

Abstract
The Dedicator of Cytokinesis (DOCK) family (DOCK1–11) of genes are essential mediators of cellular migration, growth, and fusion in a variety of cell types and tissues. Recent advances in whole genome sequencing (WGS) of patients with undiagnosed genetic disorders have identified several rare pathogenic variants in DOCK genes. We conducted a systematic review and performed a patient database and literature search of reported DOCK pathogenic variants that have been identified in association with clinical pathologies such as global developmental delay, immune cell dysfunction, muscle hypotonia, and muscle ataxia among other categories. We then categorized these pathogenic DOCK variants and their associated clinical phenotypes under several unique categories: developmental, cardiovascular, metabolic, cognitive, or neuromuscular. Our systematic review of DOCK variants aims to identify and analyze potential DOCK-regulated networks associated with neuromuscular diseases and other disease pathologies, which may identify novel therapeutic strategies and targets. This systematic analysis and categorization of human associated pathologies with DOCK pathogenic variants is the first report to the best of our knowledge for a unique class in this understudied gene family that has important implications in furthering personalized genomic medicine, clinical diagnoses, and improve targeted therapeutic outcomes across many clinical pathologies.
Keywords: DOCK, hypotonia, skeletal muscle, intellectual disability
Background
Dedicator of Cytokinesis (DOCK) proteins function as guanine nucleotide exchange factors (GEFs) that promote the release of GDP and GTP binding to small GTPases of the Rho protein family. The activation of these small GTPases requires the utilization of specific enzymes called guanine nucleotide exchange factors (GEFs), which serve as a molecular switch using the exchange of GDP for GTP on the Rho GTPase with the requirement of magnesium (Mg2+) (Laurin & Côté, 2014). The DOCK proteins can be further classified into subfamilies (DOCKs A-D) based on evolutionary conservation of key protein-protein interaction domains. Many DOCK proteins have vital functions throughout the central nervous system and musculoskeletal system. Additionally, more recent analyses of the DOCK gene family have revealed key contributions to vascular biology, development, and health (Benson & Southgate, 2021).
Clinical and Diagnostic Implications
Currently in the literature, a number of animal models involving loss-of-function or gene-dosage studies on the DOCK gene family correlate to musculoskeletal, neurodegenerative disease, and neurodevelopmental disorders. For example, there are a multitude of animal studies related to understanding the role of in Dock3 as a secondary modulator of musculoskeletal pathology and neurodegenerative disease, such as Duchenne muscular dystrophy (DMD) and Alzheimer’s disease (AD) (Chen et al., 2009; Namekata et al., 2004; Reid et al., 2020; Tachi et al., 2012). Similarly, studies of human patients with DOCK3 pathogenic variants, have been correlated with neurodevelopmental disorders, such as global developmental delay, most notably in young children (Helbig, 2017,Iwata-Otsubo, 2018). However, studies specifically on pathogenic DOCK variants and how they impact human disease remain limited and require further exploration. DOCK variants also appear with a high degree of phenotypic heterogeneity in relation to a multitude of clinical symptoms associated with a vast set of human pathologies ranging from metabolic and immune disorders to neuromuscular disorders and developmental disorders. This presents a challenge in investigating the molecular drivers and clinical pathologies caused by these rare DOCK gene pathogenic variants for clinicians and researchers. Therefore, this systematic review aims to provide a comprehensive analysis of human pathogenic DOCK variants and organize their wide range of clinical phenotypes into several unique categories, which we collectively refer to as ‘DOCKopathies’.
The high degree of phenotypic heterogeneity, specifically in the clinical phenotypes observed in pathogenic DOCK variants is common among rare diseases, providing challenges in diagnosis and interpretation for clinicians and researchers. This also leads to challenges in classification, particularly in the case of rare missense variants (Wu et al., 2021). The efficacy and utility of personalized genomic medicine relies on the ability to assess pathogenicity. In our systematic review, we are the first to provide a comprehensive and structured clinical and molecular analysis of all 11 DOCK genes as well as categorize pathogenic variants in correlation with other clinical phenotypes and outcomes.
Methods
Databases for Patient Record Variant Search Strategy
Variants of interest in DOCK genes were extracted from PubMed and ClinVar (Landrum et al., 2015). For our DOCK variant search strategy in PubMed, we used the following search terms, relating to phenotypes often presented by pathogenic variants in DOCK genes such as “immune”, “muscle”, “metabolic”, and “intellectual disability”. In addition we reviewed all DOCK variants reported in ClinVar and classified pathogenic and likely pathogenic variants by variant type and molecular consequence as classified by ClinVar. We further limited our search to the phenotypes associated with variants noted to be pathogenic or likely pathogenic associated with the terms “developmental”, “cardiovascular”, “nervous system”, “immune system”, “metabolic”, and “2 or more” for those that presented with more than one category of pathophysiology. A complete summary of all DOCK genes, their chromosomal positions, and any overlapping genes are shown in Supp. Table S1. A full list of all DOCK pathogenic variants identified in this study are available in Supp. Table S2. These categories were determined by consulting the International Classification of Disorders-11 (ICD-11) and Human Phenotype Ontology (HPO) terms. A detailed flow diagram of these methods is available (Figures 1A and 1B).
Figure 1: Summary of Methodology.
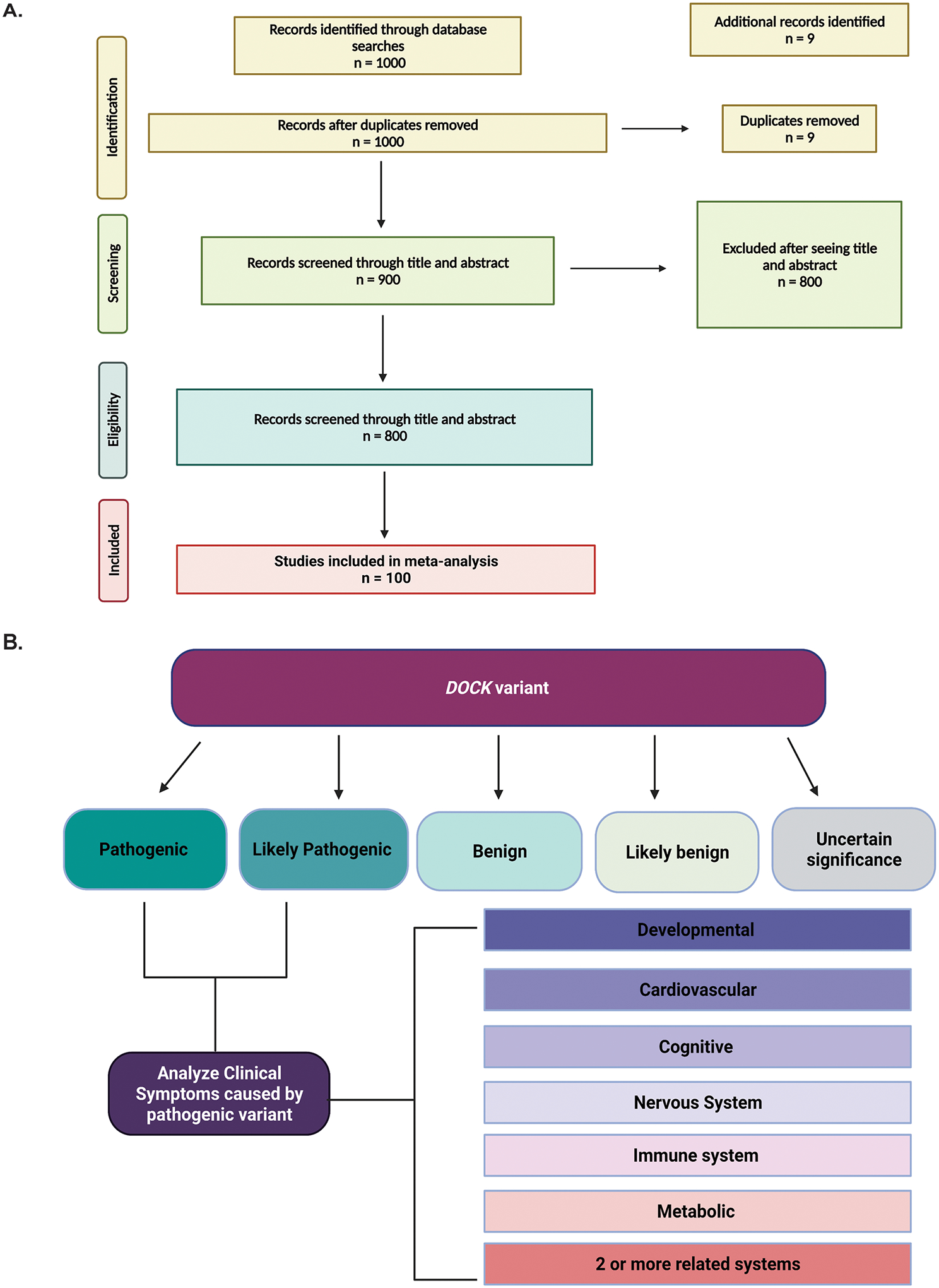
Meta-analysis was conducted using two databases, PubMed and ClinVar. Initial inquiry began with PubMed (A) in which we reviewed 1000 available primary articles on DOCK variants. We continued our search analyzing reported variants in ClinVar (B) in which we sorted each variant as reported by ClinVar in their respective categories.
Inclusion and exclusion criteria
Publications returned from the databases were imported into the EndNote™ reference manager, and duplicate citations were removed. Articles were included if 1) a DOCK variant was noted to be pathogenic or likely pathogenic for a disease or syndrome 2) if the DOCK variant described was associated to a developmental defect, muscle defect, neurological defect, alterations in social behavior, or other pathophysiology. Publications were excluded if the DOCK variant discussed had been identified in an individual where a more clearly pathogenic DOCK variant was seen or where the DOCK variant discussed was associated with only common disorders. We used the American College of Medical Genetics and Genomics (ACMG) derived ClinVar classification system to classify each variant by pathogenicity (pathogenic, likely pathogenic, benign, likely benign, risk factor, and uncertain significance), variation type (deletion, duplication, indel, insertion, single nucleotide polymorphism), and molecular consequence (frameshift, missense, nonsense, copy number gain, and copy number loss) (Richards et al., 2015). Differences in classification were studied across variants in all 11 DOCK genes.
Results
Organizing DOCK variants by pathogenicity, molecular consequences, and variation types
We identified and reviewed approximately 1,000 manuscripts that referenced DOCK variants. Our main goal was to focus on clinical data so we excluded 800 studies on exploring phenotypes in animal models for which no patient DOCK pathogenic variant had been identified. Following exclusions, we were left with 100 papers detailing patient cases published from 2011 to 2020 (Figure 1). In our ClinVar analysis across all 11 of the DOCK genes, we found that 23.40% of variants were classified as pathogenic, 2.35% were likely pathogenic, and 44.5% of were classified as variants of uncertain significance (VUS) (Figure 2A). The vast majority of variants were missense variants (Figure 2B). Approximately 62% of all variants were single nucleotide polymorphisms and approximately 20% were DNA duplications (Figure 2C).
Figure 2: All DOCK variants identified and sorted in ClinVar.
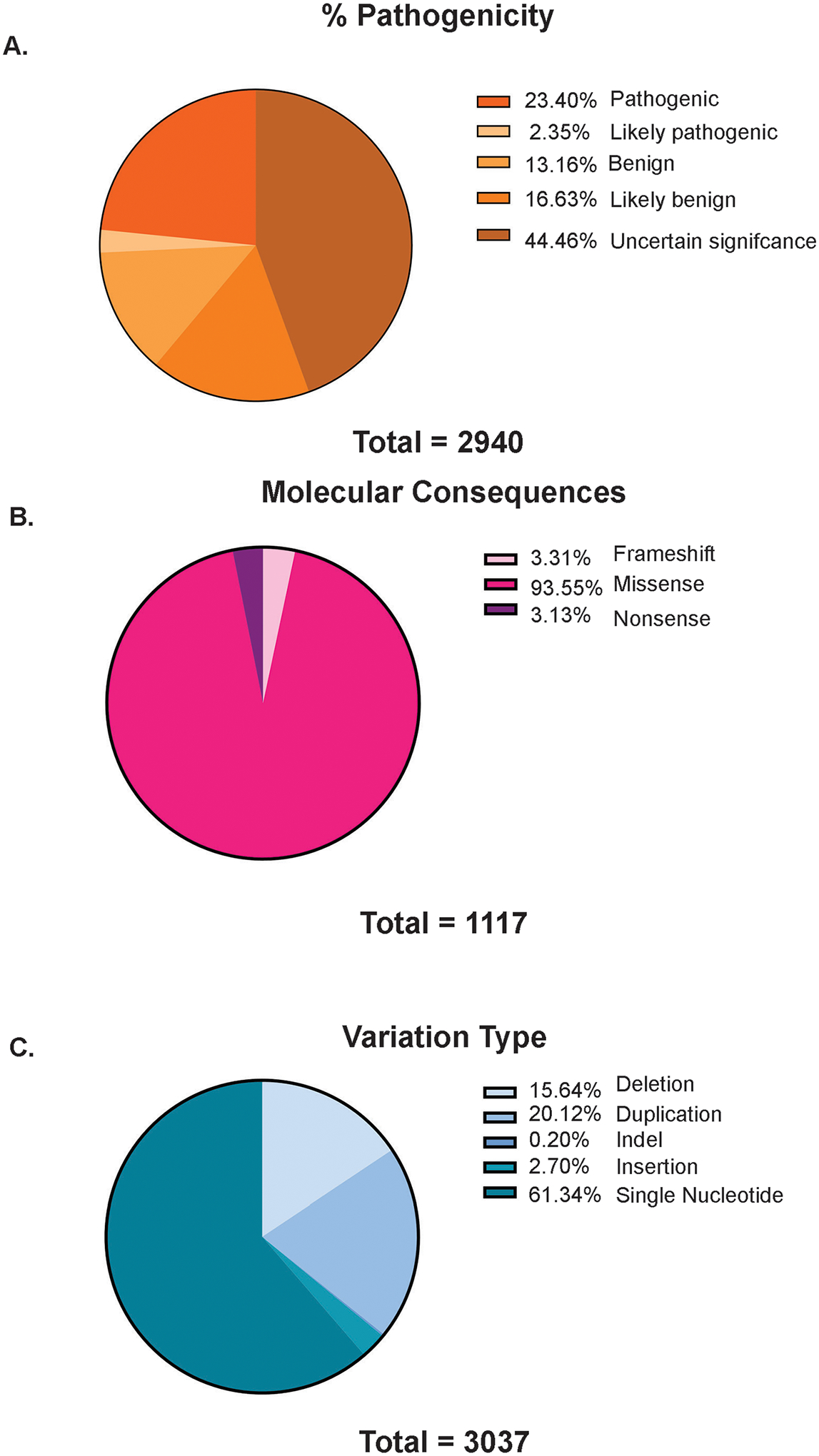
Comprehensive analysis at all DOCK variants identified on ClinVar. We sorted variants for their percentage (%) pathogenicity (A), by molecular consequence as either frameshift, nonsense, or missense (B) and sorted by variant type, as either deletion, duplication, indel, insertion, or single nucleotide (C).
Organizing pathogenic DOCK variants by molecular consequences
We then studied the nature, impact, and likely molecular consequences for each pathogenic or likely pathogenic DOCK variant across each gene. Quantification of molecular consequences is shown in Supp. Table S3. In our analysis, a wide variety of DOCK gene mutation types and impacts were observed, often with distinct trends in different genes (Figure 3). In DOCK1, pathogenic variants were reported as copy number gains or losses. In DOCK2 variants 41.38% were copy number gains, while only a small percentage were reported as being missense and nonsense variants (3.45% and 17.24%). In DOCK3, pathogenic variants were identified as copy number gains and primarily as duplications. DOCK3 pathogenic variants also presented with missense and nonsense mutations (20.00% and 10.00%, respectively). DOCK4 and DOCK5 pathogenic variants were mainly represented by copy number gains and losses. Frameshifts represented the largest classification of DOCK6 variants (~33.33%). A variety of impacts of DOCK7 patients had a large percentage were frameshifts (22.92%) and copy number losses (25.00%). DOCK8 variants were mainly copy number losses (39.33%) while few were due to nonsense, missense, frameshift mutations or deletions (2.81%, 1.69%, 4.49%, and 12.36%). Lastly, DOCK9, DOCK10, and DOCK11 pathogenic variant cases were mainly copy number gains or losses. However, both DOCK9 and DOCK10 variants among all 11 had the highest reported pathogenic mutations due to copy number gains (73.33% for DOCK10 and 47.41% for DOCK11).
Figure 3: DOCK variant molecular consequences.
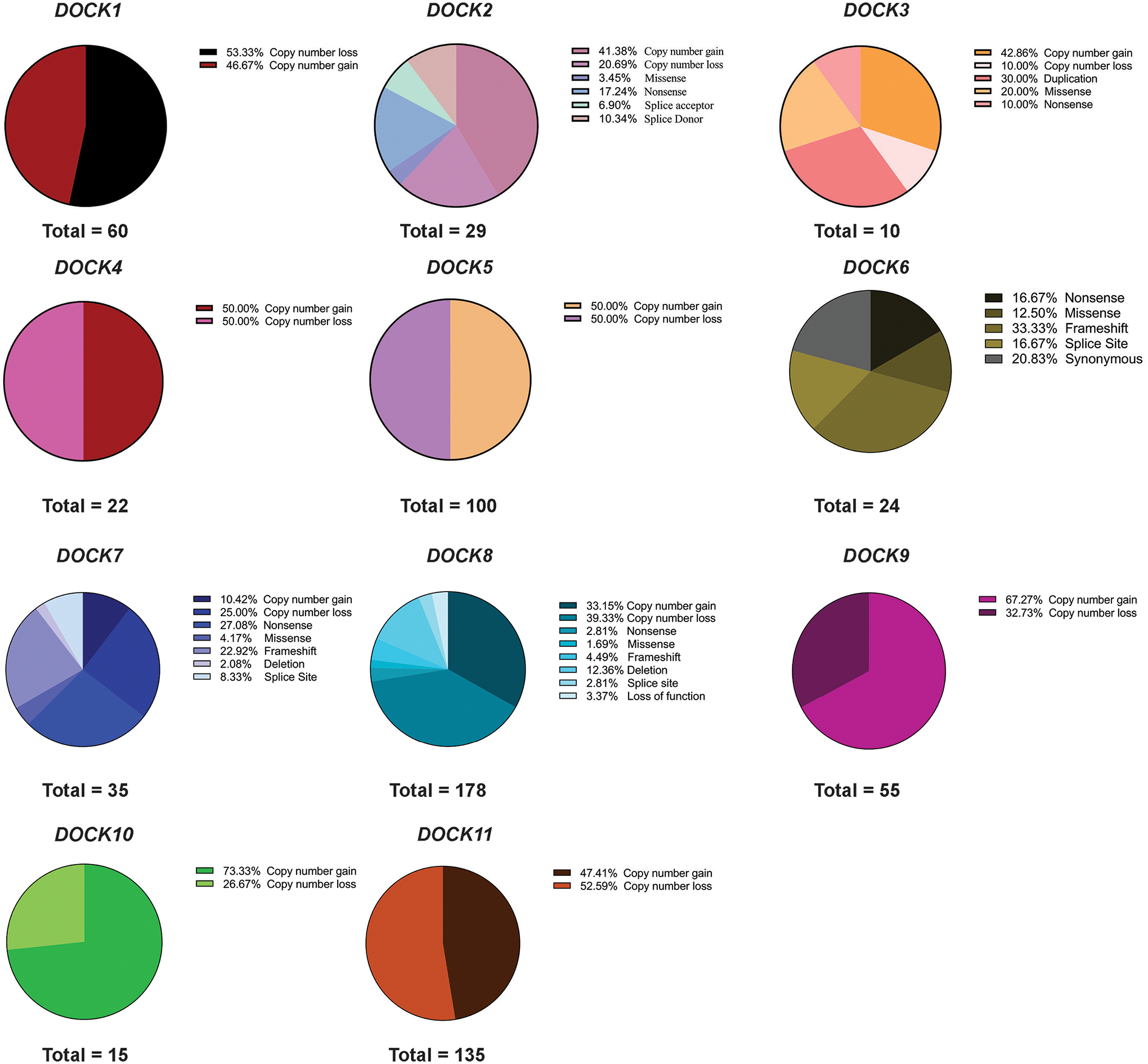
Each pathogenic DOCK variant for each subclass (1–11) were analyzed by molecular consequence as reported by ClinVar.
Organizing pathogenic DOCK variants by variation and clinical phenotype.
In DOCK1 patients, duplications (46.67%) and deletions predominated (53.33%). By contrast, the bulk of DOCK2, DOCK4, DOCK9, DOCK10, and DOCK11 variants were duplications. Some variant types were seen predominantly in some genes. For example, DOCK6 had a large amount of single nucleotide variants at nearly 54% of variants (Figure 4). We further analyzed each individual DOCK pathogenic variant and its reported pathophysiology by grouping all reported symptoms under seven categories: developmental, cardiovascular, immune system, nervous system, cognitive, metabolic, and neuromuscular or if the variant presented with multiple related pathophysiologies, it was classified as 2 or more (Figure 5). Specific pathologies attributed to each category are listed (Table 1) and specification of each pathogenic variant per category are shown in Supp. Table S2. Additionally, a numerical summary of each DOCK’s variant’s classifications is provided in Supp. Table S3. While many DOCK patient variants impacted multiple systems, the presence of pathogenic variants in nearly every DOCK gene resulted in a developmental phenotype (e.g. craniofacial defects, syndactyly etc.). Nearly 60% of all DOCK genes when impacted by a deleterious variant resulted in a developmental defect.
Figure 4: DOCK Mutation Type.
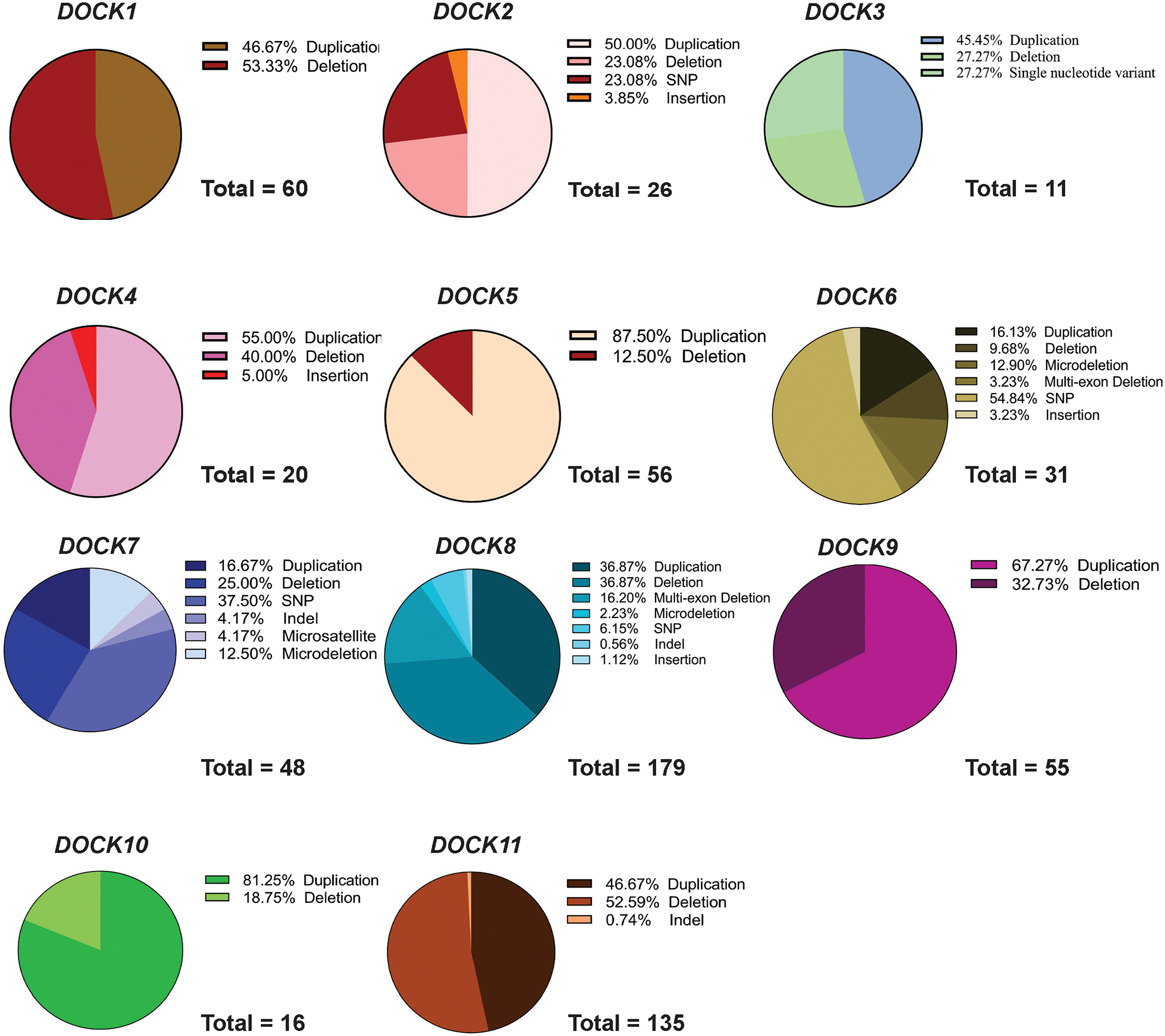
Each pathogenic DOCK variant for each subclass (1–11) was analyzed by variant type, either duplication, deletion, indel, insertion etc. as reported by ClinVar.
Figure 5: Clinical Symptoms of DOCK pathogenic variants.
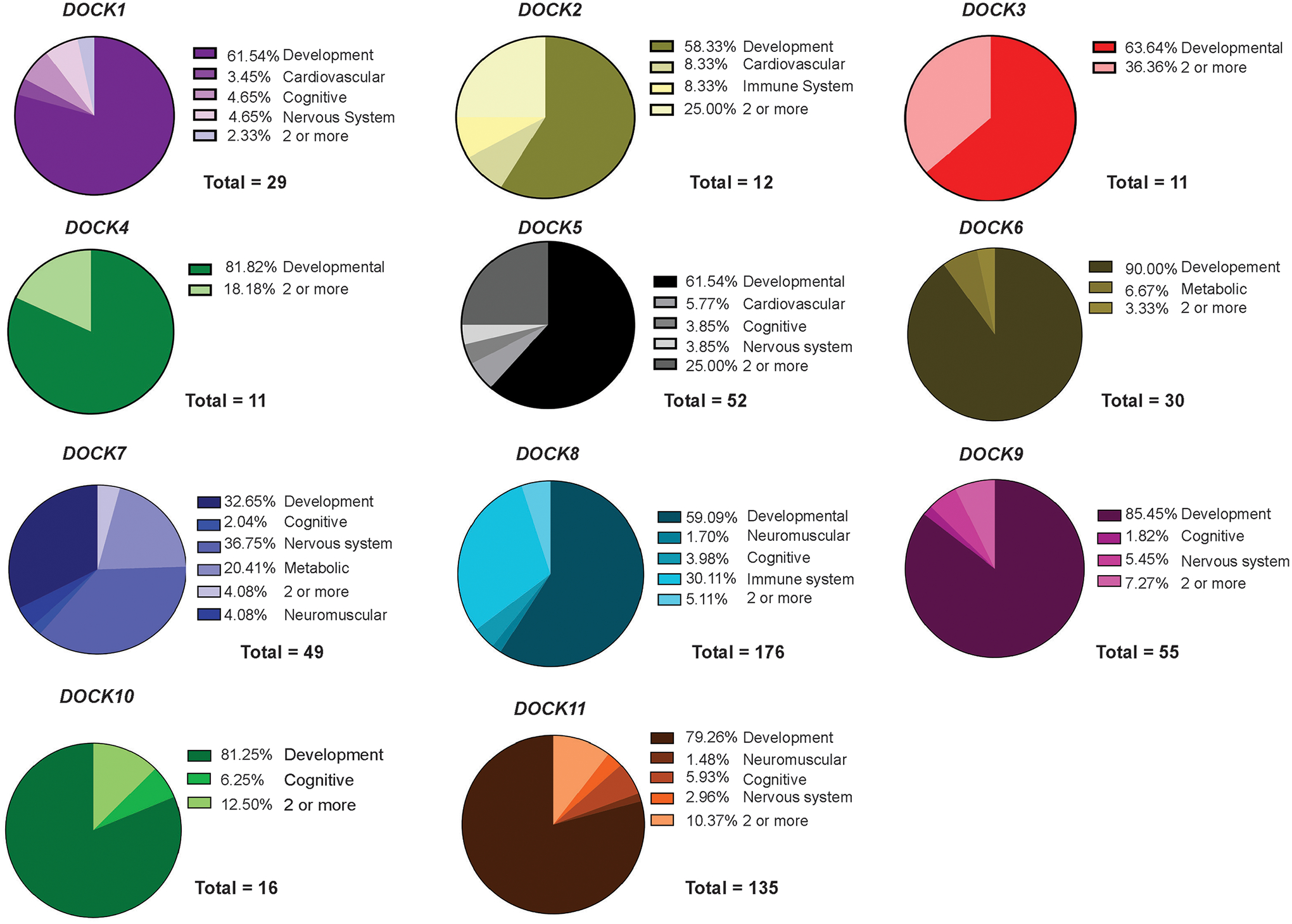
DOCK pathogenic variants were identified on ClinVar and were collectively categorized by several classifications: ‘Developmental’, ‘Cardiovascular’, ‘Cognitive’, ‘Metabolic’, ‘Nervous system’, and if there were multiple conditions associated with each category they were sorted as ‘2 or more’.
Table 1. Clinical Presentations of Pathogenic DOCK Variants.
Clinical symptoms as reported by ClinVar for each pathogenic DOCK variant and their respective categorization in Figure 5 are listed.
| Development | |
|---|---|
| DOCKs Presenting | |
| abnormal facial shape | DOCK1, DOCK2, DOCK3, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| abnormality of limb and bone morphology | DOCK5 |
| abnormality of the ear | DOCK1, DOCK2, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| abnormality of the skull | DOCK4 |
| abnormality of the vertebrae | DOCK5, DOCK11 |
| Adams‐Oliver Syndrome | DOCK6 |
| Adams‐Oliver Syndrome 2 | DOCK6 |
| agenesis of the corpus callosum | DOCK1, DOCK5, DOCK8, DOCK9 |
| ambiguous genitalia | DOCK4, DOCK8, DOCK11 |
| Arnold‐Chiari malformations | DOCK5, DOCK8 |
| bilateral hyperplasia of choroid plexus | DOCK8 |
| bilateral single tranverse palmar creases | DOCK5, DOCK9 |
| blepharophimosis | DOCK8, DOCK11 |
| camptodactyly | DOCK2 |
| cerebellar hypoplasia | DOCK8 |
| cerebral white matter hypoplasia | DOCK1 |
| cleft palate | DOCK1, DOCK2, DOCK4, DOCK5, DOCK8, DOCK9, DOCK10, DOCK11 |
| clinodactyly | DOCK8, DOCK10 |
| coarse facial features | DOCK2, DOCK10 |
| coloboma | DOCK2, DOCK5, DOCK8, DOCK9, DOCK10 |
| craniosynostosis | DOCK2, DOCK8, DOCK11 |
| cryptorchidism | DOCK1, DOCK5, DOCK8, DOCK10, |
| Dandy‐Walker malformation | DOCK9 |
| delayed fine motor development | DOCK1, DOCK2, DOCK5, DOCK8, DOCK9, DOCK11 |
| delayed gross motor development | DOCK1, DOCK2, DOCK4, DOCK5, DOCK8, DOCK9 |
| delayed speech and language development | DOCK1, DOCK4, DOCK5, DOCK8, DOCK9, DOCK10, DOCK11 |
| developmental delay | DOCK1, DOCK2, DOCK3, DOCK4, DOCK5, DOCK6, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| distal urethral duplication | DOCK1 |
| dolichocephaly | DOCK2, DOCK8 |
| dysmorphic features | DOCK2, DOCK3 |
| esotropia | DOCK9 |
| failure to thrive | DOCK5, DOCK6, DOCK8, DOCK9, DOCK10, DOCK11 |
| fetal cystic hygroma | DOCK1 |
| gastroesophageal reflux | DOCK2 |
| genu recurvatum | DOCK8 |
| global developmental delay | DOCK1, DOCK2, DOCK3, DOCK4, DOCK5, DOCK6, DOCK7, DOCK8, DOCK9, DOCK11 |
| gonadal dysgenesis | DOCK11 |
| holoprosencephaly | DOCK2, DOCK9, DOCK11 |
| hydrocephaly | DOCK8 |
| hydroureter | DOCK5 |
| hypertelorism | DOCK9, DOCK11 |
| hypospadias | DOCK10 |
| hypotelorism | DOCK1 |
| increased nuchal translucency | DOCK5, DOCK8, DOCK11 |
| intrauterine growth retardation | DOCK1, DOCK2, DOCK3, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| macrocephaly | DOCK2, DOCK8, DOCK9, DOCK11 |
| microcephaly | DOCK1, DOCK5, DOCK8, DOCK9, DOCK11 |
| microglossia | DOCK2 |
| micrognathia | DOCK1, DOCK2, DOCK3, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| micropenis | DOCK1, DOCK8 |
| multiple congenital anomalies | DOCK1 |
| nevus flammeus | DOCK9 |
| omphalocele | DOCK8, DOCK9 |
| Pierre‐Robin sequence | DOCK2 |
| platybasia | DOCK5 |
| polydactyly | DOCK1, DOCK2, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| ptosis | DOCK11 |
| csoliosis | DOCK2, DOCK5, DOCK8, DOCK10, DOCK11 |
| short stature | DOCK2, DOCK3, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| smooth philtrum | DOCK1, DOCK9 |
| strabismus | DOCK1 |
| syndactyly | DOCK1, DOCK2, DOCK3, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| talipes equinovarus | DOCK8, DOCK9, DOCK11 |
| tapered fingers | DOCK5 |
| tracheomalachia | DOCK8 |
| trigonocephaly | DOCK8 |
| urinary tract malformations | DOCK7 |
| ventriculomegaly | DOCK8, DOCK9, |
| vitiligo | DOCK5 |
| wide nasal bridge | DOCK5, DOCK8, DOCK9, DOCK10, DOCK11 |
| Cardiovascular | |
| DOCKs Presenting | |
| abnormality of cardiac morphology | DOCK5, DOCK8, DOCK11 |
| abnormality of the fetal cardiovascular system | DOCK5 |
| aortic valve stenosis | DOCK11 |
| atria septal defect | DOCK4, DOCK5, DOCK8, DOCK9, DOCK10, DOCK11 |
| bicuspid aortic valve | DOCK8 |
| coarctation of the aorta | DOCK8, DOCK9, DOCK10 |
| esophageal atresia | DOCK8, DOCK11 |
| heart murmur | DOCK8 |
| hypertrophic cardiomyopathy | DOCK11 |
| hypoplastic aortic arch | DOCK9 |
| hypoplastic left heart | DOCK5, DOCK8, DOCK11 |
| mitral valve prolapse | DOCK1 |
| obsolete malformation of the heart and great vessels | DOCK1, DOCK2, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| patent ductus arteriosis | DOCK1, DOCK2, DOCK8, DOCK11 |
| pericardial effusion | DOCK5 |
| pulmonic stenosis | DOCK8 |
| right bundle branch block | DOCK1 |
| supraventricular tachycardia | DOCK2 |
| tetrology of Fallot | DOCK8, DOCK9, DOCK10 |
| ventricular septal defect | DOCK1, DOCK2, DOCK3, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| Cognitive Behavioral | |
| DOCKs Presenting | |
| ADHD | DOCK1, DOCK8 |
| aggressive behavior | DOCK1 |
| anxiety | DOCK8 |
| autistic behavior | DOCK1, DOCK5, DOCK8, DOCK10, DOCK11 |
| bipolar affective disorder | DOCK9, DOCK11 |
| intellectual disability | DOCK1, DOCK2, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| learning disability | DOCK5, DOCK8, DOCK11 |
| polyphagia | DOCK11 |
| schizophrenia | DOCK11 |
| Nervous System Disorder | |
| DOCKs Presenting | |
| bilateral sensorineural hearing impairment | DOCK9, DOCK11 |
| cerebral palsy | DOCK1, |
| encephalopathy | DOCK2, DOCK11 |
| epileptic encephalopathy | DOCK7 |
| seizures | DOCK1, DOCK5, DOCK6, DOCK8, DOCK9, DOCK11 |
| Immune System | |
| DOCKs Presenting | |
| hyper‐IgE recurrent infection sydrome, autosomal recessive | DOCK8 |
| hypogammaglobulinemia | DOCK10 |
| immunodeficiency | DOCK2, DOCK8 |
| Metabolic | |
| DOCKs Presenting | |
| familial hypercholesterolemia 1 | DOCK6 |
| hypobetalipoproteinemia, familial, 2 | DOCK7 |
| Neuromuscular | |
| DOCKs Presenting | |
| ataxia | DOCK1, DOCK3 |
| dystonia | DOCK1, |
| flexion contracture | DOCK4, DOCK5, DOCK10 |
| gait ataxia | DOCK1, |
| gait disturbance | DOCK8, DOCK11 |
| generalized amyotrophy | DOCK5 |
| hypotonia | DOCK3, DOCK4, DOCK5, DOCK7, DOCK8, DOCK9, DOCK10, DOCK11 |
| iIncoordination | DOCK5, DOCK11 |
| involuntary movements | DOCK4 |
| severe neonatal hypotonia in males | DOCK5 |
| spasticity | DOCK2, DOCK8, DOCK11 |
| torticollis | DOCK5 |
Notably, the plethora of developmental clinical phenotypes attributed to defects in these DOCK genes included micrognathia, intrauterine growth retardation, developmental delay, intellectual disability and a series of craniofacial defects (such as cleft palate or abnormal face shape) were associated with 10 of the 11 DOCK genes. DOCK6 was reported to correlate with Adams-Oliver disease, a rare disorder characterized by defects of the scalp and abnormalities of the upper and lower limbs such as fingers, arms, toes, and legs (Shaheen et al., 2011). Many reported DOCK3 compound heterozygous and homozygous missense variants were associated with neurodevelopmental syndromes in children including autism, attention-deficit hyperactive disorder (ADHD), global developmental delay, and neurodevelopmental disability. Interestingly, pathogenic DOCK11 variants were associated with, more than any other DOCK gene, patients presenting with cognitive behavioral phenotypes including ADHD, bipolar disorder, schizophrenia, and polyphagia.
Pathogenic variants in DOCKs 1, 2, 5, 7, 10, and 11 had commonalities in symptoms of muscle weakness such as muscle hypotonia, ataxia, or defect in coordination. This has been previously reported as associated defects in the central nervous system with patients presenting with abnormal ataxic gait and muscle hypotonia (de Silva et al., 2003; K.L. Helbig et al., 2017; Iwata-Otsubo et al., 2018; Wiltrout et al., 2019). We previously demonstrated that DOCK3 is a dosage-sensitive regulator of pathologies in normal and DMD patient muscle, and thus identified as a novel secondary biomarker for the disease (Reid et al., 2020). Taken together with the findings presented here, this highlights the importance of appropriate regulation of this DOCK family of proteins in biological processes related to muscle, and that further exploration of these variants are needed to understand their impact in disease.
Immune system defects were associated with pathogenic DOCK2 and DOCK8 variants only, where distinct childhood immunological deficiencies were observed in 8.33% and 30.11% of cases. We identified case reports from several pediatric patients with compound heterozygous DOCK2 variants and associated T- and B-cell combined immunodeficiencies lead to severe bacterial and viral infections (Dobbs et al., 2015). Similarly, DOCK8 has important roles in dendritic cell transmigration, T-cell survival, and NK cytotoxicity. Pathogenic compound DOCK8 heterozygous variants have also been identified in patients with combined immunodeficiency disease with elevated IgE, atopy, and recurrent viral infections (Biggs et al., 2017; Dimitrova & Freeman, 2017; Engelhardt et al., 2015). Many DOCK variants appeared to impact normal heart development and morphology. With the exception of DOCK6, DOCKs1–10 reported pathogenic variant patients with phenotypes including abnormal cardiac morphology, ventricular septal defects, and malformation of the heart and blood vessels.
Commonly reported nervous system disorders were seizures, specifically in DOCKs 1, 5, 6, 7, 8, 9, and 11. Among these, only DOCK2 and DOCK11 variants were usually associated with encephalopathy, while DOCK7 variants were associated with conditions such encephalopathy and epilepsy. We also defined a phenotypic category involving any metabolic defects in these groups. Interestingly, only patients with deleterious DOCK6 and DOCK7 variants correlated with pathologies pertaining to metabolic deficiencies. DOCK6 had a reported associated case of familial hypercholesterolemia, leading to significantly elevated low-density lipoprotein (LDL). DOCK7 was reported in a patient with hypobetalipoproteinemia (FHBL), a disorder that impairs the absorption and transport of lipids. Interestingly, introns of both DOCK6 and DOCK7 appear to modulate the expression of Angiopoietin-like (ANGPTLs) genes located on the opposite DNA strand of the introns of the DOCK6 and DOCK7 genes which have important roles in the trafficking and metabolism of lipids (Quagliarini, 2012). This highlights their novel role and the identification of these rare DOCK variants may play in the pathophysiology of known clinical disease, as well as their identification leading to the development of potential therapeutic targets.
Future Prospects
DOCK family members have been shown to modulate or activate downstream effectors such as Rho GTPases like RAC1, RHOA, WAVE/WASF1, and N-WASP. Many of these pathways are involved in the rearrangement of the actin cytoskeleton, metabolism, and cell migration. They are strongly expressed in the brain and spinal cord (Côté & Vuori, 2002; Makihara et al., 2018). Recently, deleterious variants in these genes have been identified as pathogenic in relation to a variety of disorders with pathologies ranging from cognitive, to developmental, to cardiovascular effects. DOCK variants have been identified in pathologies related to cognitive function in terms of behavior such as attention deficit hyperactive disorder (ADHD), autism, or global developmental delay (GDD) (K. L. Helbig et al., 2017; Iwata-Otsubo et al., 2018). Other pathogenic DOCK variants are identified in relation to developmental disorders, including craniofacial defects, and neurological defects such as ataxia and hypotonia. In this systematic review, we sought to analyze, interpret, and classify these DOCK variants by molecular consequence, mutation type, and phenotypic association. In doing so we have generated a comprehensive review of clinical outcomes related to pathogenic DOCK variants. Our findings highlight the more than 3,000 DOCK pathogenic variants identified in patients to date span copy number gains and losses, frameshifts, missense and nonsense variants, and other types of small and large deletions and duplications. Copy number gains predominate, ranging as the cause of disease from as low as 10% of variants to nearly 67% in particular DOCK genes. This number is undoubtedly impacted by biases related to the testing methodology and initial discovery of causation for this type of variant.
The over-representation of larger duplications or deletions can present a challenge in therapeutic solutions if using gene editing technologies such as clustered regularly interspaced short palindromic repeats (CRISPR) or exon-skipping drugs to restore multi-exon deletions (Aartsma-Rus et al., 2009; Aartsma-Rus et al., 2012; Adikusuma et al., 2017; Adkin et al., 2012; Akcakaya et al., 2018). As previously noted, nearly 60% of cases involving pathogenic DOCK variants had a developmental phenotype. This can be a challenge to therapeutic approaches, as our findings indicate that there may be limitations in effectiveness for specific DOCK-associated developmental pathologies, as well as complications in assuring that therapeutic approaches can cross the blood brain barrier to achieve efficacy. Drug targeting involving Rho GTPases, such as RAC, may be a more attractive alternative for variants with large multi-exon deletions, where gene editing technologies would be unusable (Guo et al., 2019).
Our results further noted that patients with deleterious variants in several of these DOCK genes present with cognitive behavioral phenotypes such as ADHD and global developmental delay (K. L. Helbig et al., 2017; Iwata-Otsubo et al., 2018). Indeed, we found that nearly every pathogenic DOCK subclass presented with a cognitive behavioral phenotype. Nearly 5% of pathogenic variants across all genes resulted in a neuromuscular disorder such as ataxia or hypotonia. Patients with DOCK6 and DOCK7 pathogenic variants present with metabolic phenotypes (Table 1 and Figure 5). Both DOCK6 and DOCK7 contain domains involved in activation of the Rho GTPase, RAC1, which is responsible for a variety of metabolic pathways such as insulin signaling and glucose processing. Additionally, both DOCKs have been shown to regulate Angiopoietin-like proteins (ANGPTLs) a gene family that has been identified as important regulators of metabolic disorders, specifically ANGPTL8 in introns 18 and 19 of DOCK6 and Angiopoietin-like 3 (ANGPTL3) within the intron of DOCK7 (Quagliarini, 2012). This highlights the importance of identifying novel gene networks and rare pathogenic variants in order to find novel and effective potential therapeutic targets.
Investigating these broad spectrum of DOCK pathogenic variants and associated pathologies holds potential for development of therapeutic targets to ameliorate disease and symptoms through the intersection of experimental and computational modeling. This analysis can be used to evaluate the plausibility for pathogenic disease and enhance therapeutic potential to treat known diseases. The investigation of rare, pathogenic DOCK variants may allow for evaluation of their impact at structural and functional levels on protein function and stability, which may be able to predict DOCK variant risk to human health or how they may worsen clinical outcomes (Petrosino et al., 2021). Defining commonalities among DOCK variants causing disease in DOCKopathies, will aid in identifying plausible therapeutic approaches such as oligo-mediated gene exon-skipping and CRISPR-mediated gene editing to restore DOCK protein function and yield treatments for this subset of patients. These strategies are being applied successfully to enhance quality of life and outcomes for patients with other rare genetic disorders and their families.
Supplementary Material
Acknowledgements:
The authors wish to thank Sophia Harnew-Spradley, Veronica Markayla Sanders, and Jeffrey Fairley for their assistance with the DOCK variant clinical case systematic review. The authors also wish to thank members of Alexander and Worthey labs for assistance and critiques with the work.
Funding Information:
Research reported in this publication was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, HHS of the National Institutes of Health under award number R01HD095897 awarded to M.S.A. M.S.A. and E.A.W. are also supported by an NIH Office of Research Infrastructure Program (ORIP) U54 grant U54OD030167. A.S. is funded by a NIH NINDS T32 training grant number 5T32NS095775. M.A.L. is funded by the National Institute of Neurological Disorders and Stroke K08NS120812.
Footnotes
Conflicts of Interests: The authors declare no conflicts of interest.
Data availability statement:
All data is full available in the aforementioned databases.
Web resources:
References:
- Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen G-J, & den Dunnen JT (2009). Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human Mutation, 30(3), 293–299. 10.1002/humu.20918 [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus A, Verschuuren J, Campion G, van Ommen G. j., & van Deutekom J (2012). Exon skipping for DMD. Orphanet Journal of Rare Diseases, 7(Suppl 2), A20. http://www.ojrd.com/content/7/S2/A20 [Google Scholar]
- Adikusuma F, Williams N, Grutzner F, Hughes J, & Thomas P (2017). Targeted Deletion of an Entire Chromosome Using CRISPR/Cas9. Molecular Therapy, 25(8), 1736–1738. 10.1016/j.ymthe.2017.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adkin CF, Meloni PL, Fletcher S, Adams AM, Muntoni F, Wong B, & Wilton SD (2012). Multiple exon skipping strategies to by-pass dystrophin mutations. Neuromuscular Disorders, 22(4), 297–305. 10.1016/j.nmd.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akcakaya P, Bobbin ML, Guo JA, Malagon-Lopez J, Clement K, Garcia SP, Fellows MD, Porritt MJ, Firth MA, Carreras A, Baccega T, Seeliger F, Bjursell M, Tsai SQ, Nguyen NT, Nitsch R, Mayr LM, Pinello L, Bohlooly-Y M, Aryee MJ, Maresca M, & Joung JK (2018). In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 10.1038/s41586-018-0500-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson CE, & Southgate L (2021). The DOCK protein family in vascular development and disease. Angiogenesis, 24(3), 417–433. 10.1007/s10456-021-09768-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs CM, Keles S, & Chatila TA (2017). DOCK8 deficiency: Insights into pathophysiology, clinical features and management. Clin Immunol, 181, 75–82. 10.1016/j.clim.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Peto CA, Shelton GD, Mizisin A, Sawchenko PE, & Schubert D (2009). Loss of Modifier of Cell Adhesion Reveals a Pathway Leading to Axonal Degeneration. The Journal of Neuroscience, 29(1), 118–130. 10.1523/jneurosci.3985-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté J-F, & Vuori K (2002). Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. Journal of Cell Science, 115(24), 4901–4913. 10.1242/jcs.00219 [DOI] [PubMed] [Google Scholar]
- de Silva MG, Elliott K, Dahl HH, Fitzpatrick E, Wilcox S, Delatycki M, Williamson R, Efron D, Lynch M, & Forrest S (2003). Disruption of a novel member of a sodium/hydrogen exchanger family and DOCK3 is associated with an attention deficit hyperactivity disorder-like phenotype. Journal of Medical Genetics, 40(10), 733–740. 10.1136/jmg.40.10.733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova D, & Freeman AF (2017). Current Status of Dedicator of Cytokinesis-Associated Immunodeficiency: DOCK8 and DOCK2. Dermatol Clin, 35(1), 11–19. 10.1016/j.det.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs K, Domínguez Conde C, Zhang SY, Parolini S, Audry M, Chou J, Haapaniemi E, Keles S, Bilic I, Okada S, Massaad MJ, Rounioja S, Alwahadneh AM, Serwas NK, Capuder K, Çiftçi E, Felgentreff K, Ohsumi TK, Pedergnana V, Boisson B, Haskoloğlu Ş, Ensari A, Schuster M, Moretta A, Itan Y, Patrizi O, Rozenberg F, Lebon P, Saarela J, Knip M, Petrovski S, Goldstein DB, Parrott RE, Savas B, Schambach A, Tabellini G, Bock C, Chatila TA, Comeau AM, Geha RS, Abel L, Buckley RH, İkincioğulları A, Al-Herz W, Helminen M, Doğu F, Casanova JL, Boztuğ K, & Notarangelo LD (2015). Inherited DOCK2 Deficiency in Patients with Early-Onset Invasive Infections. N Engl J Med, 372(25), 2409–2422. 10.1056/NEJMoa1413462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt KR, Gertz ME, Keles S, Schäffer AA, Sigmund EC, Glocker C, Saghafi S, Pourpak Z, Ceja R, Sassi A, Graham LE, Massaad MJ, Mellouli F, Ben-Mustapha I, Khemiri M, Kilic SS, Etzioni A, Freeman AF, Thiel J, Schulze I, Al-Herz W, Metin A, Sanal Ö, Tezcan I, Yeganeh M, Niehues T, Dueckers G, Weinspach S, Patiroglu T, Unal E, Dasouki M, Yilmaz M, Genel F, Aytekin C, Kutukculer N, Somer A, Kilic M, Reisli I, Camcioglu Y, Gennery AR, Cant AJ, Jones A, Gaspar BH, Arkwright PD, Pietrogrande MC, Baz Z, Al-Tamemi S, Lougaris V, Lefranc G, Megarbane A, Boutros J, Galal N, Bejaoui M, Barbouche MR, Geha RS, Chatila TA, & Grimbacher B (2015). The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol, 136(2), 402–412. 10.1016/j.jaci.2014.12.1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Peng Y, Wang L, Sun X, Wang X, Liang C, Yang X, Li S, Xu J, Ye W-C, Jiang B, & Shi L (2019). Autism-like social deficit generated by Dock4 deficiency is rescued by restoration of Rac1 activity and NMDA receptor function. Molecular Psychiatry. 10.1038/s41380-019-0472-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig KL, Mroske C, Moorthy D, Sajan SA, & Velinov M (2017). Biallelic loss-of-function variants in DOCK3 cause muscle hypotonia, ataxia, and intellectual disability. Clinical Genetics, 92(4), 430–433. 10.1111/cge.12995 [DOI] [PubMed] [Google Scholar]
- Helbig KL, Mroske C, Moorthy D, Sajan SA, & Velinov M (2017). Biallelic loss-of-function variants in DOCK3 cause muscle hypotonia, ataxia, and intellectual disability. Clinical Genetics, n/a–n/a. 10.1111/cge.12995 [DOI] [PubMed] [Google Scholar]
- Iwata-Otsubo A, Ritter AL, Weckselbatt B, Ryan NR, Burgess D, Conlin LK, & Izumi K (2018). DOCK3-related neurodevelopmental syndrome: Biallelic intragenic deletion of DOCK3 in a boy with developmental delay and hypotonia. American Journal of Medical Genetics Part A, 176(1), 241–245. 10.1002/ajmg.a.38517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, & Maglott DR (2015). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Research, 44(D1), D862–D868. 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin M, & Côté J-F (2014). Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes & Development, 28(6), 533–547. 10.1101/gad.236349.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin M, Fradet N, Blangy A, Hall A, Vuori K, & Côté J-F (2008). The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proceedings of the National Academy of Sciences, 105(40), 15446–15451. 10.1073/pnas.0805546105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makihara S, Morin S, Ferent J, Côté J-F, Yam PT, & Charron F (2018). Polarized Dock Activity Drives Shh-Mediated Axon Guidance. Developmental Cell, 46(4), 410–425.e417. 10.1016/j.devcel.2018.07.007 [DOI] [PubMed] [Google Scholar]
- Namekata K, Enokido Y, Iwasawa K, & Kimura H (2004). MOCA Induces Membrane Spreading by Activating Rac1. Journal of Biological Chemistry, 279(14), 14331–14337. 10.1074/jbc.M311275200 [DOI] [PubMed] [Google Scholar]
- Petrosino M, Novak L, Pasquo A, Chiaraluce R, Turina P, Capriotti E, & Consalvi V (2021). Analysis and Interpretation of the Impact of Missense Variants in Cancer. Int J Mol Sci, 22(11). 10.3390/ijms22115416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AL, Wang Y, Samani A, Hightower RM, Lopez MA, Gilbert SR, Ianov L, Crossman DK, Dell’Italia LJ, Millay DP, van Groen T, Halade GV, & Alexander MS (2020). “DOCK3 is a dosage-sensitive regulator of skeletal muscle and Duchenne muscular dystrophy-associated pathologies”. bioRxiv, 2020.2003.2027.010223. 10.1101/2020.03.27.010223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, & on behalf of the, A. L. Q. A. C. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Faqeih E, Sunker A, Morsy H, Al-Sheddi T, Shamseldin Hanan E., Adly N, Hashem M, & Alkuraya Fowzan S. (2011). Recessive Mutations in DOCK6, Encoding the Guanidine Nucleotide Exchange Factor DOCK6, Lead to Abnormal Actin Cytoskeleton Organization and Adams-Oliver Syndrome. The American Journal of Human Genetics, 89(2), 328–333. 10.1016/j.ajhg.2011.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachi N, Hashimoto Y, & Matsuoka M (2012). MOCA is an integrator of the neuronal death signals that are activated by familial Alzheimer’s disease-related mutants of amyloid β precursor protein and presenilins. Biochemical Journal, 442(2), 413–422. 10.1042/bj20100993 [DOI] [PubMed] [Google Scholar]
- Wiltrout K, Ferrer A, van de Laar I, Namekata K, Harada T, Klee EW, Zimmerman MT, Cousin MA, Kempainen JL, Babovic-Vuksanovic D, van Slegtenhorst MA, Aarts-Tesselaar CD, Schnur RE, Andrews M, & Shinawi M (2019). Variants in DOCK3 cause developmental delay and hypotonia. European Journal of Human Genetics, 27(8), 1225–1234. 10.1038/s41431-019-0397-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li R, Sun S, Weile J, & Roth FP (2021). Improved pathogenicity prediction for rare human missense variants. Am J Hum Genet, 108(10), 1891–1906. 10.1016/j.ajhg.2021.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is full available in the aforementioned databases.