

Abstract
Background
Defective Cernunnos gene in nonhomologous end‐joining (NHEJ) pathway of the DNA repair is responsible for radiosensitive severe combined immunodeficiency (SCID). Herein, presented a new patient with Cernunnos deficiency and summarized the clinical, immunological, and molecular features of reported patients in the literature.
Case
The patient was a 6‐month‐old female born to consanguineous parents. She presented with long‐lasting fever, diarrhea, poor feeding, and restlessness. She had suffered from recurrent fever of unknown origin and multiple episodes of oral candidiasis. In the physical examination, microcephaly, failure to thrive, oral candidiasis, pustular rash on fingers, and perianal ulcers, but no dysmorphic feature were observed. The immunologic workup revealed lymphopenia, neutropenia, normocytic anemia, low T‐ but normal B‐ and natural killer (NK)‐ cells, low immunoglobulin (Ig)G, and normal IgA, IgM, and IgE. The T‐cell receptor excision circle (TREC) was low and the lymphocyte transformation test (LTT) was abnormal to mitogens and antigens. She was diagnosed with T− B+ NK+ SCID and improved by intravenous immunoglobulin along with antimicrobials. A homozygous splice site variant, c.390 + 1G > T, at the intron 3 of the NHEJ1, was identified and the diagnosis of Cernunnos deficiency was established. However, while a candidate for hematopoietic stem cell transplantation, she developed sepsis and died at 11 months of age.
Conclusions
Cernunnos deficiency should be considered as a differential diagnosis in patients with microcephaly, growth retardation, recurrent infections, T‐cell defects, and hypogammaglobulinemia. The normal B‐cell level in the index patient is an unexpected finding in Cernunnos deficiency which requires further evaluation.
Keywords: BCG, Cernunnos deficiency, inborn errors of immunity, NHEJ1, SCID, severe combined immunodeficiency
This study presents a novel patient with Cernunnos deficiency and reviews the clinical, immunological, and molecular features of similarly affected patients in the literature. Defective Cernunnos gene in nonhomologous end‐joining (NHEJ) pathway of the DNA repair is responsible for radiosensitive severe combined immunodeficiency (SCID), manifesting as microcephaly, growth retardation, recurrent infections, T cell defects, and hypogammaglobulinemia.

1. INTRODUCTION
Cernunnos, which is encoded by NHEJ1 gene (OMIM number: 611290) on chromosome 2q35, is involved in the nonhomologous end‐joining (NHEJ) pathway of the DNA repair. Other components of the pathway include DNA ligase IV, XRCC4, Artemis, and DNA‐PKcs (DNA‐dependent protein kinase catalytic subunit), deficiency of which contribute to the development of well‐described chromosomal instability syndromes (Slatter & Gennery, 2020). Cernunnos is involved in the junctional diversity of the antigen receptors and plays important role in DNA double‐strand break repair and V(D)J recombination (Menon & Povirk, 2017).
In 2003, a patient (2BN patient) with severe combined immunodeficiency (SCID) phenotype, short stature, and increased radiosensitivity was identified. Her cells showed impaired V(D)J recombination and defects in rejoining DNA strand breaks, however, at the time she did not have defects in any of the known NHEJ factors (Dai et al., 2003). Two years later, Ahnesorg et al. (2006) used a yeast two‐hybrid screen in 2BN patient's cells for XRCC4 interactors and identified a conserved 33 kDa protein with structural similarities to XRCC4, thus named it XRCC4‐like factor or XLF. They showed that XLF directly interacts with XRCC4‐Ligase IV complex in vitro and in vivo. They also produced XLF‐complemented cells by introducing the wild‐type XLF cDNA to 2BN cells and showed that they are far more susceptible to radiation exposure.
Another research team independently introduced five patients with similar features and defined Cernunnos gene defect through cDNA functional complementation cloning. These shared features included growth retardation, microcephaly, dysmorphic features, recurrent infections, and combined immunodeficiency (with low B and T cells but normal natural killer (NK) cells), hypogammaglobulinemia, and increased sensitivity of their fibroblast to gamma ionizing radiation (Buck et al., 2006).
In the latest International Union of Immunological Societies (IUIS) classification of human inborn errors of immunity (IEI), Cernunnos deficiency is categorized as T−B−NK+ severe combined immunodeficiency (Bousfiha et al., 2020). According to the Iranian National Registry of Primary Immunodeficiencies (Abolhassani et al., 2018), CID/SCID compromises 12% of 3056 registered IEI patients and the lowest survival rate among different categories of IEI is observed in patients with a CID (mainly severe CID) in the first 5 years of life, which could have been prevented by earlier diagnosis and treatment.
Although most of the chromosomal instability syndromes have clinically indistinguishable shared features, introducing newly identified patients with novel pathogenic variants can help characterize each syndrome in a more precise way, prevent some of the iatrogenic complications such as radioactive‐related damages, and reduce the diagnosis delay in similar patients, which itself has a considerable impact in therapeutic decision‐making (such as early application of hematopoietic stem cell transplantation [HSCT]) and prognosis. In this viewpoint, we aim to present an Iranian patient with SCID phenotype, who was found to have defect in the NHEJ1 gene, however, with unexpected immunologic profile. We also reviewed all patients with Cernunnos deficiency reported in the literature.
2. CASE REPORT
The patient was a 6‐month‐old female who was presented to our hospital with fever (for 2 weeks), diarrhea (for 5 days), poor feeding, and restlessness. She was born to consanguineous parents (first cousins). The family history was non‐contributary and her 8‐year‐old brother was healthy (Figure 1).
FIGURE 1.
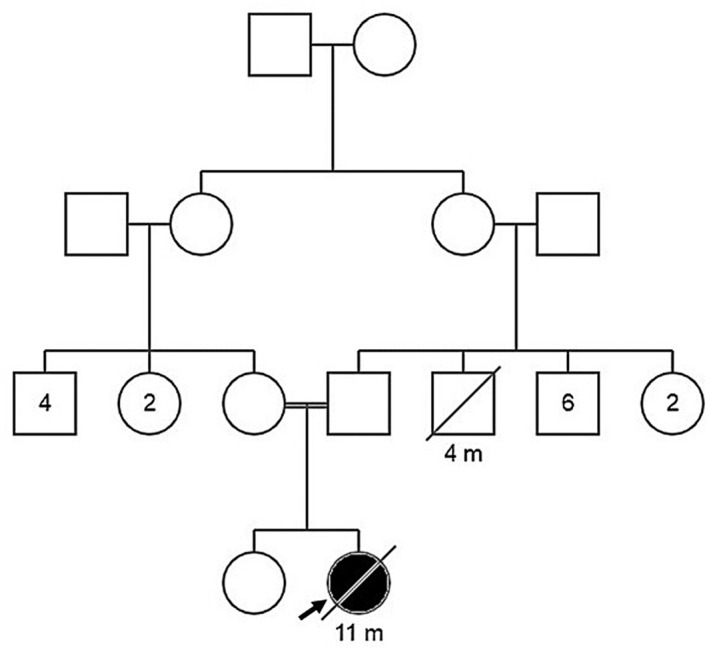
The proband's Pedigree.
The fever had been recurrent with unknown origin and she was hospitalized three times for fever workup. She had a history of urinary tract infection (UTI). She suffered from multiple episodes of oral candidiasis, controlled by Nystatin suspension.
On physical examination, the patient was toxic and febrile. She had microcephaly (head circumference: 36.5 cm, <3rd percentile) and failure to thrive (58 cm (−3.0 SD) height and 4.7 Kg (−3.7 SD) weight, <3rd percentile). No facial dysmorphism was observed. She had hoarseness and in the lung auscultation, rhonchi, and wheezing were noted. She had developed erythema and swelling at the site of the BCG vaccine injection, resolved after 10 days. Oral candidiasis, pustular rash on fingers, and multiple perianal punch out ulcers were found. No hepatosplenomegaly or lymphadenopathy was detected.
At this time the differential diagnoses included combined immunodeficiency, severe combined immunodeficiency, chronic granulomatous disease, congenital neutropenia, hemophagocytic lymphohistiocytosis, metabolic disorders, bacterial sepsis, and viral or fungal infections.
The chest x‐ray and chest computed tomography (CT) scan were normal. In the head and neck CT scan mastoid opacification was reported. The echocardiography and abdominal ultrasound were also normal. Bronchoscopy evaluation, performed due to hoarseness, showed tracheomalacia in the lower one‐third of the trachea. Bronchoalveolar lavage (BAL) culture and smear for bacteria and fungi were negative.
In the initial laboratory result, leukopenia (WBC: 1100 cell/μl), lymphopenia (ALC: 1089 cell/μl), neutropenia (ANC: 11 cell/μl), normocytic anemia (Hb: 7.5 g/dL), high inflammatory markers (ESR: 44 mm/hour, CRP: 153 mg/L) and lactate dehydrogenase (LDH: 444 IU/L), and abnormal liver enzymes (AST: 178 IU/L, ALT: 110 IU/L) were found. The blood culture was positive for gram positive cocci. The EBV and CMV viral load, HIV antibody (Ab), HBS antigen, HBS Ab, and HCV Ab were all negative. The COVID‐19 polymerase chain reaction (PCR) test was negative. TORCH study and metabolic disorder workup were already performed in another center and yielded normal results. The bone marrow aspiration was hypocellular without any maturation arrest. In the immunologic workup, she had low T CD3+ (141 cell/μl), low T CD4+ (119 cell/μl), low T CD8+ (22 cell/μl), normal B CD19+ (555 cell/μl) and B CD20+ (544 cell/μl), and normal NK CD16+CD56+ (272 cell/μl) cell counts. The immunologic profile revealed low IgG (83 mg/dL), while normal IgA (195 mg/dL), IgM (203 mg/dL), and IgE (0.2 IU/ml). The nitroblue tetrazolium (NBT) test was normal. The T‐cell receptor excision circle (TREC) was low. The lymphocyte transformation test (LTT) was abnormal to mitogens and antigens (PHA: 1.5, BCG: 2, Candida: 1.5) (Supporting Information ESM_ 1).
She was placed on vancomycin (10 mg/kg/dose/QID) and meropenem (20 mg/kg/dose/TDS), voriconazole (30 mg/kg/BD), and G‐CSF (10 mcg/kg/day due to persistent neutropenia). However, the fever persisted and the neutropenia was non‐responsive to G‐CSF.
She was diagnosed with T− B+ NK+ severe combined immunodeficiency (SCID) and received intravenous immunoglobulin (IVIg) along with previously prescribed antimicrobials, which resulted in considerable clinical improvement. She was discharged with prophylactic treatment cotrimoxazole (10 mg/kg daily), voriconazole (5 mg/kg), acyclovir (15 mg/kg daily), isoniazid (25 mg daily), G‐CSF (10 mcg/day), and monthly IVIg (3 gr/month).
Later, the genetic study using Whole Exome Sequencing showed a homozygous canonical splice site variant, c.390 + 1G > T, at the intron 3 of NHEJ1 gene (NM_024782.3); based on the American College of Medical Genetics (ACMG) guidelines this variant can be classified as likely pathogenic and the diagnosis of Cernunnos deficiency was established (CADD score: 34). This variant has not been reported previously, but Sheikh et al. (Sheikh et al., 2017) have reported another nucleotide change (c.390 + 1G > C) at the same position in a Saudi Arabian family with three affected siblings. They showed that this change results in two aberrantly spliced mRNA isoforms, both causing in‐frame deletions at the RNA level (skipping full length of exon 3 containing 71 amino acids and skipping of 69 bp at the end of exon 3 including 23 amino acids). They did not find any protein expression using immunoblotting. Considering their results, we can confer that our detected variant would have a similar effect and act as a loss of function variant.
Our patient mildly improved and was candidate for the HSCT. However, in the meanwhile, her clinical condition deteriorated and she unfortunately died at 11 months of age due to sepsis.
3. DISCUSSION
NHEJ pathway, in which Cernunnos (XLF) is a core player, is active throughout the cell cycle (particularly in G1) and mediates ligation of broken DNA double strands without any need of homologous template. Moreover, NHEJ functions in the generation of T‐cell receptor and immunoglobulin repertoire, making it a ubiquitous pathway in DNA repair and immune system. Therefore, loss of function mutations in genes involved in NHEJ lead to CID or SCID in both human and mice with often shared clinical phenotypes (Menon & Povirk, 2017; Sharma et al., 2020).
We reviewed the literature published from 2003 to date on a series of 49 patients with autosomal recessive hypomorphic mutations in NHEJ1 including the present case report. The literature search and evaluation were performed using “NHEJ1”, “nonhomologous end‐joining factor 1”, “XLF’, “XRCC4‐like factor”, and “Cernunnos” keywords. In addition, reference lists of major reviews and case series were manually searched for additional studies. Table 1 summarizes demographic, clinical, and molecular findings of reported patients.
Table 1.
Summary of demographic, clinical, and molecular findings in Cernunnos deficiency
| Pt | Sex | Country of origin | Parental Consanguinity | FH of IEIs, Early death, or miscarriage | AOD (month) | Infectious complications | Growth failure | Neurologic/learning disorders | Anomalies | Cytopenia | Other Clinical features | Zygosity/variant type/affected exon(s) or intron(s) | c. DNA change (NM_024782.3) | Treatments | Life status at the time of study | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | USA | No | Yes | 72 | Immunodeficiency | Yes | None | – | – | – | Homozygous/Frameshift dup/Exon 2 | c.11dupT (p.E5Gfs*43) | BMT | Alive | Dai et al. (2003) |
| 2 | – | France | No | No | 168 | Recurrent RTI, Invasive warts, Severe cholangitis | Yes | Microcephaly, Mental retardation | Urogenital and bone malformation, Facial dysmorphia | AIHA, ITP | Chromosomal abnormalities | Compound heterozygous/Missenses; missense/Exon 2; exon3 | c.169C > G (p.R57G); c.367 T > C (p.C123R) | IS, IGRT, Splenectomy | Deceased (18 y, Septic Shock) | Buck et al. (2006) |
| 3 | – | Turkey | Yes | Yes | 24 | Recurrent RTI and GI tract infections | Yes | Microcephaly | None | None | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | IGRT | Deceased (4 y, Septic Shock) | Buck et al. (2006) |
| 4* | M | Turkey | Yes | Yes | 156 |
Pneumocystis carinii pneumonia, chronic Giardia lamblia enteritis, Salmonella and Campylobacter enteritis, molluscum contagiosum, and warts |
Yes | Microcephaly | Facial dysmorphia, Bone malformation | AIHA, ITP | – | Homozygous/Indel/Intron 2 | c.177 + 1_177 + 3delGTAinsTT | IGRT, AB prophylaxis | Alive | Buck et al. (2006) |
| 5* | F | Turkey | Yes | Yes |
24 |
Recurrent RTI | Yes | Microcephaly | Facial dysmorphia, Bone malformation | None | – | Homozygous/Indel/Intron 2 | c.177 + 1_177 + 3delGTAinsTT | IGRT, AB prophylaxis | Alive | Buck et al. (2006) |
| 6 | M | Italy | Yes | No | 84 | Recurrent RTI | Yes | Microcephaly | Facial dysmorphia | Bone marrow aplasia, Pancytopenia | Chromosomal abnormalities | Homozygous/Missense/Exon 2 | c.169C > G (p.R57G) | IGRT | Alive | Buck et al. (2006), Faraci et al. (2009) |
| 7 | M | Spain | Yes | No | 144 | Recurrent RTI | No | Microcephaly, developmental delay | Facial dysmorphia | None | – | Homozygous/Missense/Exon 2 | c.169C > G (p.R57G) | – | Alive | Dutrannoy et al. (2010) |
| 8* | M | Germany | No | Yes | 72 | Chronic diarrhea, Recurrent RTI, Adenoviral arthritis, UTI | Yes | Microcephaly | None | Neutropenia | Hepatosplenomegaly, Abdominal lymphadenopathy | Compound heterozygous/ Frame shift dup; large deletion/Exon 4; exons 2 and 3 |
c.495dupA (p.D166Rfs*20); 1.9 kb deletion |
IGRT | Alive | Dutrannoy et al. (2010), Meyer‐Bahlburg et al. (2014) |
| 9* | M | Germany | No | Yes | 24 |
Recurrent RTI |
No | None | None | None | None | Compound heterozygous/ Fame shift dup; large deletion/Exon 4; exons 2 and 3 |
c.495dupA (p.D166Rfs*20); 1.9 kb deletion |
None | Alive | Dutrannoy et al. (2010) |
| 10 | M | Malaysia | No | No | 96 | – | No | Microcephaly | Facial dysmorphia, Clinodactyly | Thrombocytopenia | Left hearing loss | Compound heterozygous/ Nonsense; large deletion/Exon 4; exons 1–3 |
c.526C > T (p. R176*); 6.9 kb deletion |
IGRT, AB prophylaxis | Alive | Dutrannoy et al. (2010) |
| 11 | M | Turkey | Yes | No | 12 | – | Yes | Microcephaly | None | AIHA | dystrophy and mouth lesions | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | Blood transfusions, BMT | Deceased (1.5 y, Septic shock) | Dutrannoy et al. (2010) |
| 12 | F | Turkey | Yes | Yes | 20 | Recurrent RTI, Mucocutaneous candidiasis, purulent otitis, Diffuse molluscum contagiosum | Yes | Microcephaly | None | None |
Wheezing attacks, clubbing, bronchiolitis Obliterans, pulmonary HTN |
Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | IGRT, AB prophylaxis | Deceased (4.5 y, Unknown) | Turul et al. (2011) |
| 13 | – | Poland | No | No | 72 | Recurrent RTI | Yes | Microcephaly, dd | None | None | – | Homozygous/Nonsense/Exon 4 | c.501C > A (p.Y167*) | – | Alive | Du et al. (2012) |
| 14 | M | Pakistan | Yes | No | 24 | Episodic diarrhea, vomiting and cough | Yes | Microcephaly, dd | None | None | – | Homozygous/Nonsense/Exon 2 | c.169C > T (p.R57*) | – | Alive | Du et al. (2012) |
| 15 | M | Turkey | Yes | No | 132 | Recurrent RTI, UTI | Yes | Microcephaly, Febrile seizure, moderate dd |
Microphthalmia, Blepharophimosis, posterior cleft palate, acral anomalies |
None | Bilateral VUR | Homozygous/Large deletion/Exons 2–5 | ex 2–5 deletion | IGRT | Alive | Du et al. (2012), Verloes et al. (2001) |
| 16 | F | Turkey | Yes | Yes | 1 | Diarrhea | Yes | Microcephaly | Facial dysmorphia | None | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | BMT | Alive | Çağdaş et al. (2012) |
| 17 | F | Turkey | Yes | Yes | 9 | Recurrent RTI, UTI, Otitis media, CMV pneumonia | Yes | Microcephaly | Facial dysmorphia | Neutropenia, Coombs positive AIHA | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | Irradiated RBC transfusions, IGRT, IS, AB prophylaxis, BMT | Alive | Çağdaş et al. (2012) |
| 18 | F | Turkey | Yes | Yes | 36 | Recurrent perianal abscess | Yes | Microcephaly, | Facial dysmorphia, polydactyly | Pancytopenia, Neutropenia | Spontaneous chromosomal breakages | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | IGRT, BMT (candidate) | Alive | Cipe et al. (2014) |
| 19 | M | Turkey | Yes | Yes | 7 |
Oral thrush, Recurrent RTI, chronic diarrhea, fungal abscess |
Yes | Microcephaly, Focal convulsion | Facial dysmorphia | Pancytopenia, Neutropenia | NHL | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | Anti‐epileptic, Chemotherapy (RTX and MTX), IGRT, AB prophylaxis | Alive | Patiroglu et al. (2015) |
| 20 | F | Turkey | Yes | No | 42 | RTI, diarrhea | Yes | Microcephaly | Facial dysmorphia | Pancytopenia | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | IGRT, AB prophylaxis | Alive | Akar et al., 2016) |
| 21 | – | Netherlands | – | – | 48 | Recurrent RTI | Yes | Microcephaly | – | – | – | Homozygous/Nonsense/Exon 4 | c.501C > A (p.Y167*) | BMT | Alive | Speert et al. (2016) |
| 22 | – | Netherlands | – | – | 1.5 | Diarrhea | Yes | Microcephaly | – | – | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | BMT | Alive | Speert et al. (2016) |
| 23 | – | Netherlands | – | – | 15 | UTI, RTI | Yes | Microcephaly | – | – | Autoimmunity | Homozygous/Nonsense/Exon 2 | c.169C > T (p.R57*) | BMT | Alive | Speert et al. (2016) |
| 24 | – | Netherlands | – | – | 120 | RTI | Yes | Microcephaly | – | – | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | BMT | Alive | Speert et al. (2016) |
| 25 | – | Netherlands | – | – | 96 | – | Yes | Microcephaly | – | – | – | Homozygous/Frameshift dup/Exon 3 | c.324dupG (p.R109Afs*3) | BMT (candidate) | Alive | Speert et al. (2016) |
| 26 | – | Netherlands | – | – | 11 | Recurrent fungal RTI | Yes | Microcephaly | – | – | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | BMT (candidate) | Alive | Speert et al. (2016) |
| 27 | – | Netherlands | – | – | 96 | Recurrent RTI | Yes | Microcephaly | – | – | – |
Homozygous/Nonsense/Exon 5 |
c.532C > T (p.R178*) | BMT (candidate) | Alive | Speert et al. (2016) |
| 28 | – | Netherlands | – | – | 108 | RTI, Diarrhea | Yes | Microcephaly | – | – | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | BMT | Alive | Speert et al. (2016) |
| 29 | – | Netherlands | – | – | 12 | BCGitis, Otitis | Yes | Microcephaly | – | – | Autoimmunity | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | BMT | Alive | Speert et al. (2016) |
| 30 | M | Spain | Yes | No | 48 | RTI | Yes | None | None | Bone marrow aplasia, Pancytopenia | Short telomere | Homozygous/Nonsense/Exon 2 | c.169C > T (p.R57*) | IGRT, Blood transfusion, BMT | Deceased (8 y, Severe complications after HSCT) | Carrillo et al. (2017) |
| 31* | M | Saudi Arabia | Yes | Yes | 180 | Chest infections, Bronchiectasis, Hepatitis B | Yes | Microcephaly, Neurological manifestations (Hyperreflexia), dd | Facial dysmorphia, Radial deviation of little fingers | Pancytopenia | – | Homozygous/Canonical splice site/Intron 3 | c.390 + 1G > C | IGRT, AB prophylaxis | Alive | Sheikh et al. (2017) |
| 32* | F | Saudi Arabia | Yes | Yes | 2 | Chest infections, Bronchiectasis, Hepatitis B | Yes |
Microcephaly, muscle spasms, mouth pulling to one side, progressive ataxia, worsening proximal muscle weakness, and inability to walk without assistance, dd |
Facial dysmorphia | Pancytopenia | – | Homozygous/Canonical splice site/Intron 3 | c.390 + 1G > C | IGRT, AB prophylaxis | Deceased (20 y, Septic shock) | Sheikh et al. (2017) |
| 33* | M | Saudi Arabia | Yes | Yes | 12 | Recurrent otitis media, Hepatitis B | Yes | Microcephaly, Neurological manifestation, dd | Facial dysmorphia | Pancytopenia | Allergic to IVIg | Homozygous/Canonical splice site/Intron 3 | c.390 + 1G > C | No IGRT due to anaphylaxis | Alive | Sheikh et al. (2017) |
| 34 | F | Iran | Yes | Yes | 36 | BCG adenitis, Mastoiditis, Otitis media, UTI, Oral candidiasis, Pansinusitis | Yes | Microcephaly | None | Autoimmune anemia and thrombocytopenia | Neonatal hypothyroidism, Splenomegaly, Lymphadenopathy | Homozygous/Nonsense/Exon 5 | c.526C > T (p.R176*) | Levothyroxine, AB prophylaxis, IGRT, BMT | Alive | Yazdani et al. (2017) |
| 35 | M | Pakistan | – | – | 256 | No | Yes | Microcephaly | Skeletal anomalies (clinodactyly) | Mild pancytopenia |
MDS (Initially monosomy 7, replaced by del (20)) |
Homozygous/Missense/Exon 3 | c.236 T > C (p.L79P) | – | Alive | Kager et al. (2018) |
| 36 | F | Spain | No | No | 4.5 | – | Yes | Microcephaly | None | None | – | Homozygous/Nonsense/Exon 2 | c.169C > T (p.R57*) | BMT | Alive | Recio et al. (2018) |
| 37 | F | Spain | No | No | 94 | RTI | Yes | Microcephaly | Facial dysmorphia | ITP | – | Homozygous/Nonsense/Exon 2 | c.169C > T (p.R57*) | BMT | Alive | Recio et al. (2018) |
| 38 | F | Iran | Yes | Yes | 30 | Axillary lymphadenitis following BCG | Yes | Microcephaly, dd | – | AIHA | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | IS | Alive | Esmaeilzadeh et al. (2019) |
| 39 | F | Turkey | Yes | Yes | 18 | – | Yes | dd | – | – | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | BMT | Alive | Firtina et al. (2020) |
| 40 | F | Turkey | Yes | Yes | 66 | – | Yes | dd | – | – | – | Homozygous/Nonsense/Exon 5 | c.532C > T (p.R178*) | BMT | Alive | Firtina et al. (2020) |
| 41 | M | Oman | Yes | Yes | 3 | Klebsiella pneumonia sepsis | Yes | Microcephaly | Facial dysmorphia, Ectopic kidney with normal function | AIHA, G6PD def. | Supraventricular tachycardia | Homozygous/Missense/Exon 3 | c.367 T > C (p.C123R) | Blood transfusion, AB, IS, adenosine and cardioversion, IGRT | Deceased | Al‐Marhoobi et al. (2020) |
| 42 | M | India | – | – | 204 | – | – | – | – | – | AML | Homozygous/Canonical splice site/Intron 4 | c.530‐2A > T | – | – | Arunachalam et al. (2021) |
| 43 | – | Italy | – | – | 108 | – | – | – | Facial dysmorphia | Aplastic anemia (BMF) | – | Homozygous/Missense/Exon 2 | c.169C > G (p.R57G) | BMT | Alive | Miano et al. (2021) |
| 44* | – | Italy | – | Yes | 144 | – | – | – | Facial dysmorphia | Aplastic anemia (BMF) | – | Homozygous/Missense/Exon 4 | c.506A > T (p.E169V) | BMT | Alive | Miano et al. (2021) |
| 45* | – | Italy | – | Yes | 180 | – | – | – | Facial dysmorphia | Aplastic anemia (BMF) | – | Homozygous/Missense/Exon 4 | c.506A > T (p.E169V) | BMT | Alive | Miano et al. (2021) |
| 46 | M | India | – | – | 10 | Recurrent gastroenteritis, Pneumonia, CMV infection | – | – | – | AIHA | – | Homozygous/Frameshift/ del/Exon 5 | c.544_545delGA (p.E182Tfs*3) | IS | – | Vignesh et al. (2020) |
| 47 | M | India | – | – | 11 | Skin pustule and abscess, Oral thrush | – | – | – | – | Generalized erythematous macular rash | Homozygous/Frameshift/ del/Exon 3 | c.221_222delGT (p.C74Sfs*4) | – | – | Vignesh et al. (2020) |
| 48 | F | Iran | Yes | Yes | 2 | Oral thrush, BCGitis, Bilateral otitis | Yes | Microcephaly, dd, bilateral mild sensorineural hearing loss | No | No | Eczema, thymus atrophy, hypothyroidism | Homozygous/Canonical splice site/Intron 3 | c.390 + 1G > T | Levothyroxine, AB prophylaxis, IGRT, BMT (candidate) | Alive | Farsi et al. (2021) |
| 49 | F | Iran | Yes | No | 6 | Oral candidiasis, Diarrhea, UTI, BCGitis, mastoiditis | Yes | Microcephaly | None | Bicytopenia | Tracheomalacia, perianal ulcers, pustular rash on fingers | Homozygous/Canonical splice site/Intron 3 | c.390 + 1G > T | IGRT, AB prophylaxis, BMT (candidate) | Deceased (11 months, Septic shock) | This report, 2022 |
Abbreviations: AB; antibiotic, AIHA; autoimmune hemolytic anemia, AOD; age of diagnosis, BCG; bacillus Calmette–Guerin, BMF; bone marrow failure, BMT; bone marrow transplant, CMV; cytomegalovirus, dd; developmental delay, FH; family history, FTT; failure to thrive, GI; gastrointestinal, HTN; hypertension, IEI; inborn error of immunity, IGRT; immunoglobulin replacement therapy, IS; immunosuppressive, ITP; immune thrombocytopenic purpura, IVIg; intravenous immunoglobulin, MDS; Myelodysplastic syndrome, MTX; methotrexate, NHL; non‐Hodgkin lymphoma, RTI; respiratory tract infection, RTX; rituximab, UTI; urinary tract infection,
Siblings.
The most common clinical manifestations are growth failure (n = 40, 81.6%), infectious disorders (n = 38%, 77.6%), and microcephaly (n = 38%, 77.6%) (Figure 2). Other less frequent manifestations included cytopenia (n = 24%, 48.9%), facial dysmorphism (n = 20%, 40.8%), neurologic/learning disorder (microcephaly, developmental delay, and seizures) (n = 14%, 28.6%), skeletal anomalies (mainly acral malformations) (n = 8%, 16.3%), organomegaly (n = 3%, 6.1%), and malignancy (n = 3%, 6.1%).
FIGURE 2.
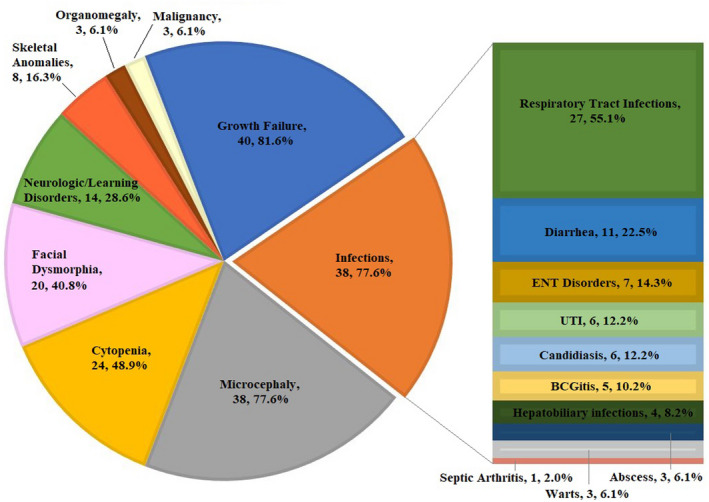
The spectrum of clinical manifestations among 49 patients with Cernunnos deficiency. The most common clinical manifestations are growth failure, microcephaly, and infectious disorders. RTI; respiratory tract infection, UTI; urinary tract infection, ENT; ear‐nose‐throat, BCG; bacillus Calmette–Guerin.
Infectious disorders mostly included respiratory tract infections (n = 27%, 55.1%) and diarrhea (n = 11%, 22.5%), followed by ear‐nose‐throat disorders (n = 7%, 14.3%), UTI (n = 6%, 12.2%), candidiasis (n = 6%, 12.2%), bacillus Calmette–Guerin (BCG) lymphadenitis (n = 5%, 10.2%), hepatobiliary disorders (Hepatitis B and cholangitis) (n = 4%, 8.2%), and less commonly warts (n = 3%, 6.1%), abscesses (n = 3%, 6.1%), and septic arthritis (n = 1%, 2.0%).
A summary of immunologic characteristics in reported patients is illustrated in Figure 3 and Supporting Information ESM_2. Most patients had lymphopenia (32 of 39%, 82.1%), low T CD3+ cells (31 of 38%, 81.6%), low T CD4+ cells (23 of 27%, 85.2%), low T CD8+ cells (20 of 27, 74.1%), low B cells (36 of 38%, 94.7%), but normal NK cells (20 of 30%, 67%). In addition, seven of 30 (23.3%) patients had elevated NK levels. The LTT was had suboptimal results in 16 out 18 (88.9%) patients. The majority of patients exhibited low IgG (27 of 35%, 77.2%) and low IgA (28 of 34, 82.0%), while the number of patients with low (15 of 32%, 46.9%) and normal (13 of 32%, 40.6%) IgM was rather equal. The interesting finding in our patient was T− B+ NK+ flowcytometry pattern, which is not commonly expected in Cernunnos deficiency. Another patient with the similar immunologic phenotype was also an Iranian patient with BCG lymphadenitis, neonatal hypothyroidism, variable infections (mastoiditis, otitis media, candidiasis, and complicated UTI), failure to thrive (FTT), microcephaly, organomegaly, and cytopenia. She had normal CD19+ B‐cell population, but different affected XLF domain (Yazdani et al., 2017). These observations may be explained by ethnic/genetic background of these two patients.
FIGURE 3.
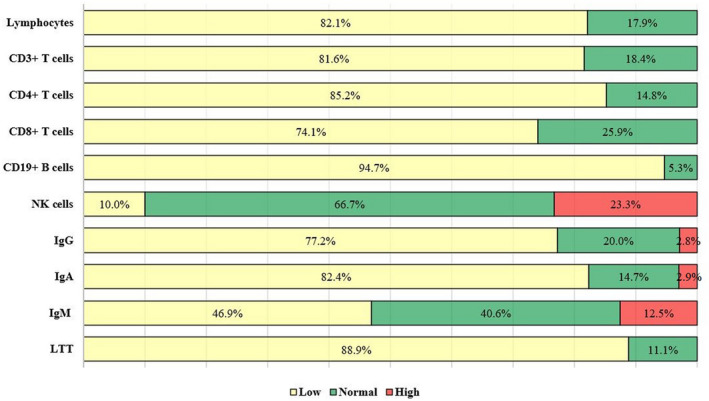
Immunological phenotype of patients with Cernunnos deficiency. The majority of patients represented lymphopenia, low T cells and its subset, low B cells, and normal natural killer (NK) cells. In addition, abnormal lymphocyte transformation test (LTT) and low serum IgG and IgA were reported in most of the reported patients, while the number of patients with low and normal IgM was rather equal.
As depicted in Figure 4, 20 different causative variants have been reported in patients affected by Cernunnos deficiency. XLF consists of a globular head domain, interacting with XRCC4, a coiled‐coil stalk domain, and interacting with another XLF to form homodimer. The protein also has a C‐terminal region that mediate its binding to DNA and also binding to Ku protein (a recruiting hub for multiple NHEJ factors) through a highly conserved Ku‐binding motif (KBM).
FIGURE 4.
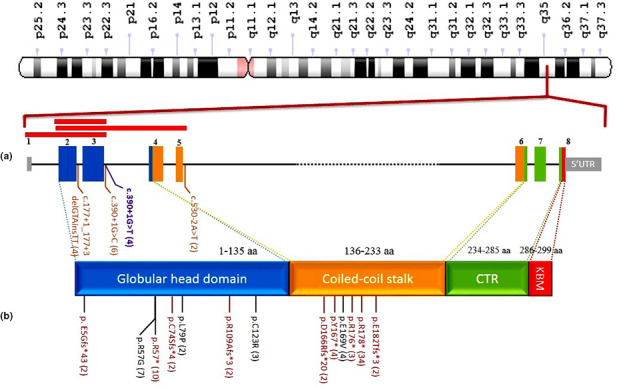
Spectrum of mutations in functional regions of XLF. (a) Schematic representation of NHEJ1 exons (the size of exons is displayed on their scale, but the scales for introns and untranslated regions have not been regarded) and functional region of XLF protein [amino acids numbers corresponding to globular head (blue) and coiled‐coil stalk (orange) domains are from Menon and Povirk (2017) and for Ku‐binding motif (KBM) is from Frit et al. (2019); CTR: C‐terminal region. (b) Reported variants. Missense variants are shown in black, frameshift and nonsense in red, splicing variant in brown and the proband's variant in purple. Red boxes on top of exons show reported large deletions. Numbers in parentheses show allele count of each variant.
All the variants have been located in globular head and coiled‐coil stalk domains, more than half of them clustered in just 16 amino acids (166 to 182) of stalk domain. Nonsense variants are the most common point mutations (52%), followed by missense and splicing (16.4%), frameshift variants (10.2%), and large deletion (5.1%). There is no clear correlation regarding type/location of the mutation and clinical presentations/severity in the patients. As illustrated in Table 1, the three siblings reported by Sheikh et al. have similar symptoms, but our patient with the same affected nucleotide had different symptoms with no dysmorphic features or neurological presentation, but having skin manifestation.
In patients with SCID, including Cernunnos deficiency, HSCT is associated with favorable outcomes and provide long‐term immune reconstitution. In this review, 17 out of 19 patients (89.5%) who received HSCT survived. Six other patients were planned for HSCT, one of whom (this report) had died while waiting for a matched donor. Finding an appropriate donor can be a general obstacle, yet patients with Cernunnos deficiency have some limitations regarding conditioning regimen. In other words, the co‐administration of chemotherapy and radiotherapy can be harmful due to the varying degrees of radiosensitivity observed in these patients (Haddad & Hoenig, 2019; Slatter & Gennery, 2020). Recently, Slack et al. in a cohort of patients with DNA double‐strand break repair disorders, reported 77 patients with DNA ligase IV deficiency, Cernunnos deficiency, or Nijmegen breakage syndrome, of whom 73 patients had received conditioning regimen, 69% were survived and the survival rate was notably higher among those with RIC regimen (p = 0.006) (Slack et al., 2018). Further specific studies are required to ascertain the role of HSCT in the survival of Cernunnos deficient patients. Besides, studies on XLF−/− mice have shown that the slope of B‐cell loss in bone marrow and spleen increases and stem cell differentiation deteriorates with aging, affecting all hematopoietic lineages (Roch et al., 2019). Therefore, timely diagnosis of Cernunnos deficiency and taking measurements for early transplantation remarkably affect patients' morbidity and mortality.
In conclusion, Cernunnos deficiency should be considered as a differential diagnosis in patients with microcephaly, growth retardation, recurrent infections, T‐cell defects, and hypogammaglobulinemia. Normal B‐cells should not rule out the diagnosis, as patients may display T− B+ NK+ phenotype.
AUTHOR CONTRIBUTIONS
M. Jamee and Z. Chavoshzadeh contributed to the conceptualization, data curation, and supervision of the study; M. Jamee wrote the original draft; Z. Chvoshzadeh, N. Khakbazan Fard, Sh. Fallah, M. Fallahi, B. Sh. Shamsian, and S. Sharafian diagnosed and managed the patient when she was alive; Z. Golchehre performed genetic analyses and draw Figure 4. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
We thank the patient and his family for their contribution to this study.
CONSENT
Informed consent was obtained from the parents of the patients prior to inclusion in the study.
Funding Information
The authors received no specific funding for this research.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ETHICS STATEMENT
This study was approved by the ethics committee of Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Supporting information
Table S1 Laboratory investigations in the proband
Table S2 Immunological Characteristics in Patients with Cernunnos Deficiency
Jamee, M. , Khakbazan Fard, N. , Fallah, S. , Golchehre, Z. , Fallahi, M. , Shamsian, B. S. , Sharafian, S. , & Chavoshzadeh, Z. (2022). Cernunnos defect in an Iranian patient with T− B+ NK + severe combined immunodeficiency: A case report and review of the literature. Molecular Genetics & Genomic Medicine, 10, e1990. 10.1002/mgg3.1990
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- Abolhassani, H. , Kiaee, F. , Tavakol, M. , Chavoshzadeh, Z. , Mahdaviani, S. A. , Momen, T. , Yazdani, R. , Azizi, G. , Habibi, S. , Gharagozlou, M. , Movahedi, M. , Hamidieh, A. A. , Behniafard, N. , Nabavi, M. , Bemanian, M. H. , Arshi, S. , Molatefi, R. , Sherkat, R. , Shirkani, A. , … Aghamohammadi, A. (2018). Fourth update on the Iranian national registry of primary Immunodeficiencies: Integration of molecular diagnosis. Journal of Clinical Immunology, 38(7), 816–832. 10.1007/s10875-018-0556-1 [DOI] [PubMed] [Google Scholar]
- Ahnesorg, P. , Smith, P. , & Jackson, S. P. (2006). XLF interacts with the XRCC4‐DNA ligase IV complex to promote DNA nonhomologous end‐joining. Cell, 124(2), 301–313. 10.1016/j.cell.2005.12.031 [DOI] [PubMed] [Google Scholar]
- Akar, H. H. , Patiroglu, T. , Hershfield, M. , & van der Burg, M. (2016). Combined Immunodeficiencies: twenty years experience from a single center in Turkey. Central‐European Journal of Immunology, 41(1), 107–115. 10.5114/ceji.2015.56168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Marhoobi, R. , Al‐Musalhi, M. , Naseem, S. U. , Wali, Y. , Alsayegh, A. , & Al‐Tamemi, S. (2020). Combined immunodeficiency, hemolytic anemia, and growth retardation secondary to a homozygous mutation in the NHEJ1 gene. Journal of Pediatric Hematology/Oncology, 42(4), 333–335. 10.1097/mph.0000000000001545 [DOI] [PubMed] [Google Scholar]
- Arunachalam, A. K. , Maddali, M. , Aboobacker, F. N. , Korula, A. , George, B. , Mathews, V. , & Edison, E. S. (2021). Primary Immunodeficiencies in India: Molecular diagnosis and the role of next‐generation sequencing. Journal of Clinical Immunology, 41(2), 393–413. 10.1007/s10875-020-00923-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousfiha, A. , Jeddane, L. , Picard, C. , Al‐Herz, W. , Ailal, F. , Chatila, T. , Cunningham‐Rundles, C. , Etzioni, A. , Franco, J. L. , Holland, S. M. , Klein, C. , Morio, T. , Ochs, H. D. , Oksenhendler, E. , Puck, J. , Torgerson, T. R. , Casanova, J. L. , Sullivan, K. E. , & Tangye, S. G. (2020). Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. Journal of Clinical Immunology, 40(1), 66–81. 10.1007/s10875-020-00758-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck, D. , Malivert, L. , de Chasseval, R. , Barraud, A. , Fondanèche, M. C. , Sanal, O. , Plebani, A. , Stéphan, J. L. , Hufnagel, M. , le Deist, F. , Fischer, A. , Durandy, A. , de Villartay, J. P. , & Revy, P. (2006). Cernunnos, a novel nonhomologous end‐joining factor, is mutated in human immunodeficiency with microcephaly. Cell, 124, 287–299. 10.1016/j.cell.2005.12.030 [DOI] [PubMed] [Google Scholar]
- Çağdaş, D. , Özgür, T. T. , Asal, G. T. , Revy, P. , De Villartay, J. P. , van der Burg, M. , Sanal, Ö. , & Tezcan, I. (2012). Two SCID cases with Cernunnos‐XLF deficiency successfully treated by hematopoietic stem cell transplantation. Pediatric Transplantation, 16(5), E167–E171. 10.1111/j.1399-3046.2011.01491.x [DOI] [PubMed] [Google Scholar]
- Carrillo, J. , Calvete, O. , Pintado‐Berninches, L. , Manguan‐García, C. , Sevilla Navarro, J. , Arias‐Salgado, E. G. , Sastre, L. , Guenechea, G. , López Granados, E. , de Villartay, J. P. , Revy, P. , Benitez, J. , & Perona, R. (2017). Mutations in XLF/NHEJ1/Cernunnos gene results in downregulation of telomerase genes expression and telomere shortening. Human Molecular Genetics, 26(10), 1900–1914. 10.1093/hmg/ddx098 [DOI] [PubMed] [Google Scholar]
- Cipe, F. E. , Aydogmus, C. , Babayigit Hocaoglu, A. , Kilic, M. , Kaya, G. D. , & Yilmaz Gulec, E. (2014). Cernunnos/XLF deficiency: A syndromic primary immunodeficiency. Case Reports in Pediatrics, 2014, 614238. 10.1155/2014/614238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. , Kysela, B. , Hanakahi, L. A. , Manolis, K. , Riballo, E. , Stumm, M. , Harville, T. O. , West, S. C. , Oettinger, M. A. , & Jeggo, P. A. (2003). Nonhomologous end joining and V(D)J recombination require an additional factor. Proceedings of the National Academy of Sciences, 100(5), 2462–2467. 10.1073/pnas.0437964100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, L. , Peng, R. , Björkman, A. , Filipe de Miranda, N. , Rosner, C. , Kotnis, A. , Berglund, M. , Liu, C. , Rosenquist, R. , Enblad, G. , Sundström, C. , Hojjat‐Farsangi, M. , Rabbani, H. , Teixeira, M. R. , Revy, P. , Durandy, A. , Zeng, Y. , Gennery, A. R. , de Villartay, J. P. , … Pan‐Hammarström, Q. (2012). Cernunnos influences human immunoglobulin class switch recombination and may be associated with B cell lymphomagenesis. The Journal of Experimental Medicine, 209(2), 291–305. 10.1084/jem.20110325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrannoy, V. , Demuth, I. , Baumann, U. , Schindler, D. , Konrat, K. , Neitzel, H. , Gillessen‐Kaesbach, G. , Radszewski, J. , Rothe, S. , Schellenberger, M. T. , Nürnberg, G. , Nürnberg, P. , Teik, K. W. , Nallusamy, R. , Reis, A. , Sperling, K. , Digweed, M. , & Varon, R. (2010). Clinical variability and novel mutations in the NHEJ1 gene in patients with a Nijmegen breakage syndrome‐like phenotype. Human Mutation, 31(9), 1059–1068. 10.1002/humu.21315 [DOI] [PubMed] [Google Scholar]
- Esmaeilzadeh, H. , Bordbar, M. R. , Hojaji, Z. , Habibzadeh, P. , Afshinfar, D. , Miryounesi, M. , Fardaei, M. , & Faghihi, M. A. (2019). An immunocompetent patient with a nonsense mutation in NHEJ1 gene. BMC Medical Genetics, 20(1), 45. 10.1186/s12881-019-0784-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci, M. , Lanino, E. , Micalizzi, C. , Morreale, G. , Di Martino, D. , Banov, L. , Comoli, P. , Locatelli, F. , Soresina, A. , & Plebani, A. (2009). Unrelated hematopoietic stem cell transplantation for Cernunnos‐XLF deficiency. Pediatr Transplant, 13, 785–789. 10.1111/j.1399-3046.2008.01028.x [DOI] [PubMed] [Google Scholar]
- Farsi, Y. , Hojabri, M. , Eslamian, G. , Shamsian, B. , Mansour Ghanaie, R. , Keramatipour, M. , Chavoshzadeh, Z. , & Eskandarzadeh, S. (2021). A case of cernunnos immunodeficiency with a novel genetic mutation. SSRN Electronic Journal. 10.2139/ssrn.3997299. Online ahead of print. [DOI] [Google Scholar]
- Firtina, S. , Yin Ng, Y. , Hatirnaz Ng, O. , Kiykim, A. , Aydiner, E. , Nepesov, S. , Camcioglu, Y. , Sayar, E. H. , Reisli, I. , Torun, S. H. , Cogurlu, T. , Uygun, D. , Simsek, I. E. , Kaya, A. , Cipe, F. , Cagdas, D. , Yucel, E. , Cekic, S. , Uygun, V. , … Sayitoglu, M. (2020). Mutational landscape of severe combined immunodeficiency patients from Turkey. International Journal of Immunogenetics, 47(6), 529–538. 10.1111/iji.12496 [DOI] [PubMed] [Google Scholar]
- Frit, P. , Ropars, V. , Modesti, M. , Charbonnier, J. B. , & Calsou, P. (2019). Plugged into the Ku‐DNA hub: The NHEJ network. Progress in Biophysics and Molecular Biology, 147, 62–76. 10.1016/j.pbiomolbio.2019.03.001 [DOI] [PubMed] [Google Scholar]
- Haddad, E. , & Hoenig, M. (2019). Hematopoietic stem cell transplantation for severe combined immunodeficiency (SCID). Frontiers in Pediatrics, 7, 481. 10.3389/fped.2019.00481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speert, I. J. , Rozmus, J. , Schwarz, K. , Warren, R. L. , van Zessen, D. , Holt, R. A. , Pico‐Knijnenburg, I. , Simons, E. , Jerchel, I. , Wawer, A. , Lorenz, M. , Patıroğlu, T. , Akar, H. H. , Leite, R. , Verkaik, N. S. , Stubbs, A. P. , van Gent, D. C. , van Dongen, J. J. , & van der Burg, M. (2016). XLF deficiency results in reduced N‐nucleotide addition during V(D)J recombination. Blood, 128, 650–609. 10.1182/blood-2016-02-701029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kager, L. , Jimenez Heredia, R. , Hirschmugl, T. , Dmytrus, J. , Krolo, A. , Müller, H. , Bock, C. , Zeitlhofer, P. , Dworzak, M. , Mann, G. , Holter, W. , Haas, O. , & Boztug, K. (2018). Targeted mutation screening of 292 candidate genes in 38 children with inborn haematological cytopenias efficiently identifies novel disease‐causing mutations. British Journal of Haematology, 182(2), 251–258. 10.1111/bjh.15389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon, V. , & Povirk, L. F. (2017). XLF/Cernunnos: An important but puzzling participant in the nonhomologous end joining DNA repair pathway. DNA Repair (Amst), 58, 29–37. 10.1016/j.dnarep.2017.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer‐Bahlburg, A. , Dressler, F. , & Baumann, U. (2014). Chronic arthritis in a boy with Cernunnos immunodeficiency. Clinical Immunology, 154(1), 47–48. 10.1016/j.clim.2014.06.003 [DOI] [PubMed] [Google Scholar]
- Miano, M. , Grossi, A. , Dell'Orso, G. , Lanciotti, M. , Fioredda, F. , Palmisani, E. , Lanza, T. , Guardo, D. , Beccaria, A. , Ravera, S. , Cossu, V. , Terranova, P. , Giona, F. , Santopietro, M. , Cappelli, E. , Ceccherini, I. , & Dufour, C. (2021). Genetic screening of children with marrow failure.The role of primary Immunodeficiencies. American Journal of Hematology, 96(9), 1077–1086. 10.1002/ajh.26242 [DOI] [PubMed] [Google Scholar]
- Patiroglu, T. , Akar, H. H. , van der Burg, M. , & Kontas, O. (2015). A case of XLF deficiency presented with diffuse large B cell lymphoma in the brain. Clinical Immunology, 161(2), 394–395. 10.1016/j.clim.2015.06.009 [DOI] [PubMed] [Google Scholar]
- Recio, M. J. , Dominguez‐Pinilla, N. , Perrig, M. S. , Rodriguez Vigil‐Iturrate, C. , Salmón‐Rodriguez, N. , Martinez Faci, C. , Castro‐Panete, M. J. , Blas‐Espada, J. , López‐Nevado, M. , Ruiz‐Garcia, R. , Chaparro‐García, R. , Allende, L. M. , & Gonzalez‐Granado, L. I. (2018). Extreme phenotypes with identical mutations: Two patients with same non‐sense NHEJ1 homozygous mutation. Frontiers in Immunology, 9, 2959. 10.3389/fimmu.2018.02959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch, B. , Abramowski, V. , Chaumeil, J. , & de Villartay, J. P. (2019). Cernunnos/Xlf deficiency results in suboptimal V(D)J recombination and impaired lymphoid development in mice. Frontiers in Immunology, 10, 443. 10.3389/fimmu.2019.00443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, R. , Lewis, S. , & Wlodarski, M. W. (2020). DNA repair syndromes and cancer: insights into genetics and phenotype patterns [Review]. Frontiers in Pediatrics, 8(683), 570084. 10.3389/fped.2020.570084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh, F. , Hawwari, A. , Alhissi, S. , Al Gazlan, S. , Al Dhekri, H. , Rehan Khaliq, A. M. , Borrero, E. , El‐Baik, L. , Arnaout, R. , Al‐Mousa, H. , & Alazami, A. M. (2017). Loss of NHEJ1 protein due to a novel splice site mutation in a family presenting with combined immunodeficiency, microcephaly, and growth retardation and literature review. Journal of Clinical Immunology, 37(6), 575–581. 10.1007/s10875-017-0423-5 [DOI] [PubMed] [Google Scholar]
- Slack, J. , Albert, M. H. , Balashov, D. , Belohradsky, B. H. , Bertaina, A. , Bleesing, J. , Booth, C. , Buechner, J. , Buckley, R. H. , Ouachée‐Chardin, M. , Deripapa, E. , Drabko, K. , Eapen, M. , Feuchtinger, T. , Finocchi, A. , Gaspar, H. B. , Ghosh, S. , Gillio, A. , Gonzalez‐Granado, L. I. , … Gennery, A. R. (2018). Outcome of hematopoietic cell transplantation for DNA double‐strand break repair disorders. The Journal of Allergy and Clinical Immunology, 141(1), 322–28.e10. 10.1016/j.jaci.2017.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatter, M. A. , & Gennery, A. R. (2020). Update on DNA‐double strand break repair defects in combined primary immunodeficiency. Current Allergy and Asthma Reports, 20(10), 57. 10.1007/s11882-020-00955-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turul, T. , Tezcan, I. , & Sanal, O. (2011). Cernunnos deficiency: a case report. Journal of Investigational Allergology & Clinical Immunology, 21(4), 313–316. [PubMed] [Google Scholar]
- Verloes, A. , Dresse, M. F. , Keutgen, H. , Asplund, C. , & Smith, C. I. (2001). Microphthalmia, facial anomalies, microcephaly, thumb and hallux hypoplasia, and agammaglobulinemia. American Journal of Medical Genetics, 101(3), 209–212. 10.1002/ajmg.1373 [DOI] [PubMed] [Google Scholar]
- Vignesh, P. , Rawat, A. , Kumrah, R. , Singh, A. , Gummadi, A. , Sharma, M. , Kaur, A. , Nameirakpam, J. , Jindal, A. , Suri, D. , Gupta, A. , Khadwal, A. , Saikia, B. , Minz, R. W. , Sharma, K. , Desai, M. , Taur, P. , Gowri, V. , Pandrowala, A. , … Singh, S. (2020). Clinical immunological, and molecular features of severe combined immune deficiency: A multi‐institutional experience from India. Frontiers Immunology, 11, 619146. 10.3389/fimmu.2020.619146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani, R. , Abolhassani, H. , Tafaroji, J. , Azizi, G. , Hamidieh, A. A. , Chou, J. , Geha, R. S. , & Aghamohammadi, A. (2017). Cernunnos deficiency associated with BCG adenitis and autoimmunity: First case from the national Iranian registry and review of the literature. Clinical Immunology, 183, 201–206. 10.1016/j.clim.2017.07.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Laboratory investigations in the proband
Table S2 Immunological Characteristics in Patients with Cernunnos Deficiency
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.