

Abstract
Rationale:
Patient-derived organoids (PDOs) are a promising technology to support precision medicine initiatives for patients with pancreatic ductal adenocarcinoma (PDAC). PDOs may improve clinical next-generation sequencing (NGS) and enable rapid ex vivo chemotherapeutic screening (pharmacotyping).
Methods:
PDOs were derived from tissues obtained during surgical resection and endoscopic biopsies and studied with NGS and pharmacotyping. PDO-specific pharmacotype is assessed prospectively as a predictive biomarker of clinical therapeutic response by leveraging data from a randomized-controlled clinical trial.
Results:
Clinical sequencing pipelines often fail to detect PDAC-associated somatic mutations in surgical specimens that demonstrate a good pathological response to previously administered chemotherapy. Sequencing the PDOs derived from these surgical specimens, after biomass expansion, improves the detection of somatic mutations and enables quantification of copy number variants. The detection of clinically relevant mutations and structural variants is improved following PDO biomass expansion. On clinical trial, PDOs were derived from biopsies of treatment naïve patients prior to treatment with FOLFIRINOX (FFX). Ex vivo PDO pharmacotyping with FFX components predicted clinical therapeutic response in these patients with borderline resectable or locally advanced PDAC treated in a neoadjuvant or induction paradigm. PDO pharmacotypes suggesting sensitivity to FFX components were associated with longitudinal declines of tumor marker, CA-19–9 and favorable RECIST imaging response.
Conclusion:
PDOs establishment from tissues obtained from patients previously receiving cytotoxic chemotherapies can be accomplished in a clinically-certified laboratory. Sequencing PDOs following biomass expansion improves clinical sequencing quality. High in-vitro sensitivity to standard-of-care chemotherapeutics predicts good clinical response to systemic chemotherapy in PDAC.
Keywords: Pancreatic cancer, precision medicine, organoids, chemosensitivity, pharmacotyping
Translational Relevance Statement
Current approaches to precision medicine in pancreatic ductal adenocarcinoma (PDAC) are limited by the absence of predictive biomarkers for therapeutic response. Despite past work investigating genome-level determinants of chemotherapeutic sensitivity, the lack of targetable genetic mutations and low tumor epithelial cellularity hamper efforts to broadly impact patient care with sequencing alone. We present an alternative vision, in which patient-derived tumor organoid models (PDOs) are studied in a clinical laboratory as predictive biomarkers of chemotherapeutic response. Herein we demonstrate this vision, showing for the first time that PDOs can be integrated into a clinical laboratory setting and that molecular characterization of PDOs generates clinically-actionable genetic data in patients with PDAC. Further, we demonstrate that PDO pharmacotype is a predictive biomarker of clinical chemotherapeutic response in PDAC. Prospective randomized controlled trial data confirming our findings will enable precision medicine and improve prognosis for patients diagnosed with PDAC.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) impacts over 50,000 people annually in the US and is the most lethal adult malignancy(1,2). Systemic cytotoxic chemotherapeutics remain a mainstay of treatment in all stages of disease. Clinical equipoise exists between two standard of care therapeutic combinations, with both FOLFIRINOX (active compounds: 5-fluorouracil, irinotecan and oxaliplatin) and gemcitabine/nab-paclitaxel having been shown to modestly improve survival in advanced disease when compared to single-agent therapies(3,4). Though the overall survival benefit is modest in advanced disease, the selection of an optimal therapeutic strategy in earlier stages of disease can be associated with excellent histopathologic response and clinical outcomes(5,6). Predictive biomarkers of clinical response to systemic chemotherapeutics are urgently needed in PDAC to enable patient stratification and optimal therapeutic selection.
Previous work demonstrates the value of next-generation sequencing (NGS) as a standard of care to identify targetable molecular alterations in PDAC(7). Patients who receive matched therapy for PDAC harboring a targetable mutation have significantly better overall survival than patients who do not receive matched therapy(8). The value of clinical NGS testing, however, is necessarily dependent upon the quality and quantity of malignant epithelium present in a given pathology specimen. A suboptimal clinical specimen impairs the sensitivity of standard NGS techniques to identify mutations with low variant allele frequency. This may be more pronounced in a paucicellular tumor like PDAC, where stromal cells outnumber the malignant epithelial component present in a tissue section.
The capacity to rapidly establish and expand 3-dimensional tissue cultures derived from PDAC has been proposed as a tractable strategy to support precision medicine. These patient-derived organoids (PDOs) reliably demonstrate the molecular characteristics of in vivo disease when genotyped in the research setting. Further, PDOs demonstrate a range of chemosensitivity when pharmacotyped that mirrors the variable response to chemotherapy seen in the clinic. Recent data support the notion that PDO pharmacotyping can be utilized as a predictive biomarker of clinical chemotherapeutic response, associating gene-expression signatures of PDO chemotherapeutic response to clinical PDAC outcomes(9).
Encouraged by the ability to pharmacotype PDOs in a clinically relevant timeframe(10), we hypothesized that the PDOs could serve to improve the performance of NGS employed in the clinical laboratory. We, additionally, set out to prospectively evaluate PDO pharmacotyping as a predictive biomarker of clinical chemotherapeutic sensitivity in the context of a multi-institutional randomized, controlled, clinical trial.
MATERIALS AND METHODS
Patients and pancreatic cancer samples
Patients with a presumed or confirmed diagnosis of PDAC were eligible for enrollment onto IRB-approved tissue acquisition protocols at Johns Hopkins Hospital and Massachusetts General Hospital (MGH). For those undergoing surgery, portions of the tumor were harvested by a research pathologist following resection. Tissue was also obtained during diagnostic endoscopic ultrasound-directed biopsy as part of a multi-institutional clinical trial (NCT03563248). Per NCT03563248 clinical trial protocol, the patients underwent baseline body CT and CA19–9 measurement, following 8 treatment cycles of FOLFIRINOX. Body CT imaging was repeated after cycles 4 and 8 for response evaluation, after which the patients underwent stereotactic body radiation therapy (SBRT) and the CT was repeated. For the retrospective registry evaluation, somatic genome sequencing results for patients undergoing surgical resection for PDAC between November 2017 and April 2020 were evaluated. Written informed consent from patients involved in the work was obtained in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule), and the studies conducted herein were approved by the institutional review boards of all participating centers.
Organoid culture
Organoids were established from surgically resected PDAC tissue harvested from the primary tumor at surgical pathological primary evaluation or from biopsy cores acquired via EUS core needle biopsy, as previously described(10). The surgical specimens were mechanically minced with a scalpel and dissociated enzymatically in a digestion media consisting of culture medium and collagenase XI (Sigma-Aldrich, cat. C9407–1G). EUS core needle biopsies were mechanically dissociated. Single cells were suspended in liquid Matrigel, plated in domes and covered with human complete feeding media. Cultures were passaged for cell line expansion or characterization biweekly. Complete protocol details are in keeping with prior work performed by our group(9). A standardized protocol was written for the CLIA-certified setting (Supplementary material section 7).
Next-generation sequencing
For the solid tumor panel in the discovery set and in the second validation set (CLIA set), DNA was isolated from FFPE specimens after macro-dissection of identified tumor regions from five to ten 4µM unstained slides using the Siemens TPS automated method, (Siemens Healthineers, Malvern, PA) with Versant Tissue Preparation reagents according to vendor specifications. Organoid cell pellets were washed with PBS and manually isolated using Qiagen QIAamp DNA Mini Kit (Qiagen, Beverly, MA). DNA concentration was assessed using the Qubit fluorometer using either DNA-HS or DNA-BR reagents according to vendor specification (Thermo Fisher Scientific, Waltham, MA). The primary tumor DNA underwent standard solid tumor panel sequencing Solid Tumor Panel-v4.0 at Hopkins Molecular Diagnostics Laboratory.
For the whole-exome sequencing, the organoids were pelleted, dissociated with TryplE express, and DNA were extracted using Qiagen QIAamp DNA Mini Kit (Qiagen, Beverly, MA). Whole-exome sequencing libraries were formed and quality controlled for DNA quantity by Picogreen (Life technologies, cat# P7589). Illumina HiSeq with an average coverage of 156 reads was performed. For the first validation set, both primary tumors and organoids were sequenced using Illumina HiSeq with an average yield of 8.87 GB per sample. For the second validation set, both primary tumor and organoids were established and cultured in a CLIA-certified Johns Hopkins Cytogenetics Laboratory facility, and sequenced using Solid Tumor Panel-v4.0 at CLIA-certified Hopkins Molecular Diagnostics Laboratory.
Organoid pharmacotyping
Organoids were dissociated into single cells and plated on a 384-well assay plate in 10% Matrigel. After a 48-hour recovery period, chemotherapeutics were administered using a semi-automated Tecan D300e dispenser (Tecan, Männedorf, Switzerland), normalized to maximum of 1% DMSO (For FOLFIRINOX synergy matrix, 2.5%). Chemotherapeutics were tested across a logarithmically designed curve: gemcitabine, paclitaxel and irinotecan (range: 8.0 × 10−12 mol/L to 2.0 × 10−6 mol/L), and 5-FU and oxaliplatin (range: 1.0 × 10−8 to 1.0 × 10−4 mol/L). Negative controls included wells with DMSO normalization alone. Cell viability was read at 5 days using CellTiter Glo (Promega Corp, Madison, WI, USA) on a luminometer (Spectra-Max, Molecular Devices, LLC, San Jose, CA, USA)(11). Pharmacotyping was performed in four technical replicates for standard pharmacotyping template of the 5 chemotherapeutics.
For the FOLFIRINOX synergy matrix, 5 concentrations (plus zero) of each compound were administered in two technical replicates of each possible combination, and adding at least 10 technical replicates for the concentrations forming the standard combination curves. The template covered three 384-well plates. On each plate, wells were randomized to even possible variation.
Population distributions of the IC50 values of each single agent chemotherapeutic were derived from pharmacotyping 50 distinct organoids. Non-linear curve fitting for data from each organoid tested independently in biological and technical triplicate across a clinically-significant range of chemotherapeutic doses were performed in a manner previously described(9,10). The ranking of each organoid on the distribution was based on the mean estimate of the IC50, simultaneously displaying the confidence interval (95%) of the estimate in the forest plot. The putative clinical chemotherapeutic response was categorized into thirds (sensitive, intermediate, resistant), correlating with the response distribution generally observed during treatment in the clinical care of patients diagnosed with pancreatic cancer. The estimates were categorized in thirds of the distribution, the lowest being the Sensitive, the highest being the Resistant, and those in between being Intermediate.
Bioinformatics
For the discovery set and the first validation set of PDO sequencing, bwa v0.7.7 (mem) was used for running the alignments against hg38 genome. The default parameters were used. Piccard-tools1.119 was used to add read groups as well as remove duplicate reads. GATK v3.6.0 base call recalibration steps were used to create a final alignment file. Copy number ideograms were formed, but only individual variant calls were considered in the report in contrast to the clinical STP pipeline. For the second validation set and STP sequencing in the discovery set, a standard clinical bioinformatic pipeline of the Hopkins Genomics were used, including both variant calls and CNV calls. Variant calls are based on an in-house variant caller (MDL VC 8.0, Johns Hopkins, Baltimore, MD), and a third-party variant caller (Haplotyper Genome Analysis TK-3.3) using the Bayesian statistical model. CNV calls are based on unique read depth of a gene relative to that of all other genes, normalized to a pool of reference samples. All the BAM files were individually reviewed in IGV (Broad Institute, Cambridge, MA, US) to identify variants that were shared or discrepant between the primary tumor and PDO to confirm the presence/absence of the variant in the sample. Viability curves for single-agent pharmacotyping were fitted using nonlinear logistic regression with Prism v8 (GraphPad Software, La Jolla, CA, US), and the corresponding report sheets were extracted as output. For optimized IC50 values, the dose-response sigmoid curves were manually fitted by removing some artefactually low luminometer values at lowest anti-cancer agent concentrations caused by spillover effect. For missing confidence intervals due to steeply vertical sigmoid curve, upper or lower limit was imputed matching the missing distance from the mean to the already existing confidence interval estimate. Population distributions by IC50 values were illustrated by collective analysis of each chemotherapeutic from the extracted Prism reports using a CRAN package forest plot or a CRAN package violin plot. For randomized templates and synergy matrices, Tecan d300E dispenser reports were joined with the luminometer reads of the corresponding wells in R studio and formatted suitable for each bioinformatic package. Combination chemotherapy synergy was analyzed using a publicly available online tool SynergyFinder version 2.0(12).
Ethical approval
The study was approved by the Johns Hopkins Institutional Review Board.
Data availability statement
The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.
RESULTS
Somatic mutations are challenging to detect in biopsies and surgical specimens obtained from patients with PDAC who were treated with prior therapy
Previous translational research has suggested that sequencing a PDO derived from expanded tumor epithelium may increase sensitivity for identification of clinically-relevant mutations(13). In an effort to identify which patient groups would benefit most from mutational profiling in PDOs, we performed a retrospective registry analysis of clinical NGS detection of somatic PDAC gene variants. Two-hundred ninety (290) patients who underwent resection for PDAC with NGS data generated in our clinical, Clinical Laboratory Improvement Amendments (CLIA) approved, laboratory were included.
The prevalence of common PDAC variants – KRAS, TP53, and SMAD4 – were examined. Mutations in these key driver genes are frequently encountered in the clinical setting and wild-type gene calls can be assessed as a surrogate for low-sensitivity of the clinical NGS pipeline in any given patient specimen. To assess for patient-specific risk factors for low NGS sensitivity, we characterized the clinical and pathological factors associated with wild-type calls in our clinical practice data. Clinicopathological characteristics of this registry-based cohort are presented in supplementary table 1. Tissue quantity, whether low as a result of needle biopsy or high when derived from surgical bulk tumor, was not associated with a statistically significant difference in the capacity to detect somatic mutations (Fig. 1A). Factors impacting tissue quality, specifically those that impair tumor epithelial fitness such as prior chemotherapeutic and radiotherapeutic stress, were associated with impaired capacity to detect somatic mutations (Fig. 1B-C). These findings were particularly striking when evaluating the association between mutation detection rate and pathological response to neoadjuvant therapy, as measured by the College of American Pathologists (CAP) grading scheme for tumor response in post-therapy specimens (Fig. 1D)(14). In those patients with a CAP grade demonstrating a favorable response to prior treatment, and therefore associated with a low ratio of malignant epithelium to surrounding normal stroma, the capacity to detect tumor-associated gene mutations was impaired. Specifically, the post-treatment tumor sections from patients with superior responses to therapy were more than twice as likely to have clinical sequencing that suggested somatic wild-type KRAS and TP53 (Fig. 1D). For SMAD4, the frequency of a wild-type call increased by nearly 15%. To ensure that a broader effect on sequencing sensitivity was not driving the detection of somatic variants, we assessed the detection of common germline variants as a negative control (data not shown). Tissue specimen type, the receipt of therapy before tissue sampling, and pathological response to therapy were not associated with the capacity to detect common germline variants in sequenced tissues.
Figure 1.
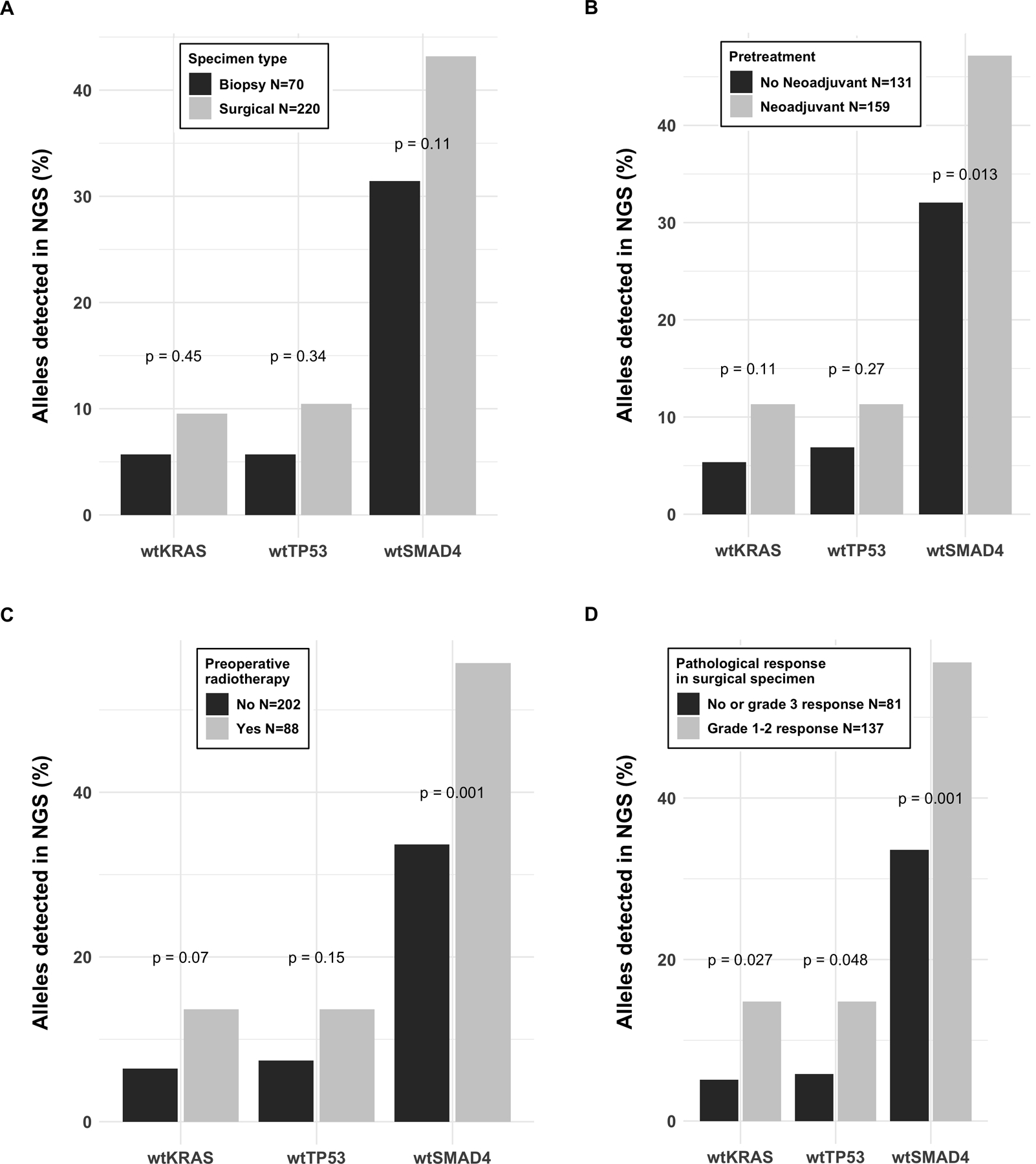
Frequency (%) of the wild type gene variant reported as a result of the clinical NGS panel of the primary pancreatic ductal adenocarcinoma in a registry cohort. Specimen type (biopsy or resection) was not significantly associated with the frequency of the wild type gene reported (A), whereas the wild type findings were more commonly reported after pretreatment by chemo- (B) or radiotherapy (C), and good response to pretreatment in the surgical pathology report (D).
PDO sequencing improves sensitivity for detection of clinically-relevant somatic mutations in PDAC
We next examined the capacity of tumor-derived PDO tissues to improve the sensitivity of clinical methods to detect PDAC-specific somatic mutations. We first evaluated the concordance between sequencing of paired primary tumor and PDO samples using both clinical laboratory protocols and research-specific methods. In a discovery set of 11 tumor-PDO pairs, the CLIA NGS results were compared to PDO whole-exome sequencing (WES). Mutations were common using both techniques with a variant co-detection rate of 80% (Fig. 2A). When the findings were manually curated from sequence reads outside the thresholds introduced in the CLIA bioinformatics pipeline, 8% of uncommon variants were identified in PDO-derived data, and 12% in the primary tumor data only. The underlying etiology for sequencing discordance between primary tumor and PDO methods, including the absence of detection of a variant, a low allele frequency not meeting threshold for mutation call in our bioinformatics pipeline, and others, are detailed in Supplementary Table 2.
Figure 2.
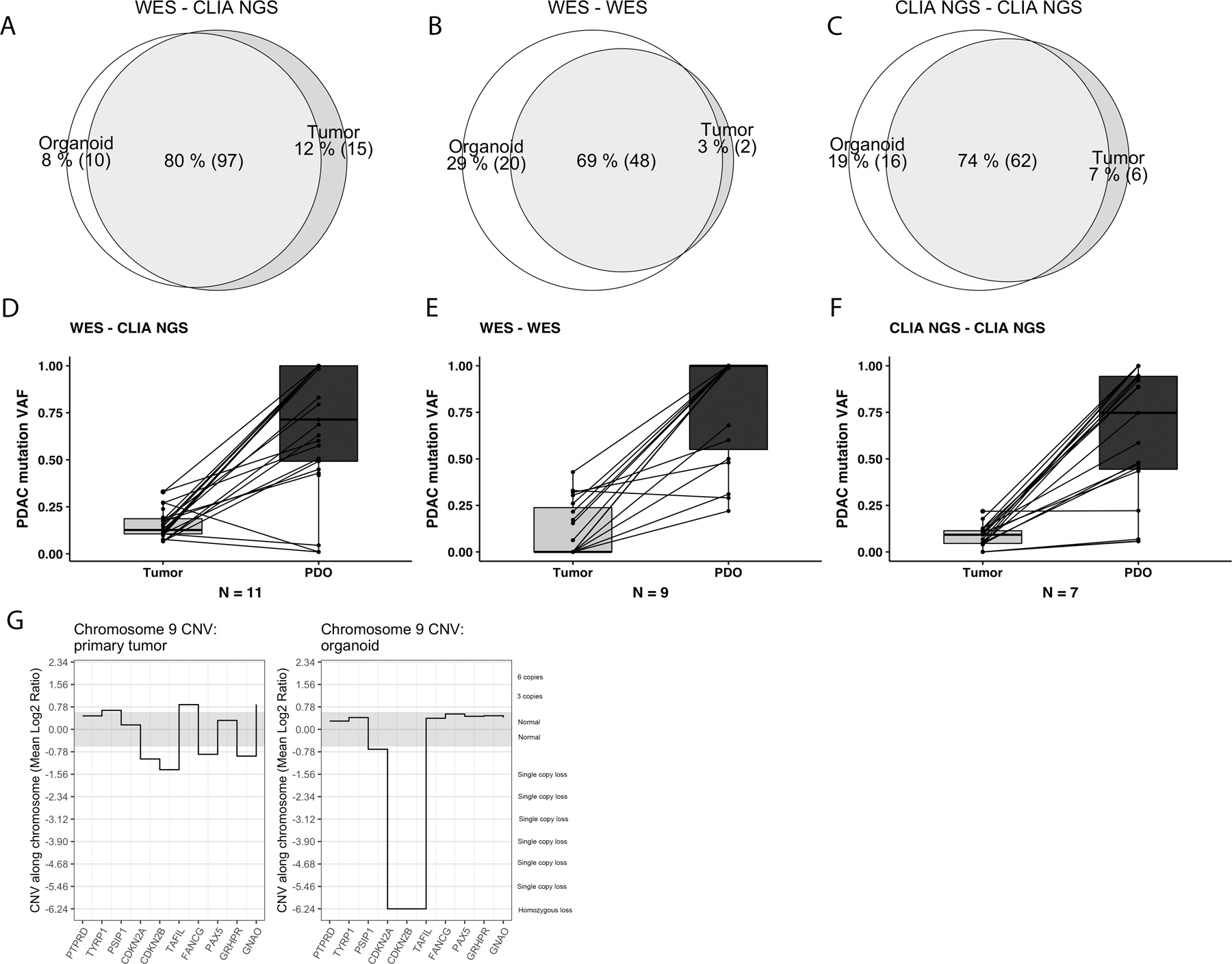
Mutated gene variants detected in three independent datasets by CLIA-approved next generation sequencing of primary tumors and patient-derived organoids (PDO) established from the same primary tumor. In discovery set (A), whole exome sequencing of the PDO was compared to CLIA solid tumor panel (STP) of the primary tumor. In validation set 1 (B), whole-exome sequencing (WES) was performed on both primary tumor and organoid, and in CLIA validation set 2 (C), the CLIA-approved NGS-STP methods were performed for both primary tumor and organoid. The detection of variant allele frequencies (VAF, %) for typical pancreatic cancer driver mutations (KRAS, TP53, SMAD4 and CDKN2A) are substantially higher in PDOs than in primary tumors in discovery dataset (D) and validation datasets 1 (E) and 2 (F). Relevant pancreatic cancer mutations detected by WES of the primary tumor and the corresponding organoids established from the same tumor in the validation set 1 (G). A homozygous copy number loss of the chromosomal area of the CDKN2A gene in the organoid compared to primary tumor. Though demonstrating a slight decrease in CNV, clinically-relevant bioinformatics pipelines would fail to call CDKN2A loss in the primary tumor.
The capacity of PDO NGS to supplement current clinical laboratory methods was validated in two separate patient cohorts. In the first (Fig. 2B), we directly compared WES performed from direct patient tumor samples with WES performed from matched PDOs. WES resulted in limited coverage (mean 36x for primary tumors; 34x for organoids) and, relative to targeted NGS, significantly hampered the sensitivity of our bioinformatics pipeline. In this first validation set, 69% of variants identified were called from both PDO and primary tumor WES data. This validation set suggested that a greater proportion of calls, 29%, were able to be detected in the PDO data alone, and only 3% from the primary tumor only (Fig. 2B). We suspect that this difference was in part due to limited sequencing depth. When the reads were manually verified using the Integrative Genomics Viewer (IGV, Broad Institute, CA, US), most of the variant calls were missed from the primary tumor data because of a low variant allele frequency that failed to meet the threshold for calling in our bioinformatics pipeline (Supplementary Table 2). These data prompted evaluation of PDO generation and molecular characterization wholly within a clinical laboratory environment.
PDOs in clinical laboratories facilitate improved, clinically-relevant, molecular characterization
With an understanding of the tissue factors impacting sensitivity of NGS in our CLIA-Laboratory (Fig. 1), and initial data to suggest organoid sequencing improves sensitivity for mutation calling in our pipeline (Fig. 2A-B), we developed standard operating procedures for direct molecular testing of PDOs in our CLIA facility. An IRB-approved pilot clinical trial of CLIA organoid establishment followed by clinical NGS was conducted for 20 patients undergoing surgical resection for PDAC. Notably, this work focused on individuals at high-risk for NGS failure and featured patients previously exposed to treatment with chemotherapy and radiotherapy (19/20; 95%).
PDO characterization followed previously established nomenclature(10). PDOs were successfully established in 18/20 (90%) in the CLIA laboratory environment. Successful passage through the expansion phase and to characterization with NGS was successful in 13 PDOs. Characterization was considered following expansion of cystic or solid organoids to a biomass threshold occupying a mean of eleven 50 µL Matrigel domes. The median time needed in culture after surgical resection to reach DNA extraction was 28 days. DNA was extracted from PDOs at the time of the first passage (14 days) in seven cases, at the time of the second passage in four cases, and at eighth passage in two cases (56 days in culture). PDO DNA was transferred to the CLIA molecular diagnostics laboratory for NGS alongside DNA extracted from macrodissected H&E sections of the primary tumor – in keeping with the protocols of our established clinical NGS pipeline. Data from the primary tumor were used as a positive control.
DNA extraction in early passage cultures (i.e., during the establishment phase, passage 1 or 2) resulted in a mean total DNA from each sample of 1710 ng. In keeping with prior data from both our experience and others, the detection of somatic mutations can be a challenge in these early cultures prior to malignant biomass expansion. Within the first two passages, seven of eleven (64%) PDOs demonstrate mutant variant allele frequencies below the bioinformatics threshold to call. In some, robust growth of large cystic organoid structures expanding throughout the Matrigel domes were apparent (Supplementary Fig. 1) and in each of these cases our control analysis verified germline concordance with primary tissues obtained from the patient. In these cases, sequencing demonstrated early and robust normal ductal epithelial growth in culture as only germline variants were readily detected. The presence of malignant tissues in these early heterogenous cultures was confirmed by manually inspecting the genomic positions of key driver mutations in NGS data. In four of the seven found to have predominantly germline signatures, somatic mutations in KRAS and TP53 were detected manually with 1–4% variant allele frequency (below the 5% call limit of our clinical bioinformatics pipeline). Repeat genetic analysis following the expansion phase of organoid growth commonly demonstrated high VAF in somatic driver mutation hotspots, corresponding to a decline in the growth rate of the normal ductal epithelial clone in culture over time.
Mutational profiles were directly compared between PDO and matched primary tumor using CLIA approved NGS. Similar to results obtained in using research-specific methods, sequencing data derived from the PDO proved more sensitive than data from standard FFPE histopathologic tissues for the detection of somatic variants of clinical interest (Fig. 2C). When specifically focused on the detection of typical somatic driver mutations associated with PDAC (KRAS, TP53, SMAD4 and CDKN2A), increased mutant VAFs are detected using standard bioinformatics pipelines for PDOs compared to primary tumor in all three datasets (Fig. 2D-F). Supporting a call for PDO-based precision medicine, we found several clinically-relevant mutations with PDO NGS that remained undetected by clinical NGS performed on primary tumor. Examples include KRAS p.Gly12Asp in JHH 214 and TP53 p.R363fs in JHH 217 (Supplementary Fig. 2).
In contrast to mutational profiling, PDO sequencing is particularly valuable in the assessment of copy number alterations. A tumor sample sequenced with high-coverage targeted NGS, for example, can fail to detect heterozygous loss of tumor suppressor genes due to the high ‘contamination’ ratio from normal cells present in portion of tumor sample harvested for analysis. In some samples with low tumor cellularity, even a homozygous loss can be difficult to call. When leveraging PDOs to enrich the malignant biomass before clinical sequencing, however, many of these tumor suppressor gene losses become easy to call by standard bioinformatics pipelines. One common region of interest in PDAC is the potential homozygous loss of CDKN2A on chromosome 9 (Fig. 2G, Supplementary Fig. 3). A homozygous loss of CDKN2A was detected in four out of seven organoids but was not apparent in the sequencing data from the corresponding primary tumors. Similar examples were seen for TP53 on chromosome 17, an amplifying gain of PALB2 on chromosome 16, and other significant structural variation throughout the cancer genomes (loss of heterozygosity of chromosome 17 p arm was present in three and 18 q arm in five PDOs out of seven). The ability to detect structural variations from NGS increased alongside increasing somatic VAF, reflecting the expansion of malignant biomass in the culture. Taken together, these data suggest that PDO-based sequencing significantly improves mutant VAF detection in clinical pipelines for PDAC. Adopting PDOs into CLIA settings may improve the clinical ability to detect mutations associated with targeted therapeutic options.
PDO pharmacotyping in precision medicine
Single agent pharmacotyping was performed for each PDO against standard-of-care chemotherapies used to treat patients with PDAC. Individual dose-response curves were generated and analyzed as previously described(9,10) (Fig. 3A, Supplementary Fig. 4). Data were aggregated to derive an empirical population distribution of chemotherapeutic sensitivity to each agent (Fig. 3B-F). Sensitivity was assessed using the mean and 95% confidence interval of the half maximal inhibitory concentration (IC50) value for each chemotherapeutic. Clinical correlation was used to estimate putative breakpoints for in vitro classification of chemotherapeutic sensitivity (sensitive, intermediate or resistant).
Figure 3.
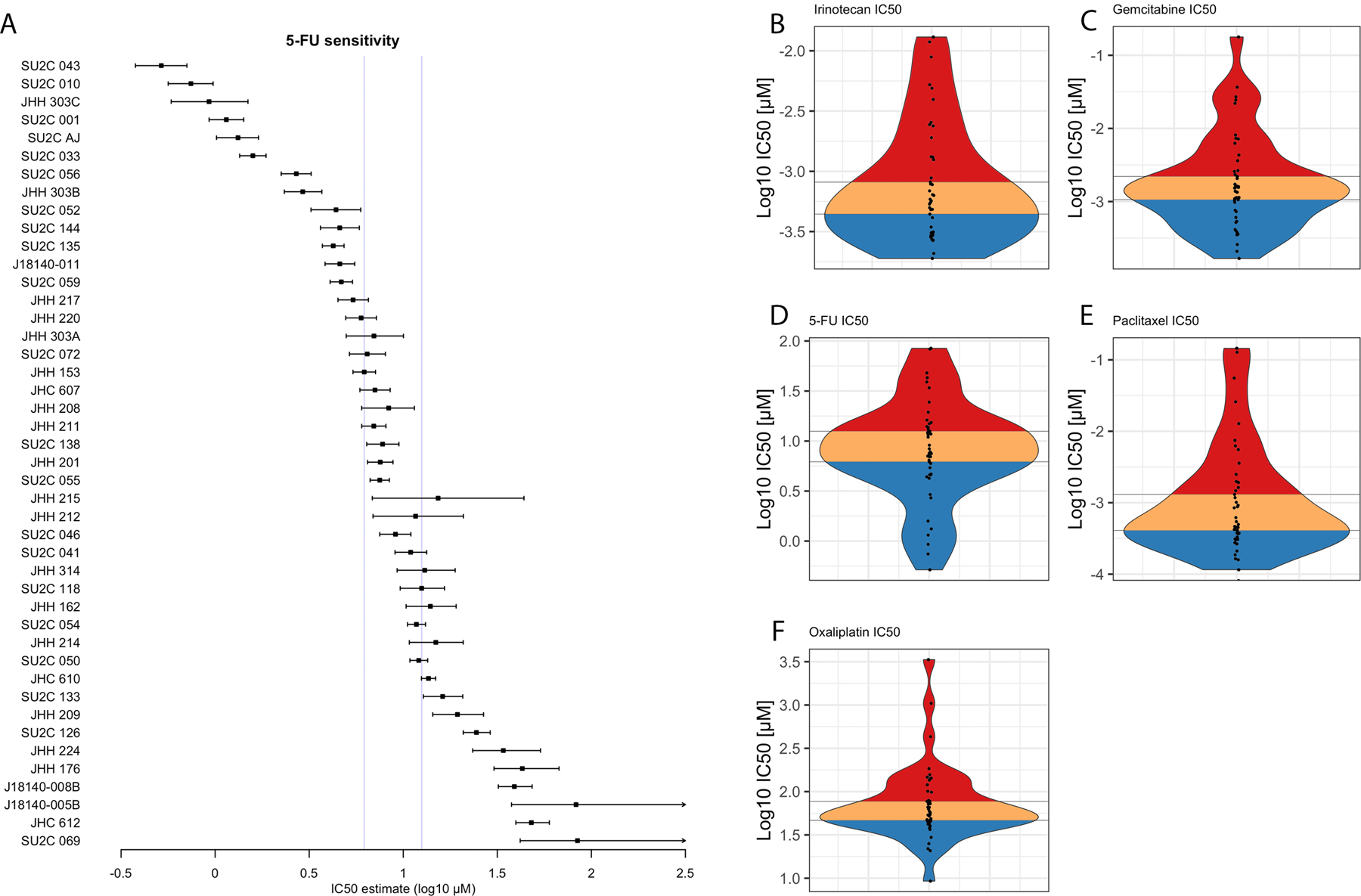
Pharmacotyping-derived population distribution of PDOs (left axis label) showing half maximal inhibitory concentration (IC50) and 95% confidence interval in dose-response testing against 5-fluorouracil (5-FU (A). A population distribution of mean PDO IC50 values presented as a violin plot for Irinotecan (B), 5-FU (C), Oxaliplatin (D), Gemcitabine (E) and Paclitaxel (F). Putative clinically-relevant cohorts are categorized in thirds as sensitive (blue), intermediate (orange) and resistant (red) to the chemotherapeutic described.
In addition to single agent pharmacotyping, we built a combinatorial pharmacotyping pipeline to evaluate for synergy and antagonism of two commonly administered combination chemotherapies, gemcitabine–nab-paclitaxel (GA) and FOLFIRINOX (5-FU, irinotecan-SN38, oxaliplatin), using Synergyfinder v2(12). With GA treatment, cellular fitness and survival closely mirrored what would be expected by a model accounting purely for the additive effect of the compounds at each dose (Fig. 4A-B, overall zip score 0.836). Delta scoring remained modest across the clinical dose ranges explored and, in some cases, would modulate between mild synergy and mild antagonism at doses that varied by less than one log concentration (Supplementary Fig. 5). Similarly, no synergistic effects were observed with coadministration of 5-FU, SN-38 and Oxaliplatin (Fig. 4C-F, zip synergy scores confined between −10 and 10).
Figure 4.
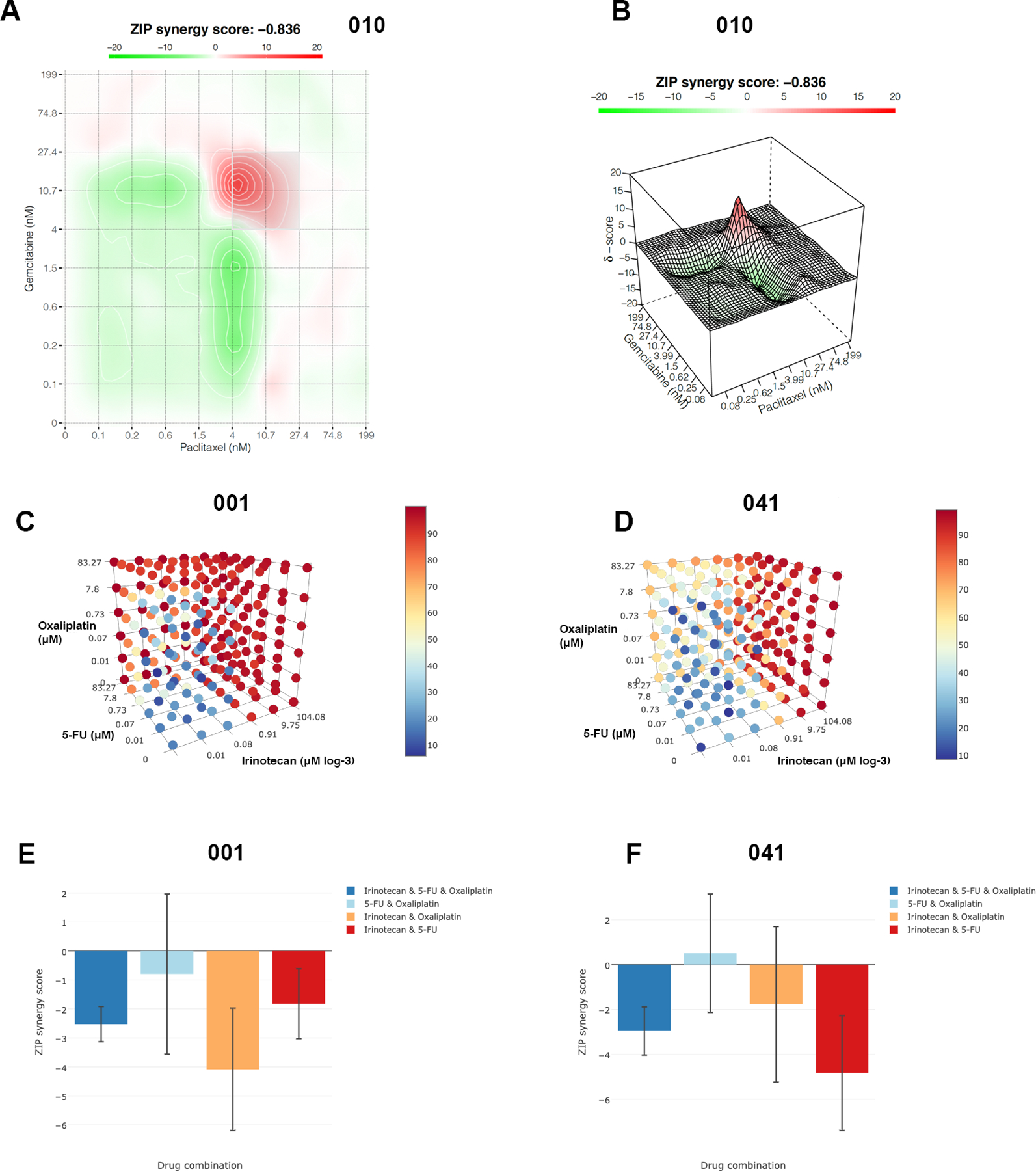
Synergy estimates of Gemcitabine and Paclitaxel combination in organoid ex vivo pharmacotyping of case 010 presented in 2D plot (A) and 3D surface plot (B). The red areas reflect synergy of the axis concentrations by ZIP synergy score. Despite the red peak, the result shows practically no synergistic effect of the selected drug combination. Synergy tensor cubes showing the viability-based inhibition after ex vivo pharmacotyping by the combinations of the three drugs (5-FU, Irinotecan and Oxaliplatin) for a more sensitive culture 001 (C) and for a more resistant culture 041 (D). ZIP synergy scores (E-F) after combination pharmacotyping of 5-FU, Irinotecan and Oxaliplatin. Negative ZIP scores throughout reflect the lack of synergy of the drugs in the combination.
To evaluate the utility of PDO pharmacotyping as a predictive biomarker of therapeutic response in PDAC, we generated organoids from endoscopic biopsies in newly-diagnosed, localized PDAC patients enrolled on a prospective, randomized controlled trial of losartan in combination with FFX (NCT03563248). These treatment-naïve specimens were used to generate PDOs and patient-specific putative chemotherapeutic sensitivity was hypothesized using pharmacotype. Given the negative results of synergy/antagonism work done prior, we utilized each of the three single compound distributions in FFX to assign putative in vivo response as being sensitive, intermediate or resistant. The clinical response to FFX for each patient was determined using longitudinal measurements of serum carbohydrate-antigen 19–9 (CA19–9) and bi-monthly cross-sectional imaging. PDO pharmacotyping was compared to the clinical treatment response to assess for the capacity for PDO pharmacotype to serve as a predictive biomarker of clinical chemotherapeutic response.
Eight of 12 patients demonstrated a favorable clinical response to chemotherapy, defined by a decrease in CA19–9 value by at least 40% of the baseline value (Fig. 5A). In keeping with our core hypothesis, PDOs grown from patients with the most robust clinical response to FFX had ex vivo pharmacotypes suggesting sensitivity to all three components of FFX (Fig. 5A). Five of the 7 most responsive CA19–9 decreases were seen in patients whose PDO was sensitive to all three components as single agents. Of the 5 patients with minimal to no CA19–9 change after starting FFX, none had a corresponding PDO suggesting FFX sensitivity. When limiting the analysis to patients with an elevated CA19–9 value (74 U/mL or greater at baseline(15)), the clinical CA19–9 response correlated directly with pharmacotype (Supplementary Fig. 6). In fact, the proportion of each clinical response mirrored the proportion of agents in FFX that the corresponding patient’s PDO was sensitive to ex-vivo. Similarly, all patients with a PDO sensitive to all three components showed a tumor volume reduction graded as partial response by the RECIST criteria (Fig. 5B). The clinical opportunity to dramatically improve patient care is highlighted by the patients in whom a PDO pharmacotype suggests sensitivity to GA, despite resistance to FFX. Our prospectively collected data would suggest these patients would have benefited from administration of an alternative systemic therapy.
Figure 5.
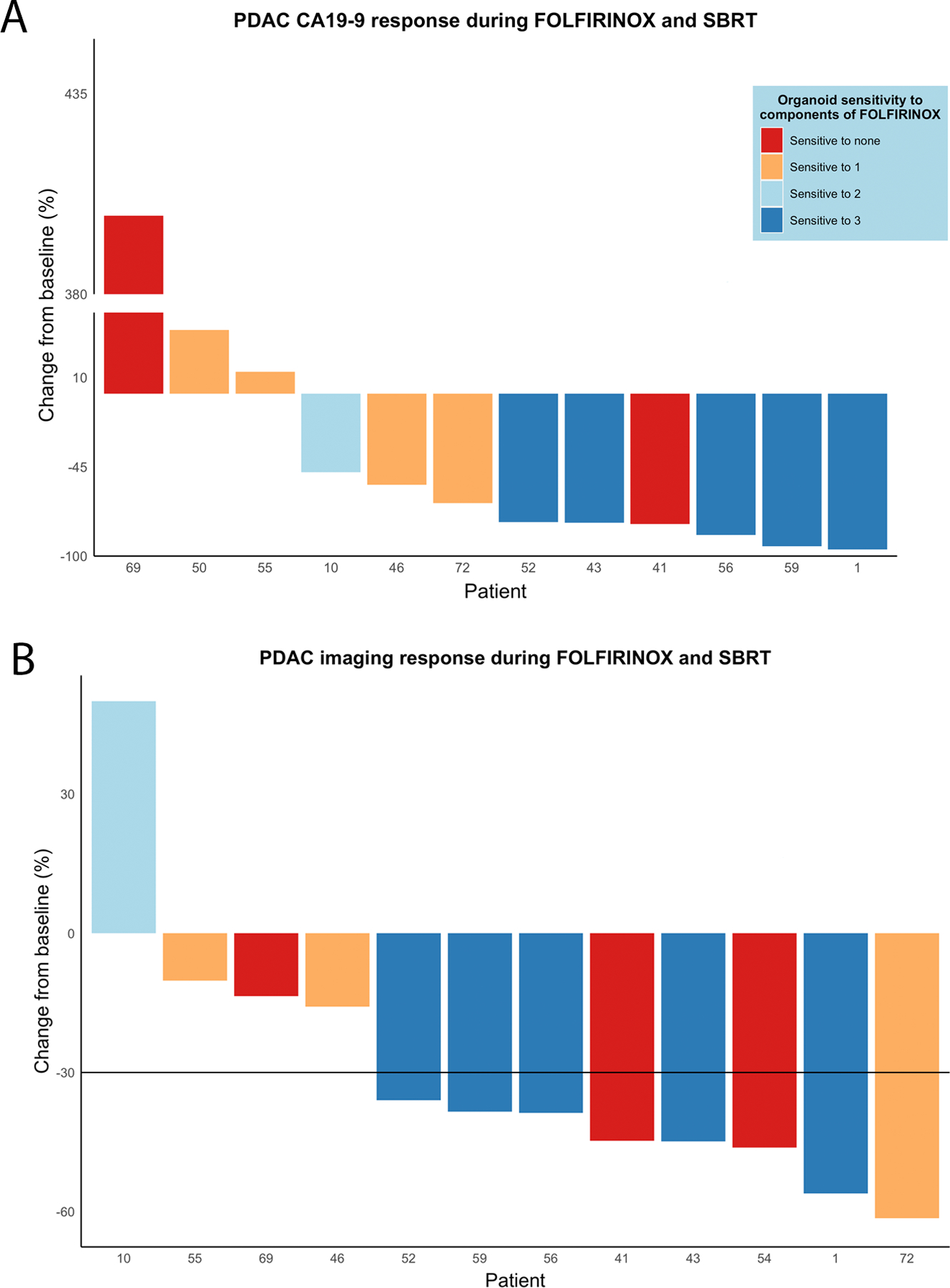
A waterfall plot showing patients’ CA19–9 responses to FOLFIRINOX as percentage change during the time period of neoadjuvant therapeutic administration (A). Only patients clearly producing plasma CA19–9 were included, whereas one patient with a CA19–9 baseline level of 3 U/mL was excluded. Sensitivity of the patients’ organoid in single agent pharmacotyping to each three compounds of FOLFIRINOX are presented by colors of the bar: dark blue = sensitive to all three drugs, light blue = sensitive to two drugs, orange = sensitive to one drug, and red = sensitive to none of the three drugs. Waterfall plot presenting the tumor volume change in computed tomography imaging (RECIST response) during the period of neoadjuvant therapy (B).
DISCUSSION
PDAC is a devastating malignancy with an urgent clinical need for breakthroughs in key areas to improve patient outcomes. Early detection and novel therapeutic development continue to be fields of great interest. Pre-malignant or early-stage tumor detection can allow for curative resection in a high-volume surgical setting with mortality rates below 1%(16). Novel therapeutic development, particularly in targeting mutant KRAS variants in a manner akin to that done for KRAS G12C, remains ongoing(17,18). Until such time as novel therapeutics are brought to market, the most favorable outcomes will continue to be limited to the cohort of surgical patients, approximately 30%, that have uniquely sensitive tumors to standard combination chemotherapy. The most tractable immediate strategy to broadly improve patient outcomes is in the identification of predictive biomarkers for clinical chemotherapeutic response.
Here, we present two novel findings arising from a multi-institutional randomized, prospective clinical trial. First, we demonstrate the capacity of rapidly established PDO models of disease to serve as reliable patient-specific predictive biomarkers of clinical chemotherapeutic response. Second, we present the first prospective experience with PDAC PDOs in a CLIA laboratory for the detection of unique clinically-actionable genetic mutations after surgical resection. Taken together, these data advocate for the rapid translation of organoid technologies, currently used in the United States only for basic science research, into the clinical environment.
Our work in studying a large living biobank of PDAC PDOs with pharmacotyping enabled the derivation of a clinically-relevant population distribution for putative clinical response. Using an ongoing clinical trial, we demonstrate that PDO pharmacotyping is an accurate predictive biomarker for each patient’s clinical response to treatment with standard-of-care cytotoxic chemotherapeutics. Specifically, the best clinical responses to FFX were seen when a patient’s PDO pharmacotyping profile indicated sensitivity to each of FFX’s single-agent compounds. Clinical chemotherapeutic response was tracked in a prospective manner consistent with best practices, using both the PDAC-specific tumor marker, CA 19–9, and well-validated imaging criteria (RECIST). In supporting the hypothesis that PDO pharmacotyping can serve as a critical predictive biomarker of chemotherapeutic response to FFX, follow-on studies are warranted to evaluate a wider variety of clinically available cytotoxic chemotherapeutics. In this model, PDO pharmacotyping can be utilized as a stratifying tool to identify an optimal therapeutic combination for each individual patient. This technique may be ideal for patients with resectable tumors electing to pursue a surgery-first approach to disease management, as the post-operative convalescence would allow time for pharmacotyping to be completed and guide adjuvant chemotherapeutic selection.
There have been multiple efforts to study correlation between PDO in vitro dose-response and observed clinical response to chemotherapy in various cancer types(19), some with encouraging retrospective results in gastrointestinal cancer(20,21). In PDAC, all previous attempts have been retrospective evaluations that included myriad chemotherapeutic types started only after initiation of an organoid culture(9,22,23). We were able to utilize a randomized clinical trial where the organoids were established from a treatment naïve primary tumor, and all patients received a standardized clinical course of chemotherapy with FFX. The highly-curated clinical RCT dataset provided an ideal setting to evaluate PDO pharmacotyping as a predictive biomarker of chemotherapeutic response. Further, the work presented opportunities to improve the processes required to successfully establish organoid models in a clinical setting. We also report the first experience with a PDAC PDO pipeline in a CLIA laboratory, highlighting a remarkably high success rate through the establishment phase of development. Currently approved CLIA-approved methods for molecular characterization in PDOs is limited to sequencing efforts. The data presented here support the notion that a CLIA pharmacotyping assay for routine use is urgently needed.
Pharmacotype-guided chemotherapeutic selection in PDAC is a tractable strategy for precision medicine and remains an area of continued research interest. In our work, we found that the cytotoxic effects of combination chemotherapies, like those used in most gastrointestinal malignancies, are likely not related to synergistic effects of the agents, but rather can be accounted for by the cumulative effects of each agent individually. These data are consistent with those found by others in prior work focused on mathematical modeling of clinical chemotherapeutic effect (24). In one case tested using combination regimens, a narrow range of synergy was identified between gemcitabine and nab-paclitaxel. Notably, the synergism was limited to less than one log in the dose range for both agents with a rapid transition to mild antagonism detected. With a particular eye towards clinical translation, it would likely pose a challenge to recapitulate this precise dosage and elicit synergy in the clinical setting. In the remainder of PDOs challenged with multiagent pharmacotyping, no synergy was elicited when testing gemcitabine and nab-paclitaxel, or the components of FFX in 2- or 3-drug combinations. We have found that full combination matrices of multiple agents are costly, time-consuming, and require substantially more organoid biomass to complete. Our work here suggests unique patient-specific sensitivities can be detected by pharmacotyping performed with traditional methods (single-agent).
Another appealing clinical application for PDOs is utilizing them in selected cases to amplify the malignant epithelial cancer cell compartment for downstream clinical molecular, or genetic, characterization (Figs. 1 and 2). This is especially appealing in paucicellular tumors such as PDAC for methods based on genetic characterization such as NGS. We catalogued KRAS, and other common somatic driver mutations in PDAC, as surrogates of sensitivity during CLIA NGS. The difference in somatic variants detected between pretreated and treatment naïve PDACs, and between poor and good chemotherapeutic response, indicates that the sensitivity of sequencing methods to detect variant may be compromised in certain cases due to malignant epithelial quantity or fitness. Our current and previous findings support the hypothesis that PDO methods enrich the ratio of extracted DNA towards a greater representation of the malignant epithelial cell compartment at the expense of non-malignant components of the tumor microenvironment. When sequenced, this enrichment results in higher somatic variant allele frequencies and a greater number of mutation calls resulting from CLIA bioinformatics pipelines. This approach is directly applicable with major clinical significance when mutation calls identify targetable genes in this era of precision medicine. Further, PDO-based sequencing methods may be the only reliable way in paucicellular tumors to detect targetable genomic changes driven by copy number variation and gene rearrangements (Fig. 2G). Despite this potential utility we are mindful that PDOs may convert clinical chemotherapeutic selection into a black box problem with enough clinical fidelity to, at times, omit NGS in the clinical pipelines that support precision medicine initiatives(25). Further, PDO-based study appears to be a tractable strategy to explore non-genetic mechanisms of heterogeneity and chemotherapeutic response.
The work here has several limitations and opportunities for continued study. Despite being the largest prospective characterization of PDO as a potential predictive biomarker of clinical response, the number of evaluable subjects was limited by enrollment and in translational research abruptly halted in response to the emergence of the COVID-19 pandemic. Opportunities for continued improvement include refinement of the technical and logistical standards needed to carry out this multi-institutional work. With ongoing work, it will be interesting to assess the degree to which the population distribution of chemotherapeutic response derived in our laboratory will be generalizable to others or, perhaps, be dependent upon a more complex tumor model that incorporates other components of the tumor microenvironment. The need to complete a large pharmacotyping effort and derive a laboratory-specific population distribution of response would significantly hinder clinical adoption of the technology across other institutions and laboratories. The patient data utilized to assess the prognostic capacity of PDOs is further limited by two things. First, CA19–9 for patients presenting with low initial values, including Lewis antigen negative patients, limited the number of patients on study who were evaluable. Secondly, there are data to suggest that, unlike in metastatic disease, imaging characteristics of patients with borderline resectable and locally-advanced disease stage do not reflect the underlying response of the tumor to chemotherapy(26,27). With this in mind, RECIST criteria may not be an ideal measure of chemotherapeutic response in this work and underlies our decision to focus predominantly on the longitudinal response of CA19–9 over time.
Finally, our data suggest that a focus on particularly early, rapid expansion examples of a PDO (i.e., first 2–3 weeks in culture) may result in normal ductal epithelial expansion prior to malignant cell expansion. With this in mind, the ideal timing for PDO-based molecular characterization for precision medicine remains to be fully explored. Nevertheless, as we have previously demonstrated, PDOs can be established, expanded, and characterized from existing tissue acquisition protocols within current frameworks of care within a clinically relevant timeframe(10). In pancreatic cancer patients undergoing surgery as a first treatment approach, for example, PDO generation and pharmacotyping can be expedited and completed during the course of a patient’s routine recovery from an operation enabling personalized chemotherapeutic treatment to be administered in the adjuvant setting.
CONCLUSION
PDOs can be generated in a CLIA-certified laboratory setting from pretreated tumors with acceptable success rate, in a clinically meaningful time span, and in a manner that facilitates precision medicine approaches to difficult to treat cancers. Further, PDOs, through pharmacotyping, can serve as a predictive biomarker for clinical therapeutic response to standard-of-care chemotherapeutics. PDO sensitivity ex-vivo predicts good prospectively observed patient clinical response to combination chemotherapy in PDAC.
Supplementary Material
Acknowledgements
The authors wish to thank Elana Fertig Ph.D., Department of Oncology, Division of Biostatistics and Bioinformatics, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, for key support in bioinformatics and statistical analysis in the completion of this work, and Professor Andrew J. Ewald, Ph.D. Department of Cell Biology at Johns Hopkins University, for a comprehensive review and suggestions for the manuscript.
TT Seppälä was supported by fellowship grants and research funding from Sigrid Juselius Foundation, Instrumentarium Science Foundation, Emil Aaltonen Foundation, Jane and Aatos Erkko Foundation, Relander Foundation, and the iCAN precision medicine flagship of the Finnish Academy. JW Zimmerman is supported by 5T32-CA009071. RA Burkhart is supported by the National Institutes of Health/National Cancer Institute (K08CA248710) and a Stand Up To Cancer–Lustgarten Foundation Pancreatic Cancer Interception Translational Cancer Research Grant (grant number SU2C-AACR-DT26–17). Stand Up To Cancer (SU2C) is a division of the Entertainment Industry Foundation and funding is administered by the American Association for Cancer Research, the scientific partner of SU2C.
Footnotes
Conflict of Interests Disclosure Statement
TT Seppälä is the CEO and co-owner of Healthfund Finland; reports an interview honorarium from Boeringer Ingelheim.
DP Ryan is a consultant/advisory board member for MPM Capital, Gritstone Oncology, Oncorus, Maverick Therapeutics, 28/7 Therapeutics, Thrive/Exact Sciences; has equity in MPM Capital, Acworth Pharmaceuticals, and Thrive/Exact Sciences; is a legal consultant for Boeringer Ingelheim; and serves as author for Johns Hopkins University Press, UpToDate, McGraw Hill.
DT Ting is a consultant/advisory board member for Pfizer, ROME Therapeutics, Merrimack Pharmaceuticals, Ventana Roche, Nanostring Technologies, Inc., Foundation Medicine, Inc., and EMD Millipore Sigma, which are not related to this work; is a founder and has equity in PanTher Therapeutics, ROME therapeutics, and TellBio, Inc., which are not related to this work; and has research funding from ACD-Biotechne, PureTech Health LLC, and Ribon Therapeutics, which was not used in this work. DT Ting’s interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict-of-interest policies.
References
- 1.Howlader N, Noone A, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review, 1975–2016 Natl. Cancer Institute. Bethesda, MD. 2019. Available from: https://seer.cancer.gov/csr/1975_2016/ [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res 2014;74:2913–21. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med 2018;379:2395–406. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He J, Blair AB, Groot VP, Javed AA, Burkhart RA, Gemenetzis G, et al. Is a Pathological Complete Response Following Neoadjuvant Chemoradiation Associated with Prolonged Survival in Patients with Pancreatic Cancer? Ann Surg 2018;268:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Golan T, Barenboim A, Lahat G, Nachmany I, Goykhman Y, Shacham-Shmueli E, et al. Increased Rate of Complete Pathologic Response After Neoadjuvant FOLFIRINOX for BRCA Mutation Carriers with Borderline Resectable Pancreatic Cancer. Ann Surg Oncol 2020;27:3963–70. [DOI] [PubMed] [Google Scholar]
- 7.Pishvaian MJ, Bender RJ, Halverson D, Rahib L, Hendifar AE, Mikhail S, et al. Molecular profiling of patients with pancreatic cancer: Initial results from the know your tumor initiative. Clin Cancer Res 2018;24:5018–27. [DOI] [PubMed] [Google Scholar]
- 8.Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol 2020;21:508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov 2018;8:1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seppälä TT, Zimmerman JW, Sereni E, Plenker D, Suri R, Rozich N, et al. Patient-derived Organoid Pharmacotyping is a Clinically Tractable Strategy for Precision Medicine in Pancreatic Cancer. Ann Surg 2020;272:427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burkhart RA, Baker LA, Tiriac H. Testing susceptibility of patient-derived organoid cultures to therapies: Pharmacotyping. Methods Mol Biol Humana Press Inc.; 2018. page 253–61. [DOI] [PubMed] [Google Scholar]
- 12.Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 2020;48:W488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romero-Calvo I, Weber CR, Ray M, Brown M, Kirby K, Nandi RK, et al. Human organoids share structural and genetic features with primary pancreatic adenocarcinoma tumors. Mol Cancer Res 2019;17:70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SM, Katz MHG, Liu L, Sundar M, Wang H, Varadhachary GR, et al. Validation of a Proposed Tumor Regression Grading Scheme for Pancreatic Ductal Adenocarcinoma After Neoadjuvant Therapy as a Prognostic Indicator for Survival. Am J Surg Pathol 2016;40:1653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai S, George B, Wittmann D, Ritch PS, Krepline AN, Aldakkak M, et al. Importance of Normalization of CA19–9 Levels following Neoadjuvant Therapy in Patients with Localized Pancreatic Cancer. Ann Surg 2020;271:740–7. [DOI] [PubMed] [Google Scholar]
- 16.Canto MI, Kerdsirichairat T, Yeo CJ, Hruban RH, Shin EJ, Almario JA, et al. Surgical Outcomes After Pancreatic Resection of Screening-Detected Lesions in Individuals at High Risk for Developing Pancreatic Cancer. J Gastrointest Surg 2020;24:1101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018;172:578–589.e17. [DOI] [PubMed] [Google Scholar]
- 18.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020;383:1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verduin M, Hoeben A, De Ruysscher D, Vooijs M. Patient-Derived Cancer Organoids as Predictors of Treatment Response. Front Oncol 2021;11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao Y, Xu X, Yang L, Zhu J, Wan J, Shen L, et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020;26:17–26.e6. [DOI] [PubMed] [Google Scholar]
- 21.Ganesh K, Wu C, O’Rourke KP, Szeglin BC, Zheng Y, Sauvé CEG, et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat Med 2019;25:1607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Driehuis E, Van Hoeck A, Moore K, Kolders S, Francies HE, Gulersonmez MC, et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc Natl Acad Sci 2019;116:26580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharick JT, Walsh CM, Sprackling CM, Pasch CA, Pham DL, Esbona K, et al. Metabolic Heterogeneity in Patient Tumor-Derived Organoids by Primary Site and Drug Treatment. Front Oncol 2020;10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer AC, Sorger PK. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017;171:1678–1691.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seppälä TT, Zimmerman JW, Burkhart RA. Solving for Chemotherapeutic Sensitivity: Adapting “Black Box” Methods to Study Patient-Derived Tumor Organoids. Ann Surg Oncol 2021;29:4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dreyer SB, Upstill-Goddard R, Paulus-Hock V, Paris C, Lampraki E-M, Dray E, et al. Targeting DNA Damage Response and Replication Stress in Pancreatic Cancer. Gastroenterology 2021;160:362–377.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmocker RK, Wright MJ, Ding D, Beckman MJ, Javed AA, Cameron JL, et al. An Aggressive Approach to Locally Confined Pancreatic Cancer: Defining Surgical and Oncologic Outcomes Unique to Pancreatectomy with Celiac Axis Resection (DP-CAR). Ann Surg Oncol 2021;28:3125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.