

Abstract
Introduction
Despite the clinical efficacy of Immune Checkpoint Inhibitors (ICI) in non-small cell lung cancer (NSCLC), only around 20% of patients remain disease free at 5 years. Here we use digital spatial profiling to find candidate biomarker proteins associated with ICI resistance.
Materials and Methods
Pre-treatment samples from 56 patients with NSCLC treated with ICI were analyzed using the NanoString GeoMx® digital spatial profiling (DSP) method. A panel of 71 photocleavable oligonucleotide-labeled primary antibodies was used for protein detection in 4 molecular compartments (tumor, leukocytes, macrophages, and immune stroma). Promising candidates were orthogonally validated with quantitative immunofluorescence (QIF). Available pre-treatment samples from 39 additional patients with NSCLC that received ICI, and 236 non-ICI treated operable NSCLC patients were analyzed to provide independent cohort validation.
Results
Biomarker discovery using the protein-based molecular compartmentalization strategy allows 284 protein variables to be assessed for association with ICI resistance by univariate analysis using continuous log-scaled data. Of 71 candidate protein biomarkers, CD66b in the CD45+CD68 molecular compartment (immune stroma) predicted significantly shorter OS (HR 1.31, p=0.016) and was chosen for validation. Orthogonal validation by QIF showed that CD66b was associated with resistance to ICI therapy but not prognostic for poor outcome in untreated NSCLC [discovery cohort (OS; HR 2.49, p=0.026), validation cohort (OS; HR 2.05, p=0.046), non-ICI treated cohort (OS; HR 1.67, p=0.06)].
Discussion
Using the DSP technique, we have discovered that CD66b expression is indicative of resistance to ICI therapy in NSCLC. Since CD66b identifies neutrophils, further studies are warranted to characterize the role of neutrophils in ICI resistance.
Keywords: immunotherapy resistance, NSCLC, digital spatial profiling, quantitative immunofluorescence, immunohistochemistry (IHC)
Introduction
Immune checkpoint inhibitors (ICI) targeting either programmed cell death protein 1 (PD-1) or its ligand (PD-L1) have been integrated into first-line therapeutic options for advanced NSCLC patients. Despite their clinical efficacy, only a minority achieves 5-year survival ranging from 20% to 32% in the unselected and high PD-L1 expressing patients, respectively [1, 2]. The majority of patients will not respond due to innate or acquired resistance to immunotherapy (ITX) [3]. Innate (primary) resistance to ICI, manifests as progressive disease (PD) by Response Evaluation Criteria in Solid Tumors (RECIST) criteria on first Computer Tomography (CT) evaluation [4], and accounts for 7–27% of first-line treatment and 20–44% of second-line treatment [3, 5]. Acquired (secondary) resistance is seen in most ICI treated lung cancer patients and is defined as PD after initial clinical benefit [3, 6], usually after 4–10 months of ICI treatment [4] [7].
Patient selection biomarkers for ICI has been a challenge. Based on the data from KEYNOTE-024, routine immunohistochemical (IHC) PD-L1 testing is used to select for pembrolizumab monotherapy in the frontline setting [8]. However, PD-L1 IHC assays are neither sensitive nor specific with an area under the curve (AUC) less than 0.7 [9, 10]. While tumor mutational burden (TMB) is another marker being evaluated in trials of ICI, its predictive value is limited [11]. In addition, although T-cell cytotoxicity is thought to be the mechanism of tumor cell killing by ICIs the assessment of T-cell infiltration in tumors has not proven to be a biomarker for response. The complexity of tumor microenvironment (TME), the heterogeneity of tumor cells, the multiple assays and scoring systems challenge further the discovery of reliable markers to inform for resistance to ICI and better stratify patients who will benefit from ICIs. Sensitive and specific biomarkers of resistance have not yet been determined and validated clinical practice [7, 12].
Thus, in this work we attempt to discover new biomarkers for resistance to ICI therapy using a spatially informed high-plex discovery tool, digital spatial profiling (DSP). This tool enables the assessment of multiple markers in a spatially informed manner [13]. The GeoMx DSP® System (NanoString Technologies) can simultaneously detect multiple proteins in single formalin fixed, paraffin embedded (FFPE) tissue sections in a quantitative and spatially resolved manner; additionally, fluorescent tissue markers can be used to guide the DSP to interrogate specific cellular/molecular compartments. In this study, we used DSP technology as a discovery tool to find spatially resolved protein markers associated with resistance to ICI in advanced NSCLC. Then, amongst candidate predictors, we further assessed CD66b expression by quantitative immunofluorescence (QIF), to validate the association with outcome by an orthogonal quantitative method and on an independent cohort.
Materials and Methods
Patient cohorts and tissue microarrays
We analyzed retrospectively collected FFPE pre-ITX tumor specimens represented in a tissue microarray (TMA) format from 58 patients with NSCLC treated with ICI in the advanced setting between 2017 and 2019 at Yale University School of Medicine, New Haven, CT (Yale Tissue MicroArray 471; YTMA471) constituting our discovery cohort. A total of 56 cases included in YTMA471 had available or adequate tissue for protein quantification. In addition, we analyzed 39 available FFPE tumor specimens, from a cohort of 53 patients with NSCLC treated with ICI between 2011 and 2017 (YTMA404) and 236 evaluable samples from 287 patients with NSCLC who did not receive ICI, between 2011 and 2016 (YTMA423), as our internal validation cohorts. The cohorts of patients with available samples are summarized in Supplementary Table 1. All tissue samples were collected and used under the approval from the Yale Human Investigation Committee protocol #9505008219 with an assurance filed with and approved by the U.S. Department of Health and Human Services. The Yale Human Investigation Committee approved the patient informed consent or in some cases waiver of consent all in accordance with the ethical guidelines of the U.S. Common Rule.
For TMA construction, tumors were reviewed by a pathologist using hematoxylin and eosin–stained preparations to select representative tumor areas. Then, two cores (diameter: 660μm each) were extracted from two tumor blocks, respectively, and arrayed in two recipient TMA master blocks, each TMA block thus containing one nonadjacent 660μm diameter tumor core per NSCLC case (Suppl. figure 1). Tumor core selection was not based on specific tumor regions or location.
For all the experiments, we assessed two slides derived from two independent blocks from YTMA471, YTMA404 and YTMA423 respectively, each block containing one nonadjacent tumor core per patient. From each block of the YTMA471 (discovery cohort) serial 5 μm sections were cut. The slides used for DSP and QIF protocols were 15 μm apart (neutrophil’s diameter ranges from 12–15 μm). This might explain some heterogeneity in the expression of CD66b when quantified by the two methods. The cases which had pre-immunotherapy specimens and received ICI, constituted our discovery and validation cohorts.
Digital Spatial Profiling
Briefly, once the slides were deparaffined and subjected to antigen retrieval procedures, we incubated them overnight with three fluorescent-labeled visualization antibodies to detect tumor cells [pancytokeratin (CK)], leukocytes (CD45), and macrophages (CD68) to define the molecular compartments. These were combined with a cocktail of 77 (71 targets and 6 controls) unique photocleavable oligonucleotide-conjugated antibodies targeting immuno-oncology markers (Suppl. Table 2) previously validated by NanoString. Once the staining was completed, slides were loaded onto the GeoMx DSP instrument, where they were scanned to produce a digital fluorescent image of the tissue.
Individual regions of interest (ROI) of a maximum diameter of 660μm covering the entire TMA core were generated, and then each ROI was segmented in three molecularly defined tissue compartments by fluorescent colocalization: tumor compartment (panCK+), leukocyte compartment (CD45+/CD68−), macrophage compartment (CD68+). An aggregate immune stromal cell compartment (IC), defined as the summation of leukocyte and macrophage expression (panCK−/CD45+/CD68+), was generated postacquisition (Figure 1).
Figure 1.
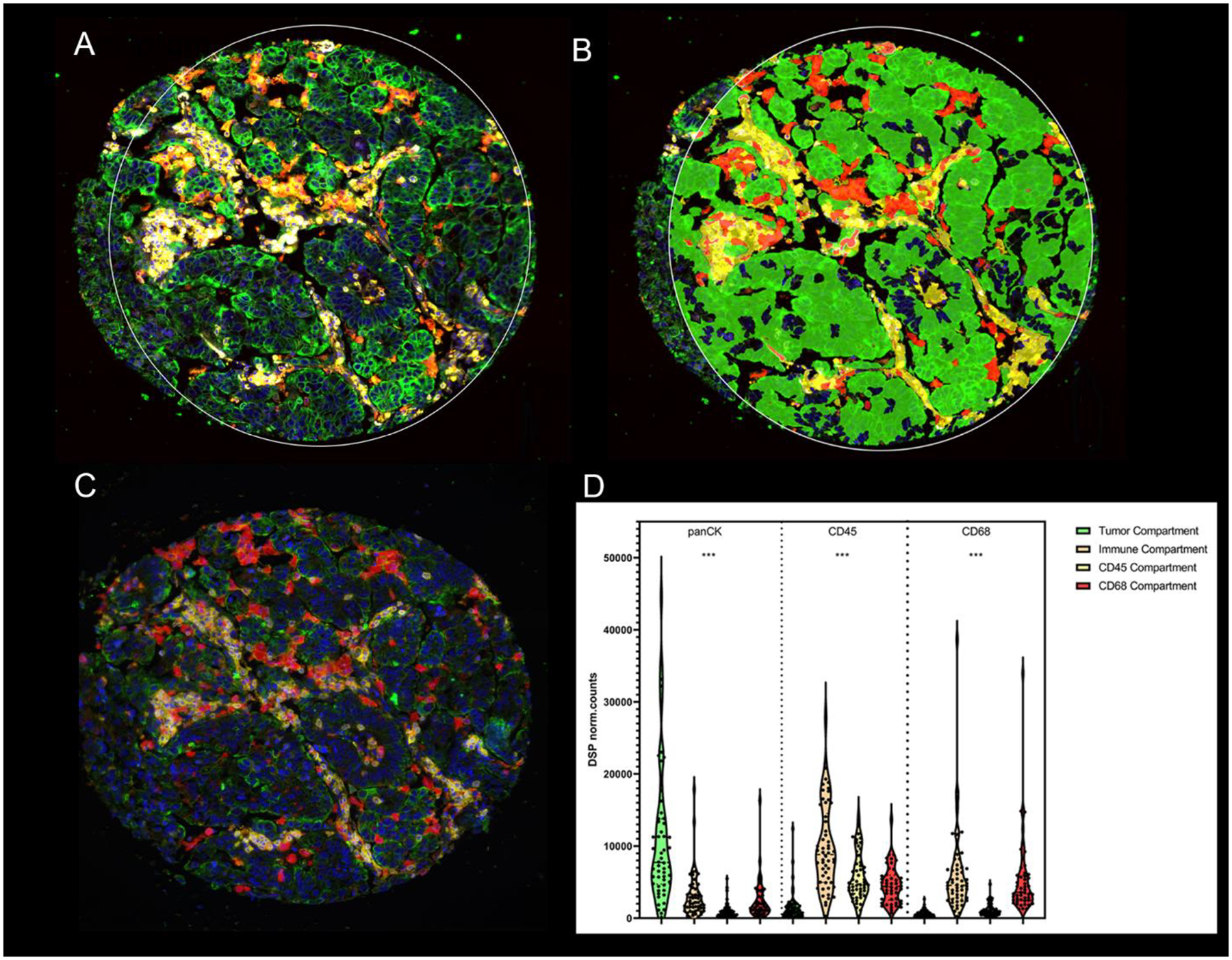
Representative TMA spot showing the fluorescence image (A) and the compartmentalized image created by fluorescence colocalization (B) using GeoMx DSP; Cytokeratin (panCK) (green), CD45 (yellow), CD68 (red), SYTO13 (blue) and (C) using QIF; Cytokeratin (green), CD45 (yellow), CD68 (red), DAPI (blue); (D) Normalized DSP counts of panCK, CD45 and CD68 in the four molecular compartments (tumor, Immune stromal, CD45 and CD68). P-values legends ***:P<0.001, Yale tissue microarray, YTMA; quantitative immunofluorescence, QIF; digital spatial profiling, DSP
Every ROI (TMA spot) was sequentially exposed to UV light to decouple the oligonucleotides from the profiling reagent (primary antibody). Photocleaved from these areas of illumination (AOI) or molecular compartments, were DNA oligos that were then collected via microcapillary aspiration and deposited into a 96-well plate, hybridized to 4-color, 6-spot optical barcodes, and finally quantitated on the nCounter® platform (NanoString Technologies). Digital counts from barcodes corresponding to protein probes were first normalized to internal spike-in controls (ERCC), and then normalized against the geometric mean of two housekeeping genes (GAPDH and ribosomal protein S6 (S6)) for each ROI compartment individually (Suppl. Figure 3). The expression level across different areas of interest for all targets, including negative isotype (Mouse IgG1, Mouse IgG2a and Rabbit IgG) and positive (GAPDH, Histone H3, S6) controls in each AOI is shown in Supplementary Figure 4.
Multiplexed QIF CD66b+ cell panel
Two multiplex panels using an orthogonal method (QIF) were performed to validate DSP observations: panel A (CD45/CD68/PD-L1), to evaluate PD-L1, was performed on YTMA471 and panel B (CD45/CD68/CD66b), to evaluate CD66b in the 4 molecular compartments analyzed in DSP, was performed simultaneously on YTMA471, YTMA404 and YTMA423. Fluorescent images were acquired using a PM-2000 system (Navigate Biopharma, Carlsbad, CA, USA) and automated quantitative analysis (AQUA™) method of QIF was used to determine different levels of expression. A QIF score was generated by dividing the sum of target pixel intensities by the area of the molecularly designated compartment, as previously described [14, 15]. The compartments generated by DSP and QIF in the discovery cohort, YTMA471, were comparable for most of the cases. (Figure 1, Suppl. Figure 5). In order to distinguish tumor from tissue stroma and other components, an epithelial tumor “mask” was created by binarizing the cytokeratin (CK) signal and creating an epithelial compartment. Similarly, CD45+ “mask” was created by the CD45+ signal subtracting the CD68+ “mask” to create a CD45+/CD68− “mask” equal to the DSP generated leukocyte (CD45+/CD68−) compartment. In addition, we created a fourth mask that included all the CD45+ cells (CK-/CD45+/CD68+) that was our immune stromal mask and represented tumor immune microenvironment. As CD66b is an activation marker for human granulocytes (predominantly neutrophil granulocytes), measurements in the tumor compartment captured part of intraepithelial protein expression, rather than reflecting true colocalization with tumor cells.
QIF scores were normalized to the exposure time and bit depth at which the images were captured, to compensate for any variability. All acquired histospots were visually assessed and cases with staining artifacts were omitted from the analysis. A sequential immunofluorescence protocol with isotypespecific primary antibodies was used to detect epithelial tumor cells (polyclonal rabbit anti- cytokeratin, wide spectrum screening, DAKO, Carpinteria, CA), CD45+ cells (mouse IgG1, clone 2B11+PD7/26, Dako), CD68+ cells (mouse IgG3, clone PG-M1, Dako), PD-L1+ cells (rabbit IgG, clone E1L3N, Cell Signaling Technology) and CD66b+ cells (mouse IgG1, clone 80H3, LSBio). Cell nuclei were visualized by the signal from 4′,6-diamidino-2-phenylindole (DAPI) stain, and the targets were visualized with species-specific secondary antibodies and Alexa 488 (cytokeratin), Cyanine 3+ (Cy3+) (lymphocytes), Alexa 750 (Cy7) (macrophages) and Cyanine 5 (Cy5) (CD66b+ cells and PD-L1).
Statistical analysis
For statistical analysis, we averaged the normalized digital counts, or AQUA scores derived from the two blocks. Continuous log-scaled data was used to identify significant associations between markers and treatment response in terms of either hazard ratio (HR) or odds ratio (OR) in Cox or logistic regression (LR) models, respectively. Benjamini–Hochberg false discovery adjustment method was performed, (padjusted=0.05) considering the number of comparisons performed per compartment (tumor, CD45, CD68 and immune stroma). All hypothesis testing was performed at a two-sided significance level of a=0.05.
Survival analysis was performed using the R packages survival and survminer. The association between target expression and durable and long-term clinical benefit was computed by LR using the R package gtsummary. To create the Kaplan–Meier plot representing the association of CD66b expression with survival, samples were categorized as either “high” or “low” by using X-tile optimal cutpoint analysis [16]. Reported p values in the plot are associations of the model (log-rank test). Spearman r was used to measure the strength and direction of association between the two different methods, DSP and QIF. Kruskal-Wallis test was used to compare the distribution of panCK, CD45 and CD68 in the 4 molecular compartments to confirm the DSP platform successfully profiled tumor and stromal compartments.
The software used for visualization of the data and statistical purposes were GraphPad™ Prism® v9.0 software for Windows (GraphPad Software, Inc., La Jolla, CA), and R studio Version 1.4.1717. The optimal survival cutpoint analysis was performed using X-tile.
Results
Our discovery cohort included 56 NSCLC cases, each represented by two TMA cores in two master blocks. 284 protein variables per ROI were generated, where 71 protein markers (excluding controls), were measured in four molecularly defined tissue compartments.
After normalization of each variable, a univariate unadjusted analysis using continuous log-scaled data was performed to validate the cohort and method by showing that high PD-L1 expression in tumor cells (panCK+) [OS; HR, 0.75; 95% confidence intervals (CI) 0.54–1.05; p=0.092, PFS; HR, 0.67; 95% CI, 0.48–0.94; p=0.017] and leukocytes (CD45+/CD68−) [OS; HR, 0.41; 95%CI 0.24–0.69; p<0.001, PFS; HR 0.52; 95%CI 0.32–0.84; p=0.008], predicted longer survival, respectively (Tables 1,2 Suppl. Figure 6). All variables significantly associated with outcome on ICI therapy are summarized in Tables 1, 2. In the immune stromal compartment (panCK−/CD45+/CD68+) 4 markers were associated significantly with PFS, and 3 with OS. Of these, S100B, a calcium binding protein, was significantly associated with favorable outcome in all 4 compartments. In NSCLC, S100B has been studied as a biomarker to detect subclinical brain metastases [17]. Brain metastasis status did not significantly influence the immunotherapy efficacy in lung cancer patients, in a recent metanalysis, but the role of S100B role in predicting benefit to immunotherapy in NSCLC is unclear [18]. In this study we focused on biomarkers of resistance and further exploration of S100B was not pursued.
Table 1.
Markers significantly associated with OS under ICI in the discovery cohort YTMA471; Overall Survival, OS; Immune Checkpoint Inhibitors, ICI; Yale tissue microarray, YTMA; AOI, Area of Illumination
| MARKERS ASSOCIATED WITH OS | ||||||
|---|---|---|---|---|---|---|
| Compartment/AOI | N | Marker | Univariate HR (95% CI) | P-value | Multivariate HR (95% CI) | P-value |
| CK (panCK+) | 54 | S100B | 0.71 (0.53, 0.96) | 0.017 | 0.70 (0.51, 0.95) | 0.041 |
| EpCAM | 1.31 (1.01,1.69) | 0.035 | 1.24 (0.91, 1.68) | 0.20 | ||
| ARG1 | 1.52 (1.06, 2.19) | 0.026 | 1.34 (0.91,1.98) | 0.14 | ||
| CD66b | 1.32 (1.13, 1.54) | 0.001 | 1.25 (1.04, 1.50) | 0.020 | ||
| Immune Stroma (panCK−/CD45+/CD68+) | 54 | S100B | 0.56 (0.40,0.78) | <0.001 | 0.56 (0.7,0.84) | 0.006 |
| EpCAM | 1.32 (1.02,1.70) | 0.034 | 1.41 (1.09, 1.82) | 0.054 | ||
| CD66b | 1.31 (1.06,1.60) | 0.016 | 1.25 (1.04, 1.52) | 0.045 | ||
| CD45 (panCK−/CD45+/CD68−) | 52 | PD-L1 | 0.41 (0.24, 0.69) | <0.001 | 0.41 (0.22, 0.75) | 0.005 |
| S100B | 0.54 (0.38,0.77) | <0.001 | 0.47 (0.33, 0.68) | <0.001 | ||
| P44/42 MAPK ERK1/2 | 0.35 (0.15,0.80) | 0.019 | 0.43 (0.17, 1.09) | 0.094 | ||
| EGFR | 0.50 (0.28, 0.90) | 0.014 | 0.48 (0.24, 0.95) | 0.023 | ||
| CD66b | 1.26 (1.03, 1.54) | 0.034 | 1.20 (0.96, 1.51) | 0.13 | ||
| CD68 (panCK−/CD68+) | 49 | S100B | 0.60 (0.43, 0.84) | 0.002 | 0.59 (0.38,0.93) | 0.020 |
| EpCAM | 1.29 (1.02, 1.64) | 0.034 | 1.40 (1.02, 1.93) | 0.041 | ||
| Phospho-JNK (T183/Y185) | 1.44 (1.02, 2.05) | 0.048 | 1.56 (1.04,0.036) | 0.13 | ||
| P44/42 MAPK ERK1/2 | 0.31 (0.13, 0.75) | 0.013 | 0.34 (0.12,0.96) | 0.047 | ||
| CD66b | 1.37 (1.12, 1.67) | 0.004 | 1.32 (1.08, 1.62) | 0.009 | ||
Note: Bold terms have P values that are significant after adjusting for multiple comparisons and/or after controlling for clinical prognostic factors in the multivariate analysis. Overall survival (OS) was calculated from the start of immunotherapy until death or censoring.
Table 2.
Markers significantly associated with PFS under ICI in the discovery cohort YTMA471; Progression Free Survival, PFS; Immune Checkpoint Inhibitors, ICI; Yale tissue microarray, YTMA; AOI, Area of Illumination
| MARKERS ASSOCIATED WITH PFS | ||||||
|---|---|---|---|---|---|---|
| Compartment/AOI | N | Marker | Univariate HR (95% CI) | P-value | Multivariate HR (95% CI) | P-value |
| CK (panCK+) | 54 | PD-L1 | 0.67 (0.48, 0.94) | 0.017 | 0.50 (0.36, 0.71) | 0.002 |
| EpCAM | 1.48 (1.17, 1.88) | <0.001 | 1.37 (1.08, 1.73) | 0.02 | ||
| Tim-3 | 0.63 (0.42,0.94) | 0.024 | 0.61 (0.39, 0.95) | 0.056 | ||
| CD66b | 1.19 (1.02,1.38) | 0.039 | 1.29 (1.11,1.51) | 0.009 | ||
| CD44 | 0.76 (0.61, 0.96) | 0.024 | 0.63 (0.50, 0.80) | 0.002 | ||
| Immune Stroma (panCK−/CD45+/CD68+) | 54 | S100B | 0.64 (0.47, 0.87) | 0.003 | 0.66 (0.48, 0.90) | 0.024 |
| EpCAM | 1.4 (1.12, 1.82) | 0.004 | 1.40 (1.09, 1.80) | 0.046 | ||
| CD66b | 1.24 (1.02, 1.51) | 0.040 | 1.31 (1.08, 1.58) | 0.023 | ||
| FOXP3 | 1.54 (1.03, 2.31) | 0.045 | 1.33 (0.87, 2.02) | 0.3 | ||
| CD45 (panCK−/CD45+/CD68−) | 52 | PD-L1 | 0.52 (0.32, 0.84) | 0.008 | 0.47 (0.29, 0.75) | 0.007 |
| S100B | 0.63 (0.46, 0.86) | 0.002 | 0.61 (0.44, 0.84) | 0.011 | ||
| CD14 | 0.63 (0.40, 0.98) | 0.037 | 0.58 (0.37, 0.93) | 0.037 | ||
| CD68 (panCK−/CD68+) | 49 | S100B | 0.69 (0.50, 0.95) | 0.018 | 0.74 (0.50,1.08) | 0.12 |
| EpCAM | 1.42 (1.12,1.81) | 0.004 | 1.49 (1.08, 2.05) | 0.015 | ||
| MART1 | 1.73 (1.08,2.76) | 0.021 | 2.25 (1.11, 4.56) | 0.016 | ||
| P44/42 MAPK ERK1/2 | 0.40 (0.17, 0.92) | 0.040 | 0.47 (0.19, 1.18) | 0.12 | ||
| CD66b | 1.28 (1.05, 1.56) | 0.019 | 1.39 (1.12, 1.73) | 0.004 | ||
| FOXP3 | 1.67 (1.12,2.48) | 0.018 | 2.03 (1.16, 3.56) | 0.013 | ||
Note: Bold terms have P values that are significant after adjusting for multiple comparisons and/or after controlling for clinical prognostic factors in the multivariate analysis. Progression-free survival (PFS) was calculated from the start of immunotherapy until progression of disease, death, or censoring.
CD66b predicted significantly shorter OS (HR, 1.31; 95%CI 1.06–1.60; p=0.016) and shorter PFS (HR, 1.24; 95%CI 1.02–1.51; p = 0.04) in immune stroma compartment. In the multivariate analysis including seven clinical prognostic factors [stage at immunotherapy, smoking history, presence of liver or/and brain metastasis, lung immune prognostic index (LIPI) score, derived neutrophils/(leukocytes-neutrophils) (dNLR) ratio and sex] [19, 20], CD66b expression remained statistically significantly associated with overall and progression free survival. Moreover, high levels of CD66b in immune stromal compartment were significantly associated with progressive disease at 12 and 24 months of ICI therapy (Suppl. Tables 3,4,5). Expression of arginase 1 (ARG1), an urea cycle enzyme that is released by activated neutrophils [21], was positively correlated with CD66b expression in immune compartment (r=0.73, p<0.01). (suppl. Table 6). No association was observed between CD66b and PD-L1 or CD8+ cells in immune compartment (Suppl. Figure 7). Thus, we chose to validate CD66b using both an orthogonal method and an independent cohort.
First, we determined its expression using a fluorescence-based QIF system (AQUA™), as previously described [14]. We used this method to assess both the discovery cohort and two further independent cohorts showing the localization and the expression range of CD66b in NSCLC (Figure 2). First, using continuous log-scaled data by QIF we confirmed that CD66b in immune stromal compartment (panCK−/CD45+/CD68+) (OS; HR 5.09,95%CI 1.29–20.1; p=0.042, PFS; HR 2.88, 95% CI 0.59–14; p=0.2), was associated with unfavorable clinical outcome to ICI therapy in the discovery cohort YTMA471 and was significantly associated with shorter OS. The difference in PFS, although favoring CD66b low cases, was not statistically significant. To validate this finding on an independent cohort, we first identified the optimal OS cutpoint for CD66b+ expression in immune stromal compartment using X-tile [16]. We determined the best single cut-point using the X-tile plot which occurs as a CD66b AQUA Score of ≥ 321 in immune stromal compartment in the discovery cohort (YTMA471). This cutpoint was then used in the validation (YTMA404) and the non-ICI treated cohort (YTMA423) included in simultaneous experiments.
Figure 2.
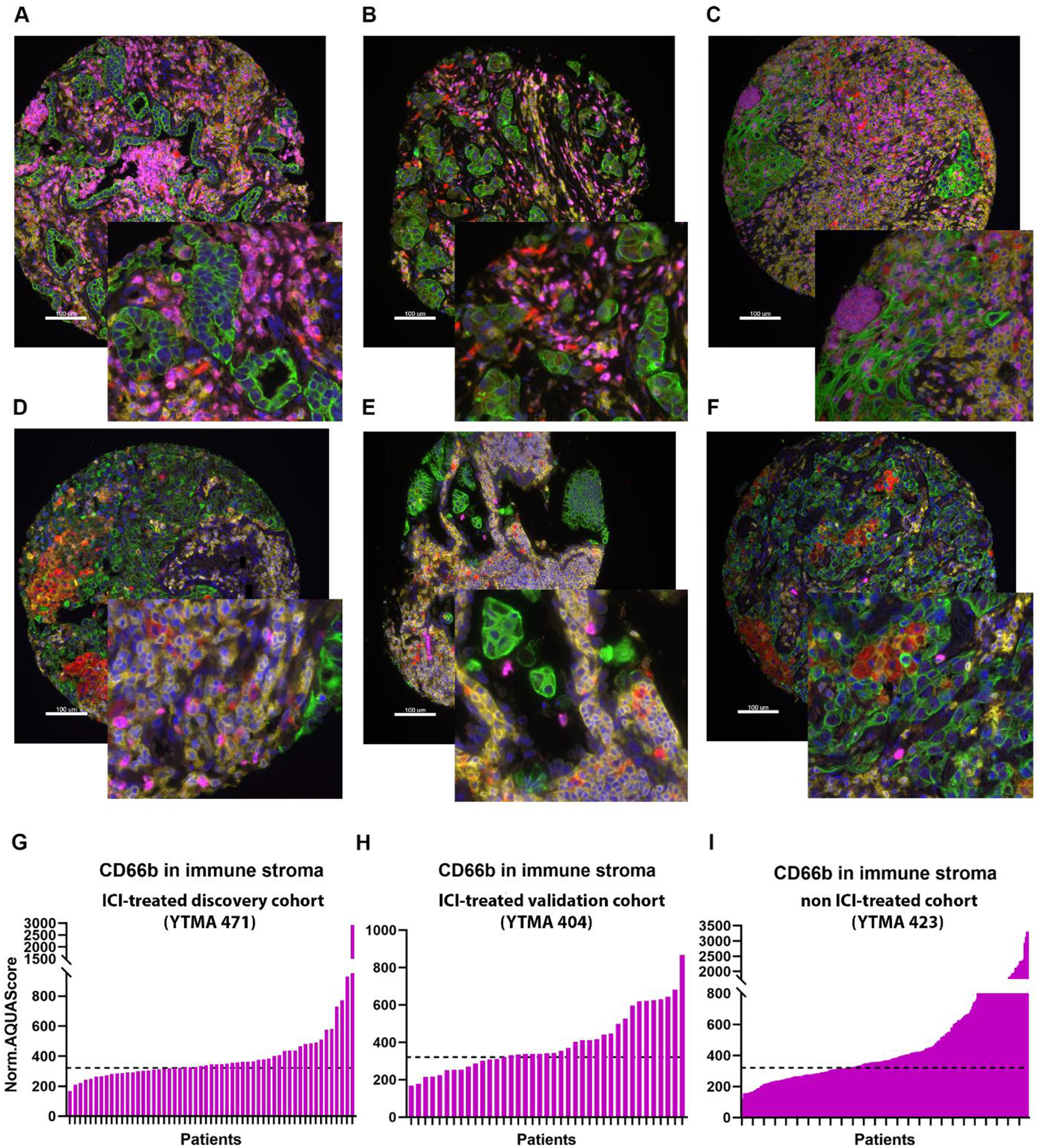
Representative images of high (≥ 321) (A, B, C) and low (<321) (D, E, F) CD66b Normalized AQUA Score immune stromal compartment (IC) in the discovery cohort YTMA471 (A,D), the validation cohort YTMA404 (B,E) and the non-ITX cohort YTMA423 (C,F) by QIF. Cytokeratin (green), CD45 (yellow), CD68 (red), CD66b (magenta), DAPI (blue). Dynamic range of CD66b expression in immune stromal compartment (IC) in the discovery cohort YTMA471 (G), validation cohort, YTMA404 (H) and in the non-ITX cohort, YTMA423 (I). The dashed line is on the level of Normalized (Norm) AQUA Score 321, calculated with X-tile, in the discovery cohort YTMA471 and applied on YTMA404 and YTMA423; Immune stroma compartment, IC; Yale tissue microarray, YTMA; quantitative immunofluorescence, QIF; automated quantitative analysis (AQUA™); digital spatial profiling, DSP; Immune Checkpoint Inhibitors, ICI
In the discovery cohort, an AQUA Score ≥ 321 could identify CD66b infiltrated tumors which were more likely to develop resistance to ICI (OS; HR 2.49, p=0.026, PFS; HR 1.50, p=0.2). When we used the same AQUA Score as a cutoff value in the two independent cohorts, high CD66b in immune stroma in ICI treated cohort (YTMA404), was associated with resistance to ICI therapy (OS; HR 2.05, p=0.046, PFS; HR 1.49, p=0.2) but was not prognostic for survival in the non-ICI treated cohort YTMA423 (OS; HR 1.67, p=0.06, DFS; HR 1.2, p=0.5)] as illustrated in Figure 3.
Figure 3.
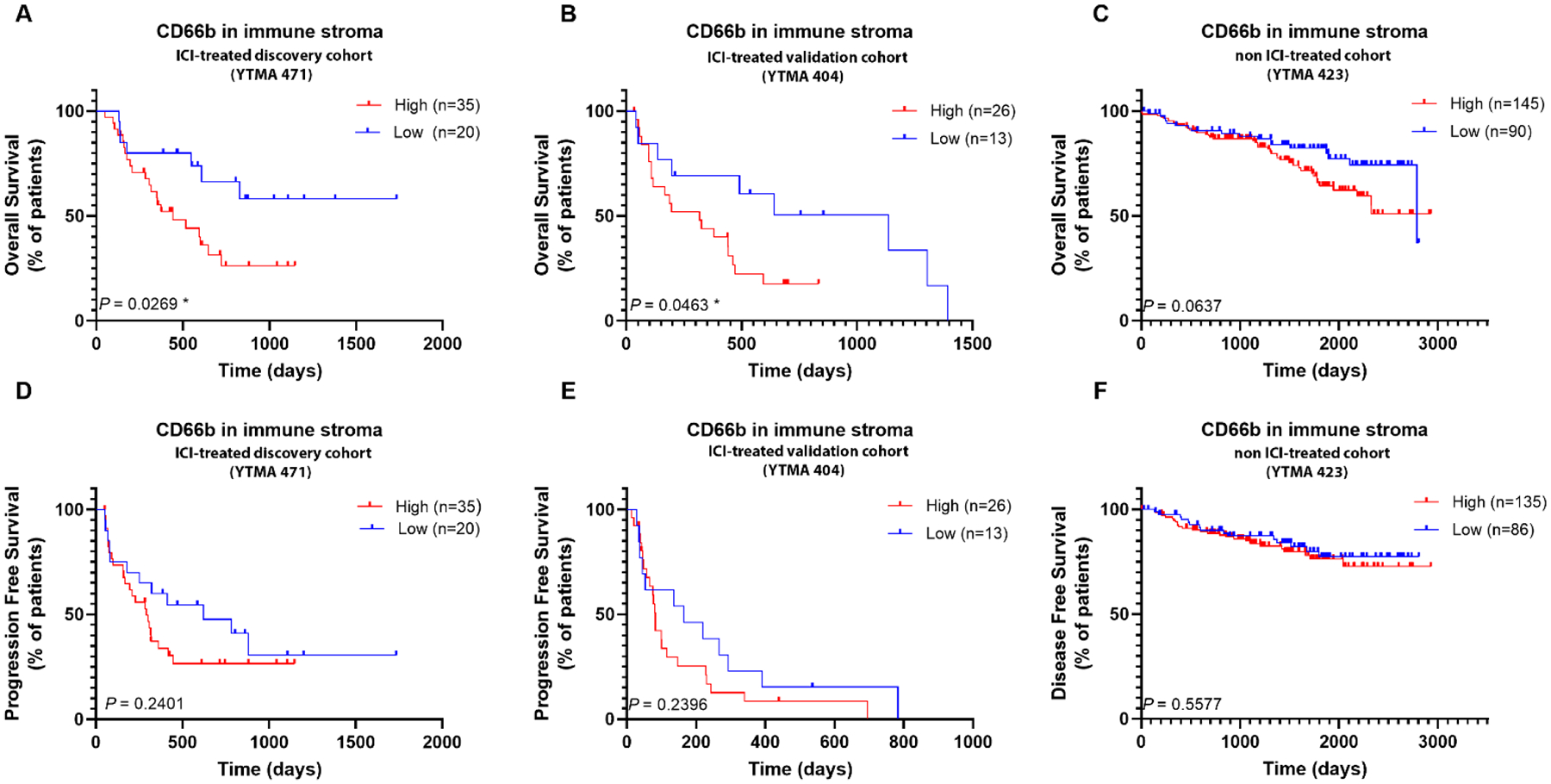
Shown are Kaplan-Meier estimates of overall (A, B, C) and progression free survival (D,E,F), according to each cohort. A, D. Discovery Cohort YTMA471, B, E. Validation Cohort YTMA404, C,F. Non-ICI treated Cohort YTMA423; non-Immune Checkpoint Inhibitors, non-ICI, Yale tissue microarray, YTMA
Discussion
In this study, we utilized DSP technology as a high-plex screening tool to identify biomarkers of resistance to ICI treatment in NSCLC. We found that CD66b expression was significantly associated with unfavorable clinical outcome. We confirmed this finding with an orthogonal method and after controlling for clinical prognostic factors. Moreover, using two independent NSCLC cohorts we validated the indicative role of CD66b expression. High CD66b cell tumor infiltration is consistent with previous findings that myeloid lineage cell population contributes to ICI treatment failures [22–25].
Several studies suggest that tumor associated neutrophils (TAN) enhance tumor cell invasion by forming neutrophil extracellular traps (NETs) in human tissue [26, 27]. NETs, consisting of extracellular DNA webs, infiltrate the TME and promote tumor growth and invasion of cancer cells through the activation of PI3K signaling pathways [28] [29] and possibly other mechanisms. Reported inverse correlation between NETs and CD8+ T cells could indicate that the NETs are inhibitory to either the chemotaxis or physical migration of CD8-mediated antitumor immunity. This observation could be therapeutically exploited to enhance immunotherapy efficiency [30]. Although using NETs as biomarker seems promising, quantification of NETs on the tissues is challenging and more studies supporting reliable measurement of NETs are needed [26, 31]. This work raises the possibility of using CD66b as a biomarker that may be more specifically defined than other methods for definition of NETs.
A recent study found that the lowest quartile of (CD8+ T cells in tumor)/(CD66b+ cells in stroma) ratio was associated with ICI treatment failure in 28 NSCLC patients [22]. The researchers quantified CD66b cells in tumor and stroma compartment separately and although high CD66b cell index was correlated with poor clinical outcome, it didn’t reach statistical significance, most likely due to small cohort size. In addition, the predictive value of pretreatment neutrophil-to-lymphocyte ratio (NLR) in serum has been demonstrated in recent studies but is inconsistent [32]. Neutrophils’ short lifespan in blood circulation (hours) vs in tissue (days) may account for the variance in the interpretation of the NLR ratio in serum [33, 34]. Here, we showed that CD66b expression in immune stroma is significantly associated with shorter survival in NSCLC patients treated with ICI, in two independent cohorts, regardless of the NLR status. Interestingly, high CD66b cell infiltration was also significantly associated with progressive disease, at 12 or 24 months of ICI therapy in our cohort [35]. Further work is required to better understand the relationship between CD66b expression and clinical benefit.
Resistance to ICI is clinically complex and biomarkers, able to identify an ICI “resistant” population, are an unmet need [3]. CD66b, a marker of neutrophil granulocytes, was associated with resistance to ICI in our study. One possible mechanism underlying CD66b role in ICI resistance is further suppression of natural killer (NK) cells-mediated immunity [36, 37]. TAN rich tumors have been also associated with lower TIL infiltration through a direct TAN-mediated L-arginine depletion in the T-cells leading to T-cell downregulation [23, 38]. Moreover, tumors that express Cathepsin C (CTSC), a protease essential in inflammation, can regulate neutrophil infiltration and promote metastasis and tumor progression [39]. Targeting CTSC or the myeloid checkpoints expressed on TANs [40, 41] may further enhance immune response by redirecting immune cells to eliminate tumor cells and ultimately overcome resistance to ICI treatment.
Assessment of CD66b alongside PD-L1 expression from pre-treatment tumor biopsies, could facilitate identification of NSCLC patients that can benefit from ICI therapy combined with a neutrophil antagonist. This is an interesting concept, since CD66b expression, can be easily assessed by immunohistochemistry (IHC) or QIF. Although CD66b infiltration has been studied in resectable NSCLC, its role remains controversial in this setting [42–44]. We didn’t find any association of TAN with survival in our non-ICI treated cohort. Thus, CD66b+ TAN is indicative but not prognostic for survival. We also do not state it is predictive since there it is no longer possible to randomize patients to placebo rather ICI, and thus it is no longer possible to calculate interaction, as would be required to claim the biomarker is predictive. Finally, the fact the neutrophils are easily identified on an H&E stain raised the question of their potential use based on routine specimens. We have not tried to assess this here, since this is a TMA based study. Furthermore, we are uncertain whether there is a 100% overlap between CD66b+ and morphologically defined neutrophils. This issues may be addressed in the future in studies validating CD66b in whole tissue sections.
This study has several limitations. Each of the cohorts we studied are single institution, retrospective collections of tumors from patients treated in routine practice. Differences depending on biopsy site (primary/locoregional vs. distant metastasis)[45] line of therapy (first line vs. later line), histology (squamous vs non-squamous)[46] or mutational status ([47, 48]) may influence biomarker performance, but we are underpowered to assess those variables. In the future, we hope to address the predictive value of CD66b in multi-institutional studies and clinical trials. Another weakness of this study is that the discovery and validation was done using tissue in the TMA format. While not used in the clinic, by assessing two nonadjacent tumor cores per case in the TMAs, we tried to address the limitation of using TMAs instead of whole-tissue sections. It can also be argued that if a biomarker is discovered and validated on a TMA, it is also likely to apply to much larger tissue samples seen in the standard tissue section. We have used the AQUA Sore of 321, identified by X-tile, as the cutpoint for optimal patient stratification. Although all the experiments were performed simultaneously, to account for batch/instrument variability, the determination of the cutpoint is experimentally specific. Future studies are required in larger cohorts to create a standardized cutpoint for CD66b quantification.
In conclusion, we identified and validated CD66b as a biomarker indicative of resistance to ICI treatment in NSCLC patients. Several other promising candidate biomarkers in spatial context were identified and are currently under investigation in our lab. Leveraging quantitative, high-plex, spatial approaches to profile tissue from immunotherapy treated patients, may help find directions for combination therapeutic strategies to overcome ICI resistance. This work suggests that neutrophil antagonism may be a provocative future target in selected advanced NSCLC patients and warrants further investigation.
Supplementary Material
Acknowledgements
Dr. Rimm, Herbst and Schalper are supported by the Yale SPORE in Lung Cancer and the Yale Cancer Center CCSG and Dr Rimm is supported by a sponsored research agreement from Konica Minolta. Myrto Moutafi is receiving a scholarship from the Hellenic Society of Medical Oncologists (HESMO).
Disclosure of Potential Conflict of Interest
David L. Rimm has served as an advisor for Astra Zeneca, Agendia, Amgen, BMS, Cell Signaling Technology, Cepheid, Danaher, Daiichi Sankyo, Genoptix/Novartis, GSK, Konica Minolta, Merck, NanoString, PAIGE.AI, Roche, and Sanofi. Amgen, Cepheid, NavigateBP, NextCure, and Konica Minolta fund research in David L. Rimm’s lab. Kurt A. Schalper received research funding from Genoptix/Navigate (Novartis), Tesaro/GSK, Moderna Therapeutics, Takeda, Surface Oncology, Pierre-Fabre Research Institute, Merck, Bristol-Myers Squibb, AstraZeneca, Ribon Theraputics and Eli Lilly; has received honoraria for consultant/advisory roles from Moderna Therapeutics, Shattuck Labs, Pierre-Fabre, AstraZeneca, EMD Serono, Ono Pharmaceuticals, Clinica Alemana de Santiago, Agenus and Takeda. Roy S. Herbst has served as Non-Executive Director for Immunocore and is a member of the Board of Directors (nonexecutive/independent) for Junshi Biosciences; is a consultant for AbbVie, Armo Biosciences, AstraZeneca, Bristol Myers Squibb, Bayer HealthCare Pharmaceuticals Inc., Bolt Biotherapeutics, Candel Therapeutics, Inc., Checkpoint Therapeutics, Cybrexa Therapeutics, DynamiCure Biotechnology, LLC, Eli Lilly and Company, eFFECTOR Therapeutics, Inc., EMD Serono, Foundation Medicine, Inc., Genentech/Roche, Genmab, Gilead, HiberCell, Inc., I-Mab Biopharma, Immune-Onc Therapeutics, Inc., Immunocore, Johnson and Johnson, Loxo Oncology, Merck and Company, Mirati Therapeutics, NextCure, Novartis, Ocean Biomedical, Inc., Oncocyte Corp, Oncternal Therapeutics, Pfizer, Refactor Health, Inc., Ribbon Therapeutics, Sanofi, STCube Pharmaceuticals, Inc, Takeda, WindMIL Therapeutics, Xencor, Inc; has received research support from AstraZeneca, Eli Lilly and Company, Genentech/Roche, and Merck and Company; is a committee chair in American Association for Cancer Research, International Association for the Study of Lung Cancer, Society for Immunotherapy of Cancer, Southwest Oncology Group
Jon Zugazagoitia has served as a consultant for Astra Zeneca, BMS, Roche, Pfizer, Novartis, and Guardant Health; reports speakers’ honoraria from BMS, Pfizer, Roche, Astra Zeneca, NanoString and Guardant Health; reports travel honoraria from BMS, Pfizer, Roche, Astra Zeneca, and NanoString; receives research support/funds from BMS, Astra Zeneca, and Roche. Prajan Divakar is an employee and stockholder of NanoString Inc. Sandra Martinez-Morilla is a current employee at Boehringer Ingelheim. Other authors have no potential conflicts of interest.
Abbreviations that are not standard in the field
- ICI
Immune Checkpoint Inhibitors
- non-ICI
non-Immune Checkpoint Inhibitors
- QIF
quantitative immunofluorescence
- AQUA™
automated quantitative analysis
- IC
Immune stroma compartment
- DSP
digital spatial profiling
- YTMA
Yale tissue microarray
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT author statement
Myrto Moutafi: Conceptualization, Investigation, Methodology, Validation, Formal analysis, Writing - Original Draft, Visualization Sandra Martinez-Morilla: Methodology, Data Curation, Writing - Review & Editing Prajan Divakar: Formal analysis, Data Curation, Writing - Review & Editing Ioannis Vathiotis, Niki Gavrielatou, Thazin Nwe Aung: Data Curation, Writing - Review & Editing Vesal Yaghoobi: Validation, Writing - Review & Editing Aileen I. Fernandez: Data Curation, Writing - Review & Editing Jon Zugazagoitia: Investigation, Writing - Review & Editing Roy Herbst: Conceptualization, Writing - Review & Editing, Funding acquisition Kurt A. Schalper: Conceptualization, Writing - Review & Editing, Funding acquisition David L. Rimm: Conceptualization, Investigation, Methodology, Validation, Writing - Original Draft, Writing - Review & Editing, Supervision, Funding acquisition
References
- 1.Topalian SL, et al. , Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol, 2019. 5(10): p. 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reck M, et al. , Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50. J Clin Oncol, 2021. 39(21): p. 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kluger HM, et al. , Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. Journal for ImmunoTherapy of Cancer, 2020. 8(1): p. e000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah S, et al. , Clinical and molecular features of innate and acquired resistance to anti-PD-1/PD-L1 therapy in lung cancer. Oncotarget, 2018. 9(4): p. 4375–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh RJ and Soo RA, Resistance to immune checkpoint inhibitors in non-small cell lung cancer: biomarkers and therapeutic strategies. Therapeutic advances in medical oncology, 2020. 12: p. 1758835920937902–1758835920937902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gettinger SN, et al. , Clinical Features and Management of Acquired Resistance to PD-1 Axis Inhibitors in 26 Patients With Advanced Non-Small Cell Lung Cancer. J Thorac Oncol, 2018. 13(6): p. 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang F, Wang S, and Zhou Q, The Resistance Mechanisms of Lung Cancer Immunotherapy. Frontiers in oncology, 2020. 10: p. 568059–568059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reck M, et al. , Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50%. Journal of Clinical Oncology, 2021. 39(21): p. 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu S, et al. , Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol, 2019. 5(8): p. 1195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu H, et al. , PD-L1 Expression in Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer, 2016. 11(7): p. 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samstein RM, et al. , Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet, 2019. 51(2): p. 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waldman AD, Fritz JM, and Lenardo MJ, A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nature Reviews Immunology, 2020. 20(11): p. 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merritt CR, et al. , Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat Biotechnol, 2020. 38(5): p. 586–599. [DOI] [PubMed] [Google Scholar]
- 14.Camp RL, Chung GG, and Rimm DL, Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med, 2002. 8(11): p. 1323–7. [DOI] [PubMed] [Google Scholar]
- 15.Yaghoobi V, et al. , Advances in quantitative immunohistochemistry and their contribution to breast cancer. Expert Review of Molecular Diagnostics, 2020. 20(5): p. 509–522. [DOI] [PubMed] [Google Scholar]
- 16.Camp RL, Dolled-Filhart M, and Rimm DL, X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res, 2004. 10(21): p. 7252–9. [DOI] [PubMed] [Google Scholar]
- 17.Kondrup M, et al. , S100B as a biomarker for brain metastases in patients with non-small cell lung cancer. Biomed Rep, 2020. 12(4): p. 204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu H, et al. , Brain Metastases Status and Immunotherapy Efficacy in Advanced Lung Cancer: A Systematic Review and Meta-Analysis. Frontiers in Immunology, 2021. 12(2756). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mezquita L, et al. , Association of the Lung Immune Prognostic Index With Immune Checkpoint Inhibitor Outcomes in Patients With Advanced Non-Small Cell Lung Cancer. JAMA Oncol, 2018. 4(3): p. 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brueckl WM, Ficker JH, and Zeitler G, Clinically relevant prognostic and predictive markers for immune-checkpoint-inhibitor (ICI) therapy in non-small cell lung cancer (NSCLC). BMC Cancer, 2020. 20(1): p. 1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobsen LC, et al. , Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood, 2006. 109(7): p. 3084–3087. [DOI] [PubMed] [Google Scholar]
- 22.Kargl J, et al. , Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI insight, 2019. 4(24): p. e130850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faget J, et al. , Neutrophils in the era of immune checkpoint blockade. Journal for ImmunoTherapy of Cancer, 2021. 9(7): p. e002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akbay EA, et al. , Interleukin-17A Promotes Lung Tumor Progression through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. Journal of Thoracic Oncology, 2017. 12(8): p. 1268–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schalper KA, et al. , Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat Med, 2020. 26(5): p. 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Andrea CE, et al. , Heterogenous presence of neutrophil extracellular traps in human solid tumours is partially dependent on IL-8. J Pathol, 2021. [DOI] [PubMed] [Google Scholar]
- 27.Rayes RF, et al. , Primary tumors induce neutrophil extracellular traps with targetable metastasis-promoting effects. JCI Insight, 2019. 4(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J, et al. , Role of neutrophil extracellular traps in regulation of lung cancer invasion and metastasis: Structural insights from a computational model. PLOS Computational Biology, 2021. 17(2): p. e1008257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houghton AM, et al. , Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med, 2010. 16(2): p. 219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masucci MT, et al. , The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Frontiers in Immunology, 2020. 11: p. 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elsherif L, et al. , Machine Learning to Quantitate Neutrophil NETosis. Sci Rep, 2019. 9(1): p. 16891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, et al. , Pretreatment Neutrophil-to-Lymphocyte Ratio (NLR) May Predict the Outcomes of Advanced Non-small-cell Lung Cancer (NSCLC) Patients Treated With Immune Checkpoint Inhibitors (ICIs). Frontiers in Oncology, 2020. 10(654). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tofts PS, et al. , Doubts concerning the recently reported human neutrophil lifespan of 5.4 days. Blood, 2011. 117(22): p. 6050–2; author reply 6053–4. [DOI] [PubMed] [Google Scholar]
- 34.Dixon G, et al. , A method for the in vivo measurement of zebrafish tissue neutrophil lifespan. ISRN hematology, 2012. 2012: p. 915868–915868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shukuya T, et al. , Relationship between Overall Survival and Response or Progression-Free Survival in Advanced Non–Small Cell Lung Cancer Patients Treated with Anti–PD-1/PD-L1 Antibodies. Journal of Thoracic Oncology, 2016. 11(11): p. 1927–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teijeira Á, et al. , CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity, 2020. 52(5): p. 856–871 e8. [DOI] [PubMed] [Google Scholar]
- 37.Zugazagoitia J, et al. , Biomarkers Associated with Beneficial PD-1 Checkpoint Blockade in Non-Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin Cancer Res, 2020. 26(16): p. 4360–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miret JJ, et al. , Suppression of Myeloid Cell Arginase Activity leads to Therapeutic Response in a NSCLC Mouse Model by Activating Anti-Tumor Immunity. Journal for ImmunoTherapy of Cancer, 2019. 7(1): p. 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao Y, et al. , Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell, 2021. 39(3): p. 423–437.e7. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura K and Smyth MJ, Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cellular & Molecular Immunology, 2020. 17(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, et al. , Signal Regulatory Protein (SIRPα), a Cellular Ligand for CD47, Regulates Neutrophil Transmigration *. Journal of Biological Chemistry, 2002. 277(12): p. 10028–10036. [DOI] [PubMed] [Google Scholar]
- 42.Ilie M, et al. , Predictive clinical outcome of the intratumoral CD66b-positive neutrophil-to-CD8-positive T-cell ratio in patients with resectable nonsmall cell lung cancer. Cancer, 2012. 118(6): p. 1726–37. [DOI] [PubMed] [Google Scholar]
- 43.Carus A, et al. , Tumor-associated neutrophils and macrophages in non-small cell lung cancer: no immediate impact on patient outcome. Lung Cancer, 2013. 81(1): p. 130–7. [DOI] [PubMed] [Google Scholar]
- 44.Jaillon S, et al. , Neutrophil diversity and plasticity in tumour progression and therapy. Nature Reviews Cancer, 2020. 20(9): p. 485–503. [DOI] [PubMed] [Google Scholar]
- 45.Moutafi MK, et al. , Comparison of programmed death-ligand 1 protein expression between primary and metastatic lesions in patients with lung cancer. J Immunother Cancer, 2021. 9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faruki H, et al. , Lung Adenocarcinoma and Squamous Cell Carcinoma Gene Expression Subtypes Demonstrate Significant Differences in Tumor Immune Landscape. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer, 2017. 12(6): p. 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skoulidis F, et al. , STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov, 2018. 8(7): p. 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ricciuti B, et al. , Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11-and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J Thorac Oncol, 2022. 17(3): p. 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.