

Abstract
Cervical cancer continues to be a significant cause of cancer-related deaths in women. The most common treatment for cervical cancer involves the use of the drug cisplatin in conjunction with other therapeutics. However, the development of cisplatin resistance in patients can hinder the efficacy of these treatments, so alternatives are needed. In this study, we found that PARP inhibitors (PARPi) can attenuate the growth of cells representing cervical adenocarcinoma and cervical squamous cell carcinoma. Moreover, a combination of PARPi with cisplatin increased cisplatin-mediated cytotoxicity in cervical cancer cells. This was accompanied by a dramatic alteration of the transcriptome. The FOS gene, which encodes the transcription factor Fos, was one of the most highly up-regulated genes in the dual treatment condition, leading to increased Fos protein levels, greater Fos binding to chromatin, and the subsequent induction of Fos target genes. Increased expression of Fos was sufficient to hinder cervical cancer growth, as shown by ectopic expression of Fos in cervical cancer cells. Conversely, Fos knockdown enhanced cell growth. Collectively, these results indicate that by inducing FOS expression, PARPi treatment in combination with cisplatin leads to inhibition of cervical cancer proliferation, likely through a Fos-specific gene expression program.
Introduction
Cervical cancer continues to be the second leading cause of death in women aged 20–39 despite progress made with vaccination against human papilloma virus (HPV), increased screening, and early diagnosis (1,2). These statistics highlight the need for better therapeutic interventions for cervical cancers. Currently, the standard treatment for early-stage cervical cancer involves surgery, while patients with locally-advanced disease are typically treated with a combination of chemotherapy and radiotherapy (3). Metastatic cervical cancer has traditionally been treated with chemotherapeutics, although recent advances have led to the use of more targeted therapies such as bevacizumab and pembrolizumab (4). Overall, most regimes that involve the use if chemotherapeutics use the drug cisplatin (CDDP) in combination with other drugs, such as paclitaxel (2,5). However, development of chemoresistance is a major hindrance in the continued use of cisplatin (5) and underscores the need to explore alternative drugs with increased efficacy for combinatorial treatment with cisplatin.
Recently, small molecule inhibitors of nuclear PARPs, primarily PARP-1, have been approved by the U.S. FDA for use as cancer therapeutics, particularly in gynecological cancers, such as ovarian cancer (6). Four PARP inhibitors (PARPi) have been approved by the FDA for the treatment of ovarian and breast cancers with BRCA1 or BRCA2 mutations (7,8), but the therapeutic potential of PARP inhibitors is likely to extend to other cancer types as well (7,9). PARP-1 is a member of the PARP family of proteins that catalyze ADP-ribosylation (ADPRylation) - the transfer of ADP-ribose from NAD+ to target proteins (10). PARP-1 promotes the synthesis of chains of ADP-ribose called poly(ADP-ribose) or PAR. PARPi are thought to act by inhibiting PARP-1 activity, ultimately inducing synthetic lethality with BRCA1 or BRCA2 mutations in cancers that are deficient in homologous recombination (HR)-mediated DNA repair (11,12). Recent studies, however, have reported that PARPi may have significant clinical benefits in patients in the absence of BRCA1/2 mutations or HR deficiency (HRD) (7,13–16).
Indeed, a growing body of evidence points towards a critical role for PARP-1 beyond DNA-damage repair, particularly in regulating gene transcription and RNA biology (17,18). In this regard, we have shown that inhibition of PARP-1 catalytic activity alters gene expression programs in a number of biological contexts, including breast cancer cells, adipocytes, and macrophages (19–22). In each of these cases, PARP-1 regulates gene expression through distinct mechanisms (e.g., RNA polymerase II activity, rDNA transcription, chromatin modifications, transcription factor activity). Together, these data make a strong argument for exploring the different mechanisms of action of PARP-1, and hence PARPi, in cancer, particularly in cervical cancer where relatively little is known about how PARP-1 regulates gene expression.
In this study, we observed that cervical adenocarcinoma cells treated with PARPi are sensitized to cisplatin treatment. Interestingly, this combination treatment promoted a dramatically altered transcriptome, including increased expression of the FOS gene, which encodes the transcription factor Fos. Enhanced expression of Fos in response to PARPi and cisplatin is sufficient to hinder growth in some cervical cancers.
Materials and Methods
Cell Culture
Human cervical cancer cell lines HeLa, Caski, SiHa, ME180, and SW756, as well as primary cervical cells, were purchased from the American Type Cell Culture (ATCC). HeLa cells were maintained in DMEM (Sigma-Aldrich, D5796); SiHa in EMEM (Corning, 10009CV); and Caski, ME180, SW756 in RPMI-1640 (Sigma-Aldrich, R8758). All media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The primary cervical cells were maintained in Cervical Epithelial Cell Basal Medium (ATCC, PCS-480–032) supplemented with the Cervical Epithelial Growth Kit (ATCC, PCS-480–042). Fresh cell stocks were regularly replenished from the original stocks and confirmed as mycoplasma-free every three months using a commercial testing kit.
Cell Treatments
Cells were pre-treated with 10 μM BYK204165 (Santa Cruz Biotechnology, sc-214642) for 30 minutes prior to additional treatments. Cells were treated with the chemotherapeutic agent 2 μM cisplatin (Sigma-Aldrich, P4394) as indicated.
Antibodies
The following antibodies were used for immunoblotting: Flag (Sigma-Aldrich, F3165); snRNP70 (Abcam, ab51266, RRID: AB_882630); GFP (Abcam, ab13970, RRID: AB_300798); Fos (Cell Signaling Technology, 4384, RRID: AB_2106617), and β-tubulin (Abcam, ab6046, RRID: AB_2210370). The custom rabbit polyclonal antiserum against PARP-1 used for immunoblotting was generated using an antigen comprising the amino-terminal half of PARP-1 (23) (now available from Active Motif; 39559, RRID: AB_2793257). The custom recombinant antibody-like anti-ADP-ribose binding reagent (WWE-Fc) was generated and purified in-house (24) (now available from Millipore, MABE103,1 RRID: AB_2665467). For detection of γH2AX lesions, FITC conjugated phospho-histone H2A.X (Ser139) antibody was used (clone JBW301; Millipore, RRID: AB_568825). For Fos ChIP-qPCR, a Fos rabbit monoclonal antibody was used (Cell Signaling Technology, 2250, RRID: AB_2247211).
Molecular Cloning and Generation of Cell Lines with Ectopic Expression
We used standard molecular cloning techniques to generate the following vectors for expressing or depleting proteins of interest. The human FOS cDNA was cloned into the pINDUCER20 lentiviral vector (Addgene, plasmid no. 44012, RRID: Addgene_44012) with the addition of a C-terminal Flag epitope tag and verified by sequencing. GFP was amplified from pEGFP-N3 (Clontech, RRID: Addgene_62043) and inserted into the pINDUCER20 vector using a Gibson Assembly kit (NEB, E2621).
HeLa cells were transduced with lentiviruses for ectopic expression. We generated lentiviruses by transfection of the pINDUCER20 constructs described above, together with: (i) an expression vector for the VSV-G envelope protein (pCMV-VSV-G, Addgene plasmid no. 8454, RRID: Addgene_8454); (ii) an expression vector for GAG-Pol-Rev (psPAX2, Addgene plasmid no. 12260, RRID: Addgene_12260); and (iii) a vector to aid with translation initiation (pAdVAntage, Promega), into 293T cells using Lipofectamine 3000 Reagent (Invitrogen, L3000015) according to the manufacturer’s protocol. The resulting viruses were collected in the culture medium, concentrated using a Lenti-X concentrator (Clontech, 631231), and used to infect HeLa cells. Stably transduced cells were selected with G418 sulfate (Sigma, A1720; 0.4 mg/mL) in cell culture medium
Primers for Molecular Cloning
The following oligonucleotide primers were used for molecular cloning:
- Primers for cloning a Flag-tagged FOS cDNA
- Forward: 5’- GCGGCTAGCATGATGTTCTCGGGCTTCAACGCA-3’
- Reverse: 5’-GCCCTCGAGTCACTTGTCATCGTCATCCTTATAATCCAGGGCCAGCAGCGTG-3’
- Primers for cloning an eGFP cDNA
- Forward: 5’-CTAGCTAGCATGGTGAGCAAGGGCGAGGAGCT-3’
- Reverse: 5’-GGGGAGCTCTTACTTGTACAGCTCGTCCATGCC-3’
Preparation of Nuclear Extracts
The cells were cultured and treated as described above. The cells were then washed with ice cold PBS, collected with ice cold PBS, and pelleted by centrifugation at 1000 RCF in a microcentrifuge. After collecting the cells, nuclear extracts were prepared according to the Sigma CelLytic NuCLEAR Extraction Kit protocol. Briefly, the cell pellets were resuspended in Isotonic Buffer (10 mM Tris-HCl pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 0.3 M sucrose) supplemented with 1 mM DTT, 250 nM ADP-HPD (Sigma, A0627; a PARG inhibitor), 20 μM PJ34 (a PARP inhibitor), and 1x complete protease inhibitor cocktail (Roche, 11697498001), incubated on ice for 15 min, and lysed by the addition of 0.6% NP-40 detergent with gentle vortexing. The nuclei from the lysed cells were collected by centrifugation in a microfuge at 11,000 RCF for 30 seconds. The pelleted nuclei were resuspended in Extraction Buffer C (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% v/v glycerol, 1 mM DTT, 250 nM ADP-HPD, 10 μM PJ34, and 1x complete protease inhibitor cocktail), and incubated for 20 minutes at 4°C with intermittent vigorous vortexing. The resuspended nuclear material was then clarified by centrifugation at 21,000 RCF in a microfuge for 15 minutes at 4°C. The supernatant was collected as nuclear extract.
Preparation of Whole Cell Extracts
Cells were cultured, treated, and collected as described above. The cell pellets were lysed in Lysis Buffer [10 mM HEPES pH 8.0, 2 mM MgCl2, 1% SDS, 250 units of Universal Nuclease (Pierce), and 1x protease inhibitor cocktail (Roche)] and incubated for 5 min at room temperature while being mixed to generate whole cell protein extracts. The extracts were clarified by centrifugation at 21,000 RCF in a microfuge for 15 minutes at 4°C. The supernatant was collected as whole cell extract.
Immunoblotting
Protein concentrations in the lysates were determined using Bradford reagent (Bio-Rad, 50000006) (for nuclear extracts) and a BCA protein assay (Pierce) (for whole cell extracts). The extracts were run on polyacrylamide-SDS gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat milk in TBST and incubated with the primary antibodies in 1% nonfat milk made in TBST followed by anti-rabbit HRP-conjugated IgG (1:5000) or anti-mouse HRP-conjugated IgG (1:5000). Immunoblot signals were detected using an ECL detection reagent (ThermoFisher, 34077, 34095).
Cell Survival Assays
Cell survival was quantified using a crystal violet dye-based assay. Cells were seeded as indicated in 6-well plates, grown overnight to ~50% confluence, and treated with PARP inhibitor, cisplatin, or both. The plates were incubated for several days as specified for each experiment. After collection, the cells were washed with PBS and then fixed with 10% formaldehyde for 10 mins. The fixed cells were stored in PBS at 4°C until all the time-points were collected. The fixed cells were stained with 0.1% crystal violet in 20% methanol solution for 30 minutes at room temperature. After washing to remove the excess stain, the crystal violet dye was extracted using 10% acetic acid and the absorbance measured at 595 nm using a spectrophotometer. Surviving fraction was calculated by dividing absorbance values from treated cells with absorbance values from vehicle (DMSO) treated cells. All experiments were done a minimum of three times with independent biological replicates to ensure reproducibility.
Flow Cytometry
Double stranded DNA breaks were measured by detection of γH2AX lesions using anti-phospho-Histone H2A.X (Ser139) antibody and co-staining the cells for DNA content using propidium iodide. For cell cycle analysis, the cells were stained with propidium iodide. After staining, cells were analyzed using a BD FACSCalibur flow cytometer (BD Biosciences). Data analysis was performed using FloJo data analysis software (Ashland, OR).
RNA Sequencing (RNA-seq)
Two biological replicates of HeLa cells from each treatment condition were used to isolate total RNA with the RNeasy Plus kit (QIAGEN). Total RNA samples were enriched for polyA+ RNA using Dynabeads Oligo(dT)25 (Invitrogen). Strand-specific RNA-seq libraries were prepared from the polyA+ RNA as described previously (22). The RNA-seq libraries were subjected to QC analyses (i.e., number of PCR cycles required to amplify each library, the final library yield, and the size distribution of the final library DNA fragments) and sequenced using an Illumina HiSeq 2000. The RNA seq reads were aligned to reference human genome (hg19) using TopHat (version 1.4.0) (25) and used to determine the steady state levels of the transcripts. We determined significantly regulated genes at 6 hours for the treatment groups: “BYK”, “cisplatin” and “BYK + cisplatin”. A list of unique oncogenes and tumor suppressors was compiled from Walker et.al. (26) and Zhao et .al. (27). Expression values for the “BYK + cisplatin” condition for this unique set of oncogenes and tumor suppressors was obtained from the RNA-seq data to create a cumulative distribution plot using the empirical Cumulative Distribution Function (eCDF) function and custom R script. The RNA-seq datasets generated for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo/) using accession number GSE176326.
Gene ontology (GO) analyses
GO analyses were performed using the DAVID (Database for Annotation, Visualization, and Integrated Discovery) tool (28). DAVID returns clusters of related ontological terms that are ranked according to an enrichment score. The inputs into the analyses were the sets of BYK + CDDP up-regulated and BYK + CDDP down-regulated genes from the RNA-seq analyses.
De novo motif analyses
De novo motif analyses were performed on a 2000 bp region surrounding the gene promoter using the command-line version of MEME (29). The following parameters were used for motif prediction: (1) zero or one occurrence per sequence (-mod zoops); (2) number of motifs (-nmotifs 12); (3) minimum, maximum width of the motif (-minw 8, -maxw 15); and (4) search for motif in given strand and reverse complement strand (-revcomp). The predicted motifs from MEME were matched to known motifs using TOMTOM.
Chromatin immunoprecipitation (ChIP)
HeLa cells were cultured and treated with DMSO or CDDP + BYK for 12 hours. ChIP was performed as described previously (22) with slight modifications. Briefly, the cells were cross-linked with 1% formaldehyde in PBS for 10 minutes at 37°C and quenched in 125 mM glycine in PBS for 5 minutes at 4°C. Crude nuclear pellets from these cells were sonicated to generate chromatin fragments of ~300 bp in length. The sonicated samples were incubated overnight at 4°C with a Fos antibody. The samples were washed and the immunoprecipitated genomic DNA was eluted in Elution Buffer (100 mM NaHCO3, 1% SDS), digested with proteinase K and RNase H to remove protein and RNA, respectively, extracted with phenol:chloroform:isoamyl alcohol, and precipitated with isopropanol. The precipitated ChIPed DNA along with input DNA was collected by centrifugation, air dried, and dissolved in nuclease-free water. Fos enrichment on the ChIPed DNA was then analyzed by qPCR using the primer sets listed below and a LightCycler 480 real-time PCR thermocycler (Roche) for 45 cycles.
- Primers for JUNB
- Forward: 5’- CGTGGAAGATCCAGCAGTCC-3’
- Reverse: 5’- GAGAGTTAGAAGGGGCCGGA-3’.
- Primers for NAGS
- Forward: 5’- CAACGGCAAGTTAAGAGCCC-3’
- Reverse: 5’- AGAACCACAGCCATCAGCG-3’.
siRNA-mediated knockdown
The siRNA oligos used for the knockdown experiments were purchased from Sigma: FOS (SASI_Hs01_00184573) and siRNA Universal Negative Control #1 (SIC001). The siRNA oligos were transfected at a final concentration of 30 nM using Lipofectamine RNAiMAX reagent (Invitrogen, 13778150) according to the manufacturer’s instructions. The cells were used for various assays 48 hours after siRNA transfection as indicated.
Data Availability
The RNA-seq datasets generated for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo/) using accession number GSE176326. All other data generated in this study are available within the article and its supplementary data files. Unique materials used in this study are available upon request from the corresponding author.
Results
Cervical cancer cells have different basal levels of ADPRylation and sensitivities to PARP inhibitors
To assess the effect of PARP inhibition on cervical cancer cells, we treated cell lines representing the different histological types of cervical cancer, including adenocarcinoma (HeLa cells), epidermoid (Caski and ME180 cells), and squamous (SiHa and SW756 cells) (Fig.1A), as well as primary cervical cells, with the PARP-1-selective inhibitor BYK204165 (BYK; (30)) (Fig. 1B). Each cell line had different levels of basal ADPRylation, with HeLa, ME180 and primary cells having low levels, and Caski, SiHa and SW756 cells having high levels (Fig. 1B). The basal ADPRylation in each cell lines was effectively inhibited by treatment with BYK (Fig. 1B), suggesting that PARP-1 is the primary mediator of ADPRylation in these cells.
Figure 1. Cervical cancer cell lines display a range of sensitivities to the PARP inhibitor BYK204165.
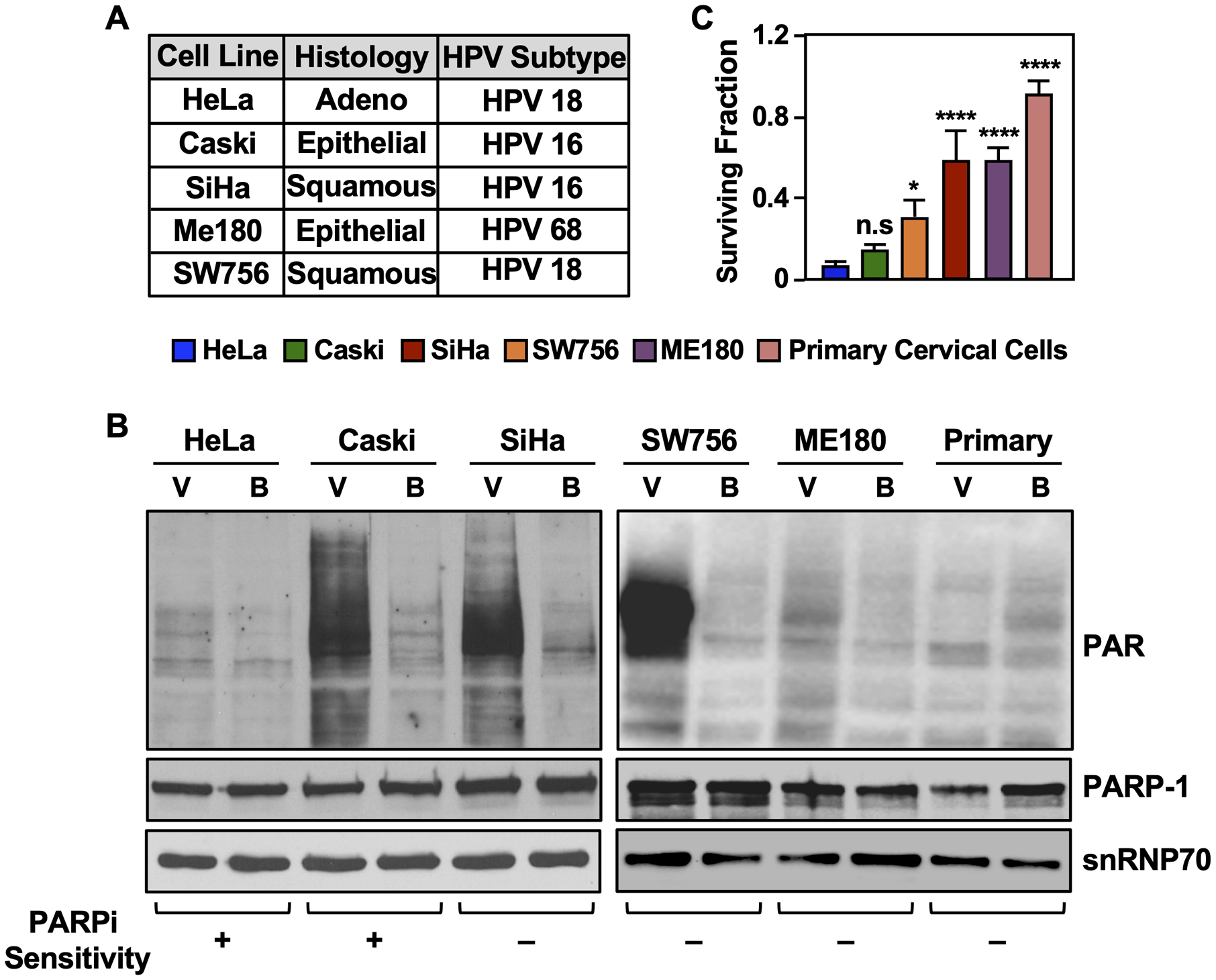
(A) Table listing the cervical cancer cell lines used in this study.
(B) Cervical cancer cell lines have different levels of nuclear ADPRylation, which is abolished by treatment with BYK204165 (BYK), a PARP-1-selective inhibitor. Immunoblots showing PAR and PARP-1 levels in cervical cancer cells as indicated. snRNP70 was used as a loading control. The cells were treated with 10 μM BYK [B] or vehicle, DMSO [V] for 6 hours. PAR levels were detected using an ADP-ribose detection reagent (WWE-Fc).
(C) Cervical cancer cell lines exhibit different levels of sensitivity to BYK. The cells were treated with 100 μM BYK for 4 days. Cell survival was quantified using crystal violet assays. Each point represents the mean ± SD (n = 3). Significance was calculated relative to cell survival in HeLa using one-way ANOVA followed by Tukey’s test (HeLa versus Caski is not significant at p < 0.05).
In cell growth and survival assays, BYK treatment had dramatically different effects on the different cell lines, with both HeLa and Caski cells exhibiting greater sensitivity to high doses of BYK (100 μM) compared to the other cell lines (Fig. 1C). Moreover, BYK treatment inhibited cell cycle progression in S-phase in HeLa cells (Supplementary Fig. S1, A and B). In previous studies with cervical and ovarian cancer cells, high ADPRylation levels were associated with greater sensitivity to PARPi (31), an effect not universally observed with the cervical cancer cell lines that we used herein. Based on the different sensitivities to PARPi observed in the different cell lines, we decided to explore the explore the therapeutic potential of PARPi in the “BYK sensitive” (HeLa and Caski) versus “BYK resistant” (SiHa, ME180, SW756) cell lines.
PARP-1 inhibition leads to increased sensitivity to cisplatin without affecting DNA damage responses in HeLa cells
We assessed the survival of the cervical cancer cells in response to treatment with BYK, cisplatin, or both in combination (Fig. 2, Supplementary Fig. S2). While cisplatin alone resulted in decreased survival in both HeLa and Caski, co-treating the cells with BYK and cisplatin led to even greater cytotoxicity (Fig. 2, A and B). Interestingly, co-treating the “BYK resistant” cells (i.e., SiHa, ME180, and SW756) with BYK and cisplatin did not enhance the sensitivity to cisplatin (Supplementary Fig. S2, A–C). These results demonstrate the different sensitivities of the cells to therapeutic mechanisms.
Figure 2. PARP inhibition enhances cisplatin-mediated cytotoxicity in HeLa cells without increasing DNA damage.
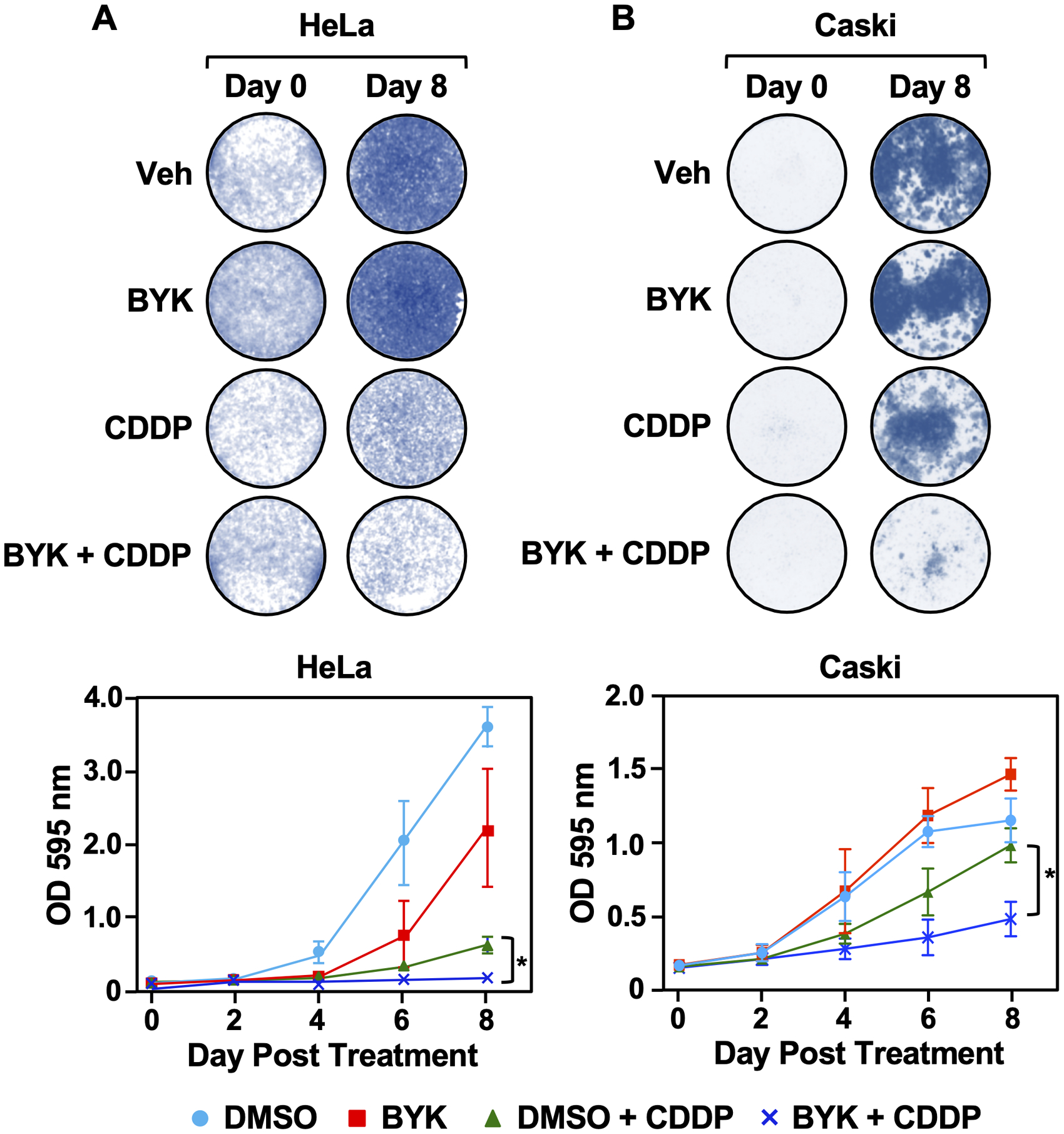
HeLa (A) and Caski (B) cells were treated with the PARP inhibitor BYK (10 μM), the chemotherapeutic drug cisplatin (CDDP; 2 μM), or both in combination as indicated. Cells were collected and stained with crystal violet at day 0, 2, 4, 6 and 8 (top panel). Cell survival was quantified for each of the time points by measuring absorbance at O.D. 595nm (bottom panel). Each point represents the mean ± SEM (n = 4; Fisher’s LSD test; * p < 0.05).
The therapeutic mechanism of action of PARP inhibitors has historically been associated with impaired DNA repair responses leading to cell death (18). To assess the effect of the combination of BYK and cisplatin on DNA damage, we treated HeLa cells with either BYK alone, cisplatin alone, or both agents in combination. The accumulation of γH2AX foci was used as a readout for the amount of double-strand DNA breaks (32). We observed that while BYK treatment led to no substantial changes in γH2AX accumulation, treating the cells with cisplatin dramatically increased the number of γH2AX foci (Supplementary Fig. S3, A and B). Cotreatment with BYK and cisplatin had no additional effect on double-strand break accumulation compared to cisplatin alone (Supplementary Fig. S3, A and B). These data suggest that while the combination of PARPi and cisplatin induces greater cell death, the effects appear to be independent of enhanced DNA damage. Taken together, these results suggest the possibility of a DNA damage-independent mechanism of action for PARPi in cervical cancer cells.
PARP-1 inhibition in the presence of cisplatin results in genome-wide transcriptional changes in HeLa cells
A number of recent studies have explored the role of PARP-1 in regulating gene expression (19–22,33,34). Given these findings, we hypothesized that PARP-1-dependent modulation of transcription could play a critical role in the cytotoxic response of cisplatin-treated HeLa cells to PARP-1 inhibition. To test this, we performed RNA-seq in HeLa cells to determine the genome-wide changes in the transcriptome in the presence of BYK, cisplatin, or BYK + cisplatin (Fig. 3A). We observed altered expression of a ~1400 genes in response to either BYK or cisplatin alone after 6 hours of treatment, relative to the vehicle control (Fig. 3B). In contrast, we observed a dramatic expansion of the transcriptome, with altered expression of an additional ~1,500 genes in response to BYK + cisplatin (Fig. 3B). These results indicate that the combined treatment induces a distinct, expanded gene expression program.
Figure 3. PARP inhibition in presence of cisplatin dramatically alters the HeLa cell transcriptome.
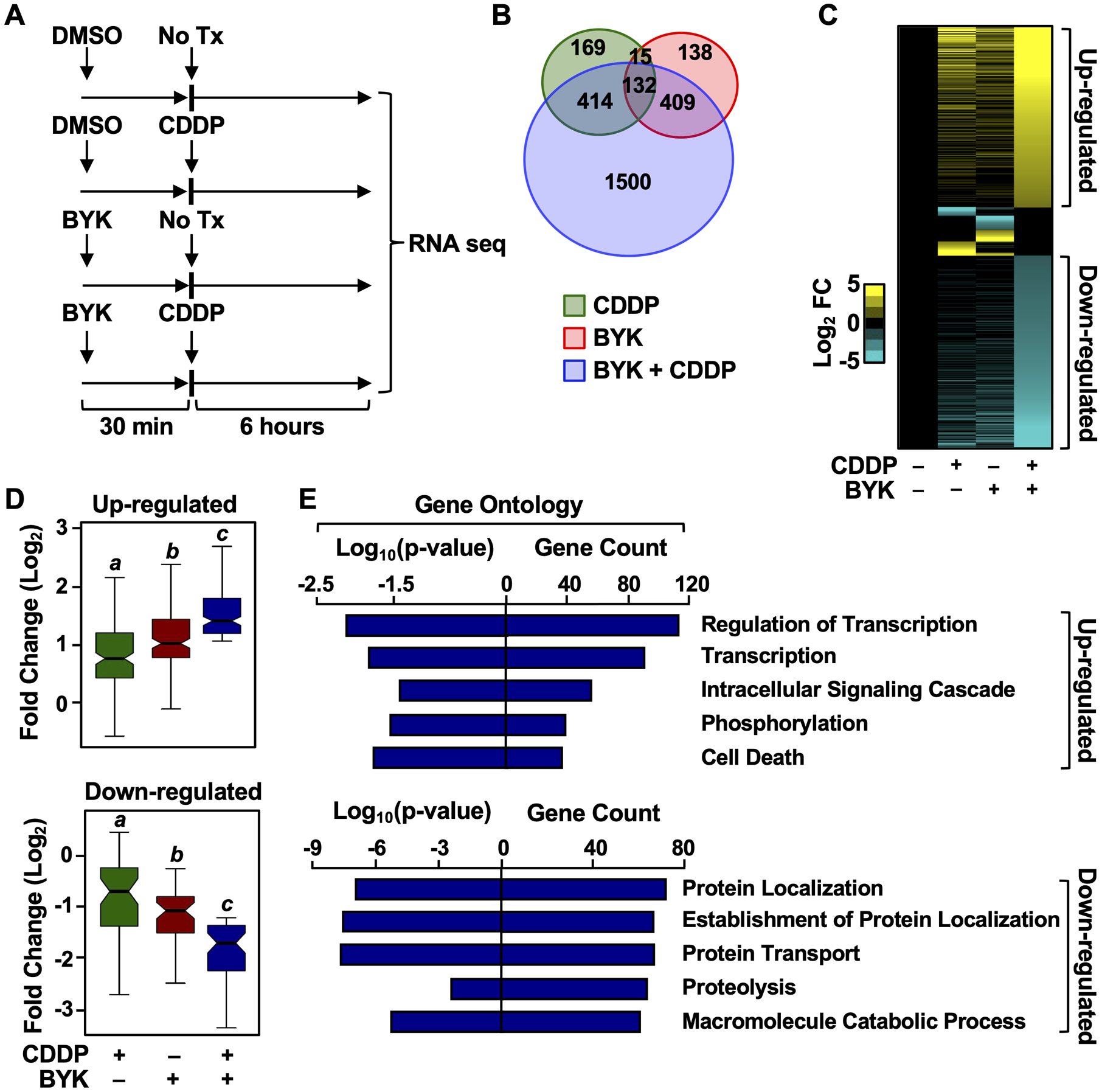
(A) Cells were treated with 10 μM BYK for 30 minutes prior to cisplatin (CDDP) treatment. 2 μM CDDP was added for 6 hours as indicated. DMSO was used as a vehicle control. RNA-seq libraries were prepared following cell collection.
(B) Venn diagram depicting differentially expressed genes from RNA-seq in HeLa cells treated with CDDP (green), BYK (red), or BYK + CDDP (blue).
(C) Heat map representation of the genes that were either up-regulated or down-regulated after treatment of HeLa cells with DMSO (vehicle), CDDP alone, BYK alone, or both.
(D) Box plots showing the fold change in expression for the up-regulated and down-regulated subsets of genes. Boxes marked with different letters are significantly different from each other (Wilcoxon Signed-Rank test; p < 2.2 × 10−16). (E) Gene ontology analysis for BYK + CDDP up-regulated and down-regulated genes. The gene counts in each term and the log10(p-value) for each term are shown.
To assess the changes in gene expression in greater detail, we focused on alterations in the relative expression of individual genes that are regulated in any one of the treatment conditions compared to the vehicle treated cells (Fig. 3C). BYK + cisplatin had the most dramatic effect on gene expression, with a distinct set of genes being either significantly up- or down-regulated in the co-treated cells (Fig. 3, C and D). Based on this observation, we hypothesized that this combination of treatments stimulated the expression of genes whose protein products mediated the increased cytotoxicity in HeLa cells. To test this, we evaluated the ontologies of the genes that were up- or down-regulated by BYK + cisplatin (Fig. 3E). We observed that ontological terms related to transcription and cell death were enriched in the up-regulated gene set (Fig. 3E, top panel). In contrast, we observed that ontological terms related to cellular maintenance, including protein localization and transport, were enriched in the down-regulated gene set (Fig. 3E, bottom panel). These results provide additional support for our hypothesis that BYK + cisplatin drives cell cytotoxicity by modulating transcriptional responses in cervical cancer cells.
Increased FOS gene transcription is associated with an altered transcriptional program in “BYK sensitive” cervical cancer cells treated with PARPi and cisplatin
To identify candidate genes in the up-regulated gene set may drive the alterations in the transcriptional program and the downstream inhibition of cell proliferation, we ranked all the genes up-regulated by BYK + cisplatin treatment based on their expression levels. Interestingly, in the set of top-ranked genes, we identified a number of genes associated with cancer-related phenotypes (both tumor suppressors and oncogenes) (Fig. 4A). One of the highest ranked genes was FOS, a known oncogene that encodes Fos, a member of the AP-1 transcription factor family (35). We observed that FOS expression is upregulated specifically in the BYK + cisplatin treatment (Fig. 4B). Importantly, Fos protein levels are elevated in the “BYK sensitive” cell lines (HeLa and Caski; Fig. 4, C and D), but not in the “BYK resistant” (SiHa, ME180, SW756, and primary cells; Fig. 4E and Supplementary Fig. S4). Based on this, we surmised that inhibition of PARP-1 catalytic activity in the sensitive cell lines in the presence of cisplatin might cause increased cell death by inducing the expression of FOS, leading to an accumulation of Fos protein, that could function as key effector in determined cellular outcomes.
Figure 4. Co-treatment of cervical cancer cells with PARP inhibitor and cisplatin induces Fos expression.
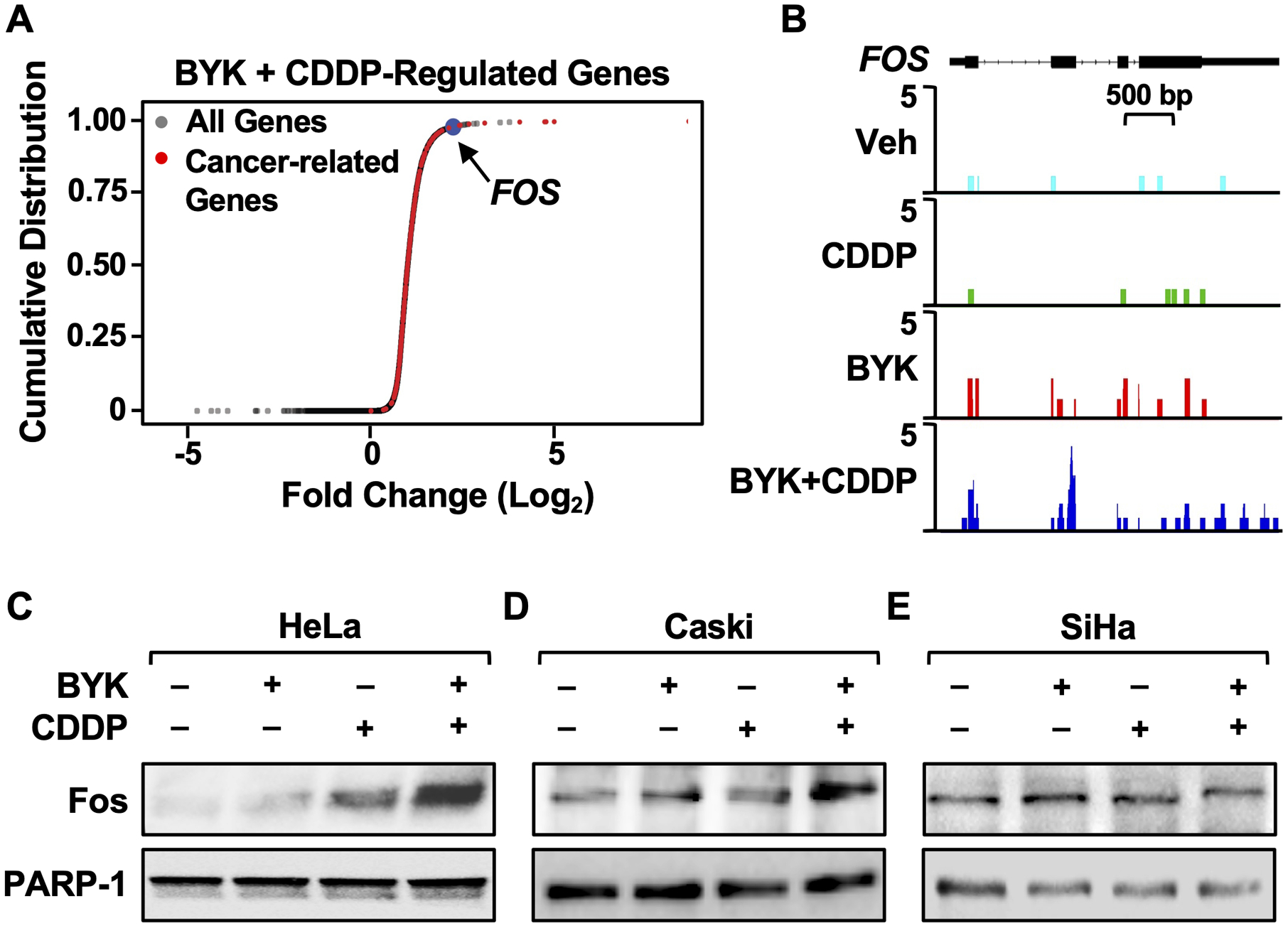
(A) PARP inhibition in the presence of cisplatin induces the expression of a distinct subset of cancer-related genes, including FOS. Cumulative frequency plots representing genes up-regulated only in BYK + CDDP-treated cells. Fold change in gene expression was measured for BYK + CDDP treatment relative to vehicle. All genes are indicated in grey, cancer-related genes are shown in red, and FOS is highlighted in blue.
(B) RNA-seq browser tracks showing BYK + CDDP-induced gene expression at the FOS locus in HeLa cells.
(C-E) BYK + CDDP treatment results in increased Fos protein levels in HeLa and Caski cells. Immunoblots showing Fos and PARP-1 in HeLa (C), Caski (D) and SiHa (E) cells treated with 10 μM BYK for 30 minutes prior to 2 μM CDDP treatment for 12 hours.
We observed an enrichment of the Fos DNA-binding motif in the gene body or upstream regulatory region (± 2 kb) of the genes up-regulated co-treatment with BYK + cisplatin (Fig. 5A). Conversely, when we selected for genes that had the Fos-binding motif in the gene body or upstream regulatory region (± 2 kb), we observed greater expression of these genes in the BYK + cisplatin treated condition (Figure 5, B and C). We validated this by assessing Fos binding to the upstream regulatory region using ChIP-qPCR. Indeed, the Fos protein was enriched at these regions upon BYK + cisplatin treatment as compared to vehicle alone (Fig. 5D). These results suggest that Fos can act as a downstream mediator of responses to BYK + cisplatin treatment by serving as a regulatory transcription factor that acts at the promoters of key target genes to drive a distinct gene expression program.
Figure 5. Co-treatment with PARP inhibitor and cisplatin induces a Fos-driven transcriptional program.
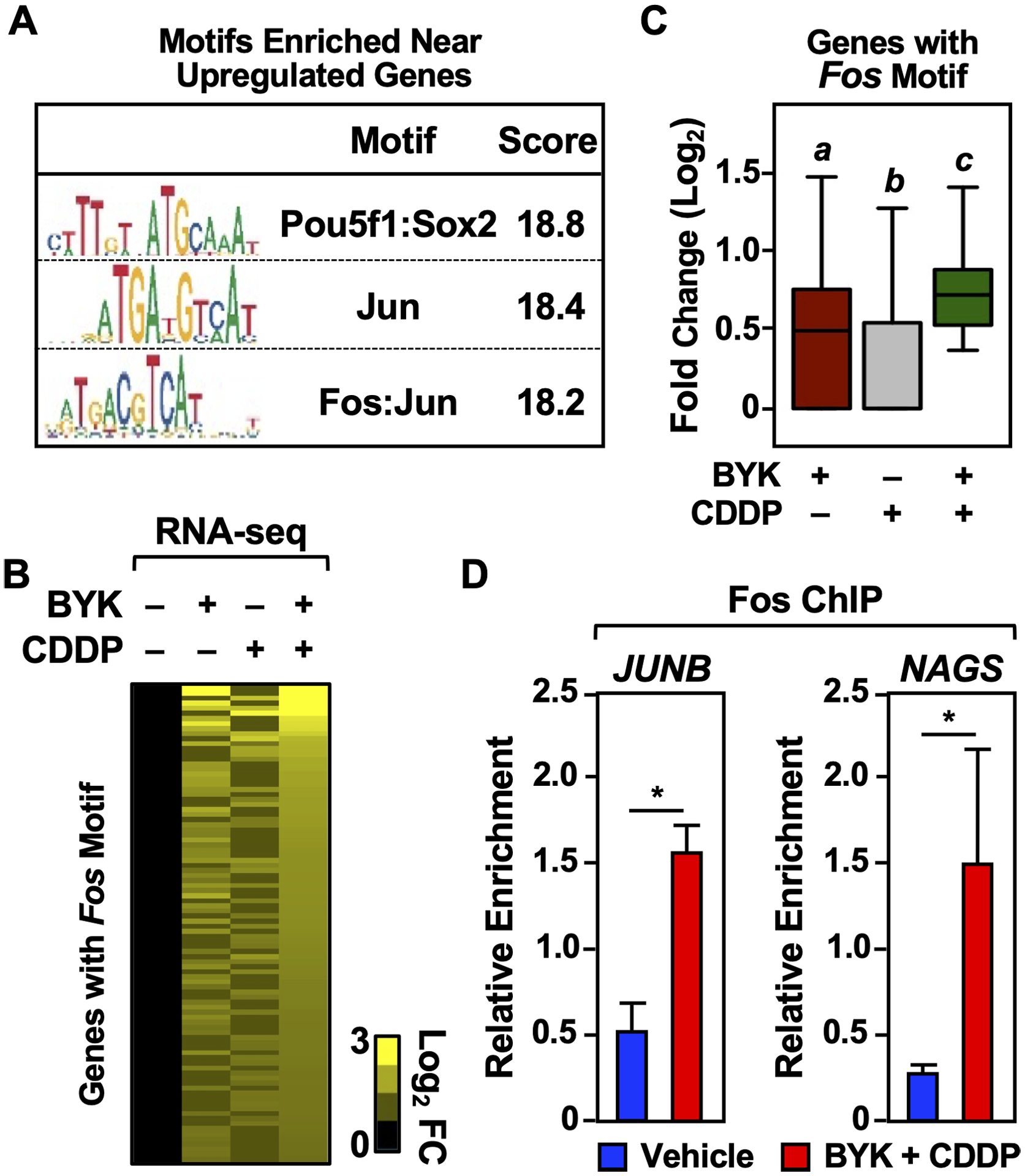
(A) The Fos-binding motif is enriched in the proximity of BYK + CDDP upregulated genes. The motifs indicated have the highest enrichment within ± 2 kb of the gene body for genes induced by BYK + CDDP.
(B and C) Fos and C-binding motif enrichment correlates to increased gene expression in BYK + CDDP treated HeLa cells. Heatmap (B) and box plot (C) representations of expression of all genes with Fos-binding motif enrichment within ± 2 kb of the gene body under the treatment conditions indicated. Boxes marked with different letters are significantly different from each other (Wilcoxon Signed-Rank test; p < 2.2 × 10−16).
(D) Increased Fos-binding is observed upon co-treatment with BYK + CDDP. ChIP-qPCR showing Fos-enrichment in Vehicle (DMSO) and BYK + CDDP treated HeLa cells. Fos-binding is analyzed in the upstream regulatory regions (<2 kb) of the genes indicated. Each bar represents the mean ± SEM (n = 3; Fisher’s LSD test; * p < 0.05).
Fos expression is a driver of BYK and cisplatin-mediated cytotoxicity cervical cancer cells.
To determine if elevated Fos protein levels might reduce the survival of cisplatin-treated HeLa cells, we ectopically expressed Fos protein from a FOS transgene in a doxycycline (Dox)-inducible manner (Fig. 6A). Dox-inducible GFP-expressing cells were used as a negative control. The cotreatment of the GFP expressing cells with BYK and cisplatin resulted in increased cell cytotoxicity, thus confirming our previous results. We then evaluated the effect of Fos expression on HeLa cell growth. Ectopic expression of Fos in HeLa cells resulted in decreased cell growth relative to the GFP-expressing control cells across a period of 6 days (Fig. 6B), thus mimicking the phenotype observed with BYK + cisplatin treatments, which act upstream, of FOS to induce its expression. However, we did not observe a significant decrease in cell growth after treating the Fos overexpressing cells with BYK + cisplatin. This corroborates our hypothesis that Fos expression alone is sufficient to drive the cytotoxicity induced by BYK and cisplatin cotreatment. Conversely, upon siRNA-mediated knockdown of Fos, we observed an increase in cell growth upon BYK + cisplatin treatment (Fig. 6C, D). This confirms that Fos is necessary for mediating the BYK + cisplatin-driven cytotoxic effects in cervical cancer.
Figure 6. Fos expression drives the cytotoxic effects of BYK + CDDP.
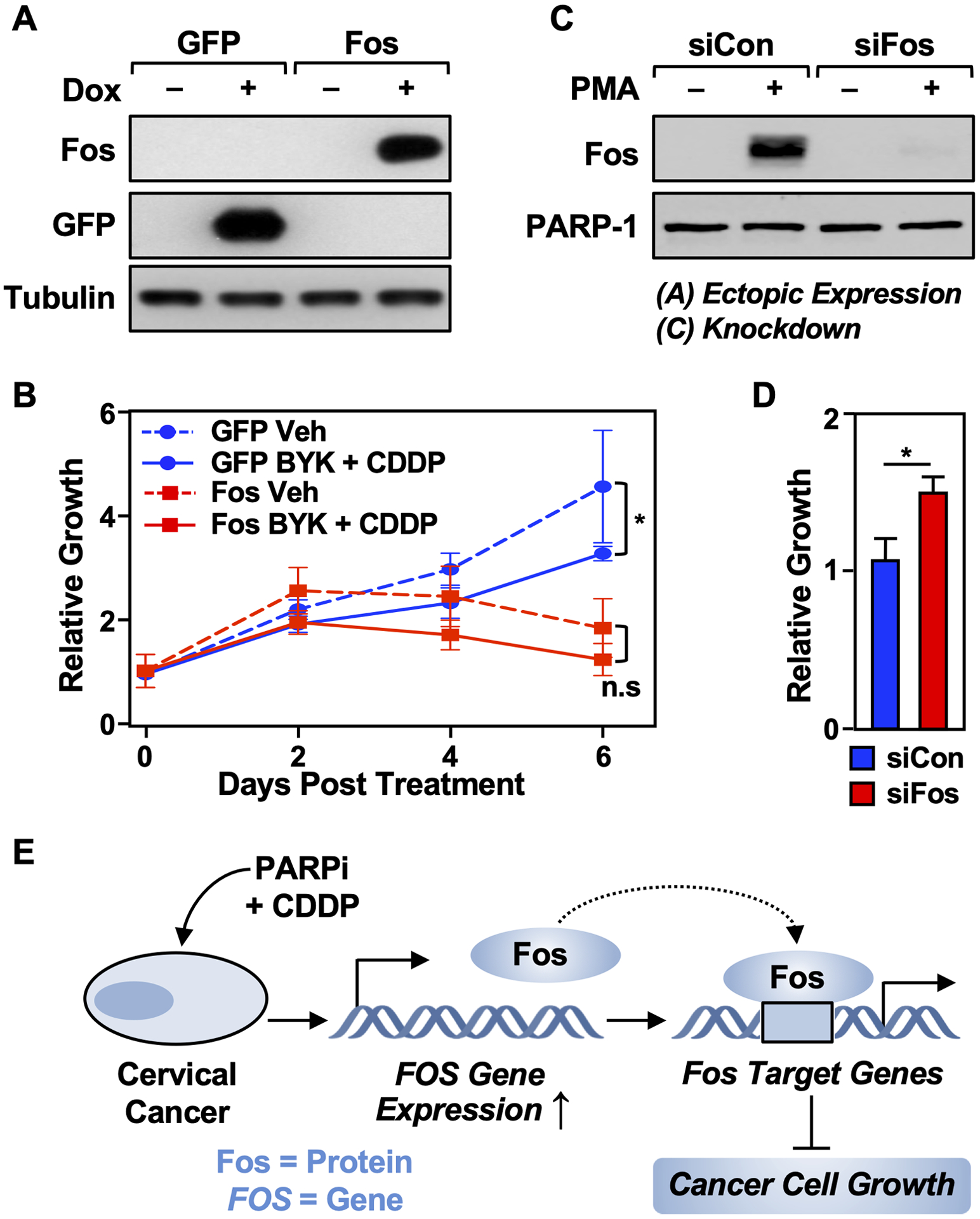
(A) HeLa cells were engineered to ectopically express either Fos or GFP. Immunoblots show the doxycycline (Dox)-inducible expression of Fos and GFP as indicated. β-Tubulin was used as a loading control. The cells were treated with 1 mg/mL of Dox for 16 hours.
(B) Ectopic expression of Fos in HeLa cells leads to reduced growth of the cells. The cells were treated with Dox as in (A) and vehicle (DMSO) or BYK + CDDP as indicated. Cell growth was quantified after 0, 2, 4, or 6 days using a crystal violet assay. Each point represents the mean ± SEM (n = 3; Sidak’s multiple comparison test; * p < 0.05; n.s. is not significant at p < 0.05).
(C) Immunoblots showing the siRNA-mediated knockdown of Fos in HeLa cells. The cells were treated with PMA to induce Fos expression for visualization.
(D) Depletion of Fos in HeLa cells leads to increased growth of the cells. HeLa cells were treated with Fos siRNA as shown in (C) and cell growth was quantified after 2 days using a crystal violet assay. Each point represents the mean ± SEM (n = 3; Student’s t-test; *< 0.05).
(E) Model showing the inhibition of cervical cancer growth by co-treatment with PARP inhibitor and cisplatin via the induction of FOS expression. See the text for details.
In conclusion, we have shown that PARP inhibition together with cisplatin treatment induces FOS expression in cervical cancer cells, which in turn causes transcriptional changes that result in diminished cell proliferation (Fig. 6E). These pre-clinical results provide a compelling argument for the use of a combination therapy of PARP inhibitors and cisplatin for treating cervical cancer patients.
Discussion
To explore potential therapeutic mechanisms that might be useful for treating cervical cancers, we performed a variety of experiments combining a PARP inhibitor with cisplatin. We used a number of cell lines that represent different cervical cancer subtypes, as well as primary cervical cells. Surprisingly, we observed that only a few of cell lines were sensitive to PARP inhibition treatment, irrespective of the basal ADPRylation levels. The factors that contribute to these differences remain to be understood. We observed that the PARPi-sensitive cervical cancer cells exhibit greater sensitivity when co-treated with cisplatin. Interestingly, this combination treatment promoted a dramatically altered transcriptome, including increased expression of the transcription factor Fos and subsequent binding of Fos to its target genes. Enhanced expression of Fos in response to PARPi and cisplatin is sufficient to hinder cervical cancer growth. Conversely, the loss of Fos correlated with greater cell growth. These results have revealed a previously unknown mechanism for the regulation of Fos-dependent transcription by PARP-1.
Alternate mechanisms of action for PARP-1-directed therapeutics
The therapeutic mechanisms of action of drugs are tied to the biological functions of the proteins that they target. The original therapeutic mechanism of action defined for PARPi was the induction of synthetic lethality in cancers that are deficient in homologous recombination (HR)-mediated DNA repair (11,12). But PARP-1 also plays important roles beyond DNA-damage repair, particularly in regulating gene transcription and RNA biology (17,18). This opens that possibility of therapeutic mechanisms of action for PARPi that extend beyond DNA repair. Indeed, recent studies have reported that PARPi may have significant clinical benefits in patients in the absence of BRCA1/2 mutations or HRD (7,9,13–16,36).
Previous work has shown that in breast cancers, PARP-1 regulates transcriptional outcomes by modifying the activity of (1) RNA polymerase II and its associated factors (37), (2) transcription factors (21,22), and (3) chromatin (20), and regulating rDNA transcription through the RNA helicase DDX21 (19). Together these examples emphasize the diversity in regulatory mechanisms impacted by PARP-1 and underscore the need to identify the distinct therapeutic mechanisms of action of PARPi in different cellular contexts.
Fos as a driver of a drug-sensitizing gene expression program
In this study, we discovered a new potential mechanism of action of for PARPi administered in combination with cisplatin, namely enhanced FOS gene expression leading to a dramatic increase in Fos protein levels. We found that a large number of genes whose expression is altered in response to BYK + cisplatin co-treatment are potential targets of Fos. The upregulated genes in this set exhibit an enrichment of the Fos binding motif, leading us to postulate that Fos might be acting by directly regulating the expression of these genes. The mechanism of Fos-dependent activation of these genes, however, remains to be determined. Together, these results extend our understanding of PARP-1-dependent transcriptional regulation.
Given that FOS is a proto-oncogene, one might expect increased Fos protein levels to be growth-promoting (38). But Fos and related members of the Fos family are known to inhibit cancer cell growth or promote apoptosis in certain contexts (38), dependent on the relative ratios of the different family members (38). Fos promotes cell cycle inhibition and cell death in hepatocellular carcinomas (39) and suppresses ovarian cancer proliferation by altering cell adhesion (40). Finally, analysis of data from The Cancer Genome Atlas datasets showed that higher FOS expression levels, along with higher FOS target gene expression, is associated with an increased survival in breast cancer patients (41). In the context of these previous studies, our results how combination therapies can alter signaling pathways that change gene expression patterns to enhance therapeutic benefits.
Use of PARPi in combination with cisplatin in cancer therapy
A number of studies in different cancer types including non-small cell lung cancer, liver cancer, oral squamous cell carcinoma, osteosarcoma and melanoma (42–46) have assessed the effect of PARP inhibition along with cisplatin treatment. Similar to our observations, in these cancers, PARP inhibition augmented the cytotoxic effects of cisplatin. While some of the studies attributed the increased cytotoxicity to greater DNA damage with PARP inhibition (42,46), none of them show a DNA-damage-independent, transcription-dependent effect of the combinatorial treatment. These data suggest that while different cancers can exhibit similar phenotypic effects to the combination of PARPi and cisplatin, the mechanism of cell death are variable and can be specific to different cancer types.
Recent work from other groups has also sought to understand the implications of using PARP inhibitors in cervical cancer. Treating HeLa cells with PARPi has been shown to induce apoptosis specifically through the modulation of PARP-1 activity (47). HeLa and SiHa cells with hyperthermia also show greater sensitivity when treated with PARP inhibitors and cisplatin together. Moreover treating HeLa cells with PARPi and cisplatin stimulates β-catenin signaling and increases cisplatin-induced cytotoxicity (48). Although the phenotype is similar to the one that we have observed, the mechanism is different. This could be due to the use of different PARPi and their respective specificities. Alternatively, these pathways could be activated concurrently; this possibility needs to be explored further.
A number of clinical trials have explored the use of PARPi in combination with chemotherapeutic agents (49). Indeed, a recently clinical trial tested a regimen of the PARPi veliparib with cisplatin in cervical cancer patients (50). They showed that adding veliparib to the chemotherapeutic regimens was safe and correlated with increased progression free survival (50). However, this work involved few patients and needs to be corroborated with a larger cohort. Collectively, our data strengthen the rationale for using PARPi with cisplatin as a therapy for cervical cancer.
Limitations of the study
While we have sought to understand the biological effects and mechanism of action of BYK and cisplatin on cervical cancer cell lines, we do not yet know the effect of this combination treatment in more complex biological settings of cervical cancers, such as xenografts in mice or patient samples. These additional data would provide valuable insight into the efficacy of the co-treatment as a therapeutic approach. We have identified cervical cancer cell types that are both sensitive and resistant to PARPi. Previous studies have shown a direct association between PARPi sensitivity and cellular ADPRylation levels (31). However, in this case no such correlation is observed and the reason for the differential sensitivities remains to be understood. We have used BYK, a PARP-1 selective inhibitor; however, FDA-approved PARPi, such as Olaparib and Niraparib, are used clinically. Thus, verifying our results in the future with other PARPi will be required to translate these studies to a clinical setting.
Supplementary Material
Implications.
Our observations, which link the gene regulatory effects of PARPi + cisplatin to the growth inhibitory effects of FOS expression in cervical cancer cells, strengthen the rationale for using PARPi with cisplatin as a therapy for cervical cancer.
Acknowledgements
We thank Dr Sridevi Challa and Dr. Cristel Camacho for critical comments on this manuscript. We thank Dr. Shrikanth Gadad for his help with the RNA-seq library preparation. We acknowledge and thank the UT Southwestern Next Generation Sequencing Core for deep sequencing services (Vanessa Schmid) and the UT Southwestern Flow Cytometry Core for the FACS analysis. This work was supported by a grant from the NIH/NICHD Reproductive Scientist Development Program to K.Y.L., and a grant from the NIH/NIDDK (R01 DK058110) and funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment to W.L.K.
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the article; or in the decision to publish the results.
Disclosure: W.L.K. is a founder, consultant, and member of the Scientific Advisory Board for Ribon Therapeutics, Inc. and ARase Therapeutics, Inc. He is also coholder of U.S. Patent 9,599,606 covering the ADP-ribose detection reagent used herein, which has been licensed to and is sold by EMD Millipore.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Cohen PA, Jhingran A, Oaknin A, Denny L. Cervical cancer. Lancet 2019;393(10167):169–82 doi 10.1016/S0140-6736(18)32470-X. [DOI] [PubMed] [Google Scholar]
- 3.Kurnit KC, Fleming GF, Lengyel E. Updates and new options in advanced epithelial ovarian cancer treatment. Obstet Gynecol 2021;137(1):108–21 doi 10.1097/AOG.0000000000004173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regalado Porras GO, Chavez Nogueda J, Poitevin Chacon A. Chemotherapy and molecular therapy in cervical cancer. Rep Pract Oncol Radiother 2018;23(6):533–9 doi 10.1016/j.rpor.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu H, Luo H, Zhang W, Shen Z, Hu X, Zhu X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des Devel Ther 2016;10:1885–95 doi 10.2147/DDDT.S106412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lightfoot M, Montemorano L, Bixel K. PARP Inhibitors in Gynecologic Cancers: What Is the Next Big Development? Curr Oncol Rep 2020;22(3):29 doi 10.1007/s11912-020-0873-4. [DOI] [PubMed] [Google Scholar]
- 7.Bitler BG, Watson ZL, Wheeler LJ, Behbakht K. PARP inhibitors: clinical utility and possibilities of overcoming resistance. Gynecol Oncol 2017;147(3):695–704 doi 10.1016/j.ygyno.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E. PARP inhibitors: clinical relevance, mechanisms of action and tumor resistance. Front Cell Dev Biol 2020;8:564601 doi 10.3389/fcell.2020.564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol 2015;33(12):1397–406 doi 10.1200/JCO.2014.58.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 2012;13(7):411–24 doi 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 11.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434(7035):913–7 doi 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 12.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434(7035):917–21 doi 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 13.Evans T, Matulonis U. PARP inhibitors in ovarian cancer: evidence, experience and clinical potential. Ther Adv Med Oncol 2017;9(4):253–67 doi 10.1177/1758834016687254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lheureux S, Lai Z, Dougherty BA, Runswick S, Hodgson DR, Timms KM, et al. Long-term responders on Olaparib maintenance in high-grade serous ovarian cancer: clinical and molecular characterization. Clin Cancer Res 2017;23(15):4086–94 doi 10.1158/1078-0432.CCR-16-2615. [DOI] [PubMed] [Google Scholar]
- 15.Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390(10106):1949–61 doi 10.1016/S0140-6736(17)32440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Martin A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2019;381(25):2391–402 doi 10.1056/NEJMoa1910962. [DOI] [PubMed] [Google Scholar]
- 17.Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 2017;31(2):101–26 doi 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim DS, Camacho CV, Kraus WL. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp Mol Med 2021;53(1):42–51 doi 10.1038/s12276-021-00557-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, Kraus WL. Activation of PARP-1 by snoRNAs Controls Ribosome Biogenesis and Cell Growth via the RNA Helicase DDX21. Mol Cell 2019;75(6):1270–85 e14 doi 10.1016/j.molcel.2019.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang D, Camacho CV, Setlem R, Ryu KW, Parameswaran B, Gupta RK, et al. Functional interplay between histone H2B ADP-ribosylation and phosphorylation controls adipogenesis. Mol Cell 2020;79(6):934–49 e14 doi 10.1016/j.molcel.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo X, Ryu KW, Kim DS, Nandu T, Medina CJ, Gupte R, et al. PARP-1 controls the adipogenic transcriptional program by PARylating C/EBPbeta and modulating is transcriptional activity. Mol Cell 2017;65(2):260–71 doi 10.1016/j.molcel.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupte R, Nandu T, Kraus WL. Nuclear ADP-ribosylation drives IFNgamma-dependent STAT1alpha enhancer formation in macrophages. Nat Commun 2021;12(1):3931 doi 10.1038/s41467-021-24225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 2004;119(6):803–14 doi 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Gibson BA, Conrad LB, Huang D, Kraus WL. Generation and Characterization of Recombinant Antibody-like ADP-Ribose Binding Proteins. Biochemistry 2017;56(48):6305–16 doi 10.1021/acs.biochem.7b00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14(4):R36 doi 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker EJ, Zhang C, Castelo-Branco P, Hawkins C, Wilson W, Zhukova N, et al. Monoallelic expression determines oncogenic progression and outcome in benign and malignant brain tumors. Cancer Res 2012;72(3):636–44 doi 10.1158/0008-5472.CAN-11-2266. [DOI] [PubMed] [Google Scholar]
- 27.Zhao M, Kim P, Mitra R, Zhao J, Zhao Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res 2016;44(D1):D1023–31 doi 10.1093/nar/gkv1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4(5):P3. [PubMed] [Google Scholar]
- 29.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 2009;37(Web Server issue):W202–8 doi 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eltze T, Boer R, Wagner T, Weinbrenner S, McDonald MC, Thiemermann C, et al. Imidazoquinolinone, imidazopyridine, and isoquinolindione derivatives as novel and potent inhibitors of the poly(ADP-ribose) polymerase (PARP): a comparison with standard PARP inhibitors. Mol Pharmacol 2008;74(6):1587–98 doi 10.1124/mol.108.048751. [DOI] [PubMed] [Google Scholar]
- 31.Conrad LB, Lin KY, Nandu T, Gibson BA, Lea JS, Kraus WL. ADP-ribosylation levels and patterns correlate with gene expression and clinical outcomes in ovarian cancers. Mol Cancer Ther 2020;19(1):282–91 doi 10.1158/1535-7163.MCT-19-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev 2002;12(2):162–9 doi 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 33.Frizzell KM, Gamble MJ, Berrocal JG, Zhang T, Krishnakumar R, Cen Y, et al. Global analysis of transcriptional regulation by poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in MCF-7 human breast cancer cells. J Biol Chem 2009;284(49):33926–38 doi 10.1074/jbc.M109.023879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gadad SS, Camacho CV, Malladi V, Hutti CR, Nagari A, Kraus WL. PARP-1 regulates estrogen-dependent gene expression in estrogen receptor alpha-positive breast cancer cells. Mol Cancer Res 2021;19(10):1688–98 doi 10.1158/1541-7786.MCR-21-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curran T, Franza BR Jr. Fos and Jun: the AP-1 connection. Cell 1988;55(3):395–7 doi 10.1016/0092-8674(88)90024-4. [DOI] [PubMed] [Google Scholar]
- 36.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med 2016;375(22):2154–64 doi 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 37.Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, et al. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016;353(6294):45–50 doi 10.1126/science.aaf7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 2003;3(11):859–68 doi 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 39.Mikula M, Gotzmann J, Fischer AN, Wolschek MF, Thallinger C, Schulte-Hermann R, et al. The proto-oncoprotein c-Fos negatively regulates hepatocellular tumorigenesis. Oncogene 2003;22(43):6725–38 doi 10.1038/sj.onc.1206781. [DOI] [PubMed] [Google Scholar]
- 40.Oliveira-Ferrer L, Rossler K, Haustein V, Schroder C, Wicklein D, Maltseva D, et al. c-FOS suppresses ovarian cancer progression by changing adhesion. Br J Cancer 2014;110(3):753–63 doi 10.1038/bjc.2013.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fisler DA, Sikaria D, Yavorski JM, Tu YN, Blanck G. Elucidating feed-forward apoptosis signatures in breast cancer datasets: Higher FOS expression associated with a better outcome. Oncol Lett 2018;16(2):2757–63 doi 10.3892/ol.2018.8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michels J, Vitale I, Senovilla L, Enot DP, Garcia P, Lissa D, et al. Synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. Cell Cycle 2013;12(6):877–83 doi 10.4161/cc.24034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang SH, Xiong M, Chen XP, Xiao ZY, Zhao YF, Huang ZY. PJ34, an inhibitor of PARP-1, suppresses cell growth and enhances the suppressive effects of cisplatin in liver cancer cells. Oncol Rep 2008;20(3):567–72. [PubMed] [Google Scholar]
- 44.Yasukawa M, Fujihara H, Fujimori H, Kawaguchi K, Yamada H, Nakayama R, et al. Synergetic Effects of PARP Inhibitor AZD2281 and Cisplatin in Oral Squamous Cell Carcinoma in Vitro and in Vivo. Int J Mol Sci 2016;17(3):272 doi 10.3390/ijms17030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng YD, Xu XQ, Peng F, Yu JZ, Wu H. The poly(ADP-ribose) polymerase-1 inhibitor 3-aminobenzamide suppresses cell growth and migration, enhancing suppressive effects of cisplatin in osteosarcoma cells. Oncol Rep 2011;25(5):1399–405 doi 10.3892/or.2011.1212. [DOI] [PubMed] [Google Scholar]
- 46.Cseh AM, Fabian Z, Quintana-Cabrera R, Szabo A, Eros K, Soriano ME, et al. PARP Inhibitor PJ34 Protects Mitochondria and Induces DNA-Damage Mediated Apoptosis in Combination With Cisplatin or Temozolomide in B16F10 Melanoma Cells. Front Physiol 2019;10:538 doi 10.3389/fphys.2019.00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghosh U, Bhattacharyya NP. Induction of apoptosis by the inhibitors of poly(ADP-ribose)polymerase in HeLa cells. Mol Cell Biochem 2009;320(1–2):15–23 doi 10.1007/s11010-008-9894-2. [DOI] [PubMed] [Google Scholar]
- 48.Mann M, Kumar S, Sharma A, Chauhan SS, Bhatla N, Kumar S, et al. PARP-1 inhibitor modulate beta-catenin signaling to enhance cisplatin sensitivity in cancer cervix. Oncotarget 2019;10(42):4262–75 doi 10.18632/oncotarget.27008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matulonis UA, Monk BJ. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: does a development pathway forward exist? Ann Oncol 2017;28(3):443–7 doi 10.1093/annonc/mdw697. [DOI] [PubMed] [Google Scholar]
- 50.Thaker PH, Salani R, Brady WE, Lankes HA, Cohn DE, Mutch DG, et al. A phase I trial of paclitaxel, cisplatin, and veliparib in the treatment of persistent or recurrent carcinoma of the cervix: an NRG Oncology Study (NCT#01281852). Ann Oncol 2017;28(3):505–11 doi 10.1093/annonc/mdw635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq datasets generated for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo/) using accession number GSE176326. All other data generated in this study are available within the article and its supplementary data files. Unique materials used in this study are available upon request from the corresponding author.