

Abstract
Enzymes are renowned for their catalytic efficiency and selectivity. Despite the wealth of carbon-carbon bond forming transformations in traditional organic chemistry and nature, relatively few C-C bond forming enzymes have found their way into the biocatalysis toolbox. Here we show that the enzyme UstD performs a highly selective decarboxylative aldol addition with diverse aldehyde substrates to make non-standard, γ-hydroxy amino acids. We increased the activity of UstD through three rounds of classic directed evolution and an additional round of computationally-guided engineering. The enzyme that emerged, UstDv2.0, is efficient in a whole-cell biocatalysis format. The products are highly desirable, functionally rich bioactive γ-hydroxy amino acids that we demonstrate can be prepared stereoselectively on gram-scale. The X-ray crystal structure of UstDv2.0 at 2.25 Å reveals the active site and provides a foundation for probing the mechanism of UstD.
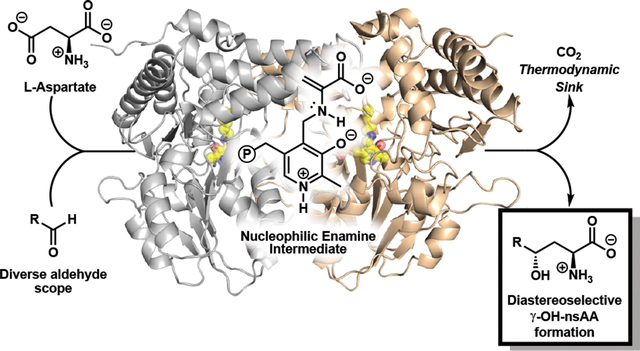
Graphical Abstract
Introduction
Major advances have been made in the practical use of enzymes for enantioselective functional group manipulations1. For example, asymmetric reduction of ketones and enantiospecific hydrolysis of racemic esters are now routine in process chemistry. There have also been impressive strides made in enzymatic C-H activation2. However, the development of enzymes to form C-C bonds on preparative scale lags far behind traditional synthetic organic methodology3. While nature is rife with C-C bond forming enzymes,4,5 these catalysts often have significant limitations, such as limited substrate scope or poor heterologous expression.6 While engineering can overcome these challenges, a more severe limitation is thermodynamic in nature: reactions that form carbon nucleophiles via C-H deprotonation, such as classic aldol transformations, are typically reversible.7 In nature, metabolic flux drives reactions and preserves the stereochemical purity of products. Laboratory approaches mimic nature by coupling reversible biocatalytic C-C bond forming reactions to a thermodynamic sink, such as a subsequent transformation or selective crystallization.8–11 While these advances are substantial, the potential of biocatalytic enzymes in assembling carbon chains is still hindered by the simple lack of high-quality, exergonic transformations.12 Hence, development of scalable and thermodynamically favorable C-C bond forming reactions may open diverse avenues of biocatalytic synthesis.
To fill this gap, we were drawn to a recently described pyridoxal phosphate (PLP) dependent enzyme involved in the biosynthesis of Ustiloxin B, an inhibitor of microtubilin polymerization (Fig. 1a)13. This enzyme, UstD, decarboxylates the side chain of l-aspartate (1), forming a putative nucleophilic enamine intermediate (Fig. 1b). This enamine then attacks an aliphatic aldehyde appended to a cyclic tetrapeptide, resulting in the formation of a γ-hydroxy amino acid side chain. The loss of CO2 renders this enantioselective C-C bond forming reaction effectively irreversible. This decarboxylative aldol addition mechanism is distinct from the classic aldolases, transketolases, and PLP-dependent Thr aldolases, which catalyse tautomerization of an imine to form an enamine nucleophile.14,15 It has been shown that the transketolase catalytic cycle can be non-natively entered through decarboxylation, and that reactions initially proceed to high conversion. However, the native proton transfer machinery eventually breaks down the product into an equilibrium mixture with starting materials.16 Although the detailed mechanism of UstD has not yet been explored, Ye et al. reported that the UstD reaction cannot be initiated from l-Ala, indicating enamine formation through tautomerization is not viable. Therefore, UstD is mechanistically distinct from classic aldolases and may have unique properties as a biocatalyst.
Figure 1.

Relevance and mechanism of enzymatic C-C bond formation. a) Bioactive molecules with a γ-hydroxy amino acid motif shown in purple. The native product of UstD is Ustiloxin B. b) The generalized decarboxylative aldol reaction of UstD showing the putative enamine nucleophilic intermediate.
The native substrate for UstD is a complex, cyclic peptide, and it was unknown if this enzyme would react promiscuously with alternative substrates. If so, the enzyme would directly produce γ-hydroxy amino acids (Fig. 1b). Such non-standard amino acids (nsAAs) are found in bioactive natural products, such as caspofungin and clavalanine (Fig. 1a)17. While nature employs side chain hydroxylation to tune bioactivity, these nsAAs are virtually absent from medicinal chemistry18 because they require multistep synthesis17. The need for multistep synthesis to prepare these nsAAs has begun to be addressed by biocatalysis, where an elegant multi-enzyme cascade was recently developed by Clapés et al. to access gamma-hydroxy nsAAs19,20. However, the ability to use a single enzyme to produce the same motif would offer greater practical utility and versatility. Beyond their use in pharmaceuticals, nsAAs can be enabling for a host of synthetic and chemical biology applications21,22. Therefore, the development of UstD for organic synthesis would introduce a valuable and much-needed enantioselective C-C bond-forming enzyme into the biocatalytic toolbox and provide direct access to a structurally complex synthon.
Here we show that the enzyme UstD performs a highly selective decarboxylative aldol addition with diverse aldehyde substrates to make non-standard, γ-hydroxy amino acids. We increased the activity of UstD through three rounds of classic directed evolution and an additional round of computationally-guided engineering. The enzyme that emerged, UstDv2.0, is efficient in a whole-cell biocatalysis format, which circumvents the need for enzyme purification, thereby facilitating its use in traditional organic settings on gram-scale. The X-ray crystal structure of UstDv2.0 at 2.25 Å reveals the active site and the molecular basis for the promiscuity of this catalyst.
Results
Initial characterization of UstD
We expressed C-His-UstD (wt-UstD) in Escherichia coli (Supplementary Figure 1), but were uncertain whether molecular recognition for the structurally complex native substrate would be required for catalytic activity. We therefore assessed the reactivity of wt-UstD with benzaldehyde (2a) and were pleased to observe a successful decarboxylative aldol addition to afford the γ-hydroxy nsAA 3a by UPLC-MS (Supplementary Figure 2). A preparative scale reaction with 0.125 mol % catalyst gave the product in 43% yield, and analysis by nuclear magnetic resonance (NMR) spectroscopy indicated a single diastereomer predominated (dr >98:2). To determine the absolute stereochemical preference for the enzyme, we analysed the product from a reaction with 4-bromobenzaldehyde (2b). The crystal structure of the product (3b) revealed the aldol addition occurred with the same stereochemical outcome as the native reaction (Supplementary Figure 2). These transformations indicated wt-UstD has potential for organic synthesis, but the comparatively modest activity (<1000 turnovers with initial reaction conditions) and low catalyst expression would hinder routine use of the natural enzyme. Given the inherent structural differences between the native tetrapeptide substrate and simpler commercially-available aldehydes (such as 2a), we hypothesized that directed evolution and reaction condition optimization could be used to increase the catalytic efficiency of UstD toward non-native substrates.
Directed evolution of UstD for improved catalytic activity
To inform our engineering process, we used a homology model of wt-UstD derived from a distantly related cysteine desulfurase (27% identity)23,24. Six residues in the predicted active site were chosen for saturation mutagenesis, and we used benzaldehyde (2a) as a model substrate for directed evolution (Fig. 2a). Mutation at positions predicted to form direct contacts with the cofactor resulted in inactivation of the catalyst, a common trend amongst PLP-dependent enzymes25. Nevertheless, these libraries yielded a single variant in a putative loop region flanking the substrate binding site, C392L, with a 2.3-fold boost in activity (Fig. 2b). Concurrently, we employed global random mutagenesis on wt-UstD to search throughout the protein sequence for activating mutations. A second activating mutation was discovered, L393M, immediately adjacent to Cys392. We combined these mutations to yield the double variant UstDC392L, L393M, which had a further increase in activity to 4.9-fold above wild-type (Supplementary Figure 3). It is common for mutation of neighboring residues to display cooperativity26,27, and we chose to test additional mutations in this region of the sequence (Fig. 2b). We used a degenerate codon mutagenesis strategy on four contiguous residues from Ile391-Ala394. We restricted the sequence space to residues commonly found among UstD homologs, which provided good structural diversity in a focused set of mutations (see Supplementary Methods for details). Screening this library revealed that mutation of Ala394 was generally deleterious. However, multiple highly active variants retained Ala394 and contained mutations at Ile391, Cys392, and Leu393. To best capture relative rate effects of mutations, catalysts were compared under dilute conditions. Variants UstDTLM and UstDFVF (the superscript refers to the identity of the residues at positions 391–393) had a 5.1-fold and 4.1-fold increase in activity relative to wt-UstD, respectively.
Figure 2.

Directed evolution of UstD and evaluation of variants. a) Lineage of activated UstD variants. Standard screening conditions: 25 mM 2a, 50 mM 1, buffer (100 mM KPi, pH 7.0, 100 mM NaCl), 5% DMSO, 37 °C, 16 h. Catalyst activity measured by total turnover number (TTN). b) Computational model of UstD bound to 3a, derived through homology modeling. Active site residues are shown as sticks and loop residues targeted for mutagenesis are coloured in orange. Potential hydrogen bonds are shown as black dashes. c) Performance evaluation of UstD and activated variants measured by Marfey’s analysis of the enzymatic products. Exact values and standard deviations are available in Supplementary Table 1 (n=3 individual experiments per substrate and variant), and error was generally below 10%. Lighter coloured bar sections represent the amount of the other Cγ epimer from which diastereomeric ratios are calculated. Absolute configuration is assigned by analogy to the product 3b and the native Ustiloxin D stereochemistry13. See Supplementary Methods for details. d) Synthesis of select products at 0.2 mmol scale with isolated yields. The different purification strategies are denoted by the different colours, free amino acid (purple), Fmoc protected amino acid (blue), lactonization with Fmoc protection (grey). Note, reactions from which 3b was purified used wt-UstD.
We next optimized reaction conditions for the most active variant, UstDTLM. Reaction mixtures were initially coloured yellow (Supplementary Figure 1) by the presence of PLP that co-purified with the enzyme but became colourless over time, suggesting the cofactor is degraded during the reaction. Gratifyingly, supplementation of PLP led to a large increase in product formation (Supplementary Figure 4). We did not observe a significant change when the concentration of 1 was increased (Supplementary Figure 4). However, we observed formation of l-alanine in reactions, indicating some 1 is lost to a non-productive protonation of the nucleophilic enamine intermediate13. We therefore used aldehyde as the limiting reagent and two equivalents of 1 for subsequent experiments, which identified an optimal initial pH of 7.0 (Supplementary Figure 4). Lastly, we varied the catalyst loading and found that UstDTLM was capable of high conversion (~70%) with just 0.01 mol % catalyst loading (Supplementary Figure 4). With these optimized conditions, we evaluated the performance of wt-UstD and both activated variants, UstDTLM and UstDFVF, with a more diverse set of aldehyde substrates. We anticipated that the striking sequence divergence in the putative loop would lead to distinct trends in substrate selectivity.
Performance analysis of UstD and its variants
Engineering enzymes for activity on a model substrate often leads to specialist catalysts with diminished activity on substrate analogs28,29. Initial comparisons among wt-UstD, UstDFVF, and UstDTLM with a small panel of aldehydes suggested that both variants had evolved towards improved overall activity (Supplementary Figure 5). We therefore expanded the substrate scope. Marfey’s reagent cleanly derivatized the diverse products, providing a uniform chromophore for quantitative measurement of turnover and selectivity via UPLC-MS30. Product formation was observed with virtually every substrate tested, from the large and hydrophobic biphenyl aldehyde (2g) to the small and hydrophilic glycolaldehyde (2p) (Fig. 2c). Generally, the variant UstDTLM performed the most turnovers and displayed excellent diastereoselectivity, typically forming a 95:5 ratio of diastereomers (dr). While UstDFVF typically performed fewer turnovers than UstDTLM with most substrates, UstDFVF generally had higher selectivity than wt-UstD or UstDTLM (Supplementary Table 1). Reactions with p-substituted aromatic aldehydes exhibited a Hammett-like reactivity trend: more product was formed as aldehyde electrophilicity increased. Activity was lowest with the electron rich p-anisaldehyde (2c), but high activity was observed for the electron deficient p-NO2-benzaldehyde (2d) with both engineered enzymes. To better capture the maximum turnover number with 2d, we repeated the reactions at lower catalyst loadings, which revealed that the engineered variants can perform ~34,000 turnovers (Supplementary Figure 6). Active site mutagenesis had little apparent impact on reactions with some highly hydrophobic substrates, such as the methoxynaphthyl (2e), 3,4-dichlorobenzyl (2f), and biphenyl (2g) aldehydes; reactivity in these cases may be limited by poor aqueous solubility (Fig. 2c). In contrast, reactivity on o-tolualdehyde (2h) and thiophene-3-carboxaldehyde (2i) increased dramatically during evolution. UstDTLM displayed a nine-fold increase in activity on 2i and a remarkable 23-fold increase in turnovers with 2h compared to wt-UstD. Activity with the imidazole substrate 2j was demonstrated and was one of the few substrates for which wt-UstD had the highest activity. To the best of our knowledge, the product is a previously unreported analog of histidine. Reactivity with the cinnamaldehyde (2k) improved with both variants relative to wt-UstD. Reactions proceeded smoothly with several aliphatic substrates, including isobutyraldehyde (2l), cyclopentylaldehyde (2m), and even 10-undecenal (2n); in this last case reactivity appeared to be limited by solubility. Pivaldehyde, however, was unreactive with all three enzymes, an observation we attribute to steric bulk near the carbonyl. The engineered UstD enzymes were active with glyoxylic acid (2o), which resulted in formation of γ-hydroxy-glutamate, an intermediate in hydroxyproline metabolism31. Lastly, we observed good reactivity with glycolaldehyde to yield the di-hydroxylated amino acid 3p. Previously, a protected form of 3p was identified as a key intermediate in the synthesis of clavalanine (Fig. 1B)17, an antibiotic that inhibits the biosynthesis of methionine32. Activity on 2p increased two-fold, with improved diastereoselectivity and pristine enantioselectivity, for UstDTLM relative to the wild-type enzyme. These substrates collectively demonstrate that the active site of UstD is remarkably permissive of diverse functional groups and that catalytic activity and selectivity can be rapidly optimized by mutation at residues 391–393.
These engineered enzymes enable a stereoselective synthesis of γ-hydroxy nsAAs in a single step from cheap, commercially available starting materials. The production of unprotected amino acids affords complete flexibility with regards to subsequent manipulation, but isolation of free amino acids themselves is challenging due to their hydrophilic, zwitterionic nature. Therefore, we selected a representative set of products to demonstrate isolation strategies (Fig. 2d). Sufficiently hydrophobic products were isolated as the free amino acid, while others utilized protection with fluorenylmethoxycarbonyl (Fmoc) to increase hydrophobicity, simultaneously adding a handle commonly used in solid phase peptide synthesis. Diverse manipulations, such as lactonization with the γ-hydroxy group, can also be employed to facilitate isolation and downstream manipulation19. Throughout these reactions a second, minor diastereomer was observed. The mixture of configurations at Cγ arises through imperfect selectivity with the aldol addition and could be aggravated by reversible retro-aldol cleavage of the major diastereomer. We tested the latter possibility by re-subjecting products 3a and 3d to reaction conditions and observed no change in the diastereomeric ratio by Marfey’s analysis (See Supplementary Figure 7). However, in the case of 3a, formation of alanine (Ala) was observed concomitant with a decrease in product peak area. This observation is consistent with slow product re-entry into the catalytic cycle via retro-aldol cleavage of 3a to reform 2a and Ala.
Linear regression guided protein engineering
The above studies relied on purified protein for preparative scale reactions. However, access to enzymes in sufficient quantity is a common and often under-appreciated limitation of biocatalysis. As is observed for many proteins, UstD had relatively low expression titers in E. coli (8 mg L−1 culture) due to poor solubility (Supplementary Figure 1). While enzyme immobilization can be used to increase the utility of purified protein catalysts33, a complementary synthetic methodology would use whole-cell preparations of UstD; this latter approach is attractive to process chemists34. Whole-cell catalysts are operationally simple to generate, stable over long periods, and obviate the need for expensive protein purification.
We sought to further engineer UstDTLM to increase soluble heterologous expression in E. coli for whole-cell biocatalysis. This enzyme contains nine Cys residues, and our homology model suggested five are surface exposed (Supplementary Figure 8). It is well known among protein crystallographers that removing surface Cys residues can increase soluble expression and increase the probability of crystallization35. However, we found that mutation of all five putative surface Cys residues to Ala eliminated catalytic activity. To identify mutations that would retain activity while increasing soluble expression, we performed sequence-similarity network analysis to identify non-Cys residues at these positions common among UstD homologs. Based on this analysis, we constructed a five-site degenerate codon library (Fig. 3a, Supplementary Figure 8).
Figure 3.

Engineering UstD for increased crystalizability and activity in whole-cell catalysis. a) Experimental process for bioinformatic and regression-guided mutagenesis of UstD. In the first stage, a small mutagenesis library is sampled to collect sequence/activity data. The second stage builds a linear regression model to correlate sequences to activity. This regression model is then used to predict activated sequences which are validated in the last stage using whole cell catalyst. The dots represent the individual measurements of triplicate technical replicates. b) Cartoon representation of the overall structure of UstD2.0. Individual monomers are coloured grey (chain A) and brown (chain B). PLP-K258 complex is shown as semitransparent yellow spheres and sticks. Inset: Active site residues superimposed on the 2mFo–DFc electron density map (blue mesh, σ = 1.2) are shown as sticks. TLMA loop residues are coloured in salmon. Hydrogen bonds are shown as black dashes.
To efficiently navigate this sequence space, we employed linear regression modelling to predict sequence-activity relationships36. We hypothesized this simple computational approach would be effective because the target residues are dispersed throughout the protein, which should make non-linear, pair-wise mutational effects unlikely. We screened and sequenced 176 random clones from this library for increased activity in lysate, which is sensitive to changes in both soluble enzyme expression and enzymatic efficiency. Although most variants in this library were inactive, we were heartened to observe several apparently improved variants (Fig. 3a). Linear regression model testing using leave-one-out cross-validation (LOOCV) of the full dataset indicated poor predictive behavior of the model for high-activity variants (Supplementary Figure 9). We suspected that the model quality was diminished by the abundance of inactive variants, for which activity measurements are indistinguishable from experimental noise. We therefore restricted our analysis to variants for which bonafide activity could be measured, leaving just 26 sequence-activity relationships. Despite the sparsity of these data (~5% of the sequence space), LOOCV showed the model was dramatically improved (See Supplementary Methods for details).
We evaluated the three most active variants predicted by the model, UstDTLM-ACASC, UstDTLM-ASCSC, and UstDTLM-ASASC. Comparisons of expression and whole cell activity were made between these variants, the parent enzyme, and most active variant identified from screening, UstDTLM-SCASC. We were delighted to find the expression titer was increased relative to UstDTLM for all variants, up to 48 mg protein L−1 culture (Supplementary Figure 10). While purified enzyme activity is slightly decreased for the new variants, their overall activity in whole cells is significantly improved (Fig. 3a, Supplementary Figure 10). Tests at analytical scale showed, at 0.25% w/v cell loading, that UstDTLM formed 3a in just 13% yield, highlighting the challenges associated with translating in vitro activity to large-scale reaction formats. In contrast, the variant with the highest whole-cell activity, the computationally-predicted UstDTLM-ACASC (designated UstDv2.0), produced 3a in 31% yield, a 2.4-fold boost over UstDTLM and a cumulative 15-fold boost over wild type. Higher conversions were achieved by increasing the cell loading of UstDv2.0to 1% w/v, which afforded 3a in 78% yield on analytical scale (Fig. 3a). To demonstrate the utility of UstDv2.0, large-scale reactions were carried out with 2a and 2d. Reaction with 2a at 0.5% w/v catalyst loading afforded 0.80 g 3a in 77% isolated yield with pristine stereoselectivity following purification by reverse-phase chromatography. Reaction with 2d at just 0.1% w/v catalyst loading provided 1.4 g 3d in 98% isolated yield with high stereoselectivity (see Supplementary Methods for details). Notably, these cell loadings are sufficient for process-scale biocatalytic reactions37, illustrating that UstDv2.0 can operate on the scale needed to meet the demands of practical organic synthesis.
Crystallography of UstDv2.0
While the engineering we report here produced a generalist variant of UstD, structural information could guide more targeted engineering for the production of specific γ-hydroxy nsAAs. Despite extensive efforts, we were unable to produce crystals of wt-UstD. In contrast, UstDv2.0 readily crystallized, which we attribute to the decrease in surface Cys residues. The 2.25-Å crystal structure of UstDv2.0 was determined using experimental phases from a Au(III) derivative (Fig. 3b, PDB ID: 7MKV). This structure revealed an active site at the dimer interface, which is common among fold-type I PLP-dependent enzymes 38. The internal aldimine involving a Schiff base linkage to Lys258 and a salt bridge between the pyridinium N1 and Asp232 were clearly resolved in the active site. The 391–393 loop harboring the activating TLM mutations projects over the top of the active site forming part of the substrate binding pocket. The remainder of the pocket appears to be solvent exposed, explaining the tolerance of UstD for diverse aldehyde substrates (Supplementary Figure 11).
In the future, we envision engineering UstD for increased activity with non-aldehyde substrates. As an initial demonstration, we showed that purified UstDv2.0 performs ~50 turnovers with the ketone substrate trifluoroacetone to produce a nsAA bearing a tertiary alcohol side chain (Supplementary Figure 12). The comparatively low turnover highlights the challenges associated with aldol addition into ketones. When nucleophilic attack is sufficiently slow, irreversible protonation of the enamine can quench the reactive intermediate and, indeed, we observed significant accumulation of l-alanine in this reaction. A similar scenario was observed with hydrolysis of an electrophilic PLP intermediate formed by TrpB and reactions with attenuated substrates were enabled by directed evolution that increased the lifetime of the reactive intermediate.39,40 Hence, future engineering to decrease the rate of enamine protonation in UstDv2.0 may further expand the substrate scope.
Discussion
Here, we improved a C-C bond forming enzyme, UstD, that catalyses a decarboxylative aldol addition using the loss of CO2 from l-aspartate as a thermodynamic driving force to produce γ-hydroxy amino acids. This mechanism of action and innate tolerance of diverse aldehydes marked UstD as a candidate for directed evolution into a versatile catalyst for organic synthesis. To screen for improved catalysts, we used a combination of globally random, site-saturation, and degenerate codon mutagenesis libraries. We illustrate the engineering potential of the active site with two variants, UstDFVF and UstDTLM, that share no mutations in common and display commensurate or superior activity to wt-UstD with the vast majority of aldehydes tested. We demonstrated how a simple regression-modeling approach to protein engineering can increase protein soluble expression and crystallizability. The evolved variant, UstDv2.0, is poised to deliver a desirable nsAA precursors for medicinal chemistry, and the crystal structure will facilitate future work to explore the mechanism and reactivity of this intriguing enzyme.
Methods
All chemicals and reagents were purchased from commercial suppliers (Sigma-Aldrich, VWR, Chem-Impex International, Alfa Aesar, Combi-blocks, Oakwood Products) at the highest quality available and used without further purification unless stated otherwise. Genes were purchased as gBlocks from Integrated DNA Technologies (IDT). E. coli cells were electroporated with an Eppendorf E-porator at 2500 V. New Brunswick I26R shaker incubators (Eppendorf) were used for cell growth. Cell disruption via sonication was performed with a Sonic Dismembrator 550 (Fisher Scientific) sonicator. UV-vis spectroscopic measurements were collected on a UV-2600 Shimadzu spectrophotometer. Optical density measurements were collected using an optical density reader (Amersham Biosciences). Ultra-high pressure liquid chromatography-mass spectrometry (UPLC-MS) data were collected on an Acquity UPLC (Waters) equipped with an Acquity PDA and QDA MS detector using either a BEH C18 column (Waters) for substituted benzaldehyde reactions, or an Intrada Amino Acid column (Imtakt) for aliphatic aldehyde reactions. All UPLC-MS data were processed using Empower 3 (Waters). Preparative column separations were performed on an Isolera One Flash Purification system (Biotage). NMR data were collected on Bruker 400 or 500 MHz spectrometers equipped with BBFO and DCH cryoprobes, respectively. All NMR chemical shifts were referenced either to a residual solvent peak or TMS internal standard. Spectra recorded using DMSO-d6 were referenced to the residual DMSO signal at 2.5 ppm for 1H and 39.52 ppm for 13C NMR. Spectra recorded using CDCl3 were referenced to the residual CHCl3 peak at 7.26 ppm for 1H NMR and 77.16 for 13C NMR. Spectra recorded using CD3OD were referenced to the CH3OD residual solvent peak at 3.31 ppm for 1H and 49.00 ppm for 13C NMR Spectra recorded using D2O:ACN-d3 as the solvent were referenced to the residual H2O signal at 4.79 ppm for 1H and absolute referenced to the 1H spectrum for 13C NMR. Signal positions were recorded in ppm with the abbreviations s, d, t, q, dd, and m, denoting singlet, doublet, triplet, quartet, doublet of doublets, and multiplet respectively. All coupling constants J are measured in Hz. High resolution mass spectrometry data were collected with a Q Extractive Plus Orbitrap (NIH 1S10OD020022–1) instrument with samples ionized by ESI.
Cloning of wild-type UstD
A codon-optimized copy of the Aspergillus flavus UstD gene was purchased as a gBlock from Integrated DNA Technologies. This DNA fragment was inserted into a pET-22b(+) vector by the Gibson Assembly method41 and transformed into electrocompetent BL21(DE3) E. coli cells via electroporation. After a 30-minute recovery period in Luria-Burtani (LB) media, cells were plated onto LB plates containing 100 μg/mL ampicillin (LBamp) and incubated overnight. A single colony was then used to inoculate 50 mL of Terrific Broth II media containing 100 μg/mL ampicillin (TBamp), which was then incubated overnight at 37 °C with shaking at 200 rpm. 500 μL of the saturated cell culture was then mixed with 500 μL of sterile 80% glycerol and snap-frozen in liquid nitrogen to generate a glycerol stock.
Plasmid Preparations
A 5-mL overnight culture of E. coli harboring the plasmid of interest was grown overnight at 37 °C with shaking at 200 rpm. The plasmid was isolated and purified using Zymo Plasmid Miniprep kits and sequenced through Functional Biosciences.
Protein Expression
An overnight culture of E. coli BL21(DE3) harboring a pET-22b(+) plasmid encoding a given UstD variant was created by inoculating 50 mL of TBamp media with a single colony. This culture was shaken at 37 °C and 200 rpm for ~16 h. 10 mL of overnight culture was then used to inoculate 1 L of TBamp, which was shaken at 37 °C and 200 rpm for approximately 1.5 h or until an optical density (OD) of 0.4–0.6 was reached. Cultures were removed from the incubator and cooled on ice for 30 min, followed by induction with 100 μM IPTG. The cultures were allowed to continue to grow for an additional ~16 h at 20 °C and shaking at 200 rpm. Cells were then harvested by centrifugation (4 °C, 30 min, 4,000 xg), and the cell pellets were stored at −20 °C overnight.
Whole Cell Preparation of E. coli harboring UstD and variants
Following protein expression, cells were harvested by centrifugation (4 °C, 30 min, 4,000 xg). The cell pellets were then resuspended in water and centrifuged two times to remove all media. The cell pellets were transferred to 50 mL conical tubes and freeze dried by lyophilization. The dried cells were stored at −80 °C until further use.
Protein purification of UstD and variants
To purify UstD, cell pellets were thawed on ice and then resuspended in lysis buffer, comprised of enzyme storage buffer (100 mM potassium phosphate buffer, pH 7.0, 100 mM sodium chloride) containing 20 mM imidazole, 1 mg/mL Hen Egg White Lysozyme (GoldBio), 0.2 mg/mL DNase (GoldBio), 1 mM MgCl2, and 150 μM pyridoxal 5’-phosphate (PLP). A ratio of 4 mL lysis buffer per gram of wet cell pellet was used. Cells lysis began by shaking for 1 h at 37 °C. The resuspended cells were subsequently sonicated (20 min, 0.8 s on, 0.2 s off, power setting 5). The resulting lysate was then spun down at 75,600 xg to pellet cellular debris. Ni/NTA beads were pre-equilibrated in storage buffer containing 20 mM imidazole. 1 mL of resin for 25 g of cells was added to the cleared lysis supernatant and incubated with nutation on ice for 1 h. The beads were then collected in a gravity column with plastic frit, and the flow-through was re-passed once to collect any remaining beads from the original vessel. The collected beads were washed with 10–20 column volumes of storage buffer containing 60 mM imidazole. Protein was eluted with 5 mL of storage buffer containing 250 mM imidazole and collecting the flow-through until the eluent was no longer yellow (color due to the enzymatically bound PLP cofactor). The eluent was then transferred to a centrifugal filter tube (Amicon® Ultra-15, 30k MWCO) and concentrated by centrifugation (4,000 xg, 15 min). Imidazole was then removed either through dialysis or through repeated dilution (with enzyme storage buffer) and concentration steps until < 1 μM imidazole.
Generation of random mutagenesis libraries
Random mutagenesis was carried out via error-prone PCR. Reaction conditions were optimized to generate 1–2 codon mutations per plasmid. Reactions were setup by adding the following to a PCR tube: 5 μL 10x Taq buffer (New England Biolabs), 1 μL 10 mM dNTP mix, 1 μL 10 μM 22b-intF, 1 μL 10 μM 22b-intR, 1 μL ~100 ng/μL parent plasmid, 5.5 μL 50 mM MgCl2, 2.5 or 5 μL 1 mM MnCl2, 1 μL DMSO, 0.5 μL Taq polymerase (New England Biolabs) and total volume was made up to 50 μL with H2O.
The PCR product was purified using a preparative agarose gel. Purified DNA fragment was inserted into a pET-22b(+) vector by the Gibson Assembly method.41 BL21 (DE3) E. coli cells were subsequently transformed with the resulting cyclized DNA product via electroporation. After 45 min of recovery in Luria-Burtani (LB) media containing 0.4% glucose at 37 °C, cells were plated onto LB plates with 100 μg/mL Ampicillin (Amp) and incubated overnight. Single colonies were used to inoculate 5 mL LB + 100 μg/mL amp (LBamp), which were grown overnight at 37 °C, 200 rpm. Colonies were sequenced and there were 1 – 2 coding mutations for both the concentrations of MnCl2.
Protein engineering (library expression, screening, and validation)
Electrocompetent BL21(DE3) were transformed with mutagenized plasmid DNA and allowed to recover for 45 min in 800 μL of Terrific Broth (TB). After recovery, the cells were plated onto LB plates containing 100 μg/mL ampicillin (LBamp) and incubated overnight. A 96-well plate containing 500 μL of TBamp per well was inoculated with single colonies. Each plate included parent positive controls (from a fresh transformation), negative controls and a sterile control that was not inoculated. The plates were grown overnight at 37 °C, 200 rpm. Expression plates were prepared with 630 μL of TBamp per well and inoculated with 20 μL of overnight culture. Glycerol stocks of each starter plate well were made from the remaining culture to ensure the sequence of any mutants of interest could be determined. The expression cultures were grown at 37 °C, 200 rpm for 2.5 h. Expression plates were then placed on ice for 30 min and induced with a final concentration of 0.1 mM IPTG in 50 μL of fresh TBamp. The expression culture was grown overnight at 20 °C, 200 rpm. Following overnight growth, the plate was centrifuged (4,000 xg, 30 min, 4 °C) and all media was removed by striking plates against a paper towel on a table. Expression plates were stored at −20 °C until further use.
A lysis buffer containing 100 mM potassium phosphate buffer (pH 7.0), 100 mM sodium chloride, 1 mg/mL Hen Egg White Lysozyme (GoldBio), 0.2 mg/mL DNase (GoldBio), 1 mM MgCl2, and 150 μM pyridoxal 5’-phosphate (PLP) was added to each well and the plate was subsequently lysed for 1 h at 37 °C. The lysate was pelleted at 4,000 xg for 30 min. Clarified lysate was added to a 96-well reaction plate where each well contained a master mix solution, such that the end reaction concentrations were 25 mM aldehyde, 25 mM L-asp, PLP, and buffer (100 mM KPi + NaCl, pH 7.0). The ratio of clarified lysate to reaction master mix was varied over the course of engineering to maintain a reasonable product measurement dynamic range. The reactions were allowed to incubate overnight at 37 °C, and were subsequently quenched with 100 μL acetonitrile and pelleted at 4,000 xg for 30 min. The cleared reaction mixture was transferred to a 0.2 μm centrifuge filter plate (PALL) and filtered at 1,500 xg for 10 min into a clean 96-well plate before being sealed prior to analysis by UPLC-MS.
The relative amount of product formed in the reactions compared to the positive control reaction was measured by absorbance at 210 nm via UPLC/MS. Given the relatively high variability in the parent signal in this assay, wells typically require an apparent 1.5-fold increase in product compared to the parent to be carried forward for validation of hits. Using the glycerol stocks from the starter culture plate (described above), wells of interest could be streaked on to a fresh LBamp plate for subsequent sequencing and validation.
Every mutant of interest was validated by heterologous expression and Ni-NTA purification, accounting for changes in soluble enzyme concentration as well as changes in activity. To study how the activity profile of UstD changed over the course of engineering, each key variant in the evolutionary lineage was expressed and purified in tandem as described above (Supplementary Figure 3). Parallel triplicate 200 μL reactions containing 25 mM benzaldehyde, 50 mM l-aspartate sodium salt monohydrate, 2.5 μM PLP, and 0.25 μM UstD variant (0.001 mol% cat., 100,000 max TON) were allowed to react at 37 °C for 16 h. After, each reaction was quenched with 200 μL of ACN containing 1 mM tryptamine as an internal standard, and the reaction mixtures were analyzed by UPLC-MS. A standard curve was made using previously purified 3a to facilitate total turnover number calculations. The variants were also trialed against several other aldehydes including: biphenyl-4-carboxaldehyde (20,000 max TON), p-anisaldehyde (20,000 max TON), and glycolaldehyde (100,000 max TON). Reactions were run using the same reaction conditions and procedure, with catalyst loading changed to match the indicated maximum turnover number. Simple fold-response measurements were used to quantify activity differences between variants (Supplementary Figure 5).
UstDTLM reaction condition optimization
All optimization reactions were conducted in triplicate on analytical scale (100 μL). PLP and l-aspartate stock solutions were made with 100 mM potassium phosphate buffer containing 100 mM sodium chloride (reaction buffer) at the indicated pH. Post-reaction quenching was done by adding 100 μL of 99:1 acetonitrile:ethanol with 1 mM tryptamine as an internal standard. Quenched reactions were then centrifuged at 15,000 xg to remove aggregated protein, and diluted with 200 μL of 1:1 water:acetonitrile. Quantification was performed by UPLC-MS analysis. Measurement of internal standard, benzaldehyde and product concentrations was done by separation on a BEH C18 column (Waters) and measurement of the corresponding 210 nm UV peak areas. Measurement of internal standard, product, l-aspartate, and l-alanine concentrations were done by separation on an Intrada amino acids column (Imtakt) using positive mode single ion readout for the M+H mass peak. Variability in injection volumes were corrected by dividing peak areas by the observed internal standard peak area for each injection. Optimization for each reaction condition component is listed below.
PLP concentration.
A reaction master mix containing 27.5 mM l-aspartate monosodium monohydrate, 27.5 mM Benzaldehyde, and 5.5% DMSO was made in 100 mM potassium phosphate buffer (pH 8.0) with 100 mM sodium chloride reaction buffer. 1.00, 0.20, 0.08, and 0.02 mM stocks of PLP were made by diluting a 20 mM PLP stock solution in reaction buffer. 0.5 dram glass vials were charged with 90.9 μL reaction master mix and 5 μL of the appropriate PLP stock (or buffer, in the case of no added PLP), and catalysis was initiated by addition of 4.1 μL of 25 μM UstDTLM (Final concentrations: 25 mM l-aspartate, 2.5 μmol; 25 mM benzaldehyde, 2.5 μmol; 1 μM UstDTLM, 0.004 mol% cat., 25,000 max TON; 50 μM, 10 μM, 4 μM, 1 μM, 0 μM PLP; 5% DMSO). Reactions were allowed to proceed in a 37 °C incubator for 16 h prior to quenching with 100 μL ACN and quantification (Supplementary Figure 4A).
l-Aspartate concentration.
A reaction master mix containing 55.6 mM benzaldehyde, 111.1 μM PLP, and 11.1% DMSO was made in 100 mM potassium phosphate buffer (pH 8.0) with 100 mM sodium chloride reaction buffer. 500, 250, 100, and 50 mM stocks of l-aspartate monosodium monohydrate were made in reaction buffer. 0.5 dram glass vials were charged with 45 μL reaction master mix and 50 μL of the appropriate l-aspartate stock, and catalysis was initiated by addition of 5 μL 5 μM UstDTLM (Final concentrations: 25 mM benzaldehyde, 2.5 μmol; 25, 50, 125, 250 mM l-aspartate, 2.5, 5.0, 12.5, 25.0 μmol, respectively; 0.25 μM UstDTLM, 0.001 mol% cat., 100,000 max TON; 2.5 μM PLP, 10 equiv. relative to UstDTLM; 5% DMSO). Reactions were allowed to proceed in a 37 °C incubator for 16 h prior to quenching with 100 μL ACN and quantification (Supplementary Figure 4B).
pH.
Five separate master mix solutions containing 25 mM benzaldehyde, 130 mM l-aspartate monosodium monohydrate, 1.3 mM PLP, and 5.2% DMSO were prepared in 100 mM potassium phosphate with 100 mM NaCl reaction buffer at pH 6.0, 6.5, 7.0, 7.5, and 8.0 (pH of buffer not altered after addition of reaction components). 0.5 dram glass vials were charged with 96.1 μL of the appropriate reaction master mix, and catalysis was initiated by addition of 3.9 μL 6 μM UstDTLM (Final concentrations: 25 mM benzaldehyde, 2.5 μmol; 125 mM l-aspartate, 12.5 μmol; 0.25 μM UstDTLM, 0.001 mol% cat., 100,000 max TON; 2.5 μM PLP, 10 equiv. relative to UstDTLM; 5% DMSO). Reactions were allowed to proceed in a 37 °C incubator for 16 h prior to quenching with 100 μL ACN and quantification (Supplementary Figure 4C).
mol% catalyst.
A reaction master mix containing 50 mM benzaldehyde, 100 mM l-aspartate monosodium monohydrate, and 10% DMSO was made in 100 mM potassium phosphate, pH 7.0, with 100 mM sodium chloride. UstDTLM stock solutions containing 50 μM, 5.0 μM, 1.7 μM, and 1 μM were made, each containing 10 equivalents of PLP. 0.5 dram glass vials were charged with 50 μL reaction master mix, and catalysis was initiated by addition of 50 μL of the appropriate UstD stocks (Final concentrations: 25 mM benzaldehyde, 2.5 μmol; 50 mM l-aspartate, 5.0 μmol; 25 μM (0.1 mol% cat., 1,000 max TON), 2.5 μM (0.01 mol% cat., 10,000 max TON), 0.83 μM (0.003 mol% cat., 30,000 max TON), 0.25 μM (0.001 mol% cat., 100,000 max TON) UstDTLM; 10 equiv. PLP relative to UstDTLM; 5% DMSO). Reactions were allowed to proceed in a 37 °C incubator for 16 h prior to quenching with 100 μL ACN and quantification (Supplementary Figure 4D).
UstD Performance Evaluation using Marfey’s Derivatization
A 0.5-dram glass vial was charged with a master mix of l-aspartate sodium salt monohydrate (0.005 mmol, 2 equiv., 50 mM final concentration), pyridoxal-5’-phosphate (10 equiv. relative to final UstD concentration), and buffer. The master mix composition was varied to ensure a uniform concentration of each UstD variant at the completion of reaction setup. To this solution the aldehydes corresponding to compounds 2a-2p (0.0025 mmol, 1 equiv., 25 mM final concentration) were added to the reaction mixtures. The reactions were initiated by the addition of UstD (0.007 mol% cat., 15,000 maximum turnover number). The reaction vessels were placed in a dark 37 °C incubator for 18 h and subsequently quenched with 200 μL of ACN. A Marfey’s derivatization reaction was then performed in order to determine ee and dr of each enzymatic reaction. In a new flat bottom glass LC vial, 6 μL of quenched reaction mix (1 equiv., 0.5 mM final total amines from unreacted l-aspartate and formed l-alanine and γ-hydroxy amino acid product) was added to a solution of 144 μL of 10.41 mM NaHCO3 (10 equiv., 5 mM final concentration) with 0.21 mM of either l-arg (0.1 mM final concentration, aldehydes 2a-2k) or tryptamine (0.1 mM final concentration, aldehydes 2l-2p), followed by addition of 150 μL 5 mM l-FDAA dissolved in ACN (5 equiv., 2.5 mM final concentration) to bring the total reaction volume to 300 μL. Each reaction vial was sealed with a pierceable LC vial cap, placed in a dark 37 °C incubator for 18 h, then quenched with 300 μL of 1:1 ACN:60 mM HCl (15 mM post-quench). Quenched reaction mixtures were analyzed by UPLC-MS no later than 24 h after quenching, results are shown in Supplementary Table 1 and Supplementary Figures 13–28.
Data availability
The structure of UstDv2.0 is available through the Protein Data Bank (ID: 7MKV). Sequence-activity data used for linear regression modelling is available through GitHub42. All other data is available from the authors upon reasonable request.
Code availability
The linear regression modeling code used during the final round of protein engineering is available through GitHub42 under the MIT License.
Supplementary Material
{kind=link}
Acknowledgments:
We thank I. Guzei for small molecule x-ray structure determination and S.H. Gellman and members of the Buller group for critical reading of the manuscript. The crystal mounting and data collection were mediated by the Collaborative Crystallography Core, Department of Biochemistry, UW-Madison and data were collected at the Life Sciences Collaborative Access Team beamline 21ID-D at the Advanced Photon Source, Argonne National Laboratory and we thank Z. Wawrzak for technical assistance during data collection. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). This work was supported by the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison, Wisconsin Alumni Research Foundation, National Institute of Health (grant DP2-GM137417, A.R.B.), Morgridge Institute for Research – Metabolism Theme Fellowship (P.K.), and the NIH Biotechnology Training Grant (T32-GM008349, J.M.E.). The Bruker AVANCE III-500 NMR spectrometers were supported by the Bender Fund. The Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. The Bruker D8 VENTURE Photon III X-ray diffractometer was partially funded by NSF Award (#CHE-1919350) to the UW–Madison Department of Chemistry
Footnotes
Competing interests: Authors have a patent pending on the use of engineered UstD for synthesis of non-standard amino acids. US20210115480A1 Inventors: Andrew R. Buller, Jonathan M. Ellis, Prasanth Kumar.
Supplementary Information is available for this paper.
Reprints and permissions information is available at www.nature.com/reprints
References
- 1.Nestl BM, Hammer SC, Nebel BA & Hauer B New generation of biocatalysts for organic synthesis. Angew. Chemie - Int. Ed 53, 3070–3095 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Zhang X et al. Divergent synthesis of complex diterpenes through a hybrid oxidative approach. Science (80-.). 369, 799–806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown DG & Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem. 59, 4443–4458 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Fesko K & Gruber-Khadjawi M Biocatalytic Methods for C-C Bond Formation. ChemCatChem 5, 1248–1272 (2013). [Google Scholar]
- 5.Schmidt NG, Eger E & Kroutil W Building Bridges: Biocatalytic C-C-Bond Formation toward Multifunctional Products. ACS Catalysis 6, 4286–4311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujii I Heterologous expression systems for polyketide synthases. Nat. Prod. Rep. 26, 155–169 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Heine A et al. Observation of covalent intermediates in an enzyme mechanism at atomic resolution. Science (80-.). 294, 369–374 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Wang ZJ et al. Improved cyclopropanation activity of histidine-ligated cytochromeP450 enables the enantioselective formal synthesis of levomilnacipran. Angew. Chemie - Int. Ed 53, 6810–6813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berkeš D, Kolarovič A, Manduch R, Baran P & Považanec F Crystallization-induced asymmetric transformations (CIAT): Stereoconvergent acid-catalyzed lactonization of substituted 2-amino-4-aryl-4-hydroxybutanoic acids. Tetrahedron Asymmetry 16, 1927–1934 (2005). [Google Scholar]
- 10.Goldberg SL et al. Preparation of β-hydroxy-α-amino Acid Using Recombinant d -Threonine Aldolase. Org. Process Res. Dev. 19, 1308–1316 (2015). [Google Scholar]
- 11.Steinreiber J et al. Overcoming thermodynamic and kinetic limitations of aldolase-catalyzed reactions by applying multienzymatic dynamic kinetic asymmetric transformations. Angew. Chemie - Int. Ed. 46, 1624–1626 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Zetzsche LE & Narayan ARH Broadening the scope of biocatalytic C–C bond formation. Nat. Rev. Chem. 4, 334–346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye Y et al. Unveiling the Biosynthetic Pathway of the Ribosomally Synthesized and Post-translationally Modified Peptide Ustiloxin B in Filamentous Fungi. Angew. Chemie Int. Ed. 55, 8072–8075 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Prier CK & Arnold FH Chemomimetic Biocatalysis: Exploiting the Synthetic Potential of Cofactor-Dependent Enzymes to Create New Catalysts. J. Am. Chem. Soc. 137, 13992–14006 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Di Salvo ML et al. On the catalytic mechanism and stereospecificity of Escherichia coli l -threonine aldolase. FEBS J. 281, 129–145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marsden SR, Gjonaj L, Eustace SJ & Hanefeld U Separating Thermodynamics from Kinetics—A New Understanding of the Transketolase Reaction. ChemCatChem 9, 1808–1814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ariza J, Font J & Ortuño RM An efficient and concise entry to (−)-4,5-dihydroxy-d-threo-l-norvaline. Formal synthesis of clavalanine. Tetrahedron Lett. 32, 1979–1982 (1991). [Google Scholar]
- 18.Blaskovich MAT Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 59, 10807–10836 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Moreno CJ et al. Synthesis of γ-Hydroxy-α-amino Acid Derivatives by Enzymatic Tandem Aldol Addition–Transamination Reactions. ACS Catal. 11, 4660–4669 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernandez K et al. Combining Aldolases and Transaminases for the Synthesis of 2-Amino-4-hydroxybutanoic Acid. ACS Catal. 7, 1707–1711 (2017). [Google Scholar]
- 21.Vargas-Rodriguez O, Sevostyanova A, Söll D & Crnković A Upgrading aminoacyl-tRNA synthetases for genetic code expansion. Curr. Op. in Chem. Biol. 46, 115–122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchand JA et al. Discovery of a pathway for terminal-alkyne amino acid biosynthesis. Nature 567, 420–424 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Yang J et al. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 12, 7–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho TH et al. Catalytic Intermediate Crystal Structures of Cysteine Desulfurase from the Archaeon Thermococcus onnurineus NA1. Archaea 2017, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P et al. L -Threonine Transaldolase Activity Is Enabled by a Persistent Catalytic Intermediate. ACS Chem. Biol. 16, 95 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reetz MT, Prasad S, Carballeira JD, Gumulya Y & Bocola M Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: Rigorous comparison with traditional methods. J. Am. Chem. Soc. 132, 9144–9152 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Romero PA & Arnold FH Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reetz MT, Bocola M, Carballeira JD, Zha D & Vogel A Expanding the Range of Substrate Acceptance of Enzymes: Combinatorial Active-Site Saturation Test. Angew. Chemie Int. Ed. 44, 4192–4196 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Romney DK, Sarai NS & Arnold FH Nitroalkanes as Versatile Nucleophiles for Enzymatic Synthesis of Noncanonical Amino Acids. ACS Catal. 9, 8726–8730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marfey P DETERMINATION OF D-AMINO ACIDS. II. USE OF A BIFUNCTIONAL REAGENT, 1,5-DIFLUORO-2,4-DINITROBENZENE. Carlsb. Res. Commun. 49, 591–596 (1984). [Google Scholar]
- 31.Wu G et al. Proline and hydroxyproline metabolism: Implications for animal and human nutrition. Amino Acids 40, 1053–1063 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MÜLLER J-C, TOOME V, PRUESS DL, BLOUNT JF & WEIGELE M Ro 22–5417, a new clavam antibiotic from Streptomyces clavuligerus. III. Absolute stereochemistry. J. Antibiot. (Tokyo). 36, 217–225 (1983). [DOI] [PubMed] [Google Scholar]
- 33.Wahab RA, Elias N, Abdullah F & Ghoshal SK On the taught new tricks of enzymes immobilization: An all-inclusive overview. React. and Func. Pol. 152, 104613 (2020). [Google Scholar]
- 34.Wachtmeister J & Rother D Recent advances in whole cell biocatalysis techniques bridging from investigative to industrial scale. Curr. Op. in Biotech. 42, 169–177 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Al-Ayyoubi M, Gettins PGW & Volz K Crystal structure of human maspin, a serpin with antitumor properties: Reactive center loop of maspin is exposed but constrained. J. Biol. Chem. 279, 55540–55544 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Fox R Directed molecular evolution by machine learning and the influence of nonlinear interactions. J. Theor. Biol. 234, 187–199 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Huffman MA et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science (80-.). 366, 1255–1259 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Eliot AC & Kirsch JF Pyridoxal Phosphate Enzymes: Mechanistic, Structural, and Evolutionary Considerations. Annu. Rev. Biochem. 73, 383–415 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Romney DK, Murciano-Calles J, Wehrmüller JE & Arnold FH Unlocking Reactivity of TrpB: A General Biocatalytic Platform for Synthesis of Tryptophan Analogues. J. Am. Chem. Soc. 139, 10769–10776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boville CE et al. Engineered Biosynthesis of β-Alkyl Tryptophan Analogues. Angew. Chemie - Int. Ed 57, 14764–14768 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Ellis JM Linear Regression Analysis of UstD-TLM. (2021). 10.5281/zenodo.5719389 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structure of UstDv2.0 is available through the Protein Data Bank (ID: 7MKV). Sequence-activity data used for linear regression modelling is available through GitHub42. All other data is available from the authors upon reasonable request.