

Abstract
Pathways regulating lung alloimmune responses differ from most other solid organs and remain poorly explored. Based on our recent work identifying the unique role of eosinophils in downregulating lung alloimmunity we sought to define pathways contributing to eosinophil migration and homeostasis. Using a murine lung transplant model, we have uncovered that immunosuppression increases eosinophil infiltration into the allograft in an IL-5 dependent manner. IL-5 production depends on immunosuppression-mediated preservation of donor-derived group 2 innate lymphoid cells (ILC2). We further describe that ischemia-reperfusion injury upregulates the expression of IL-33, which functions as the dominant and non-redundant mediator of IL-5 production by graft-resident ILC2. Our work thus identifies unique cellular mechanisms that contribute to lung allograft acceptance. Notably, ischemia-reperfusion injury, widely considered to be solely deleterious to allograft survival, can also downregulate alloimmune responses by initiating unique pathways that promote IL-33/IL-5/eosinophil-mediated tolerance.
1 ∣. Introduction
Long-term survival of lung allografts lags significantly behind that of other transplanted organs1. It has been postulated by us as well as others that this may be due to the fact that pulmonary immune responses differ from those of grafts not continuously exposed to the external environment2,3. To this end, we have recently described that eosinophils, long considered deleterious to the survival of other solid organs such as livers and kidneys, downregulate CD8+ T cell-mediated lung alloimmune responses4. In turn, proinflammatory mediators elaborated by alloreactive CD8+ T cells polarize eosinophils towards a regulatory phenotype5. This eosinophil/T cell “inflammatory” feedback loop is critical for long-term lung allograft survival as depletion of either cell type prevents tolerance induction6,7.
While we have previously defined pathways controlling CD8+ T cell trafficking and differentiation in the pulmonary allograft7,8, mechanisms controlling eosinophil migration into the transplanted lung remain unexplored. Here, we demonstrate that ischemia-reperfusion injury-mediated IL-33 production by donor-derived stromal cells facilitates eosinophil recruitment and infiltration into transplanted lung allografts. This is accomplished in an indirect manner through ILC2s that produce IL-5 in response to IL-33. Our data further define transplant-related cytokine-dependent cellular interactions that contribute to lung allograft acceptance. We also bring to light the concept that ischemia-reperfusion injury, widely considered to be uniformly deleterious to graft function, can contribute to graft acceptance as well. Our data show that, surprisingly, lung allograft tolerance may actually be facilitated by pro-inflammatory pathways that have been widely considered to be deleterious to graft survival.
2 ∣. Brief Methods
For full methods please see supplemental data. Male Balb/c, C57BL/6 (B6), B6(C)Il5tm1.1(icre)Lky/J (IL-5tdTomato), B6.SJL/BoyJ CD45.1 congenic and B6.129S7-Il7rtm1Imx/J (IL-7R−/−) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All animal procedures were performed in compliance with and approved by the Institutional Animal Care and Use Committee at The University of Maryland, Baltimore. For histologic analysis mouse lungs were harvested and fixed in 10% formalin with a lung pathologist blinded to the experimental condition graded acute rejection according to the International Society for Heart and Lung Transplantation (ISHLT). Student’s t-test was used for continuous variable comparisons while the Mann-Whitney U test was used for categorical variable comparisons.
3 ∣. Results
3.1. Both Steroid and Co-Stimulatory Blockade-Based Immunosuppression Increases the Infiltration of Eosinophils into Lung Allografts
Having recently described that eosinophils can downregulate alloimmune responses after lung transplantation4,5 we set out to quantify eosinophils within Balb/c (H2d) lungs after engraftment into fully MHC-mismatched C57BL/6 (H2b) (B6) recipients. We initially wanted to test the hypothesis that conventional steroid-based immunosuppression decreases eosinophil migration to the lung due to deleterious effects of corticosteroids on this cell population9. Surprisingly, we observed higher relative numbers of eosinophils within allografts after transplantation into mice that were treated with cyclosporine and methylprednisolone (CSA/MP) than in resting lungs or in allografts after transplantation without immunosuppression. The relative proportion of eosinophils was similar between lung allografts treated with CSA/MP compared to peri-operative co-stimulatory blockade (CSB) (Figure 1A bottom left panel). We have previously reported that the ratio of eosinophils to T lymphocytes within lung allografts is critical for the downregulation of alloimmunity4. To this end a higher relative ratio of eosinophils was evident in lung allografts treated with CSA/MP or CSB compared to resting lungs or those without immunosuppression (Figure 1A bottom right panel).
Figure 1: Eosinophil infiltration of Lung Transplants.

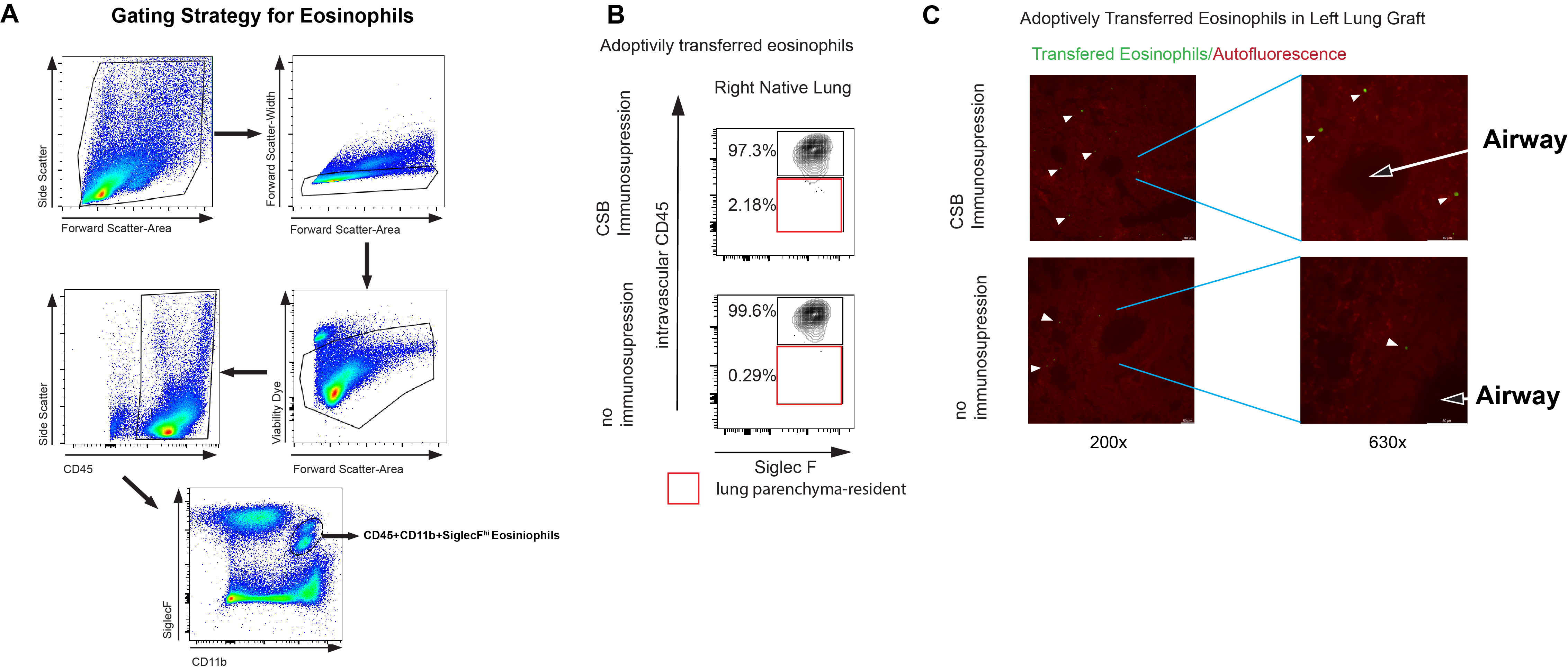
(A) Graphic representation of a left single lung transplant in the mouse (top). Relative ratio (bottom left) and eosinophil/T cell ratio (bottom right) in resting B6 lungs as well as Balb/c lungs engrafted into B6 mice in the presence or absence of immunosuppression at post-engraftment day 7. (B) Representative Ki-67 expression and viability dye exclusion in eosinophils from lungs in the presence or absence of immunosuppression at post-engraftment day 7. Representative of two separate experiments. (C) Adoptively transferred CD45 congenic eosinophils in the lung or spleen of recipients treated with CSB or not receiving immunosuppression. (D) Tissue infiltration of adoptively transferred eosinophils in the left lung graft of Balb/c to B6 CD45.1 recipients via flow cytometry (left panel) and immunofluorescence (right panel). Intravascular vs. parenchymal identification of SiglecF+CD11bhigh eosinophils performed by intravascular administration of fluorescently labeled pan-CD45 antibody followed by identification of transferred eosinophils by CD45.2+CD45.1− staining (left panel) or by cell trace violet labeling of eosinophils prior to transfer and visual inspection by fluorescent microscopy (right panel). White arrow points to pulmonary vessel with visible intravascular eosinophils in lungs with (top) or without (bottom) immunosuppression. Representative of two separate experiments. For (C) and (D), eosinophils were injected on post-engraftment day 4 and tissue was collected 18 hours later. CSB=co-stimulatory blockade, CsA/MP=cyclosporine/methylprednisolone. ns=p>0.05; *=p<0.05; **=p<.01; ***=p<.001
We next evaluated whether the observed higher relative numbers of eosinophils are the result of increased proliferation or altered viability. We noted no differences in Ki-67 expression10 or viability in lung-resident eosinophils in any experimental group (Figure 1B). To examine if eosinophil recruitment and/or tissue infiltration were responsible for this relative increase in abundance we injected 5x106 B6 CD45.2 eosinophils into Balb/c CD45.2→ B6 CD45.1 lung transplants treated with either CSB or no immunosuppression. Since resting Balb/c donor lungs contain less than 50,000 eosinophils and we have previously demonstrated that donor-derived migratory hematopoietic cells such as eosinophils are rapidly replaced by those of the recipient host11, we envisioned that such an adoptive transfer system would allow us to track transferred eosinophils using the CD45.1 vs. CD45.2 congenic markers. We noted a higher ratio of CD45.2 eosinophils in lung allografts, but not spleens, of B6 CD45.1 recipients treated with CSB immunosuppression (Figure 1C). In addition, a higher percentage of CD45.2 eosinophils were located within tissue parenchyma of immunosuppressed compared to non-immunosuppressed allografts. Anatomically eosinophils seemed to be dispersed throughout both the alveolar tissue, pulmonary interstitium, as well as peri-bronchial tissue in the presence or absence of immunosuppression (Figure 1D, Supplemental Figure 1A, B, C). Taken together, our data indicate that immunosuppression alters the homing and tissue infiltration of eosinophils into lung allografts.
3.2. IL-5 Contributes to the Relative Increase of Eosinophils in Immunosuppressed Allografts
We next evaluated chemokines and cytokines known to affect eosinophil migration and/or homeostasis12. Proinflammatory chemokines Mip-1α, Mip-1β as well as RANTES were higher in lung grafts from non-immunosuppressed animals Figure 2A) while eotaxin-1 (CCL11), eotaxin-2 (CCL24) were comparable between resting lungs and CSB-treated or non-immunosuppressed lung allografts (Figure 2B. However, interleukin-5 (IL-5) was more prevalent at both mRNA (Figure 2B) and protein levels (Figure 2C in immunosuppressed grafts. IL-5 neutralization blunted eosinophil recruitment and left lung allograft tissue infiltration in immunosuppressed hosts (Figure 2D, E) with minimal effect on the right native lungs in the same recipients (Supplemental Figure 2 A, B).
Cytokine and chemokine levels in lung allografts.

(A) MIP-1α, MIP-1β and RANTES levels in lung digests of resting B6 lungs or Balb/c to B6 lung grafts with or without CSB immunosuppression by Multiplex Assays. (B) Relative expression of eotaxin-1, eotaxin-2 or IL-5 in lung grafts by quantitative RT-PCR. (C) IL-5 production in Balb/c to B6 lung grafts with or without immunosuppression by flow cytometric evaluation of lung digests. (D) Recipient-derived (top) or adoptively transferred CD45 congenic eosinophils (bottom) in the left lung graft in the presence of IL-5 neutralization or Control IgG treatment. (E) Tissue infiltration of adoptively transferred eosinophils in the left lung graft of Balb/c to B6 recipients after IL-5 neutralization or Control IgG treatment. Representative of two separate experiments. (F) Eosinophil transwell migration in the presence or absence of eotaxin-1 and graded concentrations of IL-5. For (A) to (C), lung tissue was collected and analyzed at post-engraftment day 4. For (D) and (E), eosinophils were injected at post-engraftment day 4 and the tissue was collected 18 hours later. CSB=co-stimulatory blockade, ns=p>0.05; * =p<0.05; **=p<.01; ***=p<.001
While early studies suggested that IL-5 may act as a chemotactic factor for eosinophils13 others have proposed that this cytokine functions primarily to potentiate the activity of other chemokines or chemotactic factors14,15. To explore these possibilities, we next isolated peripheral blood eosinophils16 and evaluated IL-5-mediated migratory properties in a transwell assay using either a combination of eotaxin-1 and IL-5 or graded concentrations of IL-5 alone. We found that, while higher numbers of eosinophils transmigrated in the presence of eotaxin-1, increasing concentrations of IL-5 was able to mediate migration of eosinophils even in the absence of eotaxin (Figure 2F). Taken together we can conclude that an increase in IL-5 in immunosuppressed grafts may contribute to the regulation of eosinophil trafficking after lung transplantation.
3.3. Donor-Derived Group 2 Innate Lymphoid Cells Play a Dominant Role in IL-5 Production and are Critical for Acceptance of Lung Allografts
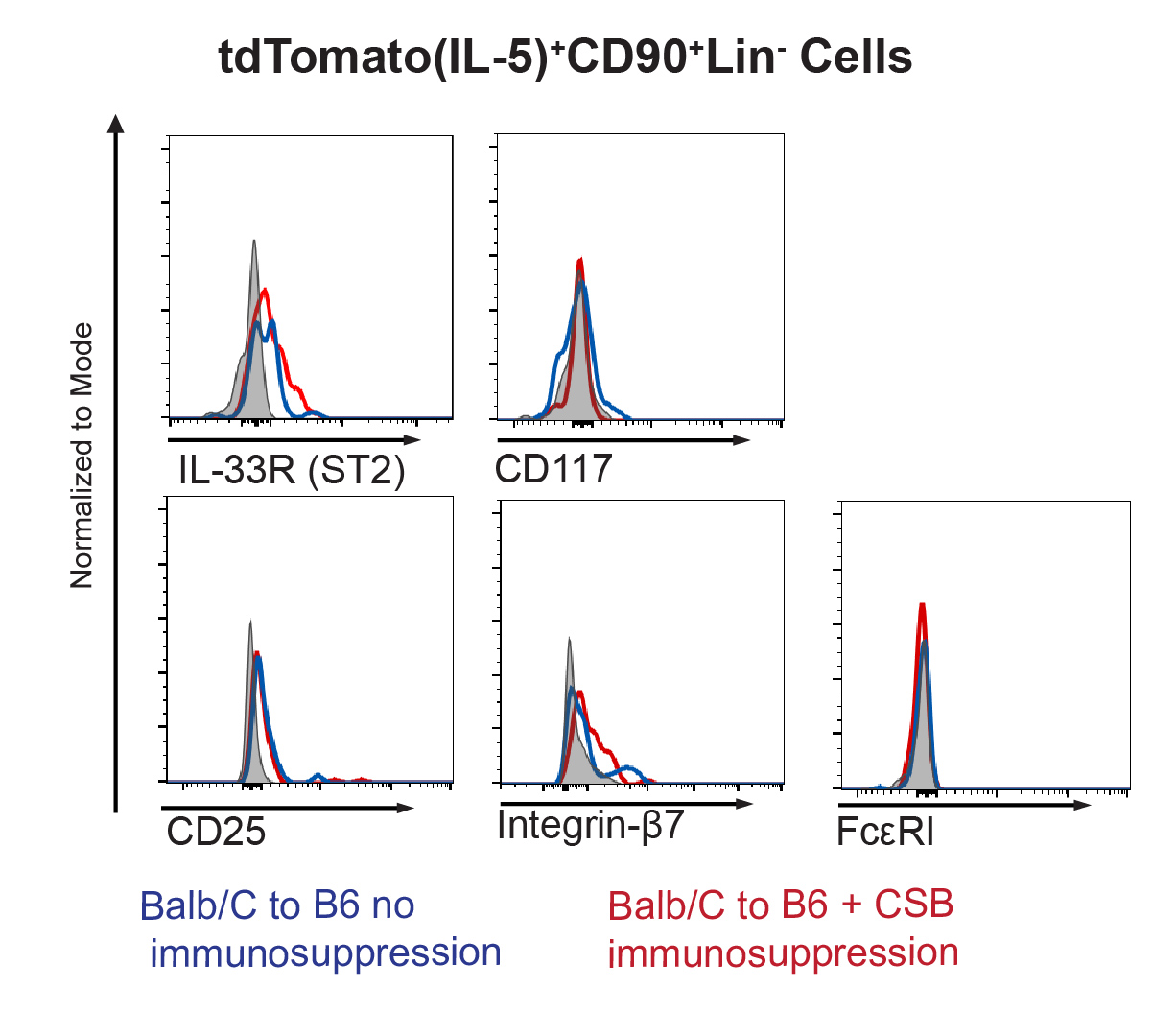
As multiple subtypes of hematopoietic cells have the ability to produce IL-517, we next compared IL-5-producing cells in Balb/c→B6 lung grafts with or without immunosuppression. In the presence of immunosuppression, we observed that among IL-5 producing cells CD45+CD90+Lin− cells underwent the greatest proportional increase (Figure 3A). This cell population expressed the serum stimulation-2 component of the IL-33 receptor (also known as ST2) as well as KLRG1, GATA-3 (Figure 3B) and had a small round appearance (Figure 3C). Similar ILC2 phenotype was evident in the Balb/c to IL-5tdTomato transgenic reporter mouse18 model (Supplemental Figure 3). Thus, phenotypically these cells were consistent with group 2 innate lymphoid cells (ILC2).
IL-5 Production in Lung Allografts.

(A) Relative abundance of IL-5 producing cells in the lung allograft in the presence or absence of immunosuppression. Top shows representative plots, bottom depicts a graphic representation of multiple experiments. Each dot represents an individual transplant. (B) Surface phenotype of IL-5+CD90+Lin− cells in the presence or absence of CSB. (C) Cytospin preparation of IL-5+CD90+Lin− cells flow cytometrically sorted from lung grafts. Tissue was collected and analyzed at post-engraftment day 4. CSB=co-stimulatory blockade, ns=p>0.05; ***=p<.001
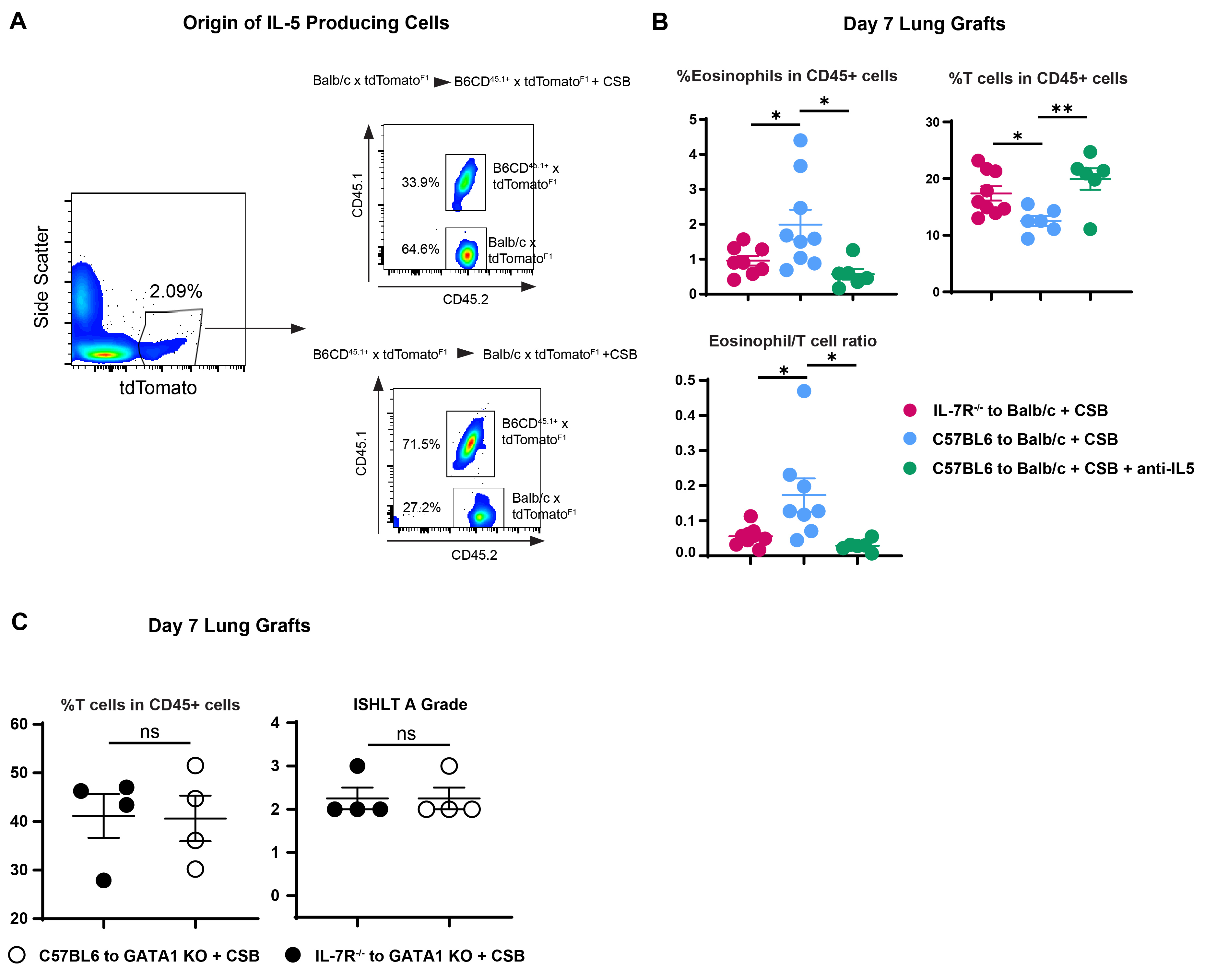
We next quantified donor and recipient-derived IL-5-producing cells in Balb/cCD45.2+→B6CD45.1+ transplants. As described above, we noted a higher relative proportion of IL-5-producing cells in the presence of CSB immunosuppression (Figure 4A). The majority of IL-5 producing cells were donor-derived and their relative ratio increased even further with immunosuppression (Figure 4A). Such dominance of donor-derived IL-5 producing cells was corroborated in IL-5tdTomato transgenic reporter mice18 where detection of IL-5 does not rely on PMA/Ionomycin stimulation and antibody staining (Supplemental Figure 4A). Since ILC2s are the dominant producers of IL-5 within the lung graft (Figure 3A) we next gated on donor (CD45.2+) vs. recipient (CD45.1+)-derived CD90+Lin−ST2+ cells in the Balb/cCD45.2+ → B6CD45.1+ strain combination. We noted that CSB immunosuppression increased the relative numbers of donor, but not recipient-derived ILC2s with the graft (Figure 4A right). Based on these data we next set out to determine the functional importance of donor-derived ILCs for IL-5-mediated graft acceptance.
Alteration of Graft-Resident ILC-2.

(A) Relative donor vs. recipient IL-5-producing cells in the presence or absence of CSB immunosuppression. Left panels demonstrate all IL-5-producing cells and the right panel is gated on CD90+Lin−ST2+ ILC2s as a percent of all lung cells. (B) ILC2 levels in B6 IL7R−/− vs. wildtype (left panel); IL-5 production in lung grafts of B6 to Balb/c + CSB vs. B6 IL7R−/− to Balb/c + CSB (right panel). (C) Relative abundance of eosinophils and T cells, eosinophil/T cell ratio and ISHLT A grade of B6 IL-7R−/− vs. B6 wildtype lungs implanted into Balb/c recipients in the presence of CSB immunosuppression. Green arrows indicate perivascular lymphocytic infiltration. (D) MHC Class I and II expression in tissue-resident ILC2s of both resting and Balb/c lungs after transplantation into B6 mice. ILC2s identified as CD90+Lin−ST2+ CD45.2+CD45.1− cells in Balb/cCD45.2+ to B6CD45.1+ graft recipients. (E) Relative numbers as well as IL-5 expression, defined as relative MFI and % positive, of graft-resident ILC2s in the presence or absence of CSB immunosuppression in Balb/c to B6 wildtype vs. B6 Rag2−/− mice. Tissue was collected and analyzed on post-engraftment day 7 for (B)(C) and on day 4 for (A)(D)(E). CSB=co-stimulatory blockade; MFI = Mean fluorescent intensity (geometric mean). ns=p>0.05; * =p<0.05; **=p<.01; ***=p<.001
The cytokine IL-7 plays a key role in the development of both T cells and ILCs19,20. However graft-resident donor-derived T cells are rapidly replaced by those of the recipient within 24-72 hours post-engraftment11, while, as described above, ILCs remain of donor origin. Thus, the use of a lung graft from an IL7 receptor-deficient donor (IL7R−/−) allows for the evaluation of donor-derived ILCs in the immune response without affecting the function of other cell types, including ILCs of recipient origin21,22. B6 IL7R−/− lungs engrafted into Balb/c recipients demonstrated lower production of IL-5, decreased eosinophil infiltration, and higher grades of acute cellular rejection (Figure 4B, C) similar to those evident after IL-5 neutralization (Supplemental Figure 4B). Interestingly in the absence of eosinophils grafts were rejected in the presence or absence of donor-derived ILCs (Supplemental Figure 4C). This indicated that, unlike the case for islet transplants23, ILCs do not in and of themselves downregulate lung alloimmunity. Taken together, our data demonstrate that donor-derived ILC2s are critical for eosinophil-mediated downregulation of alloimmune responses and lung allograft acceptance. However, how immunosuppression affects the number of IL-5 producing ILC2s in the donor graft remained unclear.
As ILC2s express both MHC Class I and II and may therefore be targets for elimination by alloreactive T cells (Figure 4D), we next considered the possibility that their lower abundance in non-immunosuppressed hosts may be the result of immunologic rejection. To test this hypothesis, we transplanted Balb/c lungs into either wild-type B6 or B6Rag2−/− recipient mice that are devoid of T and B cells24. While lower levels of ILC2s and IL-5 were noted in Balb/c grafts after transplantation into non-immunosuppressed B6 mice, B6Rag2−/− recipients demonstrated high levels of IL-5 producing ILC2s in engrafted Balb/c lungs irrespective of CSB (Figure 4E). Taken together, these data support the notion that immunosuppression preserves graft-resident donor-derived ILC2s, which contribute to graft acceptance through their production of IL-5.
3.4. Recipient-Derived ILC2s Gradually Migrate into the Lung Allograft and Increase IL-5 Production Over the Course of Three Weeks
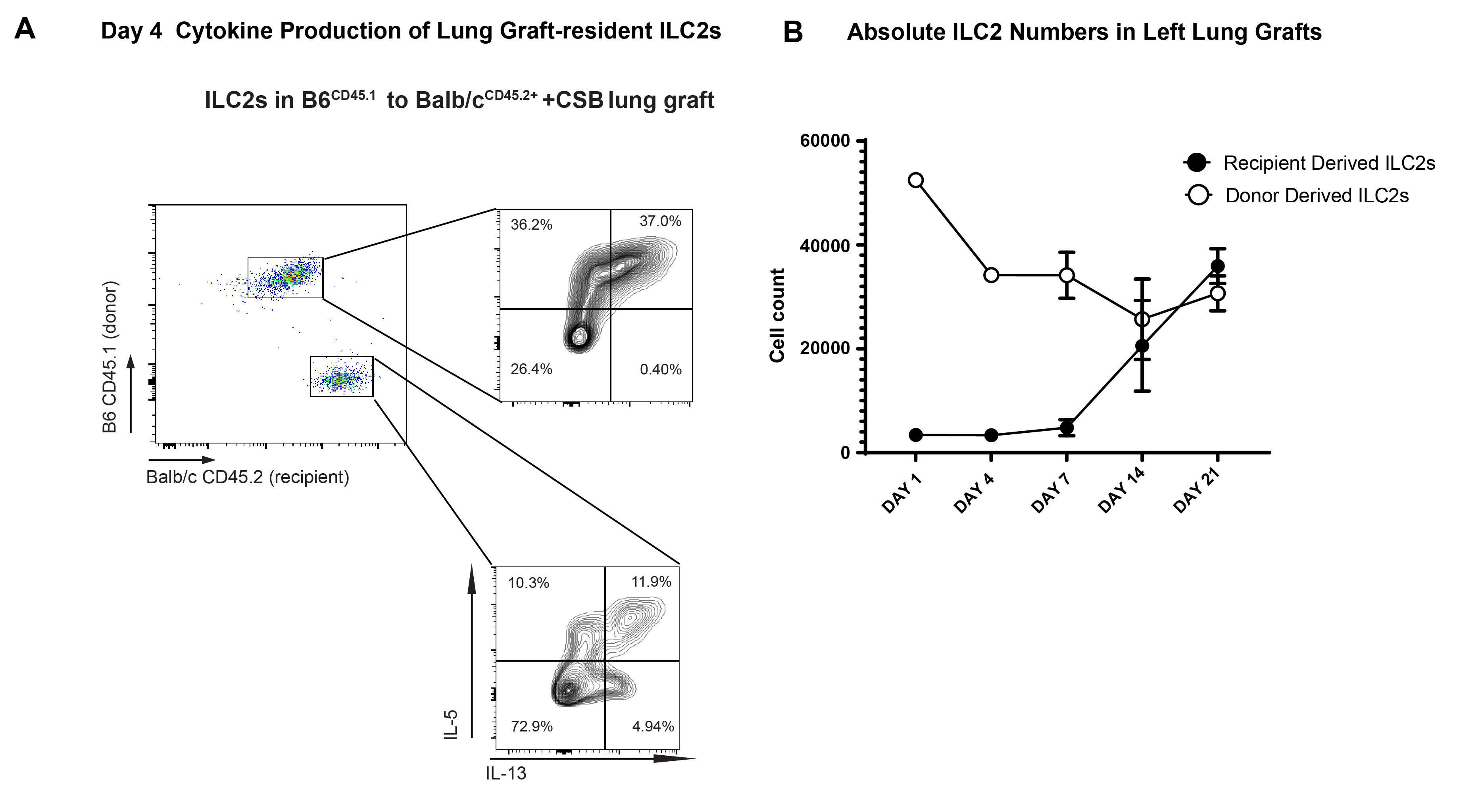
In addition to their lower relative abundance (Figure 4A), recipient-derived ILC2 also produced less IL-5 than their donor-derived counterparts four days after transplantation (Figure 5A). In fact, graft-infiltrating recipient-derived ILC2s produced less IL-5 than those residing in resting lungs (Figure 5A). This was reflective of a more global reduction in their activation as evidenced by lower levels of IL-13 as well (Supplemental Figure 5A). As ILC2s are considered tissue-resident cells that populate various organs as early as embryogenesis25 we next considered the possibility that recipient-derived ILC2 progenitors infiltrating lung allografts might require a period of maturation to reach full cytokine production capacity. To test this hypothesis, we engrafted B6CD45.1+ donor lungs into Balb/cCD45.2+ recipients and evaluated the grafts 1, 4, 7, 14 and 21 days after transplantation. As demonstrated in Figure 5B and Supplemental Figure 5B we noted a gradual increase in recipient-derived CD45.2+ ILC2s with enhanced migration into the lung parenchyma (Figure 5C). We also observed a progressive increase in their IL-5 production (Figure 5D) that reached comparable levels to donor-derived ILC2s 21 days after engraftment. Taken together, these data support the notion that recipient-derived ILC2s do not play an important functional role in the local regulation of alloimmune responses early after transplantation.
IL-5 Production by Donor and Recipient Derived ILC2.

(A) IL-5 production, defined as relative IL-5 MFI or % IL-5+ in donor vs. recipient-derived ILC2s of Balb/cCD45.2+ to B6CD45.1+ lung transplants (left side of graphs) vs. resting non-transplanted Balb/cCD45.2+ and B6CD45.1+ lungs (right side of graphs). Relative MFI of 1 was set as level defined by B6CD45.2+ ILC2 from resting non-transplanted lungs which were run with every experiment as an internal control. Tissue was collected and analyzed at post-engraftment day 4. (B) Relative ratio of donor vs. recipient-derived ILC2s in grafts of Balb/cCD45.2+ to B6CD45.1+ lung transplants on day 1,4,7,14 and 21 days post-engraftment. Graft recipients were treated with CSB immunosuppression. (C) Relative percentage of intraparenchymal vs. intravascular donor and recipient-derived ILC2s in Balb/cCD45.2+to B6CD45.1+ lung grafts as identified by flow cytometry. (D) Relative alteration in IL-5 expression in donor vs recipient-derived ILC2s in Balb/cCD45.2+ to B6CD45.1+ lung grafts on day 1,4,7,14 and 21 days post-engraftment. Data representative of three separate experiments. CSB=co-stimulatory blockade; MFI = Mean fluorescent intensity (geometric mean). ns=p>0.05; * =p<0.05; ***=p<.001
3.5. IL-33 Induced by Ischemia Reperfusion Injury Activates Donor-Derived ILC2s and Plays a Critical Role in Lung Allograft Acceptance
Interestingly, in addition to detecting lower IL-5 production in recipient-derived cells we noted that donor derived ILC2s produce more IL-5 in freshly transplanted lung grafts compared to those derived from resting, non-transplanted lungs (Figure 5A). In order to determine if this increased activation was the result of an alloimmune response, we next transplanted B6CD45.1+ lungs into MHC-identical B6CD45.2+ congenic recipients without any immunosuppression and evaluated IL-5 production in donor and recipient ILC2s 4 days after engraftment. Notably, donor-derived ILC2s in transplanted syngeneic lungs also demonstrated higher production of IL-5 than ILC2s from resting lungs (Figure 6A). Thus, alloimmunity was not a prerequisite for the activation of ILC2s.
ILC2 activation by ischemia-reperfusion injury-induced IL-33.

(A) IL-5 production, defined as relative IL-5 MFI or % IL-5+ in donor vs. recipient-derived ILC2s of B6CD45.1+ to B6CD45.2+ lung transplants. (B) Relative IL-5 MFI of ILC2s and tissue concentrations of IL-25, TSLP and IL-33 of resting vs. hilar-clamped lung (C) Expression of full-length and cleaved mature forms of IL-33 in brain dead donor prior to harvest and Perfadex®-preserved brain-dead donor lungs after ≃ 6 hours of cold storage as identified by Western Blot and immunohistochemistry. Immunohistochemistry in Balb/c to B6 transplants with CSB immunosuppression shown on day 4 post-implantation and human immunohistochemistry was performed on Perfadex® preserved donor lung after ≃6 hours of cold storage but prior to implantation. Red arrows: cytoplasmic staining of IL-33 in bronchial epithelium. (D) Relative IL-5 MFI of ILC2s in resting vs. hilar-clamped lungs of IL-33-deficient mice. For (A), (B) and (D), lung tissue was collected and analyzed on post-operative day 4. BLEO = bleomycin; MFI = Mean fluorescent intensity (geometric mean). ns=p>0.05; ***=p<.001
Ischemia reperfusion injury (IRI) associated with the temporary interruption of blood flow during donor harvest and eventual restoration during reimplantation is an unavoidable consequence of solid organ transplantation. To test the role of IRI in ILC2 activation we next utilized a model of temporary pulmonary hilar clamping. We and others have previously used this model as a reductionist form of IRI in the absence of other confounding factors associated with organ transplantation26. Indeed, a 2-hour period of vascular occlusion resulted in an increase in IL-5 production by ILC2s compared to those from resting lungs (Figure 6B) with other subtle changes in phenotype (Supplemental Figure 6A). As the cytokines IL-25 (also known as IL-17E), thymic stromal lymphopoietin (TSLP) and IL-33 have been previously described to activate ILC2s and upregulate IL-5 production27-29 we next evaluated their levels in the lung tissue. Post-hilar clamp levels of IL-25 and TSLP remained below the level of detection, despite their upregulation by other stimuli such as intratracheal bleomycin (Figure 6B). By contrast, however, IL-33 levels increased significantly in lungs after IRI (Figure 6B).
To investigate the physiology of IL-33 in donor lungs we next utilized human lung tissue obtained during the process of transplantation. We and others have reported that IL-33 can exist as a full length precursor (flIL-33), which is nearly exclusively a nuclear protein30-33, or as a proteolytically cleaved shorter mature forms (mIL-33) with progressive cleavage increasing activity33-36. Full length IL-33 does not engage surface cytokine receptors and is known to induce only mild lymphocytic and neutrophilic inflammation while the cleaved mature form signals through the canonical T1/ST-2 receptor with robust activity34. While we have previously described that resting human lungs contain substantial amounts of the full-length IL-3334,37, in the setting of brain death followed by preservation and cold-storage in Perfadex® solution, as is customary post organ harvest, we were able to document an increase in the cleaved isoform which progressed post organ preservation (Figure 6C, left). IL-33 was detected in the cytoplasm of bronchial epithelial cells of human lungs by immunohistochemistry after cold storage (Figure 6C, middle). Mouse lung allografts showed similar results 4 days after engraftment (Figure 6C, right).
To evaluate the role of IL-33 in IRI-mediated IL-5 production by ILC2s we performed transient hilar clamping of lungs in IL-33-deficient mice. Unlike the case in wildtype animals (Figure 6B) we noted no increase in IL-5 compared to resting lungs (Figure 6D), indicating that IL-33 is critical for the activation of lung-resident ILC2s by IRI. Taken together, we can conclude that processes related to lung donation, including brain death as well as cold storage, augment the production and activation of IL-33 within the graft which is critical for IL-5 production following transplantation. Interestingly pretreatment of murine donor grafts by IL-33 prior to implantation increased the number of eosinophils in the graft but did not alter T cell numbers or effector differentiation (Supplemental Figure 6B). Such data suggests that the timing, amount and anatomic niches of IL-33 production may influence its effect on alloimmunity. However, the role of the IL-33/IL-5 pathway on lung allograft acceptance remained unknown.
To directly examine the IRI/IL-33/IL-5 axis in pulmonary allograft acceptance we next transplanted lungs from B6 IL-33−/− or wildtype mice into Balb/c recipients in the presence of CSB immunosuppression and evaluated the immunologic response one week later. In the absence of donor IL-33, IL-5 production in the graft was reduced with a decrease in graft-resident eosinophils, an increase in the relative number of graft-resident T cells and a decrease in the eosinophil/T cell ratio (Figure 7A). Most importantly, an increase in the ISHLT A grade of rejection was evident in the absence of donor IL-33 (Figure 7B). Such changes were consistent with lower activation level of graft-resident ILC2s (Supplemental Figure 7). Taken together with our data demonstrating that IL-33 is predominantly expressed in bronchial epithelial cells these findings suggest that IL-33 may downregulate alloreactivity at defined anatomic sites.
Graft acceptance depends on donor IL-33 expression in donor.

(A) Relative abundance of eosinophils and T cells as well as eosinophil/T cell ratio in B6 IL-33−/− vs. wildtype grafts transplanted into CSB-treated Balb/c recipients. (B) Rejection grade of B6 IL-33−/− vs. wildtype donor grafts transplanted into CSB-treated Balb/c recipients. Top panel shows representative histology (200x, H&E) and bottom panel shows ISHLT A grade. Blue arrows indicate perivascular lymphocytic infiltration. Green arrows indicate obliterative arteritis observed in some B6 IL-33−/− to Balb/c lung grafts. Lung tissue was collected and analyzed on post-engraftment day 7. Each dot represents a single mouse lung transplant. CSB=co-stimulatory blockade. ns=p>0.05; * =p<0.05; **=p<.01; ****=p<.0001
4 ∣. Discussion
We have recently described that eosinophils play a critical role in lung allograft acceptance based on inducible nitric oxide synthase mediated disruption of CD8+ T cell effector differentiation and graft rejection4,5. Thus the importance of eosinophils for lung transplant tolerance4,5, combined with the known deleterious role of corticosteroids on their physiology9, led us to hypothesize that “traditional” steroid-based immunosuppression may be detrimental for lung allograft survival. In fact, we have postulated that the reliance on conventional immunosuppression is the reason for inferior survival of the lung allograft when compared to other solid organs38,39. However, both steroid-based as well as CSB-based immunosuppression increase graft-resident eosinophils to downregulate alloimmunity. The increase in eosinophils depends on IL-5, which is primarily produced by donor-derived activated ILC2s. Donor-derived ILC2s are preserved in the presence of immunosuppression due to amelioration of their rejection by the recipient. Interestingly, ischemia-reperfusion injury (IRI) contributes to activation of ILC2s due to elaboration of IL-33 by donor-derived stromal cells. Collectively, such data define a novel pathway that contributes to immunosuppression-mediated lung graft acceptance by preserving a tolerogenic population of donor-derived passenger leukocytes.
ILCs are a recently described immunocytes that reside at barrier surfaces and play a key role in protection against pathogens. Based on their location and long-term residence in grafted organs ILCs are poised to regulate the alloimmune response but their precise contribution to graft acceptance or rejection is still being defined40. Work from our group has demonstrated that cytokine production by ILC3s plays a critical role in establishing tolerogenic tertiary lymphoid structures in lung allografts41 while others have demonstrated the association of ILC1 and ILC2 with amelioration of lung ischemia-reperfusion injury42. The protective role of ILC3 has been extended to intestinal transplantation where Kang and colleagues have demonstrated that ILC3s play a protective role in long-term graft survival while ILC1s are associated with rejection43.
The existence of ILC2s was initially described by Fort, who demonstrated that IL-25 administration induced the production of IL-5 and IL-13 in Rag2−/− mice lacking both T and B cells44. Activation of ILC2s has been demonstrated to occur directly by cytokines as well as indirectly through exposure to environmental allergens such as house dust mites and fungus45,46. Based on their anatomic location and function they have been implicated in potentiating multiple pulmonary disease processes including asthma, rhinitis and lung fibrosis47-49. However, in other disease models ILC2s serve a protective function by regulating post-influenza repair of damaged epithelium and restoring barrier integrity in allergy50,51. Our findings add to their functional repertoire the downregulation of deleterious alloimmune responses after lung transplantation. Our data thus extend the role of ILC2s in pulmonary immunoregulation and point out how innate cellular and cytokine networks, that have evolved to respond to pathogens and environmental stressors, can also play a role in alloimmunity. Our findings further expand on the concept of tolerogenic passenger leukocytes and demonstrate that donor-derived ILC2s play a critical and non-redundant role in lung tolerance induction. Thus, our data support the notion that indiscriminate elimination of all donor-derived leukocytes may be detrimental to long-term lung allograft survival. It is also noteworthy that immunosuppression-mediated inhibition of alloreactive T cells contributes to tolerance not only by ameliorating direct graft destruction but also by preserving tolerogenic donor-derived passenger leukocytes.
Huang and colleagues have recently reported that IL-33 induced ILC2s in islet graft are critical for maintaining graft survival and proper function independent of regulatory T cells23. Such protective effects of ILC2s were attributed to their ability to produce IL-10. This observation supports our hypothesis that recipient-derived ILC2s migrate into lung allograft in response to local IL-33 elaborated based on ischemia-reperfusion injury. However, in our model, the mechanism of ILC2-mediated graft acceptance seemed to function in a different fashion as donor, rather than recipient-derived ILC2s, are critical for graft acceptance. This is demonstrated in Figures 4B and C where ILC-deficient lung grafts are rejected even in the presence of CSB immunosuppression despite the presence of recipient ILC2. Furthermore, in our model ILC2-mediated graft acceptance completely depended on the presence of eosinophils as eosinophil-deficient but ILC2-sufficient GATA1KO mice reject allogenic grafts (Supplemental Figure 4C). Such data demonstrates that, unlike the case for islet transplantation, ILCs promote lung allograft acceptance in an eosinophil-dependent manner.
It is interesting that donor-derived ILC2 play a dominant role in IL-5 production early post-engraftment but are gradually supplanted by recipient-derived cells over the course of three weeks. The majority of ILCs populate their respective organs during the neonatal period with limited production and organ seeding during postnatal life52. Recent reports, however, have challenged this notion as circulating ILC2s have been detected in peripheral blood and draining lymph nodes during inflammation in both humans and mice46,53. Also, recruitment of ILC3s to lung allografts points to their migratory potential41,54, the origin of While specific mechanistic fate mapping studies in a helminth model of infection demonstrated that circulating ILC2s can originate from both lung and intestine based on the stage of infection recipient-derived ILC2s populating the donor lung have yet to be determined. Their recruitment might be mediated by local or systemic inflammatory cascades initiated by transplant related immunity and IL-33 may be critical to this process23,55.However, as described in Figure 5B and Supplemental Figure 5B both the total number and relative ratio of donor vs. recipient ILC2s equilibrate over the course of 3 weeks with equal contribution at the end of that period. Such data may suggest that accelerating and strengthening ILC2 migration and maturation may play a role in developing lung-specific tolerogenic protocols.
It is interesting that both eosinophil migration and tolerance depend on IL-5 production by ILC2s in our model. While such data support our previous demonstration that eosinophils play a dominant and non-redundant role in lung allograft acceptance4-6 we recognize that other aspects of IL-5 production and/or ILC2 activation may play a beneficial role on graft survival. For example, IL-5 can downregulate immune responses in an eosinophil-independent manner through induction of CD4+CD25+Foxp3+ regulatory T cells56. ILC2s are also known to produce amphiregulin, which can facilitate repair of damaged epithelium and restore pulmonary homeostasis50. Thus, efforts to facilitate activation of lung-resident ILC2s may play a very important role in allograft tolerance based on multiple mechanisms.
Experimental and clinical efforts focus on amelioration of IRI due to its deleterious effects through the activation of neutrophils57, monocytes26, as well as other cells of the innate and adaptive immune system. The work presented here demonstrates that in addition to deleterious effects IRI may also downregulate inflammation in an IL-33 dependent manner. While traditionally considered to promote type-2 immunity36 recent work has demonstrated its function to be highly context-dependent as in some models IL-33 can result in IFN-γ dependent type-1 immune response as well58. Some studies have demonstrated that IL-33 is an alarmin, functioning to alert the immune system to tissue damage rather than participating in canonical cytokine signaling59. Interestingly, recent work in heterotopic heart transplant models has demonstrated that therapeutic administration of IL-33 can extend graft survival through a variety of mechanisms including skewing the immune response toward Th-2 polarization and increasing the generation and function of immunosuppressive myeloid and regulatory cell populations60-63. Data presented here extends these findings by furthering the notion that IL-33 can facilitate allograft acceptance, albeit through an IL-5/eosinophil-dependent mechanism that may be unique to mucosal barrier organs such as lungs6.
T cell and eosinophil numbers, ratio, as well as the ISHLT grade of rejection were similarly increased in both ILC and IL-33 deficient compared to wild-type donor lungs, supporting the IL-33/ILC/IL-5 axis described above (Figure 4 vs. Figure 7). Nevertheless the histologic pattern of rejection differed somewhat allografts deficient in either ILC2s or IL-33. Specifically some, but not all, IL-33-deficient donor lungs demonstrated a unique histologic pattern of aortitis with obliteration of small pulmonary arterial branches by inflammatory cells (green arrows in Figure 7B). Perhaps one reason for such differences involves the intrinsic chemotactic properties of IL-33, which can affect lymphocyte migration independently of its effects on ILC activation64,65. It is thus possible that IL-33 directly controls specific anatomic niches of lymphocyte accumulation, and its deficiency may alter lymphocytic patterns of rejection. In fact, data demonstrating that indiscriminate IL-33 administration to the donor does not ameliorate CD8+ T cell differentiation despite increasing the number of graft-resident eosinophils supports this notion (Supplemental Figure 6B). Taken together data presented in this manuscript expand on the complexity of lung allograft immunoregulation. A better understanding of such patterns may shed light on immunologic strategies that can be employed to facilitate graft acceptance in the absence of traditional non-specific global immunosuppression.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by VA Health System I01BX002299, NIH NHLBI: R01HL094601, R01HL151078 NIH NIAID: P01 AI116501, R01 AI145108-01
Abbreviations:
- CSB
co-stimulatory blockade
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- ILC2
group 2 innate lymphoid cells
- H&E
haemotoxylin and eosin
- ISHLT
International Society for Heart and Lung Transplantation
- MFI
Mean fluorescent intensity
- CsA
cyclosporine
- MP
methylprednisolone
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclosure as described by the American Journal of Transplantation
Supporting Information
Additional supporting information may be found online in the Supporting Information section.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Rana A, Gruessner A, Agopian VG, et al. Survival benefit of solid-organ transplant in the United States. JAMA Surg. 2015;150(3):252–259. [DOI] [PubMed] [Google Scholar]
- 2.Yeung JC, Keshavjee S. Overview of clinical lung transplantation. Cold Spring Harb Perspect Med. 2014;4(1):a015628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shepherd HM, Gauthier JM, Li W, Krupnick AS, Gelman AE, Kreisel D. Innate immunity in lung transplantation. J Heart Lung Transplant. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onyema OO, Guo Y, Mahgoub B, et al. Eosinophils downregulate lung alloimmunity by decreasing TCR signal transduction. JCI Insight. 2019;4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Onyema OO, Guo Y, Wang Q, et al. Eosinophils promote inducible NOS-mediated lung allograft acceptance. JCI Insight. 2017;2(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onyema OO, Guo Y, Hata A, et al. Deciphering the role of eosinophils in solid organ transplantation. Am J Transplant. 2020;20(4):924–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krupnick AS, Lin X, Li W, et al. Central memory CD8+ T lymphocytes mediate lung allograft acceptance. J Clin Invest. 2014;124(3):1130–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi T, Hsiao HM, Tanaka S, et al. PD-1 expression on CD8(+) T cells regulates their differentiation within lung allografts and is critical for tolerance induction. Am J Transplant. 2018;18(1):216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altman LC, Hill JS, Hairfield WM, Mullarkey MF. Effects of corticosteroids on eosinophil chemotaxis and adherence. J Clin Invest. 1981;67(1):28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun X, Kaufman PD. Ki-67: more than a proliferation marker. Chromosoma. 2018;127(2):175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krupnick AS, Lin X, Li W, et al. Orthotopic mouse lung transplantation as experimental methodology to study transplant and tumor biology. Nat Protoc. 2009;4(1):86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elsner J, Escher SE, Forssmann U. Chemokine receptor antagonists: a novel therapeutic approach in allergic diseases. Allergy. 2004;59(12):1243–1258. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi Y, Suda T, Suda J, et al. Purified interleukin 5 supports the terminal differentiation and proliferation of murine eosinophilic precursors. J Exp Med. 1988;167(1):43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mould AW, Ramsay AJ, Matthaei KI, Young IG, Rothenberg ME, Foster PS. The effect of IL-5 and eotaxin expression in the lung on eosinophil trafficking and degranulation and the induction of bronchial hyperreactivity. J Immunol. 2000;164(4):2142–2150. [DOI] [PubMed] [Google Scholar]
- 15.Mould AW, Matthaei KI, Young IG, Foster PS. Relationship between interleukin-5 and eotaxin in regulating blood and tissue eosinophilia in mice. J Clin Invest. 1997;99(5):1064–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, Lee JJ. Expression of IL-5 in thymocytes/T cells leads to the development of a massive eosinophilia, extramedullary eosinophilopoiesis, and unique histopathologies. J Immunol. 1997;158(3):1332–1344. [PubMed] [Google Scholar]
- 17.Greenfeder S, Umland SP, Cuss FM, Chapman RW, Egan RW. Th2 cytokines and asthma. The role of interleukin-5 in allergic eosinophilic disease. Respir Res. 2001;2(2):71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nussbaum JC, Van Dyken SJ, von Moltke J, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502(7470):245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheikh A, Abraham N. Interleukin-7 Receptor Alpha in Innate Lymphoid Cells: More Than a Marker. Front Immunol. 2019;10:2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patra AK, Avots A, Zahedi RP, et al. An alternative NFAT-activation pathway mediated by IL-7 is critical for early thymocyte development. Nat Immunol. 2013;14(2):127–135. [DOI] [PubMed] [Google Scholar]
- 21.Vonarbourg C, Diefenbach A. Multifaceted roles of interleukin-7 signaling for the development and function of innate lymphoid cells. Semin Immunol. 2012;24(3):165–174. [DOI] [PubMed] [Google Scholar]
- 22.Yagi R, Zhong C, Northrup DL, et al. The transcription factor GATA3 is critical for the development of all IL-7Ralpha-expressing innate lymphoid cells. Immunity. 2014;40(3):378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Q, Ma X, Wang Y, et al. IL-10 producing type 2 innate lymphoid cells prolong islet allograft survival. EMBO Mol Med. 2020;12(11):e12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869–877. [DOI] [PubMed] [Google Scholar]
- 25.Saluzzo S, Gorki AD, Rana BMJ, et al. First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep. 2017;18(8):1893–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsiao HM, Fernandez R, Tanaka S, et al. Spleen-derived classical monocytes mediate lung ischemia-reperfusion injury through IL-1beta. J Clin Invest. 2018;128(7):2833–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leyva-Castillo JM, Galand C, Mashiko S, et al. ILC2 activation by keratinocyte-derived IL-25 drives IL-13 production at sites of allergic skin inflammation. J Allergy Clin Immunol. 2020;145(6):1606–1614 e1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toki S, Goleniewska K, Zhang J, et al. TSLP and IL-33 reciprocally promote each other's lung protein expression and ILC2 receptor expression to enhance innate type-2 airway inflammation. Allergy. 2020;75(7):1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han M, Rajput C, Hong JY, et al. The Innate Cytokines IL-25, IL-33, and TSLP Cooperate in the Induction of Type 2 Innate Lymphoid Cell Expansion and Mucous Metaplasia in Rhinovirus-Infected Immature Mice. J Immunol. 2017;199(4):1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopach P, Lockatell V, Pickering EM, et al. IFN-gamma directly controls IL-33 protein level through a STAT1- and LMP2-dependent mechanism. J Biol Chem. 2014;289(17):11829–11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clerman A, Noor Z, Fishelevich R, et al. The full-length interleukin-33 (FLIL33)-importin-5 interaction does not regulate nuclear localization of FLIL33 but controls its intracellular degradation. J Biol Chem. 2017;292(52):21653–21661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luzina IG, Clerman A, Fishelevich R, Todd NW, Lockatell V, Atamas SP. Identification of the IL-33 protein segment that controls subcellular localization, extracellular secretion, and functional maturation. Cytokine. 2019;119:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luzina IG, Fishelevich R, Hampton BS, et al. Full-length IL-33 regulates Smad3 phosphorylation and gene transcription in a distinctive AP2-dependent manner. Cell Immunol. 2020;357:104203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luzina IG, Pickering EM, Kopach P, et al. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J Immunol. 2012;189(1):403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong J, Bae S, Jhun H, et al. Identification of constitutively active interleukin 33 (IL-33) splice variant. J Biol Chem. 2011;286(22):20078–20086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lott JM, Sumpter TL, Turnquist HR. New dog and new tricks: evolving roles for IL-33 in type 2 immunity. J Leukoc Biol. 2015;97(6):1037–1048. [DOI] [PubMed] [Google Scholar]
- 37.Luzina IG, Kopach P, Lockatell V, et al. Interleukin-33 potentiates bleomycin-induced lung injury. Am J Respir Cell Mol Biol. 2013;49(6):999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madariaga ML, Kreisel D, Madsen JC. Organ-specific differences in achieving tolerance. Curr Opin Organ Transplant. 2015;20(4):392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witt CA, Puri V, Gelman AE, Krupnick AS, Kreisel D. Lung transplant immunosuppression - time for a new approach? Expert Rev Clin Immunol. 2014;10(11):1419–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prosser AC, Kallies A, Lucas M. Tissue-Resident Lymphocytes in Solid Organ Transplantation: Innocent Passengers or the Key to Organ Transplant Survival? Transplantation. 2018;102(3):378–386. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka S, Gauthier JM, Fuchs A, et al. IL-22 is required for the induction of bronchus-associated lymphoid tissue in tolerant lung allografts. Am J Transplant. 2020;20(5):1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monticelli LA, Diamond JM, Saenz SA, et al. Lung Innate Lymphoid Cell Composition Is Altered in Primary Graft Dysfunction. Am J Respir Crit Care Med. 2020;201(1):63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang J, Loh K, Belyayev L, et al. Type 3 innate lymphoid cells are associated with a successful intestinal transplant. Am J Transplant. 2021;21(2):787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fort MM, Cheung J, Yen D, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15(6):985–995. [DOI] [PubMed] [Google Scholar]
- 45.Klein Wolterink RG, Kleinjan A, van Nimwegen M, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. 2012;42(5):1106–1116. [DOI] [PubMed] [Google Scholar]
- 46.Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen-Experienced Group 2 Innate Lymphoid Cells Acquire Memory-like Properties and Enhance Allergic Lung Inflammation. Immunity. 2016;45(1):198–208. [DOI] [PubMed] [Google Scholar]
- 47.Christianson CA, Goplen NP, Zafar I, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. 2015;136(1):59–68 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dhariwal J, Cameron A, Trujillo-Torralbo MB, et al. Mucosal Type 2 Innate Lymphoid Cells Are a Key Component of the Allergic Response to Aeroallergens. Am J Respir Crit Care Med. 2017;195(12):1586–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao Y, De Los Santos FG, Wu Z, Liu T, Phan SH. An ST2-dependent role of bone marrow-derived group 2 innate lymphoid cells in pulmonary fibrosis. J Pathol. 2018;245(4):399–409. [DOI] [PubMed] [Google Scholar]
- 50.Monticelli LA, Sonnenberg GF, Abt MC, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12(11):1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Golebski K, Layhadi JA, Sahiner U, et al. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity. 2021;54(2):291–307 e297. [DOI] [PubMed] [Google Scholar]
- 52.Schneider C, Lee J, Koga S, et al. Tissue-Resident Group 2 Innate Lymphoid Cells Differentiate by Layered Ontogeny and In Situ Perinatal Priming. Immunity. 2019;50(6):1425–1438 e1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. 2014;134(3):671–678 e674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ricardo-Gonzalez RR, Schneider C, Liao C, Lee J, Liang HE, Locksley RM. Tissue-specific pathways extrude activated ILC2s to disseminate type 2 immunity. J Exp Med. 2020;217(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang Y, Mao K, Chen X, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science. 2018;359(6371):114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tran GT, Hodgkinson SJ, Carter NM, et al. IL-5 promotes induction of antigen-specific CD4+CD25+ T regulatory cells that suppress autoimmunity. Blood. 2012;119(19):4441–4450. [DOI] [PubMed] [Google Scholar]
- 57.Kreisel D, Sugimoto S, Zhu J, et al. Emergency granulopoiesis promotes neutrophil-dendritic cell encounters that prevent mouse lung allograft acceptance. Blood. 2011;118(23):6172–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith DE. The biological paths of IL-1 family members IL-18 and IL-33. J Leukoc Biol. 2011;89(3):383–392. [DOI] [PubMed] [Google Scholar]
- 59.Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A. 2009;106(22):9021–9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin H, Li XY, Jin XB, et al. IL-33 prolongs murine cardiac allograft survival through induction of TH2-type immune deviation. Transplantation. 2010;89(10):1189–1197. [DOI] [PubMed] [Google Scholar]
- 61.Turnquist HR, Zhao Z, Rosborough BR, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol. 2011;187(9):4598–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brunner SM, Schiechl G, Falk W, Schlitt HJ, Geissler EK, Fichtner-Feigl S. Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int. 2011;24(10):1027–1039. [DOI] [PubMed] [Google Scholar]
- 63.Matta BM, Lott JM, Mathews LR, et al. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. 2014;193(8):4010–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puttur F, Denney L, Gregory LG, et al. Pulmonary environmental cues drive group 2 innate lymphoid cell dynamics in mice and humans. Sci Immunol. 2019;4(36). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan W, Zhang B, Liu X, Zhang C, Liu J, Miao Q. Interleukin-33-Dependent Accumulation of Regulatory T Cells Mediates Pulmonary Epithelial Regeneration During Acute Respiratory Distress Syndrome. Front Immunol. 2021;12:653803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.