

Abstract
Lysophosphatidic acid (LPA) is a bioactive lipid enriched in the tumor microenvironment of immunosuppressive malignancies such as ovarian cancer (OvCa). While LPA enhances the tumorigenic attributes of cancer cells, the immunomodulatory activity of this phospholipid messenger remains largely unexplored. Here, we report that LPA operates as a negative regulator of type-I interferon (IFN) responses in OvCa. Ablation of the LPA-generating enzyme autotaxin in OvCa cells reprogrammed the tumor immune microenvironment, extended host survival, and improved the effects of therapies that elicit protective responses driven by type-I IFN. Mechanistically, LPA sensing by dendritic cells triggered PGE2 biosynthesis that suppressed type-I IFN signaling via autocrine EP4 engagement. Moreover, we identified an LPA-controlled, immune-derived gene signature associated with poor responses to combined PARP inhibition and PD-1 blockade in OvCa patients. Controlling LPA production or sensing in tumors may therefore be useful to improve cancer immunotherapies that rely on robust induction of type-I IFN.
INTRODUCTION
Lysophosphatidic acid (LPA) is a bioactive lipid overproduced by aggressive tumor types such as ovarian, pancreatic, and breast cancer (1–3). This phospholipid messenger has been demonstrated to act as a potent mitogen that stimulates the proliferation, migration, invasiveness, and chemoresistance of malignant cells (4–8). LPA is highly enriched in the ascites of epithelial ovarian cancer (OvCa) patients compared with effusions from healthy individuals and women with benign ovarian neoplasms or non-malignant ascitic transudate (9–12). Importantly, overexpression of cell adhesion-related genes induced by this phospholipid correlates with poor prognosis in OvCa patients (13). Autotaxin (ATX), encoded by ENPP2 and frequently overexpressed by OvCa cells (14), is a secreted enzyme that generates LPA by cleaving the choline group from the abundant plasma phospholipid lysophosphatidylcholine (LPC) (15,16). LPA sensing occurs via six G protein-coupled receptors (LPA1–6) that are variably expressed in multiple cell types, and signaling through these receptors induces diverse intracellular responses that can alter cell metabolism, migration, and proliferation (17). Of note, while the direct protumoral role of ATX-LPA in cancer cells has been well established (18), whether this pathway facilitates malignant progression and resistance to therapy by suppressing anti-tumor immunity remains largely unexplored.
The vast majority of metastatic OvCa patients are refractory to standard treatments and current forms of immunotherapy (19–21), indicating that potent, yet unidentified, immunosuppressive mechanisms are actively engaged in these aggressive tumors. Type-I interferons (IFN), mainly IFNα and IFNβ, have been demonstrated to be required for the development of robust anti-cancer immune responses and for the optimal efficacy of immunotherapy in several malignancies (22,23). Nonetheless, the precise mechanisms by which OvCa disables type-I IFN responses to hinder anti-tumor immunity have not been determined. Here, we report that LPA operates as a negative regulator of type-I IFN production by dendritic cells (DCs). Genetic ablation of the LPA-generating enzyme ATX in malignant cells, enhanced DC function, reprogrammed the immune contexture of metastatic OvCa, prolonged overall host survival, and enhanced the immunotherapeutic effects of multiple anti-cancer interventions by boosting protective type-I IFN responses.
RESULTS
Loss of ATX in malignant cells delays OvCa progression and extends host survival
We sought to define the immunoregulatory role of LPA in the setting of metastatic OvCa. To this end, we used the orthotopic ID8-Defb29/Vegf-A OvCa model, which engenders a highly chemoresistant and immunosuppressive peritoneal carcinomatosis that recapitulates the advanced stages of human OvCa (24–26). ELISA and lipidomic analyses determined that the ascitic fluid generated by these mouse peritoneal tumors contained LPA levels and species similar to those present in the ascites of OvCa patients (Fig. 1, A and B). Consistent with prior reports (9–12), we found high concentrations of diverse LPA species in the ascites of OvCa patients compared with plasma samples from OvCa patients or from cancer-free women (Supplementary Fig. S1A). Similarly, the ascites generated by ID8-Defb29/Vegf-A ovarian tumors contained increased levels of multiple LPA species in comparison with peritoneal wash samples obtained from naïve mice (Supplementary Fig. S1B). Female mice bearing established ID8-Defb29/Vegf-A OvCa also showed higher circulating levels of ATX than their tumor-free counterparts (Supplementary Fig. S1C). Of note, the Enpp2 mRNA (encoding ATX) was primarily expressed by cancer cells at tumor locations, compared with diverse infiltrating leukocyte populations (Fig. 1C). Thus, we used CRISPR/Cas9 to abrogate Enpp2/ATX in malignant cells and further understand the function of this enzyme in the OvCa microenvironment. Enpp2 expression and ATX production were markedly reduced in Enpp2-targeted OvCa cells (Enpp2 sgRNA), compared with their isogenic counterparts harboring a control sgRNA that does not target the mouse genome (Supplementary Fig. S2A and S2B). Ablating Enpp2 in malignant cells did not compromise their in vitro proliferation (Supplementary Fig. S2C) or early orthotopic establishment in vivo (Supplementary Fig. S2D), yet it drastically reduced the concentrations of ATX and LPA in the peritoneal cavity of mice bearing metastatic OvCa for 5–6 weeks (Fig. 1, D and E). Decreased tumor-induced splenomegaly and a ~50% reduction in the amount of hemorrhagic ascites was concurrently observed in the same mice developing ATX/LPA-deficient OvCa (Supplementary Fig. S2E–S2H). Notably, ablation of Enpp2/ATX in OvCa cells diminished malignant ascites development and accumulation over time (Fig. 1F), and markedly extended overall host survival (Fig. 1G). Similar effects were observed when cancer cell-intrinsic Enpp2/ATX was ablated in the PPNM mouse model of high-grade serous tubo-ovarian carcinoma (HGSC) that carries the most common genetic abnormalities found in human HGSCs (27) (Supplementary Fig. S2I; Fig. 1, H and I). Hence, malignant cells are the main source of ATX-LPA at tumor locations and this axis is crucial for the aggressive behavior and progression of metastatic OvCa in two independent mouse models of disease.
Figure 1. Genetic loss of ATX in malignant cells compromises metastatic OvCa progression.
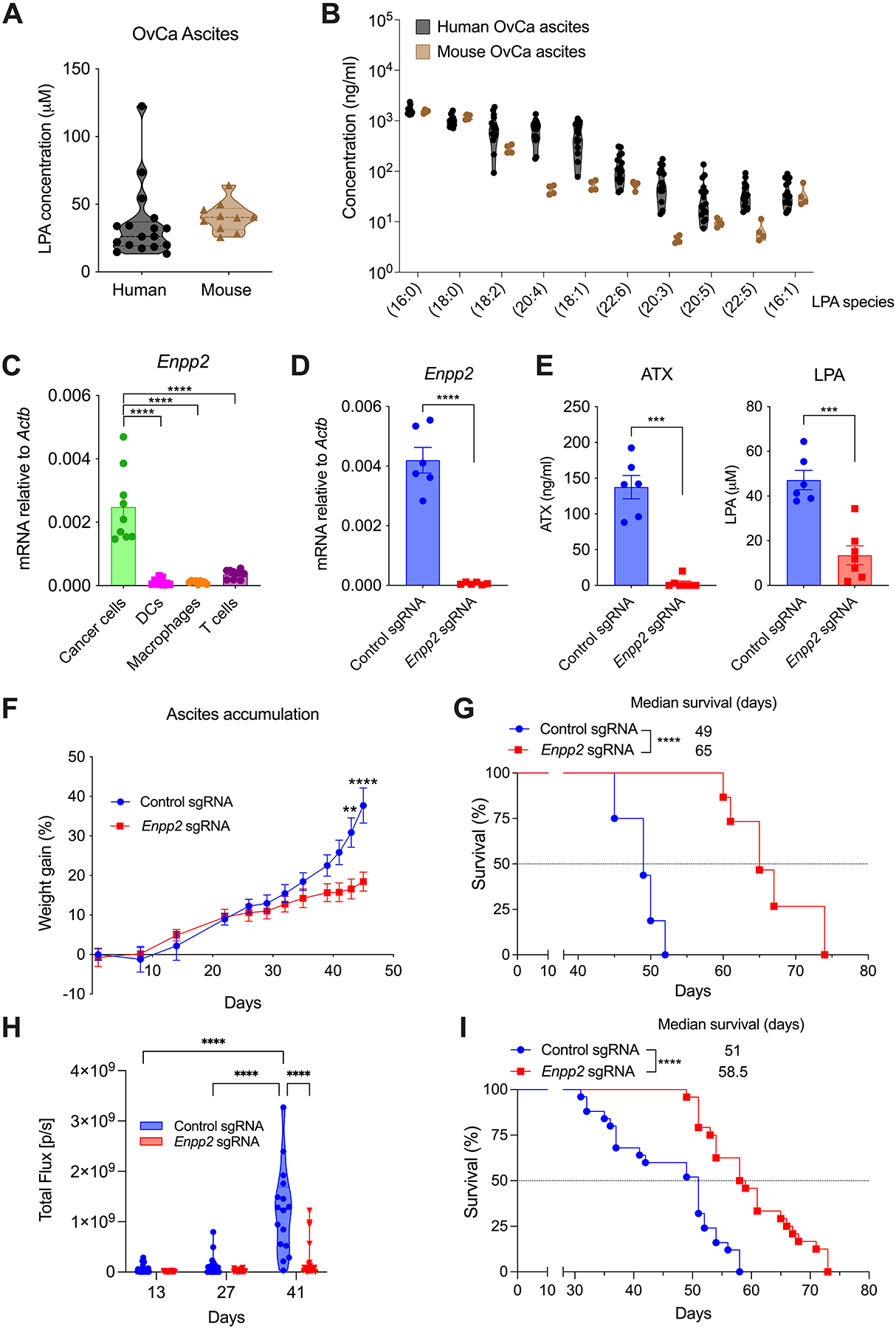
(A) LPA concentration was determined by ELISA in malignant ascites from human OvCa patients (n = 17) and mice bearing ID8-Defb29/Vegf-A OvCa for 5–6 weeks (n = 10). (B) Lipidomic analyses were performed to evaluate specific LPA species in malignant ascites from human OvCa patients (n = 15) and mice bearing advanced OvCa (n = 4). (C) Cancer cells, DCs, macrophages, and CD3+ T cells were sorted from peritoneal wash samples of mice bearing metastatic OvCa for 3–4 weeks (n = 9). Expression of the Enpp2 transcript was determined by RT-qPCR, and data were normalized to endogenous levels of Actb. (D) Expression of the Enpp2 transcript was determined in cancer cells sorted from the ascites of mice bearing control or Enpp2-null OvCa for 40 days (n = 6/group). (E) Autotaxin (ATX) and LPA concentrations were quantified in ascites from mice bearing control or Enpp2-null OvCa for 40 days (n = 6–7/group). (F) Ascites accumulation and (G) overall host survival in mice developing control or Enpp2-null ID8-Defb29/Vegf-A OvCa (n = 15–16 mice/group). (H) Quantification of peritoneal carcinomatosis in mice bearing luciferase-expressing control or Enpp2-null PPNM tumors for 13, 27 and 41 days (n = 24–25/group). (I) Overall survival rates for the same mice described in panel H (n = 24–25 mice/group). Data in C, D, E and F are shown as mean ± SEM. (C), One-way ANOVA (Tukey’s multiple comparisons test). (D, E), Two-tailed Student’s t-test. (F), Two-way ANOVA (Šídák’s multiple comparisons test). (H), Two-way ANOVA (Tukey’s multiple comparisons test). (G, I), Log-rank test for survival. **P<0.01, ***P<0.001, ****P<0.0001. Control sgRNA, scrambled single-guide RNA. Enpp2 sgRNA, autotaxin-targeting single-guide RNA.
Tumor-intrinsic ATX controls the OvCa immunoenvironment
We next evaluated whether loss of ATX in malignant cells, and the ensuing reduction of LPA production in the tumor milieu, altered the immune contexture of metastatic OvCa. We focused our analyses on the middle stages of disease progression (days 35–40), when mice developing control or ATX-deficient OvCa had similar numbers and proportions of SSChiCD45− cancer cells in the peritoneal cavity (Supplementary Fig. S3A; Fig. 2, A and B). At this stage, mice developing ATX-null OvCa demonstrated superior CD3+ T cell infiltration with higher proportions of CD8+ cytotoxic T cells at tumor locations (Fig. 2, A and C–E). Importantly, we found increased antigen-experienced (CD44+) CD4+ and CD8+ T cells expressing IFNγ and TNFα in the peritoneal cavity of mice developing ATX-deficient OvCa, compared with their ATX-sufficient counterparts (Fig. 2, F–J; Supplementary Fig. S3B–S3H). These changes were accompanied by reduced proportions of both CD11bhiF4/80+ macrophages and total CD11c+MHC-II+ DCs (Supplementary Fig. S3I–S3K), whereas no alterations were observed in the proportion of CD11b+Gr1+ myeloid cells (Supplementary Fig. S3I and S3L). Importantly, loss of ATX in OvCa cells further augmented NK cell infiltration into tumor sites (Supplementary Fig. S3I and S3M). These data indicate that tumor-derived ATX-LPA controls the immune composition of OvCa and suggest that this phospholipid messenger might operate as a global immunomodulatory mediator.
Figure 2. Ablation of ATX reprograms the immune microenvironment of metastatic OvCa.
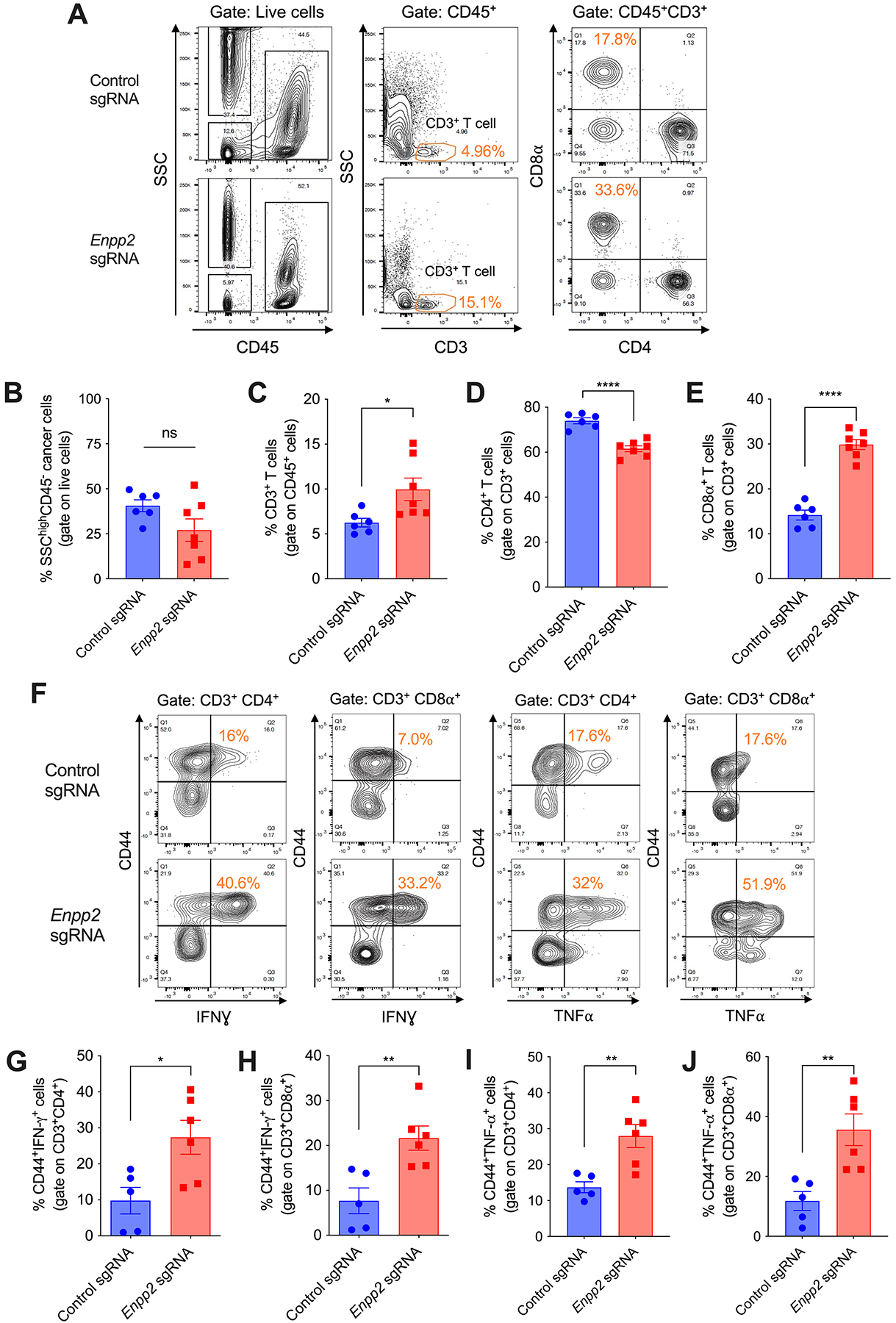
(A-E) Peritoneal wash samples were collected from mice developing control or Enpp2-null ID8-Defb29/Vegf-A OvCa for 35–40 days, and cells were analyzed by flow cytometry (n = 6–7/group). (F-J) The proportion of IFNγ− and TNFα−expressing cells within CD3+CD4+CD44+ and CD3+CD8α+CD44+ T cells was determined in the ascites of mice bearing control or Enpp2-null OvCa (n = 5–6/group). Data in B-E, G-J are shown as mean ± SEM. (C-E, G-J), Two-tailed Student’s t-test. *P<0.05, **P<0.01, ****P<0.0001.
LPA inhibits type-I IFN production by multiple DC subsets
We hypothesized that LPA could directly alter immune cell functions in the tumor microenvironment. We conducted single-cell RNA-seq analyses of total CD45+ leukocytes sorted from the ascites of mice developing metastatic OvCa to evaluate the biodistribution and expression levels of Lpar1-Lpar6 genes encoding LPA receptors 1–6 (LPA1–6). These experiments revealed preferential expression of Lpar1, Lpar2, and Lpar6 in multiple OvCa-associated immune cells (Supplementary Fig. S4A and S4B), with diverse intratumoral DC subsets, including type 1 conventional DCs (cDC1), type 2 conventional DCs (cDC2), and plasmacytoid DCs (pDCs) demonstrating the highest expression of Lpar6 (Supplementary Fig. S4B). RT-qPCR analyses using total CD45+CD11c+MHC-II+ tumor-associated DCs (tDCs) confirmed that these cells robustly expressed Lpar6, followed by Lpar2 and Lpar5 (Supplementary Fig. S4C). Of note, total splenic DCs (sDCs) and bone marrow-derived DCs (BMDCs) from naïve mice also showed high Lpar6 levels, accompanied by moderate expression of Lpar3 and Lpar5 (Supplementary Fig. S4D and S4E). We therefore surmised that LPA could alter DC functions.
Mouse BMDCs exposed to LPA concentrations similar to those found in the ascites of OvCa patients exhibited drastic changes in global gene expression, with 1,264 genes demonstrating significant downregulation and 1,155 genes showing significant upregulation (fold change > 1.5; P val < 0.05; FDR < 5%). These altered gene subsets were analyzed to identify potential upstream regulators mediating the observed transcriptional changes. Strikingly, LPA-exposed BMDCs showed marked downregulation of gene networks induced by type-I IFN signaling, with IFN-β, IFNAR1, IRF3, IRF7, and STAT1 emerging as the top predicted upstream regulators (Fig. 3, A and B). Conversely, immunosuppressive gene programs controlled by PTGER4/EP4 signaling, SOCS1, and STAT3 were significantly activated in BMDCs exposed to LPA (Fig. 3, A and C). Subsequent RT-qPCR analyses confirmed that LPA treatment downregulated multiple type-I IFN-stimulated genes (ISGs), such as Ddx58, Ifit1, Ifit2, and Isg15, in a time- and dose-dependent manner (Supplementary Fig. S5A and S5B). Hence, we hypothesized that LPA might suppress type-I IFN expression by diverse DC types. Mouse BMDCs, sDCs, and pDCs stimulated with various Toll-like Receptor (TLR) agonists demonstrated a dose-dependent decrease in IFNβ production upon LPA exposure (Fig. 3, D–G). Similar effects were observed when human monocyte-derived DCs (moDCs) and pDCs obtained from peripheral blood of cancer-free donors were stimulated with TLR agonists in the presence of LPA (Fig. 3H; Supplementary Fig. S5C). In LPS- or Poly (I:C)-stimulated BMDCs, LPA treatment inhibited the phosphorylation of TBK1 and IRF3 (Fig. 3, I and J), which are signaling events required for optimal type-I IFN expression (28). Hence, LPA acts as a negative regulator of type-I IFN production by diverse human and mouse DC subsets. Consistent with these in vitro findings, tDCs sorted from mice developing Enpp2-null OvCa with diminished LPA (Fig. 1E) demonstrated marked upregulation of Ifna and Ifnb1 transcripts (Fig. 3K), as well as overexpression of multiple type-I ISGs (Fig. 3L), compared with their counterparts isolated from mice bearing ATX-sufficient tumors. Similar effects were observed in cancer cells and neutrophils sorted from the peritoneal cavity of mice growing Enpp2-null OvCa (Supplementary Fig. S5D–S5G), indicating that tumor-derived LPA can further repress type-I IFN production and sensing by additional cell types present in the same microenvironment.
Figure 3. LPA blunts type-I IFN production by DCs.
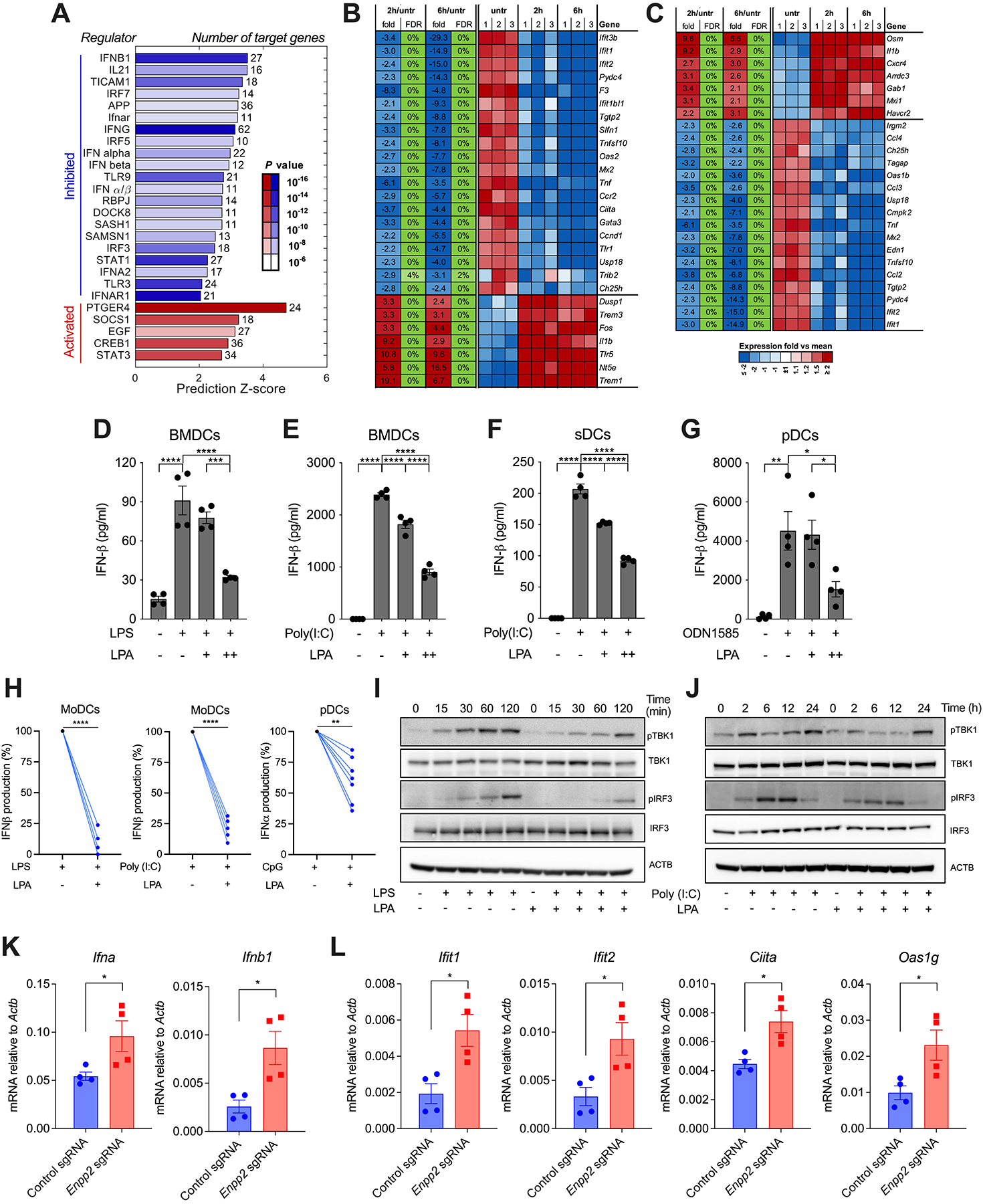
BMDCs were left untreated or stimulated with LPA (100 μM) for 2 or 6 hours, and global transcriptional profiles were analyzed by RNA-seq (n = 3 independent biological replicates per group). (A) Ingenuity Pathway Analysis (IPA) for predicted upstream regulators of differentially expressed genes. (B-C) Heatmap representation of type-I IFN (B) and PTGER4/EP4 (C) target genes. (D-G) BMDCs, sDCs, or pDCs were left untreated or pretreated for 2 hours with LPA at 10 μM (+) or 100 μM (++), and cells were then stimulated with LPS, Poly (I:C), or CpG ODN1585, as described in the methods. Production of IFN-β in culture supernatants was determined by ELISA (n = 4). (H) MoDCs or purified pDCs from cancer-free donors were treated with LPA (100 μM) for 2 hours and cells were then stimulated with LPS, Poly (I:C), or CpG-C274 as described in the methods (n = 5–7). Production of IFN-α or IFN-β in culture supernatants was determined by ELISA. (I-J) Representative immunoblot analysis for phospho-TBK1 (pTBK1), total TBK1, phospho-IRF3 (pIRF3), and total IRF3 in LPA-exposed BMDCs stimulated with either LPS (100 ng/ml) (I) or Poly (I:C) (10 μg/ml) (J) for the indicated times. (K-L) RT-qPCR analysis of type-I IFN transcripts (K) and type-I ISGs (L) in tDCs sorted from the ascites of mice bearing control or Enpp2-null ID8-Defb29/Vegf-A OvCa. Data were normalized to Actb in all cases (n = 4 independent mice/group). FDR, false discovery rate; fold/average, fold change of expression relative to average normalized reads of all samples; Expr, log2 value of normalized reads. Data in D-G and K-L are shown as mean ± SEM. (D-G), One-way ANOVA (Tukey’s multiple comparisons test). (H), Two-tailed paired Student’s t-test. (K, L), Two-tailed Student’s t-test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
LPA-induced PGE2 suppresses type-I IFN responses in DCs via PTGER4/EP4
Next, we sought to determine how LPA blunts type-I IFN expression in DCs. We examined the involvement of prostaglandin E2 (PGE2) in this process since our transcriptomic analyses indicated that gene programs induced upon engagement of the PGE2 receptor PTGER4 (also known as EP4) were enriched in DCs exposed to LPA (Fig. 3, A and C), and also because PGE2 overproduction in the tumor microenvironment can suppress anti-cancer immunity via multiple mechanisms (29,30). LPA treatment rapidly induced expression of Ptgs2/Cox-2 (encoding prostaglandin-endoperoxide synthase 2) and triggered PGE2 production by BMDCs in a time- and dose-dependent manner (Fig. 4A; Supplementary Fig. S6A). This process was mediated by activation of p38 MAPK (Fig. 4B), whereas other factors previously implicated in prostanoid generation such as PPARδ, PPARα, CREB, JNK, NFκβ, or AP-1 did not play role (Supplementary Fig. S6B) (31–34). To ascertain whether LPA-induced PGE2 inhibited autocrine type-I IFN responses, we treated BMDCs with the PTGER4/EP4 antagonist PGN 1531 (35) and then stimulated them with bacterial LPS in the presence or absence of LPA. Blocking EP4 engagement by PGE2 fully restored the expression of multiple type-I ISGs in LPS-treated BMDCs exposed to LPA (Fig. 4C). These data reveal that the LPA-PGE2-EP4 axis disables autocrine type-I IFN signaling in DCs.
Figure 4. LPA-induced PGE2 suppresses type-I IFN responses in DCs via EP4.
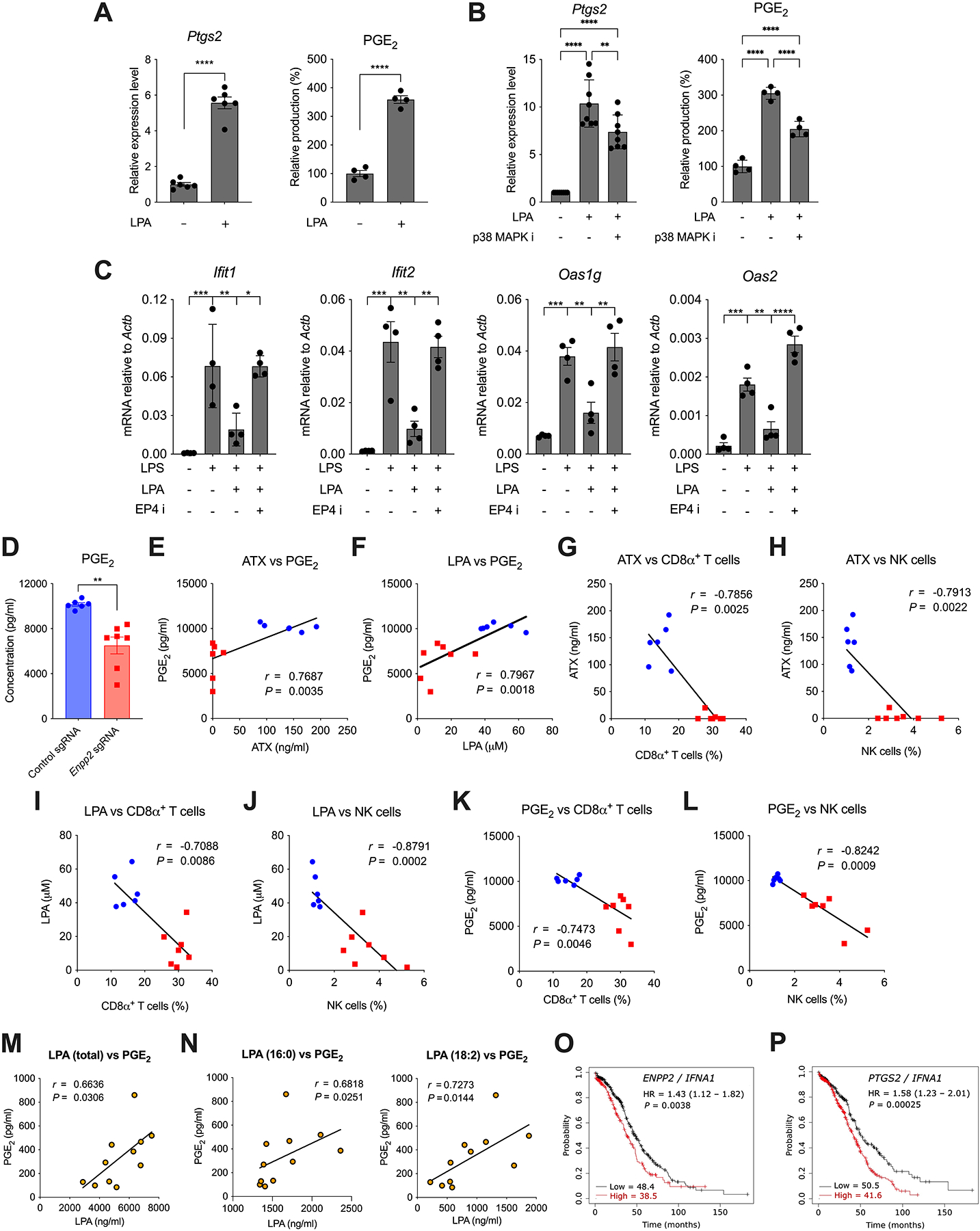
(A, left) BMDCs were left untreated or incubated with LPA (100 μM) for 2 hours and expression of Ptgs2 was determined by RT-qPCR (n = 6). (A, right) BMDCs were left untreated or incubated with LPA (100 μM) for 6 hours and PGE2 was determined in culture supernatant by ELISA (n = 4). (B, left) BMDCs were pretreated with the p38 MAPK kinase inhibitor SB203580 for 1 hour and then stimulated with LPA for 2 hours. Expression of Ptgs2 was determined by RT-qPCR (n = 8). (B, right) BMDCs were pretreated with the p38 MAPK kinase inhibitor SB203580 for 1 hour and then stimulated with LPA for 6 hours. PGE2 levels were determined in culture supernatant by ELISA (n = 4). (C) BMDCs were pretreated with the EP4 antagonist PGN 1531 (5 μM) for 1 hour and then stimulated with LPA and LPS for 4 hours. Expression of type-I ISGs were quantified by RT-qPCR. Data were normalized to Actb (n = 4). (D) PGE2 was quantified in ascites fluid from mice bearing control or Enpp2-null ID8-Defb29/Vegf-A OvCa for 40 days (n = 6–7/group). Correlation of PGE2 levels with ATX (E) or LPA (F) concentration in the same samples. (G-L) Proportion of infiltrating CD8α+ T cells or NK cells vs. levels of ATX (G-H), LPA (I-J), or PGE2 (K-L) in the peritoneal cavity of mice bearing control (blue dots) or Enpp2-null (red dots) OvCa (n = 13). (M-N) PGE2 concentration vs. levels of LPA (total), LPA (16:0), and LPA (18:2) in ascites from human OvCa patients (n = 11). (O-P) Overall survival curves for HGSOC patients in TCGA cohorts classified by the expression ratios of ENPP2/IFNA1 (O) or PTGS2/IFNA1 (P); HR, Hazard ratio. Numbers in the bottom of the graph denote the median overall survival (months) for each group. Data in A-D are shown as mean ± SEM. (A, D), Two-tailed Student’s t-test. (B, C), One-way ANOVA (Tukey’s multiple comparisons test). (E-N), Spearman’s rank correlation coefficient (r). (O and P), Log-rank test. (A-D), *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. (E-P), exact P-values are shown.
Mice bearing Enpp2-null OvCa demonstrated lower PGE2 levels in the ascites than hosts bearing control ATX-sufficient OvCa (Fig. 4D). In this setting, intrinsic ATX expression and LPA production correlated with the amount of PGE2 at tumor sites (Fig. 4, E and F), and high levels of ATX, LPA, or PGE2 were associated with reduced CD8+ T cell and NK cell infiltration (Fig. 4, G–L). Importantly, the concentration of PGE2 in cell-free ascites samples from patients with high-grade serous OvCa (HGSOC) positively associated with the level of total LPA in the same milieu (Fig. 4M). Among the individual LPA species analyzed, only 16:0 and 18:2 demonstrated a significant positive association with PGE2 expression (Fig. 4N; Supplementary Fig. S6C). Furthermore, analysis of OvCa patient cohorts from The Cancer Genome Atlas (TCGA) determined that high expression ratios of ENPP2 or PTGS2 to IFNA1 (encoding IFNα1) in human tumor specimens correlated with decreased overall survival (Fig. 4, O and P).
ATX deficiency enhances the anti-tumor effects of interventions that elicit type-I IFN
We hypothesized that the ATX-LPA arm could operate as a major mechanism of resistance to therapies that induce type-I IFN responses against cancer. To test this new concept, female mice bearing ATX-deficient or sufficient ID8-based OvCa were treated with the TLR3 agonist Poly (I:C), a prototypical type-I IFN inducer (36,37), and malignant progression and host survival were monitored over time. Consistent with our initial results (Fig 1, F and G), vehicle-treated mice developing Enpp2-null OvCa showed reduced ascites accumulation and prolonged survival, compared with their counterparts bearing ATX-sufficient OvCa (Fig. 5, A–C). Notably, while Poly (I:C) increased the median survival of mice bearing control tumors by only one week (~15%), treatment with this TLR3 agonist induced superior therapeutic effects in mice bearing Enpp2-null OvCa devoid of ATX, leading to an extended delay in ascites accumulation (Fig. 5, A and B) and a ~60% increase in overall survival in comparison with vehicle-treated mice bearing control OvCa (Fig. 5C). Validating these effects in an independent model of HGSC (27), mice bearing ATX-deficient PPNM tubo-ovarian tumors also demonstrated delayed metastatic progression and a drastic increase in overall survival upon Poly (I:C) administration, while their ATX-sufficient counterparts did not respond to this treatment (Fig. 5, D–F). Of note, antibody-mediated blockade of the Interferon α/β receptor 1 (IFNAR1) fully abrogated the therapeutic effects of Poly (I:C) in mice bearing Enpp2-null OvCa (Fig. 5G; Supplementary Fig. S7A and S7B), demonstrating that lack of tumor-derived LPA enhances protective type-I IFN signaling elicited by this treatment.
Figure 5. Therapeutic effects of Poly (I:C) administration in mice bearing ATX-null OvCa.
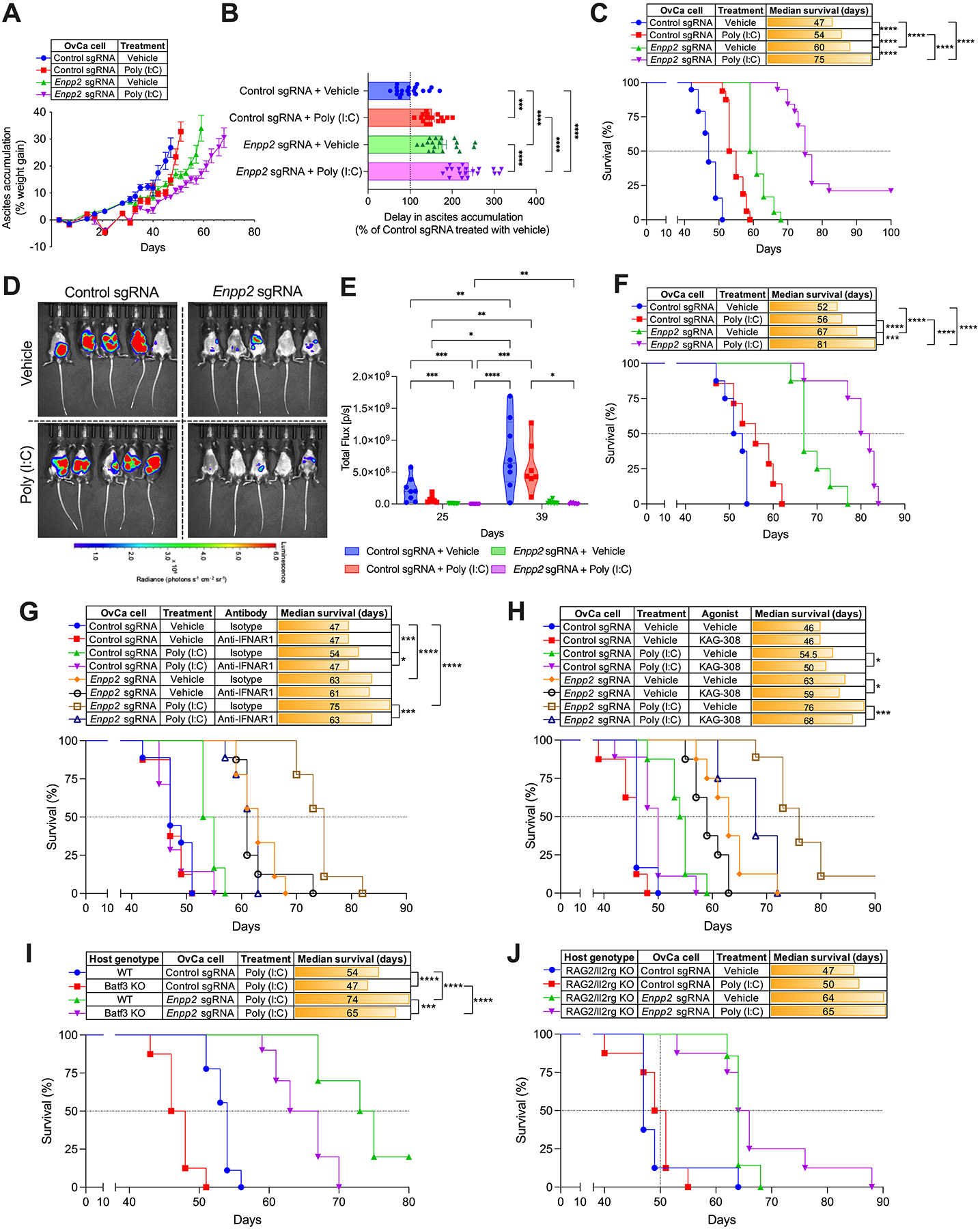
Wild type C57BL/6J mice were challenged i.p. with control or Enpp2-null ID8-Defb29/Vegf-A (A-C; n = 16–19 mice/group) or PPNM (D-F; n = 7–8 mice/group) cancer cells. After 10 days, mice were treated with vehicle control or Poly (I:C) as described in the methods. (A) Ascites accumulation denoted as percent weight gain over time. (B) Changes in ascites development were analyzed by calculating the area above the curve in each experimental group starting on day 21 and a cutoff of 35% of weight gain. Data are represented as percent change compared with the control sgRNA group treated with vehicle. (C) Overall survival curves for the same mice described in panels A and B. (D) Representative bioluminescent imaging at day 39 and quantification of peritoneal carcinomatosis (E) in mice bearing luciferase-expressing control or Enpp2-null PPNM tumors for 25 and 39 days with or without Poly (I:C) treatment. (F) Overall survival curves for the same mice described in panels D and E. (G) Experiments were repeated as in A-C, but 3 days after tumor implantation mice (n = 6–9/group) were treated with isotype control or anti-IFNAR1 blocking antibodies, as described in the methods. Host survival was monitored over time. (H) Experiments were repeated as in A-C, but 9 days after tumor implantation mice (n = 6–9/group) were orally treated with vehicle control or the EP4 agonist KAG-308 as described in the methods. Host survival was monitored over time. (I) Overall survival in mice of the indicated genotypes implanted with control or Enpp2-null ID8-Defb29/Vegf-A OvCa treated with Poly (I:C) (n = 8–10/group). (J) Overall survival in Rag2/Il2rg double knockout mice implanted with control or Enpp2-null ID8-Defb29/Vegf-A OvCa receiving the indicated treatments (n = 7–8/group). Data in A and B are shown as mean ± SEM. (B), One-Way ANOVA with Tukey’s multiple comparisons test. (E), Two-way ANOVA (Tukey’s multiple comparisons test). (C, F, G, H, I and J), Log-rank test for survival. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To functionally determine whether impaired PGE2 production (Fig. 4D) improved the therapeutic effects of Poly (I:C) in mice bearing ATX-deficient OvCa, we carried out rescue experiments using the selective EP4 agonist KAG-308 that is suitable for in vivo dosing via oral administration (38–40). Direct engagement of EP4 using KAG-308 bypassed the PGE2 production defects observed in tumors devoid of ATX/LPA and significantly reduced the survival benefit induced by Poly (I:C) treatment in this genetic context (Fig. 5H; Supplementary Fig. S7C and S7D). The anti-cancer effects elicited by TLR3 agonism in ATX-deficient tumors were mediated by DCs, as Batf3-deficient mice devoid of cDC1s (41) demonstrated impaired therapeutic responses to Poly (I:C) administration (Fig. 5I). Furthermore, immunodeficient (RAG2/IL2rg double knockout) mice lacking NK, T, and B cells implanted with ATX-null OvCa were totally refractory to this treatment (Fig. 5J). Hence, tumor-intrinsic ATX-LPA functions as a negative regulator of protective type-I IFN in OvCa and ablating this pathway could be used to evoke therapeutic anti-tumor immunity via TLR3 stimulation.
Treatment with Poly(ADP-Ribose) Polymerase (PARP) inhibitors has been shown to evoke beneficial type-I IFN responses in the tumor microenvironment through the DNA-sensing pathway cGAS-STING (42–44). Indeed, previous reports indicated that OvCa cells treated with PARP inhibitors accumulate cytosolic DNA that can subsequently induce immunostimulatory type-I IFN by neighboring antigen-presenting cells via STING activation (42). We reasoned that the ATX-LPA axis could also limit the therapeutic effects of PARP inhibition in OvCa. We prioritized talazoparib as it is the most potent PARP inhibitor currently in the clinic (45) and because its immunotherapeutic effects have been proposed to be independent of the BRCA status of the cancer cell (43). Notably, LPA exposure blocked the induction of Ifna, Ifnb1, and type-I ISGs in BMDCs cocultured with OvCa cells pretreated with talazoparib (Fig. 6, A and B). In vivo, oral talazoparib administration diminished ascites accumulation and extended survival by ~30% in mice bearing ATX-sufficient OvCa (Fig. 6, C–E). However, the therapeutic effects of talazoparib drastically improved in female mice bearing Enpp2-null OvCa devoid of ATX-LPA, eliciting a further delay in ascites accumulation and a ~63% increase in their median survival (Fig. 6, C–E). Next, to ascertain whether the enhanced therapeutic effects observed were mediated by the host STING pathway, we performed similar survival experiments in wild type (WT) or STING knockout mice. STING ablation did not affect the survival of talazoparib-treated mice bearing control ATX-sufficient OvCa, yet it significantly curtailed the therapeutic effects of this PARP inhibitor in host developing Enpp2-null ovarian tumors (Fig. 6F). Hence, ATX-LPA production by OvCa cells can limit STING-driven type-I IFN responses evoked by talazoparib administration.
Figure 6. Therapeutic effects of the PARP inhibitor talazoparib in mice bearing ATX-null OvCa.
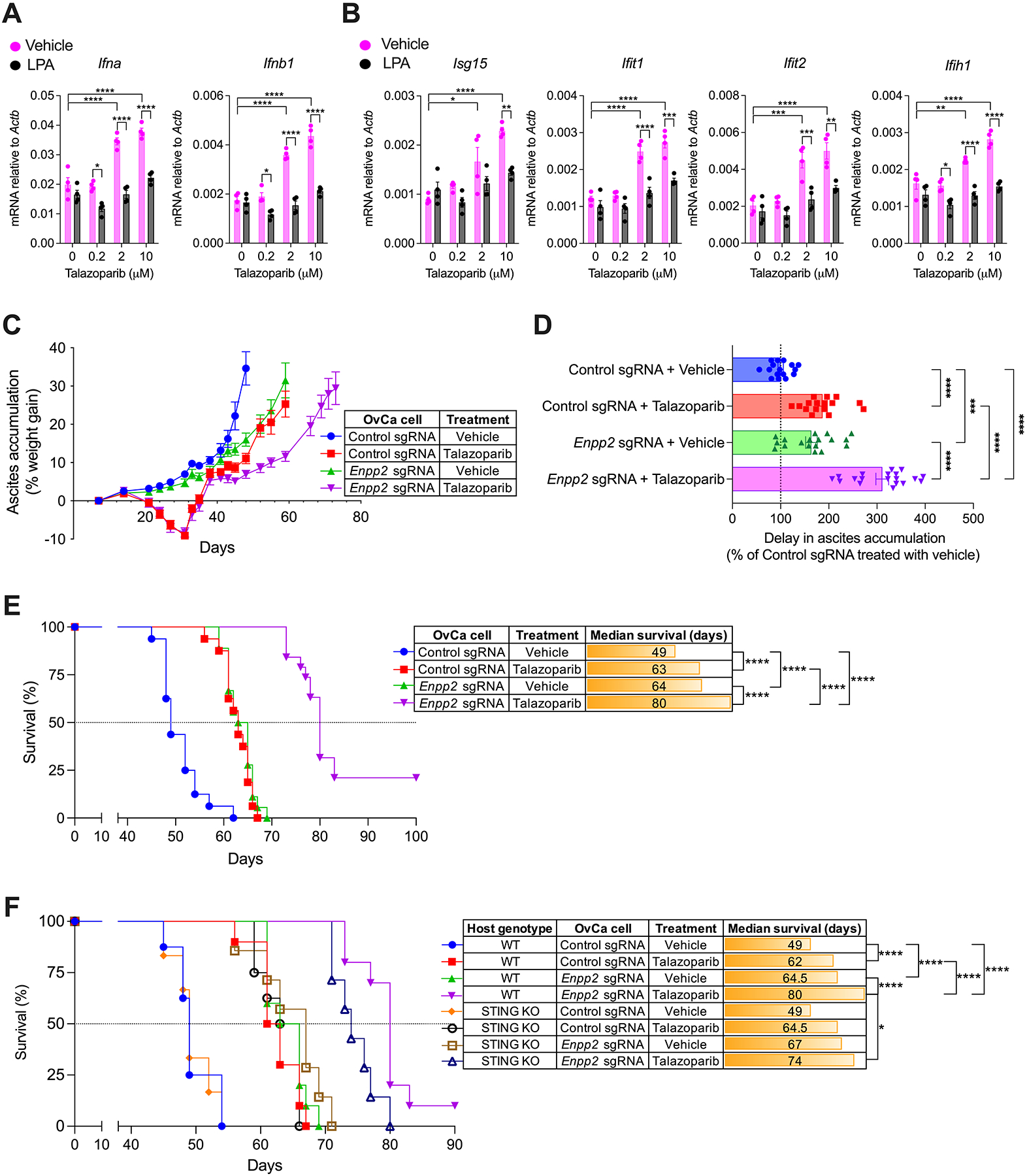
(A and B) RT-qPCR analysis of type-I IFN transcripts (A) and type-I ISGs (B) in BMDCs cocultured with talazoparib-treated OvCa cells in the presence or absence of LPA. Data were normalized to Actb in all cases (n = 4). (C-E) C57BL/6J mice (n = 16–19/group) were challenged via i.p. injection with 1.5 × 106 control or Enpp2-null ID8-Defb29/Vegf-A OvCa cells. After 7 days, mice were treated once daily with vehicle or talazoparib (0.33 mg/kg) by oral gavage for up to 28 days. (C) Ascites accumulation denoted as percent weight gain over time. (D) Changes in ascites development were analyzed by calculating the area above the curve in each experimental group starting on day 35 and a cutoff of 35% of weight gain. Data are represented as percent change compared with control sgRNA group treated with vehicle. (E) Overall survival curves for the same mice described in C and D. (F) Survival experiments were repeated as described in C-E but including STING-deficient (KO) hosts (n = 6–10/group). Data in A-D are shown as mean ± SEM. (A and B), Two-way ANOVA (Tukey’s multiple comparisons test). (D), One-Way ANOVA with Tukey’s multiple comparisons test. (E and F), Log-rank test analysis for survival. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
An LPA-controlled gene signature predicts resistance to combined PARP and PD-1 inhibition in the clinic
OvCa patients are resistant to multiple forms of cancer immunotherapy, especially to the blockade of typical immune checkpoints such as PD-1 (19–21). Nonetheless, recent reports indicate that the immunostimulatory effects of PARP inhibition, mainly mediated by type-I IFN, can enhance responses to anti-PD-1 therapy in OvCa patients and mouse models of this disease (42,46). We hypothesized that tumor-intrinsic ATX-LPA could represent a mechanism of resistance to this combination treatment. Mice bearing ATX-sufficient OvCa failed to respond to PD-1 blockade (Fig. 7A). While these mice demonstrated a modest increase in survival upon treatment with talazoparib, addition of anti-PD-1 antibodies did not improve the observed therapeutic benefit (Fig. 7A). Of note, PD-1 blockade enhanced the effects of talazoparib only in mice bearing ATX-deficient ovarian tumors devoid of LPA (Fig. 7A). Hence, we next examined whether the expression status of LPA-regulated genes that we identified (Fig. 3, A-C) are associated with response to combined immunotherapy with a PARP inhibitor plus checkpoint blockade in OvCa patients. To this end, we analyzed NanoString mRNA expression data from 44 tumor specimens within the TOPACIO/Keynote-162 trial (NCT02657889), in which HGSOC patients were treated with a combination of the PARP inhibitor Niraparib and the PD-1 blocker Pembrolizumab (46). Importantly, we identified seven LPA-controlled genes whose expression was significantly decreased in tumors from the non-responder vs. the responder patients in this trial (Fig. 7, B and C). These genes were not differentially expressed due to prior chemotherapy exposure (Fig. S8A), and further correlation analyses indicated that they form a co-regulated module selectively found in tumors from the non-responders (Fig. 7B; Supplementary Fig. S8B and S8C). Indeed, downregulation of the LPA-controlled gene module was observed in 77% of the non-responder group in unsupervised hierarchical clustering (Fig. 7D). We generated an LPA signature score denoting a summary of the gene module expression and found that, regardless of their homologous recombination deficiency (HRD) status, non-responders demonstrated significantly higher scores than responder patients in this trial (Fig. 7E). Hence, the status of this LPA-controlled gene signature might be useful for predicting resistance to combined PARP and PD-1 inhibition in HGSOC patients.
Figure 7. Expression of LPA-controlled genes and clinical response to combined PARP and PD-1 inhibition.
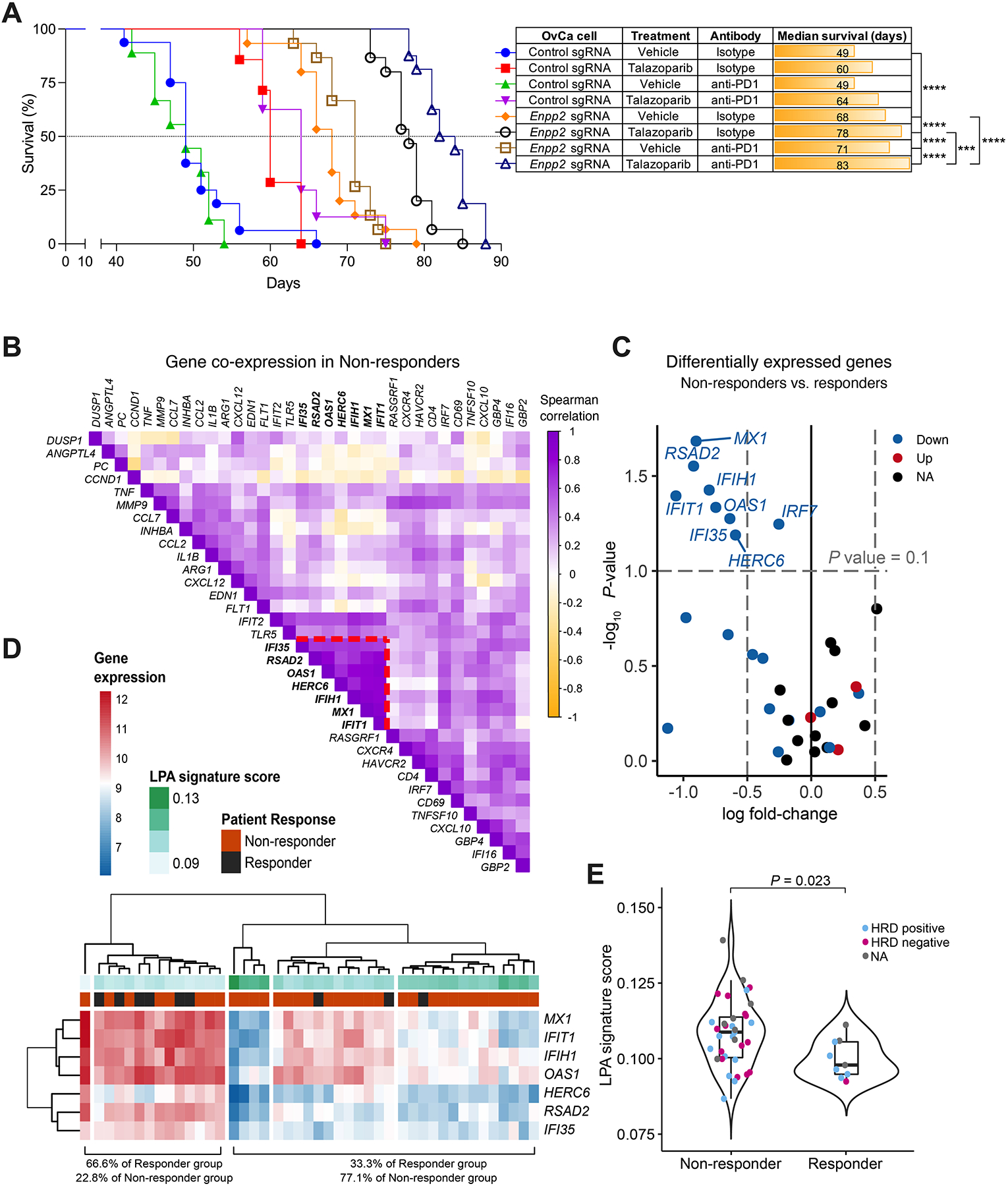
(A) C57BL/6J female mice (n = 7–16/group) were challenged i.p. with control or Enpp2-null ID8-Defb29/Vegf-A OvCa cells. After 7 days, mice were treated with talazoparib alone or in combination with isotype control or anti-PD-1 antibodies as described in the methods, and overall survival was monitored. (B) Correlation analysis within the non-responder group uncovering a distinct module of co-regulated genes (highlighted with red dashed lines). (C) Differential mRNA expression analysis between the groups showing that the 7 co-regulated genes are significantly decreased in non-responder patients. (D) Hierarchical clustering analysis of patient groups and LPA signature score, defined as the inverse of the median expression. (E) LPA signature score distribution as a function of the response category of each sample (Kruskal-Wallis test). (A), Log-rank test for survival. ***P<0.001, ****P<0.0001
DISCUSSION
Type-I IFNs are required for optimal cancer immunosurveillance and for the effective immunological control of malignant progression by diverse therapeutic modalities (23). However, the dominant mechanisms through which aggressive tumors inhibit protective type-I IFN remain incompletely understood. Our study uncovers that ATX-LPA is a major immunoregulatory axis that curtails protective type-I IFN responses in metastatic OvCa.
We found that malignant cells in the ovarian tumor microenvironment are the main source of ATX, and that LPA locally generated by this enzyme can readily control the immune contexture of metastatic OvCa, modulating DC function and preventing the accumulation and activation of T and NK cells at tumor locations. While recent reports suggest that LPA can directly block the activation and intratumoral infiltration of cytotoxic CD8+ T cells (47,48), the role of this bioactive lipid as a major regulator of DC function and type-I IFN responses in immunosuppressive tumors such as OvCa had not been established.
We determined that multiple DC types present at tumor locations and lymphoid tissue exhibited high expression of genes encoding various LPA receptors, suggesting that these myeloid cells could readily sense and respond to this bioactive lipid. Indeed, LPA-exposed DCs demonstrated marked transcriptomic alterations and produced copious amounts of PGE2 via p38 MAPK activation, which inhibited autocrine type-I IFN responses through the prostanoid receptor EP4. Defining the main LPA receptor(s) and the precise downstream signaling events mediating the observed modulation of type-I IFN responses in DCs exposed to this phospholipid will be of significant interest. Whether tumor-derived LPA can simultaneously alter the function of other OvCa-infiltrating leukocytes to evade immune control and promote malignant progression also warrants further investigation.
Loss of ATX in OvCa cells decreased LPA and PGE2 production at tumor sites, enhanced type-I IFN responses in tumor-associated DCs, and augmented the anti-OvCa effects of treatment with canonical inducers of type-I IFNs such as Poly (I:C) or the PARP inhibitor talazoparib. The improved survival benefit conferred by these treatments in mice bearing ATX-deficient OvCa was reduced upon IFNAR1 blockade or STING ablation, respectively. Nonetheless, untreated mice bearing ATX-deficient OvCa still demonstrated a significant increase in overall survival that was not mediated by type-I IFN signaling or STING activation. While these results are consistent with the reported direct protumorigenic role of LPA in cancer cells, they also suggest that this bioactive lipid may coordinate additional immunoregulatory mechanisms beyond type-I IFN suppression, which deserve further exploration.
Increasing evidence demonstrates the potent immunosuppressive role of tumor-derived PGE2 in multiple cancers (29,49–51). This prostanoid has also been shown to inhibit type-I IFN responses in macrophages stimulated with bacterial LPS, and in the setting of viral infections (35,52). Yet, a role for tumor-derived LPA as a direct inducer of PGE2 that inhibits global type-I IFN responses and limits the efficacy of OvCa immunotherapies had not been established.
A recent Phase II TOPACIO trial investigated the combination of PARP inhibition plus PD-1 blockade in relapsed, platinum resistant OvCa patients (53). The correlative analyses indicated that HRD status was associated with prolonged progression-free survival but not to the proportion of patients with response (46), indicating that additional factors contribute to the response/resistance to this combination treatment. Indeed, our study identified a DC-derived, LPA-controlled gene module whose decreased expression in tumor specimens from HGSOC patients was associated with resistance to combined PARP and PD-1 inhibition in this trial (46). Whether non-responder patients demonstrate superior ATX-LPA production in the tumor, or increased expression of LPA receptors on infiltrating immune cells, deserves further investigation as it could lead to novel actionable biomarkers predicting response or resistance to this combination therapy. Whether other mechanisms of immunoregulation predominant in ovarian tumors, such as persistent endoplasmic reticulum stress responses (54), cooperate with detrimental LPA signaling to exacerbate tumor growth, immune escape, and resistance to combination immunotherapy also deserves additional research.
Various ATX small-molecule inhibitors are currently being tested in human clinical trials, particularly in the setting of pulmonary fibrosis (55), raising the possibility that these compounds might be repurposed to improve the anti-cancer effects of interventions that rely on potent type-I IFN responses. Beyond ovarian tumors, ATX-LPA is commonly overproduced in pancreatic, breast, and prostate cancers, implying that targeting this axis may also be useful to unleash type-I IFN and enhance the efficacy of immune-activating treatments in these aggressive malignancies.
METHODS
Patient-derived specimens
Plasma samples from cancer-free women were obtained from the New York Blood Center. Plasma and ascites samples from patients with stage III-IV HGSOC were obtained under written informed consent, following appropriate institutional biospecimen collection and use protocols established at Weill Cornell Medicine and Memorial Sloan-Kettering Cancer Center. All human specimens were deidentified for subsequent experimental analyses. The ascites was centrifuged at 4°C for 10 min at 1,300 rpm. Supernatants were then collected, depleted of cells by passing through 0.22-μm filters, and stored frozen at −80°C in small aliquots until use.
Mice and experimental OvCa models
C57BL/6J, B6.129S(C)-Batf3tm1Kmm/J (BATF3 KO), and Tmem173gt/J (STING KO) female mice were obtained from The Jackson Laboratory. C57BL/6NTac.Cg-Rag2tm1Fwa Il2rgtm1Wjl (Rag2/Il2rg double knockout) female mice were obtained from Taconic Biosciences. Female mice were housed in pathogen-free microisolator cages at the animal facilities of Weill Cornell Medicine and used at 8–12 weeks of age. Mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Weill Cornell Medicine under protocol number 2011–0098. Parental ID8 cells expressing luciferase (ID8-Luc) and the aggressive ID8-Defb29/Vegf-A derivate were cultured and used as previously described (24,56). Both cell lines were obtained under MTA from Drs. K. Roby and J. Conejo-Garcia, respectively. The PPNM cell line (Trp53−/−R172HPten−/−Nf1−/−MycOE) was generously provided by Dr. R. Weinberg under MTA (27). Briefly, 1.5 × 106 ID8-based OvCa cells suspended in 200 μl of sterile PBS were intraperitoneally (i.p.) injected into WT or transgenic mice. Alternatively, 1.5 × 106 PPNM cells were suspended in PBS containing Matrigel (Corning Matrigel matrix, Cat# 47743–716) at a 1:1 ratio, and 200 μl of the mix were administered i.p. into WT mice, as reported (27). Metastatic progression, ascites accumulation, and host survival were monitored over time. Tumor burden in the peritoneal cavity was assessed by live bioluminescent imaging. Briefly, mice were given a single i.p. injection of VivoGlo luciferin (2 mg/mouse, Promega) and then imaged on a Xenogen IVIS Spectrum In Vivo imaging system at the Weill Cornell Research Animal Resource Center.
Generation of ATX-deficient OvCa cell lines using CRISPR/Cas9
The 20-nucleotide crRNA targeting murine Enpp2 (Mus musculus chromosome 15, 15; 15 D1, NC_000081.7) was directed at the genomic sequence TCTCCATGGACCAACACATCTGG, (the 3 additional nucleotides highlighted in bold represent the protospacer adjacent motif, or PAM). This target sequence corresponds to exon 3 of the murine Enpp2 transcript and was manually chosen by identifying a 20-nucleotide fragment immediately upstream of the highlighted PAM (57). The on- and off-target effects of the manually selected CRISPR sequence were then analyzed using the Broad Institute’s Genetic Perturbation Platform (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). To validate the genomic editing capacity of the crRNA, RT-qPCR was performed on total RNA isolated from cells transfected with sgRNA-Cas9 complexes containing the Enpp2 crRNA described above. The primer for Enpp2 quantification via RT-qPCR anneals to the same nucleotides as the Enpp2 crRNA target site. The primers for evaluating deletion efficacy are listed in Table S1. The scrambled crRNA contains a 20-nucleotide sequence that is computationally designed to be non-targeting within the human or murine genomes (http://sfvideo.blob.core.windows.net/sitefinity/docs/default-source/user-guide-manual/alt-r-crispr-cas9-user-guide-ribonucleoprotein-transfections-recommended.pdf?sfvrsn=1c43407_12). The sequence for this non-targeting sgRNA control was CGUUAAUCGCGUAUAAUACG. Since ATX expression in cancer cells is commonly repressed in vitro via epigenetic mechanisms (58), ID8-Defb29/Vegf-A or PPNM cancer cells were treated for 4 hours with the HDAC inhibitor TSA at 250 nM (Sigma-Aldrich Cat# T1952–200UL) and then transfected with ATTO-550-labeled sgRNA-Cas9 complexes using the Neon transfection system, following the manufacturer’s protocol (Thermo Fisher Scientific; https://assets.thermofisher.com/TFS-Assets/LSG/manuals/neon_device_man.pdf). All materials for sgRNA-Cas9 complex generation were purchased from Integrated DNA Technologies (IDT), and prepared as instructed (http://sfvideo.blob.core.windows.net/sitefinity/docs/default-source/user-guide-manual/alt-r-crispr-cas9-user-guide-ribonucleoprotein-transfections-recommended.pdf?sfvrsn=1c43407_12). Twenty-four hours post-transfection, ATTO-550+ cells were individually sorted, expanded as clones, and screened for ATX ablation for subsequent experiments.
Primary cell isolation and analysis
Cancer cells (CD45−SSChigh), total DCs (CD45+CD11c+MHC-II+CD11b+), macrophages (CD45+CD11b+F4/80high), T cells (CD45+CD3+), and neutrophils (CD45+CD11b+F4/80−Ly6G+) were sorted from single-cell suspensions of malignant ascites or peritoneal lavage samples obtained from mice bearing metastatic OvCa. Murine BMDCs were generated from bone marrow precursor cells isolated from the tibias and femurs of mice. Bone marrow cells were flushed and plated on bacteriological plates at 3 × 106/plate in 10 ml of complete RPMI media (RPMI + L-glutamine + 10% FBS + HEPES + Sodium Pyruvate + non-essential amino acids + β mercaptoethanol + Pen/strep) containing 20 ng/ml of recombinant granulocyte macrophage colony-stimulating factor (GM-CSF, Peprotech Cat# 315–03). Three days later, an equal volume of the media described above was added to the culture, and non-adherent cells were harvested 3–4 days thereafter (day 6–7). BMDCs were further enriched with UltraPure CD11c MicroBeads (Miltenyi Biotech Cat# 130-125-835) and used directly for in vitro profiling and functional assays. Murine sDC and pDCs were magnetically purified from spleens (Miltenyi Biotech Cat# 130-125-835, 130-107-093) and used directly for subsequent in vitro functional assays. Human monocyte-derived DCs were generated by isolating CD14+ cells (Miltenyi Biotech Cat# 130-050-201) from blood/buffy coats using Ficoll-gradient centrifugation and plated in complete RPMI media containing human recombinant GM-CSF at 1000 IU/ml and IL-4 at 500 IU/ml (both from Peprotech Cat# 300–03 for GM-CSF, 200–04 for IL-4) for 7 days. Cells were then harvested and used for in vitro assays (59). Human pDCs from healthy donors were purified as previously described (60) using negative selection (Miltenyi Biotech Cat# 130-090-509).
RNA extraction and quantitative RT-PCR analysis
Total RNA was isolated using the RNeasy Mini kit or QIAzol lysis reagent (Qiagen) according to the manufacturer’s instructions. 0.1–1 μg of RNA was reverse-transcribed to generate cDNA using the qScript cDNA synthesis kit (Quantabio). Quantitative RT-PCR was performed using PerfeCTa SYBR green fastmix (Quantabio) on a QuantStudio 6 Flex real-time PCR system (Applied Biosystems). Normalized gene expression was calculated by the comparative threshold cycle method using ACTB for human or Actb for mouse as endogenous controls.
Flow cytometry
Analyses were conducted using fluorochrome-conjugated antibodies purchased from BioLegend, unless stated otherwise. Cells were washed with PBS, Fc-gamma receptor-blocked using TruStain fcX™ (anti-mouse CD16/32, clone 93, Cat# 101319) and then stained for surface markers at 4°C in the dark for 30 minutes using the following antibodies: anti-CD45 (clone 30-F11, Cat# 103115), anti-CD3 (clone 17A2, Cat# 100216), anti-CD4 (clone RM4–5 Cat# 100547), anti-CD8α (clone 53–6.7 Cat# 100725), anti-CD44 (clone IM7 Cat# 103032), anti-CD11c (clone N418 Cat# 117309), anti-I-A/I-E (Tonbo biosciences, clone M5/114.15.2 Cat# 35–5321), anti-CD11b (clone M1/70 Cat# 101228), anti-F4/80 (clone BM8 Cat# 123110), anti-Gr1 (clone RB6–8C5 Cat# 108416), anti-Ly6G (Tonbo biosciences, clone 1A8 Cat# 25–1276) and anti-NK1.1 (clone PK136 Cat# 108748). Cells were then washed and stained with DAPI for live/dead discrimination. For intracellular cytokine staining, cells from malignant peritoneal wash samples were stimulated for 5 hours in complete RPMI containing cell activation cocktail with brefeldin A (Biolegend Cat# 423304). Cells were collected and stained for surface markers and intracellular cytokines using anti-IFN-γ (clone XMG1.2 Cat# 505826) and anti-TNF-α (clone MP6-XT22 Cat# 506308), following the Foxp3/Transcription Factor Staining buffer set (Thermo Fisher Scientific Cat# 00-5523-00). Flow cytometry was performed on LSRII or Fortessa-X20 instruments (BD Biosciences). Cell populations were sorted from peritoneal lavage or ascites samples from OvCa-bearing mice using a FACSAria sorter (BD Biosciences), and flow cytometry data were analyzed using FlowJo (TreeStar).
Immunoblot analysis
BMDCs were washed twice in 1X cold PBS and cell pellets were lysed using RIPA lysis and extraction buffer (Thermo Fisher Scientific) supplemented with a protease and phosphatase inhibitor tablet (Roche). Homogenates were centrifuged at 14,000 rpm for 30 min at 4°C, and the supernatants were collected. Protein concentrations were determined using a BCA protein assay kit (Thermo Fisher Scientific). Equivalent amounts of protein were separated via SDS-PAGE and transferred onto PVDF membranes following standard protocols. The following antibodies were used: anti-beta actin (Cell Signaling Technologies Cat# 4967), anti-pTBK1 (Cell Signaling Technologies Cat# 5438), anti-TBK1 (Cell Signaling Technologies Cat# 3504), anti-pIRE3 (Cell Signaling Technologies Cat# 29047), anti-IRE3 (Cell Signaling Technologies Cat# 4302), and goat anti-rabbit secondary antibodies conjugated with HRP (Thermo Fisher Scientific Cat# G-21234). SuperSignal West Pico and Femto chemiluminescent substrates (Thermo Fisher Scientific) were used to image blots in a FluorChemE instrument (ProteinSimple).
ELISA
Total LPA, ATX and PGE2 concentrations in malignant ascites from human OvCa patients and mice bearing advanced OvCa were measured by ELISA (Echelon Biosciences, Cat# K-2800S for LPA; Echelon Biosciences, Cat# K-5600 for ATX; Enzo Lifesciences, Cat# ADI-900–001 for PGE2). BMDCs were stimulated with the indicated concentration of LPA (Cayman Chemical Company Cat# 65528-98-5) for various time points, and PGE2 levels in the supernatant were measured using the PGE2 ELISA kit described above. OvCa cells were stimulated with TSA (Sigma-Aldrich Cat# T1952) for 24 hours and secreted ATX was quantified in the supernatant using the ATX ELISA kit. Murine BMDCs, sDCs, and pDCs were left untreated or pretreated for 2 hours with LPA (10 or 100 μM), and cells were then stimulated for 4 hours with LPS (100 ng/ml) (Invivogen Cat# tlrl-eklps), 24 hours with Poly (I:C) (50 μg/ml for BMDCs or 10 μg/ml for sDCs) (Invivogen Cat# vac-plc), or 24 hours with CpG ODN1585 (1 μg/ml) (Invivogen Cat# tlrl-1585). Production of IFN-β in culture supernatants was determined by ELISA (PBL Assay Science, Cat# 42400–2). Human moDCs or purified pDCs from healthy donors were pretreated with LPA (100 μM) for 2 hours and cells were then stimulated for 4 hours with LPS (100 ng/ml), 24 hours with Poly (I:C) (10 μg/ml), or 24 hours with CpG-C274 (0.075 μM). Production of IFN-β was quantified in culture supernatants by ELISA (PBL Assay Science, Cat# 41435–1). IFN-α production by human pDCs was determined by ELISA (Mabtech Cat# 3425–1H-6) according to the manufacturer’s protocol. Plates were read using a Vairoskan Instrument (Thermo Fischer Scientific).
Treatment of BMDCs with inhibitors and antagonist in vitro
BMDCs were independently pretreated for 1 hour with the p38 MAPK inhibitor SB203580 (10 μM; Invivogen Cat# tlrl-sb20), the PPAR alpha antagonist GW6471 (50 μM; Selleck Chemical Cat# S2798), the PPAR delta antagonist GSK3787 (10 μM; Selleck Chemical Cat# S8025), the CREB inhibitor 666–15 (10 μM; MedChem Express, Cat# HY-128686); the JNK inhibitor JNK-IN-8 (10 μM; Selleck Chemical Cat# S4901); the NF-κB inhibitor BAY 11–7821 (50 μM; MedChem Express Cat# HY-13453), or the NF-κB/AP1 dual inhibitor SP 100030 (5 μM; Tocris Cat# 5309) and then stimulated with LPA (100 μM) for 2 hours. Expression of Ptgs2 was determined by RT-qPCR. For in vitro assessment of EP4 signaling, BMDCs were pretreated for 1 hour with the EP4 antagonist PGN 1531 (5 μM; Tocris Cat#5327) and then stimulated with LPA (100 μM) and LPS (100 ng/ml) for 4 hours. Expression of type-I ISGs was subsequently quantified by RT-qPCR.
BMDC RNA-sequencing and bioinformatic analyses
Purified CD11c+ BMDCs were cultured overnight in complete RPMI media (described above) lacking FBS to promote LPA sensing (61). Cells were then left untreated or exposed to LPA (100 μM) for 2 or 6 hours, and total RNA was subsequently isolated using the RNeasy MinElute kit (Qiagen). All samples passed the RNA quality control examined by Agilent Bioanalyzer 2100, and mRNA libraries were generated and sequenced at the Weill Cornell Epigenomics Core Facility. RNA-seq data was aligned using bowtie2 (62) against mm10 genome and RSEM v1.2.12 software (63) was used to estimate gene-level read counts using Ensemble transcriptome information. DESeq2 (64) was used to estimate significance of differential expression difference between any two experimental groups. Gene expression changes were considered significant if passed the FDR < 5% threshold at both 2 and 6 hours of LPA treatment against the untreated condition. Gene set enrichment analysis was done using Ingenuity Pathway Analysis (IPA, Qiagen) using “Upstream regulators” option. Regulators at P < 10−6 with significant predicted activation/inhibition states (Z-score > 2) were reported. Data were deposited under NCBI GEO Accession number GSE182062.
Single cell RNA-seq analysis
Viable leukocytes (DAPI−CD45+) were isolated by FACS from the ascites of mice bearing ID8-Defb29/Vegf-A OvCa for 25 days (n = 4 independent mice) and cells (~8,000) were then processed for single-cell RNA sequencing at the Genomics Resources Core Facility of Weill Cornell Medicine. Raw gene expression matrices were generated for each sample by the Cell Ranger (v.3.0.2) Pipeline coupled with mouse reference version GRCm38 (mm10). The output filtered gene expression matrices were analyzed by R software (v.3.5.3) with the Seurat package (v.3.0.0). In brief, genes expressed at a proportion >0.1% of the data and cells with >200 genes detected were selected for further analyses. Low-quality cells were removed if they met the following criteria: (1) <800 unique molecular identifiers (UMIs); (2) <500 genes; or (3) >5% UMIs derived from the mitochondrial genome. Further, gene expression matrices were normalized by the NormalizeData function and 2,000 features with high cell-to-cell variation were calculated using the FindVariableFeatures() function. To reduce dimensionality of the datasets, the RunPCA() function was conducted with default parameters on linear-transformation scaled data generated by the ScaleData() function. At the end, we clustered cells using the FindNeighbors() and FindClusters() functions and performed nonlinear dimensional reduction with the RunUMAP() function with default settings. All details regarding the Seurat analyses performed in this work can be found in the website tutorial (https://satijalab.org/seurat/v3.0/pbmc3k_tutorial.html). Data were deposited under NCBI GEO Accession number GSE182047.
In vitro coculture of cancer cells and BMDCs
ID8-Defb29/Vegf-A cancer cells were treated with DMSO (vehicle) or talazoparib at 0.2, 2, or 10 μM for 24 hours. Cells were then washed twice with PBS and cocultured with BMDCs at a 1:1 ratio in the presence or absence of LPA (100 μM) for 24 hours. Nonadherent cells, corresponding to >90% BMDCs by FACS, were then collected for gene expression analyses.
NanoString mRNA expression analysis from human tissue
Forty-four formalin-fixed, paraffin embedded HGSOC samples from the TOPACIO trial (n = 9 responders and 35 non-responders) were analyzed with a NanoString assay of 780 genes, as previously described (46). The normalized log-transformed NanoString expression scores were filtered to keep only the genes (n = 36) that were differentially regulated by LPA exposure in BMDCs (Fig. 3, B and C). Genes significantly associated with chemo-experience status of the samples were removed from downstream analysis (VEGFA, CCL4, HAVC2) using Kruskal-Wallis Rank test (P-value < 0.1 and |fold-change| > 2) (Supplementary Fig. S8A). Spearman correlation within each response group was then used to identify co-expressed genes specific to response category that are not shaped by imbalance of categories. The LPA signature score for each sample was defined as the inverse of the median expression of the 7 downregulated genes identified. Heatmap visualization utilizes hierarchical clustering of euclidean distances with complete linkage. The HRD status of the tumors analyzed has been previously described (46).
Lipidomics
LPA species in plasma, malignant ascites, or peritoneal wash samples were analyzed and quantified using LC-MS at the Lipidomics Core Facility of Wayne State University School of Medicine.
Measurement of cancer cell proliferation and viability by MTT assay
Control or Enpp2-null OvCa cells were seeded in 96-well plates at a density of 6,000/well and allowed to adhere overnight. Cells were then exposed to LPA for 24 and 48 hours. Proliferation and viability of cancer cells was measured by MTT (Sigma-Aldrich, Saint Louis, MO).
Analysis of OvCa Patient Survival
Kaplan-Meier Plotter (65) was used to evaluate the correlation between ENPP2/IFNA1 and PTGS2/IFNA1 expression ratios in tumor specimens and the overall survival of OvCa patients in the TCGA dataset. Analyses were performed using only JetSet validated probes and the auto select cut-off feature in all tumors from the serous histology, irrespective of p53, debulking, or chemotherapy status (n = 557 patients).
In vivo treatments
Wild type C57BL/6J, C57BL/6NTac.Cg-Rag2tm1Fwa Il2rgtm1Wjl (Rag2/Il2rg double knockout) or B6.129S(C)-Batf3tm1Kmm/J (BATF3 KO) mice were implanted via i.p. injection with 1.5 × 106 control or Enpp2-null OvCa cells. After 10 days, mice were i.p. treated every 5 days for a total of 5 times with vehicle control (distilled water) or Poly (I:C) at 100 μg/mouse (Invivogen Cat# vac-plc). For in vivo assessment of type-I IFN signaling, mice were i.p. treated every 3 days for 15 times with isotype control antibodies (Bioxcell Cat# BE0083) or anti-IFNAR blocking antibodies (Bioxcell Cat# BE0241) at 200 μg/mouse, starting 3 days after tumor implantation. For in vivo stimulation of EP4 signaling, mice were orally administered every day for a total of 25 days with vehicle control or the EP4 agonist KAG-308 (3mg/kg; MedChem Express, Cat# HY-128686), starting 9 days after tumor implantation. Wild type C57BL/6J mice or Tmem173gt/J (STING KO) mice were challenged i.p. with 1.5 × 106 control or Enpp2-null OvCa cells. After 7 days, mice were treated daily via oral gavage with vehicle control (10% N,N-Dimethylacetamide (DMAc, Sigma, 1% Solutol, 89% PBS) or talazoparib at 0.33 mg/kg (MedChem Express, Cat# HY-16106) for the indicated times. For in vivo assessment of checkpoint blockade using anti-PD1antibodies, mice were i.p. treated every 3 days for 7 times with isotype control (Bioxcell Cat# BE0089) or anti-PD1 blocking antibodies (Bioxcell Cat# BE0146) at 200 μg/mouse, starting 10 days after OvCa challenge.
Statistical analyses
All statistical analyses were performed using the GraphPad Prism software (version 9). Significance for pairwise correlation analyses was calculated using the Spearman’s correlation coefficient (r). Comparisons between two groups were assessed using unpaired or paired (for matched comparisons) two-tailed Student’s t-test. Multiple comparisons were assessed by one-way ANOVA including Tukey’s multiple comparisons test. Grouped data were analyzed by two-way ANOVA including the Tukey or Šídák multiple comparisons test. Host survival rates were compared using the Log-rank test. Data are presented as mean ± SEM or violin plots. Unless otherwise stated, P-values < 0.05 were considered statistically significant.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
This study uncovers that ATX-LPA is a central immunosuppressive pathway in the ovarian tumor microenvironment. Ablating this axis sensitizes OvCa hosts to various immunotherapies by unleashing protective type-I IFN responses. Understanding the immunoregulatory programs induced by LPA could lead to new biomarkers predicting resistance to immunotherapy in cancer patients.
ACKNOWLEDGEMENTS
We thank the members of the Flow Cytometry Core Facility, the Epigenomics Core Facility, and the Genomics Resources Core Facility at Weill Cornell Medicine for their excellent assistance with cell sorting, bulk RNA-seq, and single-cell RNA-seq experiments, respectively. This research was supported by U.S. Department of Defense Ovarian Cancer Research Program grants W81XWH-16-1-0438, OC190443, OC200166 and OC200224 (J.R.C-R.); a Stand Up To Cancer Innovative Research Grant, Grant Number SU2C-AACR-IRG-03-16 (J.R.C-R.), a Stand Up To Cancer Phillip A. Sharp Innovation in Collaboration Award, Grant Number, SU2C-AACR-PS-24 (J.R.C-R. and A.D.D,A.); National Institutes of Health grants R01NS114653 (J.R.C-R. and E.A.R-S.), R21CA248106 (J.R.C-R. and E.A.R-S.), 1R01AI132446 (F.J.B.), and F31CA257631 (A.E.); the Pershing Square Sohn Cancer Research Alliance grant (J.R.C-R.); the Mark Foundation for Cancer Research ASPIRE award (J.R.C-R.); the Wade F. B. Thompson/Cancer Research Institute CLIP grant (J.R.C-R.); the Ann Schreiber Mentored Investigator Award of the Ovarian Cancer Research Alliance (C.S.); the Irvington Institute-Cancer Research Institute Postdoctoral Fellowship Award (C.S. and C-S.C); National Research Foundation of Korea (NRF) grant 2020R1C1C1010303 (M.S.) and National Cancer Center of Korea grant NCC-203175 (M.S.); Cancer Foundation Finland, Instrumentarium, and Maud Kuistila Memorial Foundation (J.C.); Academy of Finland, Sigrid Juselius Foundation, and Cancer Foundation Finland (A.F.). Stand Up To Cancer is a division of the Entertainment Industry Foundation. The indicated Stand Up To Cancer Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Conflicts of Interest Statement:
F.J.B. is a founder of IpiNovyx Bio. D.Z. reports institutional grants from Genentech, AstraZeneca, and Plexxikon, as well as personal fees from Genentech, AstraZeneca, Xencor, Memgen, Synthekine, Immunos, Accurius, Mana Therapeutics, Celldex, and Calidi Biotherapeutics, outside of the submitted work. D.Z. is also an inventor on a patent related to the use of oncolytic Newcastle Disease Virus for cancer therapy. A.D.D’A. is a consultant/advisory board member for Lilly Oncology, Merck-EMD Serono, Cyteir Therapeutics, Third Rock Ventures, AstraZeneca, Ideaya Inc., Cedilla Therapeutics Inc., a stockholder in Ideaya Inc., Cedilla Therapeutics Inc., and Cyteir, and reports receiving commercial research grants from Lilly Oncology and Merck-EMD Serono. J.R.C-R. is a member of the scientific advisory board of NextRNA Therapeutics, Inc., and scientific consultant for Autoimmunity Biologic Solutions, Inc. J.R.C-R. and C-S.C. hold a patent on the targeting of ATX-LPA for the treatment of disease.
Data availability
Data generated in this study are available in the Gene Expression Omnibus (GEO) database under the accession numbers GSE182062 for bulk RNA-seq and GSE182047 for scRNA-seq. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Juan R. Cubillos-Ruiz (jur2016@med.cornell.edu).
REFERENCES
- 1.Benesch MG, Tang X, Maeda T, Ohhata A, Zhao YY, Kok BP, et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J 2014;28(6):2655–66 doi 10.1096/fj.13-248641. [DOI] [PubMed] [Google Scholar]
- 2.Chen J, Li H, Xu W, Guo X. Evaluation of serum ATX and LPA as potential diagnostic biomarkers in patients with pancreatic cancer. BMC Gastroenterol 2021;21(1):58 doi 10.1186/s12876-021-01635-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li YY, Zhang WC, Zhang JL, Zheng CJ, Zhu H, Yu HM, et al. Plasma levels of lysophosphatidic acid in ovarian cancer versus controls: a meta-analysis. Lipids Health Dis 2015;14:72 doi 10.1186/s12944-015-0071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009;15(6):539–50 doi 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamada T, Sato K, Komachi M, Malchinkhuu E, Tobo M, Kimura T, et al. Lysophosphatidic acid (LPA) in malignant ascites stimulates motility of human pancreatic cancer cells through LPA1. J Biol Chem 2004;279(8):6595–605 doi 10.1074/jbc.M308133200. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi M, Okabe K, Kato K, Okumura M, Fukui R, Fukushima N, et al. Differential function of lysophosphatidic acid receptors in cell proliferation and migration of neuroblastoma cells. Cancer Lett 2012;316(1):91–6 doi 10.1016/j.canlet.2011.10.030. [DOI] [PubMed] [Google Scholar]
- 7.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov 2019;9(5):617–27 doi 10.1158/2159-8290.CD-18-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minami K, Ueda N, Ishimoto K, Tsujiuchi T. Lysophosphatidic acid receptor-2 (LPA2)-mediated signaling enhances chem-resistance in melanoma cells treated with anticancer drugs. Mol Cell Biochem 2020;469(1–2):89–95 doi 10.1007/s11010-020-03730-w. [DOI] [PubMed] [Google Scholar]
- 9.Fang X, Gaudette D, Furui T, Mao M, Estrella V, Eder A, et al. Lysophospholipid growth factors in the initiation, progression, metastases, and management of ovarian cancer. Ann N Y Acad Sci 2000;905:188–208 doi 10.1111/j.1749-6632.2000.tb06550.x. [DOI] [PubMed] [Google Scholar]
- 10.Fang X, Schummer M, Mao M, Yu S, Tabassam FH, Swaby R, et al. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim Biophys Acta 2002;1582(1–3):257–64 doi 10.1016/s1388-1981(02)00179-8. [DOI] [PubMed] [Google Scholar]
- 11.Xiao YJ, Schwartz B, Washington M, Kennedy A, Webster K, Belinson J, et al. Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: comparison of the lysophospholipid contents in malignant vs nonmalignant ascitic fluids. Anal Biochem 2001;290(2):302–13 doi 10.1006/abio.2001.5000. [DOI] [PubMed] [Google Scholar]
- 12.Klymenko Y, Bos B, Campbell L, Loughran E, Liu Y, Yang J, et al. Lysophosphatidic acid modulates ovarian cancer multicellular aggregate assembly and metastatic dissemination. Sci Rep 2020;10(1):10877 doi 10.1038/s41598-020-67565-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murph MM, Liu W, Yu S, Lu Y, Hall H, Hennessy BT, et al. Lysophosphatidic acid-induced transcriptional profile represents serous epithelial ovarian carcinoma and worsened prognosis. PLoS One 2009;4(5):e5583 doi 10.1371/journal.pone.0005583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seo EJ, Kwon YW, Jang IH, Kim DK, Lee SI, Choi EJ, et al. Autotaxin Regulates Maintenance of Ovarian Cancer Stem Cells through Lysophosphatidic Acid-Mediated Autocrine Mechanism. Stem Cells 2016;34(3):551–64 doi 10.1002/stem.2279. [DOI] [PubMed] [Google Scholar]
- 15.Perrakis A, Moolenaar WH. Autotaxin: structure-function and signaling. J Lipid Res 2014;55(6):1010–8 doi 10.1194/jlr.R046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Li M, Yin N, Zhang J. The Expression Regulation and Biological Function of Autotaxin. Cells 2021;10(4) doi 10.3390/cells10040939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geraldo LHM, Spohr TCLS, Amaral RFD, Fonseca ACCD, Garcia C, Mendes FA, et al. Role of lysophosphatidic acid and its receptors in health and disease: novel therapeutic strategies. Signal Transduct Target Ther 2021;6(1):45 doi 10.1038/s41392-020-00367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brindley DN. Lysophosphatidic Acid Signaling in Cancer. Cancers (Basel) 2020;12(12) doi 10.3390/cancers12123791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chae CS, Teran-Cabanillas E, Cubillos-Ruiz JR. Dendritic cell rehab: new strategies to unleash therapeutic immunity in ovarian cancer. Cancer immunology, immunotherapy : CII 2017;66(8):969–77 doi 10.1007/s00262-017-1958-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nature reviews Disease primers 2016;2:16061 doi 10.1038/nrdp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol 2019;30(7):1080–7 doi 10.1093/annonc/mdz135. [DOI] [PubMed] [Google Scholar]
- 22.Borden EC. Interferons α and β in cancer: therapeutic opportunities from new insights. Nat Rev Drug Discov 2019;18(3):219–34 doi 10.1038/s41573-018-0011-2. [DOI] [PubMed] [Google Scholar]
- 23.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol 2015;15(7):405–14 doi 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 24.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med 2004;10(9):950–8. [DOI] [PubMed] [Google Scholar]
- 25.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015;161(7):1527–38 doi 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 2018;562(7727):423–8 doi 10.1038/s41586-018-0597-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iyer S, Zhang S, Yucel S, Horn H, Smith SG, Reinhardt F, et al. Genetically Defined Syngeneic Mouse Models of Ovarian Cancer as Tools for the Discovery of Combination Immunotherapy. Cancer Discov 2021;11(2):384–407 doi 10.1158/2159-8290.CD-20-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol 2013;34(2):67–73 doi 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015;162(6):1257–70 doi 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang D, DuBois RN. The Role of Prostaglandin E(2) in Tumor-Associated Immunosuppression. Trends in molecular medicine 2016;22(1):1–3 doi 10.1016/j.molmed.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singer CA, Baker KJ, McCaffrey A, AuCoin DP, Dechert MA, Gerthoffer WT. p38 MAPK and NF-kappaB mediate COX-2 expression in human airway myocytes. Am J Physiol Lung Cell Mol Physiol 2003;285(5):L1087–98 doi 10.1152/ajplung.00409.2002. [DOI] [PubMed] [Google Scholar]
- 32.Xu L, Han C, Lim K, Wu T. Cross-talk between peroxisome proliferator-activated receptor delta and cytosolic phospholipase A(2)alpha/cyclooxygenase-2/prostaglandin E(2) signaling pathways in human hepatocellular carcinoma cells. Cancer Res 2006;66(24):11859–68 doi 10.1158/0008-5472.CAN-06-1445. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Zhou J, Wei X, Chen R, Geng J, Zheng R, et al. miR-144 and targets, c-fos and cyclooxygenase-2 (COX2), modulate synthesis of PGE2 in the amnion during pregnancy and labor. Sci Rep 2016;6:27914 doi 10.1038/srep27914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai ZZ, Yang HL, Ha SY, Chang KK, Mei J, Zhou WJ, et al. Cyclooxygenase-2 in Endometriosis. Int J Biol Sci 2019;15(13):2783–97 doi 10.7150/ijbs.35128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perkins DJ, Richard K, Hansen AM, Lai W, Nallar S, Koller B, et al. Autocrine-paracrine prostaglandin E. Nat Immunol 2018;19(12):1309–18 doi 10.1038/s41590-018-0243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 2009;206(7):1589–602 doi 10.1084/jem.20090247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, et al. In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res 2009;69(18):7329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murahashi Y, Yano F, Chijimatsu R, Nakamoto H, Maenohara Y, Amakawa M, et al. Oral administration of EP4-selective agonist KAG-308 suppresses mouse knee osteoarthritis development through reduction of chondrocyte hypertrophy and TNF secretion. Sci Rep 2019;9(1):20329 doi 10.1038/s41598-019-56861-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishimura R, Shirasaki T, Tsuchiya K, Miyake Y, Watanabe Y, Hibiya S, et al. Establishment of a system to evaluate the therapeutic effect and the dynamics of an investigational drug on ulcerative colitis using human colonic organoids. J Gastroenterol 2019;54(7):608–20 doi 10.1007/s00535-018-01540-y. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe Y, Murata T, Amakawa M, Miyake Y, Handa T, Konishi K, et al. KAG-308, a newly-identified EP4-selective agonist shows efficacy for treating ulcerative colitis and can bring about lower risk of colorectal carcinogenesis by oral administration. Eur J Pharmacol 2015;754:179–89 doi 10.1016/j.ejphar.2015.02.021. [DOI] [PubMed] [Google Scholar]
- 41.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008;322(5904):1097–100 doi 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell reports 2018;25(11):2972–80 e5 doi 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res 2019;79(2):311–9 doi 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov 2019;9(6):722–37 doi 10.1158/2159-8290.CD-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boussios S, Abson C, Moschetta M, Rassy E, Karathanasi A, Bhat T, et al. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R D 2020;20(2):55–73 doi 10.1007/s40268-020-00301-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farkkila A, Gulhan DC, Casado J, Jacobson CA, Nguyen H, Kochupurakkal B, et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat Commun 2020;11(1):1459 doi 10.1038/s41467-020-15315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SC, Dacheux MA, Norman DD, Balazs L, Torres RM, Augelli-Szafran CE, et al. Regulation of Tumor Immunity by Lysophosphatidic Acid. Cancers (Basel) 2020;12(5) doi 10.3390/cancers12051202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matas-Rico E, Frijlink E, van der Haar Avila I, Menegakis A, van Zon M, Morris AJ, et al. Autotaxin impedes anti-tumor immunity by suppressing chemotaxis and tumor infiltration of CD8(+) T cells. Cell reports 2021;37(7):110013 doi 10.1016/j.celrep.2021.110013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, DuBois RN. Role of prostanoids in gastrointestinal cancer. J Clin Invest 2018;128(7):2732–42 doi 10.1172/JCI97953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018;172(5):1022–37 e14 doi 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pelly VS, Moeini A, Roelofsen LM, Bonavita E, Bell CR, Hutton C, et al. Anti-Inflammatory Drugs Remodel the Tumor Immune Environment to Enhance Immune Checkpoint Blockade Efficacy. Cancer Discov 2021;11(10):2602–19 doi 10.1158/2159-8290.CD-20-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J, et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity 2014;40(4):554–68 doi 10.1016/j.immuni.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 53.Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol 2019;5(8):1141–9 doi 10.1001/jamaoncol.2019.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer 2021;21(2):71–88 doi 10.1038/s41568-020-00312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zulfikar S, Mulholland S, Adamali H, Barratt SL. Inhibitors of the Autotaxin-Lysophosphatidic Acid Axis and Their Potential in the Treatment of Interstitial Lung Disease: Current Perspectives. Clin Pharmacol 2020;12:97–108 doi 10.2147/CPAA.S228362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000;21(4):585–91. [DOI] [PubMed] [Google Scholar]
- 57.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8(11):2281–308 doi 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li S, Wang B, Xu Y, Zhang J. Autotaxin is induced by TSA through HDAC3 and HDAC7 inhibition and antagonizes the TSA-induced cell apoptosis. Mol Cancer 2011;10:18 doi 10.1186/1476-4598-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nair S, Archer GE, Tedder TF. Isolation and generation of human dendritic cells. Curr Protoc Immunol 2012;Chapter 7:Unit7 32 doi 10.1002/0471142735.im0732s99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med 2018;10(423) doi 10.1126/scitranslmed.aam8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao P, Aoki Y, Badri L, Walker NM, Manning CM, Lagstein A, et al. Autocrine lysophosphatidic acid signaling activates beta-catenin and promotes lung allograft fibrosis. J Clin Invest 2017;127(4):1517–30 doi 10.1172/JCI88896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9(4):357–9 doi 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 2011;12:323 doi 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer 2012;19(2):197–208 doi 10.1530/ERC-11-0329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are available in the Gene Expression Omnibus (GEO) database under the accession numbers GSE182062 for bulk RNA-seq and GSE182047 for scRNA-seq. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Juan R. Cubillos-Ruiz (jur2016@med.cornell.edu).