

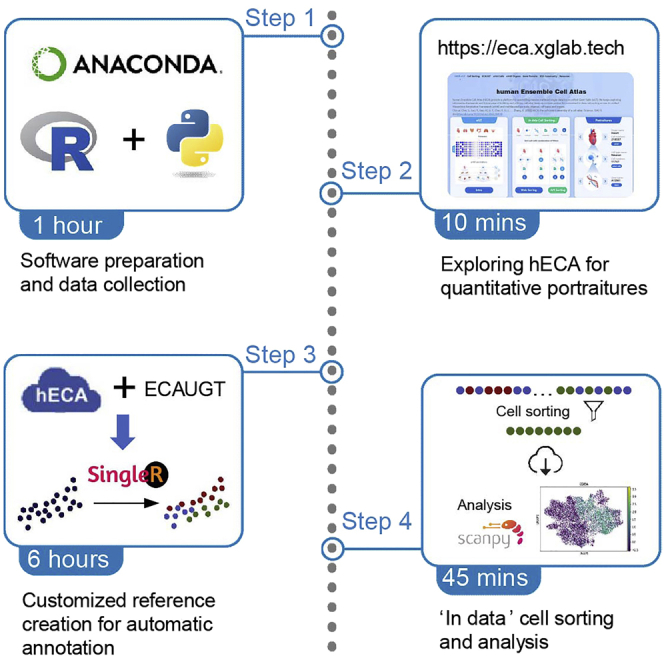
Summary
Human Ensemble Cell Atlas (hECA) provides a unified informatics framework and the cell-centric-assembled single-cell transcriptome data of 1,093,299 labeled human cells from 116 published datasets. In this protocol, we provide three applications of hECA: “quantitative portraiture” exploration with websites, customizable reference creation for automatic cell type annotation, and “in data” cell sorting with logical conditions. We provide detail steps of connecting to the database, searching cell with conditions, downloading data, and annotating new datasets with customized reference.
For complete details on the use and execution of this protocol, please refer to Chen et al. (2022).
Subject areas: Bioinformatics, Single Cell, RNAseq, Gene Expression, Computer sciences
Graphical abstract
Highlights
-
•
Utilizing cell-centric-assembled single-cell transcriptome data from hECA
-
•
Exploring multiview quantitative descriptions of biological entities
-
•
Customizing reference data for automatic cell type annotation
-
•
In data cell sorting with a combination of conditions on gene expression and metadata
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Human Ensemble Cell Atlas (hECA) provides a unified informatics framework and the cell-centric-assembled single-cell transcriptome data of 1,093,299 labeled human cells from 116 published datasets. In this protocol, we provide three applications of hECA: “quantitative portraiture” exploration with websites, customizable reference creation for automatic cell type annotation, and “in data” cell sorting with logical conditions. We provide detail steps of connecting to the database, searching cell with conditions, downloading data, and annotating new datasets with customized reference.
Before you begin
Hardware preparation
For the website tools of hECA, an individual computer with any web browser (Chrome is recommended) and network connection is required.
For the command line tools for ‘in data’ sorting and automatic cell-type annotation, a computer with a Linux operation system and network connection is required. The RAM requirement is according to the number of cells for analysis. A 16 GB RAM is sufficient for the experiments in this protocol and additional 200 MB RAM is required for each 1k cells.
Software preparation
Timing: <1 h
This section is only required for applications with command line tools such as customizable reference creation for cell-type annotation and ‘in data’ cell sorting. More detailed versions of the involved packages can be found in our GitHub repository.
-
1.
Python package installation.
‘In data’ cell sorting application of hECA requires the cell sorting tool ECAUGT in Python3.
In this step, we install the Python 3.9.12 and the dependencies with Anaconda (conda=4.10.3).-
a.Anaconda can be downloaded from https://www.anaconda.com/products/distribution according to the computer operating system.
-
b.Once the Anaconda is installed, we recommend creating a new conda environment for the hECA tools in case of the influence of the installed packages on the computer. Run the following commands in the command line:>conda create -n hECA pip>conda activate hECA
-
c.Install ECAUGT from PyPI:>pip install ECAUGT(The dependencies of ECAUGT will be installed automatically).
-
d.Any single-cell analysis tool can be used to conduct downstream analysis on the downloaded data. In this pipeline, we take scanpy as an example. Install scanpy with conda:>conda install -c conda-forge scanpy python-igraph leidenalg
-
a.
-
2.
R package installation.
For customized reference creation and automatic cell-type annotation, preprocessing tools and annotation tools in the R platform are required.-
a.Install the latest version of R.>conda install -c conda-forge r-base=4.1.3
 CRITICAL: The channel parameter and the version parameter are essential because the following software package requires R with a version greater than 4.0.
CRITICAL: The channel parameter and the version parameter are essential because the following software package requires R with a version greater than 4.0. -
b.Install R package Seurat and its essential dependencies. Notice the r-curl and r-rgeos are installed with conda.>conda install -c conda-forge r-curl>conda install -c conda-forge r-rgeos>R>install.packages('Seurat')Then Seurat can be installed in the R command line:The other dependencies of Seurat will be installed automatically in this command and this process may take over 30 min for a newly-created environment.
-
c.Install the BiocManager, SingleR, and scran in the R command line:>install.packages("BiocManager")>BiocManager::install("SingleR")>BiocManager::install("scran")
-
d.GeneSymbolUniform_Rtoolkit is the data preprocessing tool of hECA which unifies the features of the query data before annotation. It can be downloaded from our GitHub repository. Install the GeneSymbolUniform_Rtoolkit and its dependency rlist in shell:>cd hECA_GeneSymbolUniform_Rtoolkit/>unzip GeneSymbolRef_SelectAll_upd0731.csv.zip>R>Install.packages('rlist')CRITICAL: Keep the names of the files in this folder unchanged. The unzip step is required.
-
a.
Data collection
This section is only required for automatic annotation with a customized reference. The query data should be collected and transferred into a gene-by-cell expression matrix. In this protocol, we use human neuron single-cell data from PsychENCODE (https://explorer.nimhgenetics.org/) as an example. The download data are a UMI matrix including 27412 cells with 17176 genes.
>wgethttp://resource.psychencode.org/Datasets/Derived/SC_Decomp/DER-22_Single_cell_expression_raw_UMI.tsv
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| hECA database | (Chen et al., 2022) | http://eca.xglab.tech/ |
| PsychENCODE data | (Wang et al., 2018) | http://resource.psychencode.org/Datasets/Derived/SC_Decomp/DER-22_Single_cell_expression_raw_UMI.tsv |
| Software and algorithms | ||
| Seurat | (Stuart et al., 2019) | https://github.com/satijalab/seurat |
| SingleR | (Aran et al., 2019) | https://www.bioconductor.org/packages/release/bioc/html/SingleR.html |
| Scanpy | (Wolf et al., 2018) | https://scanpy.readthedocs.io/en/stable/index.html |
| ECAUGT | (Chen et al., 2022) | https://pypi.org/project/ECAUGT/ |
| GeneSymbolUniform_Rtoolkit | (Chen et al., 2022) | https://github.com/XuegongLab/hECA_GeneSymbolUniform_Rtoolkit |
| The repository of this protocol | This work | https://github.com/XuegongLab/hECA_protocol_case |
Step-by-step method details
Here we describe step-by-step how to use hECA tools for exploring ‘quantitative portraiture’ of different biological entities, annotating single-cell data with customized reference data from hECA, and sorting cells from hECA database with a complex logical combination of conditions.
Exploring hECA database and quantitative portraiture
In this section, we describe the usage of the website tools of hECA. Users can follow these steps in any operating system with a web browser. Users can gain a multi-view quantitative description of their interested biological entities or sort the subgroup of cells in hECA database without coding.
-
1.There is no strict order to explore the quantitative portraiture in hECA. Users can switch their exploring focus at any time by clicking the navbar.
-
a.To Explore portraits of organs in hECA, users can click ‘uHAF Organs’ to browse the page (http://xglab.tech/#/organGallery).
-
i.Users can select their interested organs. Here we use bone marrow as an example.
-
ii.A brief introduction of the selected organ, cell number and cell-type composition of the collected data, UMAP embedding, and anatomical relationships with other organs are presented on this page (Figure 1).
-
i.
-
b.To explore the portraits of cell types in hECA, users can click ‘uHAF Cells’ to browse the page (http://xglab.tech/#/cellTypeList?viewType=tree) and select the cell type of interest by exploring the uHAF tree or searching by the name.
-
i.Here we present fibroblast as an example. The description of fibroblast as well as the link to Wikipedia and Cell Ontology is provided (Figure 2A).
-
ii.Click the “View details” button. The abundance of fibroblast in different organs, the marker genes of fibroblasts, and the UMAP embedding of all fibroblasts in hECA data are presented on the new page (Figure 2B).
-
i.
-
c.To explore gene portraits in hECA, users can click the ‘Gene Portraits’ in the navbar to browse the page (http://eca.xglab.tech/#/genePortrait). Users can enter their interested gene symbols and view the distributions across the selected organ or cell types (Figure 3).
-
a.
-
2.Our online cell sorting tool based on hECA database and ECAUGT is available by clicking ‘Cell Sorting’ to browse the page (http://eca.xglab.tech/#/cellSorting). Users can sort hECA data in multiple steps, where each step can be a combination of conditions on both metadata and gene expression. Subsequent filtering steps are based on the previous step results. Here we present an example of sorting the cells expressing CD19 across all organs.
-
a.Add a step with a gene filter that CD19 expression should be larger than 0.1, and press the ‘Apply’ button.
-
b.The website takes 2–3 s to finish sorting and present the results. Users can view basic statistics including cell number, cell-type composition and original organs of these cells (Figure 4).
-
c.Press the ‘Analysis’ button to view more detailed information about the sorted cells such as the expression distribution of any interested gene across different cell types and organs. Users can also press ‘Download Data’ to gain the IDs of sorted cells in a txt file and then use ECAUGT to download data (the method is mentioned in step 6).
-
a.
Figure 1.

The portrait of bone marrow on the hECA website
Figure 2.

The portrait of fibroblast on the hECA website
(A) Fibroblast on the uHAF tree.
(B) Portrait of fibroblast.
Figure 3.

The portrait of ACTB gene on the hECA website
Figure 4.

The web tool of cell sorting on the hECA databases
Customized reference creation for automatic annotation
The steps in the following sections are designed for more professional usages of hECA. A common application of the single-cell atlas is using data as the reference of automatic annotation for new datasets. Here we give an example of creating a customized neuron cells reference from hECA and using it to annotate the dataset from PsychENCODE (Wang et al., 2018).
-
3.We use ECAUGT, the cell sorting tool of hECA, to sort and download neuron cells from hECA as the customized reference.
-
a.Import required packages in Python.>import sys>import pandas as pd>import ECAUGT>import time>import multiprocessing>import numpy as np
-
b.Connect to the hECA database.>endpoint = "https://HCAd-Datasets.cn-beijing.ots.aliyuncs.com">access_id = "LTAI5t7t216W9amUD1crMVos">access_key = "ZJPlUbpLCij5qUPjbsU8GnQHm97IxJ">instance_name = "HCAd-Datasets">table_name = "HCA_d">ECAUGT.Setup_Client(endpoint, access_id, access_key, instance_name, table_name)CRITICAL: Ensure that the network connection is stable and these parameters are unchanged. The output in Figure 5A means a successful connection.
-
c.Get the IDs of neuron cells for download. Here ‘include_children’ parameter is set as True which means that all subtypes of neuron cells are included in the result. The number of cells will be printed when sorting is finished.>rows_to_get = ECAUGT.search_metadata("cell_type == Neuron", include_children=True)Note: ‘search_metadata’ function receives a string parameter as the metadata condition and returns the IDs of sorted cells. We provide an example to sort cells with complex conditions on both metadata and gene expression in step 6. More details about the usage of ECAUGT can be found in the document of ECAUGT (http://eca.xglab.tech/ecaugt/index.html).
-
d.Download the expression value of all genes and the metadata of the sorted cells.It will take about 5 h to download all the data containing 180 thousand neuron cells for this example, while the network fluctuation may cause errors. So, we separate sorted cells into chunks, each of which contains 10000 cells. The following codes generate both expression matrix csv file and metadata csv file in pair.>for i in range((len(rows_to_get)//10000)+1):> st = i∗10000> ed = (i+1)∗10000> print(f'start to download cells from id {st} to {ed}')> result = ECAUGT.get_columnsbycell_para(rows_to_get = rows_to_get[st:ed], do_transfer = True, thread_num = 95)> print(f'saving cells from id {st} to {ed}')> genes = result.columns[:43878]> metaCols = result.columns[43878:43878+16]> expr = result.loc[:,genes]> meta = result.loc[:,metaCols]> expr.to_csv(f"hECA_exprs_{i}.csv", index=True)> meta.to_csv(f"hECA_metadata_{i}.csv", index=True)
-
e.Merge all files to create the customized reference in R:>library(Seurat)>library(data.table)>for (i in 1:18){> message(paste0('/home/test2/hECA_data/hECA_exprs_',i,'.csv'))> tmpexpdf = as.data.frame(t(fread(paste0('/home/test2/hECA_data/hECA_exprs_',i,'.csv'))))> tmpexpdf = tmpexpdf[-1,]> tmpmetadf = as.data.frame(fread(paste0('/home/test2/hECA_data/hECA_metadata_',i,'.csv')))> expdf = cbind(expdf,tmpexpdf)> metadf = rbind(metadf,tmpmetadf)>}>row.names(metadf) <- metadf$cid>metadf = metadf[,1:16]>colnames(expdf) = rownames(metadf)>ref_obj <- CreateSeuratObject(expdf, meta.data = metadf)>saveRDS(ref_obj, file = "Neuron_hECA_reference.rds")
-
a.
-
4.Preprocess the collected query data.
-
a.Unify the gene number and symbol of the collected data with GeneSymbolUniform_Rtoolkit in the command line.>cd hECA_GeneSymbolUniform_Rtoolkit/>Rscript Rtoolkit_GeneSymbolUniform.R ../DER-22_Single_cell_expression_raw_UMI.tsv ../CRITICAL: GeneSymbolUniform_Rtoolkit takes a gene-by-cell expression matrix in a csv file as input and generates two files: the uniformed expression matrix with 43878 genes and the modified gene report. The first parameter is the path of the raw expression matrix as the input, and the second parameter is the path of the output folder. The gene names in the input matrix should be gene symbols instead of Ensemble IDs. The users should use the Rscript command in the terminal instead of running the r file in RStudio.
-
b.Preprocess the uniformed expression matrix in R.
-
i.Load the uniformed expression matrix:>data.matrix <- as.data.frame(fread("∼/UniformedExpression.csv"))>row.names(data.matrix) <- data.matrix$V1>data.matrix = data.matrix[,-1]>cellid = colnames(data.matrix)>samp.devStage <- data.frame(samp.devStage = ifelse(grepl("ˆFetal",cellid),"Fetal","Adult"))>rownames(samp.devStage) = cellid
-
ii.Preprocess the query data with Seurat.We follow the Seurat pipeline to conduct quality control and normalization on the uniformed data. More detail about the usage of Seurat can be found in https://satijalab.org/seurat/index.html.>query_obj <- CreateSeuratObject(counts = as.matrix(data.matrix), meta.data = samp.devStage)>selected_c <- WhichCells(query_obj, expression = nFeature_RNA > 200)>selected_f <- rownames(query_obj)[Matrix::rowSums(query_obj) > 3]>query_obj.filt <- subset(query_obj, features = selected_f, cells = selected_c)>query_obj.filt <- NormalizeData(query_obj.filt)>saveRDS(query_obj.filt, file = "Neuron_query.rds")
-
i.
-
a.
-
5.Automatic annotation with SingleR.
-
a.Load the reference data and query data:>refrds = readRDS('∼/Neuron_hECA_reference.rds')>query_obj.filt = readRDS('∼/Neuron_query.rds')>gene.use = intersect(row.names(refrds),row.names(query_obj.filt))>refrds.filt <- subset(refrds, features = gene.use)>ct.ref <- refrds.filt$cell_type>trainedR <- trainSingleR(refrds.filt@assays$RNA@counts, ct.ref, de.method = "wilcox")
-
b.Train the singleR model.>trainedR <- trainSingleR(refrds@assays$RNA@data, ct.ref, de.method = "wilcox")
-
c.Annotate query data.To speed up this protocol, we subset the first 1000 cells in the query data to be annotated as an example.>query_sample <- query_obj.filt[,1:1000]>predict <- classifySingleR(query_sample@assays$RNA@data,trainedR)The output of SingleR model is a DataFrame with 5 columns: scores, first labels, tuning.scores, labels, and pruned.labels. Pruned.labels is the automatic annotation result we need and the tuning.scores present the similarity between the annotated cells and the target cell types (Figure 5B).
-
a.
Figure 5.

Customized reference creation for automatic annotation
(A) Printed message when successfully connected to the hECA database.
(B) The output of the SingleR annotation model on the query neuron cell data.
‘In data’ cell sorting
ECAUGT is designed for sorting cells with a logical combination of any conditions on both metadata and gene expression. This ‘in data’ cell sorting enables complex experiment design in the digital space, which may be difficult to conduct in wet experiments. Here we present an example of sorting T cells from human lung without CD4 expression.
-
6.‘In data’ cell sorting with ECAUGT.
-
a.Open the Python command line and import the required packages:>import sys>import pandas as pd>import ECAUGT>import time>import multiprocessing>import numpy as np
-
b.Connect to the hECA database with following codes:>endpoint = "https://HCAd-Datasets.cn-beijing.ots.aliyuncs.com">access_id = "LTAI5t7t216W9amUD1crMVos">access_key = "ZJPlUbpLCij5qUPjbsU8GnQHm97IxJ">instance_name = "HCAd-Datasets">table_name = "HCA_d">ECAUGT.Setup_Client(endpoint, access_id, access_key, instance_name, table_name)
-
c.Sort T cells satisfying the conditions.
-
i.Sort cells that are: (1) from the Lung organ, (2) sequenced by 10× platform, and (3) annotated as T cell or its subtypes (The unified hierarchical annotation framework (uHAF) which defines the subtypes of T cell can be found on http://xglab.tech/#/cellTypeList).> metadata_condition = "cell_type == T cell && organ== Lung && seq_tech==10×">cid_label = ECAUGT. search_metadata (metadata_conditions=metadata_condition, include_children=True)Note: Different conditions should be combined in a string with logistical operators ‘&&’, ‘||’, and ‘!’. The condition string can include brackets and is not sensitive to spaces.
-
ii.Sort cells with CD4 expression value lower than 0 from the former result.>gene_condition = ECAUGT.seq2filter("CD4<=0")>df_result_tcell = ECAUGT.get_columnsbycell_para( rows_to_get = cid_label, cols_to_get=['CD4'], col_filter=gene_condition, do_transfer = False, thread_num = 40)Note: The grammar of the gene condition string is similar to the metadata condition string, and genes existed in the gene condition string should be included in the ‘cols_to_get’ parameter. Users can adjust the ‘thread_num’ parameter to decide the number of threads used in data downloading.Here we reorganize the sorted cell IDs to a ‘rows_to_get’ variable for data downloading in the next step and print the number of cells.>rows_to_get = [[x[0]] for x in df_result_tcell]>print(len(rows_to_get))
-
i.
-
d.Download the expression values of all genes and metadata of the sorted cells, and save the downloaded data in a cell-by-feature matrix in pandas.DataFrame.> result = ECAUGT.get_columnsbycell_para(rows_to_get = rows_to_get, cols_to_get = None, col_filter = None, do_transfer = True, thread_num = 40)Note: This download process costs about 36 min (about 3 min/1000 cells for 40 threads) so ensure that the network connection is stable. More threads can accelerate this process.Optional: Users can save the download result into a pickle file.>import pickle>pickle.HIGHEST_PROTOCOL>result.to_pickle("sorted_tcells_raw.pk")
-
a.
-
7.A downstream analysis is conducted with scanpy as an example. The usage of scanpy can be found in its document (https://scanpy.readthedocs.io/en/stable/index.html).
-
a.Separate the downloaded data into an expression matrix and a metadata matrix, and create a scanpy object from the matrices.>import scanpy as sc>expr = result.iloc[:,:43878]>meta = result.iloc[:,43878:43878+16]>meta.reset_index(inplace=True)>expr.reset_index(inplace=True)>expr.drop(['cid'],axis=1,inplace=True)> adata = sc.AnnData(X = expr, obs = meta)>adata.var_names_make_unique()
-
b.Conduct quality control:>sc.pp.filter_genes(adata, min_counts=5)>sc.pp.filter_genes(adata, min_cells=3)
-
c.Find variable genes and conduct dimension reduction with principal component analysis (PCA):>sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)>sc.pp.scale(adata, max_value=10)>sc.tl.pca(adata, svd_solver='arpack')
-
d.Generate UMAP embedding and visualize some marker genes of CD8 T cells:>sc.pp.neighbors(adata, n_neighbors=10, n_pcs=20)>sc.tl.umap(adata)>sc.pl.umap(adata,color=['CD8A','CD8B','GZMK','CD3E','GZMH','GZMB'],ncols=3)Here we check the marker genes of Memory CD8 T cells (CD8A, CD8B, and GZMK) and the marker genes of Naïve CD8 T cells (CD3E, GZMH, and GZMB). In the UMAP, we can see CD8 T cells locate in the right part and these two subtypes of CD8 T cells form two clusters in the UMAP (Figure 6).Optional: Users can conduct further analysis according to their experiment design or use other single-cell analysis tools like Seurat to conduct the downstream analysis.
-
a.
Figure 6.

Expression pattern of the marker genes of two CD8 T cell subtypes
Expected outcomes
‘Exploring quantitative portraitures’ provides multi-view information and visualization of users’ interested biological entities. ‘Customized reference creation and automatic annotation’ provides data of sorted cells as the reference for automatic annotation methods as well as the annotation results with prediction scores. ‘In data cell sorting’ sorts the hECA database with complex conditions and provides analysis results in scanpy.
Limitations
Now hECA database contains 1,093,299 cells from 38 organs, which only covers parts of the published human single-cell data. More datasets and organs will be included in the future version of our database. We plan to add the single-cell data from other omics, like scATAC-seq, into our unified information framework. For the quantitative portraitures, we will keep trying state-of-art analysis tools to improve the annotation and add new analysis results such as cell-cell communication.
ECAUGT is sensitive to network fluctuation and may cost a long time when processing a large number of cells. We have constructed a new database framework and succeeded to accelerate the process by about 10 times. Next, we will update ECAUGT based on this framework to save time cost.
Troubleshooting
Problem 1
Input error when using GeneSymbolUniform_Rtoolkit to unify the gene symbols in step 4(a), including matrix format error and original gene names error, leads to an all-zero output matrix.
Potential solution
Ensure that the input matrix is a gene-by-cell expression matrix in a csv file. The gene names in the input matrix should be gene symbols instead of Ensemble IDs or full gene names. The printed information during processing, including the shape and first 5 genes of the query data, can help users to locate the problem.
Problem 2
ECAUGT fails to connect to the hECA database in step 3(b) and step 6(b).
Potential solution
Check the network connection of the computer and ensure that the connection parameters are the same as those in the protocol.
Problem 3
Errors of the metadata and gene condition during cell sorting in the step 3(c) and step 6(c).
Potential solution
Users should set condition combination on metadata in ‘search_metadata’ interface and condition combination on genes in ‘seq2filter’ interface. Metadata in the condition string must exist in the database (the list of searchable metadata columns is available in the ECAUGT document). Genes in the condition string must exist in the 43878 unified gene symbols and be included in the ‘cols_to_get’ parameter of the ‘get_columnsbycell_para’ interface.
Problem 4
Connection breaks during cell download in step 3(d).
Potential solution
Split cell IDs into chunks after the sorting step and download one chunk each time. Increasing the number of download threads helps to speed up the process and avoid network error.
Problem 5
Error of working path when using GeneSymbolUniform_Rtoolkit in step 4(a).
Potential solution
The users should use the Rscript command in the terminal to perform step 4. Running GeneSymbolUniform_Rtoolkit in RStudio requires additional settings and may lead to error.
Problem 6
Failure to install rlist package from CRAN.
Potential solution
If so, the users can install this package from GitHub:
>install.packages("devtools")
>devtools::install_github("renkun-ken/rlist")
Problem 7
Failure to install Seurat package.
Potential solution
If finding difficulties to install the R package Seurat, we suggest the users to follow the tutorial on their website https://satijalab.org/seurat/articles/install.html and check the issues in their GitHub repository https://github.com/satijalab/seurat/issues.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Xuegong Zhang (zhangxg@tsinghua.edu.cn).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work is supported in part by National Key R&D Program of China grant 2021YFF1200900, NSFC grants 62050178, 61721003, 32000453, and 42050101. The work is also supported by the Tsinghua-Fuzhou Institute of Data Technologies (TFIDT2021003).
Author contributions
X.Z. and L.W. conceptualized and designed the project. X.Z., Y.C., M.H., J.L., H.G., S.C., F.L., and L.W. designed the protocol. Y.C. and M.H. designed and conducted the example experiments in the protocol. X.Z., Y.C., M.H., H.G., and L.W. wrote the manuscript, with inputs from all authors. All authors read and approved the manuscript.
Declaration of interests
The database technology behind the data storage used in hECA is being applied for a patent.
Contributor Information
Lei Wei, Email: weilei92@tsinghua.edu.cn.
Xuegong Zhang, Email: zhangxg@tsinghua.edu.cn.
Data and code availability
HECA database and quantitative portraits is available on http://eca.xglab.tech/. Code of ECAUGT is available on https://github.com/XuegongLab/ECAUGT. Code of GeneSymbolUniform_Rtoolkit is available on https://github.com/XuegongLab/hECA_GeneSymbolUniform_Rtoolkit. All codes and jupyter notebooks are available at https://zenodo.org/record/6703333 (https://doi.org/10.5281/zenodo.6703333).
References
- Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Luo Y., Gao H., Li F., Chen Y., Li J., You R., Hao M., Bian H., Xi X., et al. hECA: the cell-centric assembly of a cell atlas. iScience. 2022;25:104318. doi: 10.1016/j.isci.2022.104318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Liu S., Warrell J., Won H., Shi X., Navarro F.C.P., Clarke D., Gu M., Emani P., Yang Y.T., et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362:eaat8464. doi: 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
HECA database and quantitative portraits is available on http://eca.xglab.tech/. Code of ECAUGT is available on https://github.com/XuegongLab/ECAUGT. Code of GeneSymbolUniform_Rtoolkit is available on https://github.com/XuegongLab/hECA_GeneSymbolUniform_Rtoolkit. All codes and jupyter notebooks are available at https://zenodo.org/record/6703333 (https://doi.org/10.5281/zenodo.6703333).