

Abstract
Chimeric antigen receptor T cell (CAR-T) therapy directed at CD19 produces durable remissions in the treatment of relapsed/refractory non-Hodgkin’s lymphoma (NHL). Nonetheless, many patients receiving CD19 CAR-T cells fail to respond for unknown reasons. To reveal changes in 4–1BB-based CD19 CAR-T cells and identify biomarkers of response, we employed single cell RNA sequencing and protein surface marker profiling of patient CAR-T cells pre- and post-infusion into NHL patients. At the transcriptional and protein levels, we note the evolution of CAR-T cells toward a non-proliferative, highly differentiated, and exhausted state, with an enriched exhaustion profile in CAR-T cells of patients with poor response marked by TIGIT expression. Utilizing in vitro and in vivo studies, we demonstrate TIGIT blockade alone improves the anti-tumor function of CAR-T cells. Altogether, we provide evidence of CAR-T cell dysfunction marked by TIGIT expression driving a poor response in NHL patients.
Keywords: Cell Therapy, Lymphoma, CAR-T cells, Immunotherapy, Checkpoint Blockade
Introduction
CAR-T cells are T cells engineered with a chimeric antigen receptor to specifically lyse tumor cells expressing the targeted antigen. The safety and efficacy of CD19 CAR-T cell products in B cell malignancies has led to FDA authorization of four products for the treatment of pediatric B cell acute lymphoblastic leukemia (B-ALL) and B cell subtypes of non-Hodgkin’s lymphoma (NHL). The CAR construct designs used in these applications contain intracellular CD3ζ with either 4–1BB (4–1BB.CAR) or CD28 (CD28.CAR). The 4–1BB intracellular domain is thought to convey resistance to exhaustion and superior CAR-T cell persistence in comparison to the CD28 intracellular domain which exhibits greater short-term activity and adverse events(1–3). Here, a 4–1BB design was applied in the generation of CAR-T cells for the treatment of relapsed/refractory non-Hodgkin’s lymphoma (NHL).
While approximately 50% of patients with NHL undergo remission after initial chemotherapy, prognosis for chemorefractory disease is poor without intervention with adoptive cell therapies(4–7). Following CD19 CAR-T cell therapy, approximately 30–40% of NHL patients will have durable remissions(8,9). Mechanisms of early relapse or refractory disease remain inconclusive, with significant variation between studies due to differences such as disease type, product design, and product manufacture(10–14). Proposed mechanisms of resistance include poor T cell quality, T cell exhaustion, antigen loss/modulation, host factors, and the tumor microenvironment(15). While several studies have focused on comparisons of infusion products, detailed single cell studies evaluating post-infusion CAR-T cell transcriptional profiles and phenotypes associated with clinical outcomes remain lacking(16,17). We address this gap by evaluating CAR-T cells from serial pre- and post-infusion samples in patients with both favorable and poor outcomes by applying methods to assess single cell transcriptomics and cell surface protein expression in individual cells.
Results
CD19 CAR-T cells demonstrate significant transcriptional heterogeneity that changes after infusion into patients
To describe the evolution of CAR-T cells after infusion into NHL patients and to identify mechanisms and biomarkers of response, our initial study examined manufactured CAR-T cell products and isolated CAR-T cells from post-infusion blood samples from patients treated for CD19+ relapsed/refractory B cell lymphoma with known outcomes (Supplemental Table 1). For this analysis, we employed the use of the single cell methods scRNA sequencing and/or flow cytometry to investigate time points after infusion that are known from previous studies to be associated with peak expansion (day 14) and contraction (day 30) and represent key changes in CAR-T cell activity(18) (study schema depicted in Figure 1A; a checklist of which assays were performed with each sample is provided in Supplemental Table 1). Overall, the study employed 14 manufactured CAR-T cell products, 14 samples from day 14, and 13 samples from day 30. This sampling represents 13 patients with favorable response (complete or partial remission (CR; PR)) and 4 patients with poor response (stable or progressive disease (SD; PD)) (details on patient clinical information in Supplemental Table 2). In addition, our data is supported by a commercial product validation dataset that consists of 4 samples from day 14 and 3 samples from day 30 from 5 individual patients.
Figure 1. CD19 CAR-T cells demonstrate significant transcriptional heterogeneity that changes after infusion into patients.
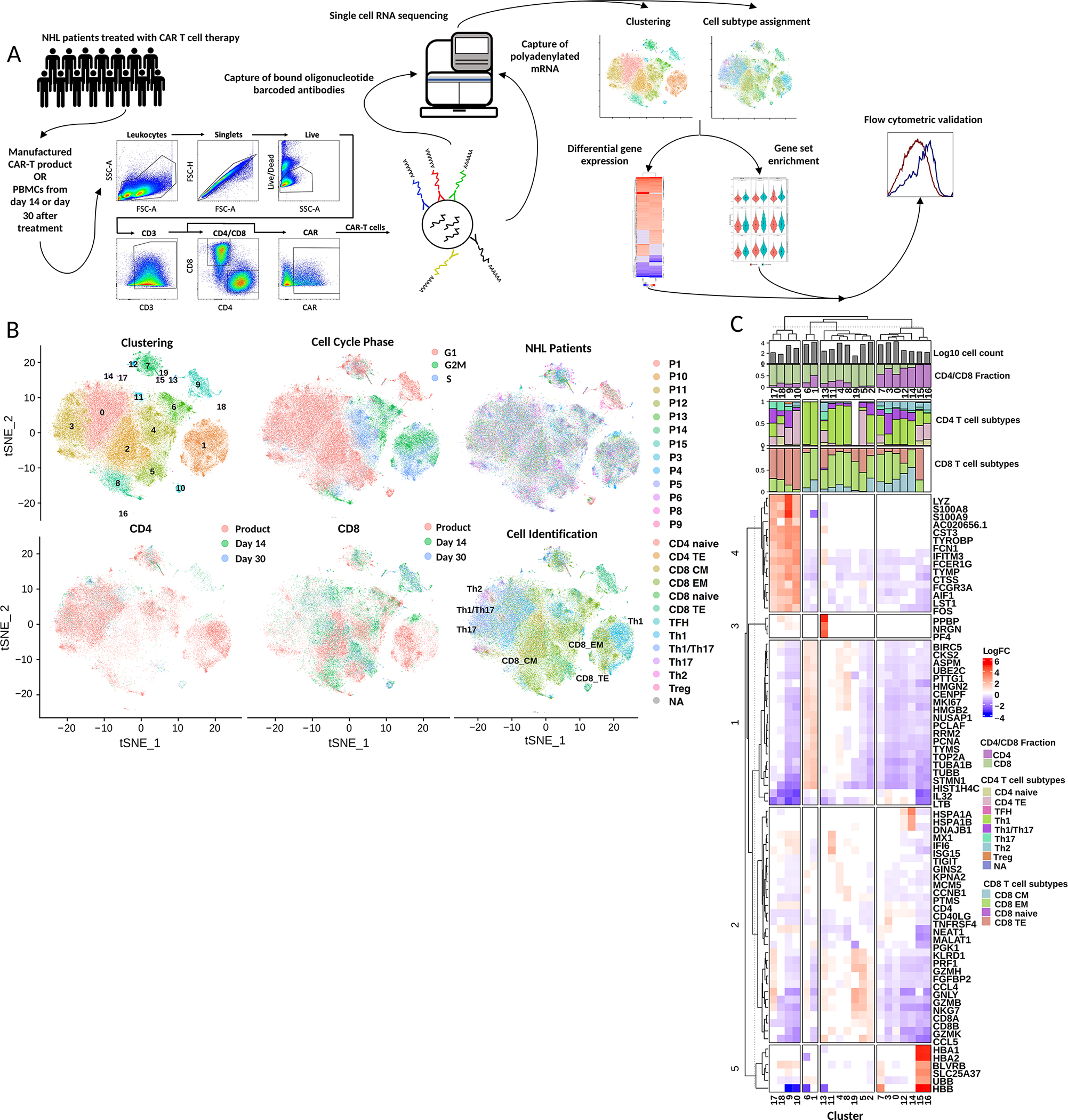
A) Study scheme. Viable CAR-T cells were sorted from the CAR-T cell products or peripheral blood mononuclear cells of NHL patients. scRNAseq and feature barcoding libraries were then prepared and subsequently sequenced. After quality control, dimension reduction was performed and analyzed by cluster or cell subtype with differential gene expression and gene set signature scoring. Validation of memory marker and exhaustion marker expression was performed with flow cytometry. B) tSNE dimension reduction of scRNA sequencing data with overlays depicting cluster assignment, cell cycle analysis, patient number, CD4 or CD8 T cell group by time point and T cell subtype. C) Heatmap of differentially expressed genes (adjusted p-value < 0.05) between all clusters of B) by log fold change. The top 3 to 10 genes with highest absolute log fold change are represented. Clusters are annotated with cell subtype proportions.
To isolate CAR-T cells for scRNA sequencing, viable CD3+CAR+ cells were sorted from cryopreserved CAR-T cell products or PBMCs. Next, libraries were generated with the 10x Genomics Chromium single cell 3’ platform with feature barcoding technology to allow simultaneous and paired quantification of transcriptional and cell surface protein expression in individual CAR-T cells(19). The inclusion of feature barcoding in addition to enabling assessments of key markers at the protein level also allowed the discrimination of the memory markers CD45RA and CD45RO that cannot be discriminated at the RNA level. The libraries were sequenced and the data stringently filtered to remove on average 8.1% mitochondrial reads and yielded 94,000 cells with an average of 3,917 cells per sample, 8,518 reads per cell, and 2,263 unique detectable genes per cell (Supplemental Table 3). Batch effect removal was applied to remove differences due to sample preparation or sequencing.
To appreciate the heterogeneity present in the CAR-T cells both among patients and time points, RNA-based dimension reduction and unbiased clustering were applied on the scRNAseq dataset to yield 19 clusters with distinct transcriptional profiles (Figure 1B;C). At pre-infusion, a consistent clustering pattern was observed across patients, with the majority of pre-infusion cells in clusters 0–4. After infusion, heterogeneity changed across patients, with additional clusters identified as well as an increased distribution of cells across clusters. For example, at day 14 the proportion of cells in clusters 5–10 increased as compared to pre-infusion while the frequency of cells in clusters 2 and 4 remained the same. From day 14 to day 30, the distribution of cells across clusters decreases, with four clusters (C0, C2, C5, C9) becoming dominant. Thus, we see an increase in heterogeneity followed by a loss by day 30 (Supplemental Figure 1A). In accordance with peak expansion expected around day 14, clusters primarily composed of pre-infusion and day 14 samples (C1, C4, and C6) contained the most actively-dividing cells as evidenced by their abundance in the S phase by cell cycle analysis (Figure 1B; Supplemental Figure 1B). Differential gene expression of individual clusters compared to other clusters demonstrated the cells belonging to these clusters were highly enriched (p.adj < 0.05) in genes associated with immature T cell types and proliferation such as MKI67, PCNA, TYMS, and TOP2A (Figure 1C)(20). In contrast, other major clusters (C2, C5, C8) which consist predominantly of CD8 CAR-T cells from day 14 and day 30 samples were found not to be actively proliferating and exhibited higher expression (p.adj < 0.05) of genes associated with effector CD8 T cell phenotypes including GZMB, GZMH, GZMK, PRF1, GNLY, CCL4, and CCL5 (Figure 1C). To investigate if the heterogeneity across clusters correlated with particular T cell subtypes, SingleR cell ID annotation was applied. SingleR utilizes predefined T cell subtype gene signatures to assign a CD4 or CD8 T cell subtype to each cell, including naive, central memory (CM), effector memory (EM) and terminal effector (TE) CD8 T cell subtypes(21,22). Application of cell identification results to the dimension reduction plot showed distinct localization of CD4 and CD8 T cells. While the predominant CD8 subtype was effector memory, two clusters were enriched with central memory transcriptional profiles (C0, C2) and three clusters enriched with terminal effector transcriptional profiles (C5, C9, C10) (Supplemental Figure 1C). Overall, CAR-T cells demonstrated significant heterogeneity across time points (product, day 14, day 30), cell cycle phase, cell type, and patient.
Circulating CD8 CAR-T cells differentiate to an effector-like state and express high levels of TIGIT post-infusion
In CAR-T cell therapy, the expansion of cytotoxic CD8 CAR-T cells is thought to drive clinical responses through direct tumor lysis. In line with this notion, we hypothesized that we would observe evidence of greater cytotoxic T cell to helper T cell expansion as well as differentiation of post-infusion CD8 CAR-T cells compared to pre-infusion CD8 CAR-T cells. Accordingly, we observed that the relative proportion of CD8 to CD4 CAR-T cells significantly increased after infusion from 52% to 87% CD8 CAR-T cells on average (p = 0.0005) (Figure 2A). The most significantly upregulated genes in post-infusion CD8 CAR-T cells from merged day 14 and day 30 samples included transcription factors (PRDM1, EOMES) and cytotoxic effector molecules (GZMB, PRF1, GZMK, CCL5) associated with differentiation into cytotoxic effector cells (DESeq2; p.adj < 0.05) (Figure 2B). Notably, transcription factors associated with exhaustion (TOX, TOX2, NR4A2, NR4A3) were also significantly upregulated post-infusion, and gene set enrichment analysis of memory/effector versus exhaustion gene sets showed post-infusion CD8 CAR-T cells were significantly enriched in exhaustion-related genes compared to pre-infusion (p < 0.05) (Supplemental Figure 2A). Notably, upregulated expression of the exhaustion markers CTLA4, LAG3, HAVCR2 (TIM3), PDCD1 (PD1), VSIR (VISTA), and TIGIT in merged day 14 and day 30 post-infusion samples compared to the product was observed in both CD4 and CD8 CAR-T cells, with TIGIT being the most significant for both cell types (pre- vs. post-infusion CD8 CAR-T; average log2FC = 2.39; p < 0.0001) (Figure 2C; Supplemental Figure 2B; Supplemental Figure 2C). Quantification of cell type proportions across time points using SingleR transcription-based cell assignments indicated a shift from an equal proportion of CD8 central memory and effector memory profiles in the product towards an effector memory profile at day 14 and a combination of effector memory and terminal effector profiles at day 30 (Figure 2D). Altogether, gene expression analyses indicate CD8 CAR-T cells undergo differentiation towards a cytotoxic effector profile and exhaustion after infusion into patients.
Figure 2. Circulating CD8 CAR-T cells differentiate to an effector-like state and express high levels of TIGIT post-infusion.
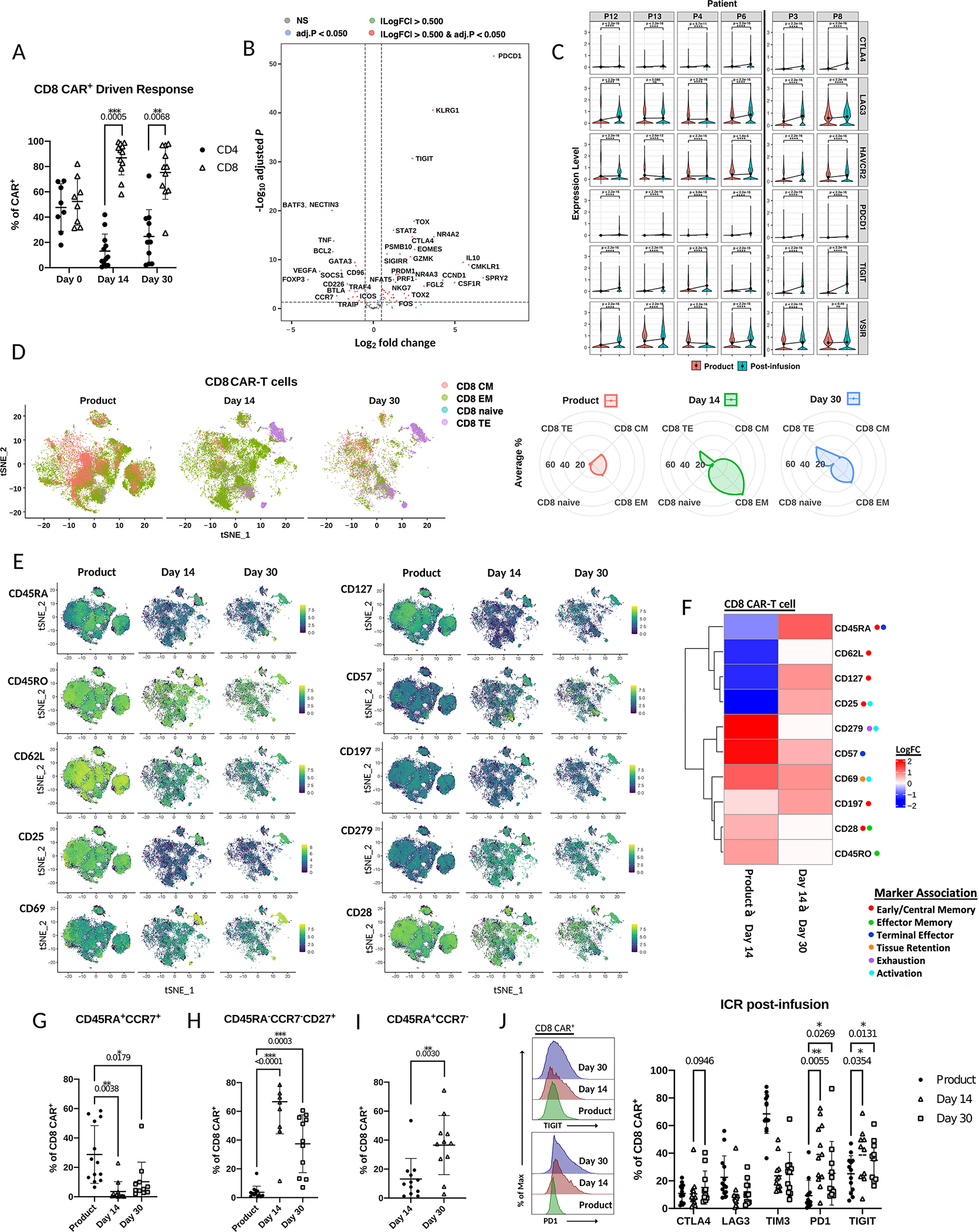
A) Percentage of CD4 or CD8 CAR-T cells of total CAR-T cells in patients at day 14 or day 30 post-infusion as measured by flow cytometry (Wilcoxon matched-pairs signed rank test). B) Volcano plot of differentially expressed genes (adjusted p-value < 0.05;DESeq2 on two merged cell groups to compare) in combined day 14 and day 30 post-infusion CD8 CAR-T cells compared to product CD8 CAR-T cells. C) Violin plots of exhaustion marker normalized RNA expression before and after infusion (combined day 14 and day 30) in individual patients. Comparisons made by Wilcoxon rank-sum test. Black line separates responders (left) and poor responders (right) D) Left – scRNA tSNE dimension reduction of all samples with CD8 cell subtype assignment overlaid across time points. Right – relative frequency of CD8 T cell subtype assignments of total CAR-T cells at each time point. E) scRNA tSNE dimension reduction plots with relative surface expression overlay for each of the indicated markers as measured by feature barcoding. Each of the indicated time points contains total CAR-T cells from that time point. F) Heatmap of differentially expressed surface markers (adjusted p-value < 0.05; zlm) from E) in CD8 CAR-T cells between time points. Color represents log fold increase/decrease in the latter time point. G-I) Comparison of percentage of CD8 CAR-T cells with the indicated memory phenotype across time points by Mixed-effects analysis as measured by flow cytometry. J) Left – Histograms of fluorescence intensity of TIGIT and PD1 across time points as measured by flow cytometry. Each curve represents a concatenation of all samples with equal proportions of CD8 CAR-T cells from each sample. Right – Comparison across time points by Mixed-effects analysis of the percentage of CD8 CAR-T cells expressing checkpoint receptors CTLA4, LAG3, TIM3, PD1, or TIGIT as measured by flow cytometry.
Given the changes in the transcriptional profile of CD8 CAR-T cells post-infusion, changes in cell surface phenotype upon infusion were expected to reflect differentiation towards effector memory or terminally differentiated phenotypes. We previously demonstrated that CD8 CAR-T cell products are enriched in an early memory phenotype with expression of CD45RA, CCR7, and TCF7(23). Here, feature barcoding markers were chosen for their association with stemness/early memory (CD45RA, CD197 (CCR7), CD127 (IL-7R), CD62L, CD25, CD28), activation/effector memory (CD25, CD69, CD45RO, CD279 (PD1)), terminal differentiation (CD45RA, CD57) and exhaustion (PD1)(24,25). Consistent with the RNA expression data, we observed greater cell surface expression of the naive and early memory markers CD45RA, CD127, CD62L, and CD25 in the product (Figure 2E;F). By day 14, a global increase in CD45RO, CD69, CD57, and PD1 (CD279) was observed suggesting differentiation towards an activated, effector-like state (Figure 2E;F). However, at the cluster-level, clusters 5, 9, and 10 were found to be enriched in CD45RA and CD57, indicative of cluster-specific enrichment with a terminally differentiated phenotype. We next compared changes from day 14 to day 30 for evidence of further differentiation and observed an additional increase in global CD45RA and CD57 expression. There was also a global increase in the memory markers CD127, CD25, and CD197 from day 14 to day 30, consistent with contraction of effector cells by day 30 after tumor resolution. Overall, the cell surface phenotypes of CD8 CAR-T cells both pre- and post-infusion as measured by feature barcoding showed high similarity to the RNA expression results indicating CD8 T cell differentiation to effector memory and terminal effector phenotypes.
Next flow cytometry was utilized to validate changes in memory status and exhaustion marker expression in CD8 CAR-T cells with additional patient samples (Supplemental Table 2). Similar to the previous analysis using surface protein feature barcoding, we observed an increased proportion of CD45RA+CCR7+ cells in the product (average 29%) as compared to post-infusion samples (average 3.6%), indicative of a greater proportion of early memory T cells (Figure 2G). At day 14, an average 83.3% of CD8 CAR-T cells were CD45RA- and the predominant phenotype (average 66.7%) was CD45RA-CCR7-CD27+, a significant shift toward an effector phenotype as compared to CAR-T cells in the product (p < 0.0001) (Figure 2H). While day 30 samples also contained high frequencies of CD45RA-CCR7-CD27+ cells, there was a significant increase in CD45RA+CCR7- cells among CD8 CAR-T cells, once again indicative of terminal differentiation (p< 0.01) (Figures 2I; Supplemental Figure 2D). Notably, at both day 14 and day 30, CD8 CAR-T cells could be distinguished from endogenous CD8 T cells by flow cytometry by expression of CD27, which may contribute to the long-term maintenance observed in 4–1BB.CAR-T cells (Supplemental Figure 2E)(26). Altogether, the data supports a shift from early memory to effector memory and terminal effector CD8 CAR-T cells in post-infusion samples.
As gene expression and feature barcoding datasets indicated higher levels of exhaustion marker expression post-infusion, we further assessed changes in checkpoint molecules by flow cytometry. In the manufactured CAR-T cell product, 68% of CD8 CAR-T cells expressed TIM3. While this could be indicative of early exhaustion or senescence, this expression was attributed to activation during CAR-T cell manufacture, as expression dropped after infusion to an average 24% TIM3+ (Figure 2J). Due to the decrease in TIM3 protein expression post-infusion, it was not considered a dominant mediator of CAR-T cell exhaustion in our patients. Among the exhaustion markers assayed, PD1 and TIGIT expression were significantly induced to an average 39.1% PD1+ and 38.7% TIGIT+ CD8 CAR-T cells at day 14 (p < 0.05) (Figure 2J). TIGIT expression was the most sustained at day 30 with an average 34.8% TIGIT+ CD8 CAR-T cells. In addition, among the exhaustion markers assayed, PD1 and TIGIT were also the most upregulated on CD4 CAR-T cells, increasing to an average 50.4% PD1+ and 42.9% TIGIT+ post-infusion (day 14 and day 30) (Supplemental Figure 2F). To determine if the observed expression of PD1 and TIGIT could be attributed to CAR signaling, we compared expression of exhaustion markers in CAR-T cells to endogenous T cells. Both CD4 and CD8 CAR-T cells showed enrichment of TIGIT compared to endogenous CD4 and CD8 T cells, with an average 9.5% and 10.4% higher expression of TIGIT on CD8 CAR-T cells at day 14 and day 30, respectively (Supplemental Figures 2G–H).
As there are a variety of different CD19 CAR-T products used clinically, we next assessed TIGIT and PD1 expression after infusion on patient T cells from patients receiving three different FDA-approved CD19 CAR-T cell products (Yescarta; Kymriah; Tescartus). Similar to the CAR-T product used for the previous studies, we observed high expression of TIGIT and PD1 on CD4 and CD8 CAR-T cells with an average 65.6% and 53.4% CD8 CAR-T cells expressing TIGIT and PD1, respectively (Supplemental Figure 2I–K). Once again, endogenous T cells also expressed TIGIT, though lower than CAR expressing cells (Supplemental Figure 2I; Supplemental Figure 2K). Altogether, our data indicates there is elevated expression of TIGIT on CD4 and CD8 CAR-T cells post-infusion and TIGIT expression in these cells is strengthened by CAR expression. Though PD1 in addition to TIGIT was also found to be consistently induced post-infusion at both the RNA and protein level, we focused further studies on TIGIT as it is comparatively less studied in this context and TIGIT expression exhibited more marked and durable changes.
CAR-T cells of poor responders are enriched in an exhaustion-like phenotype post-infusion that includes high TIGIT expression
With evidence of exhaustion in post-infusion CAR-T cells, we next investigated for differences in the exhaustion profile in CAR-T cells from favorable and poor responders. We found CD8 CAR-T cells of poor responders exhibited significantly decreased expansion and persistence compared to responders by flow cytometry (p < 0.05) (Figure 3A). A similar but not statistically significant result was observed for CD4 CAR-T cells (Supplemental Figure 3A). At the RNA cluster-level, cell frequencies across responder groups were comparable (Supplemental Figure 1A, Figure 3B). Across assigned CD8 T cell subtypes, CD8 CAR-T cells of poor responders contained a significantly greater proportion of effector memory cells, likely indicative of greater CAR signaling in proportion and magnitude in poor responders (p = 0.044) (Supplemental Figure 3B). We next compared differentially expressed genes of post-infusion CD8 CAR-T cells between poor and favorable response groups to identify dysregulated genes in the CD8 CAR-T cells of poor responders. Of note, significantly upregulated transcription factors included FOS, JUNB, JUND, FOSB, JUN, NR4A2, NFKBIA, and PRDM1 (MAST; p.adj < 0.0001) (Figure 3C). To confirm a dysfunctional profile in post-infusion CD8 CAR-T cells from poor response patients, we performed dysfunction scoring by applying three different exhaustion signatures(1,27,28). Consistently, we observed significantly higher dysfunctional scores in CD8 CAR-T cells of poor responders both globally (p < 0.0001) and within the most predominant clusters (Figure 3D). Higher dysfunction scores were also consistent across CD8 CAR-T cell subtypes (Supplemental Figure 3C). Upon direct investigation of exhaustion marker expression within individual patients, upregulation of TIGIT was the most prominently observed in the CD8 CAR-T cells of poor responders (logFC = 2.8) as compared to responders (logFC = 2.25) (Figure 3E). This was also even more evident in the percentage of CD8 CAR-T cells expressing TIGIT at the RNA level, where we observed ~62% (range 56–67%) of post-infusion CD8 CAR-T cells of poor responders expressed TIGIT compared to ~27% (range 12–37%) of cells from responders (Figure 3E).
Figure 3. CAR-T cells of poor responders are enriched in an exhaustion-like phenotype post-infusion that includes high TIGIT expression.
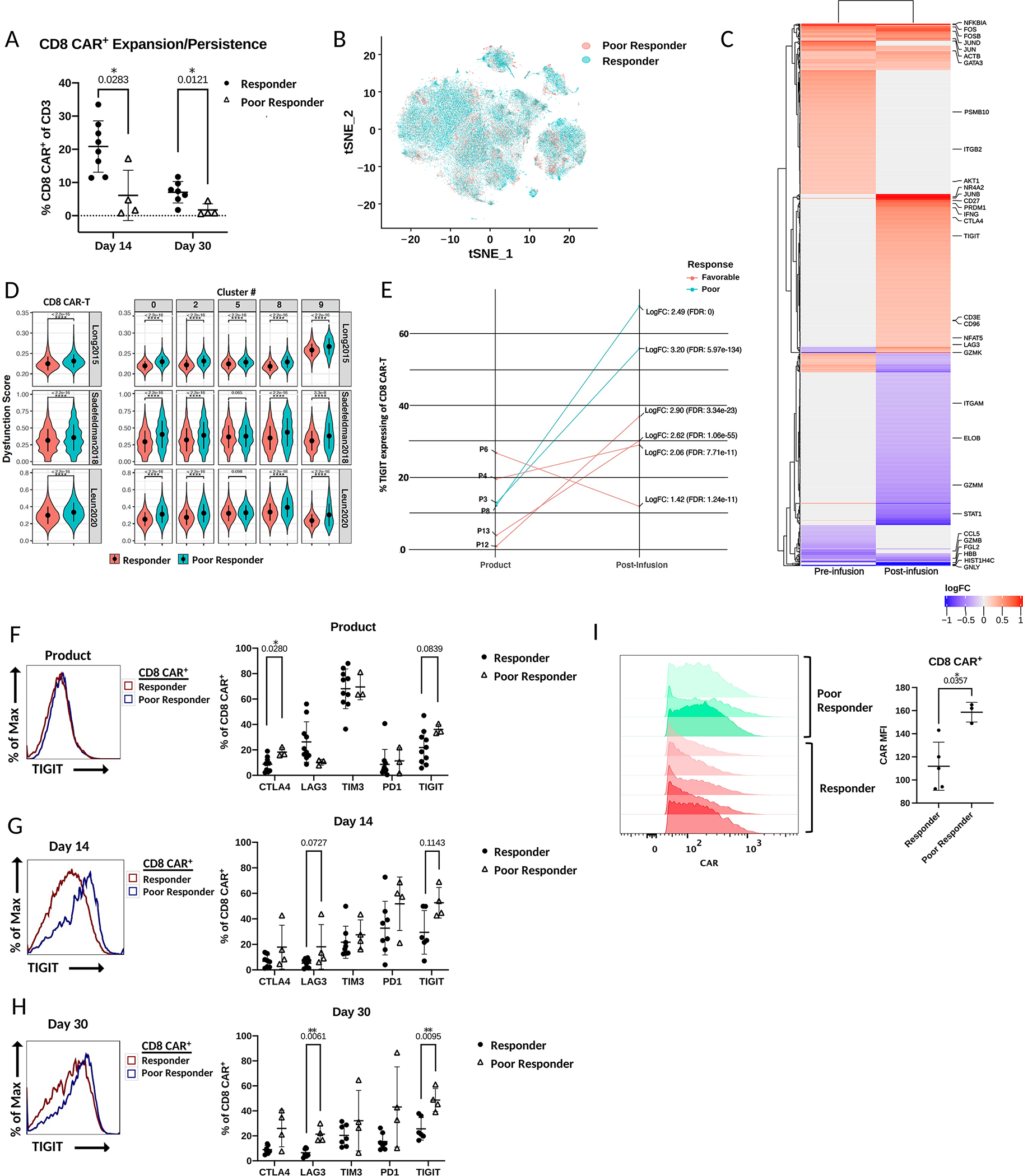
A) Percentage of CD8 CAR-T cells of total T cells between patient response groups at day 14 and day 30 post-infusion as measured by flow cytometry (comparison by Wilcoxon matched-pairs signed rank test). B) scRNA tSNE dimension reduction with patient response group overlay. C) Heatmap of immunoregulatory genes that are differentially expressed (adjusted p-value < 0.05; zlm) between CD8 CAR-T cells of the different response groups before infusion and after infusion. Color represents log fold increase/decrease in poor responders. D) Violin plot comparison of CAR-T cell dysfunction scores between response groups with three exhaustion gene sets by Wilcoxon rank-sum test. Left – comparison of total CD8 CAR-T cells between response groups. Right – comparison of CD8 CAR-T cells between response groups within the most predominant clusters. E) Comparison between individual patients and response groups with zlm by R package MAST of the percentage of TIGIT expressing CD8 CAR-T cells before and after infusion. Average log fold change refers to read counts. F-H) Left – Histograms of fluorescence intensity of TIGIT between response groups as measured by flow cytometry. Each curve represents a concatenation of all samples with equal proportions of CD8 CAR-T cells from each sample. Right – Comparison between patient response groups of the percentage of CD8 CAR-T cells expressing the checkpoint receptors CTLA4, LAG3, TIM3, PD1, or TIGIT by Mann-Whitney tests as measured by flow cytometry. I) Comparison of CAR expression in CD8 CAR-T cells of the product between responders and poor responders. Statistical comparison by Mann-Whitney test.
Consistent with the transcriptional analysis of TIGIT by scRNAseq, measurement of exhaustion marker expression by flow cytometry showed exhaustion markers were consistently expressed at higher levels in post-infusion poor responder CD8 CAR-T cells compared to the cells of responders (Figure 3F–H). Among the exhaustion markers, TIGIT was once again the most prominent, with a greater than 20% increase in the average percentage of TIGIT+ CD8 CAR-T cells in the post-infusion poor responder samples (Figure 3F–H). A similar but non-significant trend was also observed in CD4 CAR-T cells (Supplemental Figure 3D). Interestingly, a comparison of TIGIT expression between endogenous CAR- CD8 T cells of poor and favorable response groups showed a similar trend at day 14 (p = 0.11) and day 30 (p = 0.17) with higher TIGIT expression in poor response groups (Supplemental Figure 3E). Consistent differences in the protein expression of memory markers CD45RA, CCR7, and CD27 between CD8 CAR-T cells of response groups were not apparent (Supplemental Figure 3F). As elevated CAR expression can induce antigen-independent signaling and accelerate CAR-T cell differentiation, exhaustion, and apoptosis, we compared CAR expression in the CD8 CAR-T cells of products across response groups to assess if this alternative mechanism to antigen-dependent signaling could contribute to our findings(29). Indeed, we observed a trend of greater CAR expression in CD8 CAR-T cells of poor responders compared to responders (Figure 3I). Overall, our data support that CD19 CAR-T cells of poor responders in our trial have an enrichment in differentiated and exhausted CD8 CAR-T cells and TIGIT is a novel prognostic marker of response.
TIGIT expression is increased in CAR-T cells with an exhaustion phenotype
TIGIT expression has been associated with a dysfunctional T cell phenotype in chronic infection and cancer but its role in CAR-T cell dysfunction has not been explored(30–32). As our previous flow cytometry analyses revealed TIGIT was the most widely expressed checkpoint receptor in post-infusion CD8 CAR-T cells, particularly in poor responder patients, we assessed if TIGIT can serve as a marker of CAR-T cell dysfunction. To address this question, we separated TIGIT+ and TIGIT- CD8 CAR-T cells in silico and performed differential gene expression. Similar to the profile observed comparing favorable and poor responders, total TIGIT+ cells overexpressed genes associated with exhaustion (Figure 4A). In pre-infusion TIGIT+ cells, upregulated genes included PDCD1 (PD1), LAG3, EOMES, and PRDM1 and downregulated genes included TCF7, SELL, and CCR7 (p.adj < 0.05), corresponding with increased differentiation and decreased stemness. Post-infusion, TIGIT+ CAR-T cells had elevated levels of exhaustion markers including TOX, PD1, and GZMK. Elevated TIGIT expression was not associated with any particular cluster or cell subtype in day 14 and day 30 post-infusion CD8 CAR-T cells (Figure 4B; Supplemental Figure 4A–B). We next applied T cell dysfunction scoring to compare TIGIT+ cells to TIGIT- cells. TIGIT+ cells had higher dysfunction scores globally (p < 0.0001) and in the most predominant clusters across the three gene sets tested (Figure 4C). Furthermore, high dysfunction scores co-localized with TIGIT expression (Figure 4B;C). Accordingly, upon comparison of exhaustion marker protein expression in TIGIT+ versus TIGIT- CD8 CAR-T cells across all three time points, we observed significantly increased expression of CTLA4, LAG3, and PD1, with average fold changes of 1.94, 1.95, and 1.48, respectively (p < 0.05) (Figure 4D–F). The same trend was observed in the CD4 CAR-T cells as well as the endogenous CD8 T cells of patients, with consistent positive fold increases for most exhaustion markers across patients and time points between TIGIT+ and TIGIT- T cells (Supplemental Figures 4C–D). Altogether, this evidence supports TIGIT as a marker of dysfunction in the context of CAR-T cell therapy and NHL.
Figure 4. TIGIT expression is increased in CAR-T cells with an exhaustion phenotype.
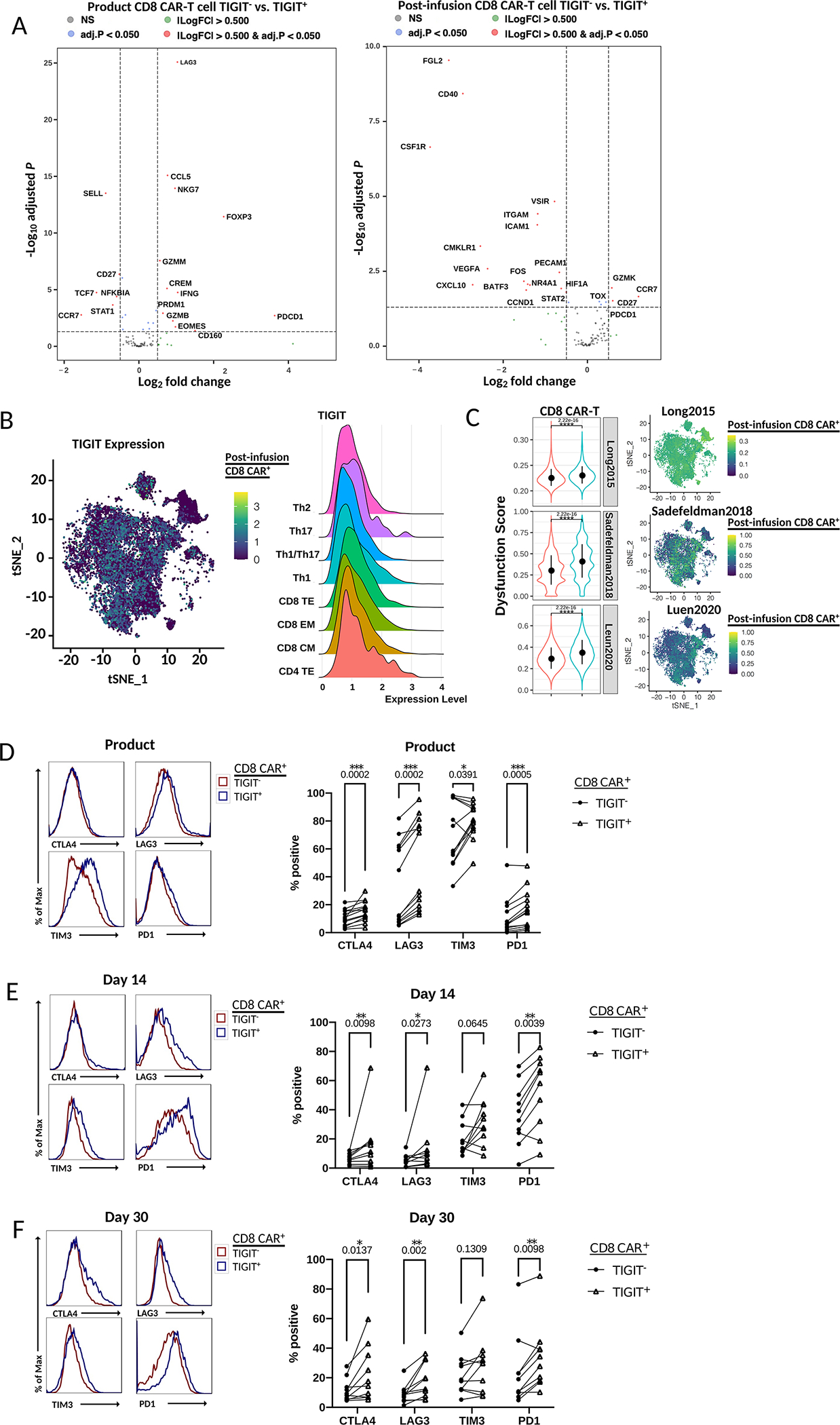
A) Volcano plot of differentially expressed genes (adjusted p-value < 0.05; DESeq2 on two merged cell groups to compare with) between TIGIT+ and TIGIT- CD8 CAR-T cells in pre-infusion or post-infusion samples by scRNA seq. B) Left - scRNA tSNE dimension reduction with TIGIT RNA expression overlay in merged day 14 and day 30 post-infusion CD8 CAR-T cells. Right – Ridge plot of TIGIT RNA expression across post-infusion CAR-T cell subtype assignments. C) Left - Violin plot comparison of dysfunction scores between TIGIT+ and TIGIT- cells of total CD8 CAR- T cells by Wilcoxon rank-sum test. Rows indicate the exhaustion gene set utilized. Right – scRNA tSNE dimension reduction plots with dysfunction scores overlaid on post-infusion CD8 CAR-T cells. Each plot corresponds to the indicated exhaustion gene set. D-F) Left – Histograms of the fluorescence intensity of checkpoint receptor expression comparing TIGIT+ and TIGIT- CD8 CAR-T cells at the indicated time point as measured by flow cytometry. Each curve represents a concatenation of all samples with equal proportions of CD8 CAR-T cells from each sample. Right – Comparison of the percentage of cells expressing checkpoint receptors CTLA4, LAG3, TIM3, or PD1 between TIGIT+ and TIGIT- CD8 CAR-T cells by Wilcoxon matched-pairs signed rank test.
TIGIT blockade improves CAR-T cell anti-tumor function
TIGIT is an immunoregulatory protein thought to inhibit T cells and NK cells by competing with the co-stimulatory receptor DNAM-1 (gene name CD226) in binding to their common ligands PVR and PVRL2(33,34). Studies also suggest that TIGIT can bind to DNAM-1 in cis, disrupting DNAM-1 homodimerization, and it may directly inhibit T cell activation by signaling through its inhibitory ITT and ITIM domains(32,35). In NHL, PVR expression has been reported on tumor cells and endothelial cells(30); other contexts have additionally reported expression on intratumoral myeloid cells(36,37). This pathway is thought to be important in immune evasion, with complex regulation of its components. For example, PVR is often expressed at higher levels in human malignancies while displaying protumorigenic properties, making it an attractive therapeutic target(38). Furthermore, in T cell lymphoma, DNAM-1 surface expression on circulating endogenous CD8 T cells is diminished compared to healthy controls, while serum levels of DNAM-1 are elevated, indicating shedding of its membrane form and the potential role of this pathway as an immune escape mechanism(39). However, in the same context, it was also observed recombinant DNAM-1 could induce cytotoxicity on PVR-expressing tumor cells(39). Together, this highlights the need to determine if TIGIT competition with DNAM-1 for PVR binding is a predominant mechanism of CAR-T cell dysfunction. To test this, we first measured DNAM-1 expression on CAR-T cells from the clinical product and after infusion in patients. In the product, CAR-T cells express high protein levels of DNAM-1 in both CD4 and CD8 subtypes with an average 97.7% and 99.2% DNAM-1+, respectively (Supplemental Figure 5A; Figure 5A). The protein expression of DNAM-1 remains high post-infusion at day 14 and day 30 in CD4 and CD8 CAR-T cells with an average 77.6% and 84.4% of CD4 CAR-T cells and 60% and 80% of CD8 CAR-T cells expressing DNAM-1, respectively. We next determined if TIGIT and DNAM-1 were co-expressed, and observed DNAM-1 expression at day 14 and day 30 post-infusion on the majority of TIGIT+ CD4 (76%; 83%) and CD8 (67%; 80%) CAR-T cells and endogenous CD4 (84%; 86%) and CD8 (68%; 86%) T cells indicating TIGIT competes with DNAM-1 for binding their common ligand PVR (Figure 5B; Supplemental Figure 5B). The percentage co-expression of TIGIT and DNAM-1 is slightly elevated in patient responders due to higher levels of TIGIT expression (Figure 5C; Supplemental Figure 5C).This data indicated TIGIT blockade in combination with CAR-T cell therapy may be a means to improve CAR-T cell therapy.
Figure 5. TIGIT blockade improves CAR-T cell anti-tumor function.
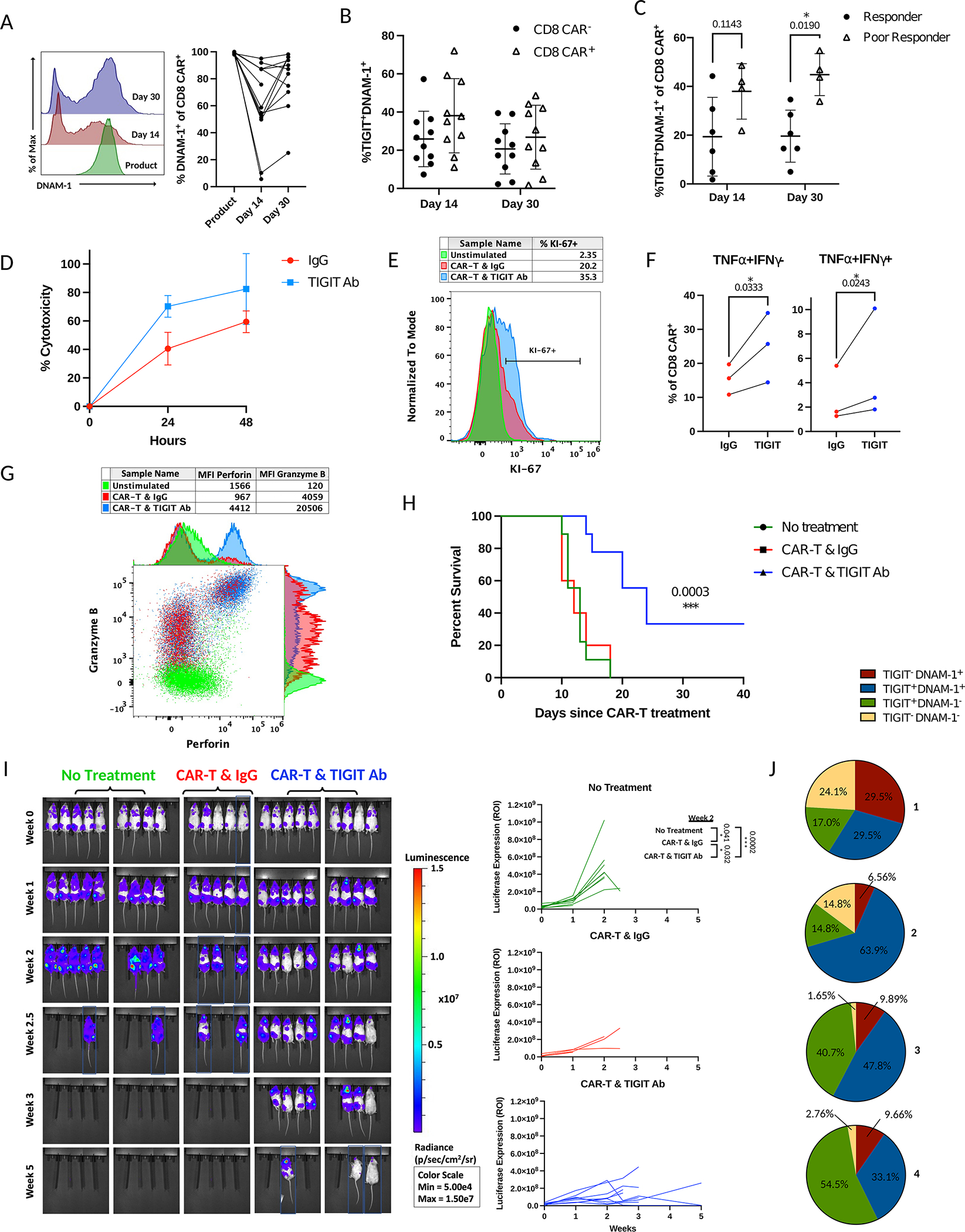
A) Left – Histograms of fluorescence intensity of DNAM-1 in CD8 CAR-T cells of patient across time points as measured by flow cytometry. Each curve represents a concatenation of all samples with equal proportions of CD8 CAR-T cells from each sample. Right – Comparison of the percentage of DNAM-1 expressing CD8 CAR-T cells between time points by Mixed-effects analysis. B) Percent co-expression of TIGIT and DNAM-1 in CD8 CAR-T cells or endogenous CD8 T cells of patients as measured by flow cytometry. C) Comparison of the percentage of TIGIT and DNAM-1 co-expressing CD8 CAR-T cells between responders and poor responders at day 14 and day 30 post-infusion as measured by flow cytometry (comparison by Mann-Whitney test). D-G) CAR-T cell products from patients (n=3) or healthy donors (n=2) were stimulated in vitro with irradiated CD19+ Raji-PVR lymphoma cells every three days at a 4:1 CAR-T cell to Raji cell ratio. D) CAR-T cells were stimulated with Raji-PVR-GFP at day 9 of the exhaustion assay and assessed for Raji cell death after 24hrs or 48hrs of co-culture with TIGIT blockade or antibody control. E) At day 12 of the exhaustion assay, CAR-T cells were stimulated with Raji-PVR for 6 hours and stained for Ki-67. Unstimulated control represents CAR-T cells that were not stimulated with Raji cells for the duration of the exhaustion assay. Histogram depicts percent expression of Ki-67 on CD8 CAR-T cells treated with TIGIT blockade or control antibody. Representative sample shown. F) At day 15 of the exhaustion assay, CAR-T cells of patients were stimulated with Raji-PVR lymphoma cells overnight and treated with monensin 4 hours prior to staining for IFNγ and TNFα for measurement by flow cytometry. Comparison of the percentage of cytokine-expressing CD8 CAR-T cells with TIGIT blockade or antibody control by ratio paired t-test. G) At day 12 of the exhaustion assay, CAR-T cells were stimulated with Raji-PVR and treated with monensin for 6 hours prior to staining with perforin, and granzyme B. A dot plot displaying fluorescence intensity of granzyme B and perforin to compare CAR-T cells treated with TIGIT blockade or antibody control from a representative sample is shown. Unstimulated control represents CAR-T cells that were not stimulated with Raji cells in the exhaustion assay. H-J) NSG mice were inoculated i.v. with 1 million luciferase-expressing CD19+ Raji-PVR lymphoma cells and left untreated (n=9) or treated i.v. three weeks later with 3 million CAR-T cells from a single healthy donor and 10mg/kg TIGIT blocking antibody (n=9) or IgG control antibody (n=5) i.p. weekly for the duration of the experiment. Blue boxes represent cropped images. H) Percent survival of mice after treatment with CAR-T cells and control antibody or TIGIT blockade by Log-rank Mantel-Cox test. I) Comparison of tumor burden by bioluminescence in NSG mice without treatment or treated with CAR-T cells and control antibody or TIGIT blocking antibody. Time represents weeks post-treatment with CAR-T cells. Statistical comparisons made by t-test. J) Pie charts of the percentage of TIGIT and DNAM-1 co-expression patterns on CD8 CAR-T cells from the blood of mice treated with CAR-T cells and control antibody at week 2 after CAR-T cell treatment as measured by flow cytometry (n=4).
To test the effect of TIGIT blockade on CAR-T cell function and recapitulate the CAR-T cell exhaustion we observed in patients, we utilized an in vitro chronic stimulation model. In this model, clinical CAR-T products used to treat NHL patients were stimulated with CD19+ Raji lymphoma cells every three days at a 4:1 ratio of CAR-T cells to Raji cells. Aliquots of cells were analyzed by flow cytometry prior to stimulation and at intervals following co-culture. In this model, CAR-T cells proliferated robustly until day 12 of culture whereupon they stopped proliferating and exhibited a failure to kill the Raji cells after stimulation, indicative of reaching an exhausted state (Supplemental Figure 5D). When comparing the proportions of CD4 and CD8 CAR-T cells, an increase in the CD8/CD4 cell ratio was observed similar to the trend found in patients (Supplemental Figure 5E). To further validate an exhausted profile, we next compared expression of the exhaustion markers CTLA4, LAG3, PD1, TIGIT, TIM3, and VISTA across time points. Akin to the clinical setting, we observed increased expression of the inhibitory receptors on CD4 and CD8 CAR-T cells, with a peak at 37% TIGIT+ for CD4 CAR-T cells and TIGIT expression increasing from 17% pre-stimulation to 54% at day 15 on CD8 CAR-T cells (Supplemental Figures 5F–H).
To evaluate the effect of TIGIT blockade in this in vitro exhaustion model, we first transduced Raji cells with the TIGIT ligand PVR as Raji cells do not express detectable PVR. We then utilized the same model with a TIGIT blocking antibody or IgG control. On day 9 after the CAR-T cells were pulsed with the fourth stimulation of Raji-PVR, we assessed the ability of the CAR-T cells to kill the Raji-PVR cells with or without TIGIT blockade. We observed an average of 30% increase in cytotoxicity with TIGIT blockade as compared to the IgG control at this time point (Figure 5D). After the next stimulation of CAR-T cells with Raij-PVR on day 12, the CAR-T anti-tumor function was further assessed by staining for the cytokines IFNγ and TNFα, cytotoxic molecules granzyme B and perforin, and the proliferation marker Ki-67. TIGIT blockade led to a fold increase in the median fluorescent intensity of TNFα, granzyme B, and perforin of 1.52, 4.56 and 5.05, respectively, and a 15% increase in Ki-67+ proliferating CD8 CAR-T cells as compared to the IgG control (Figure 5E–G). In CD4 CAR-T cells, a 1.42 fold increase in TNFα expression was observed (Supplemental Figure 5I). As with the CAR-T primary patient samples, TIGIT and DNAM-1 co-expression was preserved in this model, suggesting TIGIT competition with DNAM-1 for PVR binding as a possible mechanism for improvement in CAR-T cell antitumor function (Supplemental Figure 5J). Overall, we were able to recapitulate TIGIT induction in CAR-T cells using a chronic stimulation model and demonstrate TIGIT blockade may enhance the anti-tumor function of the CAR-T cells.
We next evaluated the effect of TIGIT blockade in vivo using a mouse model of human NHL. Since CD19 CAR-T cells alone have significant efficacy in traditional mouse NHL models, we first used a high tumor burden model where the CAR-T cells alone have significantly less efficacy to assess if TIGIT antibody could improve overall survival. Mice were injected i.v. with luciferase expressing Raji-PVR lymphoma cells and treatment was initiated after three weeks with CAR-T cells and weekly injections of TIGIT blocking antibody or IgG control. Importantly, the combination of TIGIT blockade and CAR-T cell therapy led to a significant survival advantage over CAR-T cell treatment alone (p = .0003) with 33% of the mice surviving long-term (Figure 5H). Reduced tumor growth was also observed as measured by bioluminescent imaging (Figure 5I). TIGIT antibody blockade as a single agent therapy had no impact on tumor burden or survival in the absence of human CAR-T cells, likely due to the absence of TIGIT expression on Raji cells and the use of a human specific antibody (Supplemental Figures 6A–C). To further evaluate the effect of TIGIT blockade we employed a conventional tumor model with CAR-T cell treatment initiated one week after Raji-PVR cell injection. Once again, TIGIT blockade led to superior control of tumor growth with near-complete elimination of tumor burden, in contrast to mice treated with CAR-T cells and control IgG (Supplemental Figure 6D). Whereas the CAR-T cells of mice without TIGIT blockade showed a dysfunctional profile in the blood, spleen, and bone marrow, mice with TIGIT blockade displayed low inhibitory checkpoint receptor expression suggestive of tumor clearance and memory formation (Supplemental Figure 6E). Similar to our observations from post-infusion CAR-T cells of patient samples and the chronic in vitro exhaustion study, high TIGIT expression was observed in the circulating CAR-T cells in the mice treated with control IgG antibody at two weeks after CAR-T cell injection and was highly co-expressed with DNAM-1 in both in vivo models used (Figure 5J; Supplemental Figure 6F).
Discussion
Due to the variability in outcomes of NHL patients receiving CD19 CAR-T therapy, it is important to better understand the changes CAR-T cells undergo after infusion in the patient and to compare profiles among favorable and poor outcome patients. Here, we compared differences in both transcriptional and phenotypic profiles of purified CAR-T cells between time points and response groups. We show that CAR-T cells isolated from the blood of NHL patients exhibit marked changes after infusion toward transcriptional and surface marker profiles of highly activated and differentiated T cells(18). Both memory marker and gene expression cell identification demonstrate heterogeneity in the memory phenotypes of post-infusion CD8 CAR-T cells, including central memory, effector memory, and terminal effector cell types. Overall, we observed a shift in the predominant profiles of CD8 CAR-T cells from CD45RAHICCR7HICD127HICD62LHICD25HI to CD45ROHICD28HICD69HICD27HIPD1HI and CD45RAHICD57HICD69HIPD1HI upon infusion, supporting a unique phenotype for CAR-T cell differentiation. Overall, we observed CAR-T cells of poor responders had a more differentiated phenotype, suggesting greater antigen exposure consistent with progressive disease and less proliferative capacity.
We also noted the upregulation after infusion of several transcription factors implicated in driving T cell exhaustion (TOX, NR4A2, EOMES, PRDM1) as well as inhibitory checkpoint receptors associated with exhaustion (TIGIT and PD1). While induction of CTLA4, TIM3, and LAG3 RNA expression was observed at the RNA level, protein expression was not apparent by flow cytometry, providing further rationale to pursue investigation of the expressed inhibitory checkpoint receptor TIGIT. Comparison of antigen exposed CAR-T cells with non-CAR expressing T cells from post-infusion samples shows similar evidence of exhaustion in CAR-T cells, with significantly higher levels of expression of TIM3 and TIGIT in CD8 CAR-T cells. Together, this supports the hypothesis that chronic stimulation drives exhaustion in post-infusion CD8 CAR-T cells. Of note, CD27 in particular distinguished CD8 CAR-T cells from non-CAR expressing CD8 T cells, making it an additional marker for identification of 4.1BB CAR-T cells in future applications.
We further demonstrate that CD8 CAR-T cells from poor responding patients with progressive or stable disease are enriched with an exhaustion profile as compared to favorable outcome patients. Among the panel of exhaustion markers tested, TIGIT was found to be the most differentially expressed among response groups. Differential gene expression between post-infusion CD8 CAR-T cells of response groups revealed overexpression of transcription factors including PRDM1, NR4A2, NFKBIA, and AP-1 family members in poor responders, many of which have been implicated in driving T cell exhaustion(40,41). In particular, the RNA expression of AP-1 members FOS, JUN, JUNB, JUND, FOSB was greater in CD8 CAR-T cells of poor responders. While historically AP-1 family member expression has been associated with greater T cell mediated anti-tumor function, with the absence of AP-1 allowing exhaustion driven by NFAT, our results show high AP-1 expression post-infusion, indicating this is not a mechanism of exhaustion in our system. Similar to our findings, Lynn et al. demonstrate that AP-1 family members JUN, JUNB, and FOSB are overexpressed and all AP-1 family member binding motifs are significantly enriched in exhausted compared to functional CD28.CAR-T cells(40). Lynn et al. proceed to evaluate the protein expression of AP-1 family members, and only then are able to hypothesize a relative deficiency of c-Jun/c-Fos heterodimers and identify c-Jun overexpression as a means of providing exhaustion resistance in CAR-T cells. Thus, while the comparisons made here and by Lynn et al. are distinct, we nonetheless observe a similar pattern of RNA expression. Further studies would be required to investigate the role of AP-1 transcription factors in this context.
The differential expression of TIGIT between response groups was particularly notable given the evidence of exhaustion in CD8 CAR-T cells from poor or favorable response groups and suggested that TIGIT may be a more suitable marker for severe exhaustion at least for 4–1BB.CAR-T cells compared to commonly associated molecules such as PD1 or TOX. Therefore, we surveyed the phenotype of TIGIT+ cells to determine its relevance as a biomarker or driver of response. CD8 TIGIT+ CAR-T cells had greater dysfunctional scores compared to TIGIT- cells, upregulated TOX as well as many of the same exhausted-related genes differentially expressed between response groups, and had higher surface expression of all exhaustion markers tested including PD1. Endogenous TIGIT+ CD8 T cells also had increased exhaustion marker expression, suggesting activity of this immunoregulatory pathway in NHL independent of CAR-T cell presence. Mechanistically, TIGIT likely drives CAR-T cell dysfunction by competing with DNAM-1 for binding the ligands PVR and PVRL2 expressed on the endothelium, surrounding immune cells, and tumor tissue(30).
Despite the promising efficacy of CD19 CAR-T therapy for NHL, a significant number of patients fail to achieve remission or exhibit early relapse. As blockade of the checkpoint inhibitor PD1 on T cells has received intense attention in cancer therapy, several groups have assessed the potential for PD1/PDL1 blockade to improve CAR-T efficacy for NHL as well as other malignancies (42–48). While investigations are still ongoing, to date there is not strong evidence that impairing the PD1/PDL1 axis alone leads to significantly improved efficacy of CD19 CAR-T therapy. In fact, in some cases early disruption of PD1 has been found to negatively impact CAR-T cell activity suggesting that its potential benefit may be at later time points as compared to TIGIT abrogation in our model systems(42). More recently, efforts have focused on combination approaches to abrogate multiple checkpoint molecules as individual molecules may have specialized functions(49). For example, it was recently reported that the expression of shRNA’s to simultaneously downregulate TIGIT and PD1 expression can enhance CAR-T activity in a mouse tumor model, however, our findings are the first to demonstrate improvement of CAR-T efficacy in mouse models with TIGIT inhibition alone as well as with a clinically relevant monoclonal blocking antibody(50).
Though the marked upregulation of TIGIT on CD19 CAR-T cells and the efficacy of the combination of CD19 CAR-T cells with a clinically used TIGIT blocking antibody have not been previously reported, TIGIT has been previously explored as an important checkpoint molecule in cancer through both preclinical and clinical studies(32,51). To date the results have been somewhat conflicting, possibly due to differences in the importance of TIGIT based upon the specific context and tumor type. For example, Wen et al. recently reported TIGIT expression in a large pan cancer gene expression study correlates with exhausted T cells(52). However, the correlation of TIGIT expression with patient outcome was highly dependent on tumor type and no difference was observed in this study for DLBCL. Meanwhile, Yang et al. found that there was an increase in TIGIT positive T cells in patients with follicular lymphoma that had poor outcomes(53). In addition, despite the fact that we observe increased efficacy of TIGIT blockade combined with CD19 CAR-T cells in our lymphoma model systems, others have observed that TIGIT primarily suppresses antitumor immunity via T regulatory cells(54). Furthermore, it is possible that similar results will not be found with CD19 based antibody therapies. For example, Roider et al. did not observe any correlation of responses to a bispecific CD3/CD19 antibody with TIGIT expression in preclinical models(55). In addition to our approach to utilize a therapeutic monoclonal TIGIT blocking antibody, Hoogi et al. has engineered T cells to express a chimeric switch receptor that fuses the TIGIT extracellular domain to an activating signal and leads to enhanced T cell efficacy in preclinical models(56). Beyond T cells, TIGIT blockade has also been shown to prevent NK cell exhaustion and improve anti-tumor function(57).
There are several noteworthy considerations to review in the current study. First, a limited number of poor responder samples were available at the time of analysis. To offset this limitation, a large number of cells (94,000) were profiled by single cell RNA sequencing and combined with protein measurements by feature barcoding. Furthermore, additional poor responder samples were incorporated in a flow cytometric validation dataset. Likewise, pseudobulk comparisons, flow cytometry, and functional analyses were used to offset the limitations that exist with single cell RNA sequencing analyses, such as a tendency for false discoveries(58). Another consideration is a focus on CAR-T cells derived from the blood in contrast to tumor tissue. While it is likely tumor-derived samples could offer great insight into relevant transcriptional profiles and phenotypes of poor responder samples, this would prohibit comparison to post-infusion favorable response samples and it is preferable to seek relevant biomarkers from minimally invasive samples. Additionally, while we have demonstrated TIGIT to be a biomarker of response with potential functional relevance, future research will need to be conducted to assess the loss of CAR-T cell functionality in patients over time and between response groups as well as the clinical potential of this approach for CAR-T therapy. While we demonstrate TIGIT is expressed at a high level after infusion on FDA-approved products including CD28.CAR-T cells (Kymriah; Tescartus), more studies are also needed to determine the functional effect of TIGIT blockade with designs and manufacturing methods beside our own. It would further be beneficial to include epigenetic studies of CAR-T cells used in the treatment of NHL to strengthen the existence of exhaustion in CAR-T cells and relate it to T cell exhaustion without CAR expression.
Methods
Description of Cohorts
For both the investigational and validation cohorts, all patients failed at least 2 previous lines of therapy and were enrolled to the study in accordance with eligibility criteria described in clinicaltrials.gov (Identifier: NCT03434769; IND 17932). Leukapheresis products for generation of CAR-T cell products were obtained from NHL patients receiving CAR-T cell therapy at University Hospitals Seidman Cancer Center under a phase I/II study and utilized within 24 hours of draw. All blood samples were processed via density gradient centrifugation and cryopreserved in the University Hospitals Biorepository and Cellular Therapy facility. The study was approved by the institutional review board and all patients gave written informed consent. Available clinical information for the cohorts are available in Supplemental Table 2. The analysis of the validation cohort is limited to Supplemental Figure 2I–J.
CAR-T cell manufacture
CAR T cell manufacture was automated with the use of the CliniMACS Prodigy® device using the TCT software program and TS520 tubing set (Miltenyi Biotec, Bergisch Gladbach, Germany). The instrument setup and technical protocol were described by Zhu et al.(59). The clinical-grade reagents applied in this process were CliniMACS Buffer, TexMACS Media, CliniMACS CD4 reagent, CliniMACS CD8 reagent, TransAct, and the cytokines IL-7 and IL-15 (Miltenyi Biotec, Bergisch Gladbach, Germany). Peripheral blood apheresis products were loaded into the machine and CD4 and CD8 T cells were isolated using CliniMACS CD4 reagent and CliniMACS CD8 reagent according to the manufacturer’s instructions. The isolated T cells were then stimulated with IL-7 and IL-15 (Miltenyi Biotec, Bergisch Gladbach, Germany) at a concentration of 25μg/2L bag of TexMACS media with 3% human AB serum (Innovative Research, Novi, MI, USA). Human AB serum was removed after the 6th day of culture. The viability, purity, and potency of the products were confirmed as previously described(23). This process was performed at the Cellular Therapy Lab of University Hospitals Cleveland Medical Center Seidman Cancer Center/Case Western Reserve University Center for Regenerative Medicine.
Lentiviral vector
The 4–1BB.CAR construct applied in the clinical trial was developed by Lentigen, a Miltenyi Biotec company (Gaithersburg, MD, USA). The vector is composed of the FMC63 scFv, a CD8-derived hinge region, TNFRSF19-derived transmembrane domain, CD3ζ intracellular domain, and 4–1BB co-stimulatory domain.
Single cell RNA sequencing and feature barcoding library preparation
Cryopreserved apheresis products were thawed and pre-labelled with antibodies for flow cytometry (described below) and a panel of TotalSeq™-B antibodies (described below) from Biolegend (San Diego, CA, USA). The panel of TotalSeq™-B antibodies included CD127, CD197, CD25, CD279, CD28, CD4, CD45RA, CD45RO, CD57, CD62L, CD69, and CD8. Information for the utilized TotalSeq ™-B antibodies are located in Supplemental Table 4. Live CD3+CAR+ T cells were sorted by FACS and preparation of single cell and TotalSeq™-B libraries was performed utilizing the 10x Genomics Chromium Single Cell 3’ Reagent kits with feature barcoding technology for cell surface protein (v3) (Pleasanton, CA, USA) according to the manufacturer’s instructions. Libraries were sequenced by Psomagen, Inc (Rockville, MD, USA).
Quality control of raw 10x scRNA sequencing data
A total of 27 CAR-T samples were sequenced. For each sequenced scRNA-Seq pool, Cell Ranger (v3.1.0) from 10x Genomics (Pleasanton, CA, USA) was used to process, align, and summarize unique molecular identifier (UMI) counts against hg38 human reference genome. For 16 of the 27 CAR-T samples, 12 T-cell surface proteins (CD28, CD57, CD69, CD62L, CD197, CD25, CD279, CD45RA, CD127, CD4, CD45RO, and CD8A) are barcoded to each cell by TotalSeq™-B antibody library (Biolegend, San Diego, CA, USA). Based on each sample mRNA assay UMI metrics, cells with too low or high UMI counts or the number of genes (i.e., 2.5 × standard deviation) were filtered out. Cells with a mitochondrial UMI count proportion higher than 15% were removed. Cells with a ratio of number of genes covered to UMI counts less than 0.1 were also filtered. Doublets, as annotated by Scrublet v.0.2.1(60), were also removed. After the comprehensive quality control procedure, we only retained 24 CAR-T samples since for 3 samples (patient 5 day 14, patient 5 day 30, patient 12 day 30) either the number of read counts or the number of genes was too small compared to the other samples. See post-quality control metrics in Supplemental Table 3.
In silico cell type prediction
CAR-T cell subtypes were assigned by SingleR(21) (v1.0.6). Gene expression profile was compared with the RNA-seq transcriptome profile of 29 immune cell types(22). The best-predicted cell type was considered. CAR-T cells were grouped into CD4 and CD8 T cells. CD4+ T cells were annotated as T follicular helper cells (Tfh), regulatory T cells (Tregs), Th1, Th1/Th17, Th17, Th2, naive, or terminal effector. CD8+ T cells were annotated as naive, central memory, effector memory, or terminal effector.
10X Genomics scRNA sequencing data analysis and adjusting batch effects
The 24 CAR-T samples available post-quality control (see above) from 13 patients include a total of 94K high-quality cells with an average of 3,917 cells per sample originally sequenced from 3 batch sequencing runs. The cells were obtained from the QC step described above (Method: Quality control of raw 10X scRNA sequencing data). Individual patient samples were merged together. Seurat R package(61) (v3.2.3) was used to log-normalize expression values for total UMI counts per cell via Seurat::NormalizeData (normalization.method=”LogNormalize”, scale.factor=10000). Then, two thousand highly variable genes were identified by fitting the mean-variance relationship via Seurat::FindVariableFeatures(selection.method=”vst”). Cell cycle was inferred by a function, Seurat::CellCycleScoring, from the Seurat package using 43 genes for the S state and 54 genes for the G2M state, respectively. For batch effect removal, low dimension visualization, and clustering procedure, we first regress out the two continuous variables, 1) mitochondrial gene expression and 2) cell cycle annotation via Seurat::ScaleData(). The merged samples batch effect signal was corrected using the Harmony (v1.0) algorithm(62), with two categorical variables via RunHarmony(group.by.vars=c(“samples”,”seq.run”)). The aligned cells were then clustered using the Louvain algorithm for modularity optimization using the kNN (k nearest neighbors) graph as input with the first 50 PCs (Principal Components) and Seurat::FindClusters(resolution = 0.8). Cell clusters were visualized using the tSNE algorithm(63) with a dimension reduction input from Harmony.
Pseudobulk mRNA analysis
Given the read count matrix after the rigorous QC procedure (See Method: Quality control of raw 10x scRNA sequencing data), we generated a pseudobulk mRNA read count matrix. In Figure 2B and Figure 3C, we summed the read counts across cells, computed the total gene counts in an individual sample. In Figure 4A–B, we summed the read count by whether TIGIT is expressed or not in each sample and generated two pseudobulk mRNA count matrices. Then, DESeq2 (with all default parameters) was applied to these pseudobulk mRNA datasets in a couple of comparisons (e.g., pre-infusion vs. post-infusion CAR T, poor vs. favorable response, or TIGIT- vs. TIGIT+). In a comparison of TIGIT expression in post-infusion with that of pre-infusion in the same patient in pseudobulk model (Figure 3E), we randomly sample cell bar codes to generate three replicates within the same sample, which is suggested in DESeq2 analysis. Statistical significance is indicated by one or more asterisks, and the number of asterisks shown correspond to p-values less than 0.05, 0.01, 0.001, or 0.0001, respectively.
Marker gene detection and differential expression analysis
For each identified cluster, we compared the cells within the clusters versus all other cells using R packages Seurat and MAST(64) (v1.16.0) for statistical testing to identify all marker genes expressed distinctly compared to the other clusters. Only differentially-expressed genes of significance less than 5% FDR were retained. Without loss of the generality, the same DEG testing is applied between the CAR-T product and post-infusion CAR-T cells, or between the cells belonging to the patients with a favorable or poor outcome within either the same cluster or T cell subtype. The gene sets used for each panel are indicated in Supplemental Table 5. Statistical significance is indicated by one or more asterisks, and the number of asterisks shown correspond to p-values less than 0.05, 0.01, 0.001, or 0.0001, respectively.
Gene set variation analysis
We used GSVA (v1.36.3)(65) to compute gene set variance scores. To prepare the scRNAseq data for GSVA, we randomly sampled cells from each time point into 3 replicates. Using the pseudobulk data we then calculated gene set variance scores for each replicate. To assess differences in gene sets across time points we used limma (v3.44.3)(66–68) with the features from GSVA as input to lmFit with the default parameters. We then computed empirical Bayes statistics using eBayes prior to generating the top table. Volcano plots were produced with the EnhancedVolcano (v1.6.0) package.
Immune regulated genes
In this study, we focused on a custom-made list of 106 genes for discussion and visualization. The genes are either directly or indirectly known to be associated with human immunology and are listed in Supplemental Table 6. The gene sets used for each panel are indicated in Supplemental Table 5.
CD8+ T cell dysfunctional score
CD8+ T cell dysfunctional scores were calculated at each cell. We computed the AUC score of a gene set showing a CD8+ T cell dysfunction phenotype using AUCell_calcAUC from R package AUCell(69) (v1.8.0). Three signature gene sets are used for the prediction. The first gene set from Sade-Feldman et al. includes LAG3, PDCD1, HAVCR2, TIGIT, CD38, and ENTPD1(27). The second gene set from van der Luen et al. includes a total of 22 genes: LAYN, ITGAE, PDCD1, CTLA4, HAVCR2, LAG3, TIGIT, CXCL13, CD38, ENTPD1, CDK1, HSPH1, CCCNB1, HSPB1, MKI67, DK4, GZMB, TOX, IFNG, MIR155HG, TNFRSF9, and RB1(28). The third signature gene set from Long et al. was sorted by the log fold change between the CD19–28 CAR vs GD2–28 CAR and the top 2,000 positive log fold change genes were selected for AUC calculation(1). Statistical significance is indicated by one or more asterisks, and the number of asterisks shown correspond to p-values less than 0.05, 0.01, 0.001, or 0.0001, respectively.
Flow cytometry
Extracellular staining for flow cytometry was performed by incubating titrated amounts of fluorescent-labelled antibodies or viability dye for 15 minutes at room temperature. Secondary staining for biotin-streptavidin conjugates was performed with a 30-minute incubation at room temperature. Acquisition was performed with a BD ARIA flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) or Attune NxT flow cytometer (Invitrogen, Waltham, MA, USA). For flow cytometric analysis of granzyme B and perforin production, cells were stimulated with Raji-PVR in the presence of GolgiStop (BD Biosciences, Franklin Lakes, NJ, USA) for 6 hours prior to staining. Intracellular staining was performed following extracellular staining with eBioscience Foxp3/Transcription Buffer Staining Set (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. See Supplemental Table 4 for a complete list of flow cytometric reagents.
Flow cytometric analyses
Flow cytometric gating was based on fluorescence minus one controls in cases where bimodal distribution was not apparent. To generate histogram comparisons of fluorescence intensity across samples run with the same cytometer settings on different days, we first performed batch correction with the function SwiftReg(70) on MATLAB_R2020b (The MathWorks, Inc., Natick, MA, USA). Comparisons were then performed with concatenations of all samples containing equal proportions of the cell type of interest. For comparison of two groups without matching, a Mann-Whitney test was performed. Comparisons of two groups with matching samples were performed with Wilcoxon matched-pairs signed rank tests. For comparisons of three or more groups, a Friedman test was performed. For paired comparisons across three or more groups, a mixed-effects analysis was performed. For all plots derived from flow cytometric data, no correction was made for multiple comparisons and all comparisons made in the statistical analysis are displayed. All statistical analyses were done using Prism 9.0.2 (GraphPad Software, San Diego, CA, USA). Statistical significance is indicated by one or more asterisks, and the number of asterisks shown correspond to p-values less than 0.05, 0.01, 0.001, or 0.0001, respectively.
Cell Lines
Raji cells and 293T cells were obtained from ATCC (Manassas, VA, USA) and were authenticated using short tandem repeat profiling. The cell lines were tested regularly for Mycoplasma using a Mycoplasma Detection Kit-QuickTest (Bimake, Houston, TX, USA). Cell lines were passaged less than 6 months and used for studies within one month of thawing.
Lentiviral Transduction
293T cells (ATCC, Manassas, VA, USA) were transfected using Lipofectamine 3000 (ThermoFisher, Waltham, MA, USA) with a luciferase construct, human PVR lentiviral construct, or green fluorescence protein construct obtained from VectorBuilder (Chicago, IL, USA) to generate lentiviral vector and Raji cells (ATCC, Manassas, VA, USA) were transduced. Luciferase-expressing cells were selected with puromycin for two weeks. PVR-expressing cells were selected with blasticidin (10μg/mL) for two weeks. GFP-expressing cells were FACS sorted.
Exhaustion assay
The media utilized here contained 10% fetal calf serum, 1% penicillin/streptomycin, and 0.1% ciprofloxacin in RPMI 1640 with 2.05mM l-glutamine (cRPMI). CAR-T cells were thawed and rested overnight in 5ng/mL IL-15 and 10ng/mL IL-7 in cRPMI. The next day, CAR products were either FACS sorted with anti-FMC63-FITC to obtain a pure CAR+ population or immediately added to culture with Raji cell line at a 4:1 CAR-T cell to Raji cell ratio in 30U/mL IL-2. Every three days, CAR-T cells were counted and the effector to target ratio restored with additional Raji cells. Aliquots were taken at the indicated days for flow cytometric analysis.
Animal Studies
Male NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice approximately 8 weeks old (NSG, The Jackson Laboratory, Bar Harbor, NE, USA) were injected i.v. with 1 million luciferase-expressing Raji-PVR cells. In the mice used to generate overall survival, 3 million CAR-T cells were injected i.v. 3 weeks following tumor inoculation. Otherwise, 5 million CAR-T cells were injected i.v. 1 week following tumor inoculation. Disease progression was monitored weekly by bioluminescence imaging using the IVIS Spectrum Imager (PerkinElmer, Waltham, MA, USA) and analyzed with Living Image (PerkinElmer, Waltham, MA, USA). Following CAR-T cell injection, mice received weekly 10mg/kg i.p. injections of TIGIT antibody (BMS-986207, Bristol Myers Squibb, New York, NY, USA) or antibody control (Bristol Myers Squibb, New York, NY, USA) for the duration of the study. Mouse kidney, spleen, and lymph nodes were isolated and homogenized for flow cytometric analysis. Blood was isolated and red blood cells were lysed prior to flow cytometric analysis. Overall survival statistical significance was calculated with a Log-rank Mantel-Cox test. Investigators were not blinded for randomization or treatment. Power analysis has been done according to publications and prior experience. Animal studies were performed under an approved institutional animal care and use committee at Case Western Reserve University.
Data Availability Statement
The source R code and required Seurat objects are available at https://github.com/hwanglab/hwanglab_2021_tigitCarT or https://codeocean.com/capsule/0626385/tree/v1. The raw unfiltered FASTQ files are deposited at EGA and available at https://ega-archive.org/studies/EGAS00001005356.
Supplementary Material
Statement of Significance.
This is the first study investigating the mechanisms linked to CAR-T patient responses based on the sequential analysis of manufactured and infused CAR-T cells using single cell RNA and protein expression data. Furthermore, our findings are the first to demonstrate improvement of CAR-T cell efficacy with TIGIT inhibition alone.
Acknowledgements
This research was supported by the Case Comprehensive Cancer Center (P30CA043703) including the Hematopoietic Biorepository and Cellular Therapy Shared Resource and the Cytometry and Microscopy Shared Resource with support from the NIH shared instrumentation grant (S10-NIH OD021559). This research was also supported by the NIH grant T32AI089474.
Footnotes
Conflict-of-Interest Disclosure
The authors declare no potential conflicts of interest.
References
- 1.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015;21(6):581–90 doi 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ying Z, He T, Wang X, Zheng W, Lin N, Tu M, et al. Parallel Comparison of 4–1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol Ther Oncolytics 2019;15:60–8 doi 10.1016/j.omto.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology 2019;8(5):e1049 doi 10.1002/cti2.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ansell SM. Non-Hodgkin Lymphoma: Diagnosis and Treatment. Mayo Clin Proc 2015;90(8):1152–63 doi 10.1016/j.mayocp.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 5.Gribben JG, O’Brien S. Update on therapy of chronic lymphocytic leukemia. J Clin Oncol 2011;29(5):544–50 doi 10.1200/JCO.2010.32.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cortelazzo S, Ponzoni M, Ferreri AJ, Dreyling M. Mantle cell lymphoma. Crit Rev Oncol Hematol 2012;82(1):78–101 doi 10.1016/j.critrevonc.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 2017;130(16):1800–8 doi 10.1182/blood-2017-03-769620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017;377(26):2531–44 doi 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kersten MJ, Spanjaart AM, Thieblemont C. CD19-directed CAR T-cell therapy in B-cell NHL. Curr Opin Oncol 2020;32(5):408–17 doi 10.1097/CCO.0000000000000668. [DOI] [PubMed] [Google Scholar]
- 10.Abramson JS, Lunning M, Palomba ML. Chimeric Antigen Receptor T-Cell Therapies for Aggressive B-Cell Lymphomas: Current and Future State of the Art. Am Soc Clin Oncol Educ Book 2019;39:446–53 doi 10.1200/EDBK_238693. [DOI] [PubMed] [Google Scholar]
- 11.Hunter BD, Rogalski M, Jacobson CA. Chimeric antigen receptor T-cell therapy for the treatment of aggressive B-cell non-Hodgkin lymphomas: efficacy, toxicity, and comparative chimeric antigen receptor products. Expert Opin Biol Ther 2019;19(11):1157–64 doi 10.1080/14712598.2019.1644316. [DOI] [PubMed] [Google Scholar]
- 12.Jacoby E, Shahani SA, Shah NN. Updates on CAR T-cell therapy in B-cell malignancies. Immunol Rev 2019;290(1):39–59 doi 10.1111/imr.12774. [DOI] [PubMed] [Google Scholar]
- 13.Chavez JC, Bachmeier C, Kharfan-Dabaja MA. CAR T-cell therapy for B-cell lymphomas: clinical trial results of available products. Ther Adv Hematol 2019;10:2040620719841581 doi 10.1177/2040620719841581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chong EA, Ruella M, Schuster SJ, Lymphoma Program Investigators at the University of P. Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N Engl J Med 2021;384(7):673–4 doi 10.1056/NEJMc2030164. [DOI] [PubMed] [Google Scholar]
- 15.Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol 2019;16(6):372–85 doi 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med 2020;26(12):1878–87 doi 10.1038/s41591-020-1061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018;24(5):563–71 doi 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheih A, Voillet V, Hanafi LA, DeBerg HA, Yajima M, Hawkins R, et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat Commun 2020;11(1):219 doi 10.1038/s41467-019-13880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 2017;14(9):865–8 doi 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee MS, Hanspers K, Barker CS, Korn AP, McCune JM. Gene expression profiles during human CD4+ T cell differentiation. Int Immunol 2004;16(8):1109–24 doi 10.1093/intimm/dxh112. [DOI] [PubMed] [Google Scholar]
- 21.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 2019;20(2):163–72 doi 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carre C, et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep 2019;26(6):1627–40 e7 doi 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson Z, Roe A, Sharma AA, Lopes F, Talla A, Kleinsorge-Block S, et al. Automated Manufacture of Autologous CD19 CAR-T Cells for Treatment of Non-hodgkin Lymphoma. Front Immunol 2020;11:1941 doi 10.3389/fimmu.2020.01941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin MD, Badovinac VP. Defining Memory CD8 T Cell. Front Immunol 2018;9:2692 doi 10.3389/fimmu.2018.02692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011;134(1):17–32 doi 10.1111/j.1365-2567.2011.03470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol 2000;1(5):433–40 doi 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- 27.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2019;176(1–2):404 doi 10.1016/j.cell.2018.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer 2020;20(4):218–32 doi 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, et al. Tonic 4–1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep 2017;21(1):17–26 doi 10.1016/j.celrep.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Josefsson SE, Beiske K, Blaker YN, Forsund MS, Holte H, Ostenstad B, et al. TIGIT and PD-1 Mark Intratumoral T Cells with Reduced Effector Function in B-cell Non-Hodgkin Lymphoma. Cancer Immunol Res 2019;7(3):355–62 doi 10.1158/2326-6066.CIR-18-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS, et al. TIGIT Marks Exhausted T Cells, Correlates with Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PLoS Pathog 2016;12(1):e1005349 doi 10.1371/journal.ppat.1005349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014;26(6):923–37 doi 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 33.Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol 2011;41(4):902–15 doi 10.1002/eji.201041136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol 2012;188(8):3869–75 doi 10.4049/jimmunol.1103627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol 2011;186(3):1338–42 doi 10.4049/jimmunol.1003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao J, Zheng Q, Xin N, Wang W, Zhao C. CD155, an onco-immunologic molecule in human tumors. Cancer Sci 2017;108(10):1934–8 doi 10.1111/cas.13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med 2016;213(2):167–76 doi 10.1084/jem.20150785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kucan Brlic P, Lenac Rovis T, Cinamon G, Tsukerman P, Mandelboim O, Jonjic S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell Mol Immunol 2019;16(1):40–52 doi 10.1038/s41423-018-0168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi N, Sugaya M, Suga H, Oka T, Kawaguchi M, Miyagaki T, et al. Increased Soluble CD226 in Sera of Patients with Cutaneous T-Cell Lymphoma Mediates Cytotoxic Activity against Tumor Cells via CD155. J Invest Dermatol 2017;137(8):1766–73 doi 10.1016/j.jid.2017.03.025. [DOI] [PubMed] [Google Scholar]
- 40.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019;576(7786):293–300 doi 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A 2019;116(25):12410–5 doi 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalinin RS, Ukrainskaya VM, Chumakov SP, Moysenovich AM, Tereshchuk VM, Volkov DV, et al. Engineered Removal of PD-1 From the Surface of CD19 CAR-T Cells Results in Increased Activation and Diminished Survival. Front Mol Biosci 2021;8:745286 doi 10.3389/fmolb.2021.745286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 2017;129(8):1039–41 doi 10.1182/blood-2016-09-738245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther 2017;25(9):2214–24 doi 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 2016;126(8):3130–44 doi 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacobson CA, Westin JR, Miklos DB, Herrera AF, Lee J, Seng J, et al. Abstract CT055: Phase 1/2 primary analysis of ZUMA-6: Axicabtagene ciloleucel (Axi-Cel) in combination With atezolizumab (Atezo) for the treatment of patients (Pts) with refractory diffuse large B cell lymphoma (DLBCL). Cancer Research 2020;80(16_Supplement):CT055–CT doi 10.1158/1538-7445.Am2020-ct055. [DOI] [Google Scholar]
- 47.Jacobson CA, Locke FL, Miklos DB, Herrera AF, Westin JR, Lee J, et al. End of Phase 1 Results from Zuma-6: Axicabtagene Ciloleucel (Axi-Cel) in Combination with Atezolizumab for the Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma. Blood 2018;132(Supplement 1):4192– doi 10.1182/blood-2018-99-111523. [DOI] [Google Scholar]
- 48.Chong EA, Melenhorst JJ, Svoboda J, Dwivedy Nasta S, Landsburg DJ, Mato AR, et al. Phase I/II Study of Pembrolizumab for Progressive Diffuse Large B Cell Lymphoma after Anti-CD19 Directed Chimeric Antigen Receptor Modified T Cell Therapy. Blood 2017;130(Supplement 1):4121– doi 10.1182/blood.V130.Suppl_1.4121.4121. [DOI] [Google Scholar]
- 49.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016;44(5):989–1004 doi 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee YH, Lee HJ, Kim HC, Lee Y, Nam SK, Hupperetz C, et al. PD-1 and TIGIT downregulation distinctly affect the effector and early memory phenotypes of CD19-targeting CAR T cells. Mol Ther 2022;30(2):579–92 doi 10.1016/j.ymthe.2021.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT, the Next Step Towards Successful Combination Immune Checkpoint Therapy in Cancer. Front Immunol 2021;12:699895 doi 10.3389/fimmu.2021.699895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen J, Mao X, Cheng Q, Liu Z, Liu F. A pan-cancer analysis revealing the role of TIGIT in tumor microenvironment. Sci Rep 2021;11(1):22502 doi 10.1038/s41598-021-01933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang ZZ, Kim HJ, Wu H, Jalali S, Tang X, Krull JE, et al. TIGIT Expression Is Associated with T-cell Suppression and Exhaustion and Predicts Clinical Outcome and Anti-PD-1 Response in Follicular Lymphoma. Clin Cancer Res 2020;26(19):5217–31 doi 10.1158/1078-0432.CCR-20-0558. [DOI] [PubMed] [Google Scholar]
- 54.Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest 2015;125(11):4053–62 doi 10.1172/JCI81187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Roider T, Brinkmann BJ, Kim V, Knoll M, Kolb C, Roessner PM, et al. An autologous culture model of nodal B-cell lymphoma identifies ex vivo determinants of response to bispecific antibodies. Blood Adv 2021;5(23):5060–71 doi 10.1182/bloodadvances.2021005400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoogi S, Eisenberg V, Mayer S, Shamul A, Barliya T, Cohen CJ. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J Immunother Cancer 2019;7(1):243 doi 10.1186/s40425-019-0721-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol 2018;19(7):723–32 doi 10.1038/s41590-018-0132-0. [DOI] [PubMed] [Google Scholar]
- 58.Squair JW, Gautier M, Kathe C, Anderson MA, James ND, Hutson TH, et al. Confronting false discoveries in single-cell differential expression. Nat Commun 2021;12(1):5692 doi 10.1038/s41467-021-25960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu F, Shah N, Xu H, Schneider D, Orentas R, Dropulic B, et al. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy 2018;20(3):394–406 doi 10.1016/j.jcyt.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 60.Wolock SL, Lopez R, Klein AM. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 2019;8(4):281–-91 e9. doi 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36(5):411–20 doi 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 2019;16(12):1289–96 doi 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van der Maaten LJPGEH. Visualizing High-Dimensional Data Using t-SNE. Journal of Machine Learning Research 2008;9:2579–605. [Google Scholar]
- 64.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 2015;16:278 doi 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43(7):e47 doi 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK. Robust Hyperparameter Estimation Protects against Hypervariable Genes and Improves Power to Detect Differential Expression. Ann Appl Stat 2016;10(2):946–63 doi 10.1214/16-AOAS920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15(2):R29 doi 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017;14(11):1083–6 doi 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rebhahn JA, Quataert SA, Sharma G, Mosmann TR. SwiftReg cluster registration automatically reduces flow cytometry data variability including batch effects. Commun Biol 2020;3(1):218 doi 10.1038/s42003-020-0938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source R code and required Seurat objects are available at https://github.com/hwanglab/hwanglab_2021_tigitCarT or https://codeocean.com/capsule/0626385/tree/v1. The raw unfiltered FASTQ files are deposited at EGA and available at https://ega-archive.org/studies/EGAS00001005356.