

The L-NAME mouse model of preeclampsia mimics key aspects of disease in pregnancy, but does not demonstrate the increased long-term risk of cardiovascular disease seen in individuals following preeclampsia.
Abstract
Preeclampsia affects ∼2–8% of pregnancies worldwide. It is associated with increased long-term maternal cardiovascular disease risk. This study assesses the effect of the vasoconstrictor N(ω)-nitro-L-arginine methyl ester (L-NAME) in modelling preeclampsia in mice, and its long-term effects on maternal cardiovascular health. In this study, we found that L-NAME administration mimicked key characteristics of preeclampsia, including elevated blood pressure, impaired fetal and placental growth, and increased circulating endothelin-1 (vasoconstrictor), soluble fms-like tyrosine kinase-1 (anti-angiogenic factor), and C-reactive protein (inflammatory marker). Post-delivery, mice that received L-NAME in pregnancy recovered, with no discernible changes in measured cardiovascular indices at 1-, 2-, and 4-wk post-delivery, compared with matched controls. At 10-wk post-delivery, arteries collected from the L-NAME mice constricted significantly more to phenylephrine than controls. In addition, these mice had increased kidney Mmp9:Timp1 and heart Tnf mRNA expression, indicating increased inflammation. These findings suggest that though administration of L-NAME in mice certainly models key characteristics of preeclampsia during pregnancy, it does not appear to model the adverse increase in cardiovascular disease risk seen in individuals after preeclampsia.
Introduction
Preeclampsia is a severe obstetric disease affecting ∼2–8% of pregnancies worldwide (1, 2, 3). Its pathogenesis stems from the dysfunctional placenta, which releases anti-angiogenic and pro-inflammatory factors into the maternal circulation, resulting in widespread endothelial dysfunction (4, 5, 6). Endothelial dysfunction is responsible for systemic vasoconstriction and chronic hypertension, limiting blood supply to major organs (7). Consequently, preeclampsia can cause major organ injury and impair fetal development, making it responsible for significant maternal and perinatal morbidity and mortality (8). In addition, preeclampsia also impacts long-term health of both the pregnant individual and child (9). It is well established that individuals whose pregnancies were complicated by preeclampsia or gestational hypertension are at higher risk of cardiovascular disease post-delivery than those with normotensive pregnancies (10, 11, 12, 13, 14).
There is currently no cure for preeclampsia, and limited treatment options. Hence, there is urgent need for development of therapeutics that directly target the disease. Animal models play an important role in understanding the pathophysiology that drives preeclampsia, and offer valuable potential in the development and testing of candidate therapeutics in an in vivo system. Established preeclampsia models have been summarised in several studies (15, 16, 17, 18), but it is well accepted that one model alone is unlikely to encompass all aspects of preeclampsia pathogenesis due to the complexity and heterogeneity of the disease. Rather, each model might mimic certain key pathways or aspects of the disease process. Henceforth, assessing the benefits of a candidate therapeutic may be best performed through multiple models examining the effects over several key pathways that underpin preeclampsia.
However, most existing models of preeclampsia do not encompass the long-term effects of the disease (19, 20, 21, 22). This is a gap in the knowledge and tools available for enhancing preeclampsia research. A part of assessment of preeclampsia therapeutics should be to examine whether they can also improve long-term outcomes. This is particularly important for long-term maternal health, as cardiovascular disease is a leading cause of death of women in Australia (23). Pregnancy provides a unique window for intervention, given pregnancy complications can impact long-term maternal health and increase risk of cardiovascular disease. Animal models of preeclampsia with long-term maternal follow-up would provide an opportunity to test potential interventions in pregnancy and after birth, to determine whether it is possible to reduce the increased cardiovascular disease risk these individuals face—particularly important because very few interventions to reduce long-term risk have been studied (24).
In this article, we aimed to assess whether a popular animal model of preeclampsia could recapitulate the long-term effects of preeclampsia on maternal cardiovascular health. The model described in this article is an adaptation of the L-NG-Nitro arginine methyl ester (L-NAME) model of preeclampsia. L-NAME works by inhibiting nitric oxide synthase, the enzyme responsible for producing nitric oxide, a molecule involved in many biological processes including endothelium-dependent vasodilation (25). Reduction of nitric oxide bioavailability may contribute to vascular dysfunction in preeclampsia (26, 27). Consequently, nitric oxide donors are being considered potential candidates to examine for the treatment of preeclampsia (28, 29).
L-NAME was initially used to model preeclampsia in rats, inducing increased blood pressure, proteinuria, thrombocytopenia and fetal growth restriction (22). Since then, L-NAME has also been used to induce a preeclampsia-like phenotype in mice, inducing increased blood pressure, proteinuria, fetal growth restriction, impairment of placental morphology and other major organ and vascular alterations (30, 31, 32, 33, 34, 35, 36). However, these murine studies using L-NAME differ in their protocols, using varied concentrations of L-NAME, routes of administration, timing of exposure, and murine strains, that could respond differently to L-NAME (37). Furthermore, though L-NAME is commonly used in animal models to induce a preeclampsia-like phenotype, not many have followed up the animals post-delivery. Most of the studies that do, follow the offspring (38, 39, 40, 41, 42, 43). Only one study has examined maternal outcomes in the context of modelling long-term outcomes of a preeclamptic pregnancy, but it focused on cognitive deficit, not cardiovascular disease (44).
Therefore, in this study, we established the L-NAME model of preeclampsia in CBA x C57BL/6 (F1) mice. We assessed whether this model could mimic key pathways central to the pathogenesis of preeclampsia, as seen in other L-NAME models. Furthermore, we examined whether this model could mimic the increased maternal risk of cardiovascular disease following delivery, which is synonymous with preeclampsia.
Results
L-NAME administration to pregnant mice increased blood pressure in pregnancy
Hypertension is a key clinical characteristic of preeclampsia. Hence, we first assessed if L-NAME administered in our model could alter blood pressure. L-NAME administration significantly increased mean arterial blood pressure at E14.5 (control 103.7 ± 20.89, L-NAME 126.5 ± 20.92; mean ± SD) (P < 0.0001; Fig 1A) and E17.5 (control 111.9 ± 22.66, L-NAME 130.1 ± 18.52; mean ± SD) (P = 0.0001; Fig 1B). Both systolic and diastolic blood pressures were significantly increased at E14.5 (P < 0.0001; Fig S1A and B) and E17.5 (P = 0.0002 and P = 0.0001, respectively, Fig S1C and D).
Figure 1. Effect of L-NAME administration on mean arterial blood pressure E14.5 and E17.5 of pregnancy.

Blood pressure was measured by tail cuff plethysmography, (A) E14.5 mean blood pressure, (B) E17.5 mean blood pressure. (A, B) L-NAME administration significantly increased mean blood pressure from control levels at both time points. Data presented as mean ± SEM. Control n = 33–40, L-NAME n = 42–43. An unpaired t test was used to assess statistical differences between L-NAME and control group at each time point; ***P < 0.001 ****P < 0.0001.
Figure S1. Effect of L-NAME administration on systolic and diastolic arterial blood pressure at E14.5 and E17.5 of pregnancy.

Blood pressure measured by tail cuff plethysmography. (A, B, C, D) L-NAME administration significantly increased systolic and diastolic blood pressure at both (A, B) E14.5 and (C, D) E17.5 of pregnancy. Data presented as mean ± SEM. Control n = 33–40, L-NAME n = 42–43. An unpaired t test was used to assess statistical differences between L-NAME and control group at each time point; ***P < 0.001 ****P < 0.0001.
L-NAME administration in pregnancy did not induce proteinuria but may impact kidney structure at microscopic level
Proteinuria is evidence of kidney damage in preeclampsia. L-NAME administration did not significantly alter albumin to creatinine ratio at either E14.5 or E17.5 (Fig 2A and B). Furthermore, L-NAME did not alter the expression of genes associated with renal function, or known to be dysregulated with dysfunction (Ccr2, Ctgf, Fn1, Hsd11b2, Il-1β, Mmp2 (expressed as ratio to expression of inhibitor, Timp1), Mmp9 (expressed as ratio to expression of inhibitor Timp1), Nhe1, Nlrp3, Nox2, Nox4, Nr3c1, Nr3c2, Sccn1a, Sgk1, Tgfβ1, Tgfβ2, Tgfβ3, Tnf, and Vcam1) (Fig S2).
Figure 2. Effect of L-NAME on urine albumin to creatinine ratio, and kidney structure in pregnancy.

Maternal urine albumin and creatinine concentrations were measured via ELISA and enzymatic assay, respectively, at (A) E14.5 and (B) E17.5 of pregnancy. (A, B) Albumin to creatinine ratio was not significantly altered between the L-NAME and control mice. (C, D) Histology images of the cortex of PBS treated control mice kidneys show relatively normal glomeruli and tubules, with uniform staining. (E, F) Pathological features of L-NAME treated mice show inflammatory cell infiltration around regions of necrosis (black arrows) with haemoglobin (dark pink in section) and hyaline (light pink) casts that are surrounded by flattened nuclei and irregular cells (white arrows). Scale bar = 10 μm. Data presented as mean ± SEM. Control n = 26–28, L-NAME n = 36–39. A Mann–Whitney test was used to assess statistical differences in albumin:creatinine between L-NAME and control group at each time point.
Figure S2. Effect of L-NAME administration on expression of genes associated with renal function and dysfunction, collected at E17.5 of pregnancy.

Gene expression was assessed with qPCR. (A, B, C, D, E, F, H, J, K, L, M, N, O, P, Q, R, S, T, U, V, W) There was no significant change to any genes assessed in the kidneys collected from the mice treated with L-NAME compared to the controls (Ccr2, Ctgf, Fn1, Hsd11b2, Il-1β, Mmp2, Mmp9, Nhe1, Nlrp3, Nox2, Nox4, Nr3c1, Nr3c2, Sccn1a, Sgk1, Tgfβ1, Tgfβ2, Tgfβ3, Tnf, and Vcam1. (G, I) Mmp2 and Mmp9 expression were also expressed in a ratio to Timp1 (matrix metalloproteinase inhibitor) expression, but showed no significant difference between the control and L-NAME groups. Data presented as mean ± SEM. n = 6–10 mice/group. An unpaired t test (if normally distributed) or Mann–Whitney test was used to assess statistical differences between L-NAME and control group.
However, histological analysis revealed features in L-NAME mice that could be pathological in nature. In regions, there appeared to be haemoglobin and hyaline casts with inflammatory infiltrate and structural changes to the surrounding tubules (45). There also appeared to be increased cellularity and swelling of several glomeruli, and narrowing of the Bowman’s space, compared with the control mice. This could be a sign of the renal pathology seen in individuals whose pregnancies are complicated by preeclampsia (46, 47). Haemoglobin-type casts were also visible in the control mouse kidneys; however, these are not surrounded by inflammatory or irregular cellular patterns and are likely to be artifacts (Fig 2C–F).
L-NAME administration impairs fetal and placental growth
Placental dysfunction in preeclampsia can be associated with fetal growth restriction. Following L-NAME administration, we recorded a significant reduction in placental weight (P = 0.0423; Fig 3B) and fetal crown-to-rump length (P = 0.017; Fig 3D) corresponding to the dams culled at E17.5. However, neither fetal weight (Fig 3A), nor the fetal-to-placental weight ratio (Fig 3C) were altered. Pups of mice administered L-NAME had significantly reduced birthweight (P = 0.0234; Fig 3E) with no significant difference in crown-to-rump length (Fig 3F) compared with control at birth. There was no significant difference in the overall litter size (data not shown). L-NAME administration did not alter gestational length—all mice gave birth by the morning of E19.5.
Figure 3. Effect of L-NAME administration on E17.5 fetal, placental, and Day 1 pup size.

(A) E17.5 fetal weight, (B) E17.5 placental weight, (C) E17.5 fetal to placental weight ratio, (D) E17.5 crown-to-rump length, (E) pup birthweight (F) pup crown-to-rump length at birth. (A, B, C, D) L-NAME administration in pregnancy significantly decreased E17.5 placental weight (B) and crown-to-rump length (D), but not fetal weight (A) or the fetal to placental weight ratio (C). (E, F) At birth, pups from mice administered L-NAME had reduced pup weight (E), with no significant change to crown-to-rump length (F). Data presented as mean ± SEM. E17.5 Litters: control n = 9, L-NAME n = 11. E17.5 fetuses: control n = 68, L-NAME n = 97. At birth, litters: Control n = 35, L-NAME n = 32. Pups: Control n = 274, L-NAME n = 246. Each column of points represents the fetuses, pups or placentas corresponding to each dam. Black line and error bars represent mean ± SEM within each litter. The red transparent line across each group represents the mean across all litters. Data were statistically analysed using a nested t test; *P < 0.05, **P < 0.01.
At E17.5, the placentas of mice administered L-NAME demonstrated significantly increased expression of the anti-angiogenic Flt1 transcript (P = 0.0049; Fig 4B), and antioxidant Hmox-1 (P = 0.0193; Fig 4G). L-NAME administration did not alter placental expression of Vegfa, Plgf, Nos3, Tnf, or Vcam1 (Fig 4A and C–F). Placental structure was also studied for a subset of placentas. The placentas collected from mice given L-NAME did not have significantly altered cross-sectional area, or changes in the blood space, junctional zone, and labyrinth zone areas (Fig S3). Mean arterial blood pressure corresponding to the subset of mice used for these analyses (culled at E17.5) is presented in Fig S4.
Figure 4. Effect of L-NAME administration on placental gene expression at E17.5.

Gene expression was assessed via qPCR. (B, G) Expression of Flt1 (B) and Hmox-1 (G) were significantly increased in placentas of mice administered L-NAME compared with control. (A, C, D, E, F) L-NAME did not alter placental expression of Vegfa, Plgf, Nos3, Tnf, or Vcam1. Data are presented as mean ± SEM. Control n = 6 mice, L-NAME n = 10 mice, 1–3 placentas were chosen at random from each. Samples with low RNA yield at extraction were excluded. Data were statistically analysed using an unpaired t test (if in normal distribution) or Mann–Whitney test; *P < 0.05, **P < 0.01.
Figure S3. Effect of L-NAME administration on placental structure.

(A, B, C, D) Effect of L-NAME administration on placental (A) cross sectional area, (B) blood space, (C) junctional zone, and (D) labyrinth zone. Placental histology was assessed using ScanScope system and Aperio ImageScope software. L-NAME did not significantly alter the cross-sectional area of the placenta. Placentas from the mice culled at E17.5 were chosen at random for analysis, each placenta from a different dam. Data presented as mean ± SEM. n = 3–5 mice/group. A Mann–Whitney test was used to assess statistical differences between L-NAME and control group.
Figure S4. Mean arterial blood pressure corresponding to subset of mice culled at E17.5 of pregnancy.

Blood pressure measured by tail cuff plethysmography. (A, B) L-NAME administration significantly increased systolic and diastolic blood pressure at both (A) E14.5 and (B) E17.5. These mice were used for further organ and blood assessments at E17.5. Data presented as mean ± SEM. Control n = 8–9, L-NAME n = 11. An unpaired t test was used to assess statistical differences between L-NAME and control group at each time point; *P < 0.05, ****P < 0.0001.
L-NAME administration in vivo increases circulating factors associated with preeclampsia in pregnancy, but does not alter ex vivo vascular reactivity
Next, we assessed levels of soluble factors known to be elevated in the maternal circulation with preeclampsia. L-NAME administration caused a significant increase in serum levels of the inflammatory marker, CRP (P = 0.0205; Fig 5A), anti-angiogenic factor, sFLT-1 (P = 0.0238; Fig 5B), and potent vasoconstrictor, ET-1 (P = 0.0486; Fig 5C).
Figure 5. Effect of L-NAME administration on circulating factors associated with preeclampsia and vascular reactivity at E17.5 of pregnancy.

Circulating factors were measured by ELISA. Vascular reactivity of mesenteric arteries was assessed by wire myography. (D, E) Vasoconstriction to phenylephrine (E) Vasodilation to acetylcholine. (A, B, C) C-reactive protein, (B) sFLT-1, and (C) ET-1 were significantly increased in the mice administered L-NAME compared with controls. There was no change in vascular reactivity with L-NAME administration. LogEC50, area under the curve and maximal response for these curves are presented in Fig S5. Data are presented as mean ± SEM. Control n = 7–9 mice, L-NAME n = 7–11 mice. Levels of circulating factors were statistically analysed using an unpaired t test (for normal distribution) or Mann–Whitney test; *P < 0.05. Vascular response at each dose of agonist was statistically analysed using mixed-effects analysis with Šidák correction for multiple comparisons.
Preeclampsia features vascular dysfunction, leading to systemic maternal hypertension. Administration of L-NAME throughout pregnancy did not alter vasoconstriction to phenylephrine (Fig 5D) or vasodilation to acetylcholine (Fig 5E), assessed in ex vivo wire myography experiments. There were no changes to LogEC50 (half-maximal response corresponding to the shift of the curve), area under the curve (total response) or maximum response (Fig S5). The arteries from each group had no significant difference in response to the high potassium solution (100 mM KPSS; Fig S6).
Figure S5. LogEC50, area under the curve and maximal response of mesenteric arteries collected at E17.5 of pregnancy to phenylephrine and acetylcholine.

(A, B, C, D, E, F) Vessels collected from mice administered L-NAME had no changes in LogEC50, area under the curve, maximal constriction (Emax) or relaxation (Rmax) compared with the controls, with addition of phenylephrine (A, B, C) or acetylcholine (D, E, F). Data presented as mean ± SEM. n = 7 mice/group. Ambiguous LogEC50 values excluded. Fig 5 presents the corresponding dose–response curves. A Mann–Whitney test was used to assess statistical differences between L-NAME and control group for each parameter.
Figure S6. Response of mesenteric arteries collected at E17.5 to high potassium physiological salt solution (100 mM).
Response was measured via wire myography. The arteries collected from mice administered L-NAME in pregnancy did not have significantly altered response to the high potassium solution compared with the arteries collected from the control mice. Data presented as mean ± SEM. n = 7 mice/group. Data were statistically analysed using an unpaired t test.
Consistent with this, L-NAME administration did not alter mesenteric artery expression of the enzyme responsible for production of nitric oxide Nos3, the endothelial dysfunction marker Vcam-1, or the receptors for vasoconstrictor endothelin-1, Ednra and Ednrb (Fig S7).
Figure S7. Effect of L-NAME administration on expression of genes associated with vascular function and endothelial dysfunction in mesenteric arteries collected at E17.5 of pregnancy.

Gene expression was assessed with qPCR. (A, B, C, D) There was no significant change in expression of nitric oxide synthase Nos3 (A), endothelial dysfunction marker Vcam-1 (B), or ET-1 receptors Ednra (C) and Ednrb (D) in the vessels collected from the mice treated with L-NAME compared with the controls. Data presented as mean ± SEM. Vessels from n = 9–10 mice/group at each time point were pooled (2–3 samples each pool). A Mann–Whitney test was used to test statistical differences between the L-NAME and control group.
L-NAME administration in pregnancy does not alter maternal blood pressure, or circulating levels of factors associated with endothelial dysfunction up to 10 wk post-delivery
We assessed whether the preeclampsia-like phenotype induced by L-NAME could alter long-term blood pressure, as individuals have a higher risk of hypertension after a pregnancy complicated by preeclampsia (48). Despite mice administered L-NAME experiencing elevated blood pressure during pregnancy, their diastolic, systolic, and mean arterial blood pressure returned to control levels following discontinuation of L-NAME treatment at 1 wk post-delivery, and remained at control levels up to 10 wk post-delivery (Figs 6A and S8). Furthermore, circulating levels of ET-1 and CRP also returned to control levels at 1 wk post-delivery, and remained so up to 10 wk post-delivery (Fig 6B and C).
Figure 6. Effect of L-NAME administration in pregnancy on mean arterial blood pressure, and serum levels of endothelin-1 and C-reactive protein at 1-,2-, 4-, and 10-wk post-delivery.

Blood pressure was measured via tail cuff plethysmography and circulating factors by ELISA. (A, B, C) L-NAME administration in pregnancy did not alter post-delivery (A) blood pressures, or circulating (B) ET-1 and (C) C-reactive protein levels at any time point. Data presented as mean ± SEM. Blood pressures–Control: 1 wk n = 35, 2 wk n = 27, 4 wk n = 15, 10 wk n = 9; L-NAME: 1 wk n = 32, 2 wk n = 24, 4 wk n = 13, 10 wk n = 8. ELISAs were carried out using serum samples from n = 6–9 mice/group at each time point. Data were statistically analysed between the L-NAME and control group at each time point with a unpaired t test (if in normal distribution) or Mann–Whitney test.
Figure S8. Effect of L-NAME administration in pregnancy on systolic and diastolic arterial blood pressure post-delivery.

Blood pressure measured via tail cuff plethysmography. (A, B) Neither systolic (A) nor diastolic (B) blood pressure of mice administered L-NAME during pregnancy were significantly different from control levels at 1, 2, 4, or 10 wk post-delivery. Data presented as mean ± SEM. Control: 1 wk n = 35, 2 wk n = 27, 4 wk n = 15, 10 wk n = 9; L-NAME: 1 wk n = 32, 2 wk n = 24, 4 wk n = 13, 10 wk n = 8. An unpaired t test (if in normal distribution) or Mann–Whitney test were used to compared blood pressure in the L-NAME and control groups at each time point.
L-NAME administration in pregnancy does not alter post-delivery urine albumin to creatinine ratio, but is associated with up-regulation of Mmp9:Timp1 at 10 wk post-delivery
Consistent with findings during pregnancy, mice administered L-NAME did not have proteinuria (altered urine albumin to creatinine ratio) at any time point post-delivery (Fig S9). However, we found that Mmp9:Timp1 expression was elevated in the mice given L-NAME, at 10 wk post-delivery (P = 0.0418; Fig 7A). Mmp2 expression was also elevated at 10 wk post-delivery (P = 0.0229; Fig S10F), along with Mmp9 expression alone (P = 0.0311; Fig S10H); however, the ratio of Mmp2 to its inhibitor Timp1 was not altered (Fig S10G). There were no other changes in expression of genes associated with renal function and dysfunction (Ccr2, Ctgf, Fn1, Hsd11b2, IL-6, Nhe1, Nlrp3, Nox2, Nox4, Nr3c1, Nr3c2, Scnn1a, Sgk1, Tgfβ1, Tgfβ2, Tgfβ3, Timp1, Tnf, or Vcam-1; Fig S10A–E and I–V).
Figure S9. Effect of L-NAME administration in pregnancy on albumin to creatinine ratio post-delivery.

Albumin and creatinine concentrations assessed via ELISA and enzymatic assay, respectively. There was no significant change in albumin:creatinine at any time point post-delivery. Data presented as mean ± SEM. n = 6–9 mice/group at each time point. A Mann–Whitney test was used for statistical analysis between the L-NAME and control group at each time point.
Figure 7. Effect of L-NAME administration in pregnancy on long-term expression of markers of dysfunction in the maternal heart and kidney.

(A) Kidney expression of Mmp9 presented as a ratio to Timp1 (its inhibitor) expression. (B) Heart expression of Tnf. Relative expression at 1-,2-,4-, and 10-wk post-delivery was assessed by qPCR. Kidney Mmp9:Timp1 and heart Tnf expression were not altered in mice administered L-NAME in pregnancy compared with control at 1-, 2-, 4-, or 10- wk post-delivery. At 10 wk post-delivery, mice administered L-NAME in pregnancy had significantly increased kidney Mmp9:Timp1 expression and increased heart Tnf expression. Data are presented as mean ± SEM. Data were statistically analysed between the L-NAME and control group at each time point with a unpaired t test (if in normal distribution) or Mann–Whitney test; *P < 0.05, **P < 0.01. n = 7–9 kidneys/group, n = 4–8 hearts/group at each time point.
Figure S10. Expression of genes associated with kidney function and dysfunction in mice administered L-NAME in pregnancy, at 1-, 2-, 4-, and 10-wk post-delivery.

Kidney gene expression assessed by qPCR. (A, B, C, D, E, I, J, K, L, M, N, O, P, Q, R, S, T, U, V) There were no changes in expression in 11Bhsd2, Ccr2, Ctgf, Fn1, IL-6, Nhe1, Nlrp3, Nox2, Nox4, Nr3c1, Nr3c2, Scnn1a, Sgk1, Tgfβ1, Tgfβ2, Tgfβ3, Timp1, Tnf, or Vcam-1 post-delivery. (F, G) Mmp2 was significantly elevated in the L-NAME group at 10 wk post-delivery (F), but Mmp2 expression as a ratio to Timp1 (its inhibitor) expression was not altered (G). (H) Mmp9 expression was significantly elevated in the L-NAME group at 10 wk post-delivery (H); expression as a ratio to Timp1 is presented in Fig 7A. Data presented as mean ± SEM. n = 7–9 mice/group at each time point. An unpaired t test (if in normal distribution) or Mann–Whitney test was used to assess statistical differences between L-NAME and control group at each time point.
L-NAME administration in pregnancy is associated with increased Tnf expression in maternal hearts at 10 wk post-delivery
We examined gene expression in hearts of the dams post-delivery to assess potential long-term effects on cardiac function. We found that Tnf expression was significantly increased in the hearts of L-NAME treated mice at 10 wk post-delivery (P = 0.0078), but not at 1-, 2-, or 4-wk post-delivery (Fig 7B). There was no change in expression of the other cardiac genes assessed (Bnp, Camk2a, Ccr2, Ctgf, Il-6, Mmp2, Mmp9 Nlrp3, Nox2, Nr3c1, Nr3c2, Tgfβ1, Tgfβ2, Tgfβ3, Timp1 Vcam-1, Mmp2:Timp1, and Mmp9:Timp1; Fig S11).
Figure S11. Effect of L-NAME administration in pregnancy on expression of functional genes in hearts 1-, 2-, 4-, and 10 wk post-delivery.

Expression was assessed via qPCR. (A, B, C, D, E, F, H, J, K, L, M, N, O, P, Q, R, S) There were no changes in Bnp, Camk2a, Ccr2, Ctgf, Il-6, Mmp2, Mmp9, Nlrp3, Nox2, Nr3c1, Nr3c2, Tgfβ1, Tgfβ2, Tgfβ3, Timp1, or Vcam-1 gene expression at any time point post-delivery. (G, I) Mmp2 and Mmp9 expression were also expressed as a ratio to Timp1 expression, but both were not altered between the groups at any time point post-delivery. Data presented as mean ± SEM. n = 4–8 mice/group. An unpaired t test (if in normal distribution) or Mann–Whitney test was used to assess statistical differences between L-NAME and control group at each time point.
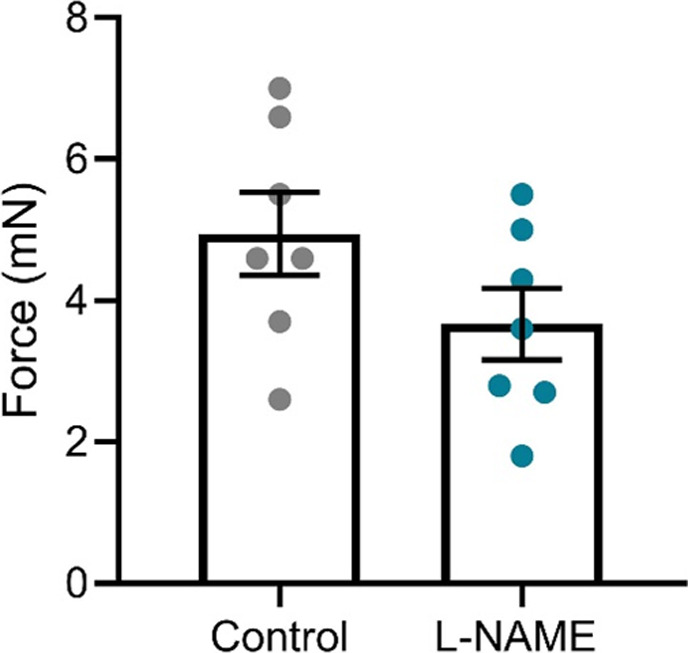
L-NAME administration in pregnancy is associated with increased mesenteric artery vasoconstriction to phenylephrine at 10 wk post-delivery
To assess the long-term effects to the vasculature, we also examined vascular reactivity of mesenteric arteries 1-, 2-, 4-, and 10-wk post-delivery. L-NAME administration in pregnancy had no effect on mesenteric artery vasoconstriction to phenylephrine at 1-, 2-, and 4-wk post-delivery (Fig 8A–C). However, at 10 wk post-delivery, the arteries from the mice treated with L-NAME in pregnancy constricted significantly more to 10−6M phenylephrine than control (P = 0.0178; Fig 8D). L-NAME administration in pregnancy did not alter vasorelaxation to acetylcholine at any time point post-delivery (Fig 8E–H). There were no changes in LogEC50, AUC or maximal response (Emax–maximum constriction; Rmax–maximum relaxation) at any time point to either phenylephrine or acetylcholine (Figs S12 and S13). The arteries from each group had no significant difference in response to the high potassium solution at each time point (100 mM KPSS; Fig S14).
Figure 8. Effect of L-NAME administration in pregnancy on vascular reactivity post-delivery.

Effect of L-NAME administration in pregnancy on vascular reactivity measured in mesenteric arteries collected at 1 wk (A, E), 2 wk (B, F), 4 wk (C, G), and 10 wk (D, H) post-delivery. Vascular reactivity was assessed using ex vivo wire myography, assessing vasoconstriction to phenylephrine and vasodilation to acetylcholine. (A, B, C) Arteries collected from the mice administered L-NAME in pregnancy did not respond differently to phenylephrine compared with controls at the 1-, 2-, and 4-wk time points post-delivery. (D) At 10 wk post-delivery, the arteries collected from the mice that were administered L-NAME in pregnancy constricted significantly more than the control vessels at 10−6M phenylephrine. (E, F, G, H) The vessels collected from the mice administered L-NAME in pregnancy did not have altered vasodilation to acetylcholine at any time point post-delivery. LogEC50, area under the curve and maximal response for these curves are presented in Figs S12 and S13. Data presented as mean ± SEM. n = 7–9 mice/group at each time point. Vascular response at each dose of agonist were statistically analysed using mixed-effects analysis with Šidák correction for multiple comparisons.*P < 0.05.
Figure S12. LogEC50, area under the curve and maximal response of mesenteric arteries to phenylephrine post-delivery.

(A, B, C, D, E, F, G, H, I, J, K, L) LogEC50, area under the curve and maximal constriction of mesenteric arteries collected at 1 wk (A, B, C), 2 wk (D, E, F), 4 wk (G, H, I), and 10 wk (J, K, L). Vessels collected from mice administered L-NAME had no changes in LogEC50, area under the curve or maximal constriction (Emax) compared with the controls with addition of phenylephrine. Data presented as mean ± SEM. n = 7–9 mice/group. Ambiguous LogEC50 values excluded. Fig 8 presents the corresponding dose–response curves. An unpaired t test (if in normal distribution) or Mann–Whitney test was used to assess statistical differences between L-NAME and control group at each time point.
Figure S13. LogEC50, area under the curve and maximal response of mesenteric arteries to acetylcholine post-delivery.

(A, B, C, D, E, F, G, H, I, J, K, L) LogEC50, area under the curve and maximal relaxation of mesenteric arteries collected at 1 wk (A, B, C), 2 wk (D, E, F), 4 wk (G, H, I), and 10 wk (J, K, L) post-delivery. Vessels collected from mice administered L-NAME had no changes in LogEC50, area under the curve or maximal relaxation (Rmax) compared with the controls with addition of acetylcholine. Data presented as mean ± SEM. n = 7–9 mice/group. Ambiguous LogEC50 values excluded. Fig 7 presents the corresponding dose–response curves. An unpaired t test (if in normal distribution) or Mann–Whitney test was used to assess statistical differences between L-NAME and control group at each time point.
Figure S14. Mesenteric artery response to high potassium salt solution post-delivery.

(A, B, C, D) Response of mesenteric arteries collected at (A) 1 wk, (B) 2 wk, (C) 4 wk, and (D) 10 wk post-delivery to high potassium physiological salt solution (100 mM). Response was measured via wire myography. The arteries collected from mice administered L-NAME did not have significantly altered response to the high potassium solution compared with the arteries collected from the control mice at any time point assessed. Data presented as mean ± SEM. n = 7–9 mice/group. An unpaired t test (if in normal distribution) or Mann–Whitney test was used to assess statistical differences between L-NAME and control group.
We also assessed expression of genes altered in vascular dysfunction. There were no changes in vascular expression of Nos3, Vcam1, Ednra, and Ednrb at any time point post-delivery (Fig S15).
Figure S15. Effect of L-NAME administration in pregnancy on post-delivery mesenteric artery expression of genes associated with vascular dysfunction.

Expression was assessed by qPCR at 1, 2, 4, and 10 wk post-delivery. (A, B, C, D) There was no change in expression of nitric oxide synthase Nos3 (A), endothelial dysfunction marker Vcam-1 (B), or ET-1 receptors Ednra (C) and Ednrb (D) in the mice administered L-NAME in pregnancy compared with the controls at any time point post-delivery. Data presented as mean ± SEM. Vessels from n = 8–9 mice/group at each time point were pooled. A Mann–Whitney test was used to assess statistical differences between L-NAME and control group at each time point.
Discussion
In this study, we established the L-NAME mouse model of preeclampsia in (CBA x C57BL/6) F1 mice, and assessed whether this model could recapitulate the long-term effects of preeclampsia on maternal cardiovascular health. During pregnancy, L-NAME administration markedly increased blood pressure, increased circulating ET-1, CRP, and sFLT-1 levels, and restricted fetal growth. Despite recapitulating key aspects of preeclampsia in the mouse, there were no major long-term cardiovascular impacts post-partum. Except, at 10 wk post-delivery, we observed increased expression of Tnf and Mmp9:Timp1 in the maternal heart and kidney, respectively, and moderately enhanced vasoconstriction.
L-NAME, a potent vasoconstrictor, enables the simulation of the systemic vasoconstriction that occurs in preeclampsia. This vasoconstriction is likely responsible for the impaired fetal and placental growth detected in our model. Ma et al have previously shown that L-NAME administration initiated in early gestation is able to induce feto-placental unit impairment, compared with administration started late in pregnancy (34). We suggest that early administration of L-NAME in our model leads to perturbations during early placental development, given we continue to observe growth restriction even after cessation of L-NAME. Moreover, we suggest the vasoconstriction caused by L-NAME, reduced blood flow to the feto-placental unit, restricting fetal growth. These early changes are especially relevant to early-onset preeclampsia, which is associated with higher rates of adverse fetal outcomes and placental insufficiency (49). Although pup and placental weight were both reduced, fetal:placental weight ratio was not altered, and placental cross-sectional area was also unchanged. This suggests that any impairments in placental capacity to pass nutrients to the fetus were not due to structural changes of the placenta. Rather, the fetal growth impairment may be due to reduced total blood supply due to vasoconstriction of the uterine arteries. Further studies are required to uncover the mechanisms behind this growth impairment, including whether there are any clinical parameters of interest including fetal brain sparing.
Interestingly, L-NAME increased placental expression of Flt1 and circulating sFLT-1. sFLT-1 is known to be increased in the circulation of individuals with pregnancies complicated by preeclampsia (50). This clearly demonstrates that administration of L-NAME caused an up-regulation of this key pathway, offering a compelling model of preeclampsia. In other studies, L-NAME rodent models demonstrate mixed results for changes in Flt1 and its soluble circulating form, although some of these discrepancies may be due to the varied L-NAME concentrations and routes of administration used (31, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62). Complex viral vector models inducing placental specific sFLT-1 overexpression have been developed to model the increased placental secretion of sFLT-1 akin to that observed in preeclampsia in women (19, 63). However, a strength of the model we demonstrate here is this model provides a cheaper, simpler and more accessible way to induce key characteristics of preeclampsia in a small animal model, with ability to study promising candidate drugs to mitigate key aspects of preeclampsia pathogenesis.
Here, we also found that mice receiving L-NAME demonstrated increased placental expression of Hmox-1, a cytoprotective antioxidant enzyme. We hypothesise that this may have been triggered by tissue hypoxia as a result of markedly reduced blood flow, as has been observed in other rat, mouse, bovine and monkey models of hypoxia (64, 65), perhaps an adaptation to mediate oxidative stress in the placenta (contrasting to in human cells, where hypoxia is a repressor of Hmox-1 (64)). However, this contrasts with other studies using L-NAME in rats, where they found placental Hmox-1 protein was reduced with L-NAME administration (54, 66, 67, 68). Studies examining the effects of L-NAME on placental oxidative stress in rats have shown mixed results (68, 69, 70, 71, 72). In L-NAME mouse models, placental Hmox-1 levels have not previously been evaluated, though one study reports that L-NAME is associated with fatty acid oxidation (32). Examination of the duration of L-NAME administration and evidence of oxidative stress levels would help determine the effect of L-NAME on placental oxidative stress and Hmox-1. This is of particular interest to our group, as we have previously demonstrated that HMOX-1 is not altered in human preeclamptic placental tissue (73).
L-NAME treatment did not induce evident signs of proteinuria in the pregnant dams, as measured by albumin to creatinine ratio (although we note this is not a clinical grade test). Increased albumin concentration in the urine is associated with kidney damage and renal injury, with normalisation to creatinine (consistently released). However, histological assessment of the kidneys demonstrated signs of pathology in the mice given L-NAME during pregnancy. It is possible that the kidney damage was not severe enough to cause proteinuria in this model. Others have reported L-NAME in mouse models demonstrate proteinuria, but these studies have based this finding on altered total urine protein (32, 35, 36), albumin alone (33), or creatinine alone (31), and not the albumin to creatinine ratio, which is a better measure, and used clinically. Furthermore, we did not find changes in expression of genes associated with kidney damage or dysfunction.
Elevated circulating levels of the potent vasoconstrictor ET-1, inflammatory marker CRP and anti-angiogenic factor sFLT-1 in our L-NAME model mirror the increase found in the circulation of individuals with preeclampsia (50, 74, 75). Intriguingly, this was not accompanied with a change in mesenteric artery vascular reactivity. We propose that this may be because L-NAME may not have a permanent effect on the vasculature, but rather works through its ability to cause vasoconstriction through decreasing production of nitric oxide. Hence, when the arteries are no longer exposed to L-NAME in an ex vivo system outside the body, nitric oxide levels can recover. Although L-NAME is commonly directly applied to vessels in ex vivo studies (used for over 20 yr (76)), few studies have explored the effect of administering L-NAME in vivo on vascular reactivity. One study showed that L-NAME administration impaired acetylcholine induced vasorelaxation in rat aortic rings (77) and another that L-NAME impaired myogenic tone of rat uterine radial arteries (78). However, it is understood that vascular adaptations in pregnancy are distinct between rats and mice (79), which may explain the discrepancy.
In this study, we chose to assess vascular reactivity in mesenteric arteries, as they are resistance arteries involved in control of blood pressure (80, 81), and allowed us to assess systemic changes in pregnancy (that can be compared with post-pregnancy responses) (79). However, future studies may benefit from investigation of uterine artery vascular reactivity to help understand the mechanisms behind the impaired placental and pup size. In addition, phenylephrine and acetylcholine were used to assess vascular reactivity as their receptors are present on mesenteric arteries, and they are commonly used to assess reactivity in the myograph field. However, it may be beneficial to assess the response to other agonists, including the vasoconstrictor, ET-1, which we know is elevated in the circulation in this model, and in individuals with preeclampsia (82).
An important part of this study was also to investigate whether the L-NAME-induced changes could impact long-term maternal cardiovascular health—an important consideration currently lacking in many established models of preeclampsia. Preeclampsia and gestational hypertension are associated with increased postpartum maternal cardiovascular disease risk (10, 11, 12, 13, 14). Hence, we expected that mice administered L-NAME would have altered cardiovascular disease risk indices post-pregnancy.
Because blood pressure returned to control levels within 1 wk post-delivery, we suspect that there could be recovery in nitric oxide levels with the cessation of L-NAME administration, permitting vasodilation and reducing blood pressure. After being elevated in pregnancy, the potent vasoconstrictor ET-1 and the inflammatory mediator CRP both returned to control levels post-delivery with L-NAME cessation. As these factors play a role in enhancing endothelial dysfunction, it is likely that their return to control levels also contribute to recovery of blood pressure. This is unlike what happens after a hypertensive pregnancy, where blood pressure often remains elevated, persisting more than 20 yr post-pregnancy (48), and where endothelial dysfunction persists (83, 84).
Proteinuria in preeclampsia can persist post-delivery, although some studies suggest this is resolved by 2 yr post-delivery (85, 86, 87). As L-NAME did not induce proteinuria in our animals during pregnancy, we did not expect long-term changes in the kidney post-delivery. However, at 10 wk post-delivery, the mice administered L-NAME in pregnancy had increased renal Mmp9:Timp1 expression. Mmp9 is a matrix metalloproteinase that (in balance with its inhibitor, Timp1) is involved in cleavage of extracellular matrix and other proteins, important for normal kidney function and structure (88). Studies have also shown Mmp9 has a role in inflammation (89, 90, 91). The elevation in Mmp9:Timp1 expression may imply an increase in inflammation in the kidneys, but further molecular studies are needed to investigate this.
Studies have observed increased cardiac dysfunction (left ventricular hypertrophy) at 1 yr post-preeclampsia (12), and an association between preeclampsia and increased risk of admission to hospital or death due to ischemic heart disease (92). Interestingly, at 10 wk post-delivery (equivalent to ∼9.5 yr in human) Tnf mRNA expression was elevated in the hearts of mice administered L-NAME in pregnancy, implying increased cardiac inflammation over time. Further studies to determine if there is an increase in Tnf protein production and other markers of inflammation are needed to confirm this. Exploration of the cardiac structure and function may be helpful in evaluating whether our model produces the same left ventricular dysfunction seen post-preeclampsia.
Because persistent endothelial dysfunction is likely to contribute to increased risk of cardiovascular disease, we assessed whether vascular reactivity may be altered post-delivery. Although there were no changes in vascular reactivity at E17.5 gestation, we found increased mesenteric artery constriction at 10 wk post-delivery at one dose of phenylephrine. This would imply that L-NAME administration may have exerted some permanent effects, either directly or indirectly (as the hypertensive phenotype may have driven other indirect changes that required time to become apparent), and further studies would be required to examine this. Increased sensitivity to vasoconstrictors would be considered detrimental; however, we do not know how functionally relevant the moderate difference we detected may be. It could be a sign of increased arterial wall stiffness as demonstrated in a study examining mice at 4 wk post-partum (93). Assessing vascular responses to other agonists may provide additional insight into other pathways that may be altered by L-NAME.
We did not observe significant changes in expression of inflammatory, or endothelial dysfunction-related genes in the mesenteric arteries post-delivery. However, there are limited data as the samples had to be pooled, leading to a low effective sample size. Increasing the sample size and examining protein levels would be valuable to discern the expression of endothelial dysfunction-relevant genes post-delivery.
The long-term changes identified here are far less adverse than what we expected, based on the increased cardiovascular risk known post-preeclampsia. In our study, we chose time points up to 10 wk postpartum, modelling the human equivalent of ∼1, 2, 4, and 9.5 yr post-delivery. Expanding the study to include later time points (beyond 10 wk post-delivery) may have uncovered further cardiovascular changes, as another model has shown (94). However, the time points used in this study were chosen to reflect the clinical phenomena, examining earlier time points akin to human clinical observations, as individuals demonstrate impaired cardiovascular function soon after a pregnancy complicated by preeclampsia (13, 48).
As the L-NAME model is thought to mainly cause reduced nitric oxide levels driving systemic vasoconstriction, the preeclampsia phenotype observed here may not be as severe as that in other models, including knockout models (95). Indeed, our study highlights that L-NAME administration started in pregnancy alone is unable to induce the severe long-term phenotype expected. In the preeclampsia field, a dogma exists to whether those who get preeclampsia already have predisposition for cardiovascular disease (96), or whether pregnancy acts as the “stress test,” exacerbating this risk (97). Here we selected mice that should not have an underlying predisposition for cardiovascular disease, nor stressors that could exacerbate risk of cardiovascular disease post-delivery. Assessment using a strain that is more sensitive to L-NAME may demonstrate a more persistent chronic effect on the cardiovascular system (37). In addition, it would be interesting to use a strain or genetic model with increased cardiovascular disease risk, potentially an obese or high salt diet mouse model, or a strain genetically modified to enhance vascular dysfunction, mimicking predisposition to preeclampsia and long-term cardiovascular disease. Some studies have also used a cardiovascular injury event post-delivery to assess whether the preeclamptic pregnancy may make the system more sensitive to injury (98). Another important aspect would be to assess whether lactation may have an effect on cardiovascular health post-hypertensive pregnancy, as studies have shown that lactation may reduce vasocontractility, enhance vasorelaxation and increase vessel distensibility (99). Here, the dams were removed from the pups following birth, and thus this would constitute a higher likelihood of hypertension and vascular dysfunction if this association held in this model.
Conclusion
Preeclampsia is a complex, multi-organ condition and is still not completely understood. We acknowledge that it is unlikely that one model alone would be able to cover all aspects of preeclampsia pathogenesis. However, the L-NAME model described here provides a relatively simple small animal model of preeclampsia that can be used to assess the potential beneficial effects of candidate therapeutics that can target and mitigate systemic vascular constriction. However, it does not amply model the long-term cardiovascular effects seen in individuals soon after a pregnancy complicated by preeclampsia. Knowing that preeclampsia is associated with increased risk of cardiovascular disease post-pregnancy, it is essential that we develop precise models of its long-term effects and develop interventions that not only improve obstetric outcomes, but also improve long-term health.
Materials and Methods
Animal studies
Animal experiments were approved by the Austin Health Animal Ethics Committee (A2018/05596) and followed the National Health and Medical Research Council ethical guidelines for the care and use of animals for scientific purposes. To have sufficient power for the final time points, a larger cohort of mice were required. 3-wk-old CBA x C57BL/6 (F1) female mice (n = 87) were sourced from the Florey Institute of Neuroscience and Mental Health (The University of Melbourne). Mice were group-housed in conventional open-top cages, on a 12-h light/dark cycle, with food and water available ad libitum (18–22°C; 50% relative humidity). Before experimentation, mice were acclimated to the CODA non-invasive blood pressure system (Kent Scientific) in a seven-stage process involving exposure to the restraining tube and tail cuff.
From 6 to 16 wk of age (equivalent to ∼15–25 yr in human equivalent (100)), F1 females were mated overnight with stud F1 male mice. Pregnancy was confirmed by the presence of a copulatory plug the following morning; designated as embryonic day (E)0.5.
L-NAME mouse model of preeclampsia
L-NAME (50 mg/kg/day; Sigma-Aldrich (30, 101)) or phosphate buffered saline (PBS; 137 mM NaCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, and 2.7 mM KCl, pH 7.4) as control was administered daily from embryonic day (E)7.5 to E17.5 of pregnancy (approximately early second trimester to term in human equivalent to model early onset disease) via 100 μl subcutaneous injection. On E14.5 and E17.5 of pregnancy, urine was collected (spot collection on/upon handling), and blood pressure measured.
After blood pressure measurement on E17.5 a subset of mice (control n = 9, L-NAME n = 11) were anesthetized with 5% isoflurane in oxygen, and cardiac puncture performed to collect maternal blood. Mice were then culled by cervical dislocation. Blood samples were allowed to coagulate at room temperature before centrifugation to separate the serum fraction, which was snap frozen and stored at −80°C. Maternal kidneys were either fixed overnight in 10% neutral buffer formalin or preserved in RNAlater. Fetuses were counted and weighed, and crown-to-rump length measured (with digital calipers). Placentas were also collected, weighed, and preserved in RNAlater (Invitrogen), or fixed in 10% neutral buffered formalin. Tissues remained in RNAlater for a minimum of 48 h (as per the manufacturer’s recommendations), then snap frozen and stored at −80°C until subsequent analysis. Formalin-fixed tissue was embedded in paraffin and sectioned on a microtome for examination of structural changes. The intestinal tract was collected in ice cold PBS for dissection of mesenteric arteries for vascular studies.
All remaining mice (control n = 35, L-NAME n = 32) were allowed to litter naturally for long-term follow-up of dams post-delivery. Day (D)1 pups were counted, weighed, and crown-to-rump length recorded before euthanasia by decapitation. Long-term maternal urine collection, blood pressure measurements and culls were performed at 1-, 2-, 4-, and 10-wk post-delivery. At cull (anaesthesia and cervical dislocation), cardiac puncture was performed and urine, maternal organs (heart and kidney collected in RNAlater) and intestinal tract collected as described above.
Vascular reactivity studies
Second order mesenteric arteries were carefully dissected from surrounding connective and adipose tissue in Krebs physiological salt solution (NaCl 120 mM, KCl 5 mM, MgSO4 1.2 mM, KH2PO4 1.2 mM, NaHCO3 25 mM, D-glucose 11.1 mM, and CaCl2 2.5 mM). Dissected arteries (2 mm length) were then mounted on the 620M Wire Myograph (Danish Myo Technology [DMT]) using 25-μm-diameter gold-plated tungsten wires (W005230; Goodfellow), and bathed in Krebs solution continuously bubbled with carbogen (95% O2, 5% CO2), and warmed to 37°C. The arteries were normalised to 100 mm Hg (13.3 kPa) pressure using the DMT normalisation module on LabChart software (ADInstruments) with IC1/1C100 = 1. Smooth muscle viability was confirmed using high potassium physiological salt solution (KPSS; NaCl 25 mM, KCl 100 mM, MgSO4 1.2 mM, KH2PO4 1.0 mM, NaHCO3 25 mM, D-glucose 11.1 mM, and CaCl2 2.5 mM). Endothelial function was assessed by pre-constricting arteries to 50–70% of maximal constriction to KPSS with phenylephrine (Sigma-Aldrich), then relaxed with the endothelial dependent dilator, acetylcholine (Sigma-Aldrich). Confirmation of response to KPSS, and greater than 80% relaxation was required for inclusion of the vessel. Constriction and relaxation dose response curves were then generated using phenylephrine and acetylcholine (10−9 to 10−4.5M).
After collection of second order mesenteric arteries for vascular studies, the remaining mesenteric arteries were excised and preserved in RNAlater as described above.
Quantitative polymerase chain reaction (qPCR)
RNA was extracted from the RNAlater preserved placenta, heart (whole), kidney, and mesenteric arteries using the GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich) and quantified with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Extracted RNA was converted to cDNA using the Applied BiosystemsTM High-Capacity cDNA Reverse Transcription Kit, as per manufacturer guidelines on the iCycler iQ5 (Bio-Rad).
Quantitative PCR with Taqman reagents was performed to quantify mRNA expression using primers purchased from Life Technologies. Primers are listed in Table 1. Quantitative PCR was performed on the CFX384 (Bio-Rad) with the following run conditions: 50°C for 2 min; 95°C for 20 s, 95°C for 3 s, 60°C for 30 s (40 cycles). All expression data were normalised to expression of reference gene (chosen based on their stability in each tissue) as an internal control and calibrated against the average Ct of the control samples, with each biological sample run in technical duplicate.
Table 1.
Primers used for Taqman PCR.
| Gene name | Gene symbol | Taqman primer assay ID |
|---|---|---|
| C–C motif chemokine receptor 2 | Ccr2 | Mm04207877_m1 |
| Calcium/calmodulin-dependent protein kinase II alpha | Cam2kα | Mm00437967_m1 |
| Connective tissue growth factor | Ctgf | Mm01192933_g1 |
| Endothelin receptor A | Ednra | Mm01243722_m1 |
| Endothelin receptor B | Ednrb | Mm00432989_m1 |
| Fibronectin 1 | Fn1 | Mm01256744_m1 |
| FMS-like tyrosine kinase 1 | Flt1 | Mm00438980_m1 |
| Hemoxygenase 1 | Hmox-1 | Mm00516005_m1 |
| Hydroxysteroid 11-beta dehydrogenase 2 | Hsd11b2 | Mm01251104_m1 |
| Interleukin-1 beta | Il-1β | Mm00434228_m1 |
| Interleukin-6 | Il-6 | Mm00446190_m1 |
| Matrix metallopeptidase 2 | Mmp2 | Mm00439498_m1 |
| Matrix metallopeptidase 9 | Mmp9 | Mm00442991_m1 |
| NADPH oxidase 2 | Nox2 | Mm01287743_m1 |
| NADPH oxidase 4 | Nox4 | Mm00479246_m1 |
| Natriuretic peptide type B | Bnp | Mm01255770_g1 |
| Nitric oxide synthase 3 | Nos3 | Mm00435217_m1 |
| NLR Family Pyrin Domain Containing 3 | Nlrp3 | Mm00840904_m1 |
| Nuclear receptor subfamily 3 group C member 1 | Nr3c1 | Mm00433832_m1 |
| Nuclear receptor subfamily 3 group C member 2 | Nr3c2 | Mm01241596_m1 |
| Placental growth factor | Plgf | Mm00435613_m1 |
| Serum/glucocorticoid regulated kinase 1 | Sgk1 | Mm00441380_m1 |
| Sodium channel epithelial 1 subunit alpha | Scnn1α | Mm00803386_m1 |
| Solute carrier family 9 (sodium/hydrogen exchanger), member 1 | Nhe1 | Mm00444270_m1 |
| TIMP metallopeptidase inhibitor 1 | Timp1 | Mm01341361_m1 |
| Transforming growth factor beta 1 | Tgfβ1 | Mm01178820_m1 |
| Transforming growth factor beta 2 | Tgfβ2 | Mm00436955_m1 |
| Transforming growth factor beta 3 | Tgfβ3 | Mm00436960_m1 |
| Tumour necrosis factor | Tnf | Mm00443258_m1 |
| Vascular cell adhesion molecule-1 | Vcam-1 | Mm01320970_m1 |
| Vascular endothelial growth factor A | Vegfa | Mm00437306_m1 |
| Ubiquitin C (reference gene for placenta, heart and kidney) | Ubc | Mm01198158_m1 |
| β-actin (reference gene for mesenteric arteries) | Actb | Mm02619580_g1 |
ELISA
Concentrations of soluble fms-like tyrosine kinase 1 (sFLT-1), endothelin-1 (ET-1), and C-reactive protein (CRP) in maternal serum were measured using the Mouse sVEGFR1/Flt-1 DuoSet ELISA kit (samples diluted 1:100), Mouse Endothelin-1 Quantikine ELISA Kit (sensitivity 0.207 pg/ml), and Mouse C-Reactive Protein/CRP Quantikine ELISA Kit (sensitivity 0.015 ng/ml) (R&D Systems), respectively, according to the manufacturer’s instructions (inter and intra assay coefficients of variation under 10%).
Urine albumin to creatinine assessment
Albumin and creatinine concentrations in urine were measured using the Mouse Albumin ELISA kit (sensitivity 5 ng/ml) and Mouse Creatinine Kit (80630, 80350; Crystal Chem) run according to manufacturer’s instructions.
Maternal kidney and placenta histology
After fixation in 10% neutral buffered formalin overnight, placentas and kidneys were washed in PBS and embedded in paraffin for sectioning at 5 μM thickness. The sections were deparaffinized in xylene and rehydrated through descending grades of ethanol before haematoxylin and eosin staining. Kidney histology was visualized and captured using a Nikon Eclipse Ci microscope and camera at 100 μm magnification (n = 3 sections/condition, whole kidneys from two dams were analysed per group).
Placental sections were imaged using the ScanScope system (Aperio Technologies). General histological appearance was assessed with the Aperio ImageScope (v12.3.0.5056) software. The total blood space (including the combined fetal and maternal blood space area), and the total cross-sectional area of the junctional zone and the labyrinth zone was measured by a blinded assessor (102).
Statistical analysis
Data were assessed for normal (Gaussian) distribution and the differences between the two groups at each time point were statistically tested either nonparametrically with a Mann–Whitney test or parametrically with an unpaired t test, as appropriate. Fetal and placental size were assessed using a linear mixed-effects model, with a fixed effect for treatment group and random effect for each litter. P-values were calculated for treatment effect using nested ANOVA. Myograph dose–response curves were produced using a nonlinear regression analysis (log[agonist] versus response–four parameters). Comparison of responses to the agonist were tested for significance using mixed-effects analysis, with Šidák correction for multiple comparisons. P-values < 0.05 were considered significantly different. Statistical analysis was performed using GraphPad Prism 8 software.
Data Availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Supplementary Material
Acknowledgements
We would like to acknowledge the staff at the Austin Health BioResources Facility (Heidelberg) for assisting with the care of the animals. Support for this work was provided by the Trevor B Kilvington Bequest. Salary support was provided by the National Health and Medical Research Council Fellowships to NJ Hannan (#1146128). The funder played no role in study design or analysis.
Author Contributions
N de Alwis: data curation, formal analysis, validation, investigation, visualization, methodology, and writing—original draft, review, and editing.
NK Binder: conceptualization, data curation, formal analysis, validation, methodology, project administration, and writing—original draft, review, and editing.
S Beard: data curation, formal analysis, investigation, and writing—review and editing.
YTM Mangwiro: conceptualization, data curation, formal analysis, investigation, methodology, project administration, and writing—original draft, review, and editing.
E Kadife: data curation, formal analysis, investigation, and writing—original draft, review, and editing.
JSM Cuffe: data curation, formal analysis, investigation, methodology, and writing—original draft, review, and editing.
E Keenan: formal analysis, methodology, and writing—review and editing.
BR Fato: data curation, formal analysis, investigation, and writing—review and editing.
TJ Kaitu’u-Lino: conceptualization, resources, supervision, and writing—review and editing.
FC Brownfoot: conceptualization, resources, supervision, and writing—review and editing.
SA Marshall: conceptualization, data curation, formal analysis, methodology, and writing—original draft, review, and editing.
NJ Hannan: conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, project administration, and writing—original draft, review, and editing.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References
- 1.Duley L (2009) The global impact of pre-eclampsia and eclampsia. Semin Perinatol 33: 130–137. 10.1053/j.semperi.2009.02.010 [DOI] [PubMed] [Google Scholar]
- 2.Kuklina EV, Ayala C, Callaghan WM (2009) Hypertensive disorders and severe obstetric morbidity in the United States. Obstet Gynecol 113: 1299–1306. 10.1097/aog.0b013e3181a45b25 [DOI] [PubMed] [Google Scholar]
- 3.Paruk F, Moodley J (2000) Maternal and neonatal outcome in early- and late-onset pre-eclampsia. Semin Neonatol 5: 197–207. 10.1053/siny.2000.0023 [DOI] [PubMed] [Google Scholar]
- 4.Roberts JM, Escudero C (2012) The placenta in preeclampsia. Pregnancy Hypertens 2: 72–83. 10.1016/j.preghy.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powe CE, Levine RJ, Karumanchi SA (2011) Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 123: 2856–2869. 10.1161/circulationaha.109.853127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harmon AC, Cornelius DC, Amaral LM, Faulkner JL, Cunningham MW, Jr, Wallace K, LaMarca B (2016) The role of inflammation in the pathology of preeclampsia. Clin Sci 130: 409–419. 10.1042/cs20150702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamarca B (2012) Endothelial dysfunction. An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva Ginecol 64: 309–320. [PMC free article] [PubMed] [Google Scholar]
- 8.Ødegård RA, Vatten LJ, Nilsen ST, Salvesen KÅ, Austgulen R (2000) Preeclampsia and fetal growth. Obstet Gynecol 96: 950–955. 10.1016/S0029-7844(00)01040-1 [DOI] [PubMed] [Google Scholar]
- 9.Bokslag A, van Weissenbruch M, Mol BW, de Groot CJM (2016) Preeclampsia; short and long-term consequences for mother and neonate. Early Hum Dev 102: 47–50. 10.1016/j.earlhumdev.2016.09.007 [DOI] [PubMed] [Google Scholar]
- 10.Irgens HU, Roberts JM, Reisæter L, Irgens LM, Lie RT (2001) Long term mortality of mothers and fathers after pre-eclampsia: Population based cohort study pre-eclampsia and cardiovascular disease later in life: Who is at risk? BMJ 323: 1213–1217. 10.1136/bmj.323.7323.1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marín R, Gorostidi M, Portal CG, Sánchez M, Sánchez E, Alvarez J (2000) Long-term prognosis of hypertension in pregnancy. Hypertens Pregnancy 19: 199–209. 10.1081/prg-100100136 [DOI] [PubMed] [Google Scholar]
- 12.Melchiorre K, Sutherland GR, Liberati M, Thilaganathan B (2011) Preeclampsia is associated with persistent postpartum cardiovascular impairment. Hypertension 58: 709–715. 10.1161/hypertensionaha.111.176537 [DOI] [PubMed] [Google Scholar]
- 13.Benschop L, Duvekot JJ, Roeters van Lennep JE (2019) Future risk of cardiovascular disease risk factors and events in women after a hypertensive disorder of pregnancy. Heart 105: 1273–1278. 10.1136/heartjnl-2018-313453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garovic VD, Bailey KR, Boerwinkle E, Hunt SC, Weder AB, Curb D, Mosley TH, Jr, Wiste HJ, Turner ST (2010) Hypertension in pregnancy as a risk factor for cardiovascular disease later in life. J Hypertens 28: 826–833. 10.1097/hjh.0b013e328335c29a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erlandsson L, Nääv Å, Hennessy A, Vaiman D, Gram M, Åkerström B, Hansson SR (2016) Inventory of novel animal models addressing etiology of preeclampsia in the development of new therapeutic/intervention opportunities. Am J Reprod Immunol 75: 402–410. 10.1111/aji.12460 [DOI] [PubMed] [Google Scholar]
- 16.Gatford KL, Andraweera PH, Roberts CT, Care AS (2020) Animal models of preeclampsia. Hypertension 75: 1363–1381. 10.1161/hypertensionaha.119.14598 [DOI] [PubMed] [Google Scholar]
- 17.Marshall SA, Hannan NJ, Jelinic M, Nguyen TPH, Girling JE, Parry LJ (2018) Animal models of preeclampsia: Translational failings and why. Am J Physiol Regul Integr Comp Physiol 314: R499–R508. 10.1152/ajpregu.00355.2017 [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Fierro ML, Hernández-Delgadillo GP, Flores-Morales V, Cardenas-Vargas E, Mercado-Reyes M, Rodriguez-Sanchez IP, Delgado-Enciso I, Galván-Tejada CE, Galván-Tejada JI, Celaya-Padilla JM, et al. (2018) Current model systems for the study of preeclampsia. Exp Biol Med (Maywood) 243: 576–585. 10.1177/1535370218755690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumasawa K, Ikawa M, Kidoya H, Hasuwa H, Saito-Fujita T, Morioka Y, Takakura N, Kimura T, Okabe M (2011) Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc Natl Acad Sci U S A 108: 1451–1455. 10.1073/pnas.1011293108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh J, Ahmed A, Girardi G (2011) Role of complement component C1q in the onset of preeclampsia in mice. Hypertension 58: 716–724. 10.1161/hypertensionaha.111.175919 [DOI] [PubMed] [Google Scholar]
- 21.Kulandavelu S, Whiteley KJ, Qu D, Mu J, Bainbridge SA, Adamson SL (2012) Endothelial nitric oxide synthase deficiency reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant mice. Hypertension 60: 231–238. 10.1161/hypertensionaha.111.187559 [DOI] [PubMed] [Google Scholar]
- 22.Molnär M, Söto T, Tóth T, Hertelendy F (1994) Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenia, and intrauterine growth retardation. Am J Obstet Gynecol 170: 1458–1466. 10.1016/s0002-9378(13)90488-9 [DOI] [PubMed] [Google Scholar]
- 23.Health Aio (2019) Welfare, Cardiovascular Disease in Women—A Snapshot of National Statistics. Canberra: AIHW. [Google Scholar]
- 24.Lui NA, Jeyaram G, Henry A (2019) Postpartum interventions to reduce long-term cardiovascular disease risk in women after hypertensive disorders of pregnancy: A systematic review. Front Cardiovasc Med 6: 160. 10.3389/fcvm.2019.00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfeiffer S, Leopold E, Schmidt K, Brunner F, Mayer B (1996) Inhibition of nitric oxide synthesis by NG-nitro-L-arginine methyl ester (L-NAME): Requirement for bioactivation to the free acid, NG-nitro-L-arginine. Br J Pharmacol 118: 1433–1440. 10.1111/j.1476-5381.1996.tb15557.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osol G, Ko NL, Mandalà M (2017) Altered endothelial nitric oxide signaling as a paradigm for maternal vascular maladaptation in preeclampsia. Curr Hypertens Rep 19: 82. 10.1007/s11906-017-0774-6 [DOI] [PubMed] [Google Scholar]
- 27.Sutton EF, Gemmel M, Powers RW (2020) Nitric oxide signaling in pregnancy and preeclampsia. Nitric Oxide 95: 55–62. 10.1016/j.niox.2019.11.006 [DOI] [PubMed] [Google Scholar]
- 28.Johal T, Lees CC, Everett TR, Wilkinson IB (2014) The nitric oxide pathway and possible therapeutic options in pre-eclampsia. Br J Clin Pharmacol 78: 244–257. 10.1111/bcp.12301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nnate DA, Mabhala M, Massey A (2020) Effectiveness of nitric oxide agents in preventing the early onset of pre-eclampsia and possible modification of metabolic factors in high-risk pregnancies: A systematic review protocol. JBI Evid Synth 18: 2658–2665. 10.11124/jbisrir-d-19-00290 [DOI] [PubMed] [Google Scholar]
- 30.Motta C, Grosso C, Zanuzzi C, Molinero D, Picco N, Bellingeri R, Alustiza F, Barbeito C, Vivas A, Romanini M (2015) Effect of sildenafil on pre-eclampsia-like mouse model induced by L-name. Reprod Domest Anim 50: 611–616. 10.1111/rda.12536 [DOI] [PubMed] [Google Scholar]
- 31.Yoshikawa K, Umekawa T, Maki S, Kubo M, Nii M, Tanaka K, Tanaka H, Osato K, Kamimoto Y, Kondo E, et al. (2017) Tadalafil improves L-NG-nitroarginine methyl ester-induced preeclampsia with fetal growth restriction-like symptoms in pregnant mice. Am J Hypertens 31: 89–96. 10.1093/ajh/hpx130 [DOI] [PubMed] [Google Scholar]
- 32.Ma R-q, Sun M-n, Yang Z (2011) Inhibition of nitric oxide synthase lowers fatty acid oxidation in preeclampsia-like mice at early gestational stage. Chin Med J 124: 3141–3147. 10.3760/cma.j.issn.0366-6999.2011.19.033 [DOI] [PubMed] [Google Scholar]
- 33.Zou Y, Li S, Wu D, Xu Y, Wang S, Jiang Y, Liu F, Jiang Z, Qu H, Yu X, et al. (2019) Resveratrol promotes trophoblast invasion in pre-eclampsia by inducing epithelial-mesenchymal transition. J Cell Mol Med 23: 2702–2710. 10.1111/jcmm.14175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma R-q, Sun M-n, Yang Z (2010) Effects of preeclampsia-like symptoms at early gestational stage on feto-placental outcomes in a mouse model. Chin Med J 123: 707–712. 10.3760/cma.j.issn.0366-6999.2010.06.013 [DOI] [PubMed] [Google Scholar]
- 35.Han Y-W, Yang Z, Ding X-Y, Yu H (2015) Differences in liver injury and trophoblastic mitochondrial damage in different preeclampsia-like mouse models. Chin Med J 128: 1627–1635. 10.4103/0366-6999.158322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Yang N, Li H, Cai W, Zhang X, Ma Y, Niu X, Yang G, Zhou X, Li Y (2018) Systemic evaluation of vascular dysfunction by high-resolution sonography in an nω-nitro-l-arginine methyl ester hydrochloride–induced mouse model of preeclampsia-like symptoms. J Ultrasound Med 37: 657–666. 10.1002/jum.14380 [DOI] [PubMed] [Google Scholar]
- 37.Buhimschi IA, Shi SQ, Saade GR, Garfield RE (2001) Marked variation in responses to long-term nitric oxide inhibition during pregnancy in outbred rats from two different colonies. Am J Obstet Gynecol 184: 686–693. 10.1067/mob.2001.110448 [DOI] [PubMed] [Google Scholar]
- 38.Abuiessa SA, Wedn AM, El-Gowilly SM, Helmy MM, El-Mas MM (2020) Pre-eclamptic fetal programming alters neuroinflammatory and cardiovascular consequences of endotoxemia in sex-specific manners. J Pharmacol Exp Ther 373: 325–336. 10.1124/jpet.119.264192 [DOI] [PubMed] [Google Scholar]
- 39.Butruille L, Mayeur S, Moitrot E, Storme L, Knauf C, Lesage J, Deruelle P (2013) Maternal hypertension induced by NO blockade does not program adult metabolic diseases in growth-restricted rat fetuses. Metabolism 62: 442–445. 10.1016/j.metabol.2012.09.006 [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Zhao W, Liu H, Kang Y, Ye C, Gu W, Hu R, Li X (2016) Developmental and functional brain impairment in offspring from preeclampsia-like rats. Mol Neurobiol 53: 1009–1019. 10.1007/s12035-014-9060-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perfilova VN, Zhakupova GA, Lashchenova LI, Lebedeva SA, Tyurenkov IN (2016) Spatial memory in the progeny of rats subjected to different types of experimental preeclampsia. Bull Exp Biol Med 161: 643–646. 10.1007/s10517-016-3475-2 [DOI] [PubMed] [Google Scholar]
- 42.Tachibana R, Umekawa T, Yoshikawa K, Owa T, Magawa S, Furuhashi F, Tsuji M, Maki S, Shimada K, Kaneda MK, et al. (2019) Tadalafil treatment in mice for preeclampsia with fetal growth restriction has neuro-benefic effects in offspring through modulating prenatal hypoxic conditions. Sci Rep 9: 234. 10.1038/s41598-018-36084-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tyurenkov IN, Perfilova VN, Lashhenova LI, Zhakupova GA, Lebedeva SA (2016) Comparison of physical development and rate of formation sensory-motor reflexes offspring of rats with different experimental model of preeclampsia. Patol Fiziol Eksp Ter 60: 10–17. [PubMed] [Google Scholar]
- 44.Ijomone OK, Shallie PD, Naicker T (2020) Oligodendrocytes death induced sensorimotor and cognitive deficit in N-nitro-l-arginine methyl rat model of pre-eclampsia. Neurochem Res 45: 902–914. 10.1007/s11064-020-02969-5 [DOI] [PubMed] [Google Scholar]
- 45.Dvanajscak Z, Cossey LN, Larsen CP (2020) A practical approach to the pathology of renal intratubular casts. Semin Diagn Pathol 37: 127–134. 10.1053/j.semdp.2020.02.001 [DOI] [PubMed] [Google Scholar]
- 46.Gaber LW, Spargo BH, Lindheimer MD (1994) 12 Renal pathology in pre-eclampsia. Baillieres Clin Obstet Gynaecol 8: 443–468. 10.1016/s0950-3552(05)80330-x [DOI] [PubMed] [Google Scholar]
- 47.Stillman IE, Karumanchi SA (2007) The glomerular injury of preeclampsia. J Am Soc Nephrol 18: 2281–2284. 10.1681/asn.2007020255 [DOI] [PubMed] [Google Scholar]
- 48.Behrens I, Basit S, Melbye M, Lykke JA, Wohlfahrt J, Bundgaard H, Thilaganathan B, Boyd HA (2017) Risk of post-pregnancy hypertension in women with a history of hypertensive disorders of pregnancy: Nationwide cohort study. BMJ 358: j3078. 10.1136/bmj.j3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Staff AC, Redman CWG (2018) The differences between early- and late-onset pre-eclampsia. In Preeclampsia: Basic, Genomic, and Clinical, Saito S (ed.), pp 157–172. Singapore: Springer Singapore. [Google Scholar]
- 50.Maynard SE, Min J-Y, Merchan J, Lim K-H, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, et al. (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111: 649–658. 10.1172/jci17189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shu W, Li H, Gong H, Zhang M, Niu X, Ma Y, Zhang X, Cai W, Yang G, Wei M, et al. (2018) Evaluation of blood vessel injury, oxidative stress and circulating inflammatory factors in an L-NAME-induced preeclampsia-like rat model. Exp Ther Med 16: 585–594. 10.3892/etm.2018.6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dong X, Shi D (2017) Simvastatin alleviates pathology in a rat model of preeclampsia involving ERK/MAPK pathway. Reprod Sci 24: 1053–1061. 10.1177/1933719116678693 [DOI] [PubMed] [Google Scholar]
- 53.Zhao Y, Zheng Y, Liu X, Luo Q, Wu D, Liu X, Zou L (2018) Inhibiting trophoblast PAR-1 overexpression suppresses sFlt-1-induced anti-angiogenesis and abnormal vascular remodeling: A possible therapeutic approach for preeclampsia. Mol Hum Reprod 24: 158–169. 10.1093/molehr/gax068 [DOI] [PubMed] [Google Scholar]
- 54.Zuo J, Jiang Z (2020) Melatonin attenuates hypertension and oxidative stress in a rat model of L-NAME-induced gestational hypertension. Vasc Med 25: 295–301. 10.1177/1358863x20919798 [DOI] [PubMed] [Google Scholar]
- 55.Possomato-Vieira JS, Gonçalves-Rizzi VH, Graça TUS, Nascimento RA, Dias-Junior CA (2016) Sodium hydrosulfide prevents hypertension and increases in vascular endothelial growth factor and soluble fms-like tyrosine kinase-1 in hypertensive pregnant rats. Naunyn Schmiedebergs Arch Pharmacol 389: 1325–1332. 10.1007/s00210-016-1296-5 [DOI] [PubMed] [Google Scholar]
- 56.Li H, Ohta H, Tahara Y, Nakamura S, Taguchi K, Nakagawa M, Oishi Y, Goto Y-I, Wada K, Kaga M, et al. (2015) Artificial oxygen carriers rescue placental hypoxia and improve fetal development in the rat pre-eclampsia model. Sci Rep 5: 15271. 10.1038/srep15271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonçalves-Rizzi VH, Possomato-Vieira JS, Sales Graça TU, Nascimento RA, Dias-Junior CA (2016) Sodium nitrite attenuates hypertension-in-pregnancy and blunts increases in soluble fms-like tyrosine kinase-1 and in vascular endothelial growth factor. Nitric Oxide 57: 71–78. 10.1016/j.niox.2016.05.004 [DOI] [PubMed] [Google Scholar]
- 58.Ramesar SV, Mackraj I, Gathiram P, Moodley J (2011) Sildenafil citrate decreases sFlt-1 and sEng in pregnant l-NAME treated Sprague–Dawley rats. Eur J Obstet Gynecol Reprod Biol 157: 136–140. 10.1016/j.ejogrb.2011.03.005 [DOI] [PubMed] [Google Scholar]
- 59.Ramesar SV, Drewes SE, Gathiram P, Moodley J, Mackraj I (2012) The effect of kraussianone-2 (Kr2), a natural pyrano-isoflavone from eriosema kraussianum, in an L-NAME- induced pre-eclamptic rat model. Phytother Res 26: 1375–1380. 10.1002/ptr.3697 [DOI] [PubMed] [Google Scholar]
- 60.Kasture V, Sundrani D, Dalvi S, Swamy M, Kale A, Joshi S (2019) Maternal omega-3 fatty acids and vitamin E improve placental angiogenesis in late-onset but not early-onset preeclampsia. Mol Cell Biochem 461: 159–170. 10.1007/s11010-019-03599-4 [DOI] [PubMed] [Google Scholar]
- 61.Gu S, Shen H, Zhou Y, Ni J, Zheng T, Mou Z, Hua X (2019) Tetramethylpyrazine reduces the consequences of nitric oxide inhibition in pregnant rats. J Cell Physiol 234: 19799–19806. 10.1002/jcp.28579 [DOI] [PubMed] [Google Scholar]
- 62.Xiao S, Zhang M, He Z, Wang D (2018) Celastrol attenuates symptoms of preeclampsia in rats by inhibiting matrix metalloproteinase-9. Eur J Pharmacol 830: 33–38. 10.1016/j.ejphar.2018.04.025 [DOI] [PubMed] [Google Scholar]
- 63.Onda K, Tong S, Beard S, Binder N, Muto M, Senadheera SN, Parry L, Dilworth M, Renshall L, Brownfoot F, et al. (2017) Proton pump inhibitors decrease soluble fms-like tyrosine kinase-1 and soluble endoglin secretion, decrease hypertension, and rescue endothelial dysfunction. Hypertension 69: 457–468. 10.1161/hypertensionaha.116.08408 [DOI] [PubMed] [Google Scholar]
- 64.Kitamuro T, Takahashi K, Ogawa K, Udono-Fujimori R, Takeda K, Furuyama K, Nakayama M, Sun J, Fujita H, Hida W, et al. (2003) Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J Biol Chem 278: 9125–9133. 10.1074/jbc.m209939200 [DOI] [PubMed] [Google Scholar]
- 65.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM (1997) Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 272: 5375–5381. 10.1074/jbc.272.9.5375 [DOI] [PubMed] [Google Scholar]
- 66.Li L, Li H, Zhou Q, Lu Y, Chen P, Wang X, Zhao H (2020) Implication of nuclear factor-erythroid 2-like 2/heme oxygenase 1 pathway in the protective effects of coenzyme Q10 against preeclampsia-like in a rat model. Microcirculation 27: e12651. 10.1111/micc.12651 [DOI] [PubMed] [Google Scholar]
- 67.Chen X, Huang J, Lv Y, Chen Y, Rao J (2021) Crocin exhibits an antihypertensive effect in a rat model of gestational hypertension and activates the Nrf-2/HO-1 signaling pathway. Hypertension Res 44: 642–650. 10.1038/s41440-020-00609-7 [DOI] [PubMed] [Google Scholar]
- 68.Yang S, Zhang R, Xing B, Zhou L, Zhang P, Song L (2020) Astragaloside IV ameliorates preeclampsia-induced oxidative stress through the Nrf2/HO-1 pathway in a rat model. Am J Physiology-Endocrinology Metab 319: E904–E911. 10.1152/ajpendo.00357.2020 [DOI] [PubMed] [Google Scholar]
- 69.Yang X, Guo L, Sun X, Chen X, Tong X (2011) Protective effects of hydrogen-rich saline in preeclampsia rat model. Placenta 32: 681–686. 10.1016/j.placenta.2011.06.020 [DOI] [PubMed] [Google Scholar]
- 70.Wang Y, Huang M, Yang X, Yang Z, Li L, Mei J (2018) Supplementing punicalagin reduces oxidative stress markers and restores angiogenic balance in a rat model of pregnancy-induced hypertension. Korean J Physiol Pharmacol 22: 409–417. 10.4196/kjpp.2018.22.4.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amaral TAS, Ognibene DT, Carvalho LCRM, Rocha APM, Costa CA, Moura RS, Resende AC (2018) Differential responses of mesenteric arterial bed to vasoactive substances in L-NAME-induced preeclampsia: Role of oxidative stress and endothelial dysfunction. Clin Exp Hypertens 40: 126–135. 10.1080/10641963.2017.1339073 [DOI] [PubMed] [Google Scholar]
- 72.Vanderlelie JJ, Perkins AV (2006) Chronic nitric oxide synthase inhibition in pregnant rats does not result in placental oxidative stress. Hypertens Pregnancy 25: 103–114. 10.1080/10641950600745483 [DOI] [PubMed] [Google Scholar]
- 73.Tong S, Kaitu’u-Lino TJ, Onda K, Beard S, Hastie R, Binder NK, Cluver C, Tuohey L, Whitehead C, Brownfoot F, et al. (2015) Heme oxygenase-1 is not decreased in preeclamptic placenta and does not negatively regulate placental soluble fms-like tyrosine kinase-1 or soluble endoglin secretion. Hypertension 66: 1073–1081. 10.1161/hypertensionaha.115.05847 [DOI] [PubMed] [Google Scholar]
- 74.Saleh L, Verdonk K, Visser W, van den Meiracker AH, Danser AHJ (2016) The emerging role of endothelin-1 in the pathogenesis of pre-eclampsia. Ther Adv Cardiovasc Dis 10: 282–293. 10.1177/1753944715624853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raio L, Bersinger NA, Malek A, Schneider H, Messerli FH, Hürter H, Rimoldi SF, Baumann MU (2019) Ultra-high sensitive C-reactive protein during normal pregnancy and in preeclampsia: A pilot study. J Hypertens 37: 1012–1017. 10.1097/hjh.0000000000002003 [DOI] [PubMed] [Google Scholar]
- 76.Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S (1990) Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol 101: 746–752. 10.1111/j.1476-5381.1990.tb14151.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takiuti NH, Carvalho MHC, Kahhale S, Nigro D, Barbeiro HV, Zugaib M (1999) The effect of chronic nitric oxide inhibition on vascular reactivity and blood pressure in pregnant rats. Sao Paulo Med J 117: 197–204. 10.1590/s1516-31801999000500004 [DOI] [PubMed] [Google Scholar]
- 78.Mukhtarova N, Ko NL, Gokina NI, Mandalá M, Osol G (2020) Enhanced vascular smooth muscle calcium sensitivity and loss of endothelial vasodilator influence contribute to myogenic tone development in rat radial uterine arteries during gestation. J Vasc Res 57: 126–135. 10.1159/000505670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cooke C-LM, Davidge ST (2003) Pregnancy-induced alterations of vascular function in mouse mesenteric and uterine arteries. Biol Reprod 68: 1072–1077. 10.1095/biolreprod.102.009886 [DOI] [PubMed] [Google Scholar]
- 80.Christensen KL, Mulvany MJ (1993) Mesenteric arcade arteries contribute substantially to vascular resistance in conscious rats. J Vasc Res 30: 73–79. 10.1159/000158978 [DOI] [PubMed] [Google Scholar]
- 81.van Drongelen J, Hooijmans CR, Lotgering FK, Smits P, Spaanderman MEA (2012) Adaptive changes of mesenteric arteries in pregnancy: A meta-analysis. Am J Physiol Heart Circ Physiol 303: H639–H657. 10.1152/ajpheart.00617.2011 [DOI] [PubMed] [Google Scholar]
- 82.Lu YP, Hasan AA, Zeng S, Hocher B (2017) Plasma ET-1 concentrations are elevated in pregnant women with hypertension -Meta-Analysis of clinical studies. Kidney Blood Press Res 42: 654–663. 10.1159/000482004 [DOI] [PubMed] [Google Scholar]
- 83.Kvehaugen AS, Dechend R, Ramstad HB, Troisi R, Fugelseth D, Staff AC (2011) Endothelial function and circulating biomarkers are disturbed in women and children after preeclampsia. Hypertension 58: 63–69. 10.1161/hypertensionaha.111.172387 [DOI] [PubMed] [Google Scholar]
- 84.Freeman DJ, McManus F, Brown EA, Cherry L, Norrie J, Ramsay JE, Clark P, Walker ID, Sattar N, Greer IA (2004) Short- and long-term changes in plasma inflammatory markers associated with preeclampsia. Hypertension 44: 708–714. 10.1161/01.hyp.0000143849.67254.ca [DOI] [PubMed] [Google Scholar]
- 85.Berks D, Steegers EAP, Molas M, Visser W (2009) Resolution of hypertension and proteinuria after preeclampsia. Obstet Gynecol 114: 1307–1314. 10.1097/aog.0b013e3181c14e3e [DOI] [PubMed] [Google Scholar]
- 86.Mangos GJ, Spaan JJ, Pirabhahar S, Brown MA (2012) Markers of cardiovascular disease risk after hypertension in pregnancy. J Hypertens 30: 351–358. 10.1097/hjh.0b013e32834e5ac7 [DOI] [PubMed] [Google Scholar]
- 87.Sandvik MK, Hallan S, Svarstad E, Vikse BE (2013) Preeclampsia and prevalence of microalbuminuria 10 years later. Clin J Am Soc Nephrol 8: 1126–1134. 10.2215/cjn.10641012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tan RJ, Liu Y (2012) Matrix metalloproteinases in kidney homeostasis and diseases. Am J Physiol Ren Physiol 302: F1351–F1361. 10.1152/ajprenal.00037.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu X, Jackson PL, Tanner S, Hardison MT, Abdul Roda M, Blalock JE, Gaggar A (2011) A self-propagating matrix metalloprotease-9 (MMP-9) dependent cycle of chronic neutrophilic inflammation. PLoS One 6: e15781. 10.1371/journal.pone.0015781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sanders J-SF, van Goor H, Hanemaaijer R, Kallenberg CGM, Stegeman CA (2004) Renal expression of matrix metalloproteinases in human ANCA-associated glomerulonephritis. Nephrol Dial Transplant 19: 1412–1419. 10.1093/ndt/gfh186 [DOI] [PubMed] [Google Scholar]
- 91.Wang H, Gao M, Li J, Sun J, Wu R, Han D, Tan J, Wang J, Wang B, Zhang L, et al. (2019) MMP-9-positive neutrophils are essential for establishing profibrotic microenvironment in the obstructed kidney of UUO mice. Acta Physiol (Oxf) 227: e13317. 10.1111/apha.13317 [DOI] [PubMed] [Google Scholar]
- 92.Smith GC, Pell JP, Walsh D (2001) Pregnancy complications and maternal risk of ischaemic heart disease: A retrospective cohort study of 129, 290 births. Lancet 357: 2002–2006. 10.1016/s0140-6736(00)05112-6 [DOI] [PubMed] [Google Scholar]
- 93.Bonney EA, Howard A, Krebs K, Begin K, Veilleux K, Gokina NI (2017) Impact of immune deficiency on remodeling of maternal resistance vasculature 4 Weeks postpartum in mice. Reprod Sci 24: 514–525. 10.1177/1933719116678691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miralles F, Collinot H, Boumerdassi Y, Ducat A, Duché A, Renault G, Marchiol C, Lagoutte I, Bertholle C, Andrieu M, et al. (2019) Long-term cardiovascular disorders in the STOX1 mouse model of preeclampsia. Sci Rep 9: 11918. 10.1038/s41598-019-48427-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garrett N, Pombo J, Umpierrez M, Clark JE, Simmons M, Girardi G (2018) Pravastatin therapy during preeclampsia prevents long-term adverse health effects in mice. JCI Insight 3: 120147. 10.1172/jci.insight.120147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Romundstad PR, Magnussen EB, Smith GD, Vatten LJ (2010) Hypertension in pregnancy and later cardiovascular risk. Circulation 122: 579–584. 10.1161/circulationaha.110.943407 [DOI] [PubMed] [Google Scholar]
- 97.Craici I, Wagner S, Garovic VD (2008) Preeclampsia and future cardiovascular risk: Formal risk factor or failed stress test? Ther Adv Cardiovasc Dis 2: 249–259. 10.1177/1753944708094227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pruthi D, Khankin EV, Blanton RM, Aronovitz M, Burke SD, McCurley A, Karumanchi SA, Jaffe IZ (2015) Exposure to experimental preeclampsia in mice enhances the vascular response to future injury. Hypertension 65: 863–870. 10.1161/hypertensionaha.114.04971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gokina NI, Fairchild RI, Bishop NM, Dawson TE, Prakash K, Bonney EA (2021) Kinetics of postpartum mesenteric artery structure and function relative to pregnancy and lactation in mice. Reprod Sci 28: 1200–1215. 10.1007/s43032-020-00402-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dutta S, Sengupta P (2016) Men and mice: Relating their ages. Life Sci 152: 244–248. 10.1016/j.lfs.2015.10.025 [DOI] [PubMed] [Google Scholar]
- 101.ShamsEldeen AM, Mehesen MN, Aboulhoda BE, Rashed LA, Elsebaie MM, Mohamed EA, Gamal MM (2021) Prenatal intake of omega-3 promotes Wnt/β-catenin signaling pathway, and preserves integrity of the blood–brain barrier in preeclamptic rats. Physiol Rep 9: e14925. 10.14814/phy2.14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Helman SL, Wilkins SJ, McKeating DR, Perkins AV, Whibley PE, Cuffe JSM, Simmons DG, Fuqua BK, Vulpe CD, Wallace DF, et al. (2021) The placental ferroxidase zyklopen is not essential for iron transport to the fetus in mice. J Nutr 151: 2541–2550. 10.1093/jn/nxab174 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.