

Abstract
Background
Because of the broad clinical spectrum, heritable autoinflammatory diseases present a management and therapeutic challenge. The most common genetic interferonopathy, Aicardi Goutières Syndrome (AGS), is associated with early onset neurologic disability and systemic inflammation. The chronic inflammation of AGS is the result of dysregulation of interferon (IFN) expression by one of nine genes within converging pathways. While each AGS subtype shares common features, distinct patterns of severity and potential for systemic complications amongst the genotypes are emerging. Multilineage cytopenias are a potentially serious, but poorly understood, complication of AGS. As immunomodulatory treatment options are developed, it is important to characterize the role of the disease versus treatment in hematologic abnormalities. This will allow for better understanding and management of cytopenia.
Methods
In total, 142 individuals with molecularly-confirmed AGS were included. Information on genotype, demographics, and all available hematologic laboratory values were collected from existing medical records. As part of a clinical trial, a subset of this cohort (n=52) were treated with a janus kinase inhibitor (baricitinib), and both pre- and post-treatment values were included. Abnormal values were graded based on Common Terminology Criteria for Adverse Events (CTCAE v5.0), supplemented with grading definitions for thrombocytosis, and were compared across genotypes and baricitinib exposure.
Results
In total, 11184 laboratory values were collected over a median of 2.54 years per subject (range 0 – 22.68 years). To reduce bias from repeated sampling within a limited timeframe, laboratory results were restricted to the most abnormal value within a month (n= 8485). The most common abnormalities were anemia (noted in 24% of subjects prior to baricitinib exposure), thrombocytopenia (9%), and neutropenia (30%). Neutropenia was most common in the SAMHD1 cohort and increased with baricitinib exposure (38/69 measurements on baricitinib versus 14/121 while not on baricitinib). Having an abnormality prior to treatment was associated with having an abnormality on treatment for neutropenia and thrombocytopenia.
Conclusion
By collecting available laboratory data throughout the lifespan, we were able to identify novel patterns of hematologic abnormalities in AGS. We found that AGS results in multilineage cytopenias not limited to the neonatal period. Neutropenia, anemia, and thrombocytopenia were common. Moderate-severe graded events of neutropenia, anemia, and leukopenia were more common on baricitinib, but rarely of clinical consequence. Based on these results, we would recommend careful monitoring of hematologic parameters of children affected by AGS throughout the lifespan, especially while on therapy, and consideration of AGS as a potential differential diagnosis in children with neurologic impairment of unclear etiology with hematologic abnormalities.
Introduction
The discovery of heritable interferonopathies, including Aicardi Goutières Syndrome (AGS), has led to new understanding of systemic impact of interferon overexpression. AGS, the most common genetic interferonopathy, results in severe neurologic disability coupled with complications of systemic inflammation.
Classic AGS is referred to as a pseudo-congenital infection because of its cerebral calcifications, hepatitis, thrombocytopenia, and anemia. These similarities may be driven by similar pathophysiology as both activate the viral sensing machinery and type I interferon (IFN) production. In the case of AGS, this is caused by pathogenic mutations in nine genes in complementary pathways related to nuclease activity [TREX1 (AGS1), RNASEH2A (AGS2), RNASEH2B (AGS3), RNASEH2C (AGS4), SAMHD1 (AGS5)], RNA processing/sensing machinery [ADAR1 (AGS6), IFIH1 (AGS7)], and histone processing [LSM11 (AGS8), and RNU7–1 (AGS9)].1–4 Irrespective of the specific pathway, all forms of AGS result in IFN activation with elevated IFN-α levels.5, 6
The association between AGS and early thrombocytopenia and anemia is well established in the neonatal period, when the manifestations of AGS are most similar to a TORCH infection.7–11 The hematologic abnormalities noted in case reports has been described as typically transient, but in rare cases this was noted to require transfusions and/or immunosupression.1, 4, 5, 12–17 The mechanism of the thrombocytopenia and anemia is less well understood, although it may be in part driven by targeted auto-antibody production.1, 13 Leukopenia has been noted in limited cases of AGS as well.14 Despite its potential clinical importance, we have limited understanding of the prevalence and extent of hematologic abnormalities in AGS, although has been reported in all known genotypes.1, 4, 5, 12–17 As part of our janus kinase studies,18 we noted baseline hematologic abnormalities affecting multiple cell lineages, which should be monitored while on treatment19.
To better characterize the association between hematologic abnormalities and age, genotype, and baricitinib exposure, we sought to characterize the extent of hematologic abnormalities using analysis of retrospective medical records in a cohort of children affected by AGS. With a better understanding of AGS-related abnormalities in these key cell lines, we can better guide clinical care and clinical trial monitoring protocols.
Methods
Study Approval
All individuals enrolled were in our retrospective natural history study [Myelin Disorders Bioregistry at Children’s Hospital of Philadelphia (IRB#14–011236; NCT01724580)]. Baricitinib was dosed in the context of either an expanded access protocol (JAGA: NCT01724580) or through a phase 2 trial (B-AGS: NCT03921554).
Cohort identification
Inclusion criteria were known AGS-related genotype and availability of complete blood counts (CBC) with available medical records. Individuals without a known genotype or with absence of medical records were excluded. This cohort represents 7 of the 9 genotypes, as LSM11 (AGS8), and RNU7–1 (AGS9) were first described after the data collection period.
Laboratory value collection and classifications
The following variables were collected for all samples as available: age at sample collection, age at disease presentation, genotype, and CBC values. The results from prior CBCs were obtained. Laboratory values were extracted from electronic medical records whenever possible and were supplemented with manual data extraction. Upper and lower limits of normal were collected where available. Laboratory abnormalities were graded as follows: anemia (moderate-severe category <10.0 g/dL, severe category <8.0 g/dL), thrombocytopenia (<75.0 × 10e9/L; <50.0 × 10e9/L), thrombocytosis (>700 × 103/μl; >900 × 103/μl), leukopenia (<3.0 × 109/L; <2.0 × 109/L); lymphopenia (<0.8 × 109/L; <0.5 × 109/L); and neutropenia (<1.5 × 109/L; <1.0 × 109/L) (Supplemental Table 1)20, 21.
Clinical context was collected as available for all laboratory abnormalities graded as severe. This included exposure to immunosuppressant medications, particularly baricitinib and corticosteroids, information surrounding major medical events (including infection), and relevant concomitant medications (eg valproic acid exposure) (Supplemental Table 3).
Each CBC parameter was considered as a categorical variable (grade) as well as a continuous variable. We grouped the graded variables as: moderate-severe (grade 2–4), severe (grade 3–4), and clinically important (grade 4 only). The grading system was based on existing guidelines using the Common Terminology Criteria for Adverse Events (CTCAE v5.0), supplemented with grading definitions for thrombocytosis, and were compared across genotypes and baricitinib exposure. A total of 11184 values were obtained. To reduce the bias from increased sampling frequency after an abnormality is found, the primary analyses were limited to the values with the greatest abnormality within a calendar month. When restricted to only the most abnormal value per month, the number of values reduced to 8485. Episodes with a duration < 1 month were classified as transient and episodes with duration ≥ 1 month were classified as persistent (Supplemental Table 5). For subjects who were treated with baricitinib, episodes were defined per lab and by treatment status (pre and post treatment initiation). The hematologic abnormalities for each parameter for each subject were classified as transient or persistent (Supplemental Table 5).
Statistical approach (See Supplemental Methodology for additional information)
We identified age at sample collection, age at disease presentation, baricitinib exposure (treatment), and genotype as being key variables. All genotypes associated with the RNASEH2-related enzymatic complex were combined due to similar mechanism and low numbers associated with RNASEH2A and RNASEH2C.
For the analyses of the impact of baricitinib, we identified 3 cohorts: never treated with baricitinib, pre-treatment, and on treatment (Supplemental Table 2). The Chi-square test was used to compare genotype and female sex between subjects who were treated versus never treated. Median (25th – 75th) percentiles were obtained for age at symptom onset for subjects who were never treated and those were ever treated. The Wilcoxon rank sum test was used to compare age at symptom onset between these two groups of subjects. Median (25th – 75th) percentiles were obtained for age at sample for all samples measured on those who were never treated and prior to treatment for those who were eventually treated. Quasi-least squares (QLS) regression was used to compare the ages at sample between the two groups, for all lab parameters combined22. The never treated and pre-treatment cohorts were not statistically different in genotype, sex, or age at onset (Supplemental Table 2). However, the median age at symptom onset was greater for those subjects who were eventually treated.
The values were compared by baricitinib exposure (−/+) at the time of laboratory collection using QLS models that included an indicator variable for being on treatment and that adjusted for genotype, age at symptom onset, and age at sample (Model 1) and genotype and age at sample (Model 2) (Figure 1; Supplemental Table 4).
Figure 1.
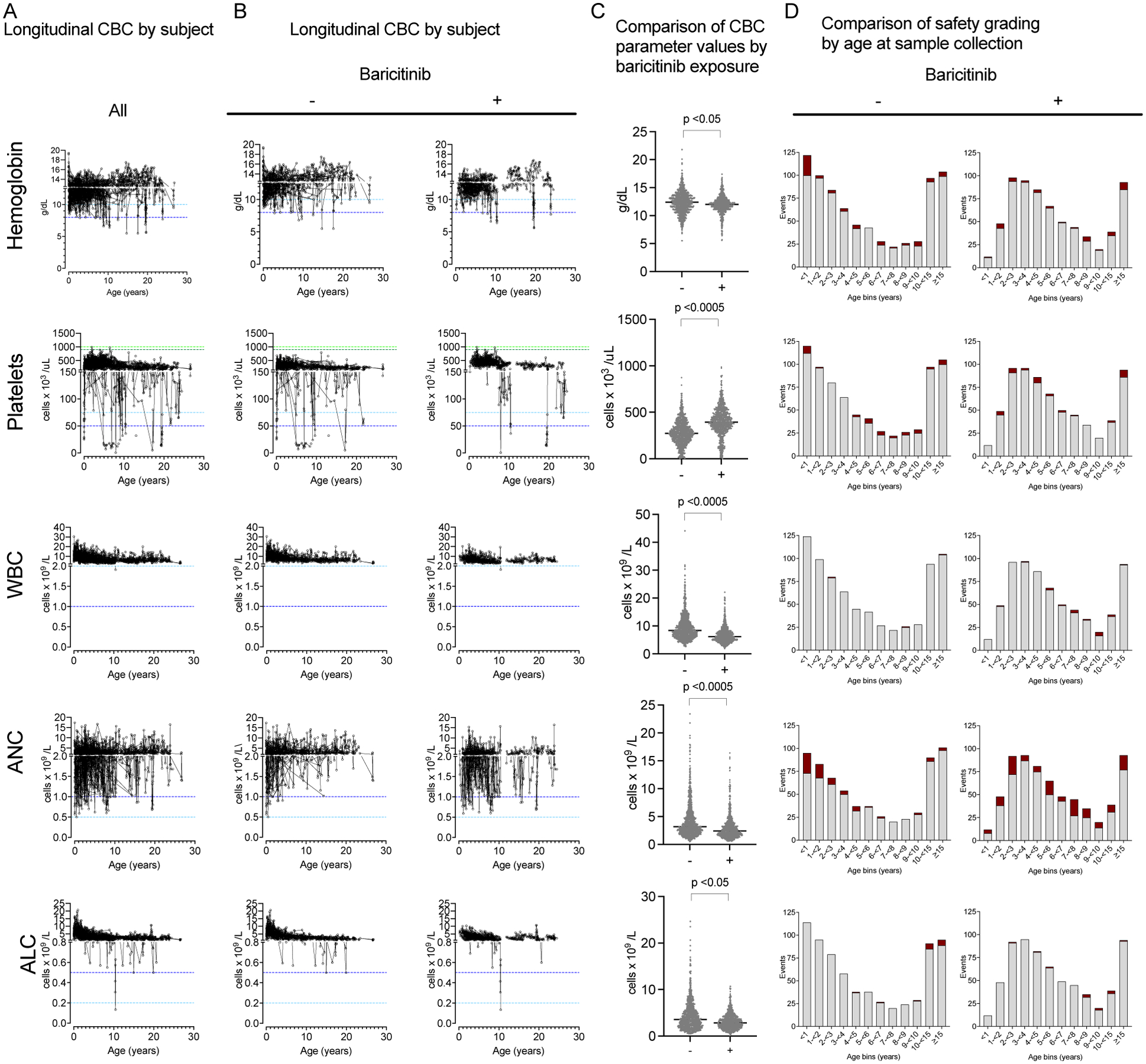
Longitudinal blood count parameters in AGS. Five CBC parameters are presented in rows: abnormalities in hemoglobin, platelets, total WBC, ANC, and ALC. A. The longitudinal samples for each subject are presented in a spaghetti plot (full cohort; A), and by (B) by baricitinib exposure (−, left; +, right). C. The values from CBC are compared by baricitinib exposure (−/+) at the time of laboratory collection. QLS adjusted models with best-fitting correlation structure were used, and the maximum p value from the unadjusted model, and 2 adjusted models are presented (See Supplemental Table 4 for the results of QLS analyses). D. The age at sample collection was divided into 1-year intervals until 10 years of age, then grouped by 10-<15 years, and ≥ 15 years. Each CBC parameter value was classified as abnormal (dark red, grade 2–4 by safety grading) or normal (light grey) as presented by baricitinib exposure (−, left; +, right). A maximum of one event per subject per month is represented.
Pairwise correlations were calculated to generate a correlation matrix for abnormalities in hematologic parameters using the safety grade for concurrently obtained values (Figure 2)23. The 95% confidence intervals (95% CI) for the odds-ratios of adverse events (AE) were calculated based on baricitinib exposure (treatment yes/no) (Forest plot A in Figure 3). The 95% CI in Figure 3 were obtained with generalized estimating equation (GEE) logistic models. Individual logistic models of AE for ANC on baricitinib exposure were then fitted for each Genotype group, with an identity correlation structure and robust sandwich covariance matrix (Forest plot B in Figure 3).
Figure 2.
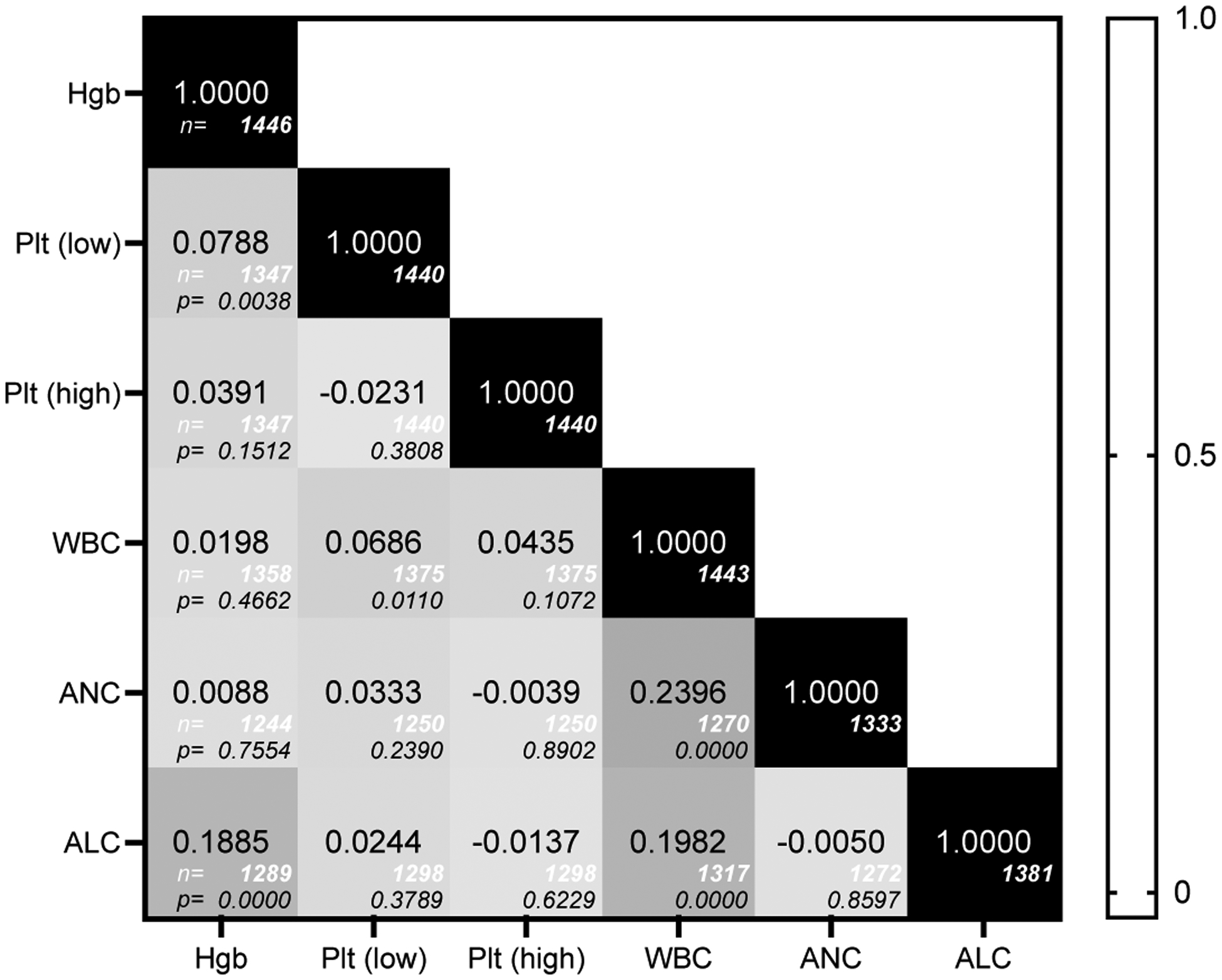
Correlation matrix for abnormalities in hematologic parameters. The safety grade for concurrently obtained parameters was compared using pairwise correlations, with the number of observations below each pairwise correlation (italics, white) and the p value for the test of Ho (italics, black).
Figure 3.
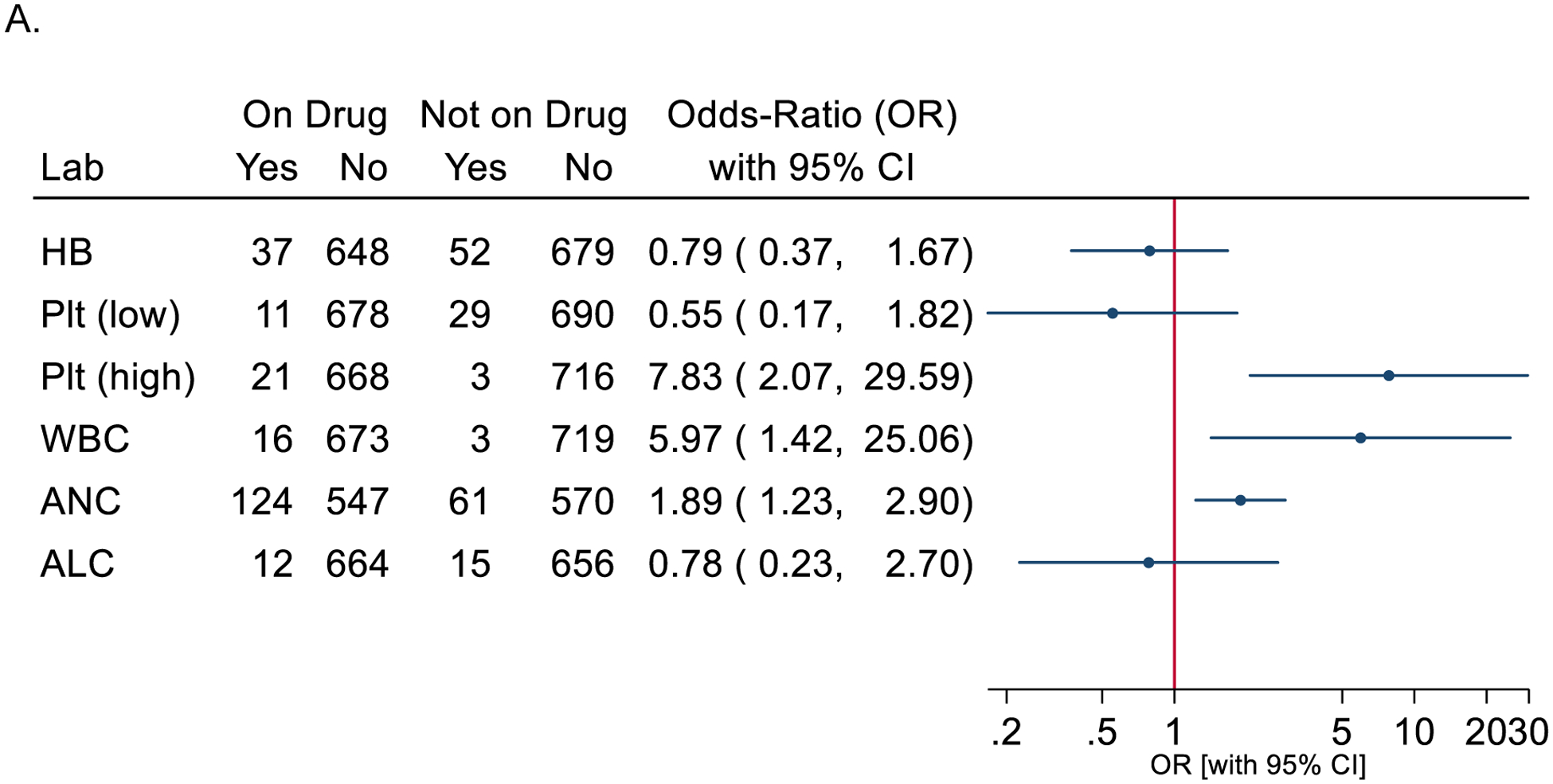
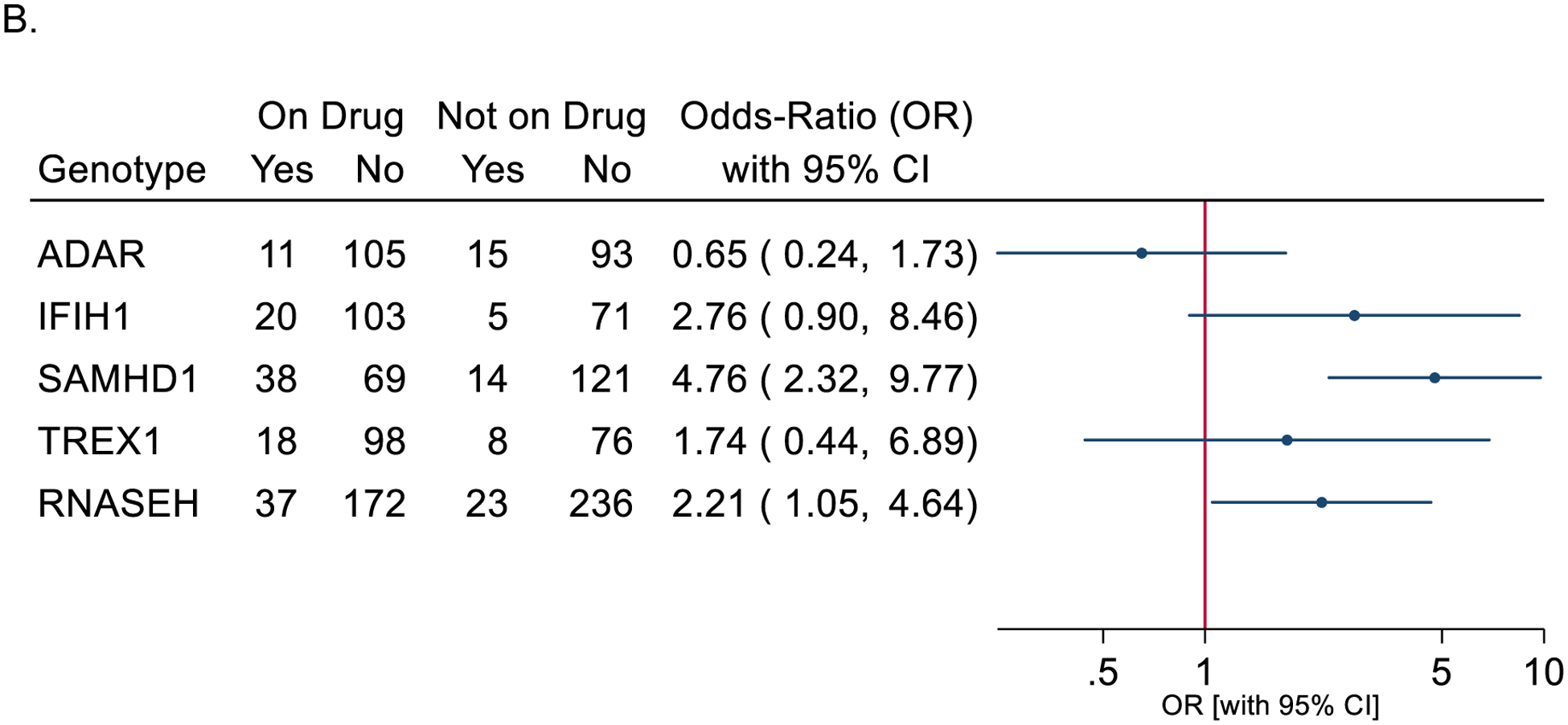
Odds ratio of hematologic abnormalities (adverse events/AE) during baricitinib exposure. Only the most abnormal value within a month was used. The 95% confidence intervals (95% CI) for the odds-ratios of AEs on-treatment versus not on-treatment are from a generalized estimating equation (GEE) logistic model. Forest plot of odds-ratio for CBC parameter abnormalities (grade 2,3,4) with bariticitinib exposure (treatment) (A), and odds-ratio plots for ANC abnormalies (grade 2,3,4) by genotype and baricitinib exposure (B).
Episodes of abnormal hematologic events for each parameter and individual were defined by considering the most abnormal values per lab per individual, sorted by parameter and draw-time within individuals. Episodes with a duration < 1 month were classified as transient and episodes with duration ≥ 1 month were classified as persistent (Supplemental Table 5). For subjects who were treated with baricitinib, episodes were defined per lab and by treatment status (pre and post treatment).
To determine if subjects who had an abnormality prior to exposure with baricitinib were more likely to have an abnormality when treated than those who did not have an abnormality prior to exposure, we next fit logistic regression models on subjects who were treated with baricitinib (Supplemental Table 6). Stata 16.0 was used for analysis with two-sided tests of hypotheses and a p-value < 0.05 as the criterion for statistical significance.
Results
In total, 8485 laboratory values were obtained from 142 individuals with molecularly-confirmed AGS (Figure 1). Data was collected over a range of patient ages ranging from 0–22.68 years with a median 2.54 years collected per individual. Abnormalities in hemoglobin (anemia), platelets (thrombocytopenia), and neutrophils (neutropenia) were the most common. We categorized each CBC parameter by degree of laboratory abnormality. While overall moderate-severe events were uncommon, we sought to characterize the role of age at sample collection, genotype, and baricitinib exposure on abnormalities. A subset of individuals were noted to have recurrent and/or persistent episodes throughout the period of monitoring (Figure 1A). Pre-baricitinib, anemia (n=31/141 subjects; 24%), platelet abnormalities (13/142; 9% with thrombocytopenia; 3/142, 2% with thrombocytosis), neutropenia (41/135; 30%), leukopenia (3/142, 2%), and lymphopenia (11/139; 8%) were noted on a population level.
Next, we hypothesized that baricitinib exposure may be associated with increased rates of abnormal CBC values. There was a shift in all parameter values with baricitinib exposure (Figure 1B), with lower levels in all parameters except for platelets, which increased on study drug (QLS adjusted model with AR(1) structure p value <0.0005 in all comparisons) (Figure 1C). When exposed to baricitinib, anemia (n=14/52 subjects; 27%), platelet abnormalities (4/52; 8% with thromobocytopenia; 9/52, 17% with thrombocytosis), leukopenia (6/52, 12%), neutropenia (35/52; 67%), and lymphopenia (6/52; 12%) were noted within the population.
We visualized the frequency of abnormal CBC parameters (grade 2 or above) by age category (Figure 1D).
Because active inflammation could be a non-specific driver of hematologic abnormalities across multiple cell lineages, we hypothesized that multiple hematologic parameters may be affected in the same individual within the same period (pairwise Pearson correlations, Figure 2). Overall, there was not a strong correlation between concurrent abnormalities across cell lineages. Anemia and thrombocytopenia (Pearson correlation 0.0788; p=0.0038), leukopenia and thrombocytopenia (Pearson correlation 0.0686, p=0.0110), and lymphopenia with anemia were weakly correlated (Pearson correlation 0.1885, p<0.0005). Two of the strongest correlations, neutropenia (ANC:WBC [0.2396]) and lymphopenia (ALC:WBC [0.1982]) with leukopenia, were expected given total WBC count includes both ANC and ALC.
To address increased sampling frequency while receiving baricitinib, we identified the most abnormal value in a one-month period to calculate odds ratios (Figure 3A). There was an increased risk of moderate-severe graded events for thrombocytosis, leukopenia, and neutropenia while on baricitinib. This increase in platelet counts on treatment is distinct from the thrombocytopenia typically reported with early onset AGS. Neutropenia was more common than either anemia or platelet abnormalities, irrespective of baricitinib exposure. Because of the increased rates of neutropenia on study medication, we next characterized the impact of genotype while on baricitinib. Individuals with SAMHD1-related disease were the most likely to have neutropenia while on baricitinib, as were individuals with RNASEH2-related disease (Figure 3B). Comparison of pre- and post- baricitinib exposure hematologic parameters (Figure 1C and 3) account for the clustering of lab values within a single subject. To determine if subjects who had an abnormality prior to exposure with baricitinib were more likely to have an abnormality when treated than those who did not have an abnormality prior to exposure, we fit logistic regression models on subjects who were treated with baricitinib (Supplemental Table 6). Baseline abnormalities (neutropenia or thromobocytopenia) prior to treatment was significantly associated with having abnormalities while on baricitinib.
Discussion
Aicardi Goutières Syndrome is a heritable interferonopathy with neurologic and systemic complications. In this report, we characterized the extent and prevalence of hematologic abnormalities in this disorder. As more children affected by Aicardi Goutières Syndrome are identified and treatment options are developed, it is critical to understand the impact of chronic inflammation on hematologic parameters versus the side effects attributable to treatment with janus kinase inhibitors. By collecting available laboratory data throughout the lifespan, we were able to identify novel patterns of abnormalities in AGS. We found that AGS can result in hematologic abnormalities that is not limited to the neonatal period. Neutropenia, anemia, and thrombocytopenia were common.
Baricitinib exposure was associated with changes in all hematologic parameters, although clinically correlations were rare in our cohort. In total, two individuals on baricitinib required transfusions not related to surgical blood loss and no individuals required antimicrobial prophylaxis due to prolonged lymphopenia or neutropenia. because of immunosuppression. Thrombocytosis was not therapy limiting, and there were no associated clinical events.
Hematologic abnormalities were independent of each other (Figure 2). This could be due to differences in the mechanism of depressed numbers, including cell line half-lives, or consumption versus production issues. In AGS, autoantibodies drive systemic complications in a subset of children, including thyroid dysfunction24. In similar pediatric interferonopathies, autoantibody production decreases with baricitinib treatment25. In our retrospective cohort, the incidence of hematologic-lineage targeting autoantibodies is unknown as it was not consistently clinically tested. Future study into the mechanism of the cell lineage changes is necessary to understand this balance.
This study includes an international cohort of children affected by AGS, but data is limited to the availability of existing medical testing. Based on standard clinical practice, we hypothesized that laboratory testing may be more likely to be repeated when abnormalities were noted. To reduce the bias from repeated testing, the most abnormal value in a month was used for analyses. It is also likely that due to intermittent testing, transient abnormalities could be potentially missed, and thus the rates of hematologic abnormalities may be underrepresented by this study.
By collecting available laboratory data throughout the lifespan, we were able to identify novel patterns of abnormalities in AGS. Neutropenia, anemia, and thrombocytopenia were common throughout the duration of monitoring. Baricitinib exposure was associated with changes in all hematologic parameters, although this was rarely clinically actionable. Abnormalities prior to baricitinib exposure correlated with abnormalities while on treatment. We would recommend monitoring of hematologic parameters of children affected by AGS throughout the lifespan consistent with recently published AGS management guidelines, particularly when baseline abnormalities have been noted19. Multilineage cytopenias are an intrinsic manifestation of AGS and thus should not limit therapeutic opportunities. Because of its neurologic heterogeneity, we hypothesize that AGS may be often underdiagnosed. Transient or persistent hematologic abnormalities may be an important clue that AGS should be considered in children with neurologic impairment of unclear etiology.
Supplementary Material
Funding
AV, LAA, JS and FG were supported by U01 NS106845 and U54TR002823 from the NIH, NINDS and NCATS. Research reported in this publication was supported by CURE Pennsylvania grant. LAA supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Award Number K23NS114113.
COI
AV receives grant and in-kind support for research from Eli Lilly, Gilead, Takeda, Illumina, Biogen, Homology, Ionis, Passage Bio, Orchard Therapeutics. AV serves on the scientific advisory boards of the European Leukodystrophy Association and the United Leukodystrophy Foundation, as well as in an unpaid capacity for Takeda, Ionis, Biogen and Illumina. The AGS scale is copyrighted by the Children’s Hospital of Philadelphia. LAA is a consultant for Takeda, Biogen, and Orchard Therapeutics. RGM received study support under a CRADA from Eli Lilly, Regeneron and SOBI and IFM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Trial registration
ClinicalTrials.gov Identifier: NCT01724580
ClinicalTrials.gov Identifier: NCT03921554
References
- 1.Uggenti C, Lepelley A, Depp M, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet 2020;52:1364–1372. [DOI] [PubMed] [Google Scholar]
- 2.Rice GI, Kitabayashi N, Barth M, et al. Genetic, Phenotypic, and Interferon Biomarker Status in ADAR1-Related Neurological Disease. Neuropediatrics 2017;48:166–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Livingston JH, Crow YJ. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutieres Syndrome and Beyond. Neuropediatrics 2016;47:355–360. [DOI] [PubMed] [Google Scholar]
- 4.Rice G, Patrick T, Parmar R, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet 2007;81:713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crow YJ, Chase DS, Lowenstein Schmidt J, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 2015;167a:296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armangue T, Orsini JJ, Takanohashi A, et al. Neonatal detection of Aicardi Goutières Syndrome by increased C26:0 lysophosphatidylcholine and interferon signature on newborn screening blood spots. Mol Genet Metab 2017;122:134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crow YJ, Shetty J, Livingston JH. Treatments in Aicardi-Goutières syndrome. Dev Med Child Neurol 2020;62:42–47. [DOI] [PubMed] [Google Scholar]
- 8.Crow YJ. Aicardi-Goutières Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © 1993–2020, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved, 1993. [Google Scholar]
- 9.Orcesi S, La Piana R, Fazzi E. Aicardi-Goutieres syndrome. Br Med Bull 2009;89:183–201. [DOI] [PubMed] [Google Scholar]
- 10.Livingston JH, Crow YJ. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutières Syndrome and Beyond. Neuropediatrics 2016;47:355–360. [DOI] [PubMed] [Google Scholar]
- 11.Sase S, Takanohashi A, Vanderver A, Almad A. Astrocytes, an active player in Aicardi-Goutières syndrome. Brain Pathol 2018;28:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amari S, Tsukamoto K, Ishiguro A, Yanagi K, Kaname T, Ito Y. An extremely severe case of Aicardi-Goutières syndrome 7 with a novel variant in IFIH1. Eur J Med Genet 2020;63:103646. [DOI] [PubMed] [Google Scholar]
- 13.Samanta D, Ramakrishnaiah R, Crary SE, Sukumaran S, Burrow TA. Multiple Autoimmune Disorders in Aicardi-Goutières Syndrome. Pediatr Neurol 2019;96:37–39. [DOI] [PubMed] [Google Scholar]
- 14.Ramantani G, Kohlhase J, Hertzberg C, et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum 2010;62:1469–1477. [DOI] [PubMed] [Google Scholar]
- 15.Marguet F, Laquerrière A, Goldenberg A, et al. Clinical and pathologic features of Aicardi-Goutières syndrome due to an IFIH1 mutation: A pediatric case report. Am J Med Genet A 2016;170a:1317–1324. [DOI] [PubMed] [Google Scholar]
- 16.He T, Xia Y, Yang J. Systemic inflammation and chronic kidney disease in a patient due to the RNASEH2B defect. Pediatr Rheumatol Online J 2021;19:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adang LA, Frank DB, Gilani A, et al. Aicardi goutières syndrome is associated with pulmonary hypertension. Mol Genet Metab 2018;125:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanderver A, Adang L, Gavazzi F, et al. Janus Kinase Inhibition in the Aicardi-Goutières Syndrome. N Engl J Med 2020;383:986–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cetin Gedik K, Lamot L, Romano M, et al. The 2021 EULAR and ACR points to consider for diagnosis and management of autoinflammatory type I interferonopathies: CANDLE/PRAAS, SAVI and AGS. Annals of the rheumatic diseases 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dame C, Sutor AH. Primary and secondary thrombocytosis in childhood. British journal of haematology 2005;129:165–177. [DOI] [PubMed] [Google Scholar]
- 21.Sarangi R, Pradhan S, Dhanawat A, Patanayak R, Benia G. Thrombocytosis in children: Clinico-hematological profile from a single centre in Eastern India. Journal of laboratory physicians 2018;10:34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shults J, Hilbe JM. Quasi-Least Squares Regression, 1st ed. New York: Chapman & Hall/CRC, 2014. [Google Scholar]
- 23.Shults J Simulating longer vectors of correlated binary random variables via multinomial sampling. Computational Statistics & Data Analysis 2017;114:1–11. [Google Scholar]
- 24.Cattalini M, Galli J, Andreoli L, et al. Exploring Autoimmunity in a Cohort of Children with Genetically Confirmed Aicardi-Goutieres Syndrome. Journal of clinical immunology 2016;36:693–699. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez GAM, Reinhardt A, Ramsey S, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 2018;128:3041–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.