

Abstract
The synthesis and subcellular localization of the proteins that comprise the Bacillus subtilis spore are under a variety of complex controls. To better understand these controls, we have identified and characterized a 31-kDa sporulation protein, called TasA, which is secreted into the culture medium early in sporulation and is also incorporated into the spore. TasA synthesis begins approximately 30 min after the onset of sporulation and requires the sporulation transcription factor genes spo0H and spo0A. The first 81 nucleotides of tasA encode a 27-amino-acid sequence that resembles a signal peptide and which is missing from TasA isolated from a sporulating cell lysate. In B. subtilis cells unable to synthesize the signal peptidase SipW, TasA is not secreted, nor is it incorporated into spores. Cells unable to produce SipW produce a 34-kDa form of TasA, consistent with a failure to remove the N-terminal 27 amino acids. In cells engineered to express sipW and tasA during exponential growth, TasA migrates as a 31-kDa species and is secreted into the culture medium. These results indicate that SipW plays a crucial role in the export of TasA out of the cell and its incorporation into spores. Although TasA is dispensable for sporulation under laboratory conditions, we find that TasA has a broad-spectrum antibacterial activity. We discuss the possibility that during the beginning of sporulation as well as later, during germination, TasA inhibits other organisms in the environment, thus conferring a competitive advantage to the spore.
In response to starvation, Bacillus subtilis constructs a highly resistant endospore during a process called sporulation (31). Soon after a cell commits to sporulation, it builds an asymmetrically positioned septum that divides the sporulating cell, called a sporangium, into two unequally sized cells with distinct developmental fates. The smaller chamber, called the forespore, ultimately becomes the spore. The larger compartment, called the mother cell, nurtures the developing spore during its formation. In the next stage of sporulation, the edge of the septum migrates toward the forespore pole of the cell and engulfs the smaller forespore compartment, resulting in a protoplast with two membrane layers that is entirely surrounded by the mother cell. The space between this double layer of membrane becomes the site of assembly of two layers of specialized peptidoglycan called the germ cell wall and the cortex. The last structure to be formed is a proteinacious shell, called the coat, that encircles and protects the spore. In its final act, the mother cell lyses, releasing the now mature spore into the environment. A series of sequentially activated transcription factors ensures that sporulation genes are activated at the proper times and in the correct compartments. Prior to the appearance of the sporulation septum, the transcription factors Spo0A and ςH direct the expression of a large group of genes (10, 12). Once the septum is formed, sigma factors ςF and ςE become active in the forespore and mother cell, respectively (20). After engulfment, the activity of ςF in the forespore is replaced by that of ςG. Similarly, the activity of ςE in the mother cell is replaced by that of ςK.
To better understand the mechanisms that guide sporulation proteins to their correct sites during spore assembly, we began to identify novel spore components and the mechanisms that control their subcellular localization. Here, we describe an abundant component of mature spores called TasA. We found that TasA synthesis occurs prior to the formation of the sporulation septum and requires the transcription factors Spo0A and ςH. Immediately after synthesis, TasA is secreted into the culture medium. Secretion appears to require the removal of the N-terminal 27 amino acids (comprising an apparent signal peptide), an event that depends on the signal peptidase SipW (32), which is encoded immediately upstream of tasA. Strikingly, the incorporation of TasA into the spore also depends on SipW. We tentatively propose that after the sporulation septum is made, the same secretory system that directs TasA into the medium also secretes TasA into the space between the septal membranes, where it becomes trapped, ultimately becoming associated with the spore peptidoglycan and possibly with the coat. These results suggest that maturation of at least one secreted sporulation protein requires SipW. SipW is not absolutely required for sporulation, since deletion of sipW (or tasA) does not have a strong effect on spore formation. However, in plate diffusion assays, TasA exhibits an antibacterial activity against a variety of gram-positive and gram-negative bacteria. Therefore, we chose the name tasA to reflect our finding that this protein is a translocation-dependent antimicrobial spore component. We speculate that this activity provides the sporulating cell with a competitive advantage during the early stages of development as well as providing protection during and after germination.
MATERIALS AND METHODS
Strains, plasmids, and recombinant DNA procedures.
Strains, plasmids, and primers for the PCR used in this study are listed in Tables 1 and 2. PCR products generated in this study are illustrated in Fig. 2B. All B. subtilis strains are congenic with the wild-type strain PY79 (34), except for RL102, which is congenic with strain 168 (Table 1). We used Luria-Bertani medium (LB) for the routine growth of B. subtilis and Escherichia coli strains, Mueller-Hinton medium for the growth of animal-pathogenic bacteria, and King’s B medium (16) for the growth of plant-pathogenic bacteria. We performed recombinant DNA techniques as described previously (28) and used E. coli DH5α as the host strain for molecular cloning. We performed PCR as specified by the instructions provided with the GeneAmp PCR core reagents (Perkin-Elmer).
TABLE 1.
Strains and plasmids used in this study
| Strains or plasmids | Genotype or description | Source or reference |
|---|---|---|
| B. subtilis | ||
| PY79 | Wild type | 34 |
| AD17 | gerE36 | 5 |
| AD28 | cotEΔ::cat | 8 |
| AD142 | cotEΔ::cat gerE36 | This study |
| RL102 | spo0HΔHindIII-EcoRI::cat trpC2 (168 background) | Losick laboratory collection |
| RL891 | spo0AΔ::erm | 21 |
| SC1159 | spoIIAC1 | 3 |
| PM806 | spoIIGAΔ17 | 21 |
| SC500 | spoIIIGΔ1 | 4 |
| SAB50 | spoIVFΔAB::cat | Losick laboratory collection |
| AGS115 | tasAΩpAGS08 | This study |
| AGS127 | gerE36 tasAΩpAGS08 | This study |
| AGS157 |
sipWΔ::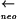
|
This study |
| AGS175 |
yqxM::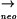
|
This study |
| AGS185 |
cotEΔ::cat gerE36 yqxM::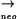
|
This study |
| AGS186 |
cotEΔ::cat gerE36 sipWΔ::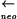
|
This study |
| AGS200 | spoIIBΔ::erm tasAΩpAGS08 | This study |
| AGS202 | spoVG::Tn917ΩHU265 tasAΩpAGS08 | This study |
| AGS207 | tasAΔ::spc | This study |
| AGS210 | cotEΔ::cat gerE36 tasAΔ::spc | This study |
| AGS215 |
cotEΔ::cat gerE36 sipWΔ::
|
This study |
| AGS219 | yqxM::neo tasAΩpAGS08 | This study |
| E. coli | ||
| DH5α | Cloning host | Laboratory collection |
| BL21(DE3) | Overproduction host | Laboratory collection |
| Plasmids | ||
| pBEST501 | Harbors the neomycin resistance gene | 14 |
| pJL74 | Harbors the spectinomycin resistance gene | 18 |
| pSL1180 | Harbors extensive multiple cloning site | Pharmacia |
| pET24b | Overexpression vector | Novagen |
TABLE 2.
Primers used in this study
| Primer | Sequencea | Restriction enzymea |
|---|---|---|
| OL58 | 5′ AAAAAAAACATATGAAGGACAGCACCATGTCTAC 3′ | NdeI |
| OL59 | 5′ AAAAACTCGAGAAAAAGAGGAGTAGTGCCTG 3′ | XhoI |
| OL72 | 5′ AAAAAAACTCGAGAGCGGTGTTTCTTCTGCGTG 3′ | XhoI |
| OL74 | 5′ AAAAAAACTCGAGTTGTGCTGACACTTGTCGTG 3′ | XhoI |
| OL75 | 5′ AAAAAAATCGATCCGAATACGATGGTCAATTAGATAG 3′ | ClaI |
| OL76 | 5′ AAAAAAACTCGAGAACGGCAGTACATTTGAGTGG 3′ | XhoI |
| OL77 | 5′ AAAAAAATCGATCCTCCAACTAAAGCTAATCCTAGTG 3′ | ClaI |
| OL91 | 5′ AAAAAAAAACTCGAGCTGATCAGCTTCATTGCT 3′ | XhoI |
| OL93 | 5′ AAAAAAACTCGAGTTTATCCTCGCTATGCGC 3′ | XhoI |
| OL94 | 5′ AAAAAAAAACATATGGCATTTAACGACATTAAA 3′ | NdeI |
| OL99 | 5′ AAAAACTCGAGCATATGTTTCGATTGTTTCAC 3′ | NdeI |
The restriction endonuclease site in each oligonucleotide is underlined, and the enzyme that cuts it is listed at the right.
FIG. 2.

(A) N terminus of TasA. The putative signal peptide and the N terminus of the mature protein are indicated. Sequence features consistent with a signal peptide are indicated: three positively charged lysines (bold) close to the N terminus, followed by a stretch of hydrophobic amino acids (underlined) and a glycine residue (italic and bold) 5 amino acids before the possible signal peptidase cleavage site. The two alanines (one at the C terminus of the putative signal peptide, and the other at the N terminus of the mature protein), consistent with a signal peptidase cleavage site, are bold. The sequenced region of the mature protein is heavily underlined. (B) tasA locus. yqxM, sipW, and tasA are indicated by boxes. The solid portions of the boxes encode putative signal peptides. The arrows in the boxes indicate the presumed direction of transcription. The brackets represent the PCR products used in constructing mutations in the locus and for overexpression. The oligonucleotides (Table 2) used to generate each PCR product are indicated. The triangles indicate the disruptions of yqxM and sipW by insertion of the neomycin resistance gene cassette. The arrows above the triangles indicate the directions of transcription of the neomycin resistance genes. Restriction enzyme recognition sites are indicated: B2, BstEII; RI, EcoRI; Ha3, HaeIII; H3, HindIII; NI, NdeI.
Manipulation of B. subtilis.
Genetic manipulation of B. subtilis was carried out as described previously (6). Strains bearing mutations in tasA, sipW, or yqxM were generated by transformation of competent cells of strain AD17, AD142, or PY79 with a DNA construct bearing the appropriate mutation, described below. We induced sporulation by nutrient exhaustion (29). We prepared free spores after 48 h of culturing, by which time >95% of the cells were spores. We performed the tetrazolium assay as described previously (13).
Preparation and electrophoresis of spore extracts.
We harvested spores by centrifugation, resuspended them in TBS (25 mM Tris-HCl [pH 7.5], 135 mM NaCl, 2.7 mM KCl), and incubated the suspension at 75°C for 10 min to reduce spore-associated protease activity. After heat treatment, we incubated the spores on ice for 5 min, pelleted them by centrifugation, washed the spore pellet once in 0.5 M NaCl, centrifuged the spores again, and then froze the pellet until use. For electrophoresis, we loaded an amount of extract corresponding to 2 ml of the original sporulating culture into each well of a sodium dodecyl sulfate (SDS)-polyacrylamide gel.
Production of cotEΔ::cat sporangial lysate.
We harvested cotEΔ::cat sporangia (from AD28) by centrifugation after 5 h of sporulation and washed the cells with TBS. We resuspended the cells in TBS, incubated them with lysozyme (2 mg/ml) for 5 min on ice, and then lysed them by sonication with a Fisher Dismembrator. We centrifuged the lysate for 10 min at 10,000 × g, filtered the supernatant through a 0.45-μm syringe filter, and froze it at −20°C until use. We then thawed the supernatant on ice and centrifuged it for 15 min at 16,000 × g. We resuspended the pellet in loading buffer and analyzed it by SDS-polyacrylamide gel electrophoresis (PAGE).
Production of cell lysates for Western blot analysis.
We harvested vegetative cells (at an optical density at 600 nm [OD600] of 0.7 to 0.9) and sporangia by centrifugation, washed them once in TBS, and froze the pellet until use. To lyse the cells, we resuspended the pellet in 75 μl of GTE (25 mM Tris-HCl [pH 7.5], 50 mM glucose, 10 mM EDTA), added lysozyme to a final concentration of 2 mg/ml, and incubated the suspension at room temperature for 5 min. We then added 25 μl of 4× loading buffer including dithiothreitol (28), and immediately boiled the samples for 5 min. The cells appeared completely lysed by light microscopy. For Western blot analysis, we centrifuged the samples for 5 min and added an amount of lysate corresponding to 0.4 OD600 unit of the original culture to each well of an SDS-polyacrylamide gel.
SDS-PAGE and Western blotting.
SDS-PAGE and Western blotting were performed as described previously (28). We transferred the proteins to a polyvinylidene difluoride membrane (Immobilon; Millipore) at constant voltage of 35 V for at least 4 h. We used the anti-TasA antiserum at a 1:5,000 to 1:10,000 dilution. After treatment with anti-TasA antibodies, we incubated the membrane with alkaline-phosphatase-conjugated secondary antibodies (Promega) and then with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (Gibco-BRL) as specified by the manufacturers.
Polyclonal antiserum.
To prepare TasA as an immunogen, we thawed an aliquot of sporangial lysate (see above) on ice and centrifuged it for 15 min at 14,000 × g. We resuspended the pellet in loading buffer and fractionated it by SDS-PAGE. We isolated the 31-kDa band from the polyacrylamide gel (11) and injected approximately 100 μg of this material intramuscularly and subcutaneously into a rabbit. We prepared antiserum from blood obtained 23 days after injection.
Amino acid sequencing.
We subjected a sporangial lysate (see above) to SDS-PAGE, transferred the protein in the gel to a polyvinylidene difluoride membrane (Immobilon) in CAPS buffer (22), stained the membrane with Coomassie brilliant blue to identify the 31-kDa band, and then destained the membrane. We then cut out a slice of the membrane containing the 31-kDa band and had Edman-degradation sequencing performed on the membrane-bound protein by B.-S. Lee at the Protein Research Laboratory, University of Illinois, Chicago, Ill.
Protein isolation from culture supernatants.
Sporulating cells were harvested after 2 h of sporulation unless otherwise noted, and vegetatively growing cells were collected at an OD600 of 0.7 to 0.9. We centrifuged the cell suspension for 5 min at 16,000 × g and filtered the supernatant through a 0.45-μm syringe filter. To precipitate proteins, we added 1 volume of supernatant to 9 volumes of absolute ethanol and incubated the mixture at −70°C overnight. We then centrifuged the sample at 14,000 × g for 10 min at 4°C, washed the pellet once with 80% ethanol, and completely dried it in a Speed-Vac concentrator (Savant) for 20 min. We electrophoresed the precipitated supernatant of 200 μl of culture in each well of a SDS-polyacrylamide gel.
Generation of deletion mutations in tasA.
We generated two deletions of tasA: one by a single-reciprocal integration (Campbell type) into the genome of a plasmid bearing an internal region of tasA (by using pAGS08) and the other by marker replacement of part of the genomic copy of tasA with a spectinomycin resistance gene (by using pAGS18b). We confirmed integration by PCR (data not shown).
First, we placed the spectinomycin resistance gene of pJL74 (in a BamHI-EcoRV fragment) into pSL1180 (Pharmacia), liberated it by digestion with SphI and BamHI, and cloned it into pET24b (Novagen) cut with SphI and BglII, resulting in pAGS05. We PCR amplified the chromosomal locus encompassing tasA with primers OL58 and OL59 (see Fig. 2B), cloned it into NdeI- and XhoI-digested pAGS05, and cut the resulting plasmid with EcoRI and HindIII to release a fragment of the tasA open reading frame (see Fig. 2B). We cloned this fragment between the EcoRI and HindIII sites of pJL74. The resulting plasmid, pAGS08, was used to transform competent cells. Transformants were selected for resistance to spectinomycin.
To generate pAGS18b, we amplified the tasA locus by PCR with primers OL74 and OL75 (see Fig. 2B), digested the PCR product with XhoI and HaeIII, and cloned it between the XhoI and EcoRV sites in pET24b. We digested this plasmid with EcoRI and HindIII, used the Klenow fragment to generate blunt termini, and cloned the spectinomycin cassette (inserted into pSL1180 and then liberated with EcoRV and SmaI) into this vector. The resulting plasmid, pAGS18b, was linearized with PvuI and used to transform competent cells to spectinomycin resistance.
Generation of deletion mutations in sipW.
First, we amplified the sipW gene by PCR with primers OL76 and OL77 (see Fig. 2B). We digested the product with XhoI and ClaI and cloned it into XhoI- and ClaI-digested pJL74. We removed a 141-bp region of the sipW open reading frame with NdeI and BstEII, generated blunt termini with Klenow fragment, and replaced the region with a neomycin resistance gene (recovered from pBEST501 [14] with SmaI). This resulted in pAGS17-1, in which the orientation of the neomycin gene-bearing insert is opposite to the orientation of sipW, and in pAGS17-2, in which the orientation of the drug resistance cassette is identical to the orientation of sipW. We linearized these plasmids with PvuI, used them to transform competent cells, and selected transformants with 3.5 μg of neomycin per ml. We used PCR amplification to confirm genomic integration by marker replacement and the orientations of the drug resistance cassette (data not shown).
Generation of an insertional mutation in yqxM.
First, we used PCR and oligonucleotides OL72 and OL77 to amplify yqxM and sipW (see Fig. 2B), digested the PCR product with XhoI and BstEII, and cloned it into similarly digested pET24b to generate pAGS21. We cut pAGS21 with HindIII, filled the overhangs with Klenow fragment, and inserted the neomycin resistance gene (see above), generating pAGS22. By PCR, we determined that the orientation of the neomycin resistance gene is the same as the putative direction of transcription of yqxM (data not shown). We amplified the yqxM open reading frame sequences flanking the inserted neomycin gene with oligonucleotides OL72 and OL77 and transformed competent cells with the PCR product. We confirmed by PCR that marker replacement had occurred (data not shown).
Overproduction of YqxM and TasA.
To insert yqxM or tasA into pAGS05 (described above), we performed PCR with primer pairs OL99 and OL91 or OL93 and OL94, respectively (see Fig. 2B). This amplified either the entire yqxM coding sequence or a portion of tasA corresponding to amino acid residues 28 through 260, resulting in a truncated version of tasA missing the sequence encoding the putative signal peptide. We digested the PCR products with XhoI and NdeI and ligated them to similarly digested pAGS05. This resulted in pAGS41, harboring the in-frame insertion of the yqxM open reading frame, or pAGS37, bearing the tasA fragment downstream of a translational start codon and upstream of a gene segment encoding six histidines. We then transformed E. coli BL21(DE3) with pAGS37 or pAGS41 as well as with the empty plasmid (pAGS05) and used these strains to inoculate 5 ml of LB supplemented with 30 μg of kanamycin per ml and incubated the cultures with shaking at 30°C overnight. The next day, we inoculated 50 ml of LB, supplemented with 30 μg of kanamycin per ml, with 2 ml of the overnight culture and grew the cells at 37°C. When the OD600 of the cultures reached 0.5 to 0.8, we added isopropyl-β-d-thiogalactopyranoside (IPTG; Gibco) to a final concentration of 1 mM. We continued incubation for 6 to 7 h, harvested the cell pellets by centrifugation at 6,000 × g for 10 min at 4°C, and then resuspended the pellets in 10 ml of TEN (10 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA). We measured the OD600 of the cell suspensions (so that equivalent amounts of overproducing lysate and control lysate could be compared in the antibacterial activity test), centrifuged the sample again, and froze the cell pellet at −20°C until use. For the antibacterial activity assay (see below), we resuspended equivalent OD600 units of the overproducing cells and cells bearing the empty vector in approximately 10 ml of TENT (TEN, 0.25% Triton X-100) and passed the samples twice through a French press at 20,000 lb/in2 to break the cells. We found that more than 99% of the cells had lysed under these conditions as judged by light microscopy. We used SDS-PAGE to verify the presence of overproduced YqxM or TasA in these lysates (data not shown).
Testing for antibacterial activity.
Animal-pathogenic bacteria and guidelines on testing were kindly provided by R. Carey, Clinical Microbiology Laboratory, Loyola University Medical Center. Plant-pathogenic bacteria were kindly provided by J. Greenberg, University of Chicago. To test bacteria for sensitivity to TasA, we first spread bacteria on agar medium plates. For plant-pathogenic strains (see Table 3), we resuspended a colony (grown on solid King’s B medium) in liquid King’s B medium and spread cells on solid King’s B medium with a cotton swab. For animal-pathogenic strains, we resuspended 0.5 MacFarland unit of culture in saline and streaked the cells on Mueller-Hinton plates with a cotton swab. Next, we used a 12-mm-diameter cork borer to punch two holes into the plates and removed the agar discs. To each well, we added 400 μl of crude E. coli lysates, prepared from strains harboring either the tasA overexpression plasmid (pAGS37) or the empty vector (pAGS05), and then incubated the plates. We grew the animal pathogens (see Table 3) at 37°C (with the exception of Micrococcus luteus, which we grew at 25°C) and the plant pathogens at 30°C. After incubation for 16 to 24 h, the diameters of the zones of clearing were measured. Each experiment was carried out at least twice, with similar results.
TABLE 3.
Inhibition of bacteria by TasA
| Bacterial strain | Response to TasA | Zone diam (mm)
|
|
|---|---|---|---|
| With TasA | Without TasA | ||
| Plant pathogensa | |||
| Agrobacterium tumefaciens GV3101 | Sensitive | 22 | 12 |
| Erwinia strain A1750 | Sensitive | 18 | 12 |
| Erwinia amylovora EG321 | Sensitive | 33/21b | 12 |
| Erwinia chrysanthemi ACH150 | Sensitive | 17 | 12 |
| Klebsiella pneumoniae A95 | Sensitive | 16 | 12 |
| Pseudomonas aeruginosa | Resistant | 12 | 12 |
| Pseudomonas aurofaciens | Sensitive | 27 | 12 |
| Pseudomonas putida | Sensitive | 17 | 12 |
| Pseudomonas syringae pv. syringae 61 | Sensitive | 18 | 12 |
| Pseudomonas syringae pv. tomato DC3000 | Sensitive | 18 | 12 |
| Animal pathogensc | |||
| Enterococcus faecalis ATCC 29212 | Sensitive | 18 | 12 |
| Enterococcus faecalis ATCC 52199d | Resistant | 12 | 12 |
| Escherichia coli ATCC 25922 | Sensitive | 22 | 12 |
| Escherichia coli ATCC 35218 | Sensitive | 21 | 12 |
| Klebsiella pneumoniae ATCC 13883 | Sensitive | 33 | 23 |
| Micrococcus luteus ATCC 4698 | Sensitive | 33 | 26 |
| Pseudomonas aeruginosa ATCC 27853 | Resistant | 12 | 12 |
| Staphylococcus aureus ATCC 29213 | Sensitive | 22 | 18 |
| Staphylococcus aureus ATCC 33591 | Resistant | 12 | 12 |
| Staphylococcus epidermis ATCC 12228 | Sensitive | 28 | 16 |
| Streptococcus bovis ATCC 9809 | Resistant | 12 | 12 |
| Clinical isolatesc | |||
| Enterobacter aerogenes T44224-1 | Resistant | 12 | 12 |
| Enterobacter aerogenes W31851-1 | Sensitive | 21 | 16 |
| Enterobacter cloacae F23604-1 | Sensitive | 19 | 16 |
| Escherichia coli 773813-2 | Sensitive | 27 | 17 |
| Escherichia coli H39494-1 | Sensitive | 17 | 12 |
| Escherichia coli T34311-1 | Sensitive | 14 | 12 |
| Klebsiella pneumoniae F23662-1 | Sensitive | 22 | 17 |
| Klebsiella pneumoniae F23673-3 | Sensitive | 19 | 14 |
| Pseudomonas aeruginosa F25235-2 | Resistant | 12 | 12 |
| Pseudomonas aeruginosa S77551-1 | Resistant | 12 | 12 |
| Otherse | |||
| Escherichia coli BL21(DE3) | Sensitive | 23 | 12 |
| Bacillus subtilis PY79 | Sensitive | 29 | 24 |
Tested on King’s B plates.
This strain exhibited an outer (33-mm) and an inner (21-mm) zone of inhibition.
Tested on Mueller-Hinton plates.
Vancomycin resistant.
Tested on LB plates.
To test for TasA activity during coculturing, we inoculated 2.5 ml of DSM medium to an OD600 of 0.1 with AGS175 or AGS157 and to an OD600 of 0.005 with either Enterobacter aerogenes (W31851-1) or Pseudomonas aeruginosa (S77551-1). After 16 h, we removed an aliquot of culture and plated it on MLS plates (6) (which killed B. subtilis but not the other strains) to determine the number of CFU. To calculate the percent survival, we divided the number of survivors after coculturing with AGS175 by the number of survivors after coculturing with AGS157 and multiplying by 100. Each percentage is the average of the results of two experiments. To measure the percent survival of B. subtilis, we incubated the culture for 48 h (so that spores would be produced), heat treated it to kill vegetative cells, and determined the number of CFU on LB plates.
RESULTS
Identification of a spore-associated protein and its gene.
To identify novel spore-associated proteins, we carried out SDS-PAGE analysis of proteins extracted from spores by boiling in the presence of SDS and dithiothreitol. To focus our search toward novel proteins, we analyzed spores missing most or all of the coat and hence missing most of the already characterized abundant coat proteins. We examined spores bearing a mutation in gerE (from AD17, in which the inner coat and much of the outer coat are missing [8, 23]), in cotE (from AD28, in which the outer coat is absent [35]), or in both genes (from AD142, in which both coat layers are lacking [8]). We identified a 31-kDa species that is present in large amounts in spores from either a cotEΔ::cat gerE36 or a gerE36 strain (Fig. 1A, lanes 1 and 2). We detected much smaller amounts of this protein in spores from a cotEΔ::cat strain or from wild-type spores (lanes 3 and 4). We also detected a 31-kDa band in a lysate of cotEΔ::cat sporangia, harvested at stage 5 of sporulation (when cortex formation was complete), from which spores and cell debris had been removed by centrifugation (Fig. 1B), indicating the presence of a non-spore-associated protein with a similar molecular mass.
FIG. 1.

SDS-PAGE analysis (15% polyacrylamide gel) of proteins extracted from wild-type and mutant spores. (A) Lanes: 1, cotEΔ::cat gerE36; 2, gerE36; 3, cotEΔ::cat; 4, wild type (WT); 5, empty lane; 6, gerE36 tasAΩpAGS08. (B) Lysate of cotEΔ::cat sporangia (sprng) 5 h after initiation of sporulation. The arrowhead indicates the 40-kDa species. The arrows indicate the position of TasA. Molecular masses are indicated in kilodaltons.
To identify the gene that encodes the 31-kDa protein, we isolated the 31-kDa protein from a sporangial lysate (Fig. 1B), carried out Edman degradation sequencing, and determined the sequence of the nine N-terminal amino acids to be NH2-AFNDIKSKD-COOH (heavily underlined in Fig. 2A). A search of the B. subtilis genome (17) revealed a perfect match of this amino acid sequence to residues 28 to 36 encoded by the putative open reading frame cotN (referred to as yqhF in previous versions of the database), encoding a hypothetical protein of 28,153.9 Da (composed of 261 amino acid residues), which we renamed tasA. tasA is located downstream from two other open reading frames, sipW and yqxM (Fig. 2B). This locus is located at 218.1° on the chromosome, between the comC locus (which encodes proteins required for competence [9]) and the sin locus (encoding proteins that govern alternate developmental pathways [1]). tasA and yqxM do not exhibit significant homology to other genes in the databases. sipW, however, is known to encode a signal peptidase (32).
To confirm that tasA encodes the 31-kDa protein and to determine if the spore-associated and non-spore-associated proteins are products of the same gene, we constructed a tasA deletion strain and generated a rabbit polyclonal antiserum against the non-spore-associated 31-kDa protein (Fig. 1B). In Western blot analysis, this antiserum reacted with both the 31-kDa band extracted from spores (Fig. 3A, lanes 1 through 4) and the band from a lysate cleared of spores by centrifugation (Fig. 3B). We did not detect the 31-kDa band when we used the preimmune serum (data not shown) or when we applied the postimmune serum to a spore extract prepared from a gerE36 tasAΩpAGS08 strain (AGS127) (Fig. 3A, lane 6). Sporangia and, to a much lesser extent, spores contained a cross-reacting species of about 55 kDa (Fig. 3B) that was present in an extract of tasA cells and therefore was not due to TasA (data not shown). We infer that the spore-associated and non-spore-associated 31-kDa proteins are products of a single gene. However, the two proteins are probably not biochemically identical, since we were unable to sequence the spore-associated protein, suggesting that the spore-associated species might possess a posttranslational modification at its N terminus that interferes with Edman degradation sequencing.
FIG. 3.

Western blot analysis (SDS–15% polyacrylamide gel) of TasA extracted from spores or a sporangial lysate. (A) Extracts of spores. Lanes: 1, cotEΔ::cat gerE36; 2, gerE36; 3, cotEΔ::cat; 4, wild type (WT); 5, empty lane; 6, gerE36 tasAΩpAGS08 (6). (B) Lysate of cotEΔ::cat sporangia (sprng) 5 h after initiation of sporulation. The arrows indicate the positions of TasA. Molecular masses are indicated in kilodaltons.
We also used SDS-PAGE to analyze proteins extracted from tasA spores. In otherwise wild-type spores, the polypeptide profile was indistinguishable from that of the wild type (data not shown). Because TasA is more readily detected in a gerE36 background, we analyzed gerE36 tasAΩpAGS08 spores. As expected, these spores lack the 31-kDa band (Fig. 1A, lane 6). In addition, several bands seen in the wild type were not apparent in the tasA mutant and a number of previously undetected bands were present, including a band of unknown identity of approximately 40 kDa (Fig. 1A, compare lanes 2 and 6).
Timing of TasA synthesis and transcription factor dependency.
To determine when TasA is synthesized, we measured the steady-state levels of TasA by Western blot analysis in cotEΔ::cat gerE36 cells (to permit easier detection of TasA) cultured in Difco sporulation medium. We did not detect any TasA until about 30 min after the initiation of sporulation (Fig. 4, lanes 2 to 4), and the signal reached a maximum at about 1.5 h (lane 6). TasA remained at approximately this level until at least 12 h after initiation (Fig. 4 and data not shown). We also investigated whether known sporulation transcription factors are required for the appearance of TasA. To do this, we prepared lysates of sporulating cells, bearing a mutation in spo0A or one of the five known sporulation sigma factor genes, at a time in development when the transcription factor would be active and tested these lysates for the presence of TasA by Western blot analysis. We examined strains mutated in the genes encoding Spo0A (from RL891, after 1 h of sporulation), ςH (from RL102, after 1 h), ςF (SC1159, after 2.5 h), ςE (from PM806, after 2.5 h), or ςG (from SC500, after 4.5 h) or in an operon required for the activation of ςK (spoIVF) (from SAB50, after 4.5 h). We detected TasA in all lysates (Fig. 5, lanes 3 through 6) except those from spo0A::erm or spo0HΔHindIII-EcoRI::cat cells (lanes 1 and 2, respectively).
FIG. 4.

Western blot analysis (SDS–15% polyacrylamide gel) of TasA steady-state levels. Lane 1 contains an extract of cotEΔ::cat gerE36 spores (to indicate the position of TasA). Extracts were prepared 30 min before sporulation (lane 2) and at 30-min intervals until the second hour of sporulation (lanes 3 to 7) and then at 1-h intervals (lanes 8 and 9). The arrow indicates the position of TasA. The numbers above the blot indicate the time at which the samples were prepared, relative to the beginning of sporulation. Molecular masses are indicated in kilodaltons.
FIG. 5.

Western blot analysis (SDS–15% polyacrylamide gel) of TasA in transcription factor mutant strains. Lanes 1 to 6 contain extracts from sporangia with the mutations spo0AΔ::erm (lane 1), spo0HΔHindIII-EcoRI::cat trpC2 (lane 2), spoIIAC1 (lane 3), spoIIGAΔ17 (lane 4), spoIIIGΔ1 (lane 5), or spoIVFΔAB::cat (lane 6). The transcription factors which are inactive in each strain are indicated above each lane. The arrow indicates the position of TasA. Molecular masses are indicated in kilodaltons.
Role of secretion in the localization of TasA.
The sequence of the N terminus of TasA (AFNDIKSKD) does not correspond to the N terminus of the predicted product of tasA but, rather, starts at amino acid residue 28 (Fig. 2A). This discrepancy would be accounted for if TasA were posttranslationally processed. We found that the 27 amino-terminal residues of the predicted gene product contain structural features characteristic of a B. subtilis signal peptide. These include three positively charged amino acids toward the N terminus of the peptide, followed by a core region composed primarily of hydrophobic amino acids and a glycine residue 5 amino acids before the putative signal peptidase cleavage site (30) (Fig. 2A). Furthermore, the last residue of the putative signal peptide and the first residue of the mature protein are alanines, also characteristic of a B. subtilis signal peptidase cleavage site (24). These observations point to the possibility that TasA is secreted by a signal peptidase-dependent mechanism (25). We suspect that both the spore-associated and the non-spore-associated forms of TasA are similarly processed, since they both migrated as 31-kDa species in SDS-PAGE (Fig. 1A, lanes 1 and 2; Fig. 1B).
If TasA is processed and secreted, it should be translocated across the cytoplasmic membrane and perhaps into the culture supernatant. To test this, we centrifuged a sporulating culture 2 h after the onset of sporulation (when TasA is present [Fig. 4, lane 7]), concentrated the proteins in the supernatant by ethanol precipitation, and then probed for TasA by Western blot analysis. We identified TasA in supernatants of sporangia of wild-type and cotEΔ::cat gerE36 strains (Fig. 6, lanes 3 and 7; lanes 2 and 6 show the position of TasA in sporangial lysates of the two strains, for comparison). We did not detect any signal in culture supernatants or sporangia of tasAΔ::spc (from AGS207) or cotEΔ::cat gerE36 tasAΔ::spc cells (from AGS210) (lanes 4, 5, 8, and 9).
FIG. 6.

Western blot (SDS–15% polyacrylamide gel) of TasA in sporangial lysates and culture supernatants. Lane 1 contains an extract of cotEΔ::cat gerE36 spores (to indicate the position of TasA). Lanes 2, 4, 6, and 8 contain lysates of sporangia of a wild-type (WT) (lane 2), tasAΔ::spc (lane 4), cotEΔ::cat gerE36 (lane 6), or cotEΔ::cat gerE36 tasAΔ::spc (lane 8) strain. Lanes 3, 5, 7, and 9 contain preparations of supernatants of wild-type (lane 3), tasAΔ::spc (lane 5), cotEΔ::cat gerE36 (lane 7), or cotEΔ::cat gerE36 tasAΔ::spc (lane 9) cells. The arrow indicates the position of TasA. sprng, sporangia; sup, supernatant. Molecular masses are indicated in kilodaltons.
If mature TasA is the result of signal peptide cleavage, there should be a signal peptidase that carries out this proteolytic event. One candidate is the product of sipW (32, 33), located immediately upstream of tasA (Fig. 2B). To test the involvement of sipW in TasA maturation, we generated a strain in which sipW was rendered nonfunctional, in a manner that would not prevent tasA expression. To build this strain, we deleted a portion of the sipW open reading frame and inserted in its place a neomycin resistance gene (14) oriented such that the constitutively active repU promoter of the resistance gene would direct tasA gene expression during sporulation as well as vegetative growth (Fig. 2B). We then used Western blotting to analyze lysates of vegetative cells, sporulating cells, or spores bearing these mutations as well as cotEΔ::cat and gerE36 (to permit easier detection of TasA). We detected a 34-kDa form of TasA in lysates of whole sporangia (Fig. 7A, lane 4). This molecular mass is consistent with that of an immature pre-form of TasA. We did not find any TasA in extracts of spores (lane 6) or in culture supernatants of sporulating cells bearing this construct (lane 5). We also detected the 34-kDa band in vegetative-cell lysates (lane 2), presumably as a result of the constitutively active repU promoter, but not in vegetative-cell supernatants from this strain (lane 3). Additionally, we constructed a strain in which the neomycin resistance gene was oriented away from tasA (Fig. 2B). In these cells, we did not detect TasA, in either spores or sporulating cells (Fig. 7B, lanes 1 and 2). These results indicate that the maturation of TasA requires SipW. Furthermore, they suggest that the association of TasA with the spore and its export into the culture medium depend on a secretory event that requires SipW. We also found a band representing a protein of 29 kDa in lysates of vegetative cells and sporangia (Fig. 7A, lanes 2 and 4) but not in spores (lane 6) or supernatants (lanes 3 and 5). We assume that this signal corresponds to a degradation product of TasA.
FIG. 7.

Western blot analysis (SDS–22% polyacrylamide gel) of TasA in the lysate of sporangia, vegetatively growing cells, and extracts of spores of cotEΔ::cat gerE36 sipWΔ::neo strains. (A) Lane 1 contains an extract of cotEΔ::cat gerE36 spores (to indicate the position of TasA). Lanes 2 through 6 contain preparations of vegetative cells (lane 2), vegetative-cell supernatant (lane 3), sporangia (lane 4), sporangial supernatant (lane 5), or spores (lane 6) from AGS215, in which the direction of transcription of the neomycin resistance gene is aligned with that of sipW. (B) Lanes 1 and 2 contain extracts of spores (lane 1) or sporangia (lane 2) of AGS186, bearing the neomycin resistance gene with the opposite orientation relative to the direction of transcription of sipW. Lane 3 contains an extract of cotEΔ::cat gerE36 spores (to indicate the position of TasA). The arrows at the side of the blot indicate the positions of TasA and pre-TasA. The arrowhead indicates the position of the putative breakdown product of TasA. sprng, sporangia; sup, supernatant; veg, vegetative. Molecular masses are indicated in kilodaltons.
If the maturation of TasA depends on SipW, the placement of the neomycin resistance gene upstream of sipW should permit the maturation and translocation of TasA. To test this, we inserted the neomycin resistance gene into yqxM, oriented to direct the expression of both sipW and tasA (Fig. 2B). We then used Western blot analysis to determine if cells of this strain produced mature TasA during sporulation and vegetative growth. Using the cotEΔ::cat gerE36 background, we identified a band corresponding to mature TasA (31 kDa) in lysates of sporangia (Fig. 8, lane 4), extracts of spores (lane 6), and culture supernatants of sporulating cells after 2 h (lane 5) and 48 h (lane 7) of sporulation. We did not detect the immature form of TasA. We also detected mature TasA in lysates and culture supernatants of vegetative cells (lanes 2 and 3). This suggests that the expression of sipW and tasA during vegetative growth is sufficient for the removal of the signal peptide and the subsequent incorporation of TasA into the spore and its secretion into the culture medium.
FIG. 8.

Western blot analysis (SDS–22% polyacrylamide gel) of TasA in preparations of sporangia, exponentially growing cells, spores, and supernatant of a cotEΔ::cat gerE36 yqxM::neo strain (from AGS185). Lanes 1 and 8 contain extracts of cotEΔ::cat gerE36 spores (to indicate the position of TasA). Lanes 2 and 3 contain extracts (lane 2) and supernatant (lane 3) of vegetative cells. Lanes 4 and 5 contain extracts of sporangia (lane 4) and supernatant of sporulating cells (lane 5) at 2 h after initiation of sporulation. Lanes 6 and 7 contain extracts of spores (lane 6) and supernatants of sporulating cultures (lane 7) at 48 h after initiation of sporulation. Lane 9 contains an extract of cotEΔ::cat gerE36 sipWΔ::neo sporangia (to indicate the position of pre-TasA). The arrows at the side of the blot indicate the positions of TasA and pre-TasA. The arrowhead indicates the position of the putative breakdown product of TasA. sprng, sporangia; sup, supernatant; veg, vegetative. Molecular masses are indicated in kilodaltons.
Role of TasA.
To learn the function of TasA, we examined mutant cells and spores by microscopy and tested their resistance properties. tasAΔ::spc cells showed no significant defect in vegetative growth and sporulated at a normal frequency as judged by light microscopy (data not shown). The spores also appeared normal by both light and electron microscopy and had wild-type levels of resistance to heat, chloroform, and lysozyme, indicating no readily detectable role for tasA in spore resistance (data not shown). However, these spores did exhibit a slight germination defect as judged by the tetrazolium overlay assay (data not shown), which measures the resumption of metabolism after germination. Wild-type spores produce a red color in this assay, whereas spores that are severely deficient in germination, e.g., gerE36, generate a white color. tasAΔ::spc spores produced a red color that was significantly lighter than the color for the wild-type spores but clearly darker than that for a typical germination mutant. It is possible, therefore, that TasA plays a role in germination. sipWΔ::neo (from AGS157) and yqxM::neo tasAΩpAGS08 (from AGS219) spores had largely the same phenotypes as tasAΔ::spc spores in all the assays described above (data not shown). In contrast, yqxM::neo (from AGS175) spores did not show a germination phenotype by the tetrazolium assay (data not shown).
To identify a function for TasA, we decided to test for an activity consistent with its production and secretion during a protective response, such as sporulation, and for which a simple assay existed. Therefore, we carried out a preliminary test for antibiotic or antibacterial activity by using a variant of the disk diffusion assay (19). We spread cells from one of a variety of strains (Table 3) on an agar plate and cut two wells into the plate. Into one, we placed a crude lysate of E. coli cells engineered to overproduce a form of TasA beginning with amino acid 28. Into the other, we placed a crude lysate of the same E. coli strain bearing a version of the overexpression plasmid that does not harbor the tasA gene. After overnight growth of the cells, we measured the clear zones surrounding the wells. This experiment revealed that TasA can kill or inhibit the growth of a variety of unrelated plant and animal pathogens. For example, we found that the plant pathogen Agrobacterium tumefaciens GV3101 is sensitive to TasA (having a zone diameter of 22 mm, compared with a diameter of 12 mm for the well containing the E. coli lysate alone) whereas Pseudomonas aeruginosa is resistant to TasA (with a zone diameter of 12 mm in both wells) (Table 3). Erwinia amylovara EG321 was also sensitive but exhibited two distinct concentric zones of clearing around the well. The inner zone (21 mm in diameter) was completely clear, whereas the outer zone (33 mm in diameter) exhibited very faint growth compared to the remainder of the growth area of the plate. We also found that a variety of animal pathogens, including clinical isolates of human pathogens, were sensitive to TasA. Intriguingly, we found that the strain used to overproduce TasA, E. coli BL21(DE3), and wild-type B. subtilis (PY79), when grown vegetatively, were sensitive to TasA. To learn whether YqxM possesses an antibacterial activity, we tested its effect on the plant pathogens listed in Table 3. We did not detect any antibacterial activity in our diffusion assay (data not shown). This indicates that the antibacterial activity seen with TasA is not a nonspecific effect of the overproduction of an ectopic histidine-tagged protein.
To confirm that the antibacterial activity of TasA is not necessarily due to a factor in the E. coli extract, we cocultured AGS175 (in which TasA is constitutively synthesized) or AGS157 (which cannot secrete TasA) with E. aerogenes (W31851-1) or P. aeruginosa (S77551-1) and determined the percent survival of each species. We found that B. subtilis producing TasA reduced the survival of E. aerogenes to 18.5%, which we interpret as indicative of sensitivity. In contrast, P. aeruginosa showed no reduction in survival, indicating resistance. The number of surviving B. subtilis spores was unaffected by the secretion of TasA or the presence of the other bacteria in the coculture experiment (data not shown). These results indicate that the toxic effect of TasA can occur in the absence of the E. coli extract.
DISCUSSION
We have identified a novel spore-associated protein, called TasA, that is also present in the cell culture fluid. Two sets of results suggest that TasA is a secreted protein. First, we were able to detect TasA in the cell culture supernatant. Second, the difference in the molecular masses of the translocated and nontranslocated species and our sequence analysis of the N terminus of secreted TasA are consistent with the possibility that a signal peptide is removed during or after translocation. Our finding that the processing and export of TasA depend on sipW suggests that TasA requires the signal peptidase SipW, directly or indirectly, for the removal of the N-terminal 27 amino acids. Two observations raise the possibility that the spore-associated TasA is translocated across a membrane, similarly to TasA found in the culture medium. First, TasA has the same mobility on SDS-PAGE whether it is extracted from spores or purified from culture supernatants. Second, localization to the spore requires sipW. Although we do not know the location of TasA within spores, it is likely to be underneath the coat (in the area of the spore peptidoglycan) or at an interior location within the coat, since we found that TasA was more readily extracted from spores in which most or all of the coat was missing (due to mutations in gerE or gerE and cotE).
The apparent interior location of TasA and the inferred role of SipW are consistent with a speculative model in which secretion plays a direct role in the localization of TasA (Fig. 9). In this view, TasA (Fig. 9A) is synthesized under the control of ςH and Spo0A in the preseptation sporangium and is processed by SipW and secreted across the cytoplasmic membrane via the general secretory pathway. Some TasA is likely to be retained in the cell envelope and presumably is the source of the non-spore-associated TasA identified in sporangial cell lysates. Because we did not detect immature TasA (except in sipW mutant cells), we infer that most TasA is translocated very soon after synthesis. After the septum is built, essentially the same secretory mechanism translocates TasA across one or both septal membranes into the intermembrane space (Fig. 9B). TasA is likely to remain trapped between the spore membrane layers for the duration of sporulation (7). Because TasA appears to be translocated immediately after synthesis, most TasA that is transported into the septum would probably have been synthesized after septum formation. We do not know what factors direct TasA synthesis at this time or whether it occurs in one or both compartments. After engulfment, TasA probably becomes associated with the spore peptidoglycan (Fig. 9C), where it may play a role in germination. The coat then assembles around the spore and, therefore, over the location of TasA (Fig. 9D). This model does not exclude the possibility that TasA is present in the coat as well as residing at a more interior location. Possibly, TasA is relocated to the coat during coat assembly but was not readily extracted in our experiments. Alternatively, TasA present in the culture supernatant could associate with spores after mother cell lysis. We are currently testing the latter possibility. In preliminary experiments, we found TasA associated with germinated spores up to 2 h after the initiation of germination (data not shown). This could have been due to TasA present in the spore cell wall prior to germination or to TasA from the culture supernatant that bound to the germinated spore.
FIG. 9.

Model of the synthesis, processing, and translocation of TasA. (A) A cell that has committed to sporulation. (B) The same cell after the formation of the sporulation septum. (C) The same cell after engulfment of the forespore. (D) The mature spore, released after mother cell lysis. The hatched layer indicates the spore peptidoglycan, and the black layer indicates the coat. See Discussion for a detailed description. Solid rectangles, TasA; solid circles, secreted TasA; shaded area, cell wall.
We do not know of any proteins other than TasA that are likely to be cleaved in vivo by SipW. However, yqxM could encode an intriguing candidate. The hypothetical yqxM gene product would possess a plausible signal peptide, and the five N-terminal residues of the hypothetical mature protein (AFHDI) share four amino acids with the N terminus of mature TasA (AFNDI). We are currently testing whether YqxM exists and whether it is processed and translocated.
Under the conditions tested, deletion of SipW has no detectable consequence for sporulation, indicating that it is not an essential signal peptidase or that other signal peptidases can substitute when SipW is absent (consistent with the work of Tjalsma et al. [32]). Many mutations in genes expressed during sporulation have only subtle or even undetectable effects under laboratory conditions. Such genes include spoIIB, spoVG (21), spoVS (26), and the majority of the known coat protein genes (27). Because spoIIB and spoVG are likely to play a role during or immediately after septum formation (21), we constructed spoIIBΔ::erm tasAΩpAGS08 and spoVG::Tn917ΩHU265 tasAΩpAGS08 strains (AGS200 and AGS202, respectively). We did not detect any difference between the tasAΩpAGS08 (AGS115) mutant and the double mutants by light microscopy or the tetrazolium overlay assay (data not shown).
We do not know the role of TasA in vivo. Its broad-spectrum antibacterial activity may inhibit the growth of competitor bacteria in nature, a trait that could be useful early in sporulation, as well as immediately upon germination (15). If so, vegetative B. subtilis cells may be among those competitor bacteria, since their growth was also inhibited by TasA. The strain of E. coli used for overproduction, BL21(DE3), was also sensitive to added TasA, but when this strain was used to overproduce TasA, no inhibition of growth was seen (data not shown). Although we have not investigated this result in detail, it is likely that the mechanism of action of TasA requires extracellular application.
Our finding that SipW and at least one of its apparent substrates are encoded in adjacent genes raises the possibility that the levels of synthesis of these two proteins are coregulated. Three observations allow us to speculate that yqxM, sipW, and tasA comprise an operon. First, in preliminary experiments with strains bearing portions of the yqxM-sipW-tasA region at the amyE locus, synthesis of TasA required sequences upstream of yqxM. Second, we could not detect any TasA when sipW was interrupted by the neomycin resistance gene, oriented away from tasA. Third, the yqxM and sipW open reading frames overlap. These results suggest that the sequences required for the synthesis of TasA are located immediately upstream of the yqxM open reading frame. If the control of TasA synthesis is largely at the level of transcription, the promoter would probably be in this region as well. Although we do not know if Spo0A actually binds anywhere within this locus, we have identified two 7-nucleotide stretches that resemble putative Spo0A binding sites (2), both of which are upstream of the yqxM open reading frame. It remains to be determined whether Spo0A binds at these or any other sites in this locus.
In summary, we have identified a sporulation protein, TasA, that is a component of the spore and is also secreted into the medium. Assembly of TasA into the spore and export out of the cell both occur concurrently with the removal of an apparent amino-terminal signal peptide and depend on sipW. Furthermore, we have identified an antibiotic activity associated with TasA, raising the possibility that TasA inhibits the growth of competitor bacteria during and after sporulation.
ACKNOWLEDGMENTS
We thank Roberta Carey for giving expert guidance in antibiotic testing and for selecting and providing animal pathogens, and we thank Alan Grossman, Richard Losick, and Patrick Stragier for providing strains. We thank Shawn Little for expert technical assistance. We are grateful to Roberta Carey, Thomas Gallagher, Alan Grossman, Shawn Little, David Popham, Orna Resnekov, Linc Sonenshein, Jan Maarten van Dijl, Chris Webb, and Alan Wolfe for helpful discussions and for critically reading the manuscript.
This work was supported by Public Health Service grant GM539898 from the National Institutes of Health and a grant from the Schweppe Foundation.
REFERENCES
- 1.Bai U, Mandic-Mulec I, Smith I. SinI modulates the activity of SinR, a developmental switch protein of Bacillus subtilis, by protein-protein interaction. Genes Dev. 1993;7:139–148. doi: 10.1101/gad.7.1.139. [DOI] [PubMed] [Google Scholar]
- 2.Baldus J M, Green B D, Youngman P, Moran C P J. Phosphorylation of Bacillus subtilis transcription factor Spo0A stimulates transcription from the spoIIG promoter by enhancing binding to weak 0A boxes. J Bacteriol. 1994;176:296–306. doi: 10.1128/jb.176.2.296-306.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cutting S, Driks A, Schmidt R, Kunkel B, Losick R. Forespore-specific transcription of a gene in the signal transduction pathway that governs pro-ςK processing in Bacillus subtilis. Genes Dev. 1991;5:456–466. doi: 10.1101/gad.5.3.456. [DOI] [PubMed] [Google Scholar]
- 4.Cutting S, Oke V, Driks A, Losick R, Lu S, Kroos L. A forespore checkpoint for mother cell gene expression during development in B. subtilis. Cell. 1990;62:239–250. doi: 10.1016/0092-8674(90)90362-i. [DOI] [PubMed] [Google Scholar]
- 5.Cutting S, Panzer S, Losick R. Regulatory studies on the promoter for a gene governing synthesis and assembly of the spore coat in Bacillus subtilis. J Mol Biol. 1989;207:393–404. doi: 10.1016/0022-2836(89)90262-3. [DOI] [PubMed] [Google Scholar]
- 6.Cutting S M, Vander Horn P B. Molecular biological methods for Bacillus. Chichester, United Kingdom: John Wiley & Sons, Ltd.; 1990. [Google Scholar]
- 7.Dijkstra A J, Keck W. Peptidoglycan as a barrier to transenvelope transport. J Bacteriol. 1996;178:5555–5562. doi: 10.1128/jb.178.19.5555-5562.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Driks A, Roels S, Beall B, Moran C P, Jr, Losick R. Subcellular localization of proteins involved in the assembly of the spore coat of Bacillus subtilis. Genes Dev. 1994;8:234–244. doi: 10.1101/gad.8.2.234. [DOI] [PubMed] [Google Scholar]
- 9.Dubnau D. Binding and transport of transforming DNA by Bacillus subtilis: the role of type-IV pilin-like proteins—a review. Gene. 1997;192:191–198. doi: 10.1016/s0378-1119(96)00804-9. [DOI] [PubMed] [Google Scholar]
- 10.Grossman A D. Genetic networks controlling the initiation of sporulation and the development of genetic competence in Bacillus subtilis. Annu Rev Genet. 1995;29:477–508. doi: 10.1146/annurev.ge.29.120195.002401. [DOI] [PubMed] [Google Scholar]
- 11.Harlow E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- 12.Hoch J A. Regulation of the phosphorelay and the initiation of sporulation in Bacillus subtilis. Annu Rev Microbiol. 1993;47:441–465. doi: 10.1146/annurev.mi.47.100193.002301. [DOI] [PubMed] [Google Scholar]
- 13.Irie R, Okamoto T, Fujita Y. A germination mutant of Bacillus subtilis deficient in response to glucose. J Gen Appl Microbiol. 1982;28:345–354. [Google Scholar]
- 14.Itaya M, Kondo K, Tanaka T. A neomycin resistance gene cassette selectable in a single copy state in the Bacillus subtilis chromosome. Nucleic Acids Res. 1989;17:4410. doi: 10.1093/nar/17.11.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz E, Demain A L. The peptide antibiotics of Bacillus: chemistry, biogenesis, and possible functions. Bacteriol Rev. 1977;41:449–474. doi: 10.1128/br.41.2.449-474.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King E O, Ward M K, Raney D E. Two simple media for the demonstration of pyocyanin and fluorescin. J Lab Clin Med. 1954;44:301–307. [PubMed] [Google Scholar]
- 17.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, Bertero M G, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell S C, Bron S, Brouillet S, Bruschi C V, Caldwell B, Capuano V, Carter N M, Choi S K, Codani J J, Connerton I F, Danchin A, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 18.LeDeaux J R, Grossman A D. Isolation and characterization of kinC, a gene that encodes a sensor kinase homologous to the sporulation sensor kinases KinA and KinB in Bacillus subtilis. J Bacteriol. 1995;177:166–175. doi: 10.1128/jb.177.1.166-175.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lennette E H, Balows A, Hausler W J Jr, Truant J P, editors. Manual of clinical microbiology. 3rd ed. Washington, D.C: American Society for Microbiology; 1980. [Google Scholar]
- 20.Losick R, Stragier P. Crisscross regulation of cell-type-specific gene expression during development in Bacillus subtilis. Nature. 1992;355:601–604. doi: 10.1038/355601a0. [DOI] [PubMed] [Google Scholar]
- 21.Margolis P S, Driks A, Losick R. Sporulation gene spoIIB from Bacillus subtilis. J Bacteriol. 1993;175:528–540. doi: 10.1128/jb.175.2.528-540.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsudaira P. Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J Biol Chem. 1987;262:10035–10038. [PubMed] [Google Scholar]
- 23.Moir A. Germination properties of a spore coat-defective mutant of Bacillus subtilis. J Bacteriol. 1981;146:1106–1116. doi: 10.1128/jb.146.3.1106-1116.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagarajan V. Protein secretion. In: Sonenshein A L, Hoch J, Losick R, editors. Bacillus subtilis and other gram-positive bacteria: biochemistry, physiology, and molecular genetics. Washington, D.C: American Society for Microbiology; 1993. pp. 3–16. [Google Scholar]
- 25.Pugsley A P. The complete general secretory pathway in gram-negative bacteria. Microbiol Rev. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Resnekov O, Driks A, Losick R. Identification and characterization of sporulation gene spoVS from Bacillus subtilis. J Bacteriol. 1995;177:5628–5635. doi: 10.1128/jb.177.19.5628-5635.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricca E, Baccigalupi L, Naclerio G, Cutting S. Spore coat differentiation in Bacillus subtilis. Res Microbiol. 1997;148:5–9. doi: 10.1016/S0923-2508(97)81894-3. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 29.Sandman K, Kroos L, Cutting S, Youngman P, Losick R. Identification of the promoter for a spore coat protein gene in Bacillus subtilis and studies on the regulation of its induction at a late stage of sporulation. J Mol Biol. 1988;200:461–473. doi: 10.1016/0022-2836(88)90536-0. [DOI] [PubMed] [Google Scholar]
- 30.Simonen M, Palva I. Protein secretion in Bacillus species. Microbiol Rev. 1993;57:109–137. doi: 10.1128/mr.57.1.109-137.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stragier P, Losick R. Molecular genetics of sporulation in Bacillus subtilis. Annu Rev Genet. 1996;30:297–341. doi: 10.1146/annurev.genet.30.1.297. [DOI] [PubMed] [Google Scholar]
- 32.Tjalsma H, Bolhuis A, van Roosmalen M L, Wiegert T, Schumann W, Broekhuizen C P, Quax W J, Venema G, Bron S, van Dijl J M. Functional analysis of the secretory precursor processing machinery of Bacillus subtilis: identification of a eubacterial homolog of archaeal and eukaryotic signal peptidases. Genes Dev. 1998;12:2318–2331. doi: 10.1101/gad.12.15.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tjalsma H, Noback M A, Bron S, Venema G, Yamane K, van Dijl J M. Bacillus subtilis contains four closely related type I signal peptidases with overlapping substrate specificities. Constitutive and temporally controlled expression of different sip genes. J Biol Chem. 1997;272:25983–25992. doi: 10.1074/jbc.272.41.25983. [DOI] [PubMed] [Google Scholar]
- 34.Youngman P, Perkins J B, Losick R. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid. 1984;12:1–9. doi: 10.1016/0147-619x(84)90061-1. [DOI] [PubMed] [Google Scholar]
- 35.Zheng L, Donovan W P, Fitz-James P C, Losick R. Gene encoding a morphogenic protein required in the assembly of the outer coat of the Bacillus subtilis endospore. Genes Dev. 1988;2:1047–1054. doi: 10.1101/gad.2.8.1047. [DOI] [PubMed] [Google Scholar]