

Abstract
Background
Cornelia de Lange syndrome (CdLS) is a genetic disorder caused by variants in cohesion genes including NIPBL, SMC1A, SMC3, RAD21, and HDAC8. According to the 2018 consensus statement, a patient with clinical scored ≥ 11 points could be diagnosed as CdLS. However, some variants in non-cohesion genes rather than cohesion genes can manifest as phenotypes of CdLS.
Objectives
This study describes six variants of non-cohesion genes (KDM6A, KMT2D, KMT2A ANKRD11, and UBE2A), and assesses the reliability of 11-points scale criteria in the clinical diagnosis of CdLS.
Methods
Whole-exome sequencing (WES) was performed on six patients with features of CdLS. Phenotypic and genotypic spectra of 40 previously reported patients with features of CdLS caused by non-cohesion genes variants and 34 previously reported patients with NIPBL variants were summarized. Clinical score comparison among patients with NIPBL variants versus those with variants in non-cohesin genes was performed.
Results
Variants in non-cohesion genes were found in six patients [KMT2A (n = 2), KMT2D, ANKRD11, KDM6A, and UBE2A]. Of them, four variants (KMT2A c.7789C > T, ANKRD11 c.1757_1776del, KDM6A c.655-1G > A, and UBE2A c.439C > T) were novel. Combining with previously reported cases, 46 patients with phenotypes of CdLS caused by variants in 20 non-cohesion genes are now reported. From this total cohort, the average clinical score of patients in ANKRD11 cohort, SETD5 cohort, and AFF4 cohort was statistically lower than those in NIPBL cohort (8.92 ± 1.77 vs. 12.23 ± 2.58, 7.33 ± 2.52 vs. 12.23 ± 2.58, 5.33 ± 1.53 vs. 12.23 ± 2.58; p < 0.05). The average clinical score of KMT2A cohort, EP300 cohort, and NIPBL cohort had not significantly different from (11 ± 2.19 vs. 12.23 ± 2.58, 10 ± 4.58 vs. 12.23 ± 2.58; p > 0.05).
Conclusion
We described 4 novel variants of non-cohesion genes in six Chinese patients with phenotypes of CdLS. Of note, three genes (KMT2D, KDM6A, and UBE2A) causing features of CdLS have never been reported. The proposed clinical criteria for CdLS needed to be updated and refined, insofar as WES was necessary to confirm the diagnosis of CdLS. Our study expanded the spectra of non-cohesion genetic variations in patients with features of CdLS.
Keywords: Cornelia de Lange syndrome, non-cohesion, clinical diagnosis, whole exome sequencing, CdLS-like phenotypes
Introduction
Cornelia de Lange syndrome (CdLS, OMIM # 122470, #614701, #610759, #300590, and #300882) is a rare genetic disorder caused by variants in cohesion complex genes including NIPBL (NM_133433.3), SMC1A (NM_006304.4), SMC3 (NM_005445.3), HDAC8 (NM_018486.2), and RAD21 (NM_006265.3) (1). Up to 70% of CdLS patients are diagnosed with NIPBL variants. Around 5% of CdLS patients carry SMC1A variants, and 5% are HDAC8 variants, less than 1% of CdLS patients are variants in SMC3 or RAD21 genes (2). The mode of inheritance of CdLS in the patient’s offspring could be either autosomal dominant (NIPBL, RAD21, or SMC3 variants) or X-linked dominant (SMC1, or HDAC8 variants) (2).
CdLS have variable phenotypes, such as microcephaly, minor facial dysmorphisms, intellectual disability, short stature, small hands, and hypertrichosis. In 2018, a diagnostic algorithm for initial evaluation of CdLS patients was proposed (3). Classic CdLS, based on clinical features, scored ≥ 11 points according to the consensus statement (3). Adopting this criteria would facilitate bedside diagnosis. However, some patients with features of CdLS were found to carry variants that were associated with other disorders, not with CdLS. Yuan et al. described a pathogenic KMT2A (NM_001197104.1) variant in one patient among 32 Turkish patients clinically diagnosed with classic CdLS (4). Cucco et al. also reported pathogenic variants in EP300 (NM_001429.3) in a patient exhibiting resemblance to the classic CdLS phenotype in 2020 (5). With the development of contemporary genetic technology, it is not surprising that more patients with features of CdLS have been identified to carry variants in non-cohesion genes and, to date, the clinical genetic characteristics of these patients are ill-defined. Adding to the existing knowledge in this field is important in the quest to characterize the extent of heterogeneity of CdLS and Cornelia de Lange syndrome-like (CdLS-like) conditions.
In this study, we analyzed six patients with features of CdLS, and identified six variants in five non-cohesion genes. To date, 40 sporadic patients with features of CdLS caused by non-cohesion variants had been reported (4–17). We also evaluated the clinical features of these patients, and calculate the clinical scores of these patients according to the 2018 consensus statement to assess the reliability of 11-points scale criteria in the diagnosis of CdLS.
Subjects and methods
Subjects
All individuals in this study were evaluated by clinical geneticists and found to have clinical signs consistent with CdLS. Clinical details were retrospectively reviewed based on recent clinical criteria (3). This study was approved by the Ethics Committee of Fuzhou Children’s Hospital of Fujian Medical University, and written informed consents were obtained from the legal guardians of the patients.
Whole-exome sequencing and variants interpretation
Genomic DNA was extracted from peripheral blood leukocytes of each patient. Blood samples from the parents were also collected. The Whole-exome sequencing (WES) was performed at Shanghai patient’s Medical Center. An adaptor-ligated library was prepared using SureSelect Human All Exon Kit (Agilent Technologies, Santa Clara, CA, America) according to the manufacturer’ s protocol. Target regions were sequenced on an Illumina Hiseq X Ten System (Illumina, San Diego, CA, America). Paired end reads were aligned to the GRCh37/hg19 human reference sequence. BAM files were generated by Picard and sequence variants were called by Genome Analysis Toolkit (GATK) Haplotype Caller. Variants were annotated by TGex and putative pathogenic variants detected in the patients by WES were validated by Sanger sequencing. Variants were classified following the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) standards and guidelines (18).
Literature review
We first searched the published literature in PUBMED, EMBASE and MEDLINE using the following search keys: (“Cornelia de Lange syndrome” OR “Cornelia de Lange syndrome-like” OR “CdLS” OR CdLS-like), without language restriction, with published data up to March 31, 2022. Then we reviewed the articles, and included the CdLS patients carrying variants in non-cohesion genes. We also used the following keys: (“Cornelia de Lange syndrome” OR “CdLS”) AND (“NIPBL”) to search the published literature in the same way. The NIPBL cases with detail clinical information were included.
Statistics analysis
Statistical analysis for phenotypic among the patients with NIPBL variants versus those with variants in non-cohesion genes was performed by (corrected) the Chi-square test or Fisher’s exact test using GraphPad prism 8.0.1 software. In addition, the mean of clinical score within each group was analyzed using one way ANOVA test or Kruskal–Wallis test in GraphPad prism 8.0.1 software. P < 0.05 was considered statistically significant.
Results
Clinical phenotype of six patients in our study
The six patients exhibited overlapping phenotypes. The main characteristics were as following: facial dysmorphism (n = 6), intellectual disability or global developmental delay (n = 6), short stature (n = 6), small hands (n = 6), short 5th finger (n = 6), and microcephaly (n = 4). Detailed clinical information of the six patients are described in Table 1 and Supplementary Table 1.
TABLE 1.
The genetic variants and clinical features of our six patients.
| ID | P1 | P2 | P3 | P4 | P5 | P6 | |
| Gender | Male | Female | Male | Male | Male | Male | |
| Age (years) | 5 | 0.83 | 3 | 1 | 2.5 | 0.58 | |
| Gene | UBE2A | KMT2D | KDM6A | ANKRD11 | KMT2A | KMT2A | |
| Variant | c.439C > T, p.Q147* | c.5845delC, p.Q1949Sfs*98 | c.655-1G > A | c.1757_1776del, p.V586Efs*41 | c.7789C > T, p.Q2597* | c.2629_2630delGA, p.D877fs*8 | |
| Inheritance | Materal | De novo | De novo | De novo | De novo | De novo | |
| Novel or reported | Novel | Reported | Novel | Novel | Novel | Reported | |
| ACMG classifcation | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | |
| Main features (2 point each if present) |
Synophrys and/or thick eyebrows | − | − | − | − | + | − |
| Short nose, concave nasal ridge and/or upturned nasal tip | + | − | − | + | + | + | |
| Long and/or smooth philtrum | − | − | − | − | − | − | |
| Thin upper lip vermilion and/or downturned corners of mouth | − | + | + | − | + | + | |
| Hand oligodactyly and/or adactyly | − | − | − | − | − | − | |
| Congenital diaphragmatic hernia | − | − | − | − | − | − | |
| Suggestive features (1 point each if present) |
Small hands and/or feet | + | + | + | + | + | + |
| Microcephaly | − | − | + | + | + | + | |
| Global developmental delay and/or intellectual disability | + | + | + | + | + | + | |
| Prenatal growth retardation | − | − | − | − | − | − | |
| Postnatal growth retardation | + | + | + | + | + | + | |
| Hypertrichosis | − | − | − | − | + | + | |
| Short fifth finger | + | + | + | + | + | + | |
| Other features | With mouth, hypertelorism, high palate, inguinal hernia, renal cyst | Long palpebral fissures with eversion of the lower lid, ventricular septal defect, single transverse palmar crease, prominent fingertip pads |
Long palpebral fissures with eversion of the lower lid, Long eyelashes, defect in the atrial septum, Coarctation of aorta, single transverse palmar crease |
Macrodontia of the upper central incisors | Ptosis, long eyelashes, hypertelorism |
Ptosis, long eyelashes, hypertelorism, cryptorchidism, patent ductus arteriosus |
|
| Clinical scored | 6 | 6 | 7 | 7 | 12 | 10 |
Molecular findings of six patients in our study
Whole-exome sequencing of the six individuals with clinically suspected CdLS identified six variants in KMT2A, KMT2D (NM_003482.3), ANKRD11 (NM_013275.4), KDM6A (NM_021140.2), and UBE2A (NM_003336.2) (Table 1).
Patient #1 was a carrier of a materal nucleotide substitution in UBE2A: c.439C > T, resulting in a premature stop codon (p.Gln147*). Patient #2 showed a de novo nucleotide deletion in KMT2D: c.5845delC, resulting in a premature stop codon (p.Q1949Sfs*98). Patient #3 had a de novo 20 nucleotides deletion in ANKRD11: c.1757_1776del, resulting in premature stop codon (p.V586Efs*41). Patient #4 showed a de novo nucleotide substitution in KMT2A: c.7789C > T, leading to premature stop codon (p.Q2597*). Patient #5 had a de novo 2 nucleotide deletion in KMT2A: c.2629_2630delGA, resulting in a premature stop codon (p.D877fs*8). Patient #6 was a carrier of a de novo splice variant in KDM6A: c.655-1G > A. The SpliceTool1 predicted to delete 2 or 94 bp causing frameshift variants and premature termination, and multiple in silico tools predict deleterious outcomes of the splice variant (scored 1 in dbscsnv11_AdaBoost and 0.937 in dbscsnv11_RandomF orest). 4 variants (UBE2A: c.439C > T, ANKRD11: c.1757_1776del, KMT2A: c.7789C > T, and KDM6A: c.655-1G > A) were novel, and the KMT2D c.5845delC and KMT2A c.2629_2630delGA were both reported (19, 20). According to the ACMG/AMP standards and guidelines, these variants could all be classified as pathogenic (For ANKRD11 c.1757_1776del, KMT2A c.7789C > T, and KDM6A c.655-1G > A: PVS1 + PS2 + PM2 + PP4; For UBE2A c.439C > T: PVS1 + PM2 + PP4; For KMT2D c.5845delC and KMT2A c.2629_2630delGA: PVS1 + PS2 + PP4).
Phenotypes and genotypes of 46 patients caused by non-cohesion genes variants
To date, 40 patients with features of CdLS caused by variants in non-cohesion genes have been comprehensively described in the literature. Including the six patients in our study, a total of 46 pathogenic/likely pathogenic variants associated with 20 epigenetic genes including ANKRD11 (n = 14), BRD4 (NM_058243.3) (n = 2), AFF4 (NM_014423.4) (n = 3), KMT2A (n = 6), EP300 (n = 3), SETD5 (NM_001080517.3) (n = 3), ARID1B (NM_001374820.1) (n = 2), SMARCB1 (NM_003073.5) (n = 1), TAF1 (NM_4606.5) (n = 1), DDX23 (NM_004818.3) (n = 1), CSNK1G1 (NM_022048.5) (n = 1), ZMYND11 (NM_006624.7) (n = 1), MED13L (NM_015335.5) (n = 1), PHIP (NM_017934.7) (n = 1), TAF6 (NM_005641.4) (n = 1), NAA50 (NM_025146.4) (n = 1), CREBBP (NM_004380.3) (n = 1), UBE2A (n = 1), KMT2D (n = 1), and KDM6A (n = 1) are described (Supplementary Table 2). Of note, although we have reported the patient with KMT2D c.5845delC before, his phenotype consistent with CdLS was not recognized (19). Therefore, this case was not included in the literature review.
We divided these patients into six groups: KMT2A group, ANKRD11 group, EP300 group, SETD5 group, AFF4 group and a remaining group consisted of a lower number of patients with features of CdLS caused by non-cohesion genes variants (BRD4, ARID1B, SMARCB1, TAF1, DDX23, CSNK1G1, ZMYND11, MED13L, PHIP, TAF6, NAA50, CREBBP, UBE2A, KMT2D, and KDM6A). The average clinical score of patients in KMT2A group was 11 ± 2.19, 8.92 ± 1.77 in ANKRD11 group, 10 ± 4.58 in EP300 group, 7.33 ± 2.52 in SETD5 group, 5.33 ± 1.53 in AFF4 group and 8.88 ± 2.62 in the remaining group (Table 2).
TABLE 2.
Clinical features of patients in CdLS-like cohort and those in NIPBL cohort.
| Clinical feature | NIPBL cohort (n = 34) | KMT2A cohort (n = 6) | ANKRD11 cohort (n = 14) | EP300 cohort (n = 3) | SETD5 cohort (n = 3) | AFF4 cohort (n = 3) | Remaining genes cohort (n = 17) | Chi-square value | P-valve |
| Synophrys | 31 | 5 | 10 | 3 | 2 | 3 | 14 | 5.049 | 0.45 |
| Thick eyebrows | 17 | 3 | 11 | 0 | 1 | 2 | 2a | 17.772 | 0.002 |
| Short nose | 19 | 2 | 0a | 1 | 0 | 1 | 3a | 19.015 | 0.001 |
| Concave nasal ridge | 23 | 4 | 5 | 0a | 2 | 0a | 5a | 14.383 | 0.011 |
| Upturned nasal tip | 22 | 2 | 11 | 2 | 1 | 1 | 7 | 8.204 | 0.192 |
| Smooth philtrum | 15 | 3 | 5 | 1 | 2 | 0 | 2 | 8.899 | 0.139 |
| Long philtrum | 28 | 3 | 13 | 1 | 1 | 1 | 10 | 14.287 | 0.012 |
| Downturned corners of mouth | 18 | 3 | 5 | 1 | 1 | 1 | 7 | 2.354 | 0.924 |
| Thin upper lip vermilion | 27 | 4 | 7 | 2 | 2 | 0 | 10 | 10.158 | 0.08 |
| Adactyly | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 4.945 | 0.754 |
| Hand oligodactyly | 7 | 0 | 0 | 0 | 0 | 0 | 2 | 2.420 | 0.944 |
| Small hands | 17 | 3 | 8 | 0 | 0 | 0 | 6 | 8.008 | 0.205 |
| Small feet | 8 | 0 | 5 | 1 | 0 | 0 | 3 | 4.438 | 0.594 |
| Microcephaly | 24 | 5 | 7 | 2 | 0 | 1 | 11 | 8.558 | 0.164 |
| Global developmental delay and/or intellectual disability | 28 | 6 | 13 | 3 | 3 | 2 | 17 | 6.197 | 0.294 |
| Prenatal growth retardation | 23 | 1a | 1a | 1 | 0a | 0a | 4a | 23.496 | 0.000 |
| Postnatal growth retardation | 23 | 6 | 1a | 3 | 1 | 1 | 10 | 24.140 | 0.000 |
| Hypertrichosis | 13 | 4 | 2 | 3 | 0 | 0 | 2 | 16.143 | 0.005 |
| Short fifth finger | 12 | 5 | 6 | 1 | 0 | 0 | 7 | 8.137 | 0.194 |
| Average clinical score (mean ± SD) | 12.23 ± 2.58 | 11 ± 2.19c | 8.92 ± 1.77b | 10 ± 4.58 | 7.33 ± 2.52b | 5.33 ± 1.53bc | 8.88 ± 2.62b | < 0.0001 |
aIndicates that the incidence of phenotype was statistically significant compared with NIPBL cohort.
bIndicates that the average clinical score was statistically significant compared with NIPBL cohort.
cIndicates that the average clinical score of the two groups were statistically significant.
Phenotypic and clinical score comparison of the non-cohesion cohort and NIPBL cohort
We collected data on a group of 34 cases of CdLS caused by NIPBL variants from the literature and summarized the phenotypes and clinical score of the total 34 patients (clinical information and reference are shown in Supplementary Tables 3, 4). This affords us the opportunity to compare the clinical features and score of patients with NIPBL variants to those with variants in non-cohesion genes (Table 2 and Figure 1).
FIGURE 1.
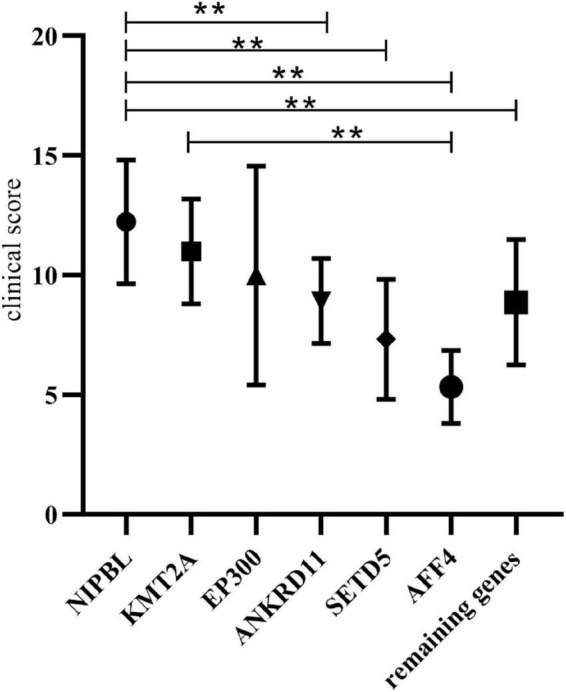
Clinical score comparison of the non-cohesion cohort and NIPBL cohort. **Indicates that the average clinical score of the two groups were statistically significant (p < 0.05).
The overall clinical characteristics are similar among these groups, however, several differences still can be observed. The patients in ANKRD11 cohort had lower frequencies of short nose, prenatal growth retardation and postnatal growth retardation than those in NIPBL cohort. The features of concave nasal ridge and prenatal growth retardation were more frequent in patients in AFF4 cohort than those in NIPBL cohort. The features of concave nasal ridge and prenatal growth retardation showed significant statistical different from the patients in the EP300 cohort and those in the NIPBL cohort. The frequencies of prenatal growth retardation in patients of KMT2A cohort showed lower than those of NIPBL cohort. Additionally, the average clinical score of patients in ANKRD11 cohort, SETD5 cohort and AFF4 cohort was statistically lower than those in NIPBL cohort, respectively (8.92 ± 1.77 vs. 12.23 ± 2.58, 7.33 ± 2.52 vs. 12.23 ± 2.58, 5.33 ± 1.53 vs. 12.23 ± 2.58; p < 0.05). The average clinical score of patients in KMT2A cohort and EP300 cohort both had no significantly difference with those in the NIPBL cohort (11 ± 2.19 vs. 12.23 ± 2.58, 10 ± 4.58 vs. 12.23 ± 2.58; p > 0.05).
Discussion
In the present study, we provided our findings on the genetic analysis of six patients referred to our clinic for short stature. Short stature can be either a component of known syndromes or occur in undefined manifold complex clinical phenotypes. It was reported that 86% of CdLS presented with short stature, a common finding occurred in other rare genetic disorders, such as Wiedemann-Steiner syndrome (WDSTS, OMIM #605135) (75%), Kabuki syndrome (KS, OMIM #147920, #300867) (57%) and KBG syndrome (KBGS, OMIM #148050) (40–77%) (19, 21–23).
Several common clinical features were observed among the six patients, all of whom had short stature, moderate to severe development delay and/or intellectual disability, small hands and short fifth fingers, partly presenting with microcephaly. Hypertrichosis was also frequent. In addition, some CdLS-specific features including synophrys, concave nasal ridge and long smooth philtrum were observed, which in combination with short stature and intellectual disability resembled CdLS. However, the six patients with clinically suspected CdLS were found to carry 6 variants in KMT2A, KMT2D, ANKRD11, KDM6A, and UBE2A, which don’t function in cohesion protein. These identified variants in the genes (KMT2A, KMT2D, KDM6A, and ANKRD11) met the pathogenicity criteria according to ACMG/AMP standards and guidelines, including de novo occurrence, absence in general population and predicted LOF effect causing diseases through this pathogenetic mechanism. The UBE2A c.439C > T variant was also deemed pathogenic given that the variant causes a premature stop codon and is absent in the general population.
KMT2A variants are associated to the WDSTS, a rare autosomal dominant condition characterized by different debilities, mainly intellectual disability, short stature, hypertrichosis, distinctive facial features (thick eyebrows, long eyelashes, narrow palpebral fissures, hypertelorism, ptosis, broad nasal tip), and skeletal abnormalities (clinodactyly, brachydactyly, advanced bone age) (24). Two patients (P5 and P6) carrying KMT2A variants in our study shared a number of clinical features of CdLS including short nose, anteverted nares, concave nasal ridge, short stature, global developmental delay, small hands, short 5th finger and microcephaly. And both had clinical scores of 12 and 10, respectively (Table 1). Previously, four patients with features of CdLS found to carry KMT2A variants have been reported. With our new two patients, the average clinical score of the six patients was no significantly different from a cohort of NIPBL patients (11 ± 2.19 vs. 12.23 ± 2.58), which infers that some patients with KMT2A variants may be misdiagnosed as CdLS without molecular test. However, the combination of ptosis and long eyelashes was more frequent in KMT2A patients than NIPBL patients (24). Four of these six patients (two patients in our study and two patients in literature) had features of ptosis and hypertelorism, which may help to distinguish WDSTS and CdLS (13, 16).
KBG syndrome is a rare condition characterized by intellectual disability, global developmental delay, short stature, skeletal anomalies, distinctive facial features, and macrodontia of the upper central incisors (23). One child (P4) in our study had a frameshift variant in ANKRD11 gene. He presented with some features of CdLS including anteverted nares, short stature, global developmental delay, microcephaly, small hands and short 5th finger (Table 1; scored 7). He also presented with macrodontia of the upper central incisors, which is a component of KBGS (25). Although KBG has clinical features overlapping with CdLS, the average clinical score is lower than the NIPBL cohort. Additionally, the features of short nose, prenatal and postnatal growth retardation had a lower frequency in ANKRD11 cohort than those in the NIPBL cohort (Table 2). More importantly, a specific combination of a triangular shaped face and a bulbous nasal tip was crucial for the accurate clinical diagnosis of KBGS and CdLS (17).
Additionally, we reported a novel patient (P1) with nonsense variant in UBE2A, involved in coding ubiquitin-conjugating enzyme E2A, with CdLS related phenotypes. Haploinsufficiency of UBE2A underlies the X-linked intellectual disability type Nascimento (XIDTN, OMIM #300860), also known as UBE2A deficiency syndrome (26). To date, about 40 patients with XIDTN had been reported in literature (26). Nascimento et al. reported that its distinct abnormalities including a myxoedematous appearance and nail dystrophies, yet these features were not observed in P1. Additionally, prominent supraorbital ridge, hypertelorism, and prominent columella/hypoplastic alae nasi and a wide mouth were also common in XIDTN (27). However, except a wide mouth, these features were not found in our patient. Interestingly, the phenotypic features of our patient with UBE2A variant shared similarities with the previously described CdLS individuals including anteverted nares, short stature, intellectual disability, small hands and short 5th finger (Table 1, scored 6). These findings suggested that individuals with UBE2A variant instead present with specific features that is only minimally overlapping with CdLS.
Our two patients (P2 and P3) found to carry KTM2D and KDM6A variants, presented with thin upper lip vermilion, downturned corners of mouth, a finding mainly described in CdLS (Table 1; score 6 and 7, respectively) (3). Pathogenic variants in the chromatin regulatory methyltransferase gene KMT2D and demethyltransferase gene KDM6A cause autosomal dominant KS, associated with specific clinical signs including arched eyebrows, long palpebral fissures with eversion of the lower lid, large protuberant ears, and prominent fingertip pads, hypoplastic left heart (28). Corresponding to it, the two patients both had long palpebral fissures with eversion of the lower lid and hypoplastic left heart, features that help to distinguish KS and CdLS. Additionally, P2 had prominent fingertip pads which are considered a distinctive of KS.
A total of 46 patients with feature of CdLS caused by variants in non-cohesion genes were summarized. We collected data on a group of 34 cases of CdLS caused by NIPBL variants from the literature and summarized the phenotypes and clinical score of those patients. This affords us the opportunity to compare the clinical features and score of patients with NIPBL variants to those with variants in non-cohesion genes. Our results suggested that KMT2A and EP300 can be included within the extended list of CdLS genes that are studied in CdLS panels. In addition, the average clinical score of ANKRD11 cohort, SETD5 cohort, and AFF4 cohort was 8.92 ± 1.77, 7.33 ± 2.52, 5.33 ± 1.53, respectively. Variants in these three genes caused limited phenotypes overlapping with CdLS. Single case with variants in some non-genes causes features of CdLS were reported in literature. The presence of one or two cases made statistical analysis impossible. Additional case accumulation is needed to further explore the relationship between these non-genetic variants and phenotypes of CdLS.
Extensive evidences showed that the cohesion complex functions in sister chromatid cohesion, as well as playing a role in the regulation of transcription. Some reports suggested that the cohesion genes including NIPBL, SMC1A, SMC3, RAD21, and HDAC8 (29–31), are involved in chromatin-mediated transcriptional regulation. Moreover, function experiments showed cell lines of individuals with CdLS displayed global transcriptional disturbances rather than cohesion defects (32). Additionally, the cohesion components and chromatin-remodeling proteins strongly interact (33). These studies provided a new perspective on the distinct roles of epigenetic mechanism of phenotypes of CdLS. As a consequence, identifying the causes of patients with features of CdLS by WES is a requisite for accurate genetic diagnosis.
Conclusion
We describe four novel variants in six Chinese patients with features of CdLS caused by variants in four non-cohesion genes (ANKRD11, KMT2D, KDM6A, and UBE2A). In addition, three genes (KMT2D, KDM6A, and UBE2A) causing phenotypes of CdLS have never been reported. Some patients carrying variants in non-cohesion genes could be misdiagnosed as CdLS solely based on the 11-points scale criteria, and WES was necessary to confirm the diagnosis of CdLS. This study expands the spectra of non-cohesion genetic variations in patients with features of CdLS.
Data availability statement
The original contributions presented in this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Fuzhou Children’s Hospital of Fujian Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
HS conducted the data analysis and interpretation and wrote the manuscript. RC contributed to the study design, helped to analyze data, and revise the first draft. Both authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank to all the participants and their families who participated in the clinical trial. We also thank Wei xia for her tolerance and companionship.
Abbreviations
- CdLS
Cornelia de Lange syndrome
- RAD21
RAD21 cohesin complex component
- NIPBL
Nipped-B-like protein
- SMC1A
Structural Maintenance of Chromosomes 1A
- HDAC8
Histone deacetylases
- SMC3
Structural Maintenance of Chromosomes 3
- KS
Kabuki syndrome
- KMT2D
lysine (K)-specific methyltransferase 2D
- KDM6A
lysine (K)-specific methylase 6A
- UBE2A
ubiquitin-conjugase E2A
- ANKRD11
Ankyrin Repeat Domain 11
- KMT2A
lysine (K)-specific methyltransferase 2A
- WES
whole-exome sequencing
- ACMG/AMP
American College of Medical Genetics and Genomics and the Association for Molecular Pathology
- BRD4
Bromodomain containing 4
- AFF4, AF4/FMR2 family
member 4
- EP300
E1A binding protein p300
- SETD5
SET domain containing 5
- SMARCB1
SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily b, member 1
- TAF1, TAF1 RNA polymerase II
TATA box binding protein (TBP)-associated factor
- DDX23
DEAD-box helicase 23
- CSNK1G1, casein kinase 1
gamma 1
- ZMYND11, zinc finger
MYND-type containing 11
- MED13L
Mediator complex subunit 13-like
- PHIP
pleckstrin homology domain interacting protein
- TAF6, TAF6 RNA polymerase II
TATA box binding protein (TBP)-associated factor
- NAA50
N-Terminal Acetyltransferase 50
- CREBBP
CREB binding protein.
Footnotes
Funding
This work was sponsored by the Key Clinical Special Discipline Construction Program of Fuzhou, Fujian, P.R.C (No. 201610191), Clinical Medical Center of Fuzhou, Fujian, P.R.C (No. 201808310), and the Basic and Clinical Research of Rare Disease of Fuzhou, Fujian, P.R.C (ZD-2019-01).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.940294/full#supplementary-material
References
- 1.Selicorni A, Mariani M, Lettieri A, Massa VJG. Cornelia de Lange syndrome: from a disease to a broader spectrum. Genes. (2021) 12:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avagliano L, Parenti I, Grazioli P, Di Fede E, Parodi C, Mariani M, et al. Chromatinopathies: a focus on Cornelia de Lange syndrome. Clin Genet. (2020) 97:3–11. 10.1111/cge.13674 [DOI] [PubMed] [Google Scholar]
- 3.Kline A, Moss J, Selicorni A, Bisgaard A, Deardorff M, Gillett P, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet. (2018) 19:649–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan B, Pehlivan D, Karaca E, Patel N, Charng W, Gambin T, et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. (2015) 125:636–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cucco F, Sarogni P, Rossato S, Alpa M, Patimo A, Latorre A, et al. Pathogenic variants in EP300 and ANKRD11 in patients with phenotypes overlapping Cornelia de Lange syndrome. Am J Med Genet A. (2020) 182:1690–6. 10.1002/ajmg.a.61611 [DOI] [PubMed] [Google Scholar]
- 6.Burns W, Bird LM, Heron D, Keren B, Ramachandra D, Thiffault I, et al. Syndromic neurodevelopmental disorder associated with de novo variants in DDX23. Am J Med Genet A. (2021) 185:2863–72. 10.1002/ajmg.a.62359 [DOI] [PubMed] [Google Scholar]
- 7.Gold NB, Li D, Chassevent A, Kaiser FJ, Parenti I, Strom TM, et al. Heterozygous de novo variants in CSNK1G1 are associated with syndromic developmental delay and autism spectrum disorder. Clin Genet. (2020) 98:571–6. 10.1111/cge.13851 [DOI] [PubMed] [Google Scholar]
- 8.Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet. (2014) 51:659–68. 10.1136/jmedgenet-2014-102573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woods SA, Robinson HB, Kohler LJ, Agamanolis D, Sterbenz G, Khalifa M. Exome sequencing identifies a novel EP300 frame shift mutation in a patient with features that overlap cornelia de lange syndrome. Am J Med Genet A. (2014) 164A:251–8. 10.1002/ajmg.a.36237 [DOI] [PubMed] [Google Scholar]
- 10.O’Rawe JA, Wu Y, Dorfel MJ, Rope AF, Au PY, Parboosingh JS, et al. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am J Hum Genet. (2015) 97:922–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, et al. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet. (2015) 47:338–44. 10.1038/ng.3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parenti I, Gervasini C, Pozojevic J, Graul-Neumann L, Azzollini J, Braunholz D, et al. Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype. Clin Genet. (2016) 89:74–81. 10.1111/cge.12564 [DOI] [PubMed] [Google Scholar]
- 13.Parenti I, Teresa-Rodrigo ME, Pozojevic J, Ruiz Gil S, Bader I, Braunholz D, et al. Mutations in chromatin regulators functionally link Cornelia de Lange syndrome and clinically overlapping phenotypes. Hum Genet. (2017) 136:307–20. 10.1007/s00439-017-1758-y [DOI] [PubMed] [Google Scholar]
- 14.Aoi H, Mizuguchi T, Ceroni J, Kim V, Furquim I, Honjo R, et al. Comprehensive genetic analysis of 57 families with clinically suspected Cornelia de Lange syndrome. J Hum Genet. (2019) 64:967–78. 10.1038/s10038-019-0643-z [DOI] [PubMed] [Google Scholar]
- 15.Tang H, Guo J, Linpeng S, Wu L. Next generation sequencing identified two novel mutations in NIPBL and a frame shift mutation in CREBBP in three Chinese children. Orphanet J Rare Dis. (2019) 14:45. 10.1186/s13023-019-1022-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demir S, Gürkan H, Oz V, Yalçıntepe S, Atlı E. Wiedemann-Steiner syndrome as a differential diagnosis of cornelia de lange syndrome using targeted next-generation sequencing: a case report. Mol Syndromol. (2020) 12:46–51. 10.1159/000511971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parenti I, Mallozzi M, Hüning I, Gervasini C, Kuechler A, Agolini E, et al. ANKRD11 variants: KBG syndrome and beyond. Clin Genet. (2021) 100:187–200. 10.1111/cge.13977 [DOI] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shangguan H, Su C, Ouyang Q, Cao B, Wang J, Gong C, et al. Kabuki syndrome: novel pathogenic variants, new phenotypes and review of literature. Orphanet J Rare Dis. (2019) 14:255. 10.1186/s13023-019-1219-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones W, Dafou D, McEntagart M, Woollard W, Elmslie F, Holder-Espinasse M, et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet. (2012) 91:358–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huisman S, Mulder PA, Redeker E, Bader I, Bisgaard AM, Brooks A, et al. Phenotypes and genotypes in individuals with SMC1A variants. Am J Med Genet A. (2017) 173:2108–25. [DOI] [PubMed] [Google Scholar]
- 22.Li N, Wang Y, Yang Y, Wang P, Huang H, Xiong S, et al. Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients. Orphanet J Rare Dis. (2018) 13:178. 10.1186/s13023-018-0909-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldenberg A, Riccardi F, Tessier A, Pfundt R, Busa T, Cacciagli P, et al. Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11. Am J Med Genet A. (2016) 170:2847–59. [DOI] [PubMed] [Google Scholar]
- 24.Castiglioni S, Di Fede E, Bernardelli C, Lettieri A, Parodi C, Grazioli P, et al. KMT2A: umbrella gene for multiple diseases. Genes. (2022) 13:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bestetti I, Crippa M, Sironi A, Tumiatti F, Masciadri M, Smeland M, et al. Expanding the molecular spectrum of ANKRD11 gene defects in 33 patients with a clinical presentation of KBG syndrome. Int. J. Mol. Sci. (2022) 23:5912. 10.3390/ijms23115912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arslan Satılmış S, Kurt E, Akçay E, Sazci A, Ceylan A. A novel missense mutation in the UBE2A gene causes intellectual disability in the large X-linked family. J Gene Med. (2021) 23:e3307. 10.1002/jgm.3307 [DOI] [PubMed] [Google Scholar]
- 27.de Oliveira J, do Prado P, da Costa S, Sforça M, Canateli C, Ranzani A, et al. Mechanistic insights revealed by a UBE2A mutation linked to intellectual disability. Nat Chem Biol. (2019) 15:62–70. 10.1038/s41589-018-0177-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adam M, Banka S, Bjornsson H, Bodamer O, Chudley A, Harris J, et al. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. (2019) 56:89–95. 10.1136/jmedgenet-2018-105625 [DOI] [PubMed] [Google Scholar]
- 29.Nolen LD, Shelagh B, Morad A, Emily P, Bickmore WA. Regional chromatin decompaction in Cornelia de Lange syndrome associated with NIPBL disruption can be uncoupled from cohesin and CTCF. Hum Mol Genet. (2013) 22:4180–93. 10.1093/hmg/ddt265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deardorff M, Wilde J, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, et al. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet. (2012) 90:1014–27. 10.1016/j.ajhg.2012.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deardorff M, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, et al. HDAC8 mutations in Cornelia de Lange syndrome provide insight into the cohesin acetylation cycle. Nature. (2012) 489:313–7. 10.1038/nature11316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, Li JR, et al. Genome-wide DNA methylation analysis in cohesin mutant human cell lines. Nucleic Acids Res. (2010) 38:5657–71. 10.1093/nar/gkq346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castronovo P, Gervasini C, Cereda A, Masciadri M, Milani D, Russo S, et al. Premature chromatid separation is not a useful diagnostic marker for Cornelia de Lange syndrome. Chromosome Res. (2009) 17:763–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.