

Abstract
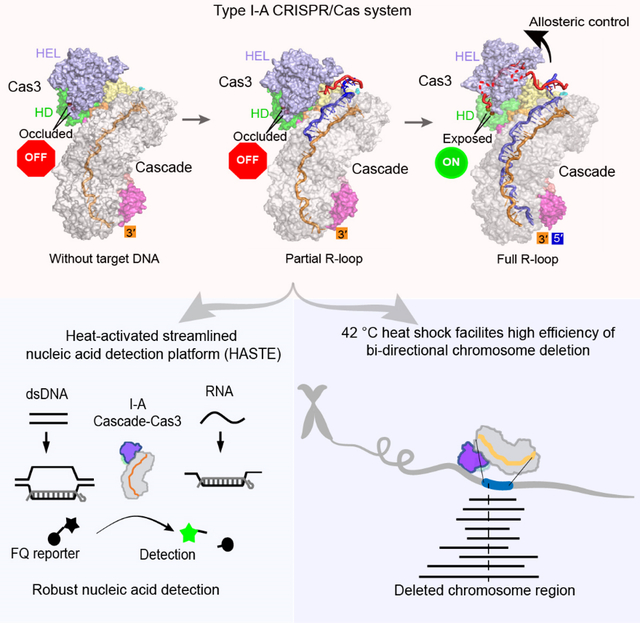
Type I CRISPR-Cas systems typically rely on a two-step process to degrade DNA. An RNA-guided complex named Cascade first identifies the complementary DNA target. The nuclease-helicase fusion enzyme Cas3 is then recruited in trans for processive DNA degradation. Contrary to this model, here we show that Type I-A Cascade and Cas3 functions as an integral effector complex. We provide four cryo-EM snapshots of the Pyrococcus furiosus Type I-A effector complex in different stages of DNA recognition and degradation. The HD nuclease of Cas3 is autoinhibited inside the effector complex. It is only allosterically activated upon full R-loop formation, when the entire targeted region has been validated by the RNA guide. The mechanistic insights inspired us to convert Pfu Cascade-Cas3 into a high-sensitivity, low-background, and temperature-activated nucleic acid detection tool. Moreover, Pfu CRISPR-Cas3 shows robust bi-directional deletion-editing activity in human cells, which could find usage in allele-specific inactivation of disease-causing mutations.
Graphical Abstract
In brief
Hu et al. show that Type I-A CRISPR-Cas3 uses an allosterically controlled mechanism to selectively cleave DNA target, which is very different from six other Type I systems. They further developed a highly sensitive nucleic acid detection platform and an efficient deletion-editing method from I-A CRISPR-Cas3.
Introduction
CRISPR-Cas systems help establish a line of RNA-based adaptive immunity against foreign genetic elements for prokaryotes (Barrangou et al., 2007; Bolotin et al., 2005; Makarova et al., 2006; Marraffini and Sontheimer, 2008; Mojica et al., 2005; Pourcel et al., 2005). The CRISPR array is transcribed and processed to RNA guides (crRNAs), which assemble with one or more of the CRISPR-associated (Cas) proteins to degrade the complementary nucleic acid targets. DNA-targeting CRISPR-Cas systems define a target by promoting R-loop formation at a dsDNA target flanked by a protospacer adjacent motif (PAM). The associated nucleases then cleave the exposed single-stranded DNAs (ssDNAs) to generate double-strand breaks (DSBs).
CRISPR-Cas systems can be classified into Class 1 and Class 2, based on whether a multi- or single-subunit Cas protein effector complex, respectively, is utilized during CRISPR interference (Makarova et al., 2020). Each Class further contains at least three types and multiple subtypes therein. Class 1 Type I is the most prevalent CRISPR system. It can be further divided into six subtypes (I-A through I-G) (Makarova et al., 2020). Among them, the most extensively studied and mechanistically understood system is the Type I-E system, based on work with Escherichia coli (Brouns et al., 2008; Hochstrasser et al., 2014; Jackson et al., 2014; Mulepati and Bailey, 2013; Mulepati et al., 2014; Rutkauskas et al., 2015; Sashital et al., 2012; Sinkunas et al., 2013; Westra et al., 2012; Wiedenheft et al., 2011; Zhao et al., 2014) and Thermobifida fusca Type I-E systems (Dillard et al., 2018; Hayes et al., 2016; Huo et al., 2014; Xiao et al., 2018; Xiao et al., 2017). A consistent mechanistic model emerges, in which the Type I-E systems shred the DNA target into pieces through a two-step process. First, a multi-subunit ribonucleoprotein (RNP) called Cascade (CRISPR associated complex for antiviral defense) utilizes the associated crRNA to recognize a complementary DNA target flanked by PAM (Brouns et al., 2008; Westra et al., 2012; Wiedenheft et al., 2011). The resulting R-loop triggers a large conformational change in Cascade (Hochstrasser et al., 2014; Huo et al., 2014; Wiedenheft et al., 2011; Xiao et al., 2017). The helicase-nuclease fusion enzyme Cas3 is then licensed to bind the R-loop-forming Cascade (Xiao et al., 2018; Xiao et al., 2017), nicks the non-target strand (NTS) DNA, and processively degrades the PAM-proximal side of the dsDNA (Dillard et al., 2018; Hochstrasser et al., 2014; Kunne et al., 2016; Mulepati and Bailey, 2013; Redding et al., 2015; Sinkunas et al., 2013). This stepwise activation mechanism is quite unique among CRISPR systems, and was rationalized as a mechanism to minimize off-targeting. At the genome editing front, the Thermobifida fusca (Tfu) (Dolan et al., 2019), Escherichia coli (Eco) (Morisaka et al., 2019) and Pseudomonas aeruginosa (Pae) (Cameron et al., 2019) Type I-E Cascade-Cas3 complexes have been shown to introduce long chromosomal deletions of up to 30–100 kilobases (kb) in human cells. Similar activity was later reported for Type I-D and I-C systems (Osakabe et al., 2020; Osakabe et al., 2021; Tan et al., 2022). Additional applications in eukaryotes include use of Cascade-FokI fusion systems to generate small indel mutations (Cameron et al., 2019) and Cascade-mediated transcriptional modulation in human (Chen et al., 2020; Pickar-Oliver et al., 2019) and plant cells (Young et al., 2019). Type I systems also find broad usage in microbial genome manipulation (Caliando and Voigt, 2015; Csorgo et al., 2020; Li et al., 2016; Luo et al., 2015; Rath et al., 2015).
Parallel work in Type I-C and I-F systems revealed a similar two-step interference mechanism, where Cas3 is trans-recruited after Cascade mediated R-loop formation (Govindarajan et al., 2020; Guo et al., 2017; Hochstrasser et al., 2016; O’Brien et al., 2020; Rollins et al., 2019). However, it remains unclear whether this mechanistic model applies to the rest of the subtypes. In fact, pulldown and in vitro reconstitution experiments suggest Cas3 may constitutively associate with Cascade in the Type I-A CRISPR-Cas system (Majumdar and Terns, 2019; Majumdar et al., 2015; Plagens et al., 2014). Focusing on the Pyrococcus furiosus (Pfu) Type I-A system (Elmore et al., 2015; Garrett et al., 2020; Majumdar and Terns, 2019), here we provide high-resolution evidence that Type I-A Cascade and Cas3 assemble into an integral effector complex; R-loop formation is inefficient in the absence of Cas3. Instead of regulating the nuclease activity of Cas3 through trans-recruitment, Type I-A system does so through allosteric activation. Cas3 is autoinhibited upon assembly into the I-A effector complex. Its nuclease activity is only activated upon full R-loop formation, by a substrate-induced conformational relay from Cascade to the HD nuclease center of Cas3. Upon activation, Cas3 further displays robust collateral single-stranded (ss)DNA cleavage activity. The findings revealed possible parallels in nuclease control mechanisms between divergent Type I and III CRISPR systems, where RNA-guided RNA binding and cleavage appears to allosterically activate the associated Cas10 HD nuclease for a follow-up DNA cleavage (Elmore et al., 2016; Foster et al., 2020; Jia et al., 2019; Wang et al., 2019; You et al., 2019). We further report a mechanism-inspired application, in which the I-A Cascade-Cas3 is converted into a robust and sensitive dsDNA detection tool. DsDNA and ssDNA target could be directly detected within 15 minutes at picomolar range without PCR amplification using fluorescence or strip paper formats; ssRNA were detected at 100-fold lower sensitivity. Moreover, we reveal that I-A CRISPR-Cas3 displays highly efficient bi-directional deletion-editing activity in human cells, which could find usage in allele-specific inactivation of disease-causing mutations and other important applications.
Results
Pfu Cascade-Cas3 forms an integral Type I-A effector complex
The Pfu Type I-A effector was previous reconstituted by mixing purified protein components with the mature crRNA (Majumdar and Terns, 2019). To obtain sufficient materials for structural studies, we resorted to a recombinant expression approach from E. coli (Figure S1). Initial attempts produced Cascade-like complexes that contained various-sized RNAs, possibly from unspecific incorporation of host RNAs. We speculated that this was due to the inefficient processing of pre-crRNA, since Cas6 from this extremophilic archaea has been shown to cleave the pre-crRNA poorly at ambient temperatures (Carte et al., 2008). Alternative pre-crRNA cleavage methods were tested. This included the use of self-cleaving ribozymes and the use of E. coli Cas6 for in vivo pre-crRNA processing (Figure S1A–E). The latter strategy made use of a chimeric CRISPR repeat containing the Pfu 5¢-handle and the E. coli 3’-handle that was efficiently cleaved by Eco Cas6 (data not shown). This approach was quite successful, producing high-quality Pfu Cascade with homogeneous chimeric crRNA (Figure S1B–E). Cas3 in I-A systems is encoded by two separate open-reading frames, Cas3¢¢ encodes the HD nuclease and Cas3¢ encodes the superfamily II helicase subunit (Figure 1A). They are renamed in this study to Cas3 HD and Cas3 HEL, respectively. We show that these two subunits assemble into a stable Cas3 complex upon mixing, suggesting that they function as a unit (Figure S1F–H) (Majumdar and Terns, 2019).
Figure 1. Pfu Cascade-Cas3 form an integral effector complex.
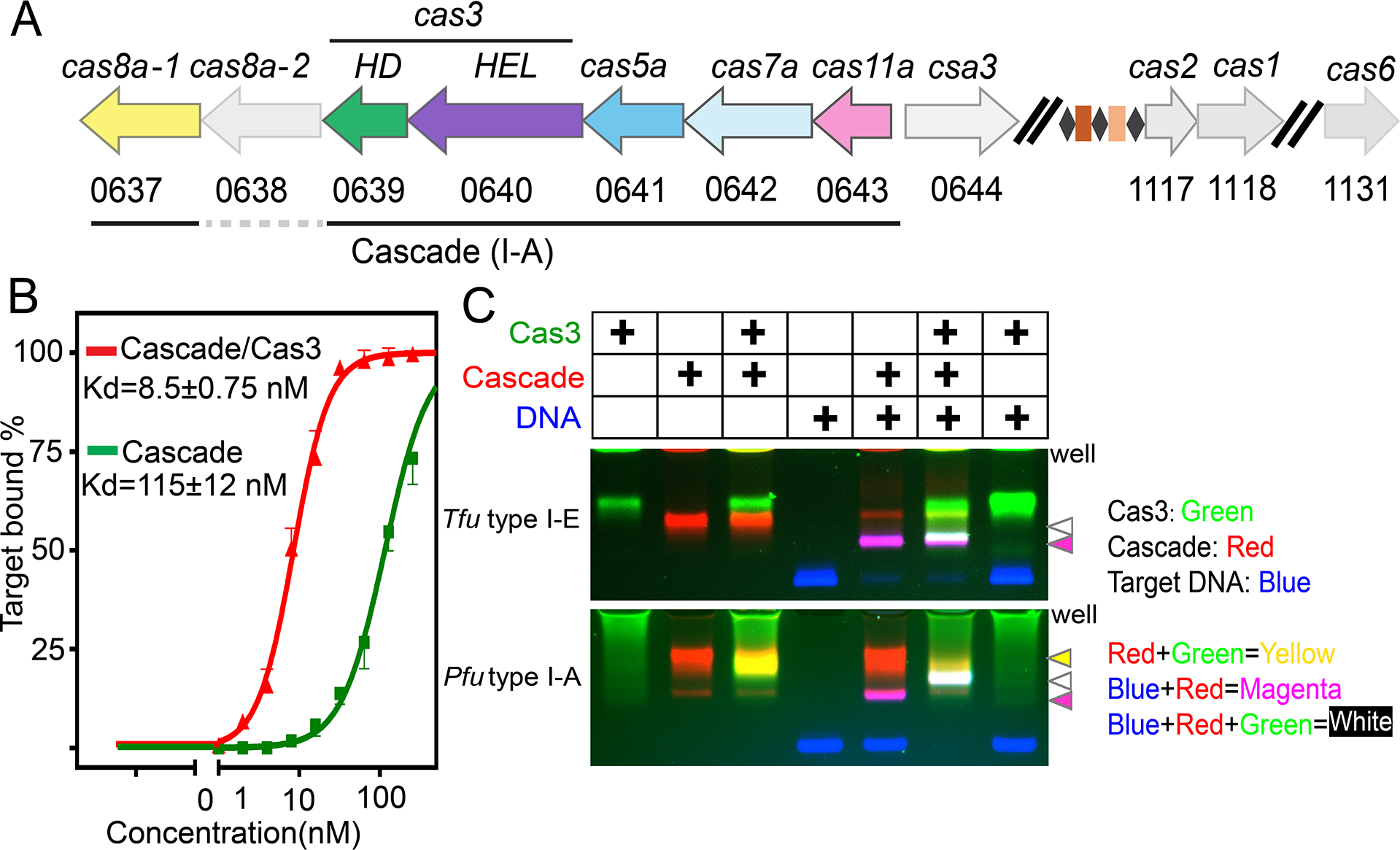
(A) Arrangement of the Pyrococcus furiosus Type I-A CRISPR-cas operon. (B) Agarose EMSA showing that Pfu Cas3 significantly improved the DNA target binding behavior of Pfu Cascade. (C) Agarose EMSA using differentially labeled binding partners to reveal that I-A Pfu Cas3 bound constitutively to Pfu Cascade. In contrast, I-E Tfu Cas3 only interacted with Tfu Cascade upon full R-loop formation. Cas3, Cascade, and dsDNA were labeled with Cy3 (rendered in green), Cy5 (red), and FAM (blue), respectively. Overlapping colors indicate complex formation that survived electrophoresis.
A surprising observation was that Pfu Cascade alone interacted with the target DNA poorly, with an apparent Kd of ~115 nM from the electrophoretic mobility shift assays (EMSAs) (Figure 1B; S1J). Further inclusion of Pfu Cas3 improved its target-searching behavior. The Kd of Pfu Cascade-Cas3 for the cognate DNA target was 8.5 nM, comparable to that of Cascades in other subtypes (Figure 1B). This observation differs from findings with the I-C, I-E and I-F systems, where Cascade was shown to bind the DNA target efficiently and independently, and Cas3 is recruited in a second step for DNA degradation (Govindarajan et al., 2020; Hochstrasser et al., 2014; Hochstrasser et al., 2016; O’Brien et al., 2020; Rollins et al., 2019; Xiao et al., 2018; Xiao et al., 2017). Further investigation revealed that the interaction between Pfu Cas3 and Pfu Cascade was constitutive rather than DNA substrate-dependent (Figure 1C), and that Pfu Cas3 recognized and stably bound to its binding partner, the Pfu Cas8a subunit, alone or in the context of the intact Pfu Cascade (Figure S1F–H). For Tfu I-E counterparts, the equivalent Cas3-Cas8 interaction was only stable in the context of the Cascade complex and after DNA binding and R-loop formation (Figures 1C; S1I) (Hochstrasser et al., 2014; Xiao et al., 2017). Collectively, our biochemical evidence suggests that Pfu Cascade and Pfu Cas3 functions as a unit by forming an integral I-A interference complex.
A thorough round of characterization was carried out for Pfu Cascade-Cas3. RNA-guided DNA cleavage was shown to be robust and PAM-dependent in vivo and in vitro (Figures S1J–N). Furthermore, Pfu Cascade-Cas3 was less salt sensitive, more thermostable, and maintained DNA-binding function better at higher temperatures than Pfu Cascade alone, again consistent with Cascade and Cas3 functioning as an integral unit (Figure S1O–P).
Overview of the four functional states in PfuCascade-Cas3 captured by cryo-EM
A series of four cryo-EM structures of Pfu Cascade and Cas3 were determined, each depicting a distinct functional state (Figures 2; S2, S3, Supplemental movie S1,). The structures of Pfu Cascade with and without Cas3 bound were determined at 3.56 and 3.37 Å resolutions, respectively (Figures 2A–B; S2A–D). The structural comparison explains why Pfu Cas3 is indispensable for efficient target searching by Pfu Cascade (explained in detail later). The overall shape and composition of Pfu Cascade agrees better with the I-C Cascade than with the I-E and I-F counterparts. Both contain seven rather than six copies of Cas7 subunit in the backbone due to the longer spacer length in the crRNA in I-A and I-C subtypes (Figures 2A–B; S4A–B). Significant differences exist, though. For example, the Cas11 subunit found in I-B/I-C/I-D subtypes is encoded as a hidden ORF overlapping the region of the cas8 gene encoding the Cas8 C-terminal domain (CTD) (McBride et al., 2020; O’Brien et al., 2020). In contrast, the Type I-A cas operon encodes a separate cas11 ORF, which is highly homologous to cas8a CTD in sequence and structure (30% sequence similarity, 4.35 Å in Cα r.m.s.d) (Figure S4C–E). An Identical set of residues are found in Cas11 and Cas8a CTD to mediate the intermolecular interactions (Figure S4D–F), therefore Cas8a CTD is a Cas11-like domain. Interestingly, the Pfu Type I-A cas operon encodes two versions of Cas8a (Figure 1A). Only Cas8a-1 assembled into Cascade; Cas8a-2 led to poor reconstitution (data not shown). In vivo knock out of Cas8a variants also indicated that only Cas8a-1 deletion caused interference deficiency in P. furiosus (Figure S4G). These two evidences suggest that Cas8a-1 is the functional component for the interference in the variants. This can be rationalized based on the sequence alignment, as Cas8a-2 lacks the key Cas11-interacting residues found in Cas8a-1 (Figure S4H). The exact function of Cas8a-2 remains unknown.
Figure 2. Overview of four cryo-EM snapshots of Pfu Cascade-Cas3 in different functional states.
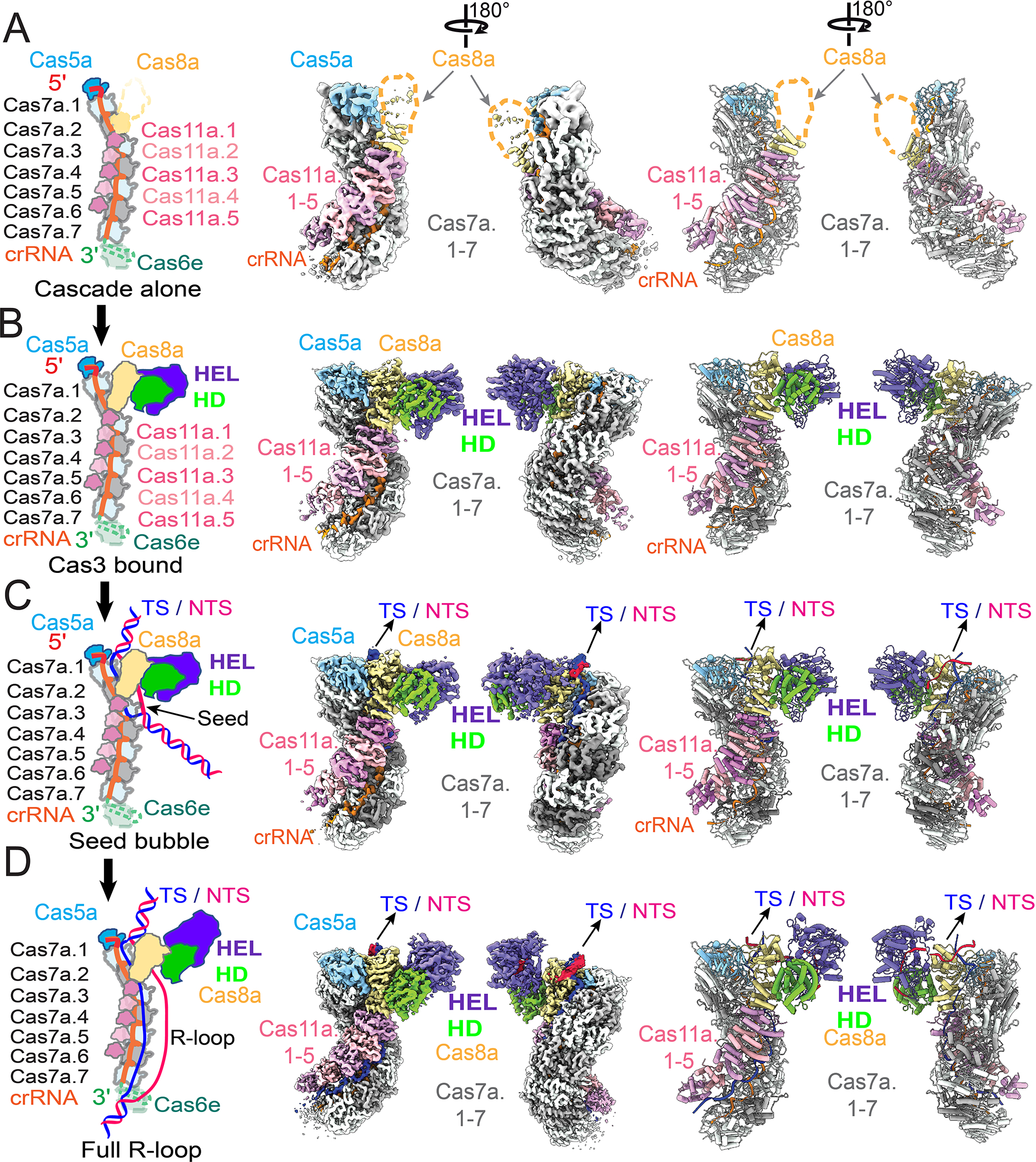
Schematics of the depicted functional state, cryo-EM density, and cartoon representation of the molecular structure of (A) apo Pfu Cascade, (B) Pfu Cascade-Cas3, (C) Pfu Cascade-Cas3 opening a partial R-loop (unwinding 17 bp of PAM-proximal dsDNA target), and (D) Pfu Cascade-Cas3 opening a full R-loop (unwinding 37 bp of PAM-proximal dsDNA target). Refer the coloring scheme from the schematics.
We further programmed Pfu Cascade-Cas3 with a protospacer-containing dsDNA and reconstructed two structures from the cryo-EM sample. A 3.88 Å R-loop intermediate was captured, in which the PAM-proximal 17 bp dsDNA is unwound, the 17 nt target-strand (TS) base-pairs with the complementary crRNA spacer, the first 4 nt non-target strand (NTS) can be traced, whereas the rest of NTS and PAM-distal dsDNA are not resolved in the cryo-EM density (Figures 2C; S2E–F). This R-loop intermediate is one-segment (6 bp) longer that the counterpart captured from the Tfu I-E Cascade/DNA sample (Xiao et al., 2017). A 3.29 Å structure of Pfu Cascade-Cas3 opening a full R-loop structure was also captured. The entire 37 nt TS DNA base-pairs with the crRNA spacer. The NTS has been nicked by Cas3 and the PAM-proximal portion re-threaded through Cas3 HEL, ready for processive degradation at Cas3 HD (Figures 2D; S2G, S3). A total of 13 nt can be traced for the PAM-proximal NTS strand; an additional 2–4 nt are inferred based on distance to connect the three segments of ssDNA (Figure 2D). This is consistent with the previous observation that Pfu Cas3 nicks the NTS strand 15–17 nt below PAM (Majumdar and Terns, 2019). The PAM-distal NTS and dsDNA remain unresolved in the map. This is the first snapshot of a Type I effector complex poised for processive degradation of NTS DNA. The two functional states share common features, such as the PAM recognition mechanism. Significant differences exist. For example, structural comparison revealed a concerted set of conformational changes taking place in Pfu Cascade-Cas3 during the partial to full R-loop transition. This observation reveals the target-dependent nuclease activation mechanism (explained in detail later).
Cas3 primes Cascade for DNA target-searching
An important structural observation is that the N-terminal domain (NTD) of Cas8a in the apo Pfu Cascade is highly mobile and largely absent from the EM density. As shown in 3D classifications, less than 10% of the apo Pfu Cascade particles contain significant Cas8a NTD densities. Even in this class, the densities are not of sufficient quality to allow unambiguous docking of Cas8a NTD (Figure 3A). The Cas8 NTD is responsible for PAM-recognition in other studied Cascades (Hayes et al., 2016). It further works with neighboring subunits to unwind the dsDNA underneath PAM, initiating R-loop formation (Xiao et al., 2017). Failing to adopt a defined conformation is expected to impede the function of Cas8a NTD to couple PAM recognition with DNA unwinding. This rationalizes the poor target-searching behavior in apo Pfu Cascade (Supplemental movie S2). Cas8a NTD is further connected to the Cas11-like Cas8a CTD and Cas11.1–5, together they form the so-called “inner belly” of the Cascade (Xiao et al., 2017). In apo Pfu Cascade, the entire inner belly has elevated conformational dynamics than the backbone subunits (Cas5a-Cas7.1–7), judged from the significantly reduced local map resolution (Figure 3B).
Figure 3. Pfu Cas3 rigidifies the PAM-recognition subunit of Pfu Cascade, enabling DNA target-binding.
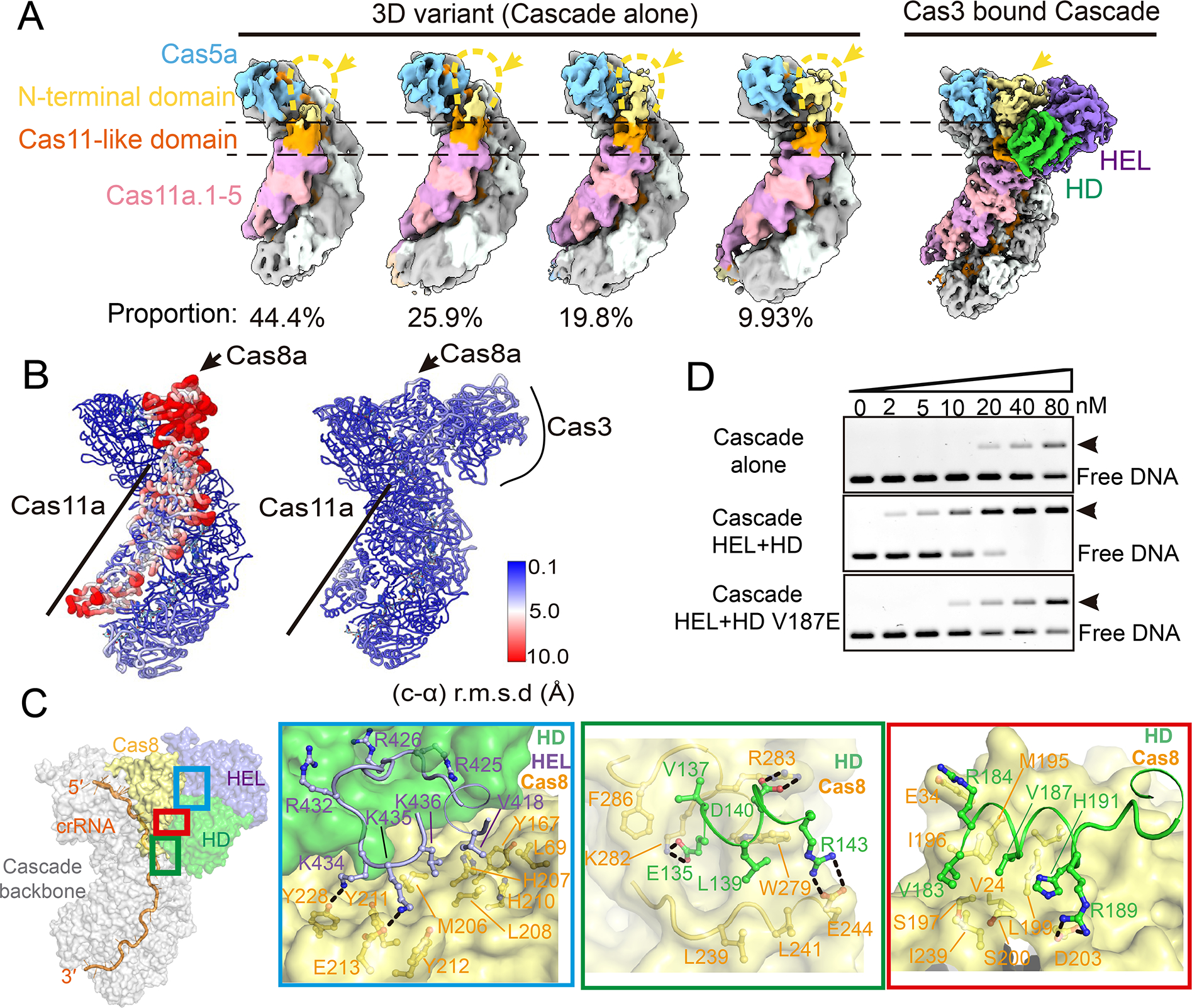
(A) Further classification revealed four 3D variants from the apo Pfu Cascade cryo-EM reconstruction, each represents the specified proportion of the total particles. They vary in the Cas8a NTD density. Only ~10% particles contain choppy densities large enough to cover entire Cas8a. In contrast, Cas8a NTD density is well defined in the Pfu Cascade-Cas3 reconstruction. (B) Local resolution estimate based on the per-residue r.m.s.d. value. Cas8a NTD and the rest of the inner belly subunits in the apo Pfu Cascade have reduced resolution and elevated motion based on this analysis. In contrast, the equivalent subunits in the Pfu Cascade-Cas3 structure are resolved at the same resolution as the rest of the structure. (C) Detailed molecular contacts between Pfu Cas3 and Pfu Cascade. An orientation view is provided to the left. The boxed regions are analyzed in zoom-in panels to the right. (D) Native-agarose EMSA showing that when the molecular contact is disrupted, by the V187E mutation to Cas3 HD, Cas3 can no longer improve the target DNA binding behavior of Cascade as the wild-type Cas3 does.
Binding of Pfu Cas3 effectively eliminates the severe domain movements in Cas8a NTD, and rigidifies the rest of the inner belly subunits in place (Figure 3A–B). The lockdown effect is achieved through an extensive Cas3-Cas8a interface that spans across the NTD and CTD of Cas8a, which prevents the hinge motion in between the two domains (Supplemental movie S2). Structural analysis revealed extensive hydrophobic and polar contacts, and the interface residues are highly conserved among Type I-A homologs (Figures 3C; S5A). To validate structural observations, we mutagenized three conserved interface residues on Cas3 individually and in combination, two from the HD nuclease (V187E, L139A) and one from the helicase subunit (V418A). In Strep-tag pull-down experiments, each mutation reduced the affinity between Cas3 and Cascade, albeit to various extents. In particular, Cas3 HD V187E and HD L139A/HEL V418A completely disrupted Cas3 interaction with Cascade (Figure S5B–C). Consistent with our conclusion that Cas3 binding enables target-searching by I-A Cascade, when Cas3 V187E instead of WT Cas3 was introduced to the EMSA assay, the DNA binding behavior of Pfu Cascade did not improve (Figure 3D). Overall, our structural and biochemical data suggests that the I-A Cascade is only functional in the context of the Cascade-Cas3 effector complex.
PAM recognition
PAM recognition promotes RNA-guided DNA unwinding in DNA-targeting CRISPR systems. in Type I CRISPR systems, the process involves PAM recognition-induced DNA bending and the insertion of a Gln-wedge into DNA duplex to initiate unwinding (Xiao et al., 2017). Here we show that the same mechanistic principles hold true inside Type I-A Cascade-Cas3, despite the lack of structural similarity between Cas8a NTD and its counterpart in I-E and I-F Cascades. Pfu Cascade-Cas3 specifies a 5’-Y−3C−2N−1 PAM, which denotes a pyrimidine at PAM-3, a cytosine at PAM-2, and any nucleotide at PAM-1; a few alternative PAMs were also found to promote interference (Elmore et al., 2015). In both the intermediate and full R-loop structures, PAM is recognized by Cas8a NTD mainly from the DNA minor groove side and towards the target-strand DNA (3’-G−1G−2G−3). Consistent with the PAM code, no sequence specific interactions are found at PAM-1. The G−2-C−2 pair at PAM-2 is strongly specified from the minor groove side by a bidentate hydrogen bond from N97 to the sugar edge of G−2 and a weak H-bond from N98 to C−2. Alternative base-pairs do not satisfy the observed H-bonding pattern, which is consistent with the strict PAM-2 code. G-3 is specified from the major groove side by a polar contact to N7 from K137 (Figure 4A). Both adenosine and guanosine suffice for this contact, which again is consistent with the PAM-3 code. Electrostatic contacts to the DNA sugar phosphate backbone are also found from Cas5a (K105, R95) and Cas8a (Y138, N149, and N98) (Figure 4B). Mutations disrupting the observed sequence-specific contacts (K137A, Y139A, K137A/Y139A, N97A, N98A, N97A/N98A) reduced the target binding affinity by at least 10-fold (Figures 4C–D; S6A–C, Supplemental movie S3). Importantly, the PAM recognition residues are highly conserved among many Cas8a and Cas5a homologs (Figure S6D–E), suggesting that a significant portion of the Type I-A CRISPR systems specify the same PAM code.
Figure 4. PAM recognition mechanism.
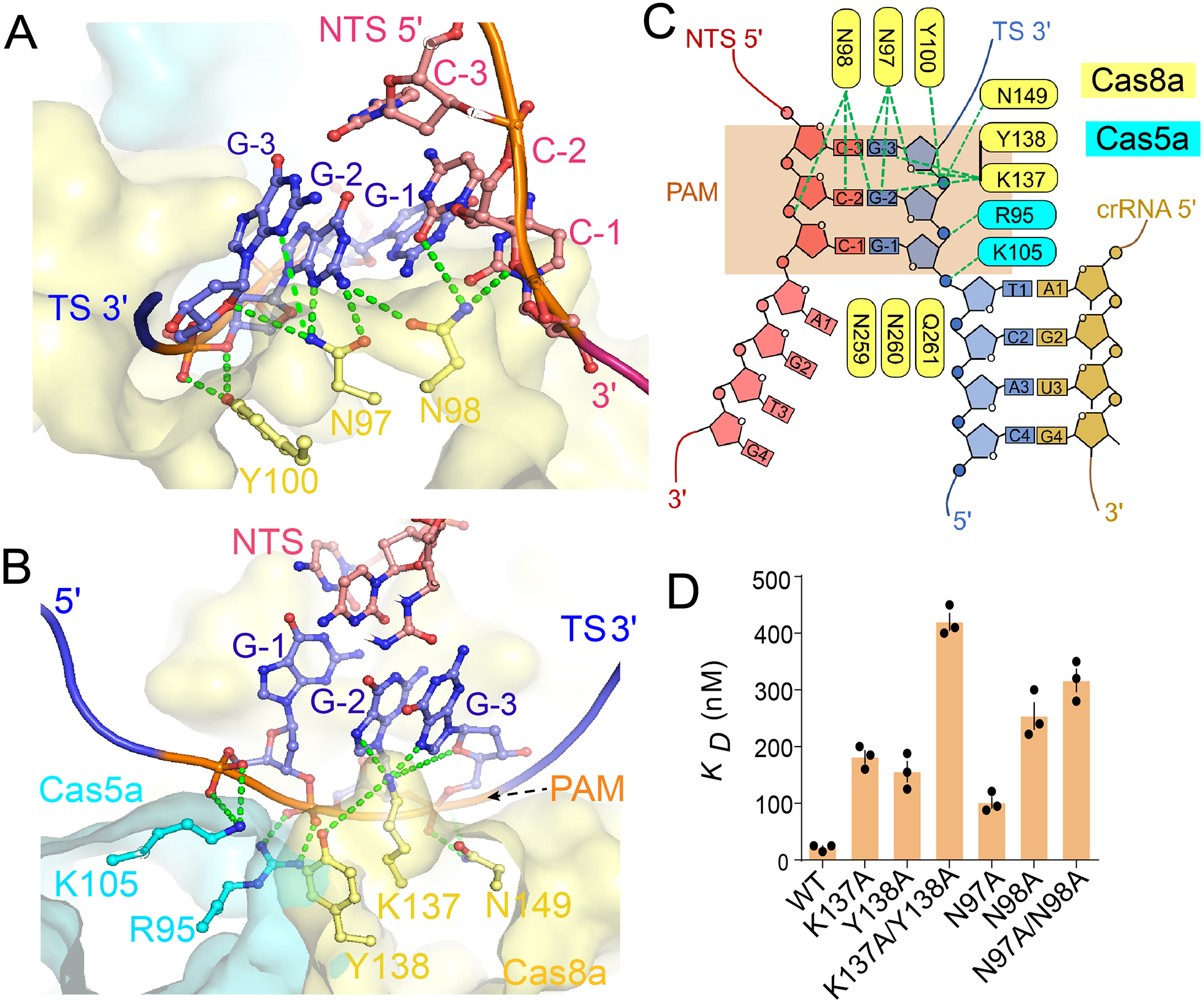
(A, B) Two zoom-in views of the PAM-recognition mechanism by Pfu Cascade-Cas3. Coloring scheme is consistent with Figure 3C. (C) Diagram of the PAM recognition contacts. (D) The impact of disrupting the observed PAM contacts on target binding affinity, evaluated using native-agarose EMSA.
Nuclease activity of Cas3 is highly regulated
Work in I-E, I-C, and I-F systems suggest that Cas3 is recruited by Cascade in trans and only after R-loop formation. This conditional recruitment mechanism is crucial in preventing off-targeting (Szczelkun et al., 2014; Xiao et al., 2018; Xiao et al., 2017). Given that Cas3 binds constitutively to Cascade in the I-A system, an alternative mechanism must be in place to prevent off-targeting. Indeed, our biochemistry suggest that the nuclease activity of Cas3 is tightly regulated, in a Cascade- and DNA target-dependent manner. Pfu Cas3 was highly active in degrading the fluorescently labeled ssDNA reporter, either by itself or in complex with Cas8a (Figure S7A). This nuclease activity against the ssDNA reporter became undetectable when Cas3 was present within the Pfu Cascade-Cas3 complex, only to be reactivated by the presence of a cognate dsDNA target (Figure S7A–C). As expected, in such cases, the target DNA was efficiently nicked at the non-target strand, and further degraded in the presence of ATP (Figure S7D–E). In contrast, noncognate DNA failed to activate Pfu Cascade-Cas3 against the ssDNA reporter (Figure S7D). Collectively, the data suggest that the Pfu Cascade silences the nuclease activity of Cas3 upon complex formation, and only reinvigorates it upon encountering the cognate DNA substrate.
Capturing snapshots of Pfu Cascade-Cas3 before, in the middle, and after R-loop formation allowed us to deduce the nuclease regulatory mechanism in high resolution. The two early snapshots revealed that, prior to full R-loop formation, the HD nuclease center of Cas3 is occluded by the nearby structural features (Figure 5A). Specifically, two loops (L1: R143-R148; and L2: L456-K474) from Cas3 HEL and one loop from the C-terminal region of Cas3 HD (Lc: L211-K237) juxtapose to form a wall structure. These loops not only block the direct entry of ssDNA into the HD active site, but also prevent the threading of ssDNA through the helicase into the HD nuclease (Figures 5B). Therefore, even though a pair of Ni2+ ions are optimally coordinated for ssDNA cleavage (Huo et al., 2014), NTS nicking does not take place due to steric hindrance. This autoinhibition is relieved in the full R-loop state - L1 restructures from a β-hairpin to a short α-helix; L2 retracts due to a rigid body movement in its residing structure; and Lc becomes disordered. Both the direct-access route and the through-helicase route become unobstructed as the result (Figures 5B).
Figure 5. Structural basis for the allosteric activation of Pfu Cas3 nuclease upon full R loop formation by Pfu Cascade-Cas3.
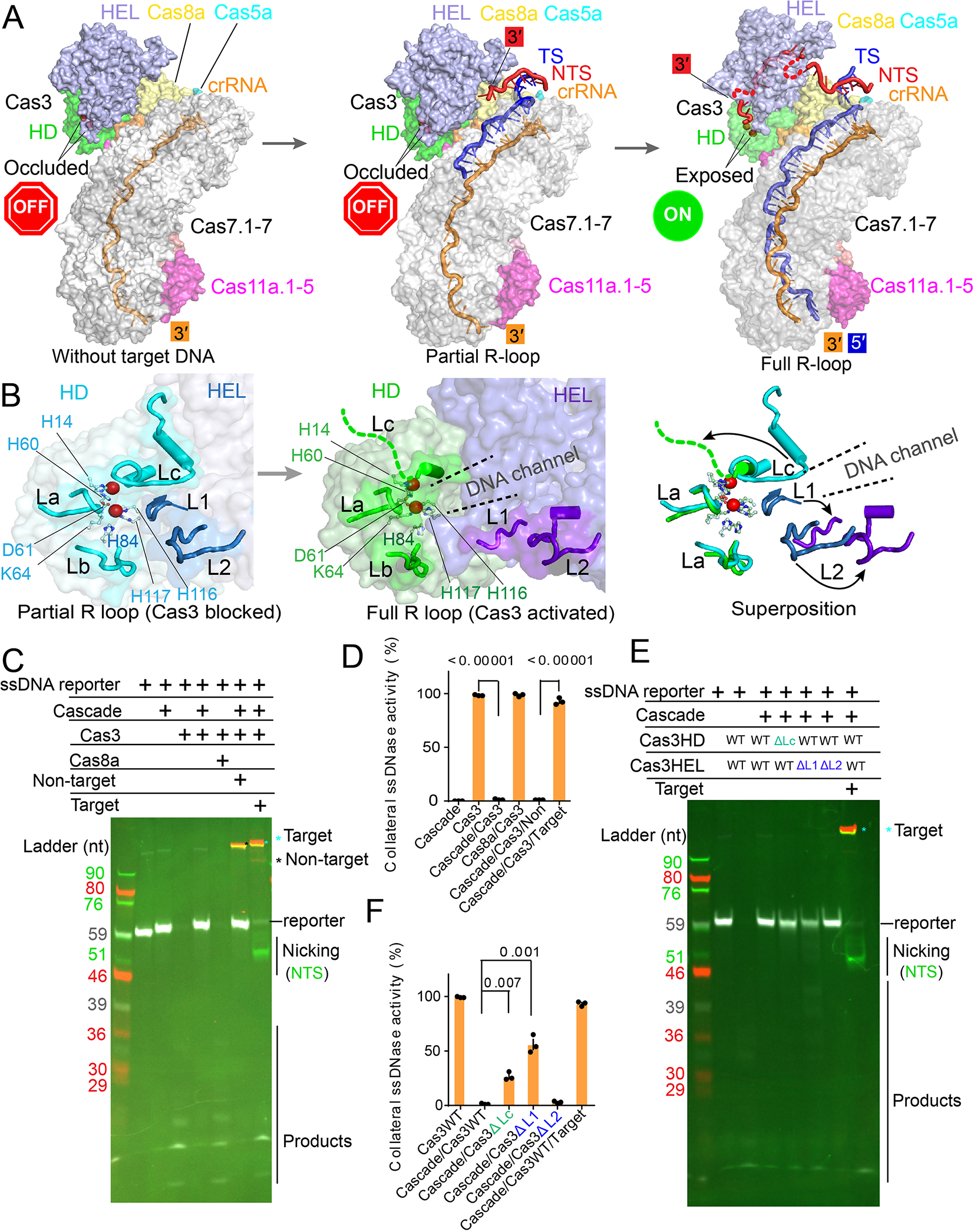
(A) Side-by-side comparison of the Pfu Cascade-Cas3 structure before, during and after R-loop formation, which reveals the timing and the nature of the conformational change during the R-loop formation process. Cas3 HD-nuclease center is only exposed upon full R-loop formation (note the accessibility of the two catalytic metal ions in dark red balls). NTS DNA has been nicked and threaded through the Cas3 helicase, ready for processive degradation by the Cas3 HD-nuclease. (B) Zoom-in of the Cas3 HD-nuclease center depicting the unlatching movements of loops L1, L2, and Lc upon R-loop formation. Together the loops form an R-loop dependent conformational switch. (C) Denaturing-PAGE revealing the R-loop dependent activation of the nuclease activity in Cas3. Upon activation, Pfu Cascade-Cas3 not only cleaves the cognate DNA target, but also the fluorescent ssDNA reporter nearby. Cy5-labeled target strand is colored in red, Cy3-labeled non-target strand in green, FAM-labeled ssDNA reporter in white color. (D) Nuclease activity changes as assayed in (C), quantified from the intensity of the full-length fluorescent ss-DNA reporter band in each assay condition. (E) Denaturing-PAGE showing the loss of autoinhibition of the Cas3 nuclease activity inside Pfu Cascade-Cas3 when the conformational switch was disrupted (ΔL1, ΔL2). Coloring scheme is the same to (C).(F) Quantification of the nuclease activity as assayed in (E).
The three conformational switches were individually deleted to evaluate their functional importance. The Cas3 nuclease activity was read out from the collateral cleavage of the fluorescent ssDNA reporter. The wild type Pfu Cascade-Cas3 showed undetectable nuclease activity in the absence of the cognate DNA target (or in the presence of a non-target dsDNA), but strong nuclease activity in the presence of a cognate DNA target (Figure 5C–D). Similar to wild-type Cas3, L2 deletion (DL2) showed little collateral ssDNA cleavage activity in the absence of the cognate substrate, suggesting the autoinhibition mechanism is still intact. In contrast, DL1 and DLc mutants appeared to have lost the autoinhibition control. They displayed strong nuclease activity even in the absence of the DNA target (Figure 5E–F). This constitutive-ON behavior would be quite detrimental because a partially matching DNA target could also be cleaved, leading to an unacceptable off-targeting scenario.
Allosteric regulation of Cas3 by Pfu Cascade, in high-resolution
Structural comparisons further revealed that the conformational state of the Cas3 nuclease center is allosterically regulated by the Pfu Cascade in an R-loop dependent manner. Formation of a full R-loop, but not a partial R-loop intermediate, triggers a relay of conformational change in the inner belly of Pfu Cascade (Figure S7F–H; Supplemental movie S4, S5). The five Cas11a subunits and the Cas11-like Cas8a CTD twist in a concerted fashion, creating interdomain movements between Cas8a NTD and CTD (Figure S7F–G). Structural changes are further relayed to Cas3 in the form of a global rigid-body motion and a local conformational change (Figure S7H). The former orients the Cas3 helicase towards the substrate, and the latter unblocks the HD nuclease center (Figure 5B). As the result, the non-target strand DNA is efficiently nicked, re-threaded through the helicase subunit, and further into the HD nuclease subunit (Figure 5A, right panel).
To establish a stringent correlation between Cas3 activity and the HD active site conformation, we further resolved a 6.6 Å cryo-EM structure of the Pfu Cas3-Cas8a complex. Despite the moderate resolution, the EM densities clearly suggest the HD nuclease center is open and accessible, similar to that in the full R-loop state of Pfu Cascade-Cas3 (Figure S7I–J). This structural observation is consistent with Pfu Cas3-Cas8a possessing high nuclease activity. It further supports the notion that the elaborate allosteric switch is set only upon complex formation with the intact Pfu Cascade.
Mechanism-inspired development of a nucleic acid detection tool
We have shown that when a cognate DNA target is present, the nuclease activity of Pfu Cascade-Cas3 is not restricted to the DNA target; a ssDNA bystander is efficiently degraded as well (Figures 5C–D, S7B–D). Since the observed cleavage activity is analogous to the collateral damage activity reported for Cas12a (Chen et al., 2018; Gootenberg et al., 2018), we further explored the possibility of converting this assay into a nucleic acid detection format. A ssDNA containing a 5’- fluorophore and a 3’-quencher was used as a reporter (Figure 6A). Cleavage of this reporter would relieve the quenching effect and produce fluorescent signals, which was indeed the case when Pfu Cascade-Cas3 was incubated with the target DNA, but not a non-target DNA (Figure 6B). As expected, Pfu Cas3 alone produced high fluorescence regardless of dsDNA presence due to the lack of autoinhibition. Further optimization revealed that a poly-dA reporter produced bigger fluorescence changes than poly-dT, poly-dC, and poly-dG reporters (Figures 6C, S8A). Virtually no background fluorescence increase was observed when a non-target DNA was used over the course of one-hour incubation, which means this platform would be less prone to reporting false positives (Figure 6D). Consistent with the fact that Pfu is a hyperthermophile, we found that the DNA cleavage reaction was heat-activatable. The signal amplification was rather small at 25 and 37 °C, but became very robust throughout the 45–85°C temperature range (Figure S8B). This is also a desirable feature because it potentially allows cell lysis, pathogen inactivation, and nucleic acid detection to be combined into a single high-temperature incubation step.
Figure 6. Mechanism-inspired development of HASTE nucleic acid detection platform.
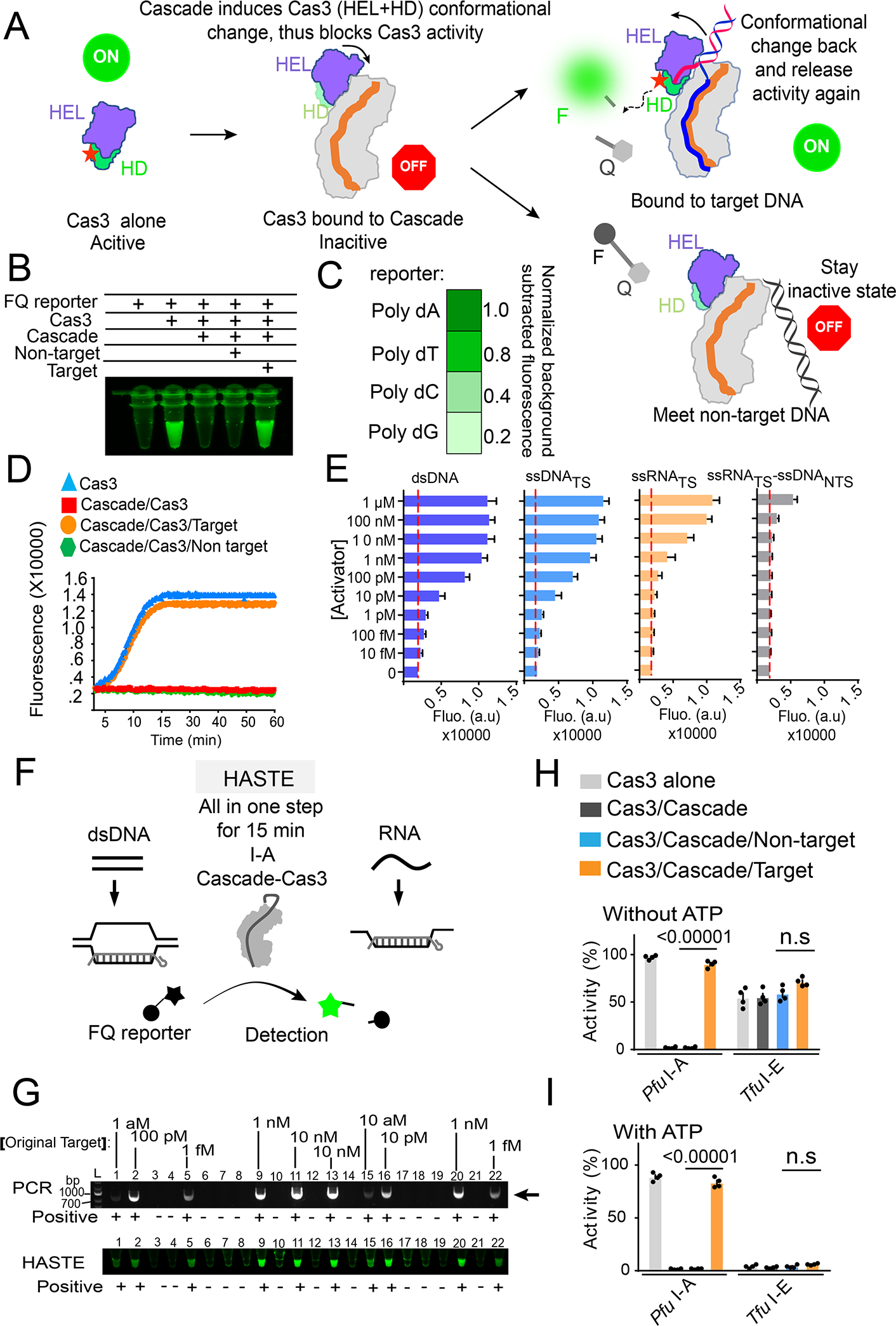
(A) Diagram of the nucleic acid detection platform, based on the nuclease activity changes inside PfuCascade-Cas3 in response to cognate and non-cognate DNA targets. F, fluorophore; Q, quencher. (B) Reagent combinations that lead to clean background and robust positive signal in test tubes. (C) Normalized fluorescence changes when four different poly-deoxynucleotide ssDNA-FQ reporters were used. (D) Real time fluorescent changes as the result of ssDNA-FQ cleavage by Pfu Cascade-Cas3 as it encounters a cognate DNA target, a non-cognate target, or none. Cas3 alone is not autoinhibited. Its strong collateral cleavage activity serves as a control. (E) Quantifications of the detection limitation of Pfu Cascade-Cas3 on dsDNA, ssDNA, ssRNA, and RNA/DNA heteroduplex. Dotted red lines indict background fluorescence. (F) Schematic for HASTE as a one-step and heat-activatable nucleic acid detection tool. (G) Excellent consistency between PCR amplification and PCR-coupled HASTE in detecting the presence of target DNA across 22 solution samples. As low as 1 attomolar target DNA was reliably detected by PCR-HASTE and no false positive was reported. (H, I) Side-by-side comparison of the collateral ssDNA cleavage activity of Pfu I-A and Tfu I-E Cascade-Cas3 in the presence or absence of a cognate DNA target, and with (I) or without (H) ATP present, respectively.
Next, we investigated the substrate scope and detection limit of the Type I-A nucleic acid detection platform. In substrate titration experiments and with the fluorescence unquenching format, Pfu Cascade-Cas3 could reliably detect the presence of 1 pM dsDNA substrate in solution without the need of PCR amplification. Interestingly, Pfu Cascade-Cas3 displayed comparable detection sensitivity towards a ssDNA target, and ~100-fold reduced sensitivity towards a ssRNA target (0.1–1 nM) (Figures 6E, S8C). These observations suggest that base-pairing to the entire spacer region of crRNA is sufficient to trigger the activation of Pfu Cas3; PAM recognition is not a prerequisite. In contrast, an RNA_TS/DNA_NTS heteroduplex target was only detected at μM range, which may reflect the combined consequence of poor PAM recognition and heteroduplex unwinding. We name our Type I-A nucleic acid detection platform HASTE (heat-activated streamlined nucleic acid detection platform) to highlight its advantage in streamlining sample inactivation and nucleic acid detection into a 15-minute high-temperature incubation step (Figure 6F). We envision that combining HASTE with isothermal amplification methods such as Loop-mediated isothermal amplification (LAMP) (Notomi et al., 2000) may further improve its detection limit. Alternatively, a 25-cycle PCR amplification step can be introduced prior to HASTE. As low as 1 aM dsDNA (single molecule level) can be robustly detected. No false positive or negative detection was observed among the twenty-two tested samples, over a wide substrate concentration range (Figure 6G).
In parallel to the fluorescent-based assay, we also developed a lateral flow strip assay, where cleavage of the ssDNA reporter will allow its FAM-labeled 5’-portion to move beyond the streptavidin-coated control line, and to carry the anti-FAM antibody coated gold nanoparticle to the secondary antibodies coated test line (Figure S8D). By systematically titrating the target DNA, we determined that the sensitivity of this detection method is in the ~10–100 fM range without amplification (Figure S8E), which is comparable with Cas12 in similar setups (Gootenberg et al., 2018). Collectively, the evidence indicates that type I-A Pfu Cascade-Cas3 has the potential to be engineered into a robust and sensitive nucleic acid detection tool.
We further investigated whether a similar nucleic acid detection platform can be developed from Thermobifida fusca Type I-E Cascade-Cas3, which has been extensively characterized in our hands (Dillard et al., 2018; Dolan et al., 2019; Huo et al., 2014; Xiao et al., 2018; Xiao et al., 2017). Results showed that Tfu Cascade-Cas3 in the same testing format suffered many problems. In the absence of ATP, Tfu Cascade-Cas3 showed high background noise in the absence of targets (unstable baseline) and low sensitivity (small dynamic range) in the presence of targets. In the presence of ATP, the collateral damage activity from Tfu Cascade-Cas3 was greatly reduced. However, very little fluorescent signal was observed in the presence of the cognate DNA target (Figures 6H–I, S8F–G). The background problem would lead to rampant false positives, and the sensitivity issue reduces the detection limit. These results suggest developing a nucleic detection platform from Type I systems similar to Tfu Cascade-Cas3 would not be straightforward. It would require significant optimizations.
Converting Pfu Cascade-Cas3 to a highly efficient and heat-inducible, gene deletion-editing tool
We further explored the potential usage of I-A CRISPR-Cas for genome editing in human cells. Nuclear localization signal (NLS) tagged Pfu Cascade-Cas3 was delivered as ribonucleoprotein complexes (RNPs) into cells through electroporation. This approach allowed us to establish a direct correlation between in vitro RNA-guided nuclease activity and in vivo editing efficacy. The experiments were carried out in a HAP1 cell line that has spontaneously reverted from haploid to the diploid forms, and with a single copy of eGFP gene inserted into the AAVS1 safe harbor in Chromosome 19. Knowing that the RNA-guided nuclease activity of Pfu Cascade-Cas3 increases significantly from 37 to 42°C (Figure S9A–B), we explored ex vivo editing at both temperatures (Figure S9C–F), and quantified the editing efficiency using Fluorescence-Activated Cell Sorting (FACS) (Figure 7A). At 37°C, ex vivo editing against the template strand of eGFP by guide 1 (G1) led to the loss of GFP signal in 36.4% of the cell population. The same experiment at 42°C led to almost quantitative GFP signal loss (92.4% editing efficiency quantified by FACS) (Figure 7B–C). To evaluate whether RNA transcription influences editing efficiency, the same experiment was repeated using Pfu Cascade-Cas3 programmed against the non-template strand of eGFP by guide 2 (G2). A similar temperature-dependent editing behavior was observed, and the editing efficiency at 42°C was comparable (90%). Combining Guide 1 and Guide 2 RNPs slightly improved the editing efficiency (Figure S9F). Shortening the 42°C incubation from 24 to 8 hours after Pfu Cascade-Cas3 delivery gave comparable editing efficiency (data not shown). This is encouraging as not all cells can withstand extended incubation at 42°C. We further expanded the GFP-editing experiments to a GFP-tagged HEK293 cell line and observed similar editing efficiency (Figures S10A–C; Supplemental movies S6, S7). This confirms that I-A Pfu Cascade-Cas3 is a highly efficient genome editing tool in human cells. Notably, delivering excess amount of Pfu Cas3 did not further improve the editing efficiency of Pfu Cascade-Cas3, which is consistent with the notion that Cascade and Cas3 function as an integral effector complex in Type I-A CRISPR systems. (Figure S10D). The high editing efficiency and temperature-tuned editing activity are two attractive traits for this deletion-editing tool.
Figure 7. I-A PfuCascade-Cas3 causes bi-directional deletion in RNA-guided fashion in human cells.
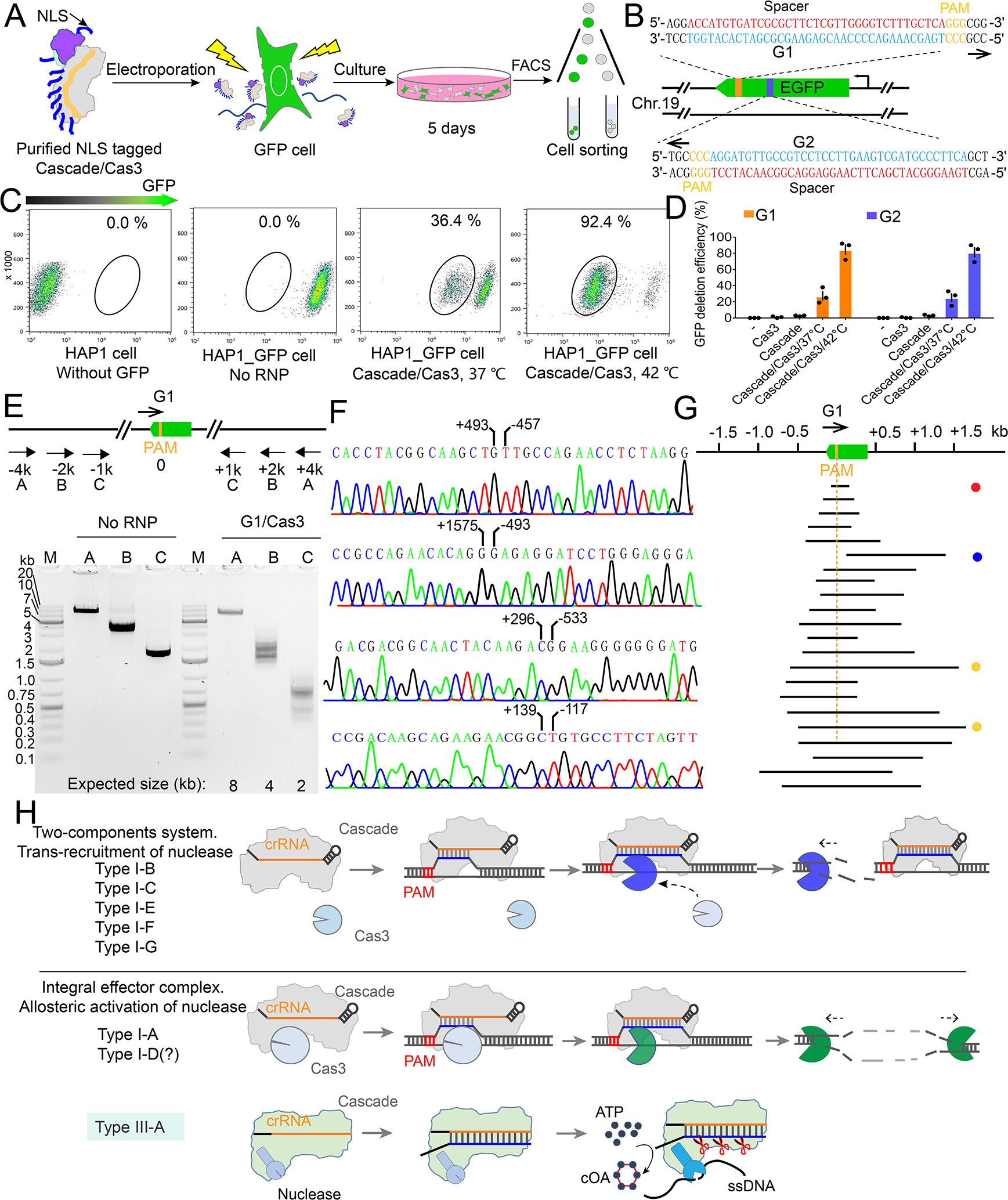
(A) Experimental procedure for Pfu Cascade-Cas3 mediated genome editing in human cells. (B) Design of the crRNA guides targeting the template (G1) and non-template (G2) strands of the GFP ORF. (C) Quantification of the editing efficiency evaluated by FACS, based on the loss of the GFP signal. An overnight incubation at 42°C immediately after RNP delivery increased the editing efficiency from 36.4% to 92.4%. (D) Quantification of G1 and G2 editing efficiency following different experimental procedures. (E) Bracketing PCR based detection and estimation of genome deletion around the targeting site. (F) Representative Sanger sequencing results revealing the deletion boundary, presumably formed by NHEJ-mediated DNA repair. (G) Distribution of the deletion range and size from the Sanger-sequencing results. Note that the vast majority are bi-directional deletions. Target site is eliminated as the result. (H) Mechanism-based classification of Type I CRISPR-Cas systems. I-A and possibly I-D systems use a distinct allosteric activation mechanism to degrade the substrate. These systems may be ancestral to the rest of the Type I systems because they share a stronger mechanistic similarity with Type III systems.
Next, we examined the editing profile of Pfu Cascade-Cas3 using PCR amplification, cloning and Sanger sequencing (Figure 7E–F). Several Type I CRISPR systems have been developed for deletion-editing in human cells (Cameron et al., 2019; Dolan et al., 2019; Morisaka et al., 2019; Osakabe et al., 2020; Osakabe et al., 2021). The majority of them are unidirectional editors, causing long-range deletions upstream of the PAM-side of the target DNA. Interestingly, our results showed that the Type I-A Pfu Cascade-Cas3 behaves as a bi-directional deletion editor, which is similar to the newly reported Type I-D editor in mammalian cells (Osakabe et al., 2020; Osakabe et al., 2021). Of the 22 clone sequences, all but one deleted a continuous region encompassing both the upstream and downstream of the target site (Figure 7E–H). Upon observing this deletion profile, we further investigated the deletion profile of Pfu Cascade-Cas3 using in vitro assays. In plasmid cleavage assays, restriction mapping revealed that both the upstream and downstream regions of the targeted plasmid were erased (Figure S10E–F). In dual-labeled linear DNA degradation assays, cleavage sites was mapped to both sides of the R-loop (Figure S10G–H), which is consistent with published observations (Majumdar and Terns, 2019). Therefore, the observed deletion-editing profile is a direct reflection of the biochemical activity of Pfu Cascade-Cas3.
Discussion
Only three out of the seven subtypes of Type I CRISPR-Cas systems (I-C, I-E, and I-F) have been well studied. The prevailing mechanistic model from these studies suggest that the effector complex Cascade uses a trans-recruitment mechanism to engage the Cas3 nuclease for target degradation. A highlight of this study is the unveiling of an alternative interference mechanism employed by the I-A CRISPR system, where Cas3 is constitutively associated with Cascade, and its activity is allosterically regulated by Cascade, in an RNA-guided and DNA substrate-dependent fashion. By capturing four functional states of the I-A Cascade-Cas3 effector complex using cryo-EM, we established strong causal relationships between structural changes and functional consequences. Binding to Cascade sets on a conformational switch in Cas3 to lock its HD nuclease center. This autoinhibition is relieved only upon full R-loop formation through a substrate-binding induced conformational relay from Cascade to Cas3. We propose that Type I systems can be classified into the allosteric-activation camp and the trans-recruitment camp (Figure 7H). This allosteric-activation mechanism likely also exists in the I-D system, where Cas3 HD is fused to the Cas8d subunit of Cascade (Makarova et al., 2020). However, the available structure-function analysis suggests the I-D cascade binds to target DNA efficiently in the absence of Cas3 HEL (McBride et al., 2020). Therefore additional studies are needed to accurately classify the mechanism in the I-D subtype. I-B and I-G have not been extensively studied. Based on the preliminary data in hand, we tentatively include them in the trans-recruitment camp. There are hints that the nuclease activity of Cas3 in the trans-recruitment camp is at least partially inhibited when it is not associated with Cascade (Gong et al., 2014; He et al., 2021; Huo et al., 2014; Yoshimi et al., 2021).
We further developed the HASTE nucleic acid detection method based on the I-A interference mechanism. This platform has many attractive features, including heat-activatable, streamlined, sensitive yet accurate, and broad-spectrum (can detect dsDNA, ssDNA, and ssRNA). HASTE may find wide usage in bacterial and viral pathogen detection where diagnostic time and biosafety is on high demand. Because this detection platform relies on the strong autoinhibition of Cas3 HD within Cascade-Cas3 complexes to maintain a low false-positive background, we predict that a robust Type I CRISPR based detection tool can only be developed from systems in the allosteric-activation camp.
Interestingly, both I-A and I-D Cascade-Cas3 produce bi-directional genome editing profile (Osakabe et al., 2020; Osakabe et al., 2021). This is a somewhat unexpected observation as the helicase activity of Cas3 is unidirectional (Dillard et al., 2018; Redding et al., 2015). Bidirectional firing of Cas3 from the Cascade-bound DNA target was reported in special circumstances, for example, in the reconstituted E coli I-E system when the acquisition complex Cas1-Cas2 is present (Redding et al., 2015). The molecular mechanism for bidirectional Cas3 firing is likely sophisticated and subtype dependent (Lin et al., 2020). Similar to the I-E story (Dolan et al., 2019), the genome editing effort using I-A Cascade-Cas3 actually helped to reveal blind spots in our mechanistic understanding, which will be investigated in the future.
Limitations of the study
The HASTE method has not been tested using real clinical samples. We anticipate further tuning to achieve optimal results in the real diagnostic setting. The deletion-editing efficiency by I-A CRISPR-Cas3 is critically dependent on temperature. A 42 °C heat shock step is required to obtain optimal results. This limitation will be overcome in follow-up studies using either structure-guided design, directed evolution or homolog screening.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Ailong Ke (ailong.ke@cornell.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The models and maps for the structures presented have been deposited in the PDB and EMDB. The accession numbers are listed in the key resources table. Uncropped original gel and microscopy images have been deposited on Mendeley Data: https://doi: 10.17632/ry9tf59h3k.1. These are all publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli Bl21(DE3) competent cells | NEB | Cat# C2527I |
| E. coli DH5a competent cells | NEB | Cat# 18265017 |
| Chemicals, peptides, and recombinant proteins | ||
| Modified and unmodified oligos | IDT | N/A |
| IPTG | GoldBio | Cat # I2481C500 |
| LB broth | Teknova | #L9145 |
| Restriction enzymes | NEB | Choose from catalog |
| iProof high fidelity PCR kit | Biorad | #1725331 |
| Acrylamide | thermal | HC2040 |
| Urea | VWR | 57-13-6 |
| Fluorescence stain Cy3 and Cy5 | Lumiprobe | 11320 and 13320 |
| SYPRO® Orange | Thermo Fisher | S6650 |
| Formamide | VWR | 75-12-7 |
| MnCl2 | Fisher Scientific | 7773-01-5 |
| Strep resin | IBA | 2-1201-025 |
| Nickel resin | QIAGEN | Cat. No. / ID: 30250 |
| lateral flow strip | HybriDetect 1 | MGDS |
| Copper Grids 1.2/1.3 200 mesh | QUANTIFOIL | Q250-CR1.3 |
| Type I-A Cascade | This study | N/A |
| Cas3 HEL | This study | N/A |
| Cas3 HD | This study | N/A |
| T.fusca type I-E Cascade | Xiao et al., 2017 | N/A |
| T.fusca type I-E Cas3 | Xiao et al., 2018 | N/A |
| Deposited data | ||
| Cascade alone | This study | PDB: 7TR6 and EMBD-26081 |
| Cascade-Cas3 | This study | PDB: 7TR8 and EMBD-26082 |
| Partial R-loop | This study | PDB: 7TR9 and EMBD-26083 |
| Full R-loop | This study | PDB: 7TRA and EMBD-26084 |
| Cas3-Cas8a alone | This study | EMBD-26097 |
| Experimental models: Cell lines | ||
| HAP1_GFP | Dolan et al. 2019 | N/A |
| HEK293_GFP | This study | N/A |
| Recombinant DNA | ||
| Complete list of plasmids | This study | Table S2 |
| Oligonucleotides | ||
| Complete list of DNA oligonucleotide sequences | This study | Table S3 |
| Miniature CRISPR array spacers | This study | Table S3 |
| Software and algorithms | ||
| EPU | Thermo Fisher Scientific | N/A |
| RELION 3 | Scheres Lab | https://www3.mrc-lmb.cam.ac.uk/relion/index.php?title%20=%20Main_PAGE |
| cryoSPARC | Ali Punjani et al., 2017 | https://cryosparc.com/ |
| Coot (v. 0.9.5.1-pre) | (Emsley and Cowtan, 2004; Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHENIX | Adams et al. (2010) | https://phenix-online.org/ |
| ChimeraX (v. 1.2) | (Goddard et al., 2018) | https://www.cgl.ucsf.edu/chimerax/ |
| ClustalW | (Goujon et al., 2010; Sievers et al., 2011) | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Other | ||
| MonoQ 5/50 GL | Cytiva | Cat# 17516601 |
| HiLoad 16/600 Superdex 200 pg | Cytiva | Cat# 28989335 |
| Quantifoil R 1.2/1.3, 200 mesh, copper | Quantifoil | Q250-CR1.3 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture
HAP1 cell culture
This GFP-tagged diploid HAP1 cell line was a gift from Yan Zhang’s lab (Tan et al., 2022). Cells were cultured in DMDM (Gibco) supplemented with 10% FBS (Gibco) at 37 °C and 5% CO2 in a humidified incubator. Cells were suspended using Trypsin-EDTA solution (GIBCO) and split every 2 to 3 days.
HEK293 cell culture
HEK293 cells were cultured in DMDM (Gibco) supplemented with 10% FBS (Gibco) at 37 °C and 5% CO2 in a humidified incubator. Cells were suspended using Trypsin-EDTA solution (GIBCO) and split every 2 to 3 days.
Bacterial strain
Escherichia coli BL21 (DE3)
E. coli BL21 (DE3) cells were used for protein production. Cells were grown in Lysogeny Broth (LB) supplemented with appropriate antibiotics.
Escherichia coli BL21 AI
E. coli BL21 AI cells were used for assaying for CRISPR interference by the Pfu I-A system. Cells were grown in Lysogeny Broth (LB) supplemented with appropriate antibiotics.
Escherichia coli DH5 alpha
E. coli DH5α was used for cloning. Cells were grown at 37 C in LB supplemented with appropriate antibiotics.
Plasmids and cloning
Plasmids, primers, and RNA guides used in this work are listed in Supplementary Tables 1, 2, and 3, respectively. Cloning was performed in E. coli DH5α. The Type I-A cas operon from Pyrococcus furiosus DSM 3638 was PCR-amplified using the iproof Polymerase (BioRad) and cloned into the pCDFDuet vector, giving rise to Plasmid pCascade/Cas3. For plasmid pCRISPR (or pcrRNA), a series of synthetic constructs composed of T7 promotor, CRISPR array (repeat-spacer-repeat with Cas6 or ribozyme derived 5’ handle-spacer) were introduced into the high copy number vector pRSFDuet. For purification, cas8a was cloned into pCDFduet with a N-terminal Twin-strep tag. cas11-cas7-cas5 operon was inserted into pETDuet with a C-terminal His tag on Cas5a. Cas3 HD and Cas3 HEL were expressed individually from the pRSFDuet vector with a N-terminal His tag. All plasmids were verified by Sanger sequencing. Bacterial transformations were carried out using chemically competent cells, and transformants were selected on LB agar plate supplemented with the appropriate antibiotics.
METHOD DETAILS
Protein expression and purification
pCDFDuet-Twin-Strep-Cas8a (StrepR), pETDuet- Cas11-Cas7-Cas5 (AmpR)and pcrRNA-Cas6 (KanR) were co-transformed into E. coli BL21 (DE3) cell under the appropriate antibiotic selection. A 4 liter cell culture was grown in LB medium at 37 °C until an optical density of 0.6 at 600 nm. The culture temperature was then reduced to 20°C and incubated for an additional 1 hour. Expression was induced with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 20°C overnight. Cells were harvested by centrifugation and resuspended in 100 mL buffer A containing 50 mM HEPES pH 7.5, and 300 mM NaCl, 10% glycerol, and 2 mM β-ME. Cells were lysed by sonication, and the debris was cleared using centrifugation at 12,000 rpm for 50 min at 4 °C. The supernatant was applied onto a pre-equilibrated 4 mL streptavidin column (Twin-strep purification). After washing with 50 ml of buffer A, the protein was eluted with 20 ml buffer B (50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 2.5 mM desthiobiotin). The sample was then concentrated to 1 ml and loaded onto a Superdex 200 16/60 size-exclusion column (GE Healthcare) equilibrated with buffer C (10 mM HEPES pH 7.5, 300 mM NaCl), the peak fractions of Cascade complex were pooled and snap-frozen in liquid nitrogen for later usage.
For Cas3 HD and Cas3 HEL purification, pRSFDuet-Cas3HD or Cas3HEL (KanR) was individually transformed into E. coli BL21 (DE3), expressed using the same procedure as described above. Cells were harvested by centrifugation and lysed by sonication in 80 ml of buffer A containing 50 mM HEPES pH 7.5, 500 mM NaCl and 20 mM imidazole, 10% glycerol, and 2 mM β-ME. The lysate was centrifuged at 12,000 rpm for 50 min at 4 °C, and the supernatant was applied to a pre-equilibrated 4 mL Ni-NTA column. After washing with 100 ml of buffer A, the protein was eluted with 20 ml buffer B (50 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol, 300 mM imidazole, and 2 mM β-ME), concentrated to 1.5 mL and further purified on Superdex 200 16/60 equilibrated with buffer C (10 mM HEPES pH 7.5, 300 mM NaCl), the peak fractions were pooled and snap-frozen in liquid nitrogen for later usage.
RNA extraction and ureal gel running
20 μL of Cascade sample at 2 μM and 20 μL phenol-chloroform solution was mixed together and vortexed vigorously for 2 min at room temperature. The aqueous and organic phases were separated by 13,000 rpm centrifuge for 15 min at room temperature. 10 μL sample was taken from the aqueous phase (top layer), mixed with 15 μL of formamide loading dye, heat-denatured at 95 °C for 10 min, and immediately loaded to a 12% ureal-polyacrylamide (PAGE) gel. After 50 minutes of electrophoresis at 25 watts, the gel was stained with EtBr to for 10 min, destained in water for 10 minutes, and scanned with the ChemiDoc imaging system (Bio-Rad) at appropriate wavelength.
In vivo assay for Cascade and Cas3–mediated interference
pCDFduet-Cascade/Cas3 (StrepR), pRSFDuet-crRNA-Cas6 (KanaR)and pETDuet-Targets (AmpR)with different PAM sequence were co-transformed into the E. coli BL21AI cell line and grown on LB agarose plates containing kanamycin (50 μg/ml), ampicillin (100 μg/ml), streptomycin (30 μg/ml). After transformation, a single colony were cultured at 37°C in nonselective LB medium (Strep + Kana) to O.D.600 of 0.3, at which point the expression of PfuCascade, Cas3, and pre-crRNA was induced for 12 h by the addition of 0.5% L-arabinose and 1 mM IPTG. Each cell culture was then divided into two equal volumes and plated onto Strep + Kana + Amp LB plates (selective for pETDuet-target) and Strep + Kan plates (non-selective for the target plasmid) in a series of dilutions. The number of colonies on each plate was counted after overnight incubation at 37°C. The CRISPR interference efficiency was reflected in the ratio of colony-forming units on the nonselective over selective plates. Each experiment was repeated three times to calculate the s.d. The experimental procedure is illustrated in Supplementary Figure 1K and Table 1.
Fluorescently labeled prespacer substrate preparation
Fluorescent DNA oligos (Supplementary Table 2) for biochemistry were synthesized (Integrated DNA Technologies) with either a 5AmMC6 or 3AmMO label, fluorescently labeled in-house by Cy3 or Cy5-NHS dye (Lumiprobe), and annealed at equimolar amount, and native PAGE purified to remove unannealed ssDNA.
Electrophoretic mobility shift assay
5 nM final concentration of fluorescently labeled target DNA was incubated with titrations of Pfu Cascade or Cas3/Cascade complex in a 20 μL total reaction volume containing 50 mM Tris pH 8.0, 150 mM NaCl, and 10% glycerol. After a 15-minute incubation at 42°C or specified condition in the figure legends,10 μL of each sample was loaded onto 1% agarose gel equilibrated in 0.5 × TBE buffer. Electrophoresis was performed at 60 V for 40 min in cold room. The fluorescent signals from the gel were recorded using a ChemiDoc imaging system (Bio-Rad).
For the fluorescence labeled protein EMSA assay in Fig. 1C., the proteins were labeled using corresponding fluorophore (Sulfo-Cy3 or Cy5, Lumiprobe) following our previously published protocol (Xiao et al., 2017). The 20 μL binding experiments used 300 nM Cy3-Cas3, 100 nM Cy5-Cascade, and 20 nM FAM-DNA in single tube or combined two or three components in one tube. Incubation and electrophoresis were carried out as above.
Plasmid transformation assay in P. furiosus
P. furiosus strains were generated via the previously described pop-in/pop-out marker technique (Elmore et al. 2016). Cultures and media were prepared as described previously (Lipscomb et al. 2011). Incubations were performed under anaerobic conditions at 95°C in defined P. furiosus media. Liquid cultures were grown to mid to late log phase, and 200 μL of culture was combined with 100 ng of plasmid DNA (in 4.0 μL). The mixtures were plated on solid defined media and incubated for ~62 h. Following incubation, colonies on each plate were enumerated, and transformation efficiency (Colony Formation Units/μg plasmid DNA) was calculated and plotted logarithmically. This assay was carried out with a minimum of three replicates.
Affinity pull-down assay
15 μg of strep-tagged Pfu Cascade and 15 μg of untagged Pfu Cas3 complex were mixed and incubated with 10 μL of strep resin at 4°C for 30 min in a binding buffer (50 mM HEPES pH7.5, 10% glycerol, 300 mM NaCl), in a total assay volume of 50 μL. The strep resin was pelleted by centrifugation at ~100 g for 30 seconds, washed 3 times with 200 μL of the corresponding binding buffer, then eluted with 70 μL of elution buffer (50 mM HEPES pH7.5, 300 mM NaCl, 2.5 mM desthiobiotin and 10% glycerol). Eluted proteins were separated on 12% SDS-PAGE and stained by Coomassie blue.
Cascade-mediated Cas3 DNA cleavage assay and collateral activity assay
The 127 bp dsDNA substrate was produced from PCR using 5′-fluorescently labeled primers (Cy3 at NTS and Cy5 at TS). The reaction mixture was prepared from 100 nM final concentration of Pfu Cascade, 100 nM Pfu Cas3 (HEL+HD) and 10 nM substrate in a cleavage buffer containing 10 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2 and 100 μM CoCl2. The reaction was incubated at 58°C (or indicated temperatures in the figures) for 30 min. The collateral cleavage activity of pfu Cas3 used 10 nM FAM-ssDNA reporter instead. After incubation, the nucleic acids were phenol-chloroform extracted and separated on a 12% denaturing polyacrylamide gel. Fluorescent signals were recorded using a Typhoon™ scanner (Amersham).
HASTE detection assay
22 samples with the corresponding concentration of target plasmid were mixed with the PCR system by introducing primers and iProof polymerase in 50 μl total reaction volume. After 25 cycles (this step is taken for 15 min), 1 μl of each sample was combined with 20 μl HASTE tool (100 nM final concentration of Pfu Cascade, 100 nM Pfu Cas3 and 100 nM ssDNA-FQ reporter) respectively, and incubated at 60 °C for 15 min. Finally, all reaction tubes were scanned using a BioRad ChemiDoc imager. For the comparison, all samples were also resolved after electrophoresis on 1% agarose gels.
Lateral flow assay for Pfu Cascade-Cas3 based nucleic acid detection
20 μL cleavage reactions were prepared and incubated at 58°C for 30 min with 1 μM final concentration of FAM-ssDNA-biotin reporter. Afterwards, 10 μL 30% PEG 6k and 30 μL of HybriDetect 1 assay buffer (Milenia) was added to the reaction and allowed to diffuse on a HybriDetect 1 lateral flow strip (Milenia) for 5 min.
RNP electroporation of GFP-HAP1 and GFP-HEK293 cells
The GFP-tagged diploid HAP1 cell line was a gift from Yan Zhang’s lab (Tan et al., 2022). The GFP-tagged HEK293 cell line was purchased from GenTarget. The two cell lines were maintained in similar fashion, in DMDM (Gibco) supplemented with 10% FBS (Gibco) at 37 °C and 5% CO2 in a humidified incubator. The cells were electroporated using the Neon Transfection system (ThermoFisher) according to the manufacturer’s instructions. Briefly, HAP1 cells were individualized with 0.05% Trypsin-EDTA solution (GIBCO), washed once with DMDM (*give description), 10% FBS and resuspended in Neon buffer R to a concentration of 5×106 cells/mL. 20–40 pmol of NLS-Pfu Cascade/NLS-Pfu Cas3 complex were mixed with approximately 5×104 cells in buffer R (Neon Transfection system) in a total volume of 14 μL. Each mixture was electroporated using a 10 μL Neon tip (1450 V, 13 ms, 4 pulses) and plated in 6-well tissue culture plates containing 2 mL IMDM, 10% FBS.
Flow cytometry analysis, FACS sorting and single cell isolation
Cells were individualized with 0.05% Trypsin-EDTA solution (GIBCO) 5 days after electroporation and resuspended in 1× PBS before experiments. For analysis, individualized cells were analyzed on a BD Biosciences FACSAria Fusion using the 488nm laser for EGFP. Data analysis was performed using FlowJo® v10.4.1 in Flow Cytometry Facility of Cornell university. Sorted cells were then cultured in tissue culture incubator with 5% CO2 at 37°C for other usage.
DNA lesion analysis by long-range PCR genotyping
Genomic DNA of HAP1 cells were isolated using Gentra Puregene Cell Kit (QIAGEN) per manufacturer protocol. Long-range PCRs were all done using iProof DNA Polymerase (Biorad). Products were resolved on 1% agarose gels, stained by EtBr dye and visualized with Chemidoc MP imager (Biorad). See Supplementary Table 2 for all primers used for long-range PCRs. To define lesion junctions shown in Figures 7, lesion PCR reactions were purified using QIAquick PCR Purification Kit (QIAGEN), cloned into pJET vector (Thermo fisher), and transformed into DH5α cells. Plasmids from randomly picked single colonies were Sanger-sequenced to define the deletion boundaries.
Cryo-EM data acquisition
4 μL of 0.6 mg/mL SEC-purified complexes were applied to a Quantifoil holey carbon grid (1.2/1.3, 400 mesh), which had been glow-discharged for 30 sec at 30 mA current. Grids were blotted at 8°C for 4 seconds with zero force setting, 100% humidity and plunge-frozen in liquid ethane using a Mark IV FEI/Thermo Fisher Vitrobot. Cryo-EM images were collected on a 200 kV Talos Arctica transmission microscope (Thermo Fisher) equipped with a K3 direct electron detector (Gatan). The total exposure time of each movie stack was ~ 3.5 s, leading to a total accumulated dose of 50 electrons per Å^2 which fractionated into 50 frames. Dose fractionated super-resolution movie stacks collected from the K3 direct electron detector were 2× binned to a pixel size of 1.23 Å. The defocus value was set between −1.0 μm to −2.5 μm.
Cryo-EM data processing
Motion correction, CTF-estimation, blob particle picking, 2D classification, 3D classification and non-uniform 3D refinement were performed in cryoSPARC v.2 (Punjani et al., 2017). Refinements followed the standard procedure, a series of 2D and 3D classifications with C1 symmetry were performed as shown in Fig. S2 to generate the final maps. A solvent mask was generated and was used for all subsequent local refinement steps. CTF post refinement was conducted to refine the beam-induced motion of the particle set, resulting in the final maps. The detailed data processing and refinement statistics for all cryo-EM structures are summarized in Fig. S2 and Table 1 for the data acquisition and structure refinement.
Supplementary Material
Supplementary Movie S1. Rotating views of four cryo-EM snapshots of Pfu Type I-A CRISPR effector complexes. Related to Figure 2.
Supplementary Movie S2. 3D variant display of the apo Cascade complex and the Cascade-Cas3 complex, analyzed by Program CryoSPARC. Related to Figure 3. This analysis reveals the extent of the inherent conformational dynamics in each complex. The Cas8 NTD and the entire ‘inner belly’ of Cascade are significantly more dynamic in the apo Cascade complex.
Supplementary Movie S3. PAM recognition mechanism illustrated from different views of the Cascade-Cas3/partial R-loop structure. Related to Figure 4.
Supplementary Movie S4. Morphing between partial and full R-loop Cascade-Cas3 structures revealing the concerted conformational changes in the ‘inner belly’ of the Cascade upon full R-loop formation. Related to Figure 5. This conformational change is further transcended from the Cas8a subunit of Cascade to Cas3, causing a rigid-body movement in Cas3 and also triggering the allosteric activation of its HD nuclease.
Supplementary Movie S5. Animation of the envisioned partial-to-full R-loop transition and the allosteric regulation at Cas3 HD nuclease center. Related to Figure 5.
Supplementary Movie S6. Time lapse of the RNA-guided GFP knock-out editing by Pfu I-A Cascade-Cas3 (right panel), done in the GFP-tagged, diploid form of the HAP1 cells. Related to Figure 7. The negative control is displayed on the left-side panel, where the non-targeting Pfu I-A Cascade-Cas3 was electroporated into the cells.
Supplementary Movie S7. Time lapse of the RNA-guided GFP knock-out editing by Pfu I-A Cascade-Cas3 (right panel), done in the GFP-tagged HEK293 cells. Related to Figure 7. The negative control is displayed on the left-side panel, where the non-targeting Pfu I-A Cascade-Cas3 was electroporated into the cells. Note that the GFP intensities among HEK293 cells are not as homogenous as those among the HAP1 cells. This property is inherent to the HEK293 cell line, and the GFP tagging information is not available from the vendor.
Highlights.
Type I-A Cascade and Cas3 form an integral effector complex.
Cas3 nuclease is allosterically activated by Cascade, upon full R-loop formation.
Type I-A is repurposed to a heat-activated streamlined nucleic acid detection platform.
Type I-A CRISPR-Cas3 is highly efficient in bi-directional DNA deletion in human cells.
ACKNOWLEDGMENTS
This work is supported by the National Institutes of Health grants GM118174 to A.K. and GM118160 to M.P.T. and by the Swiss National Science Foundation, Grant NCCR TransCure to D.N. and H.S. This work made use of the Cornell Center for Materials Research Shared Facilities which are supported through the NSF MRSEC program (DMR-1719875). We thank Zhonggang Hou, Yan Zhang, Andrew Grimson, Elizabath Fogarty, Mariena Ramos, and Katherine Spoth for technical assistance and Gabriel Schuler for helpful discussions. We thank Ryan J. Catchpole for generation of the mini-CRISPR array plasmid used for the Pfu in vivo interference assays and for technical advice over the course of this work. We thank Yan Zhang for sharing the GFP-tagged HAP1 cell line. The authors declare competing financial interests on I-A related applications. Correspondence should be addressed to A.K. (ailong.ke@cornell.edu). Reprints and permissions information are available upon request.
Footnotes
DECLARATION OF INTERESTS
The authors have no interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, and Ehrlich SD (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliando BJ, and Voigt CA (2015). Targeted DNA degradation using a CRISPR device stably carried in the host genome. Nat Commun 6, 6989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron P, Coons MM, Klompe SE, Lied AM, Smith SC, Vidal B, Donohoue PD, Rotstein T, Kohrs BW, Nyer DB, et al. (2019). Harnessing type I CRISPR-Cas systems for genome engineering in human cells. Nat Biotechnol 37, 1471–1477. [DOI] [PubMed] [Google Scholar]
- Carte J, Wang R, Li H, Terns RM, and Terns MP (2008). Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev 22, 3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, and Doudna JA (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu J, Zhi S, Zheng Q, Ma W, Huang J, Liu Y, Liu D, Liang P, and Songyang Z (2020). Repurposing type I-F CRISPR-Cas system as a transcriptional activation tool in human cells. Nat Commun 11, 3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csorgo B, Leon LM, Chau-Ly IJ, Vasquez-Rifo A, Berry JD, Mahendra C, Crawford ED, Lewis JD, and Bondy-Denomy J (2020). A compact Cascade-Cas3 system for targeted genome engineering. Nat Methods 17, 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillard KE, Brown MW, Johnson NV, Xiao Y, Dolan A, Hernandez E, Dahlhauser SD, Kim Y, Myler LR, Anslyn EV, et al. (2018). Assembly and Translocation of a CRISPR-Cas Primed Acquisition Complex. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan AE, Hou Z, Xiao Y, Gramelspacher MJ, Heo J, Howden SE, Freddolino PL, Ke A, and Zhang Y (2019). Introducing a Spectrum of Long-Range Genomic Deletions in Human Embryonic Stem Cells Using Type I CRISPR-Cas. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore J, Deighan T, Westpheling J, Terns RM, and Terns MP (2015). DNA targeting by the type I-G and type I-A CRISPR-Cas systems of Pyrococcus furiosus. Nucleic Acids Res 43, 10353–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore JR, Sheppard NF, Ramia N, Deighan T, Li H, Terns RM, and Terns MP (2016). Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev 30, 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster K, Gruschow S, Bailey S, White MF, and Terns MP (2020). Regulation of the RNA and DNA nuclease activities required for Pyrococcus furiosus Type III-B CRISPR-Cas immunity. Nucleic Acids Res 48, 4418–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett S, Shiimori M, Watts EA, Clark L, Graveley BR, and Terns MP (2020). Primed CRISPR DNA uptake in Pyrococcus furiosus. Nucleic Acids Res 48, 6120–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Shin M, Sun J, Jung CH, Bolt EL, van der Oost J, and Kim JS (2014). Molecular insights into DNA interference by CRISPR-associated nuclease-helicase Cas3. Proc Natl Acad Sci U S A 111, 16359–16364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, and Zhang F (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan S, Borges A, and Bondy-Denomy J (2020). Distinct subcellular localization of a Type I CRISPR complex and the Cas3 nuclease in bacteria. bioRxiv, 2020.2009.2029.318501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, Eng ET, Raczkowski AM, Fox T, Earl LA, et al. (2017). Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell 171, 414–426 e412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes RP, Xiao Y, Ding F, van Erp PB, Rajashankar K, Bailey S, Wiedenheft B, and Ke A (2016). Structural basis for promiscuous PAM recognition in type I-E Cascade from E. coli. Nature 530, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Matosevic ZJ, Mitic D, Markulin D, Killelea T, Matkovic M, Bertosa B, Ivancic-Bace I, and Bolt EL (2021). A Tryptophan ‘Gate’ in the CRISPR-Cas3 Nuclease Controls ssDNA Entry into the Nuclease Site, That When Removed Results in Nuclease Hyperactivity. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser ML, Taylor DW, Bhat P, Guegler CK, Sternberg SH, Nogales E, and Doudna JA (2014). CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc Natl Acad Sci U S A 111, 6618–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser ML, Taylor DW, Kornfeld JE, Nogales E, and Doudna JA (2016). DNA Targeting by a Minimal CRISPR RNA-Guided Cascade. Mol Cell 63, 840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Nam KH, Ding F, Lee H, Wu L, Xiao Y, Farchione MD Jr., Zhou S, Rajashankar K, Kurinov I, et al. (2014). Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat Struct Mol Biol 21, 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Golden SM, van Erp PB, Carter J, Westra ER, Brouns SJ, van der Oost J, Terwilliger TC, Read RJ, and Wiedenheft B (2014). Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science 345, 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia N, Mo CY, Wang C, Eng ET, Marraffini LA, and Patel DJ (2019). Type III-A CRISPR-Cas Csm Complexes: Assembly, Periodic RNA Cleavage, DNase Activity Regulation, and Autoimmunity. Mol Cell 73, 264–277 e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunne T, Kieper SN, Bannenberg JW, Vogel AI, Miellet WR, Klein M, Depken M, Suarez-Diez M, and Brouns SJ (2016). Cas3-Derived Target DNA Degradation Fragments Fuel Primed CRISPR Adaptation. Mol Cell 63, 852–864. [DOI] [PubMed] [Google Scholar]
- Li Y, Pan S, Zhang Y, Ren M, Feng M, Peng N, Chen L, Liang YX, and She Q (2016). Harnessing Type I and Type III CRISPR-Cas systems for genome editing. Nucleic Acids Res 44, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Fuglsang A, Kjeldsen AL, Sun K, Bhoobalan-Chitty Y, and Peng X (2020). DNA targeting by subtype I-D CRISPR-Cas shows type I and type III features. Nucleic Acids Res 48, 10470–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ML, Mullis AS, Leenay RT, and Beisel CL (2015). Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res 43, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, and Terns MP (2019). CRISPR RNA-guided DNA cleavage by reconstituted Type I-A immune effector complexes. Extremophiles 23, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Zhao P, Pfister NT, Compton M, Olson S, Glover CV 3rd, Wells L, Graveley BR, Terns RM, and Terns MP (2015). Three CRISPR-Cas immune effector complexes coexist in Pyrococcus furiosus. RNA 21, 1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, and Koonin EV (2006). A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nature reviews Microbiology 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride TM, Schwartz EA, Kumar A, Taylor DW, Fineran PC, and Fagerlund RD (2020). Diverse CRISPR-Cas Complexes Require Independent Translation of Small and Large Subunits from a Single Gene. Mol Cell 80, 971–979 e977. [DOI] [PubMed] [Google Scholar]
- Mojica FJ, García-Martínez J, and Soria E (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. Journal of molecular evolution 60, 174–182. [DOI] [PubMed] [Google Scholar]
- Morisaka H, Yoshimi K, Okuzaki Y, Gee P, Kunihiro Y, Sonpho E, Xu H, Sasakawa N, Naito Y, Nakada S, et al. (2019). CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat Commun 10, 5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulepati S, and Bailey S (2013). In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J Biol Chem 288, 22184–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulepati S, Heroux A, and Bailey S (2014). Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science 345, 1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, and Hase T (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28, E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RE, Santos IC, Wrapp D, Bravo JPK, Schwartz EA, Brodbelt JS, and Taylor DW (2020). Structural basis for assembly of non-canonical small subunits into type I-C Cascade. Nat Commun 11, 5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakabe K, Wada N, Miyaji T, Murakami E, Marui K, Ueta R, Hashimoto R, Abe-Hara C, Kong B, Yano K, et al. (2020). Genome editing in plants using CRISPR type I-D nuclease. Commun Biol 3, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakabe K, Wada N, Murakami E, Miyashita N, and Osakabe Y (2021). Genome editing in mammalian cells using the CRISPR type I-D nuclease. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickar-Oliver A, Black JB, Lewis MM, Mutchnick KJ, Klann TS, Gilcrest KA, Sitton MJ, Nelson CE, Barrera A, Bartelt LC, et al. (2019). Targeted transcriptional modulation with type I CRISPR-Cas systems in human cells. Nat Biotechnol 37, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagens A, Tripp V, Daume M, Sharma K, Klingl A, Hrle A, Conti E, Urlaub H, and Randau L (2014). In vitro assembly and activity of an archaeal CRISPR-Cas type I-A Cascade interference complex. Nucleic Acids Res 42, 5125–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, and Vergnaud G (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rath D, Amlinger L, Hoekzema M, Devulapally PR, and Lundgren M (2015). Efficient programmable gene silencing by Cascade. Nucleic Acids Res 43, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding S, Sternberg SH, Marshall M, Gibb B, Bhat P, Guegler CK, Wiedenheft B, Doudna JA, and Greene EC (2015). Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell 163, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins MF, Chowdhury S, Carter J, Golden SM, Miettinen HM, Santiago-Frangos A, Faith D, Lawrence CM, Lander GC, and Wiedenheft B (2019). Structure Reveals a Mechanism of CRISPR-RNA-Guided Nuclease Recruitment and Anti-CRISPR Viral Mimicry. Mol Cell 74, 132–142 e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkauskas M, Sinkunas T, Songailiene I, Tikhomirova MS, Siksnys V, and Seidel R (2015). Directional R-Loop Formation by the CRISPR-Cas Surveillance Complex Cascade Provides Efficient Off-Target Site Rejection. Cell reports. [DOI] [PubMed] [Google Scholar]
- Sashital DG, Wiedenheft B, and Doudna JA (2012). Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol Cell 46, 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkunas T, Gasiunas G, Waghmare SP, Dickman MJ, Barrangou R, Horvath P, and Siksnys V (2013). In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J 32, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, and Seidel R (2014). Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci U S A 111, 9798–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan R, Krueger RK, Gramelspacher MJ, Zhou X, Xiao Y, Ke A, Hou Z, and Zhang Y (2022). Cas11 enables genome engineering in human cells with compact CRISPR-Cas3 systems. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Mo CY, Wasserman MR, Rostol JT, Marraffini LA, and Liu S (2019). Dynamics of Cas10 Govern Discrimination between Self and Non-self in Type III CRISPR-Cas Immunity. Mol Cell 73, 278–290 e274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Erp PB, Kunne T, Wong SP, Staals RH, Seegers CL, Bollen S, Jore MM, Semenova E, Severinov K, et al. (2012). CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell 46, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJJ, van der Oost J, Doudna JA, and Nogales E (2011). Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477, 486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo M, Dolan AE, Liao M, and Ke A (2018). Structure basis for RNA-guided DNA degradation by Cascade and Cas3. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo M, Hayes RP, Kim J, Ng S, Ding F, Liao M, and Ke A (2017). Structure Basis for Directional R-loop Formation and Substrate Handover Mechanisms in Type I CRISPR-Cas System. Cell 170, 48–60 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimi K, Takeshita K, Kodera N, Shibumura S, Yamauchi Y, Omatsu M, Kunihiro Y, Yamamoto M, and Mashimo T (2021). Dynamic mechanisms of CRISPR interference by Escherichia coli CRISPR-Cas3. bioRxiv, 2021.2007.2018.452824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L, Ma J, Wang J, Artamonova D, Wang M, Liu L, Xiang H, Severinov K, Zhang X, and Wang Y (2019). Structure Studies of the CRISPR-Csm Complex Reveal Mechanism of Co-transcriptional Interference. Cell 176, 239–253 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JK, Gasior SL, Jones S, Wang L, Navarro P, Vickroy B, and Barrangou R (2019). The repurposing of type I-E CRISPR-Cascade for gene activation in plants. Commun Biol 2, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sheng G, Wang J, Wang M, Bunkoczi G, Gong W, Wei Z, and Wang Y (2014). Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature 515, 147–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie S1. Rotating views of four cryo-EM snapshots of Pfu Type I-A CRISPR effector complexes. Related to Figure 2.
Supplementary Movie S2. 3D variant display of the apo Cascade complex and the Cascade-Cas3 complex, analyzed by Program CryoSPARC. Related to Figure 3. This analysis reveals the extent of the inherent conformational dynamics in each complex. The Cas8 NTD and the entire ‘inner belly’ of Cascade are significantly more dynamic in the apo Cascade complex.
Supplementary Movie S3. PAM recognition mechanism illustrated from different views of the Cascade-Cas3/partial R-loop structure. Related to Figure 4.
Supplementary Movie S4. Morphing between partial and full R-loop Cascade-Cas3 structures revealing the concerted conformational changes in the ‘inner belly’ of the Cascade upon full R-loop formation. Related to Figure 5. This conformational change is further transcended from the Cas8a subunit of Cascade to Cas3, causing a rigid-body movement in Cas3 and also triggering the allosteric activation of its HD nuclease.
Supplementary Movie S5. Animation of the envisioned partial-to-full R-loop transition and the allosteric regulation at Cas3 HD nuclease center. Related to Figure 5.
Supplementary Movie S6. Time lapse of the RNA-guided GFP knock-out editing by Pfu I-A Cascade-Cas3 (right panel), done in the GFP-tagged, diploid form of the HAP1 cells. Related to Figure 7. The negative control is displayed on the left-side panel, where the non-targeting Pfu I-A Cascade-Cas3 was electroporated into the cells.
Supplementary Movie S7. Time lapse of the RNA-guided GFP knock-out editing by Pfu I-A Cascade-Cas3 (right panel), done in the GFP-tagged HEK293 cells. Related to Figure 7. The negative control is displayed on the left-side panel, where the non-targeting Pfu I-A Cascade-Cas3 was electroporated into the cells. Note that the GFP intensities among HEK293 cells are not as homogenous as those among the HAP1 cells. This property is inherent to the HEK293 cell line, and the GFP tagging information is not available from the vendor.
Data Availability Statement
The models and maps for the structures presented have been deposited in the PDB and EMDB. The accession numbers are listed in the key resources table. Uncropped original gel and microscopy images have been deposited on Mendeley Data: https://doi: 10.17632/ry9tf59h3k.1. These are all publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli Bl21(DE3) competent cells | NEB | Cat# C2527I |
| E. coli DH5a competent cells | NEB | Cat# 18265017 |
| Chemicals, peptides, and recombinant proteins | ||
| Modified and unmodified oligos | IDT | N/A |
| IPTG | GoldBio | Cat # I2481C500 |
| LB broth | Teknova | #L9145 |
| Restriction enzymes | NEB | Choose from catalog |
| iProof high fidelity PCR kit | Biorad | #1725331 |
| Acrylamide | thermal | HC2040 |
| Urea | VWR | 57-13-6 |
| Fluorescence stain Cy3 and Cy5 | Lumiprobe | 11320 and 13320 |
| SYPRO® Orange | Thermo Fisher | S6650 |
| Formamide | VWR | 75-12-7 |
| MnCl2 | Fisher Scientific | 7773-01-5 |
| Strep resin | IBA | 2-1201-025 |
| Nickel resin | QIAGEN | Cat. No. / ID: 30250 |
| lateral flow strip | HybriDetect 1 | MGDS |
| Copper Grids 1.2/1.3 200 mesh | QUANTIFOIL | Q250-CR1.3 |
| Type I-A Cascade | This study | N/A |
| Cas3 HEL | This study | N/A |
| Cas3 HD | This study | N/A |
| T.fusca type I-E Cascade | Xiao et al., 2017 | N/A |
| T.fusca type I-E Cas3 | Xiao et al., 2018 | N/A |
| Deposited data | ||
| Cascade alone | This study | PDB: 7TR6 and EMBD-26081 |
| Cascade-Cas3 | This study | PDB: 7TR8 and EMBD-26082 |
| Partial R-loop | This study | PDB: 7TR9 and EMBD-26083 |
| Full R-loop | This study | PDB: 7TRA and EMBD-26084 |
| Cas3-Cas8a alone | This study | EMBD-26097 |
| Experimental models: Cell lines | ||
| HAP1_GFP | Dolan et al. 2019 | N/A |
| HEK293_GFP | This study | N/A |
| Recombinant DNA | ||
| Complete list of plasmids | This study | Table S2 |
| Oligonucleotides | ||
| Complete list of DNA oligonucleotide sequences | This study | Table S3 |
| Miniature CRISPR array spacers | This study | Table S3 |
| Software and algorithms | ||
| EPU | Thermo Fisher Scientific | N/A |
| RELION 3 | Scheres Lab | https://www3.mrc-lmb.cam.ac.uk/relion/index.php?title%20=%20Main_PAGE |
| cryoSPARC | Ali Punjani et al., 2017 | https://cryosparc.com/ |
| Coot (v. 0.9.5.1-pre) | (Emsley and Cowtan, 2004; Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHENIX | Adams et al. (2010) | https://phenix-online.org/ |
| ChimeraX (v. 1.2) | (Goddard et al., 2018) | https://www.cgl.ucsf.edu/chimerax/ |
| ClustalW | (Goujon et al., 2010; Sievers et al., 2011) | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Other | ||
| MonoQ 5/50 GL | Cytiva | Cat# 17516601 |
| HiLoad 16/600 Superdex 200 pg | Cytiva | Cat# 28989335 |
| Quantifoil R 1.2/1.3, 200 mesh, copper | Quantifoil | Q250-CR1.3 |