

Abstract
Arhinia is a rare congenital anomaly that is not typically associated with known genetic mutations and is usually discovered after an affected infant is born. Prenatal diagnosis is important because neonates with arhinia often require specialized respiratory support with creation of an artificial airway. We present a case of isolated arhinia diagnosed on second-trimester ultrasound. A patient presented for routine ultrasound at 18 weeks gestation, and nasal tissues were absent in an otherwise morphologically normal appearing fetus. Cell free fetal DNA was unremarkable. The patient elected to undergo termination of pregnancy by dilation and evacuation. Subsequent genetic analysis confirmed a normal fetal karyotype and microarray, and no examination of fetal structural anatomy was possible. Antenatal diagnosis of arhinia is important to guide maternal–fetal care decisions and requires methodical sonographic evaluation to identify this malformation prior to delivery.
Keywords: arhinia, Bosma arhinia microphthalmia syndrome, prenatal ultrasound, 3D ultrasound, prenatal genetic testing
Arhinia is an extremely rare congenital malformation characterized by total absence of the soft tissues of the nose, 1 2 with 84 reported cases in the literature. 3 4 5 6 7 Perhaps due to its rarity, this diagnosis is seldom made in the prenatal setting. The first reported case of a prenatal diagnosis of arhinia occurred in 2000. 2 With the absence of nasal passages, affected neonates are at increased risk of respiratory compromise at birth and usually require invasive methods of airway support. Additionally, knowledge of this condition at an early gestational age allows for the option of prenatal genetic testing and for discussions regarding pregnancy termination. We present a case of arhinia that was discovered during routine second-trimester imaging with the use of real-time three-dimensional (3D) ultrasound technology.
Case
The patient was a 22-year-old previously healthy primigravida whose pregnancy was complicated by acute cystitis and first trimester nausea and vomiting during pregnancy. Her daily medications consisted of a prenatal vitamin in addition to as needed use of pyridoxine-doxylamine, promethazine, and prochlorperazine. She and the father reported no family history of intellectual disabilities, congenital malformations, or genetic conditions and the patient denied use of alcohol or illicit substances.
In her first trimester, a subchorionic hematoma was noted on ultrasound examination after an episode of vaginal bleeding. She subsequently presented to care at our institution for her routine second-trimester ultrasound at 18 weeks gestational age. Her ultrasound revealed absence of the nasal bone and external soft tissues of the nose. Nares could not be identified with the use of two-dimensional (2D) real-time coronal imaging, 3D multiplanar reconstruction, or 3D surface rendering of the face ( Figs. 1 2 3 ). Detailed anatomical survey was otherwise unremarkable except for a marginal umbilical cord insertion. No other midline anomalies were observed, with any evidence of cleft lip or palate. Orbital spacing, ear placement, and brain structure were sonographically normal.
Fig. 1.
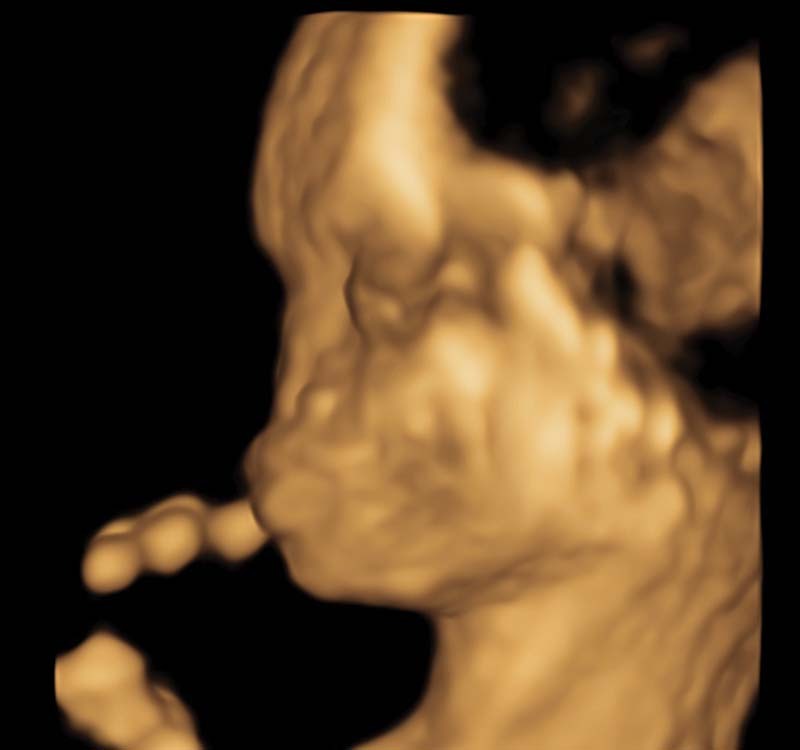
Three-dimensional surface rendering of fetal facial profile that demonstrates absence of nasal soft tissues.
Fig. 2.
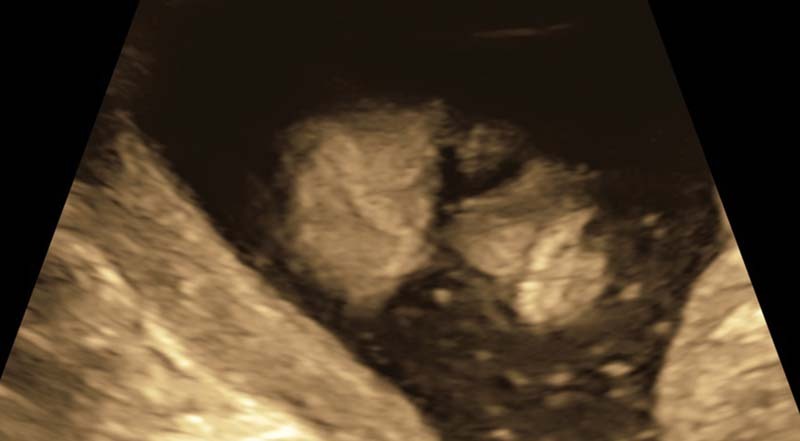
Two-dimensional coronal image of fetal face that highlights flat midface architecture and absent nares.
Fig. 3.
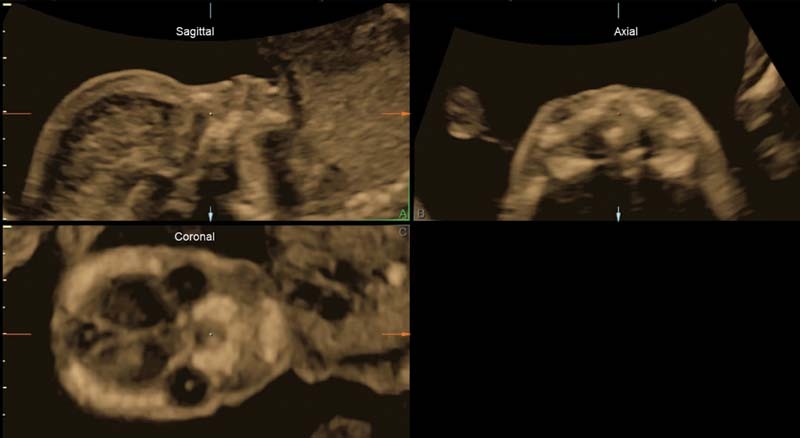
Three-dimensional multiplanar reconstruction that clearly depicts absence of the nasal bone as well as lack of external nasal structures.
Cell free fetal DNA was normal for the common aneuploidies and common microdeletions (15q11.2, 1p36, 22q11.2, 4p, and 5p). The patient elected to undergo therapeutic dilation and evacuation, which was performed without complications. The products of conception were sent for genetic evaluation and were deemed free from maternal tissue contamination. Chromosomal analysis revealed a 46, XY karyotype consistent with the cfDNA findings and a normal microarray. An assessment of the fetal face could not be performed.
Discussion
Arhinia is an extremely rare inborn error of craniofacial development defined as the absence of external nasal tissues and nasal passages. When arhinia is combined with complete absence of the olfactory system, it is termed total arhinia. 1 3 The largest series reported 80 cases of arhinia worldwide, 7 and review of the literature revealed four additional case reports published subsequently. 3 4 5 6 The majority of reported cases were diagnosed after delivery and occurred in the context of otherwise uncomplicated pregnancies. 8 Most cases are sporadic, but there are reports of families that have two affected individuals. 8 9 Neonates with arhinia typically have associated anatomic anomalies including microphthalmia or anophthalmia, cataracts, coloboma of iris, high arched or cleft palate, absent paranasal sinuses, hypoplastic maxilla, nasolacrimal duct stenosis or atresia, choanal atresia, or microtia. 7 8 This condition frequently coexists with hypogonadotropic hypogonadism (97% in one series), 7 and when arhinia is paired with microphthalmia, coloboma, and hypogonadotropic hypogonadism, it is termed Bosma Microphthalmia Syndrome. 7 10 Cognitive development is typically normal. 5 7 10 There is an increased risk of adverse neonatal outcomes. In a series of 46 cases, three infants died in the first 2 months of life: one due to respiratory insufficiency 2 hours after birth, one due to sepsis secondary to nasal reconstruction surgery at 29 days, and one due to sepsis at 10 weeks. 8
Given that neonates are usually considered obligate nasal breathers in the absence of nasal obstruction, the majority of infants with arhinia require placement of an oral airway with eventual tracheostomy formation to facilitate adequate respiration. 2 8 Other infants lack respiratory difficulty at rest but may require an orogastric tube or gastrostomy tube for enteric alimentation due to cyanosis with feeds. 11 Although cases have been reported in which affected individuals do not require surgical correction, 8 most patients undergo complex surgical reconstructions to create functional nasal passages and achieve an improved aesthetic. 5 8 12 Once the mouth breathing response is developed, usually by 6 months of age, the artificial airway can be reversed and nasal reconstruction delayed until the child is school aged. 5 8
Until recently, no genetic mutation for this condition was known. In three instances, there were identifiable aneuploidies or translocations (mos 46,XX/47,XX, + 9; XY,inv(9); and XX,t(3;12) (q13.2;p11.2)). 8 9 In-depth analysis of the patient with the translocation between chromosomes 3 and 12 uncovered a strong candidate region to contain the putative “arhinia gene” in the deleted 3q segment. Disappointingly, comparison of this region with four other patients with arhinia revealed no copy number abnormalities. Two promising genes in the deleted region, COL8A1 and CPOX, were likewise unaffected in the other individuals. 13
In 2017, a comprehensive study was published in which investigators performed genetic sequencing on 40 subjects with arhinia. The authors discovered that 84% of individuals harbored a missense mutation localized to a region of the SMCHD1 gene, which is responsible for epigenetic regulation of autosomal and X-linked genes. 7 This is interestingly the same gene responsible for an uncommon variant of muscular dystrophy termed facioscapulohumeral muscular dystrophy type 2 (FSHD2). One individual in this study met full diagnostic criteria for both arhinia and FSHD2. Whereas mutations in FSHD2 span the entire gene, those associated with arhinia were clustered tightly around the gene's ATPase domain. In zebrafish, a CRSIPR/Cas9-mediated alteration of SMCHD1 resulted in an arhinia-relevant phenotype, which affords further evidence toward this gene's role in the development of arhinia. 7 Nonetheless, the downstream alterations in gene products that culminate in the arhinia phenotype remain elusive. 7 14
The embryologic mechanisms underlying arhinia likewise remain speculative. Plausible causes for arhinia include maldevelopment of the nasal placodes, 2 the lack of development of nasal processes, or overgrowth of the medial nasal process that forms an atretic plate. 8 In the case of nasal placode failure, the medial and lateral nasal prominences are unable to grow. As a result, the soft tissues of the external nose and the nasal sac do not form. Secondarily, the lack of development of the nasal sac precludes growth of the olfactory bulb and tract. 2
Conclusion
Video 1 (A–C) Two-dimensional real-time coronal imaging and three-dimentional surface rendering of the face.
Arhinia is an uncommon condition that has historically been diagnosed in the neonatal period. Despite widespread use of prenatal ultrasound in resource-rich nations, the first documented report of arhinia diagnosed prenatally was reported in 2000. 2 Even among cases published in the last five years, the diagnosis was not made until delivery and not identified on antenatal ultrasound. 3 6 11
Establishing this diagnosis prior to delivery allows for multidisciplinary planning with neonatology, pediatric otolaryngology, and plastic surgery subspecialists to optimize the postnatal management of affected infants. Furthermore, prenatal genetic testing can be undertaken to rule out more commonly encountered genetic abnormalities associated with midline facial defects such as trisomy 13. 2 Although this test was not performed with our patient due to planned termination, single-gene sequencing of SMCHD1 with methylation profiling can be considered.
Perhaps most importantly, the early diagnosis of arhinia allows the mother to take ownership of her health care and decide how she wishes to proceed with the pregnancy. In our case, the use of 3D ultrasonography confirmed our findings on 2D imaging and provided a more direct and easily interpretable representation of the anomaly that guided our counseling with the patient. Indeed, the patient and her partner had difficulty conceptualizing the fetal anomaly until presented with 3D reconstructions and surface rendered images of the fetal face. Our case therefore serves to exemplify the utility of detailed anatomic assessment via fetal ultrasound for facial anomalies broadly and arhinia more specifically. It additionally showcases a novel application of real-time 3D and post-processing of 3D volume datasets to augment clinical decision making and provide more easily digestible information to patients unfamiliar with 2D ultrasound imaging.
Footnotes
Conflict of Interest A.E.S. reports personal fees from GE Healthcare outside the submitted work.
References
- 1.Cohen D, Goitein K J. Arhinia revisited. Rhinology. 1987;25(04):237–244. [PubMed] [Google Scholar]
- 2.Cusick W, Sullivan C A, Rojas B, Poole A E, Poole D A. Prenatal diagnosis of total arhinia. Ultrasound Obstet Gynecol. 2000;15(03):259–261. doi: 10.1046/j.1469-0705.2000.00081.x. [DOI] [PubMed] [Google Scholar]
- 3.Altuntas Z, Uyar I, Yildirim M EC. A rare congenital facial abnormality: complete arhinia. Facial Plast Surg. 2021;37(02):274. doi: 10.1055/s-0040-1715620. [DOI] [PubMed] [Google Scholar]
- 4.Borghi A, Ruggiero F, Tenhagen M. Design and manufacturing of a patient-specific nasal implant for congenital arhinia: case report. JPRAS Open. 2019;21:28–34. doi: 10.1016/j.jpra.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuller A K, McCrary H C, Graham M E, Skirko J R. The case of the missing nose: congenital arhinia case presentation and management recommendations. Ann Otol Rhinol Laryngol. 2020;129(07):645–648. doi: 10.1177/0003489420909415. [DOI] [PubMed] [Google Scholar]
- 6.Ng R L, Rajapathy K, Ishak Z. Congenital arhinia—first published case in Malaysia. Med J Malaysia. 2017;72(05):308–310. [PubMed] [Google Scholar]
- 7.Shaw N D, Brand H, Kupchinsky Z A. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat Genet. 2017;49(02):238–248. doi: 10.1038/ng.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang M M, Hu Y H, He W, Hu K K. Congenital arhinia: a rare case. Am J Case Rep. 2014;15:115–118. doi: 10.12659/AJCR.890072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou J W. Congenital arhinia with de novo reciprocal translocation, t(3;12)(q13.2;p11.2) Am J Med Genet A. 2004;130A(02):200–203. doi: 10.1002/ajmg.a.30268. [DOI] [PubMed] [Google Scholar]
- 10.Brasseur B, Martin C M, Cayci Z, Burmeister L, Schimmenti L A. Bosma arhinia microphthalmia syndrome: clinical report and review of the literature. Am J Med Genet A. 2016;170A(05):1302–1307. doi: 10.1002/ajmg.a.37572. [DOI] [PubMed] [Google Scholar]
- 11.Mondal U, Prasad R. Congenital arhinia: a rare case report and review of literature. Indian J Otolaryngol Head Neck Surg. 2016;68(04):537–539. doi: 10.1007/s12070-016-1009-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung J W, Ha D H, Kim B Y. Nasal reconstruction using a customized three-dimensional-printed stent for congenital arhinia: three-year follow-up. Laryngoscope. 2019;129(03):582–585. doi: 10.1002/lary.27335. [DOI] [PubMed] [Google Scholar]
- 13.Sato D, Shimokawa O, Harada N. Congenital arhinia: molecular-genetic analysis of five patients. Am J Med Genet A. 2007;143A(06):546–552. doi: 10.1002/ajmg.a.31613. [DOI] [PubMed] [Google Scholar]
- 14.Gurzau A D, Blewitt M E, Czabotar P E, Murphy J M, Birkinshaw R W. Relating SMCHD1 structure to its function in epigenetic silencing. Biochem Soc Trans. 2020;48(04):1751–1763. doi: 10.1042/BST20200242. [DOI] [PMC free article] [PubMed] [Google Scholar]