

Summary
The tumor microenvironment (TME) is a unique metabolic niche that can inhibit T cell metabolism and cytotoxicity. To dissect the metabolic interplay between tumors and T cells, we establish an in vitro system that recapitulates the metabolic niche of the TME and allows to define cell-specific metabolism. We identify tumor-derived lactate as an inhibitor of CD8+ T cell cytotoxicity, revealing an unexpected metabolic shunt in the TCA cycle. Metabolically fit cytotoxic T cells shunt succinate out of the TCA cycle to promote autocrine signaling via the succinate receptor (SUCNR1). Cytotoxic T cells are reliant on pyruvate carboxylase (PC) to replenish TCA cycle intermediates. In contrast, lactate reduces PC-mediated anaplerosis. Inhibition of pyruvate dehydrogenase (PDH) is sufficient to restore PC activity, succinate secretion, and activation of SUCNR1. These studies identify PDH as a potential drug target to allow CD8+ T cells to retain cytotoxicity and overcome a lactate-enriched TME.
eTOC Blurb
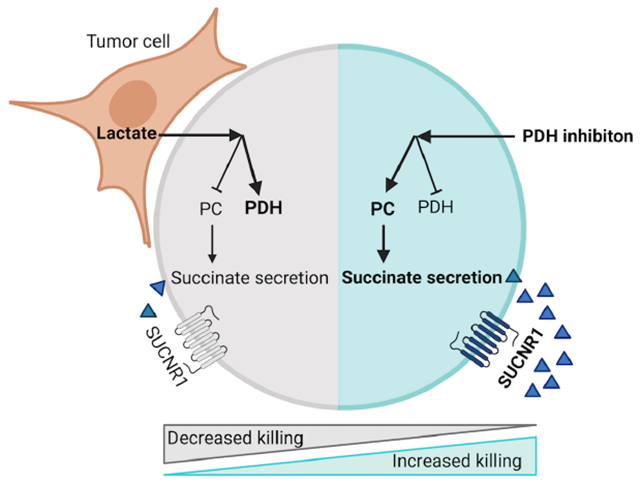
Elia et al. report tumor-derived lactate alters T cell metabolism and inhibits T cell cytotoxicity. Lactate changes the entry of pyruvate into the TCA cycle from PC towards PDH. Inhibition of PDH is sufficient to increase PC activity and succinate secretion, which activates the receptor SUCNR1 and increases the anti-tumor cytotoxicity of T cells.
Graphical Abstract
Introduction
The immune system provides a critical defense against tumor progression (Chen and Mellman, 2013). However, many tumors evolve to evade eradication by the immune system, and the tumor microenvironment (TME) suppresses anti-tumor immunity. Immune checkpoint blockade (ICB) targets pathways that inhibit anti-tumor immunity. There are FDA-approved treatments targeting the inhibitory receptors cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), and programmed death-ligand 1 (PD-L1). Despite these ntable successes, most patients with metastatic tumors still do not experience durable responses to ICB (Dougan et al., 2019; Farkona et al., 2016; Shah and Fry, 2019). Thus, a deeper understanding of pathways that regulate immune cell function and limit anti-tumor immunity is needed.
In the past few years, a wealth of evidence has emerged illustrating how metabolism is central to many aspects of CD8+ T cell biology. Metabolic reprogramming is initiated upon T cell activation and is crucial for sustaining CD8+ T cell proliferation, survival, and function (Buck et al., 2015; Delgoffe and Powell, 2015). Several studies have characterized the metabolic signatures that follow the activation and differentiation of naïve CD8+ T cells. These studies have shown that CD8+ T cells upregulate glycolysis and mitochondrial metabolism upon T cell receptor (TCR) stimulation and that disruption of either metabolic pathway leads to a failure in T cell activation (Cham and Gajewski, 2005; Chang et al., 2013; Ho et al., 2015; Johnson et al., 2018; Ron-Harel et al., 2016). However, how the TME affects these metabolic shifts and CD8+ T cell function is not completely understood (Elia and Haigis, 2021; Ringel et al., 2020; Sugiura and Rathmell, 2018). This feature is particularly relevant given that the TME has a unique metabolic niche in which key nutrients may be limiting, while immunosuppressive metabolic by-products accumulate (Elia and Haigis, 2021).
In this study, we test the hypothesis that tumor cells secrete metabolites that inhibit CD8+ T cell cytotoxicity. We identify an unexpected tumor-T cell metabolic circuit in which tumor-derived lactate in the TME rewires CD8+ T cell pyruvate metabolism, leading to a loss of cytotoxic function, but not proliferation. We discover cytotoxic CD8+ T cells rely on pyruvate carboxylase (PC), which converts pyruvate into mitochondrial oxaloacetate. This reaction sustains the TCA cycle and succinate secretion, which initiates autocrine signaling via the succinate receptor 1 (SUCNR1), a pro-inflammatory G-protein coupled receptor. We find that SUCNR1 signaling initiates the production of cytotoxic molecules in T cells to promote tumor killing. TME-derived lactate reverts this program to a more conventional TCA cycle where carbons enter through the activity of pyruvate dehydrogenase (PDH) rather than PC, and where succinate is converted to fumarate rather than secreted.
By modeling the metabolic dependencies of CD8+ T cell cytotoxicity within the TME, we show that pharmacological inhibition of PDH is sufficient to reestablish CD8+ T cell cytotoxicity through PC activity, succinate secretion, and autocrine activation of SUCNR1. Thus, this study highlights the therapeutic potential of targeting T cell metabolism to promote cytotoxicity even in a hostile metabolic environment.
Results
Nutrient conditions mimicking the TME impair CD8+ T cell cytotoxicity and PC activity
To identify the metabolic alterations in CD8+ T cells that accompany cytotoxicity, we activated naïve TCR transgenic OT-1 CD8+ T cells that recognize the chicken ovalbumin (OVA)257-264 peptide with anti-CD3/anti-CD28, IL-2, and IL-12 for 72 hours. We cultured these activated OT-1 cells with or without GFP+-B16 melanoma or GFP+-MC38 colon adenocarcinoma cells constitutively expressing the OVA protein (Figure 1A). When cultured with OVA-expressing tumor cells in RPMI, these activated T cells showed an antigen-specific increase in interferon γ (IFNγ), granzyme B (GzmB), and Ki67 expression (Figure S1A–C).
Figure 1. TME mimicking nutrient availability impairs CD8+ T cell cytotoxicity and PC activity.
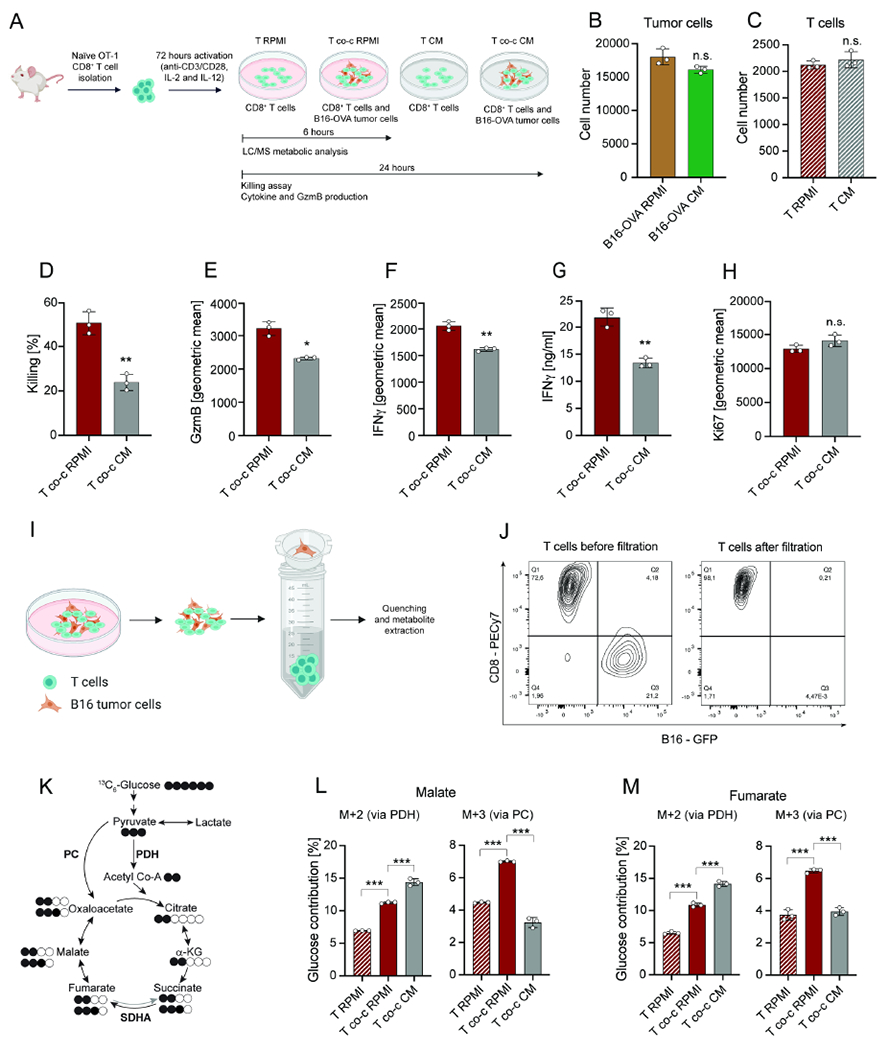
(A) Schematic representation of CD8+ T cell activation and co-culture with tumor cells in RPMI and CM. T indicates T cells in mono-culture and T co-c indicates T cells in co-culture with tumor cells. (B) Cell number of B16-OVA tumor cells in RPMI and CM. Analysis was performed after 24 hours. (C) Cell number of CD8+ T cells in mono-culture in RPMI and CM. Analysis was performed after 24 hours. (D-F) Killing and geometric mean of GzmB and IFNγ expression by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI and CM. Analysis was performed after 24 hours. (G) Secreted IFNγ levels by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI and CM. Analysis was performed after 24 hours. (H) Geometric mean of Ki67 expression by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI and CM. Analysis was performed after 24 hours. (I) Schematic representation of cell-size-based separation of B16 tumor cells and CD8+ T cells. (J) Flow cytometric measurement of T cell populations before and after filtration. (K) Schematic representation of 13C6-glucose contribution to TCA cycle intermediates via PDH or PC activity. Grey arrow depicts low activity. White circles indicate carbon-12 (12C) while black circles indicate 13C atoms from glucose. (L-M) Contribution of 13C6-glucose to malate and fumarate M+2 (derived from PDH activity) and M+3 (derived from PC activity) in CD8+ T cells in mono-culture and in co-culture with B16-OVA tumor cells in RPMI and CM. Analysis was performed after 6 hours. The number of biological replicates for each experiment was n=3. Error bars represent s.d. Two-tailed unpaired Student’s T-test was performed. *P<0.05; **P<0.01; ***P<0.001. See also Figure S1.
As the TME uniquely reflects the net metabolite consumption and secretion of cells, including abundant tumor cells, we compared the cytotoxicity of T cells cultured in fresh RPMI media or in conditioned media (CM) obtained after 24 hours of culture with tumor cells (Figure 1A). There was no significant difference in the number of tumor cells (Figure 1B) or activated CD8+ T cells (Figure 1C) after 24 hours of mono-culture in CM or RPMI, indicating that CM does not affect cell viability. However, the media composition significantly influenced CD8+ T cell activity when co-cultured with tumor cells. Co-cultures in CM significantly decreased CD8+ T cell cytolytic activity, as well as IFNγ and GzmB production (Figure 1D–G; Figure S1D–G). CM also had a significant inhibitory effect on the non-specific killing when CD8+ T cells were co-cultured with B16 cells not expressing OVA (Figure S1H). This result indicates that the inhibitory effect of CM is not directly dependent on antigen signaling. Furthermore, in accordance with the cell number measurements, the proliferation marker Ki67 was unchanged in T cells cultured with tumor cells in RPMI or CM, suggesting the effects of CM dissociate CD8+ T cell effector function (IFNγ, GzmB production, and cytolysis) from cellular proliferation (Figure 1H; Figure S1I).
To date, no method has been able to rapidly isolate T cells and tumor cells from co-cultures for further metabolic assessment. We overcame this limitation by developing a technique based on a rapid cell-size-based filtration method to separate these cell populations from co-cultures within seconds. Flow cytometry analysis verified that tumor and T cell populations were >95% pure (Figure 1I–J; Figure S1J).
We next applied rapid isolation to assess the metabolic changes that correspond with cytolytic activity in co-culture, and to examine whether T cell metabolism is altered in CM conditions. To map the metabolic profile linked to cytolytic activity, we measured 13C6-glucose utilization in activated CD8+ T cells cultured in the presence or absence of B16-OVA or MC38-OVA tumor cells in both RPMI and CM conditions as described above. When comparing the metabolic profile of CD8+ T cells grown alone or in co-culture with tumor cells, we observed that CD8+ T cells in co-culture exhibited an increase in glucose-derived carbons in central carbon metabolites, including amino acids and TCA cycle intermediates (Figure S1K). Although CD8+ T cells in co-culture in CM exhibited a similar induction in glucose contributions, we detected a significant difference in the entry of these carbons into the TCA cycle. Glucose-derived pyruvate can contribute two carbons to the TCA cycle through the PDH-dependent generation of acetyl-CoA, which combines with oxaloacetate to form citrate. Alternatively, pyruvate can contribute its full 3 carbons to the TCA cycle by generating oxaloacetate through the activity of PC (Figure 1K). Interestingly, both enzymes have been linked to fueling mitochondrial metabolism, cellular proliferation, and energy maintenance in a variety of model systems (Cardaci et al., 2015; Christen et al., 2016; Luengo et al., 2021; Sellers et al., 2015; Woolbright et al., 2019), but their role in CD8+ T cell behavior has not been determined. We observed differential activity of the two pyruvate converting enzymes, PC and PDH, in cytolytic T cell co-cultures compared with suppressed T cell co-cultures in CM. While CD8+ T cells co-cultured in RPMI displayed increased contribution through both PC (M+3) and PDH (M+2) to the TCA cycle intermediates malate and fumarate, CD8+ T cells in co-culture in CM only exhibited an increase in carbons derived from PDH (Figure 1L–M; Figure S1M–N). To estimate the relative change in PC activity, we directly compared malate M+3 with succinate M+3. While malate M+3 can derive from either an oxidative TCA cycle or PC activity, succinate M+3 is mainly generated by an oxidative TCA cycle, as reverse succinate dehydrogenase (SDH) flux is minimal or absent (Buescher et al., 2015). We confirmed CM inhibited PC activity in CD8+ T cells in co-culture with tumor cells (Figure S1L; 1O). Consistent with the effect of CM on cytotoxicity, the inhibitory effect of CM on PC activity was driven through a mechanism that was not linked to antigen-dependent signaling, as the same phenotype was observed in CD8+ T cells in co-culture with B16 cells not expressing OVA (Figure S1P).
In summary, these results demonstrate that the presence of tumor cells can stimulate antigen-specific CD8+ T cell IFNγ and GzmB production and alter CD8+ T cell metabolism but not affect Ki67 expression. However, CM inhibits IFNγ and GzmB production in CD8+ T cells as well as PC activity in an antigen-independent fashion, demonstrating that nutrient composition shaped by tumor cells can inhibit CD8+ T cell effector function and lead to metabolic reprogramming.
Lactate secretion by tumor cells rewires CD8+ T cell metabolism
To identify specific metabolite changes that may account for suppressed CD8+ T cell activity in CM, we directly compared the metabolite composition of CM versus RPMI (Figure 2A). While most of the nutrients detected in RPMI were lower in CM, the levels of lactate were significantly enriched in CM and reached 10 mM (Figure 2A–B). To identify whether lactate would accumulate to the same extent in the in vivo TME, tumor interstitial fluid (TIF) and plasma were isolated from B16-OVA tumor implanted mice (Figure 2C). Lactate levels accumulated to 11 mM in the TIF, while they were 1 mM in plasma. These TIF lactate levels were similar to those observed in CM (Figure 2D). These data confirm that lactate accumulation is a feature of the immunosuppressive TME and suggest tumor-derived lactate as a possible driver of CD8+ T cell metabolic rewiring.
Figure 2. Lactate secretion by tumor cells rewires CD8+ T cell metabolism.
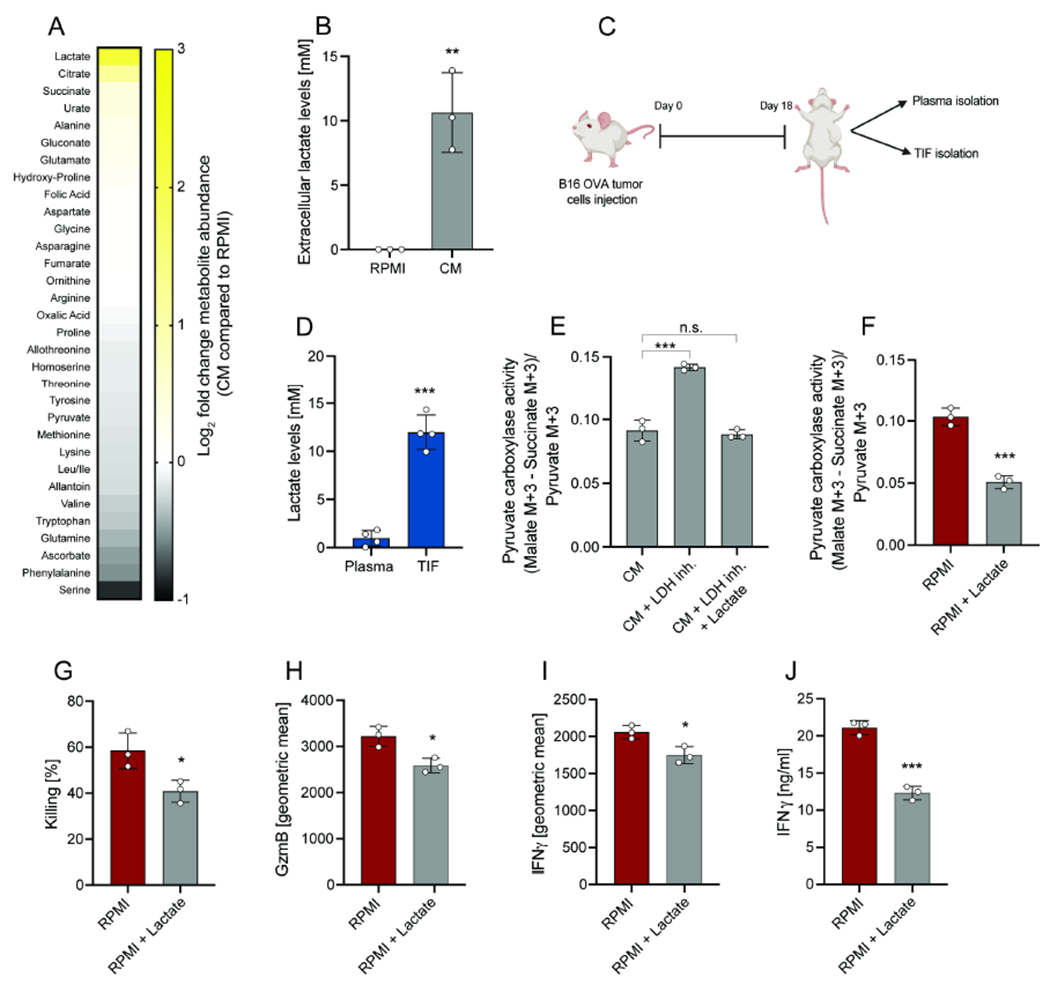
(A) Heatmap representing metabolite changes in tumor CM compared to RPMI. CM was obtained after 24 hours culture of tumor cells. (B) Absolute extracellular lactate levels in RPMI and CM. CM was obtained after 24 hours culture of tumor cells. (C) Schematic representation of plasma and TIF isolation from mice bearing B16-OVA tumors. Analysis was performed at day 18 after tumor implantation. (D) Absolute lactate levels in TIF of mice with B16-OVA tumors. (E) PC activity of CD8+ T cell in co-culture with B16-OVA tumor cells in CM, in CM obtained from tumor cells cultured with LDH inhibitor or in CM obtained from tumor cells cultured with LDH inhibitor + supplementation of lactate. Analysis was performed after 6 hours. (F) PC activity of CD8+ T cell in co-culture with B16-OVA tumor cells in RPMI and RPMI + lactate (10mM). Analysis was performed after 6 hours. (G-I) Killing and and geometric mean of GzmB and IFNγ expression by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI or RPMI + lactate (10mM). Analysis was performed after 24 hours. (J) Secreted IFNγ levels by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI or RPMI + lactate (10mM). Analysis was performed after 24 hours. The number of biological replicates for each experiment was n≥3. Error bars represent s.d. Two-tailed unpaired Student’s T-test was performed. *P<0.05; **P<0.01; ***P<0.001. See also Figure S2.
Thus, we treated the tumor cells with the lactate dehydrogenase (LDH) inhibitor sodium oxamate, which resulted in a block in lactate generation and secretion. CM from LDH-inhibitor treated cells lacked lactate and did not inhibit PC activity in T cell co-cultures (Figure 2E). However, the addition of lactate to CM or RPMI was sufficient to impair PC activity in CD8+ T cell co-cultures (Figure 2E–F). Notably, CD8+ T cell killing, as well as GzmB and IFNγ production, were all reduced in high lactate media (Figure 2G–J; Figure S2A).
Taken together, these data suggest that tumor-derived lactate leads to decreased PC activity and diminished cytolytic activity in CD8+ T cells.
Inhibition of PDH increases PC activity and CD8+ T cell cytotoxicity through TCA cycle anaplerosis
To test a possible causal link between the activities of PC and PDH and CD8+ T cell effector functions, we next modulated both branches of pyruvate oxidation (Figure 3A). First, we used a shRNA targeting PC (Figure S2B) and found that PC knockdown (KD) in CD8+ T cells was sufficient to decrease CD8+ T cell killing, GzmB, and IFNγ production (Figure 3B–E; Figure S2C–D), indicating that PC activity is a requisite for maximal CD8+ T cell function.
Figure 3. Inhibition of PDH increases PC activity and CD8+ T cell cytotoxicity through TCA cycle anaplerosis.
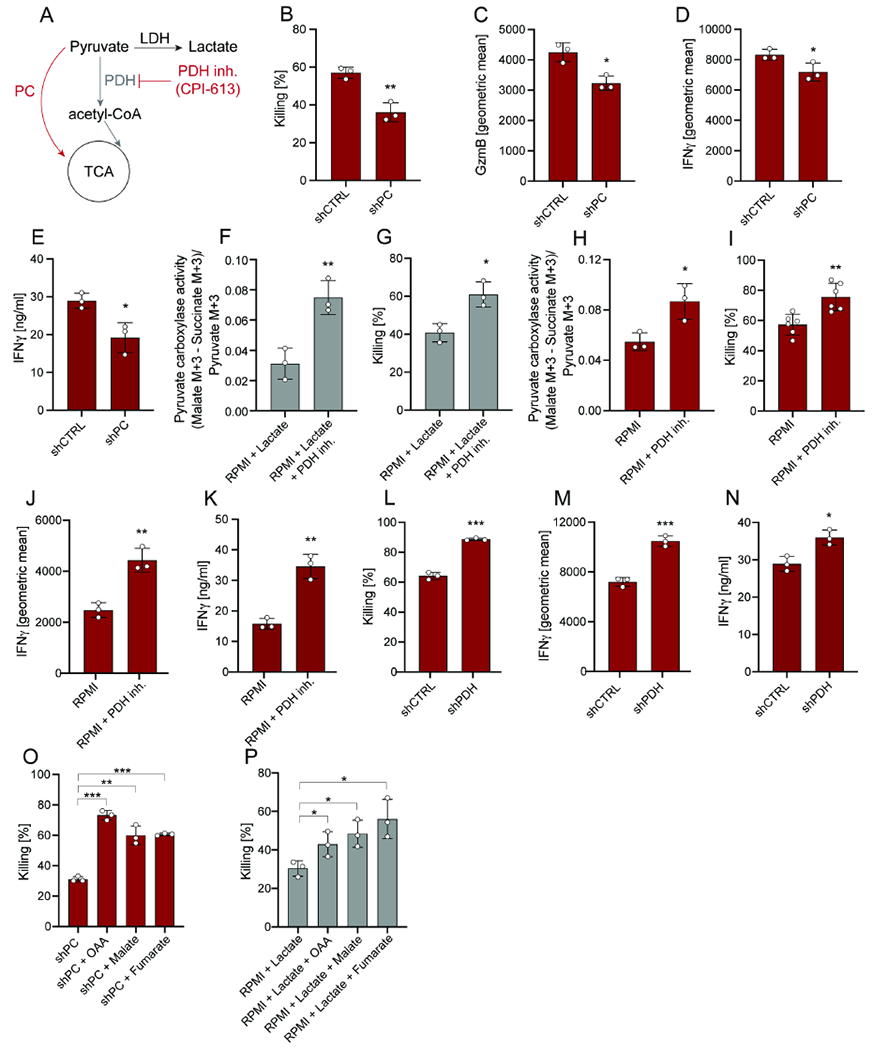
(A) Schematic representation of PC and PDH activity. Inhibition of PDH is achieved by using the inhibitor CPI-613 at the concentration of 100μM. (B-D) Killing and geometric mean of GzmB and IFNγ expression by CD8+ T cells transduced with shRNA for PC or CTRL and cultured with B16-OVA tumor cells in RPMI. Analysis was performed after 24 hours. (E) Secreted IFNγ levels by CD8+ T cells transduced with shRNA for PC or CTRL and cultured with B16-OVA tumor cells in RPMI. Analysis was performed after 24 hours. (F) PC activity of CD8+ T cells in co-culture with B16-OVA tumor cells pre-treated with or without the PDH inhibitor CPI-613 in RPMI + lactate (10mM). CD8+ T cells were pre-treated with the PDH inhibitor for 24 hours (prior co-culture). Analysis was performed after 6 hours. (G) Killing by CD8+ T cells in co-culture with B16-OVA tumor cells pre-treated with or without the PDH inhibitor CPI-613 in RPMI + lactate (10mM). CD8+ T cells were pre-treated with the PDH inhibitor for 24 hours (prior co-culture). Analysis was performed after 24 hours. (H) PC activity of CD8+ T cells in co-culture with B16-OVA tumor cells pre-treated with or without the PDH inhibitor CPI-613 in RPMI. CD8+ T cells were pre-treated with the PDH inhibitor for 24 hours (prior co-culture). Analysis was performed after 6 hours. (I-J) Killing and geometric mean of IFNγ expression by CD8+ T cells in co-culture with B16-OVA tumor cells pre-treated with or without the PDH inhibitor CPI-613 in RPMI. CD8+ T cells were pre-treated with the PDH inhibitor for 24 hours (prior co-culture). Analysis was performed after 24 hours. (K) Secreted IFNγ levels by CD8+ T cells in co-culture with B16-OVA tumor cells pre-treated with or without the PDH inhibitor CPI-613 in RPMI. CD8+ T cells were pre-treated with the PDH inhibitor for 24 hours (prior co-culture). Analysis was performed after 24 hours. (L-M) Killing and and geometric mean of IFNγ expression by CD8+ T cells transduced with shRNA for PDH or CTRL in co-culture with B16-OVA tumor cells in RPMI. Analysis was performed after 24 hours. (N) Secreted IFNγ levels by CD8+ T cells transduced with shRNA for PDH or CTRL in co-culture with B16-OVA tumor cells in RPMI. Analysis was performed after 24 hours. (O) Killing by CD8+ T cells transduced with shRNA for PC and in co-culture with B16-OVA tumor cells in RPMI +/− supplementation with OAA, malate, or fumarate. Analysis was performed after 24 hours. (P) Killing by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI + lactate (10mM) +/− supplementation of OAA, malate, or fumarate. Analysis was performed after 24 hours. The number of biological replicates for each experiment was n≥3. Error bars represent s.d. Two-tailed unpaired Student’s T-test was performed. *P<0.05; **P<0.01; ***P<0.001. See also Figure S2 and S3.
Because PC and PDH activities control the nodal branchpoint of pyruvate entry into mitochondria, we tested whether PDH inhibition would increase pyruvate availability for PC, thus promoting CD8+ T cell cytotoxicity even in the presence of lactate. To test this idea, we utilized the PDH inhibitor CPI-613 (Pardee et al., 2018; Philip et al., 2019) (Figure 3A; Figure S3A). We performed 13C6-glucose tracing in the presence or absence of CPI-613 and measured PC activity. Interestingly, inhibition of PDH was sufficient to rescue PC activity and CD8+ T cell killing in co-cultures supplemented with 10 mM lactate (Figure 3F–G). Furthermore, treatment with CPI-613 further increased PC activity, CD8+ T cell killing, and IFNγ, but not GzmB, production in RPMI co-cultures (Figure 3H–K, Figure S3B–C). Similar to our earlier observations, PDH inhibition did not affect proliferation as measured by Ki67 expression (Figure S3D). The effect of PDH inhibition on CD8+ T cell killing and IFNγ production was independently validated in the MC38-OVA model. Furthermore, genetic inhibition of PDH using shRNA yielded similar results (Figure 3L–N; Figure S3E–K).
We next probed whether high levels of pyruvate resulting from PDH inhibition increased lactate production through LDH in CD8+ T cells. 13C6-glucose tracing demonstrated that the inhibition of PDH does not increase glucose-derived pyruvate flux to lactate (Figure S3L), and that the LDH inhibitor sodium oxamate had no effect on killing efficiency of CD8+ T cells in either absence or presence of the PDH inhibitor (Figure S3M).
Next, we sought to understand how PC activity supports CD8+ T cell cytotoxicity and IFNγ production. A functional TCA cycle requires the continuous and balanced replenishment of its nine core metabolites, as many exit the mitochondrion and act as precursors for biosynthetic or signaling reactions. Since the citrate synthase reaction, which converts oxaloacetate to citrate, is irreversible and succinate dehydrogenase A (SDHA) flux is minimal or not present, the activity of PC has been mainly linked to the replenishment of fumarate, malate, and oxaloacetate (Figure 1K). In accordance, we demonstrate that in CD8+ T cells PC-derived carbons label these metabolites (Figure 1L–M). Supplementation of culture media with oxaloacetate, malate, or fumarate fully rescued CD8+ T cell cytotoxicity upon inhibition of PC by KD or tumor-derived lactate (Figure 3O–P). Finally, CD8+ T cells with PC KD or cultured in CM could not be rescued by supplementation with α-ketoglutarate (Figure S3N–O). Thus, these results indicate that PC activity is needed to replenish TCA cycle intermediates to sustain CD8+ T cell cytotoxicity.
PC activity enables succinate secretion and CD8+ T cell function
As CD8+ T cell cytolytic activity hinged on PC-dependent anaplerosis of TCA cycle intermediates, we next investigated whether these metabolites might exit actively cytolytic CD8+ T cells. Surprisingly, when analyzing the media composition of CD8+ T cells grown in mono- and co-culture with tumor cells, we found succinate to be actively secreted in the presence of tumor cells, whereas this effect was inhibited by CM (Figure 4A, Figure S4A). As PC can generate all TCA cycle intermediates that are normally derived from succinate, we next measured the ability of CD8+ T cells to convert succinate into fumarate, malate, and oxaloacetate through the activity of SDHA (Figure 1K). As expected, we observed a strong decrease in SDHA activity in CD8+ T cells engaged in cytolysis of tumor cells compared to T cells grown as monocultures (Figure 4B).
Figure 4. PC activity enables succinate secretion and CD8+ T cell function.
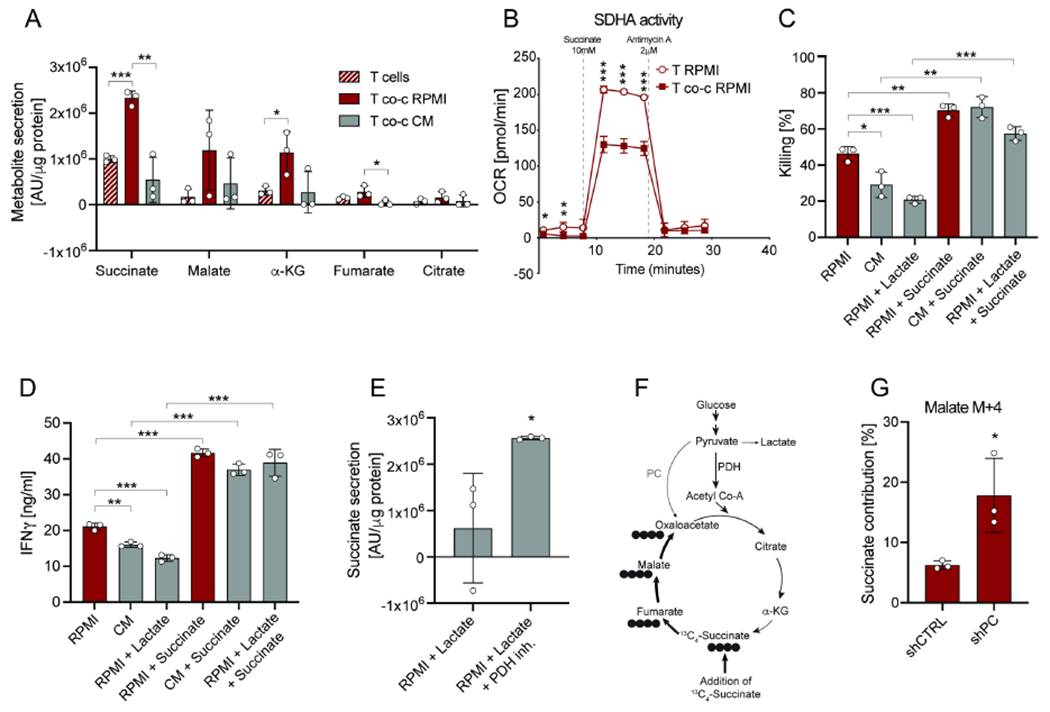
(A) Metabolite secretion in RPMI and CM from CD8+ T cells in mono-culture or in co-culture with B16-OVA tumor cells. Analysis was performed after 6 hours. (B) SDHA activity of CD8+ T cells in mono-culture and CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI. Analysis was performed after 6 hours. To determine SDHA activity, oxygen consumption rate was measured after pre-treatment with rotenone and treatment with succinate. (C) Killing by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI, CM and RPMI + lactate (10mM) +/− supplementation of dimethyl-succinate. Analysis was performed after 24 hours. (D) Secreted IFNγ levels by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI, CM and RPMI + lactate (10mM) +/− supplementation of dimethyl-succinate. Analysis was performed after 24 hours. (E) Succinate secretion by CD8+ T cells in RPMI + lactate (10mM) upon pre-treatment with the PDH inhibitor. CD8+ T cells were pre-treated with the PDH inhibitor for 24 hours (prior co-culture). Analysis was performed after 6 hours. (F) Schematic of 13C4-succinate labeling into the TCA cycle. (G) 13C4-succinate contribution to malate M+4 in CD8+ T cells transduced with shRNA for PC or CTRL and co-cultured with B16-OVA tumor cells in RPMI. Analysis was performed after 6 hours. The number of biological replicates for each experiment was n≥3. Error bars represent s.d. Two-tailed unpaired Student’s T-test was performed. *P<0.05; **P<0.01; ***P<0.001. See also Figure S4.
The downregulation of SDHA activity is in line with our data showing the crucial role of PC replenishing the left arm of the TCA cycle (Figure 3O–P, Figure S3N–O). As we did not observe significant differences in intracellular succinate levels (Figure S4A), we next probed the effect of this metabolite by supplementing it in the extracellular media. We found that exogenous succinate significantly increased CD8+ T cell killing and IFNγ, but not GzmB, production in the absence or presence of tumor-derived lactate or CM (Figure 4C–D; Figure S4B–F). Strikingly, we found PDH inhibition also rescued succinate secretion rates in the presence of lactate (Figure 4E). These results identify extracellular succinate as a metabolite signal regulating CD8+ T cell activity.
Finally, we probed the fate of succinate inside CD8+ T cells by using 13C4-succinate (Figure 4F). CD8+ T cells grown in RPMI-based co-cultures only partially incorporated 13C4-succinate into the TCA cycle intermediate malate (Figure 4G). As KD of PC significantly increased succinate incorporation (Figure 4G), these data suggest that PC activity increases CD8+ T cell cytotoxicity by allowing for the secretion of succinate into the extracellular space while maintaining TCA cycle flux through anaplerosis.
Extracellular succinate activates SUCNR1 and promotes CD8+ T cell cytotoxicity
As succinate was not primarily utilized to replenish the TCA cycle, we investigated the potential of this metabolite to serve as an autocrine signal to support CD8+ T cell cytotoxicity. Extracellular succinate binds to the succinate receptor SUCNR1 on the cell surface and promotes a G-protein coupled signaling cascade that ultimately culminates in transcriptional changes (Gilissen et al., 2016). Although SUCNR1 has been implicated in inflammatory responses (Keiran et al., 2019; Krzak et al., 2021), a link between SUCNR1 and CD8+ T cell cytotoxicity has not been previously reported. To identify whether succinate secretion by CD8+ T cells is important for the autologous activation of this receptor, we generated a shRNA for SUCNR1 and evaluated CD8+ T cell cytotoxicity. SUCNR1 KD significantly impaired CD8+ T cell killing, as well as IFNγ production (Figure 5A–C; Figure S5A–C). Additionally, succinate supplementation was no longer able to increase CD8+ T cell killing (Figure 5A) in SUCNR1 KD T cells.
Figure 5. Extracellular succinate activates SUCNR1 and promotes CD8+ T cell cytotoxicity.
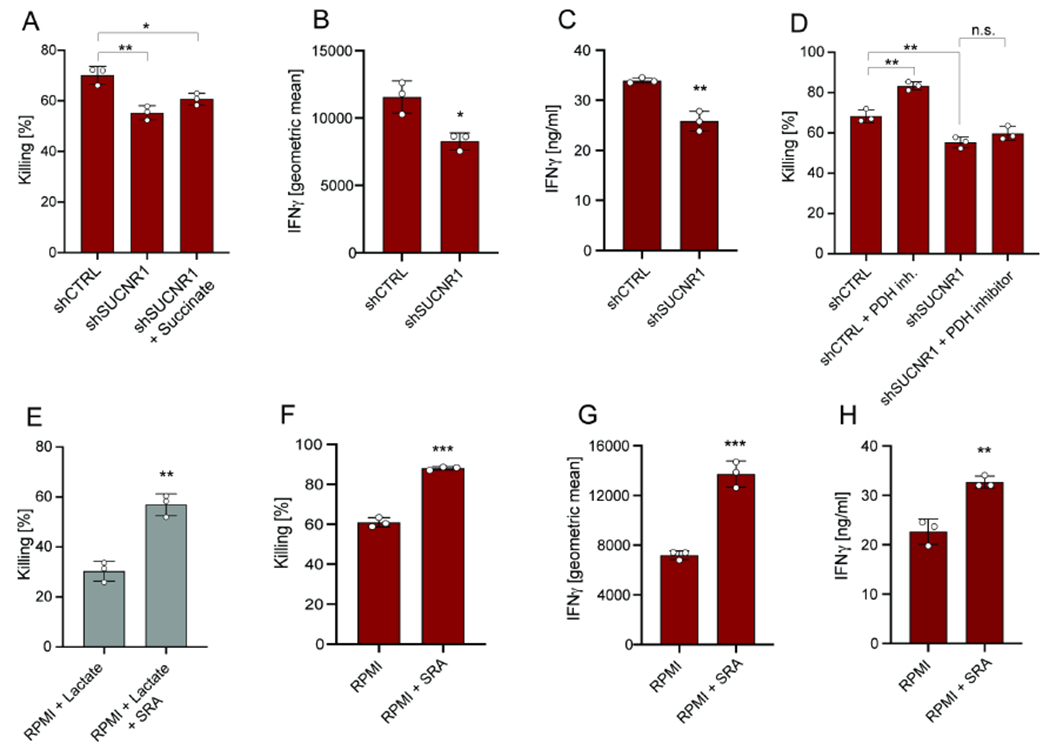
(A) Killing by CD8+ T cells transduced with shRNA for SUCNR1 or CTRL and co-cultured with B16-OVA tumor cells in RPMI +/− addition of dimethyl-succinate. Analysis was performed after 24 hours. (B) Geometric mean of IFNγ expression by CD8+ T cells transduced with shRNA for SUCNR1 or CTRL and co-cultured with B16-OVA tumor cells in RPMI. Analysis was performed after 24 hours. (C) Secreted IFNγ levels by CD8+ T cells transduced with shRNA for SUCNR1 or CTRL and co-cultured with B16-OVA tumor cells in RPMI. Analysis was performed after 24 hours. (D) Killing by CD8+ T cells transduced with shRNA for SUCNR1 or CTRL and co-cultured with B16-OVA tumor cells in RPMI upon pre-treatment with the PDH inhibitor. CD8+ T cells were pre-treated with the PDH inhibitor for 24 hours (prior co-culture). Analysis was performed after 24 hours. (E) Killing by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI + lactate (10mM) upon pre-treatment with the succinate receptor agonist (SRA) cis-epoxysuccinic acid. CD8+ T cells were pre-treated with SRA for 24 hours (prior co-culture). Analysis was performed after 24 hours. (F-G) Killing and geometric mean of IFNγ expression by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI following pre-treatment with the SRA cis-epoxysuccinic acid. CD8+ T cells were pre-treated with SRA for 24 hours (prior co-culture). Analysis was performed after 24 hours. (H) Secreted IFNγ levels by CD8+ T cells in co-culture with B16-OVA tumor cells in RPMI following pre-treatment with the SRA cis-epoxysuccinic acid. CD8+ T cells were pre-treated with SRA for 24 hours (prior to co-culture). Analysis was performed after 24 hours. The number of biological replicates for each experiment was n=3. Error bars represent s.d. Two-tailed unpaired Student’s T-test was performed. *P<0.05; **P<0.01; ***P<0.001. See also Figure S5.
We next tested whether PDH inhibition could rescue the function of SUCNR1 KD CD8+ T cells. As shown in Figure 5D, treatment with the PDH inhibitor was unable to rescue CD8+ T cell killing in SUCNR1 KD cells. Furthermore, treatment with the succinate receptor agonist (SRA) cis-epoxysuccinic acid promoted CD8+ T cell-specific killing and IFNγ production in the presence or absence of tumor-derived lactate (Figure 5E–H; Figure S5D–H). Taken together, these data are consistent with the model that SUCNR1 signaling occurs downstream of lactate–induced metabolic rewiring in CD8+ T cells.
Inhibition of PDH in vivo increases succinate secretion in the TME and promotes anti-tumor immunity
Next, we examined whether PDH inhibition increases succinate secretion within the TME in vivo using the syngeneic B16 melanoma mouse model. We implanted B16 tumor cells in 8-week old C57BL/6 mice. On day 8, when the tumor was established and palpable, we administered the PDH inhibitor CPI-613 at 25mg/kg intraperitoneally (Figure 6A). At day 15, we compared succinate levels in the TIF (representative of TME) of tumors from mice given either vehicle or PDH inhibitor. PDH inhibition increased succinate levels specifically in the TIF, but did not affect succinate levels in plasma (Figure 6B; Figure S6A). Notably, the effect of PDH inhibition on succinate secretion was abrogated by treatment with CD8+ T cell-depleting antibodies (Figure 6B). These data indicate that the effect of the PDH inhibitor impacts succinate levels in the TME in a CD8+ T cell-dependent manner.
Figure 6. Inhibition of PDH in vivo increases succinate levels in the TME and promotes anti-tumor immunity.
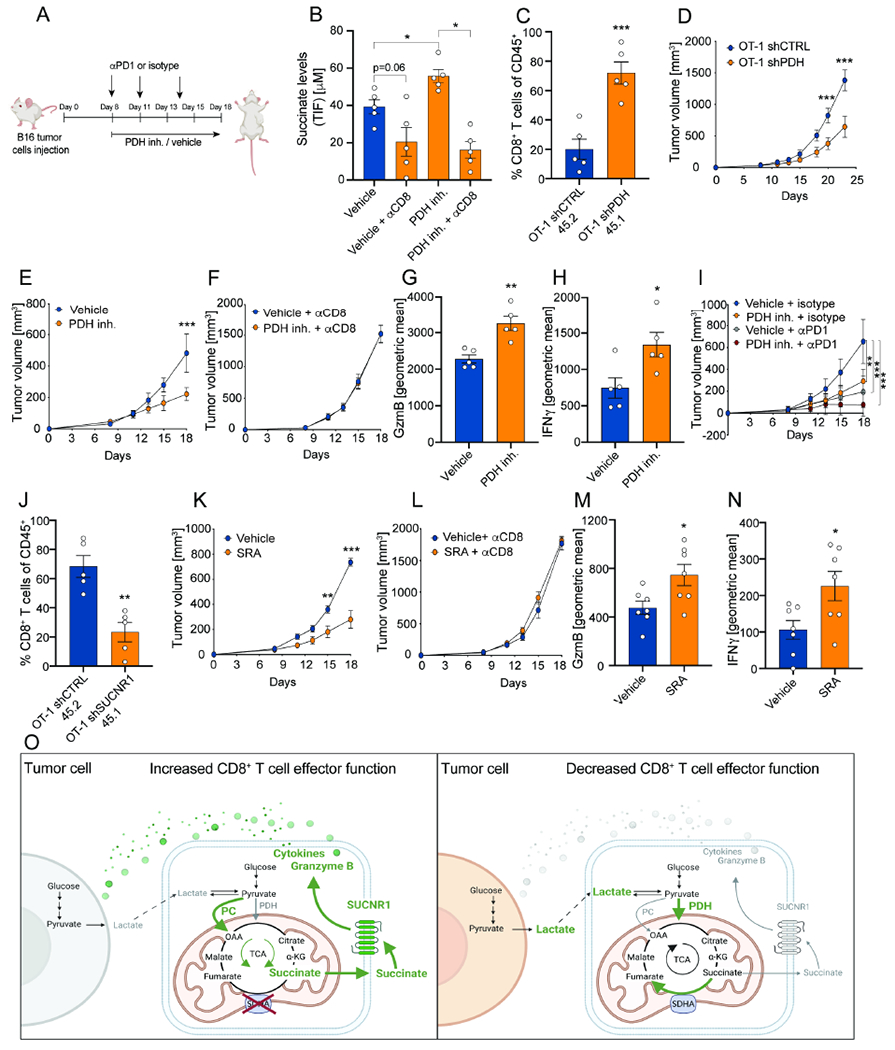
(A) Schematic representation of treatment with the PDH inhibitor CPI-613 (25mg/kg) or vehicle +/− combination with isotype control or αPD-1 in mice with B16 OVA tumors. Isotype and αPD-1 were used at 100μg dose (three doses in total). (B) Absolute succinate levels in tumor interstitial fluid from mice treated with either vehicle or the PDH inhibitor CPI-613 with or without antibodies to specifically deplete CD8+ T cells. Analysis was performed at day 18. n=5. Two-tailed unpaired Student’s T-test was performed. (C) % of OT-1 CD8+ T cells of CD45+ with the congenic markers 45.1 and 45.2 transduced with a shRNA for PDH and CTRL in B16-OVA tumors. OT-1 shPDH 45.1 and OT-1 shCTRL 45.2 were adoptively transferred using a ratio 1:1, one day prior B16-OVA tumor implantation. Analysis was performed at day 16. n=5. Two-tailed unpaired Student’s T-test was performed. (D) Average tumor growth curves showing tumor volume in mice upon adoptive transfer of OT-1 CD8+ T cells transduced with a shRNA for PDH or CTRL. Adoptive transfer was performed 9 days after tumor implantation. n=8. A two-way Anova with Sidak’s multiple comparison test was performed. (E) Average tumor growth curves showing tumor volume in mice treated with either vehicle or the PDH inhibitor CPI-613. Vehicle: n=7; PDH inh.: n=7. A two-way Anova with Sidak’s multiple comparison test was performed. (F) Average tumor growth curves showing tumor volume in mice treated with either vehicle or the PDH inhibitor CPI-613 with antibodies to specifically deplete CD8+ T cells. Vehicle αCD8: n=9; PDH inh.: n=9. A two-way Anova with Sidak’s multiple comparison test was performed. (G-H) Geometric mean of GzmB and IFNγ expression by CD8+ T cells of tumors in mice treated with either vehicle or the PDH inhibitor CPI-613. Analysis was performed at day 15. n=10. Two-tailed unpaired Student’s T-test was performed. (I) Average tumor growth curves showing tumor volume in mice treated with either vehicle or the PDH inhibitor CPI-613 and isotype control or αPD-1. Vehicle + isotype: n=9; PDH inh. + isotype: n=8; Vehicle + αPD1 : n=8; PDH inh. + αPD1: n=9. A two-way Anova with Sidak’s multiple comparison test was performed. (J) % of OT-1 CD8+ T cells of CD45+ with the congenic markers 45.1 and 45.2 transduced with a shRNA for SUCNR1 and CTRL in B16-OVA tumors. OT-1 shSUCNR1 45.1 and OT-1 shCTRL 45.2 were adoptively transferred using a ratio 1:1, one day prior B16-OVA tumor implantation. Analysis was performed at day 16. n=5. Two-tailed unpaired Student’s T-test was performed. (K) Average tumor growth curves showing tumor volume in mice treated with either vehicle or the SRA cis-epoxysuccinic acid. Vehicle: n=5; SRA: n=5. A two-way Anova with Sidak’s multiple comparison test was performed. This is a representative experiment of two independent experiments. (L) Average tumor growth curves showing tumor volume in mice treated with either vehicle or the SRA cis-epoxysuccinic acid with antibodies to specifically deplete CD8+ T cells. Vehicle αCD8: n=5; PDH inh.: n=5. A two-way Anova with Sidak’s multiple comparison test was performed. This is a representative experiment of two independent experiments. (M-N) Geometric mean of GzmB and IFNγ expression by CD8+ T cells of tumors in mice treated with either vehicle or the SRA cis-epoxysuccinic acid. Analysis was performed at day 15. n=7. Two-tailed unpaired Student’s T-test was performed. (O) Schematic representation CD8+ T cell-tumor cell metabolic circuit. On the left the metabolic activity of a metabolically fit CD8+ T cell is depicted. On the right, tumor cell metabolism impairs CD8+ T cell activity causing decreased CD8+ T cell effector function. Overall, the figure shows extracellular lactate to modulate TCA cycle entry of pyruvate through either PC or PDH, and to dictate the capacity of T cells to secrete succinate to promote cytolytic activity through the SUCNR1. Green indicates up-regulation. Grey and red indicate down-regulation. Error bars represent s.e.m. *P<0.05; **P<0.01; ***P<0.001. See also Figure S6.
Next, we tested whether PDH activity impacted CD8+ T cell tumor infiltration. To directly compare the infiltration of CD8+ T cells with or without PDH KD within the same TME, we transduced congenic marked OT-1 CD8+ T cells with shRNA for PDH or CTRL. One day prior to B16-OVA tumor implantation, we adoptively co-transferred OT-1 CD8+ T cells shCTRL 45.2 and OT-1 CD8+ T cells shPDH 45.1 and, after 16 days, we measured their numbers in B16-OVA tumors. First, we found that PDH KD OT-1 CD8+ T cells have a higher tumor-infiltrating capacity compared to control T cells (Figure 6C, Figure S6B). Second, we found that the sole transfer of OT-1 CD8+ T shPDH inhibited tumor growth when compared to the sole transfer of OT-1 CD8+ T shCTRL (Figure 6D, Figure S6C).
Finally, we validated these results using pharmacological PDH inhibition after tumors were established. Treatment with the PDH inhibitor CPI-613 significantly decreased tumor growth (Figure 6E, Figure S6D). This reduction in tumor growth was accompanied by increased GzmB and IFNγ production by CD8+ T cells (Figure 6G–H; Figure S6F–G). Furthermore, we evaluated the effect of PDH inhibition on other immune populations (Figure S6H–I). To test if PDH inhibition decreases tumor growth by a mechanism dependent solely on CD8+ T cells, we depleted CD8+ T cells using specific antibodies. Depletion of CD8+ T cells abrogated the effect of CPI-613 treatment, indicating that tumor reduction in vivo was mediated by CD8+ T cells (Figure 6F, Figure S6E). Thus, these findings suggest the PDH inhibitor metabolic treatment operates through the activation of specifically CD8+ T cells.
Next, we asked whether PDH inhibition would synergize with αPD-1 immunotherapy. We implanted B16-OVA tumors and gave three suboptimal doses of αPD-1 (100μg) or isotype with or without PDH inhibitor treatment (Figure 6A). Strikingly, we found that the combination of αPD-1 and CPI-613 further reduced tumor growth (Figure 6I, Figure S6K). We recapitulated this phenotype in a MC38 syngeneic mouse model (Figure S6L–M). Because during αPD-1 immunotherapy, progenitor T cell populations (Slamf6+) proliferate and generate terminally exhausted T cell populations (Tim-3+) (Im et al., 2016), we measured whether PDH inhibition would affect this balance. We found that PDH inhibition doesn’t significantly affect Slamf6/Tim-3 populations (Figure S6J). These data support a model in which PDH inhibitors specifically potentiate effector T cells while at the same time synergizing with immunotherapy treatment through distinct but yet complementary mechanisms.
To test the essentiality of SUCNR1 in CD8+ T cells in vivo, we transduced congenic marked OT-1 CD8+ T cells with shRNA for SUCNR1 or CTRL. One day prior to B16-OVA tumor implantation, we adoptively co-transferred OT-1 CD8+ T cells shCTRL 45.2 and OT-1 CD8+ T cells shSUCNR1 45.1 and, after 16 days, we measured their numbers in B16-OVA tumors. Notably, we found that SUCNR1 KD OT-1 CD8+ T cells demonstrated lower tumor-infiltrating capacity compared to CTRL OT-1 CD8+ T cells (Figure 6J, Figure S6B). To test the hypothesis that SUCNR1 activity limits anti-tumor cytotoxic activity of CD8+ T cells in vivo, we tested the effect of the SRA. We implanted B16 tumor cells and on day 8 we started administering either the SRA at 1mg/kg or vehicle intraperitoneally. Mice treated with the SRA had reduced tumor growth compared to mice treated with the vehicle (Figure 6K, Figure S6N). To demonstrate that the SRA targets CD8+ T cells specifically, we depleted CD8+ T cells using specific antibodies. We found that upon CD8+ T cell depletion, the effect of the SRA on tumor growth was completely abolished (Figure 6L, Figure S6O). Thus, these data demonstrate that the SRA effect is dependent on T cells. Additionally, activation of SUCNR1 was accompanied by increased CD8+ T cell IFNγ and GzmB expression (Figure 6M–N, Figure S6P).
In conclusion, we have demonstrated that the nutrient composition of the TME regulates the metabolic fitness of CD8+ T cells. In actively cytolytic CD8+ T cells, pyruvate fuels the TCA cycle through both PC and PDH. Specifically, the activity of PC is needed to replenish TCA cycle intermediates to allow for the secretion of succinate. Secreted succinate acts as a signaling molecule and activates SUCNR1 in an autocrine fashion. SUCNR1 then increases T cell cytotoxicity and thus directly links the internal metabolic program of T cells to their cytotoxic function (Figure 6O, left panel). However, the metabolic fitness of CD8+ T cells is compromised within the TME. Tumor-derived lactate changes the balance of pyruvate entry into the TCA cycle away from PC activity towards the reaction catalyzed by PDH, leading to a TCA cycle with minimal anaplerosis and inability to sustain the secretion of succinate to activate SUCNR1 (Figure 6N, right panel). Notably, our results demonstrate that TME-induced metabolic rewiring of CD8+ T cells and their cytolytic function can be restored by inhibiting PDH.
Discussion
In this study, we demonstrated that the TCA cycle exhibits a dual function in metabolically fit CD8+ T cells, not only generating metabolic intermediates, but also secreting succinate as an autocrine signaling molecule. Succinate activates the pro-inflammatory SUCNR1 receptor to drive the synthesis of inflammatory cytokines and T cell cytotoxicity. We show that metabolic stress driven by high levels of tumor-derived lactate present in the TME can reprogram CD8+ T cell pyruvate metabolism, ultimately leading to the inability to activate SUCNR1. As lactate redirects pyruvate flux from PC to PDH, CD8+ T cells operating in conditions with high lactate reactivate SDHA, leading to succinate oxidation rather than secretion. Importantly, we identify several metabolic nodal points that can be targeted to restore CD8+ T cell activity even in conditions of high lactate. Direct activation of SUCNR1 by its agonist cis-epoxysuccinic acid, or by increased PC activity and succinate secretion through inhibition of PDH can promote CD8+ T cell metabolic fitness and function in the TME. This study thus illustrates the potential of metabolic intervention to potentiate immunotherapy and lead to a better therapeutic outcome.
Previous studies have shown T cell metabolism to be essential for T cell activation and function, by providing metabolic intermediates responsible for fueling T cell proliferation and influencing the CD8+ T cell epigenome (Buck et al., 2015; Delgoffe and Powell, 2015). However, identifying the metabolic requirements for cytotoxic activity, especially in complex multicellular systems, has proven difficult (Elia and Haigis, 2021). Here, we developed a platform that allows for rapid isolation of tumor and T cell populations. This technique allowed us to separate the cell type-specific metabolic events that occur upon T cell-tumor cell interaction and to change the media composition to test the effect of certain microenvironmental factors. By using this system, we identified high levels of tumor-derived lactate present in the TME to be solely responsible for the inhibitory effect of CM on T cells. Moreover, this co-culture system allowed us to utilize T cell-dependent killing and cytokine production as a readout for CD8+ T cell effector function. As we show specifically these factors to be induced upon co-culture and inhibited by lactate, the co-culture system offers the advantage to look at the metabolic requirements of the killing process rather than activation.
Previous reports have demonstrated that lactate accumulation in B16 melanoma impairs the survival and function of CD8+ T cells and natural killer cells. Mechanistically, lactate prevents the upregulation of Nuclear Factor of Activated T cell (NFAT) and production of IFNγ (Brand et al., 2016). Another study has shown how lactate rich environments inhibit glycolysis, forcing CD8+ T cells to rely on serine biosynthesis to sustain T cell proliferation (Quinn et al., 2020). Together with our work showing an inhibitory effect of lactate on CD8+ T cell effector function, these findings suggest that highly glycolytic tumors may promote immune escape via several lactate-mediated mechanisms.
SUCNR1 and its downstream signaling targets have been studied as a critical regulator of several physiological processes, including immune responses (Gilissen et al., 2016; Keiran et al., 2019; Krzak et al., 2021; Reddy et al., 2020). Despite numerous studies of SUCNR1 in inflammation, very little attention has been given to the role of this receptor in the context of cancer. Here, we show that cytotoxic CD8+ T cells secrete succinate to activate their SUCNR1 receptor and promote cytotoxicity. These results thus further explain the role of SUCNR1 as a pro-inflammatory receptor in CD8+ T cells. It is however not known whether other immune cell types rely on succinate secretion for the autocrine activation of the receptor. Furthermore, given that high lactate concentrations also inhibit macrophage activity (Keiran et al., 2019), it will be interesting to test whether the observed lactate-induced switch from PC to PDH also modulates SUNCR1 signaling in other immune cell types. Finally, further studies will be necessary to elucidate the signaling pathways downstream of SUCNR1 in cancer. Investigating how these pathways modulate cytotoxic activity will not only shed light on how cellular metabolism fulfills the distinct requirements of proliferation and cytotoxicity but also help understand how metabolic receptors can be used as sensors for the metabolic fitness of CD8+ T cells.
As we identified specific nodal points linking the nutrient microenvironment to CD8+ T cell activity through a metabolic regulation of SUCNR1 signaling, we also validated metabolic targets that can potentiate CD8+ T cell fitness within this hostile environment and thus override the inhibitory effect of the TME. Notably, redirecting pyruvate flux from PDH to PC or direct activation of SUCNR1 can overcome lactate-induced CD8+ T cell inhibition. Additionally, we show that PDH inhibition synergizes with αPD-1 immunotherapy by specifically potentiating T effector cells. These findings have immediate therapeutic implications as the PDH inhibitor we used is currently in clinical trials (Pardee et al., 2018; Philip et al., 2019). Finally, targeting the production of lactate might be an alternative therapeutic approach (Yuan et al., 2021).
In summary, we conclude that mapping T cell metabolism within conditions that mimic the TME can provide valuable insights and identify new targets to improve T cell function within this hostile environment.
Limitations of the study
In this study, we demonstrated metabolic rewiring of CD8+ T cells in conditions mimicking the TME in vitro. However, in vivo tracing of tumor-infiltrating lymphocytes still presents several technical challenges in the field, mainly due to the long isolation process of T cells from tumors that inevitably affects metabolic readouts. Furthermore, it is a technical challenge to obtain high numbers of T cells from tumors for conventional biochemistry. Thus, to probe the relevance of our findings in vivo, we modulated the most important metabolic nodes in vivo and measured T cell effector functions.
Additionally, we identified SUCNR1 as a sensor for the metabolic fitness of CD8+ T cells responsible for balancing CD8+ T cell cytotoxic output with nutrient availability. In the future, to determine how this receptor affects CD8+ T cell function in cancer, a focused characterization of the signaling pathways downstream of SUCNR1 will be required.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marcia C. Haigis (marcia_haigis@hms.harvard.edu).
Materials availability
Plasmids generated in this study will be made available by request to the lead contact.
Data and code availability
All relevant data are available from the corresponding author on request. This paper does not report original codes.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
B16 cells and MC38 cells expressing chicken ovalbumin (B16-OVA and MC38-OVA) and a GFP marker were generated by retroviral transduction as previously described (Drijvers et al., 2020). Both cell lines were grown in RPMI-1640 (Thermo Fisher Scientific; cat. 11875-119) supplemented with 10% dialyzed FBS (Thermo Fisher Scientific; cat. 1743489), 100 units/ml penicillin and 100 mg/ml streptomycin (Thermo Fisher Scientific; cat. 5140-122), 10 mmol/l HEPES (Thermo Fisher Scientific; cat. 15630-130), 1 mmol/l sodium pyruvate (Thermo Fisher Scientific; cat. 11360-070), and 55 mmol/l 2-mercaptoethanol (Thermo Fisher Scientific; cat. 21985–023). B16-OVA cells and MC38 cells without GFP were used for in vivo experiments (Drijvers et al., 2020). Platinum-E cells were selected in 1 μg/ml puromycin and 10 μg/ml blasticidin (Thermo Fisher Scientific; cat. A11139-03). B16-OVA, MC38 and Platinum-E cells were cultured in DMEM (Thermo Fisher Scientific; cat. 11965-118) supplemented with 10% FBS (MilliporeSigma; cat. F2442), 100 units/ml penicillin and 100 mg/ml streptomycin (Thermo Fisher Scientific; cat. 5140-122). All cells were confirmed Mycoplasma negative in the year of submission of the manuscript using PCR-based assays.
Mice
8 weeks WT C57BL/6J (cat. 000664) and OT-1 (C57BL/6J-Tg(Tcra/Tcrb)1100Mjb/J) (cat. 003831) mice were purchased from The Jackson Laboratory or bred in our animal facility. These mice were used to generate congenially marked (CD45.1/2) OT-1 mice. All mice were maintained in a pathogen-free facility and used according to institutional and National Institutes of Health guidelines. Harvard Medical School is accredited by the American Association of Accreditation of Laboratory Animal Care.
METHOD DETAILS
CD8+ T cell isolation and cultures
Primary naïve CD8+ T cells were purified from spleens of OT-1 mice using a naïve CD8+ T cell isolation kit (Miltenyi Biotec; cat. 130-096-543). The isolated naïve CD8+ T cells were activated for 72 hours at 37 °C in plates coated with 2 μg/ml anti-CD3 (clone 145-2C11; Bio X Cell; cat. BE0001-1) and 2 μg/ml anti-CD28 (clone 37.51; Bio X Cell; cat. BE0015-1) in the presence of 100 units/ml IL-2 (R&D Systems; cat. 202-IL-050) and 10 ng/ml IL-12 (PeproTech; cat. 210-12-50 mg). CD8+ T cells were cultured either in RPMI-1640 (Thermo Fisher Scientific, cat. 11875-119) supplemented with 10% FBS (MilliporeSigma; cat. F244), 100 units/ml penicillin and 100 mg/ml streptomycin (Thermo Fisher Scientific; cat. 15140-122), 10 mmol/l HEPES (Thermo Fisher Scientific; cat. 15630-130), 1 mmol/l sodium pyruvate (Thermo Fisher Scientific; cat. 11360-070), and 55 mmol/l 2-mercaptoethanol (Thermo Fisher Scientific; cat. 21985–023) or CM. CM was obtained from tumor cells cultured for 24 hours and filtered using 0.2 μm filters. Sodium lactate (Sigma; cat. 867-56-1) was added at the concentration of 10 mM; dimethyl succinate (Sigma; cat. 106-65-0) and dimethyl α-ketoglutarate (Sigma; cat. 349631) were added at the concentration of 500 μM; sodium fumarate (Sigma; cat. F1506), malic acid (Sigma; cat. M7397) and oxaloacetic acid (Sigma; O4126) were added at the concentration of 4 mM.
Killing, proliferation, IFNγ and GzmB assays
For killing, proliferation (Ki67), IFNγ and GzmB assays 15,000 tumor cells were plated in 96-well plates and transferred to a 37 °C incubator to let them attach. 24 hours later, a test plate was counted and pre-activated OT1 CD8+ T cells were plated on top using a 1:5 ratio (CD8+ T cells : tumor cells). Antigen-specific killing was evaluated by co-culturing pre-activated CD8+ T cells with B16-OVA while non antigen-specific killing was evaluated by co-culturing pre-activated CD8+ T cells with B16 not expressing OVA. All assays were performed after co-culture for 24 hours. For killing assays, plates were trypsinized, tumor cells and CD8+ T cells were resuspended in PBS (Invitrogen; cat. 14190-250) and analyzed by flow cytometry to determine the numbers of cancer cells and T cells. The percentage of killing was calculated as previously described(Drijvers et al., 2020). For proliferation (Ki67), IFNγ and GzmB analyses, single cell suspensions of CD8+ T cells were prepared by mechanical dissociation, resuspended in staining buffer (PBS containing 1 % FBS and 2 mM EDTA) with the indicated antibodies, and analyzed by flow cytometry (see flow cytometry and flow sorting).
Inhibitor assay
For studies using inhibitors, after 48 hours of activation with anti-CD3/anti-CD28, IL-2, and IL-12, CD8+ T cells were pre-treated with specific inhibitors for 24 hours prior to co-culture (Figure S4A). CPI-613 (APExBIO; cat. A4333) was used at the concentration of 100 μM. Cis-epoxysuccinic acid (Thermo Fisher Scientific; cat. AC372640050) was used at the concentration of 50 μM. Sodium oxamate (Sigma; cat. 02751) was used at the concentration of 10 mM.
shRNA knockdowns
Platinum-E cells were transfected with pMKO.1 plasmids containing a GFP selection marker and specific shRNA. Transfection was achieved by using Fugene 6 (VWR/Promega; cat. E2691) and DNA mixture at a 1:3 DNA : Fugene ratio in Opti-MEM I reduced serum medium (Life Technologies; cat. 31985-062) for 8 hours. Retroviral supernatants were collected after 24 and 48 hours transfections and filtered through 45 μm filters. Naïve CD8+ T cells were isolated using a naive CD8αβ T-cell isolation kit (Miltenyi Biotec; cat. #130-096-543) and activated on plates coated with 2 μg/ml anti-CD3 and 2 μg/mL anti-CD28 in the presence of 200 units/ml IL-2 (R&D Systems; cat. #202-IL-050) for 24 hours at 37 °C prior to transduction. Transductions were performed by spinfecting T cells at 300 x g for 120 minutes at 37 °C in presence of the retroviral supernatants and polybrene (Santa Cruz, cat. Sc-255611). The cells were incubated at 37 °C, 5% CO2 for 2 hours before washing off the virus. The T cells were expanded for 72 hours in media containing 100 units/ml IL-2 (R&D Systems; cat. 202-IL-050) and 10 ng/ml IL-12 (PeproTech; cat. 210-12-50 mg). Transduced CD8+ T cells were selected by flow-cytometric sorting based on GFP expression.
RNA isolation and quantitative real-time PCR
Total RNA was isolated with the Direct-zol RNA Miniprep Kit (BD Biosciences; cat. R2050). RNA (500 ng) was reverse transcribed into cDNA using a High-Capacity cDNA Reverse Transcription Kit (Genesee Scientific; cat. 11-327). The analyzed genes were: PC (Fw: GCCCAGAAGTTGCTACATTACCT; Rv: CTCACATTGACAGGGATTGGA), PDH (Fw: TGTGACCTTCATCGGCTAGAA; Rv: TGATCCGCCTTTAGCTCCATC), and SUCNR1 (Fw: CCATCTCTGACTTTGCTTTCCTG; Rv: GTGTAGAGGTTGGTGTGAAGCAC). The relative levels of gene transcripts compared to the control gene RPL-19 (Fw: CAGGCATATGGGCATAGGGAA; Rv: TGCCTTCAGCTTGTGGATGT) were determined by quantitative real-time PCR using SYBER Green PCR Master Mix (Quanta BioSciences; cat. 101414-270). Amplification was performed at 95 °C for 10 minutes, followed by 40 cycles of 15 s at 95 °C and 1 minute at 60 °C. The fold change in gene expression was calculated as: Fold change= 2−DDCt.
Flow cytometry and flow sorting
Single-cell suspensions of tumor infiltrating lymphocytes and CD8+ T cells were prepared by mechanical dissociation and resuspended in staining buffer (PBS containing 1 % FBS and 2 mM EDTA) and stained with the indicated antibodies. Antibodies: BUV395 anti-mouse CD45.2 (BD Biosciences; cat. 564616); APC anti-mouse CD3 (BioLegend; cat. 100236); BV421 anti-mouse CD8b (BioLegend; cat. 126629); PE-Cy7 anti-mouse CD8b (BioLegend; cat. 126616); BV605 anti-mouse CD11b (BioLegend; cat. 101237); PE/Cy7 anti-mouse IFNγ (BioLegend; cat. 505826); APC anti-mouse IFNγ (BioLegend; cat. 505809); FITC anti-mouse GzmB (BioLegend; cat. 515403); APC anti-mouse GzmB (BioLegend; cat. 396407); PerCP/Cy5.5 anti-mouse Ki67 (BioLegend; cat. 652424); BV605 anti-mouse Ki-67 (BD Biosciences; cat. 567122); BV711 anti-mouse Tim3 (Biolegend; cat. 119727), BUV563 anti-mouse Slamf6 (Biolegend, cat. 134610); BUV496 anti-mouse CD4 (BD Biosciences, cat. 612952); eF50 anti-mouse Foxp3 (ThermoFisher, cat. 48-5773-82). APC/Cy7 LIVE/DEAD fixable near-IR dead cell stain kit (Thermo Fisher Scientific; cat. L34976) was used to determine cell viability. Flow cytometry was performed on BD LSR II and BD FACS Symphony machines. FACS sorting was conducted on a BD Aria II or MoFlo Astrios EQ. Flow analyses were performed using FlowJo 10.6.1 software (TreeStar). All flow antibodies were used at a 1:200 dilution. UltraComp beads (Thermo Fisher Scientific; cat. 01-2222-42) were used for compensation.
Elisa assay
CD8+ T cells supernatants were utilized to measure IFNγ. The concentration of IFNγ was assessed with an ELISA kit (Biolegend, cat. 430804) following the manufacturer’s instructions.
SDHA activity assay
SDHA activity assay was performed after 6 hours of (co-)culture upon activation. The assay was performed as previously described (Salabei et al., 2014).
Rapid separation of CD8+ T cells and tumor cells from co-culture plates
CD8+ T cells were transferred with a 1,000 ml pipette from the co-culture plates to a cell strainer of 10 μm size (PluriSelect USA; cat. 43-50010-50) to retain any tumor cells detached from the plate. Cells were collected in 50 ml falcon tubes and pelleted by centrifugation (300 x g, 2 minutes, room temperature) to remove excess media. Six wells of one six-well plate were pooled into one falcon tube (one replicate). Medium was aspirated by using a vacuum line and the remaining cell pellet was immediately quenched as previously described (Elia et al., 2017).
Metabolite extraction and liquid chromatography-mass spectrometry analysis
Metabolites were extracted from the pellet by adding 800 μl 60% methanol (Fisher Scientific, cat. A456-500) containing 6.67 μg/ml glutaric acid (Sigma-Aldrich, cat. G3407) in LC/MS grade water (Fisher Scientific, cat. W6500). After addition of 500 μl chloroform (Sigma-Aldrich, cat. C2432), samples were vortexed for 10 minutes at 4 °C and then spun at 17,000 x g for 10 minutes at 4 °C. The upper phase was transferred into a fresh tube and dried down in a Vacufuge plus speed-vac at 4 °C. The metabolite extract was separated using an iHILIC-(P) Classic column (2.1 μm, 150 mm × 2.0 mm I.D., The Nest Group) coupled to a Thermo Scientific SII UPLC system. The autosampler and column oven were held at 4 °C and 25 °C, respectively. The iHILIC-(P) Classic column was used with buffer A (0.1% ammonium hydroxide, 20 mM ammonium carbonate) and buffer B (100% acetonitrile). The chromatographic gradient was run at a flow rate of 0.150 ml/minute as follows: 0–20 minutes: linear gradient from 80% to 20% B; 20–20.5 minutes: linear gradient from 20 % to 80 % B; 20.5–28 minutes: hold at 80% B. The mass spectrometer was operated in full scan, negative ion model. Mass spectrometry detection was carried out on a Q Extractive HF-X orbitrap mass spectrometer with a HESI source. For metabolite quantification TraceFinder software (ThermoFisher) was used. The lower phase after chloroform extraction was also dried down in a Vacufuge plus speed-vac at 4 °C and proteins were resuspended in 0.2M NaOH and heated at 90 °C for 15 minutes. The protein concentration was determined using the Pierce BCA Protein Assay Kit Assay (ThermoFisher, cat. 23227). The metabolite levels were normalized by the total protein amount per sample in μg. GraphPad Prism 9 software was used for statistical analysis.
In vivo drug treatment
8 week old C57BL/6J female WT mice were anesthetized with 2.5% 2,2,2-tribromoethanol (MilliporeSigma; cat. T48402-25 g) and 250,000 B16/MC38-OVA tumor cells without fluorescent label were injected in the flank subcutaneously. For inhibition of PDH, mice were injected intraperitoneally with the inhibitor CPI-613 at 25 mg/kg dose. CPI-613 was dissolved in a mix containing 1.5% DMSO, 60% PBS, 35% polyethylene glycol (PEG) and 5% Tween. The same mix without drug was injected as a vehicle to control animals. For activation of SUCNR1, mice were injected with the SRA cis-epoxysuccinic acid at 1mg/kg dose. SRA was dissolved in PBS. PBS was injected as a vehicle to control animals. Treatment started at day 8 after tumor implantation and continued until tumors reached 2000 mm3. Depletion with CD8+ T cells was achieved by administering anti-mouse CD8β antibodies (BioXCell; cat. BE0223) at 300 μg dose at day 0 then 200 μg dose at day 2, 5, 8 and 12. αPD-1 (CD279) (clone 29F.1A12, BioXCell; cat. BE0273) and isotype IgG2a (clone 2A3, Bio X Cell; cat. BP0089) were administered at day 8, day 11 and day 14 at 100 μg dose for B16 melanoma mice and at day 11 for MC38 mice. Tumor volume was measured with a caliper every 2 to 3 days and calculated using the formula ½ D x d2, where D is the longer diameter and d is the shorter diameter. Tumor volume was measured in all animals until day 31 after tumor implantation. In the figures, average growth curves are displayed until one of the animals exhibited a tumor volume of 2000 mm3 upon which it was sacrificed. Metabolic composition of tumor interstitial fluid and plasma were analyzed at day 18 as previously described (Elia et al., 2017). Tumor dissociation and isolation of tumor infiltrating lymphocytes were performed as previously described (Drijvers et al., 2020).
In vivo adoptive transfer
OT-1 CD8+ T cells were isolated from the spleen of congenic marked 45.1 and 45.2 mice. The use of congenic marked mice allows to distinguish, through flow cytometry, adoptively transferred CD8+ T cells from endogenous CD8+ T cells, and distinguish between CTRL and specific KD. OT-1 CD8+ T cells were transduced as described above. To evaluate whether specific genetic manipulation affects the capacity of CD8+ T cells to infiltrate the tumor, 10,000 OT-1 CD8+ T cells shCTRL 45.2 and 10,000 OT-1 CD8+ T cells shPDH 45.1 or shSUCNR1 45.1 were adoptively co-transferred in 8 week old C57BL/6J female WT mice prior tumor implantation. The day after, 250,000 B16-OVA tumor cells without fluorescent label were injected in the flank subcutaneously. 16 days after tumor implantation, tumor dissociation and isolation of tumor infiltrating lymphocytes were performed as previously described (Drijvers et al., 2020). As they were co-transferred using a 1:1 ratio, we then used the CD45.1 and CD45.2 markers as a proxy to determine the relative contribution of both populations to the total amount infiltrated in the tumor. The sum of both thus leads to +/− 100%. To evaluate tumor growth, OT-1 CD8+ T cells were transduced with shRNA for CTRL and PDH and adoptively transferred in mice bearing B16-OVA tumors. 1,000,000 OT-1 CD8+ T cells were transferred per condition. Tumor volume was measured with a caliper every 2 to 3 days and calculated using the formula ½ D x d2, where D is the longer diameter and d is the shorter diameter. In the figures, average growth curves are displayed until one of the animals exhibited a tumor volume of 2000 mm3 upon which it was sacrificed.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were perfomed in GraphPad Prism 8.3.1. A two-tailed unpaired Student t test was used for comparison of two unpaired groups. A two-way ANOVA with Sidak’s multiple comparison test was used for comparison of multiple groups. Error bars display standard deviation (SD) unless otherwise noted. P values are noted as *P<0.05; **P<0.01; ***P<0.001.
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb anti-mouse CD3ε, Clone #145-2C11 | BioXCell | cat. BE0001-1 |
| InVivomAb anti-mouse CD28, Clone #37.51 | BioXCell | cat. BE0015-5 |
| anti-mouse CD8b antibodies | BioXCell | cat. BE0223 |
| αPD-1 (CD279), clone 29F.1A12 | BioXCell | cat. BE0273 |
| Isotype IgG2a, clone 2A3 | Bio X Cell | cat. BP0089 |
| BUV395 anti-mouse CD45.2 | BD Biosciences | cat. 564616 |
| APC anti-mouse CD3 | BioLegend | cat. 100236 |
| BV421 anti-mouse CD8b | BioLegend | cat. 126629 |
| PE-Cy7 anti-mouse CD8b | BioLegend | cat. 126616 |
| BUV496 anti-mouse CD4 | BD Biosciences | cat. 612952 |
| eF50 anti-mouse Foxp3 | ThermoFisher | cat. 48-5773-82 |
| BV605 anti-mouse CD11b | BioLegend | cat. 101237 |
| PE/Cy7 anti-mouse IFNγ | BioLegend | cat. 505826 |
| APC anti-mouse IFNγ | BioLegend | cat. 505809 |
| FITC anti-mouse GzmB | BioLegend | cat. 515403 |
| APC anti-mouse GzmB | BioLegend | cat. 396407 |
| PerCP/Cy5.5 anti-mouse Ki67 | BioLegend | cat. 652424 |
| BV605 anti-mouse Ki-67 | BD Biosciences | cat. 567122 |
| BV711 anti-mouse Tim3 | Biolegend | cat. 119727 |
| BUV563 anti-mouse Slamf6 | Biolegend | cat. 134610 |
| Bacterial and virus strains | ||
| Stbl3 Chemically Competent E. coli | ThermoFisher | cat. C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI-1640 | ThermoFisher | cat. 11875-119 |
| DMEM | ThermoFisher | cat. 11965-118 |
| Penicillin-Streptomycin | ThermoFisher | cat. 15140122 |
| Fetal Bovine Serum (FBS) | Sigma | cat. F2442 |
| dialyzed FBS | Thermo Fisher | cat. 1743489 |
| 2-mercaptoethanol | ThermoFisher | cat. 21985023 |
| EDTA (0.5 M) | ThermoFisher | cat. 15575020 |
| HEPES | ThermoFisher | cat. 15630130 |
| Sodium pyruvate | ThermoFisher | cat. 11360-070 |
| Opti-MEM I reduced serum medium | Life Technologies | cat. 31985-062 |
| Fugene 6 | VWR/Promega | cat. E2691 |
| Hexadimethrine bromide (Polybrene) | Santa Cruz | cat. sc-255611 |
| Collagenase, Type I | Worthington Biochemical | cat. LS004194 |
| Percoll density gradient media | GE Healthcare LifeSciences | cat. 17089101 |
| Blasticidin | Thermo Fisher | cat. A11139-03 |
| Puromycin | InvivoGen | cat. ant-pr-1 |
| PerfeCTa SYBR® Green FastMix | Quantabio | cat. 101414-270 |
| TRIzol Reagent | ThermoFisher | cat. 15596018 |
| High-Capacity cDNA Reverse Transcription Kit | Genesee Scientific | cat. 11-327 |
| LB Broth LB | Sigma-Aldrich | cat. L7275 |
| Ionomycin from Streptomyces conglobatus | Sigma Aldrich | cat. I9657-1MG |
| GolgiStop Protein transport inhibitor | BD Biosciences | cat. 554724 |
| Quick Ligation Kit | NEB BioLabs | cat. M2200S |
| BamH I-HF Restriction Endonuclease | NEB BioLabs | cat. R3136S |
| Interleukin-2 | R&D Systems | cat. 202-IL-050 |
| Interleukin-12 | PeproTech | cat. 210-12-50 |
| Sodium lactate | Sigma | cat. 867-56-1 |
| Dimethyl succinate | Sigma | cat. 106-65-0 |
| Dimethyl a-ketoglutarate | Sigma | cat. 349631 |
| Sodium fumarate | Sigma | cat. F1506 |
| Malic acid | Sigma | cat. M7397 |
| Oxaloacetic acid | Sigma | cat. O4126 |
| CPI-613 | APExBIO | cat. A4333 |
| Cis-epoxysuccinic acid | Thermo Fisher | cat. AC372640050 |
| Sodium oxamate | Sigma | cat. O2751 |
| Glutaric acid | Sigma | cat. G3407 |
| LC/MS grade water | Fisher Scientific | cat. W6500 |
| Chloroform | Sigma | cat. C2432 |
| Methanol | Fisher Scientific | cat. A456-500 |
| Critical commercial assays | ||
| LIVE/DEAD Fixable Near-IR stain | ThermoFisher | cat. L34976 |
| Naive CD8a+ T Cell Isolation Kit, mouse | Miltenyi Biotec | cat. 130-096-543 |
| eBioscience Foxp3 / Transcription Factor Staining Buffer | ThermoFisher | cat. 00-5523-00 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | cat. 554714 |
| Direct-zol RNA Miniprep Kit | Zymo Research | cat. R2050 |
| Pierce BCA Protein Assay Kit | ThermoFisher | cat. 23227 |
| UltraComp beads | Thermo Fisher | cat. 01-2222-42 |
| ELISA Standard Set Mouse IFN-gamma | Biolegend | cat. 430801 |
| Experimental models: Cell lines | ||
| B16.F10 Melanoma | Gift from G. Dranoff (Novartis Institutes for Biomedical Research) | RRID: CVCL_0159 |
| MC38 colorectal adenocarcinoma | Laboratory of D. Vignali, University of Pittsburgh School of Medicine, Pittsburgh, PA | RRID: CVCL_B288 |
| Platinum-E cells | Cell Biolabs | cat. RV-101 |
| Experimental models: Organisms/strains | ||
| C57BL6/J | The Jackson Laboratory | cat. 000664 |
| OT-1 (C57BL/6J-Tg(Tcra/Tcrb)1100Mjb/J) | The Jackson Laboratory | cat. 003831 |
| Oligonucleotides | ||
| PC qPCR primer (mouse): Fw: GCCCAGAAGTTGCTACATTACCT Rv: CTCACATTGACAGGGATTGGA |
IDT | N/A |
| PDH qPCR primer (mouse): Fw: TGTGACCTTCATCGGCTAGAA Rv: TGATCCGCCTTTAGCTCCATC |
IDT | N/A |
| SUCNR1 qPCR primer (mouse): Fw: CCATCTCTGACTTTGCTTTCCTG Rv: GTGTAGAGGTTGGTGTGAAGCAC |
IDT | N/A |
| RPL-19 qPCR primer (mouse): Fw: CAGGCATATGGGCATAGGGAA Rv: TGCCTTCAGCTTGTGGATGT |
IDT | N/A |
| Cloning PC KD vector (mouse): Fw:CCGGCCCTTCAGCTATTTGTCCTTTCTCGAGAAAGGACAAATAGCTGAAGGGTTTTTG Rv:AATTCAAAAACCCTTCAGCTATTTGTCCTTTCTCGAGAAAGGACAAATAGCTGAAGGG |
This manuscript | N/A |
| Cloning PDH KD vector (mouse): Fw:CCGGGCTCAAGTACTACAGGATGATCTCGAGATCATCCTGTAGTACTTGAGCTTTTTG Rv:AATTCAAAAAGCTCAAGTACTACAGGATGATCTCGAGATCATCCTGTAGTACTTGAGC |
This manuscript | N/A |
| Cloning SUCNR1 KD vector (mouse): Fw:CCGGAGAGAGATGCTGATTAGTAAGCTCGAGCTTACTAATCAGCATCTCTCTTTTTTG Rv:AATTCAAAAAAGAGAGATGCTGATTAGTAAGCTCGAGCTTACTAATCAGCATCTCTCT |
This manuscript | N/A |
| Recombinant DNA | ||
| pMKO.1 GFP retroviral vector | Addgene | cat. 10676 |
| pCL-Eco | Addgene | cat. 12371 |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com |
| FlowJo 10.6.1 | FlowJo LLC | https://www.flowjo.com |
| Tracefinder 3.2 | Thermo Fisher | https://www.thermofisher.com/order/catalog/product/OPTON-31001 |
| Other | ||
| Cell strainer 10 mm | PluriSelect USA | cat. 43-50010-50 |
| GentleMACS C Tubes | Miltenyi | cat. 130-093-237 |
| Lithium Heparin Tubes (2 mL) | VWR | cat. 454237 |
| Nylon Net Filters | EMD Millipore | cat. NY2004700 |
Highlights.
Tumor-derived lactate induces a switch from PC to PDH
PDH inhibition increases PC activity and succinate secretion
Extracellular succinate activates SUCNR1 and promotes T cell cytotoxicity
PDH inhibition synergizes with immunotherapy
Acknowledgments
The authors thank the entire Haigis and Sharpe labs for productive discussions. I.E. was supported by European Molecular Biology Organization (ALTF-1078-2017) and Cancer Research Institute/Merck Fellow (CRI Award 3352) post-doctoral fellowships. G.N. was supported by the NSF Graduate Research Fellowship Program (GE1745303). K.K. was supported by a Gilead Sciences Fellow of the Life Sciences Research Foundation. This study was supported by a grant from the Ludwig Center at Harvard (M.C. Haigis and A.H. Sharpe), the NIH grant U54-CA224088 (M.C. Haigis and A.H. Sharpe), NIH grant R01CA213062 (M.C. Haigis) and P50CA101942 (G.J. Freeman). The graphical abstract and the schematic figures were generated using BioRender.com.
Declaration of Interests
MCH and AHS received research funding from Roche Pharmaceuticals. MCH received funding from Agilent Technologies. MCH and AHS are advisors to Guided Clarity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. (2016). LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, and Pearce EL (2015). T cell metabolism drives immunity. J. Exp. Med 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, Clish CB, DeBerardinis RJ, Feron O, Frezza C, Ghesquiere B, et al. (2015). A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardaci S, Zheng L, Mackay G, Van Den Broek NJF, Mackenzie ED, Nixon C, Stevenson D, Tumanov S, Bulusu V, Kamphorst JJ, et al. (2015). Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol 10.1038/ncb3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cham CM, and Gajewski TF (2005). Glucose Availability Regulates IFN-γ Production and p70S6 Kinase Activation in CD8 + Effector T Cells . J. Immunol 10.4049/jimmunol.174.8.4670. [DOI] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, Huang SCC, Van Der Windt GJW, Blagih J, Qiu J, et al. (2013). XPosttranscriptional control of T cell effector function by aerobic glycolysis. Cell 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DS, and Mellman I (2013). Oncology meets immunology: The cancer-immunity cycle. Immunity 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, Elia I, Buescher JM, Orth MF, Davidson SM, et al. (2016). Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 10.1016/j.celrep.2016.09.042. [DOI] [PubMed] [Google Scholar]
- Delgoffe GM, and Powell JD (2015). Feeding an army: The metabolism of T cells in activation, anergy, and exhaustion. Mol. Immunol 10.1016/j.molimm.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan M, Dranoff G, and Dougan SK (2019). Cancer immunotherapy: Beyond checkpoint blockade. Annu. Rev. Cancer Biol 10.1146/annurev-cancerbio-030518-055552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, LaFleur MW, Ringel AE, Yao C-H, Kurmi K, et al. (2020). Pharmacologic Screening Identifies Metabolic Vulnerabilities of CD8+ T Cells. Cancer Immunol. Res 10.1158/2326-6066.cir-20-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia I, and Haigis MC (2021). Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat. Metab 10.1038/s42255-020-00317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, Verfaillie C, Grünewald TGP, and Fendt SM (2017). Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun 10.1038/ncomms15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkona S, Diamandis EP, and Blasutig IM (2016). Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen J, Jouret F, Pirotte B, and Hanson J (2016). Insight into SUCNR1 (GPR91) structure and function. Pharmacol. Ther 10.1016/j.pharmthera.2016.01.008. [DOI] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, MacIntyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, et al. (2018). Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 10.1016/j.cell.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiran N, Ceperuelo-Mallafré V, Calvo E, Hernández-Alvarez MI, Ejarque M, Núñez-Roa C, Horrillo D, Maymó-Masip E, Rodríguez MM, Fradera R, et al. (2019). SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat. Immunol, 10.1038/s41590-019-0372-7. [DOI] [PubMed] [Google Scholar]
- Krzak G, Willis CM, Smith JA, Pluchino S, and Peruzzotti-Jametti L (2021). Succinate Receptor 1: An Emerging Regulator of Myeloid Cell Function in Inflammation. Trends Immunol. 10.1016/j.it.2020.11.004. [DOI] [PubMed] [Google Scholar]
- Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, Ferreira R, Naamati A, Ali A, Lewis CA, et al. (2021). Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 10.1016/j.molcel.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee TS, Anderson RG, Pladna KM, Isom S, Ghiraldeli LP, Miller LD, Chou JW, Jin G, Zhang W, Ellis LR, et al. (2018). A phase i study of cpi-613 in combination with high-dose cytarabine and mitoxantrone for relapsed or refractory acute myeloid leukemia. Clin. Cancer Res 10.1158/1078-0432.CCR-17-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip PA, Buyse ME, Alistar AT, Lima CM, Luther S, Pardee TS, and Cutsem E. Van (2019). A phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Futur. Oncol 10.2217/fon-2019-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn WJ, Jiao J, TeSlaa T, Stadanlick J, Wang Z, Wang L, Akimova T, Angelin A, Schäfer PM, Cully MD, et al. (2020). Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 10.1016/j.celrep.2020.108500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Bozi LHM, Yaghi OK, Mills EL, Xiao H, Nicholson HE, Paschini M, Paulo JA, Garrity R, Laznik-Bogoslavski D, et al. (2020). pH-Gated Succinate Secretion Regulates Muscle Remodeling in Response to Exercise. Cell 10.1016/j.cell.2020.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringel AE, Drijvers JM, Baker GJ, Catozzi A, García-Cañaveras JC, Gassaway BM, Miller BC, Juneja VR, Nguyen TH, Joshi S, et al. (2020). Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 10.1016/j.cell.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer M, Spinelli JB, et al. (2016). Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. 10.1016/j.cmet.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salabei JK, Gibb AA, and Hill BG (2014). Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Ii MB, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, et al. (2015). Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Invest 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NN, and Fry TJ (2019). Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, and Rathmell JC (2018). Metabolic Barriers to T Cell Function in Tumors. J. Immunol 10.4049/jimmunol.1701041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolbright BL, Rajendran G, Harris RA, and Taylor JA (2019). Metabolic flexibility in cancer: Targeting the pyruvate dehydrogenase kinase:pyruvate dehydrogenase axis. Mol. Cancer Ther 10.1158/1535-7163.MCT-19-0079. [DOI] [PubMed] [Google Scholar]
- Yuan L, Tatineni J, Mahoney KM, and Freeman GJ (2021). VISTA: A Mediator of Quiescence and a Promising Target in Cancer Immunotherapy. Trends Immunol. 10.1016/j.it.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the corresponding author on request. This paper does not report original codes.