyl groups flanked by fully substituted sp3-hybridized carbons constitutes a carbonyl deletion that can stereoselectively forge contiguous all-carbon quaternary centres39. Irradiation of cyclic ketones affords diradical intermediates 30 that can undergo a Norrish type I fragmentation, loss of CO and subsequent radical recombination (31) to produce ring-contracted products wherein the original carbonyl carbon is excised from the cyclic skeleton. An early report of this kind of decarbonylative ring contraction was discussed by Nicolaou et al. on the observation of an unexpected ring-contracted side product during their total syntheses of several hamigeran natural products40. Subsequently, this chemistry was developed as a solid-state photodecarbonylation method by Garcia-Garibay and co-workers, and then applied in the total syntheses of (±)-herbertenolide41, (+)- and (−)-α-cuparenone42, and various piperidinoindoline bis(cyclotryptamine) alkaloids, which included psychotriadine43,44. In a similar vein, Xie and co-workers reported an oxidative deformylation ring contraction (not shown) that affords related ring-contracted products on the treatment of α-formyl cyclic ketones with hydrogen peroxide39.
Efforts to develop skeletal editing tools have extended the ability to delete an atom from cyclic systems beyond carbocyclic frameworks that contain a carbonyl functional handle. In 2018, Sarpong and co-workers developed a method that formally achieves the deletion of a single carbon from saturated nitrogen heterocycles, such as piperidines (Fig. 5a)45. In the proposed mechanism, persulfate oxidizes a ligand-bound Ag(I) to Ag(II) and subsequently disproportionates into a sulfate dianion and sulfate radical anion. Saturated N-Bz protected cyclic amines then undergo hydrogen-atom transfer with the sulfate radical anion to afford an α-amino radical that is further oxidized to the iminium ion by Ag(II). This iminium ion intermediate is then trapped by H2O to afford a hemiaminal species, which, on equilibration to the aldehyde and oxidation to the carboxylic acid, undergoes silver-catalysed decarboxylative bromination to afford an acyclic bromoamide that possesses one less carbon atom. In a subsequent step, this ω-bromoamide can undergo base-promoted intramolecular cyclization to furnish ring-contracted amine products (a lower-yield one-pot process is also possible). This formal carbon deletion strategy was demonstrated on a series of simple cyclic amines, which ranged from substituted piperidines (to yield 34) to azepanes (to yield 35)45.
Fig. 5 |. Select developments in single-atom deletions from nitrogenous heterocycles.
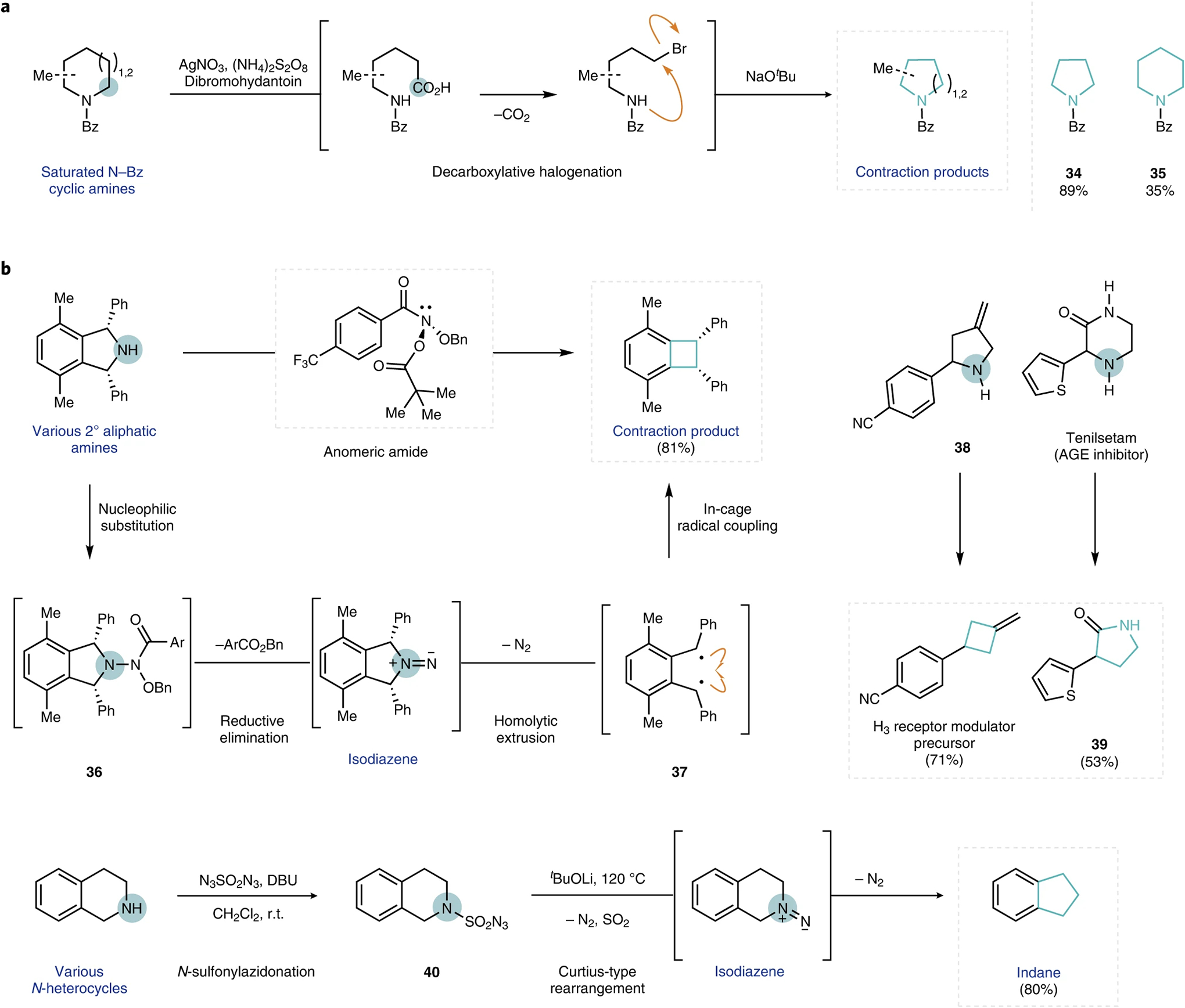
a, Representative examples of a carbon deletion reaction in nitrogen heterocycles45. b, Representative examples of nitrogen deletion reactions applied to azacyclic frameworks47,48. Deleted atoms are circled, and contracted rings are highlighted in blue for clarity. AGE, advanced glycation end product; DBU, diazabicycloundecene. r.t., room temperature.
Single-atom nitrogen deletion from saturated azacycles is also highly desirable, particularly because such deletion transformations would turn ubiquitous C–N bonds into surrogates for the formation of C–C bonds46. Levin and co-workers recently disclosed a method that deletes nitrogen from secondary aliphatic amines under mild conditions in a single step to yield ring-contracted, intramolecular carbon–carbon coupling products (Fig. 5b, top)47. In this system, nucleophilic substitution of electrophilic N-pivaloyloxy-N-alkoxyamides (anomeric amides) by secondary amine substrates followed by reductive elimination of ArCO2Bn affords isodiazene intermediates that extrude dinitrogen to produce highly reactive diradical species 37 that couple intramolecularly to form a new C–C bond between the carbons originally bound to nitrogen. Not only does this transformation exhibit a high functional group tolerance, nitrogen deletion was also successfully applied towards the synthesis of the marine metabolite polysiphenol, and utilized in the late-stage skeletal editing of several bioactive compounds, which included 38 (giving rise to an H3 receptor modulator precursor) and the advanced glycation endpoint inhibitor tenilsetam (to afford 39)47.
Lu and co-workers have also reported a method for N-atom deletion from azacycles, wherein treatment of secondary amines with N3SO2N3 produces sulfamoyl azide intermediates (Fig. 5b, bottom)48. After purification, these isolated sulfamoyl azide species 40 undergo Curtius-type rearrangements on treatment with t-BuOLi at 120 °C to generate 1,2-diazene intermediates that subsequently extrude dinitrogen to afford the ring-contracted products over two steps. More recently, Antonchick and co-workers reported an approach to access the same intermediates through in situ generated iodonitrenes at 80 °C49. Notably, the complementary oxygen deletion under reductive conditions was reported by Cao and Shi in 2017, but this transformation, to date, has only been demonstrated on acyclic dibenzyl ethers50.
Methods that accomplish ring contractions are powerful for manipulating heterocycles. Although we have highlighted selected examples that involve nitrogen, general transformations that involve other heteroatoms and carbocyclic systems hold substantial promise moving forward.
n + 1 single-atom editing
Expansions.
Exocyclic substituents have been widely employed in carbonyl chemistry to achieve ring expansions. Common reaction pathways involve semi-pinacol-like expansions, wherein cyclic tertiary carbinols with exocyclic leaving groups undergo the incorporation of the exocyclic methylene and formation of a carbonyl group51. Related to carbonyl-specific transformations, the Dowd–Beckwith rearrangement (Fig. 6a, top) homologates cyclic ketones through the generation of the exocyclic β-radical 42 from the corresponding halomethyl compound 41, which typically requires an additional ester substituent for high yields52,53. As noted in the introduction, the classification of this transformation depends on the defined starting point. Starting from α-halomethylated compound 41, the ring-expanded γ-keto ester product constitutes a ring expansion, whereas defining the beginning of this transformation as the β-keto ester prior to halomethylation renders the overall transformation an insertion (see below). Both six- and seven-membered β-keto esters can readily undergo ring expansions to the seven- and eight-membered γ-keto esters, albeit with somewhat limited functional group tolerance. The Dowd–Beckwith rearrangement was notably applied as a key retrosynthetic disconnection in the total synthesis of salvileucalin C, wherein the [5,6]-bicycle framework 45 was prepared by expanding the five-membered β-keto ester precursor 44 (Fig. 6a, bottom)54.
Fig. 6 |. Ring expansions that leverage carbonyl chemistry.
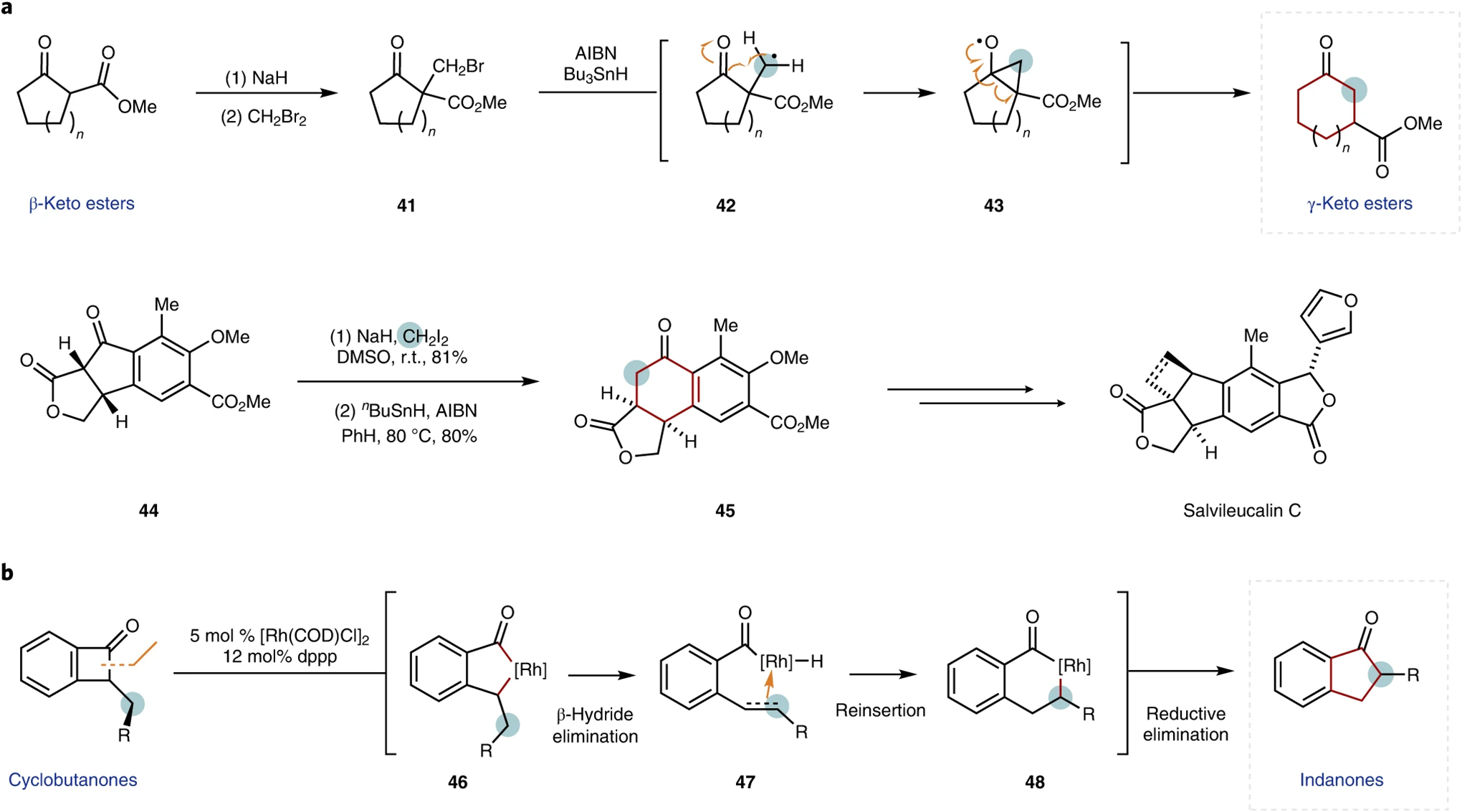
a, General reaction mechanism for the Dowd–Beckwith rearrangement reaction52 (top) with application demonstrated in the synthesis of salvileucalin C54 (bottom). b, Representative example of a rhodium-catalysed cyclobutanone rearrangement55. Expanded rings are highlighted in red for clarity. AIBN, azobisisobutyronitrile; COD, 1,5-cyclooctadiene; dppp, 1,3-bis(diphenylphosphino) propane.
Dong and co-workers developed an analogous ring expansion that does not require a halogen atom for initiation (Fig. 6b)55. Insertion of a rhodium catalyst into benzocyclobutenones 46 and subsequent β-hydride elimination generates the key metal hydride–olefin complex 47, which undergoes migratory reinsertion of the hydride to afford the expanded metallocycle 48 and ultimately a ring-expanded product55. This transformation facilitates an exo-to-endocyclic carbon atom incorporation, allows for ring expansion to occur with a net 1,2-hydrogen migration and also demonstrates an excellent regioselectivity for the formation of α-substituted indanones.
The template exemplified by the carbonyl-based transformations was expanded to other classes of heterocycles. The Wengryniuk group reported the ring expansion of benzylic tertiary alcohols through the action of bis(pyridine)-ligated I(III) dications56. Subsequent treatment of the resulting hexafluoroisopropanol ketals 50 with triethylsilane under Lewis acid conditions affords the ring-expanded cyclic ethers over two steps (Fig. 7a). Similar rearrangements for primary benzylic amines were reported by Murai and co-workers—providing access to cyclic amines (Fig. 7a, bottom)—and for aminal derivatives on treatment with N-halosuccinimides to afford cyclic amidines through nitrogen-insertion rearrangements (not shown)57.
Fig. 7 |. Select advances in single-atom ring expansions.
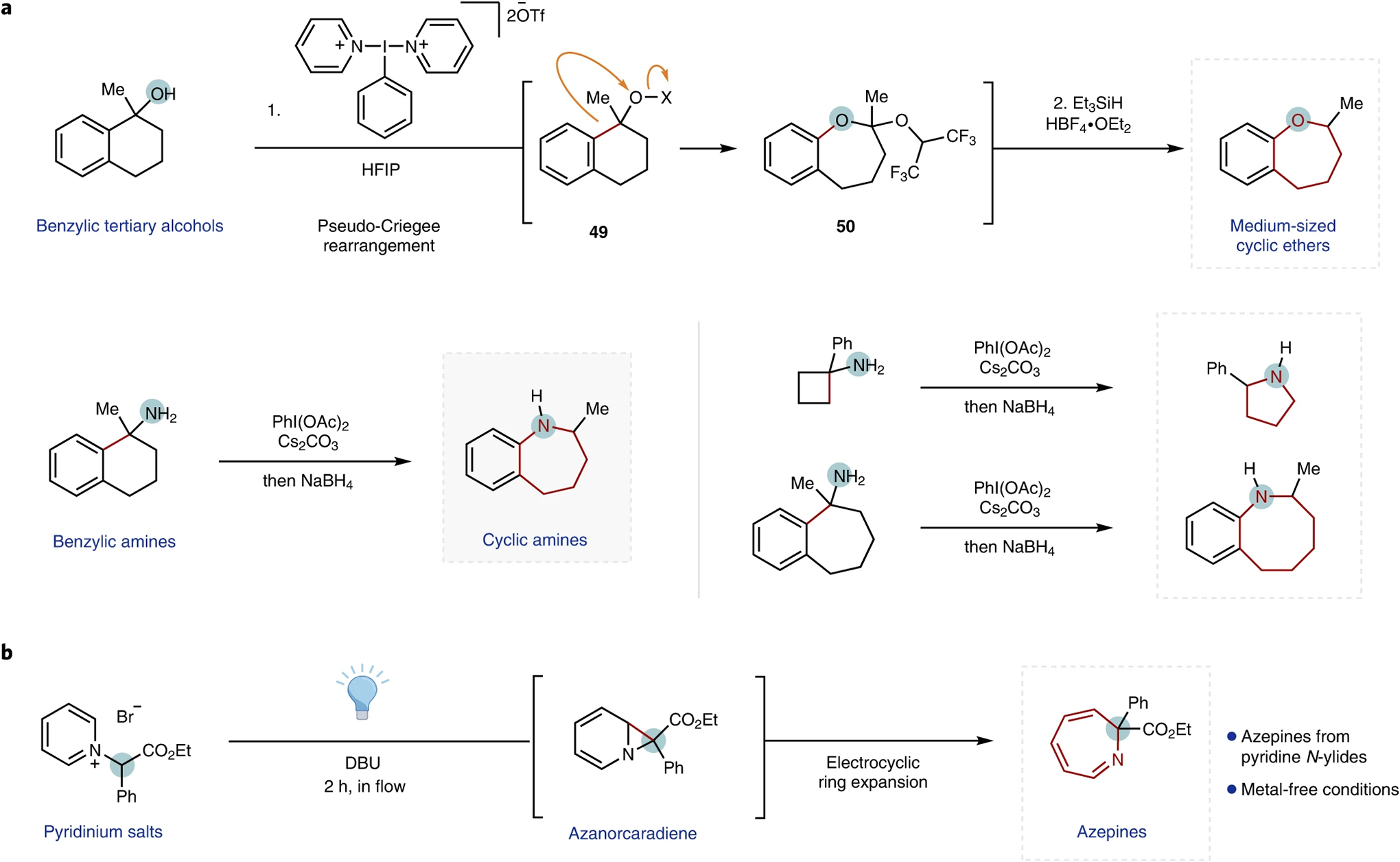
a, Representative examples of oxidative ring expansions in benzylic alcohols56 (top) and amines57 (bottom). b, Photochemical ring expansion of pyridinium salts58. Expanded rings are highlighted in red for clarity. HFIP, hexafluoroisopropanol; Tf, trifluoromethanesulfonyl.
Aromatic systems can also undergo ring expansion through migrations that involve reactive unsaturated species, such as carbenes and nitrenes. Beeler and co-workers recently reported a photochemical Stevens-like rearrangement of in situ generated pyridinium ylides to afford azepine derivatives in flow (Fig. 7b)58. This transformation is part of a larger class of related aromatic nitrene and carbene chemistry, some of which proceeds thermally, and includes the formation of related azepines from arylnitrenes59–63.
Insertions.
Perhaps the widest assortment of classical carbonyl rearrangements falls into this last category, atom insertions, with notable entries such as the Bayer–Villiger, Tiffaneau–Demjanov, and Schmidt reactions. The Beckmann rearrangement (Fig. 8a), reported in 1886, is particularly notable given its modern applications64. Indeed, the classical synthesis of caprolactam from cyclohexanone is still employed on a multiton scale (it accounts for around 90% of the global demand for Nylon)65. At the opposite end of the complexity scale, another notable example is the synthesis of azithromycin, the first 15-membered macrolide antibiotic. In 1980, azithromycin was synthesized from erythromycin A by PLIVA Laboratories using a Beckmann rearrangement followed by reduction and concomitant reductive methylation, which led to an increase in the overall potency and half-life of the resulting compound (azithromycin) compared with that of the parent macrolide66.
Fig. 8 |. Single-atom insertions that leverage carbonyl chemistry.
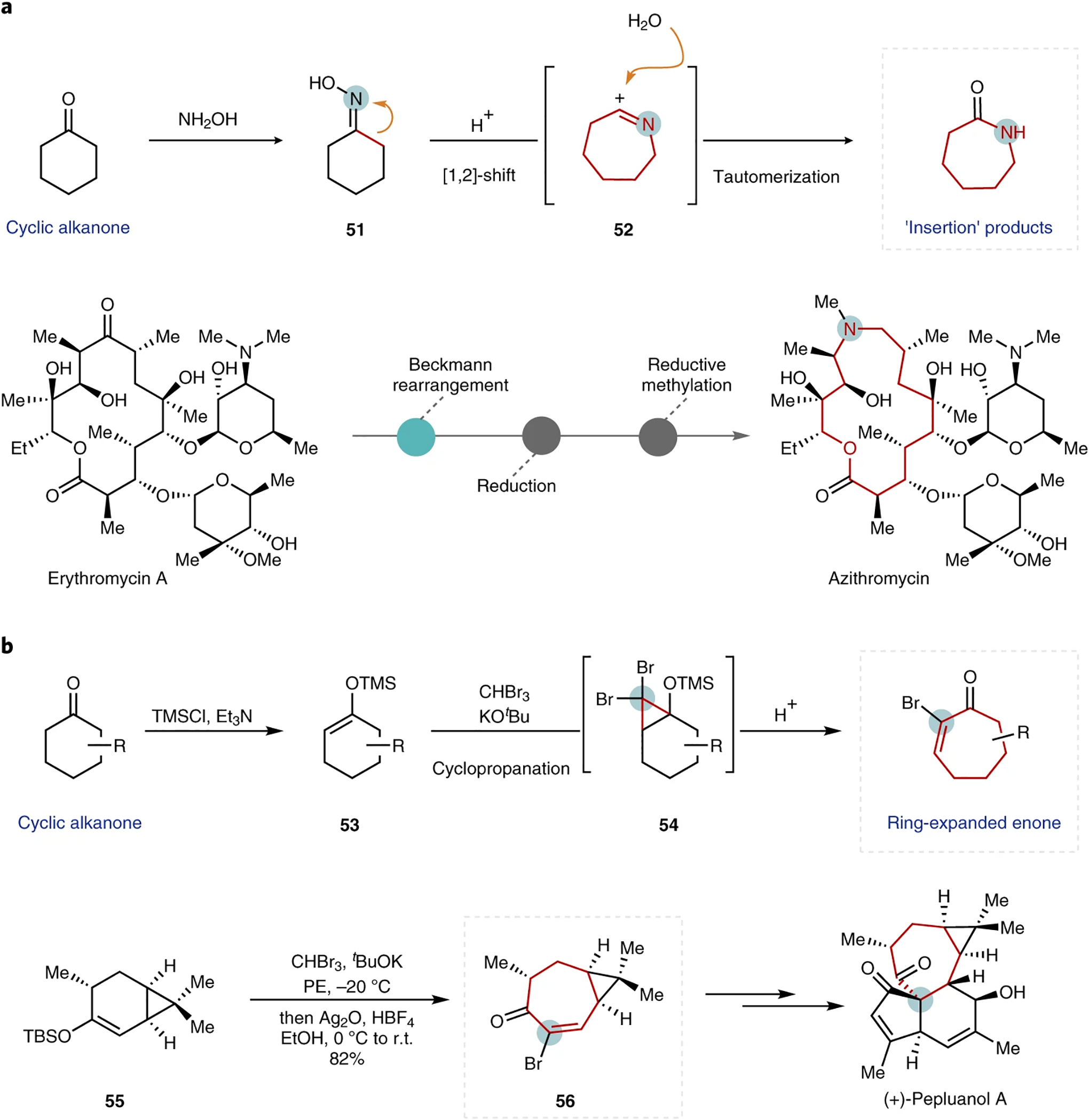
a, General reaction mechanism for a Beckmann rearrangement reaction (top) with the application demonstrated in the synthesis of azithromycin66 (bottom). b, Representative example of a cyclopropanation reaction used to achieve ring expansion (top) and a synthetic application shown in the synthesis of (+)-pepluanol A72 (bottom). Inserted atoms are circled, and expanded rings are highlighted in red for clarity. TMS, trimethylsilyl; TBS, tert-butyldimethylsilyl.
Again, here the classification of this transformation is dependent on the defined starting and ending points. If the oxime 51 is isolated and defined as the beginning of the transformation, the rearrangement process can be construed as an expansion rather than as an insertion process of the cyclic alkanone.
Cyclopropanation with halocarbenes offers an alternative approach for bimolecular insertion. In this vein, Conia and co-workers reported in 1976 the ring expansion of cyclohexanone-derived silyl enol ethers 53 using dibromocarbene generated in situ from bromoform and potassium tert-butoxide67. Ring opening of the intermediate dibromocyclopropane 54 under acidic or thermal conditions led to the formation of α-bromo-α,β-unsaturated ketones (Fig. 8b, top). Dibromocarbene was also reported to react with enol ethers68,69, enamines70 and enol acetates71 to produce ring-expanded products in varying yields. Recently, Gaich and co-workers employed a similar approach en route to an asymmetric synthesis of (+)-pepluanol A72. Synthesis of the key seven-membered enone 56 was realized from the carene-derived silyl enol ether 55.
In contrast to these carbonyl-based transformations, single-atom ring expansions in other settings remain relatively rare. In 1881, Ciamician and Dennstedt reported the ring expansion of pyrroles to give 3-halo-pyridines using haloform as a carbynyl cation equivalent73. Despite its potential, the reaction is limited by poor yields and a low functional group tolerance, due in part to the competitive Reimer–Tiemann formylation74. Recently, a variant of this transformation was reported by Levin and co-workers in which aromatic chlorodiazirines were used as alternative, isolable and stable carbynyl cation equivalents, which gave direct access to 3-arylpyridine and quinoline motifs from the corresponding pyrroles and indoles, respectively, in moderate-to-good yields and with a wide functional group tolerance (Fig. 9a, top)75. The requisite chlorodiazirines can be accessed in a single step by Graham oxidation of commercially available amidinium salts76.
Fig. 9 |.
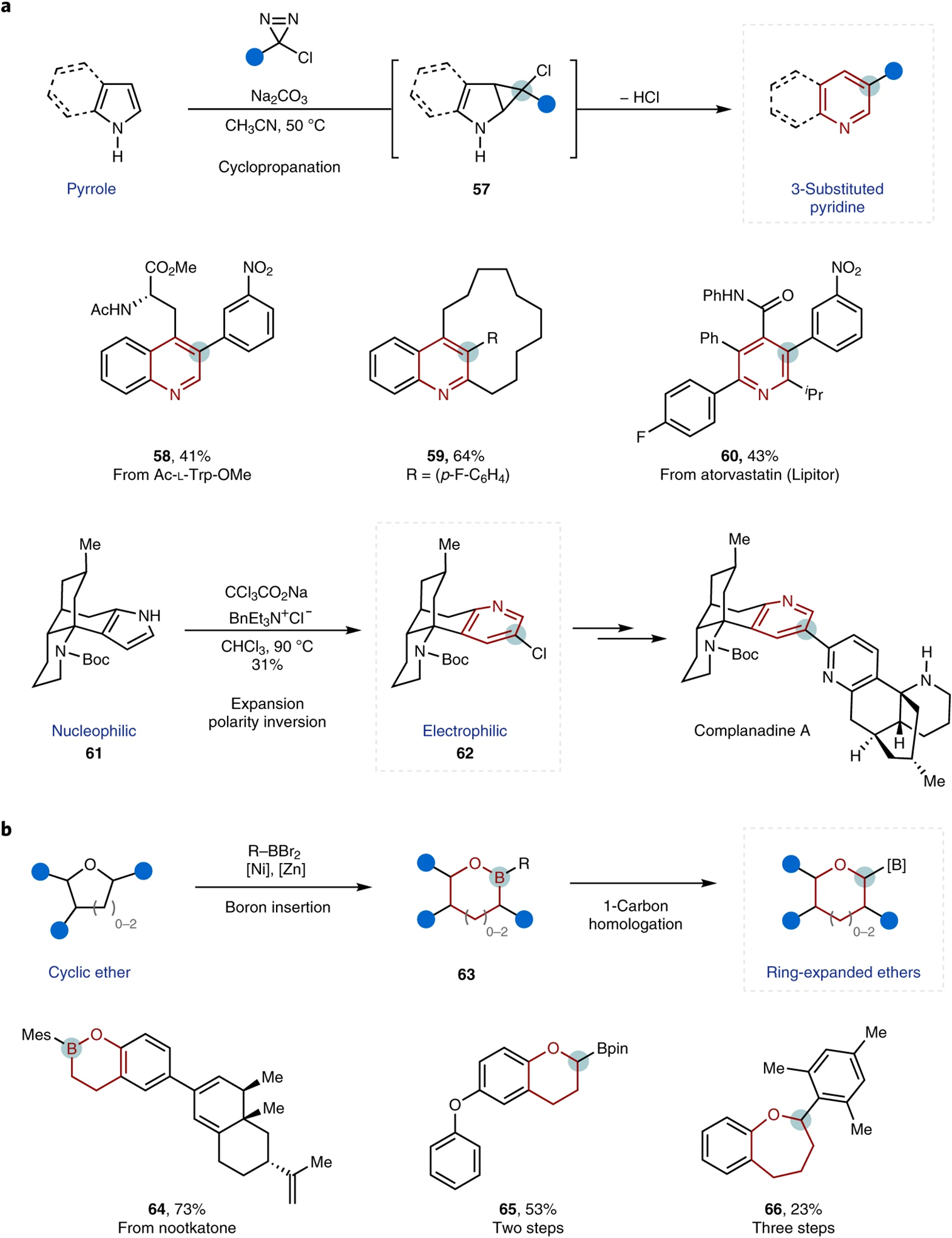
Recent advances in single-atom insertions into heterocycles. a, Representative azole-carbon insertion reaction75 (top), and applications of a related insertion reaction in the synthesis of complanadine A77 (bottom). b, Ether–boron insertion reactions78. The inserted atoms are circled, and the expanded rings are highlighted in red for clarity. Boc, tert-butoxycarbonyl group; Pin, pinacol; Mes, mesityl group.
The potential of this transformation was further demonstrated by ring expansion of a protected tryptophan (to yield 58), and a 2,3-fused indole cyclophane (to afford 59), as well as late-stage skeletal editing of N-des-alkyl Lipitor (to provide 60). Dai and co-workers employed the classical Ciamician–Dennstedt ring expansion as a key step in their synthesis of complanadine A, transforming a pyrrole 61 into pyridine 6277. Notably, this tactical decision took an electron-deficient (electrophilic) pyridine back to an electron-rich (nucleophilic) pyrrole in their retrosynthesis and enabled transformations that would have been impossible (for example, Mannich-type cyclization) in the forward direction with the electron-deficient heterocycle (Fig. 9a, bottom).
Cleavage of alkyl ether bonds typically requires harsh and strongly acidic conditions (for example, BBr3, HBr or HI). Recently, Dong and co-workers described an elegant approach for boron insertion into C(sp3)−O bonds in the presence of a nickel catalyst and zinc reductant through a ‘cleavage-then-rebound’ pathway78. Notably, the versatility of the resulting boronic esters 63 was leveraged for subsequent one-carbon homologation (with a net two steps, Fig. 9b).
Outlook
For single-atom logic to reach its full potential, each of the four classes of transformations detailed above must be developed to a level of maturity that enables context-independent deployment. That is, the toolbox will only be complete once chemists have control of editing at the single-atom level in both aromatic and aliphatic ring systems at any given position and with complete selectivity. Although carbonyl chemistry has served, and continues to serve, as a useful guide in the development of such reactions, non-traditional reactivity manifolds will clearly need to be embraced if this collective outcome is to be realized (Fig. 10).
Fig. 10 |. Dream reactions in single-atom skeletal editing and promising precedents.
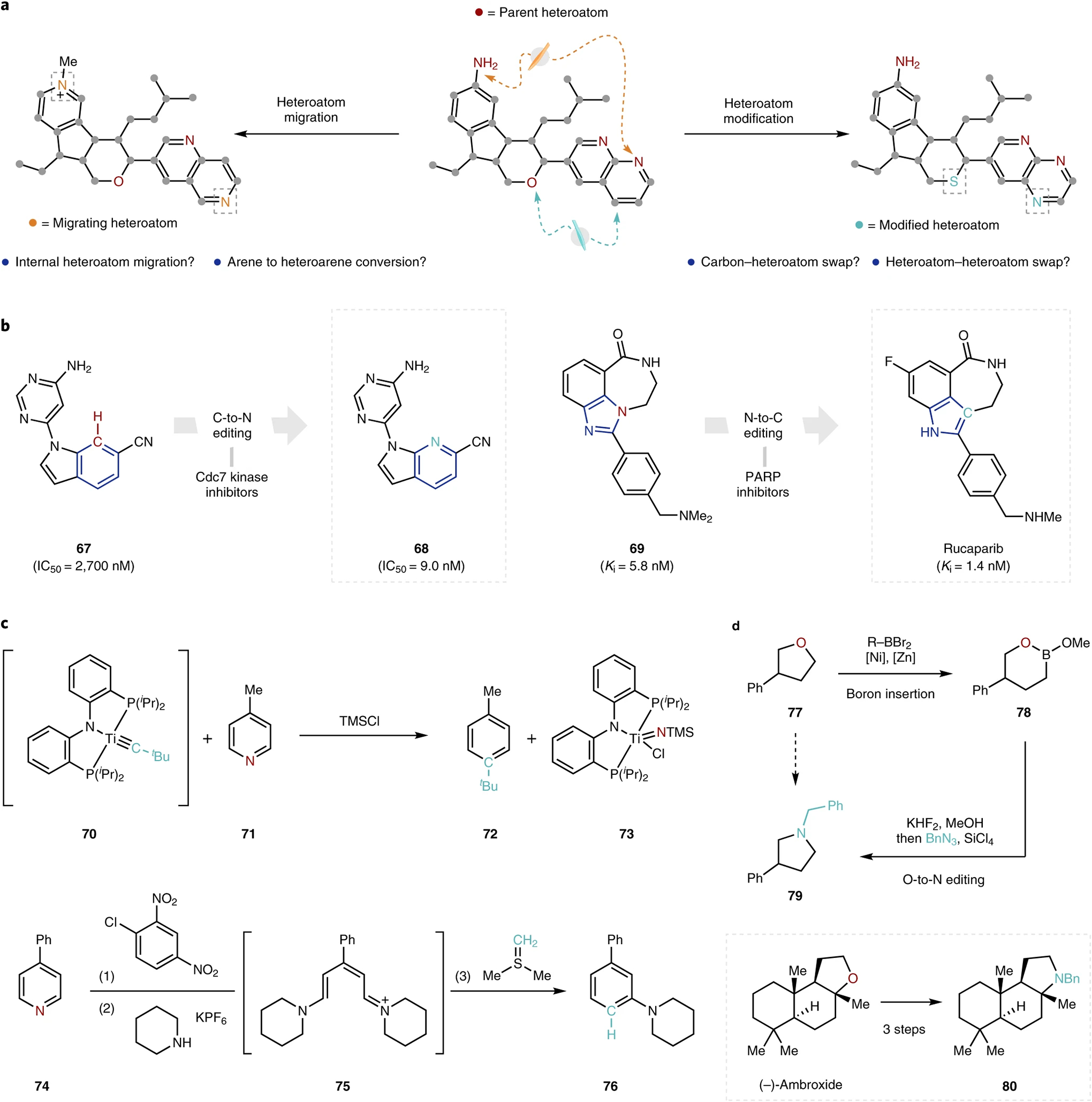
a, Wishlist of single-atom editing reactions applied to a complex molecule. b, Examples of the idealized single-atom editing logic in drug discovery shown through carbon-to-nitrogen (left) and nitrogen-to-carbon (right) atom swaps. c, Selected examples of carbon–nitrogen transmutations from Mindola and co-workers81 (top) and Kano and co-workers82 (bottom). d, Representative example of oxygen–nitrogen transmutation78. ‘Edited’ atoms are highlighted for clarity. Cdc7, Cell division cycle 7; PARP, polyadenosine diphosphate ribose polymerase; IC50, half maximal inhibitory concentration; Ki, inhibitory constant.
It is also evident in many of the above examples that the challenge of skeletal editing to date has often required resorting to multistep sequences. Although some of these pioneering efforts could not have been accomplished without such a concession, the development of single-step (or single-operation) transformations is imperative for the broad adoption of single-atom editing. The deletion of carbon atoms from N-benzoyl piperidines discussed above, for example, suffered substantial yield losses when telescoped to a single operation due to competitive reactivity with by-products of the first step45. There is no doubt that continued development will overcome this challenge.
Finally, in addition to n + 1 or n − 1 skeletal editing, the ability to maintain the ring size while replacing one atom in a ring system with another represents, in many ways, the ultimate goal of skeletal editing. Such transmutations, once more broadly developed, would enable the direct interrogation of shape-conserving structure–activity relationships of the kind central to modern medicinal chemistry (Fig. 10b). Indeed, the so-called ‘necessary nitrogen effect’ has prompted lead optimization campaigns to conduct ‘nitrogen scans’, wherein carbon atoms of a lead scaffold are replaced by nitrogen (for example, 67 to 68); this endeavour requires tedious resynthesis (often re-engineering the synthetic route altogether) to access the aza-analogues of the lead molecule79. The reverse nitrogen-to-carbon transmutation has also been employed successfully in drug discovery, as exemplified by the discovery of the indole-containing polyadenosine diphosphate ribose polymerase inhibitor rucaparib from benzimidazole precursor 6980.
Although such transmutations have yet to be achieved in a broadly applicable way, the literature holds promising examples of this chemistry using stoichiometric organometallic complexes (such as 70), as well as some multistep approaches (for example, 74 to 76) that achieve such transformations in a formal sense (Fig. 10c)81,82. In a similar vein, the boron insertion developed by the Dong group, discussed above, also enables a three-step net oxygen-to-nitrogen replacement (Fig. 10d, 77 to 79). Although these initial reports are extremely exciting and provide a foundation for future investigation, substantial work remains to be done: we highlight in Fig. 10 a small selection of ‘dream reactions’, which embody the spirit of our call-to-action here. These transformations (which include transmutations and heteroatom scans) can all, in principle, be accomplished through a combination of the transformation classes discussed above or, more tantalizingly, in a single transformation. At that point, single-atom logic will have truly reached maturity.
acknowledgements
R.S. is grateful to the National Institutes of Medical Science (R35 GM13045) and Merck Research Laboratories for funding portions of the work described herein. M.D.L. thanks the Packard Foundation and National Institutes of Health (R35 GM142768) for funding. S.F.K. is grateful for a National Science Foundation Fellowship.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Blakemore DC et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Campos KR et al. The importance of synthetic chemistry in the pharmaceutical industry. Science 363, eaat0805 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Hann MM & Keserü GM Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov 11, 355–365 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Vitaku E, Smith DT & Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals. J. Med. Chem 57, 10257–10274 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Taylor RD, MacCoss M & Lawson ADG Rings in Drugs. J. Med. Chem 57, 5845–5859 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Majumdar K & Chattopadhyay S Heterocycles in Natural Product Synthesis (John Wiley & Sons, Ltd, 2011). [Google Scholar]
- 7.Antermite D & Bull JA Transition metal-catalyzed directed C(sp3)–H functionalization of saturated heterocycles. Synthesis 51, 3171–3204 (2019). [Google Scholar]
- 8.Campos KR Direct sp3 C–H bond activation adjacent to nitrogen in heterocycles. Chem. Soc. Rev 36, 1069–1084 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Ogba OM, Warner NC, O’Leary DJ & Grubbs RH Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev 47, 4510–4544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albright H et al. Carbonyl–olefin metathesis. Chem. Rev 121, 9359–9406 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang B, Perea MA & Sarpong R Transition metal-mediated C−C single bond cleavage: making the cut in total synthesis. Angew. Chem. Int. Ed 59, 18898–18919 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engle KM et al. in Organic Chemistry – Breakthroughs and Perspectives (eds Ding K & Dai L-X) 279–333 (John Wiley & Sons, Ltd, 2012). [Google Scholar]
- 13.Bhawal BN & Morandi B Shuttle catalysis—new strategies in organic synthesis. Chem. Eur. J 23, 12004–12013 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Qiu X et al. Cleaving arene rings for acyclic alkenylnitrile synthesis. Nature 597, 64–69 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Aube J & Milligan GL Intramolecular Schmidt reaction of alkyl azides. J. Am. Chem. Soc 113, 8965–8966 (1991). [Google Scholar]
- 16.Silva LF Construction of cyclopentyl units by ring contraction reactions. Tetrahedron 58, 9137–9161 (2002). [Google Scholar]
- 17.Harmata M in Molecular Rearrangements in Organic Synthesis (ed. Rocas CM) 183–226 (John Wiley & Sons, Ltd, 2015). [Google Scholar]
- 18.Furniss BS, Hannaford AJ, Smith PWG & Tatchell AR Vogel’s Textbook of Practical Organic Chemistry 5th edn, 1111–1114 (Longman, 1989). [Google Scholar]
- 19.Jamison TF, Shambayati S, Crowe WE & Schreiber SL Tandem use of cobalt-mediated reactions to synthesize (+)-epoxydictymene, a diterpene containing a trans-fused 5−5 ring system. J. Am. Chem. Soc 119, 4353–4363 (1997). [Google Scholar]
- 20.Wolinsky J, Gibson T, Chan D & Wolf H Stereospecific synthesis of iridomyrmecin and related iridolactones. Tetrahedron 21, 1247–1261 (1965). [DOI] [PubMed] [Google Scholar]
- 21.Stoltz BM & Wood JL The stereoselective ring contraction of a pyranosylated indolocarbazole. A biosynthetic link between K252a and staurosporine? Tetrahedron Lett 37, 3929–3930 (1996). [Google Scholar]
- 22.Xiao M et al. Total synthesis of (−)-isatisine A via a biomimetic benzilic acid rearrangement. Tetrahedron 71, 3705–3714 (2015). [Google Scholar]
- 23.Tehrani KA et al. Boron(III) bromide-induced ring contraction of 3-oxygenated piperidines to 2-(bromomethyl)pyrrolidines. Tetrahedron Lett 41, 2507–2510 (2000). [Google Scholar]
- 24.Feraldi-Xypolia A, Gomez Pardo D & Cossy J Ring Contraction of 3-hydroxy-3-(trifluoromethyl)piperidines: synthesis of 2-substituted 2-(trifluoromethyl)pyrrolidines. Chem. Eur. J 21, 12876–12880 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Álvarez-Dorta D, León EI, Kennedy AR, Riesco-Fagundo C & Suárez E Sequential Norrish Type II photoelimination and intramolecular aldol cyclization of 1,2-diketones in carbohydrate systems: stereoselective synthesis of cyclopentitols. Angew. Chem. Int. Ed 47, 8917–8919 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Alvarez-Dorta D et al. Sequential Norrish Type II photoelimination and intramolecular aldol cyclization of α-diketones: synthesis of polyhydroxylated cyclopentitols by ring contraction of hexopyranose carbohydrate derivatives. Chem. Eur. J 19, 10312–10333 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Jurczyk J et al. Photomediated ring contraction of saturated heterocycles. Science 373, 1004–1012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling R & Mariano PS A demonstration of the synthetic potential of pyridinium salt photochemistry by its application to a stereocontrolled synthesis of (+)-mannostatin A1. J. Org. Chem 63, 6072–6076 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Wang S, Wang H, Tian N & Yan H Insights for diastereoselectivity in synthesis of 2,3-dihydropyrroles by photochemical ring contraction of 1,4-dihydropyridines. Tetrahedron Lett 65, 152797 (2021). [Google Scholar]
- 30.Bellamy F, Martz P & Streith L An intriguing copper salt effect upon the photochemistry of pyridine-N-oxides. Specific photoinduced syntheses of 3-substituted 2-formylpyrroles. Heterocycles 3, 395–400 (1975). [Google Scholar]
- 31.Weber H & Rohn T 2H-Azirine und 2-(ω-Cyanalkyl)furane als neuartige Photoprodukte aus [n](2,6)Pyridinophan-N-oxiden und ihre Bedeutung für den Reaktionsverlauf. Chem. Ber 122, 945–950 (1989). [Google Scholar]
- 32.Feng Z, Allred TK, Hurlow EE & Harran PG Anomalous chromophore disruption enables an eight-step synthesis and stereochemical reassignment of (+)-marineosin A. J. Am. Chem. Soc 141, 2274–2278 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Hurlow EE et al. Photorearrangement of [8]-2,6-pyridinophane N-oxide. J. Am. Chem. Soc 142, 20717–20724 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Padwa A, Smolanoff J & Tremper A Intramolecular photochemical and thermal cyclization reactions of 2-vinyl substituted 2H-azirines. Tetrahedron Lett 15, 29–32 (1974). [Google Scholar]
- 35.Padwa A, Smolanoff J & Tremper A Photochemical transformations of small ring heterocyclic systems. LXV. Intramolecular cycloaddition reactions of vinyl-substituted 2H-azirines. J. Am. Chem. Soc 97, 4682–4691 (1975). [Google Scholar]
- 36.Portillo M, Maxwell MA & Frederich JH Synthesis of nitrogen heterocycles via photochemical ring opening of pyridazine N-oxides. Org. Lett 18, 5142–5145 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Eaton PE & Cole TW The cubane system. J. Am. Chem. Soc 86, 962–964 (1964). [Google Scholar]
- 38.Eaton PE & Cole TW Cubane. J. Am. Chem. Soc 86, 3157–3158 (1964). [Google Scholar]
- 39.Yu X et al. Stereospecific construction of contiguous quaternary all-carbon centers by oxidative ring contraction. Angew. Chem. Int. Ed 156, 350–353 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Nicolaou KC, Gray DLF & Tae J Total synthesis of hamigerans and analogues thereof. Photochemical generation and Diels–Alder trapping of hydroxy-o-quinodimethanes. J. Am. Chem. Soc 126, 613–627 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Ng D, Yang Z & Garcia-Garibay MA Total synthesis of (±)-herbertenolide by stereospecific formation of vicinal quaternary centers in a crystalline ketone. Org. Lett 6, 645–647 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Natarajan A, Ng D, Yang Z & Garcia-Garibay MA Parallel syntheses of (+)- and (−)-α-cuparenone by radical combination in crystalline solids. Angew. Chem. Int. Ed 46, 6485–6487 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Dotson JJ, Bachman JL, Garcia-Garibay MA & Garg NK Discovery and total synthesis of a bis(cyclotryptamine) alkaloid bearing the elusive piperidinoindoline scaffold. J. Am. Chem. Soc 142, 11685–11690 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dotson JJ et al. Taming radical pairs in the crystalline solid state: discovery and total synthesis of psychotriadine. J. Am. Chem. Soc 143, 4043–4054 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roque JB, Kuroda Y, Göttemann LT & Sarpong R Deconstructive diversification of cyclic amines. Nature 564, 244–248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zippel C, Seibert J & Bräse S Skeletal editing—nitrogen deletion of secondary amines by anomeric amide reagents. Angew. Chem. Int. Ed 60, 19522–19524 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kennedy SH, Dherange BD, Berger KJ & Levin MD Skeletal editing through direct nitrogen deletion of secondary amines. Nature 593, 223–227 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Qin H et al. N-atom deletion in nitrogen heterocycles. Angew. Chem. Int. Ed 60, 20678–20683 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Hui C, Brieger L, Strohmann C & Antonchick AP Stereoselective synthesis of cyclobutanes by contraction of pyrrolidines. J. Am. Chem. Soc 143, 18864–18870 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao Z-C & Shi Z-J Deoxygenation of ethers to form carbon–carbon bonds via nickel catalysis. J. Am. Chem. Soc 139, 6546–6549 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Song Z-L, Fan C-A & Tu Y-Q Semipinacol rearrangement in natural product synthesis. Chem. Rev 111, 7523–7556 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Dowd P & Choi SC A new tributyltin hydride-based rearrangement of bromomethyl β-keto esters. A synthetically useful ring expansion to ɣ-keto esters. J. Am. Chem. Soc 109, 3493–3494 (1987). [Google Scholar]
- 53.Beckwith ALJ, O’Shea DM & Westwood SW Rearrangement of suitably constituted aryl, alkyl, or vinyl radicals by acyl or cyano group migration. J. Am. Chem. Soc 110, 2565–2575 (1988). [Google Scholar]
- 54.Fu C et al. Diastereoselective total synthesis of salvileucalin C. Org. Lett 16, 3376–3379 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Chen P, Sieber J, Senanayake CH & Dong G Rh-catalyzed reagent-free ring expansion of cyclobutenones and benzocyclobutenones. Chem. Sci 6, 5440–5445 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelley BT, Walters JC & Wengryniuk SE Access to diverse oxygen heterocycles via oxidative rearrangement of benzylic tertiary alcohols. Org. Lett 18, 1896–1899 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Murai K, Komatsu H, Nagao R & Fujioka H Oxidative rearrangement of spiro cyclobutane cyclic aminals: efficient construction of bicyclic amidines. Org. Lett 14, 772–775 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Mailloux MJ, Fleming GS, Kumta SS & Beeler AB Unified synthesis of azepines by visible-light-mediated dearomative ring expansion of aromatic N-ylides. Org. Lett 23, 525–529 (2021). [DOI] [PubMed] [Google Scholar]
- 59.Odum RA & Brenner M Rearrangement on deoxygenation of nitrosobenzene. J. Am. Chem. Soc 88, 2074–2075 (1966). [Google Scholar]
- 60.Wentrup C Flash vacuum pyrolysis of azides, triazoles, and tetrazoles. Chem. Rev 117, 4562–4623 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Gritsan NP et al. Ring-expansion reaction of cyano-substituted singlet phenyl nitrenes: theoretical predictions and kinetic results from laser flash photolysis and chemical trapping experiments. J. Am. Chem. Soc 123, 1425–1433 (2001). [Google Scholar]
- 62.Inui H, Sawada K, Oishi S, Ushida K & McMahon RJ Aryl nitrene rearrangements: spectroscopic observation of a benzazirine and its ring expansion to a ketenimine by heavy-atom tunneling. J. Am. Chem. Soc 135, 10246–10249 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Li G et al. An improved PIII/PV=O-catalyzed reductive C–N coupling of nitroaromatics and boronic acids by mechanistic differentiation of rate- and product-determining steps. J. Am. Chem. Soc 142, 6786–6799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beckmann E Zur Kenntniss der Isonitrosoverbindungen. Ber. Dtsch. Chem. Ges 19, 988–993 (1886). [Google Scholar]
- 65.Tinge J et al. in Ullmann’s Encyclopedia of Industrial Chemistry (American Cancer Society, 2018); 10.1002/14356007.a05_031.pub3 [DOI] [Google Scholar]
- 66.Banić Tomišić Z The story of azithromycin. Kem. Ind 60, 603–617 (2011). [Google Scholar]
- 67.Amice P, Blanco L & Conia JM Enol silyl ethers and their use for the synthesis of α-halo-α,β-unsaturated carbonyl compounds. Synthesis 1976, 196–197 (1976). [Google Scholar]
- 68.Parham WE, Soeder RW, Throckmorton JR, Kuncl K & Dodson RM Reactions of enol ethers with carbenes. V. Rearrangements of dihalocyclopropanes derived from six-, seven-, and eight-membered cyclic enol ethers. J. Am. Chem. Soc 87, 321–328 (1965). [Google Scholar]
- 69.Skattebøl L Chemistry of gem-dihalocyclopropanes. IV. Ring opening of gem-dichlorocyclopropyl ethers. J. Org. Chem 31, 1554–1559 (1966). [Google Scholar]
- 70.Ohno M A new ring-expansion reaction from enamine and dichlorocarbene. Tetrahedron Lett 4, 1753–1755 (1963). [Google Scholar]
- 71.De Selms RC A new method of ring expansion. Tetrahedron Lett 7, 1965–1968 (1966). [Google Scholar]
- 72.Yuan P, Gerlinger CKG, Herberger J & Gaich T Ten-step asymmetric total synthesis of (+)-pepluanol A. J. Am. Chem. Soc 143, 11934–11938 (2021). [DOI] [PubMed] [Google Scholar]
- 73.Ciamician GL & Dennstedt M Ueber die Einwirkung des Chloroforms auf die Kaliumverbindung Pyrrols. Ber. Dtsch. Chem. Ges 14, 1153–1163 (1881). [Google Scholar]
- 74.Wynberg H The Reimer–Tiemann reaction. Chem. Rev 60, 169–184 (1960). [Google Scholar]
- 75.Dherange BD, Kelly PQ, Liles JP, Sigman MS & Levin MD Carbon atom insertion into pyrroles and indoles promoted by chlorodiazirines. J. Am. Chem. Soc 143, 11337–11344 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Graham WH The halogenation of amidines. I. Synthesis of 3-halo- and other negatively substituted diazirines. J. Am. Chem. Soc 87, 4396–4397 (1965). [Google Scholar]
- 77.Ma D, Martin BS, Gallagher KS, Saito T & Dai M One-carbon insertion and polarity inversion enabled a pyrrole strategy to the total syntheses of pyridine-containing lycopodium alkaloids: complanadine A and lycodine. J. Am. Chem. Soc 143, 16383–16387 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lyu H, Kevlishvili I, Yu X, Liu P & Dong G Boron insertion into alkyl ether bonds via zinc/nickel tandem catalysis. Science 372, 175–182 (2021). [DOI] [PubMed] [Google Scholar]
- 79.Pennington LD & Moustakas DT The necessary nitrogen atom: a versatile high-impact design element for multiparameter optimization. J. Med. Chem 60, 3552–3579 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Golding BT in Successful Drug Discovery (eds Fischer J, Klein C & Childers WE) 201–223 (John Wiley & Sons, Ltd, 2019). [Google Scholar]
- 81.Fout AR, Bailey BC, Tomaszewski J & Mindiola DJ Cyclic denitrogenation of N-heterocycles applying a homogeneous titanium reagent. J. Am. Chem. Soc 129, 12640–12641 (2007). [DOI] [PubMed] [Google Scholar]
- 82.Morofuji T, Inagawa K & Kano N Sequential ring-opening and ring-closing reactions for converting para-substituted pyridines into meta-substituted anilines. Org. Lett 23, 6126–6130 (2021). [DOI] [PubMed] [Google Scholar]