

Abstract
Purpose:
Although local tissue-based immune responses are critical for elucidating direct tumor–immune cell interactions, peripheral immune responses are increasingly recognized as occupying an important role in anticancer immunity. We evaluated serial blood samples from patients with advanced epithelial ovarian cancer (EOC) undergoing standard-of-care neoadjuvant carboplatin and paclitaxel chemotherapy (including dexamethasone for prophylaxis of paclitaxel-associated hypersensitivity reactions) to characterize the evolution of the peripheral immune cell function and composition across the course of therapy.
Experimental Design:
Serial blood samples from 10 patients with advanced high-grade serous ovarian cancer treated with neoadjuvant chemotherapy (NACT) were collected before the initiation of chemotherapy, after the third and sixth cycles, and approximately 2 months after completion of chemotherapy. T-cell function was evaluated using ex vivo IFNγ ELISpot assays, and the dynamics of T-cell repertoire and immune cell composition were assessed using bulk and single-cell RNA sequencing (RNAseq).
Results:
T cells exhibited an improved response to viral antigens after NACT, which paralleled the decrease in CA125 levels. Single-cell analysis revealed increased numbers of memory T-cell receptor (TCR) clonotypes and increased central memory CD8+ and regulatory T cells throughout chemotherapy. Finally, administration of NACT was associated with increased monocyte frequency and expression of HLA class II and antigen presentation genes; single-cell RNAseq analyses showed that although driven largely by classical monocytes, increased class II gene expression was a feature observed across monocyte subpopulations after chemotherapy.
Conclusions:
NACT may alleviate tumor-associated immunosuppression by reducing tumor burden and may enhance antigen processing and presentation. These findings have implications for the successful combinatorial applications of immune checkpoint blockade and therapeutic vaccine approaches in EOC.
Translational Relevance.
Epithelial ovarian cancer (EOC) remains one of the few malignancies where immune checkpoint inhibitors exhibit only modest activity as monotherapy and currently have no FDA-approved indication. We evaluated the effects of standard carboplatin and paclitaxel chemotherapy (including prophylactic dexamethasone) on the peripheral immune system of patients with EOC undergoing neoadjuvant chemotherapy (NACT). Our observations of increased T-cell responses to viral antigens and of increased monocytes with concomitant elevation of HLA class II expression and antigen presentation suggest that NACT may be a promising platform for building combinatorial immunotherapy strategies in EOC. Furthermore, our finding of restored T-cell responses in all patients 2 months after completing chemotherapy supports the application of therapeutic vaccine approaches in ovarian cancer and provides guidance on the optimal timing of vaccine administration. Finally, our study highlights that monitoring peripheral immune cell responses may provide unique insights into the immune effects of systemic therapy as well as mirror its antitumor efficacy.
Introduction
Surgical cytoreduction and adjuvant or neoadjuvant chemotherapy (NACT) has long been the mainstay of frontline treatment for approximately 75% of patients with epithelial ovarian cancer (EOC). Chemotherapy for advanced EOC typically consists of carboplatin and paclitaxel administered intravenously every 3 weeks for a minimum of 6 cycles (1). EOC is, at least initially, a very chemosensitive disease with approximately 70% of patients achieving a complete clinical response after first-line chemotherapy (1, 2). This striking chemosensitivity is partly related to underlying deficiencies in homologous recombination DNA repair, which are prevalent especially among tumors of high-grade serous histology (∼75% of all EOCs), rendering them susceptible to platinum-based chemotherapy (3, 4). However, besides their direct cytotoxic effects, chemotherapy agents such as carboplatin and paclitaxel may also exhibit immune effects, which, depending on the context, may enhance or impede their overall treatment effect. Similarly, steroids and growth factors administered together with chemotherapy as prophylaxis of paclitaxel-associated hypersensitivity reactions and treatment of carboplatin-associated nausea (2) may also affect the immune system and thus impact the overall efficacy of antitumor therapy.
A limited number of previous studies have evaluated the immune effects of chemotherapy in EOC, but they have focused almost exclusively on local immune responses within the ovarian tumor microenvironment (5–9). On the other hand, the systemic immune landscape is increasingly recognized as occupying a critical role in effective natural and therapeutically induced anticancer immune responses, and intact peripheral immunity is required for immunotherapeutic efficacy (10–12). Therefore, an evaluation of the peripheral immune system across all immune cell lineages can provide a more complete understanding of the effects of conventional chemotherapy on the immune response against ovarian tumors.
To this end, we selected the setting of NACT for EOC to evaluate the effects of chemotherapy on the peripheral immune system, whereby patients receive 3 cycles of carboplatin/paclitaxel chemotherapy followed by interval cytoreductive/debulking surgery and 3 more cycles of chemotherapy. This provides a unique opportunity to dissect the relative impact of chemotherapy alone (after the first 3 cycles of NACT), as well as after the combination of surgery and adjuvant chemotherapy. Accordingly, we evaluated serial blood samples from 10 patients with advanced EOC undergoing NACT and used immunogenomic approaches to characterize the evolution of peripheral immune cell function and composition across the course of therapy. To validate these results, we subsequently evaluated an independent confirmatory cohort of 5 patients with advanced EOC before treatment and after NACT and interval surgery.
Materials and Methods
Patient peripheral blood mononuclear cell samples
Heparinized blood samples were obtained from 10 patients undergoing NACT at Dana-Farber Cancer Institute and stored in vapor-phase liquid nitrogen until the time of analysis. For validation, we obtained blood samples at Dana-Farber Cancer Institute from an additional cohort of 5 patients undergoing NACT, as well as from 12 healthy adult volunteers. Written informed consent was obtained from patients or guardians before enrollment in the study, and all procedures involving human participants were carried out in accordance with the Declaration of Helsinki. Human investigations were performed after approval by the Dana-Farber Cancer Institute institutional review board (IRB-approved protocol 02–051) and in accordance with an assurance filed with and approved by the U.S. Department of Health and Human Services. Serum CA125 concentrations and complete peripheral blood counts were determined as per standard clinical care. Ficoll density gradient centrifugation (GE Healthcare) was performed to isolate patient peripheral blood mononuclear cells (PBMC). Isolated PBMC samples were cryopreserved with 10% DMSO in FBS (Sigma-Aldrich) until the time of analysis.
IFNγ ELISpot assay
The ELISpot assay was performed as detailed previously (13, 14). In brief, cryopreserved PBMCs were thawed in complete DMEM media supplemented with 10% human serum and rested overnight. Ex vivo IFNγ ELISpot assays were performed by stimulating PBMCs with FluA viral lysate (Zeptometrix; ref. 15) or CEF (Mabtech) for 18 hours at 37°C in DMEM complete media supplemented with 10% FBS, HEPES, 5% penicillin–streptomycin, β-mercaptoethanol (Gibco), sodium pyruvate, and nonessential amino acids (Corning). Stimulation with OVA peptide provided a negative control, whereas stimulation with anti-CD3 OKT (BioLegend) served as a positive control. Responses were scored as positive if they were quantified as at least 3-fold over the average of the control (OVA for the patients and no peptide for the healthy donors) for each time point before normalization. Data for each antigen peptide (FluA or CEF) were normalized by subtracting the average of the negative control from each of the 3 replicate pool measurements at the corresponding time. Values were set to zero if the normalized value would be smaller than zero, which applied to 22% of the values. P values were calculated using the repeated-measures mixed-effect model. Fold changes were calculated at each time point relative to baseline. Values were set to 3x the control (OVA) if the average of 3 replicates was smaller than this limit before normalization.
RNA extraction
After thawing cryopreserved patient PBMCs, total RNA was isolated from 5 × 106 PBMCs following the manufacturer's recommendations (RNeasy Mini Kit, Qiagen). Eluates were evaluated for quality and concentration (Nanodrop 2000, Thermo Fisher Scientific).
Bulk TCR sequencing and analysis of bulk and single-cell TCR data
We adapted RNase H-dependent T-cell receptor (TCR) sequencing to perform α and β TCR repertoire analysis on bulk RNA specimens (16, 17). Twenty-ng bulk RNA was used in each reverse transcriptase reaction, and 4 replicates were done for each sample. Exonuclease digestion was performed to eliminate excess reverse transcriptase primers before running RNase H-dependent PCR. Sequencing libraries were sequenced using the MiSeq 300 cycle reagent kit v.2 according to the manufacturer's protocol (Illumina). The sequencing data analysis was performed using R. Fisher's exact tests with correction for multiple hypothesis testing (Benjamini–Hochberg FDR procedure) were performed to detect T-cell clonotypes with statistically significant changes in frequency. T-cell diversity was calculated using a Shannon Index (18). To reduce the variation caused by sequencing depth, we normalized the Shannon index through division by the natural log of the total number of unique molecular identifiers (UMI) in each sample. Changes in TCR clonotype frequency were considered significant if P values (Fisher's exact test, FDR corrected) were <10–4 and relative change was >10%. Novel and disappeared clonotypes were defined as those that were no longer detectable at baseline or the follow-up timepoint, respectively. For single-cell analyses, T-cell clonotypes were annotated by matching individual T-cell CDR3 amino acid sequences for the T-cell α and β chains of each patient, to CDR3 amino acid sequences for annotated paired T-cell α and β chains in the VDJdb (19) and MCPAS-TCR (20) databases.
Single-cell RNA-sequencing analysis
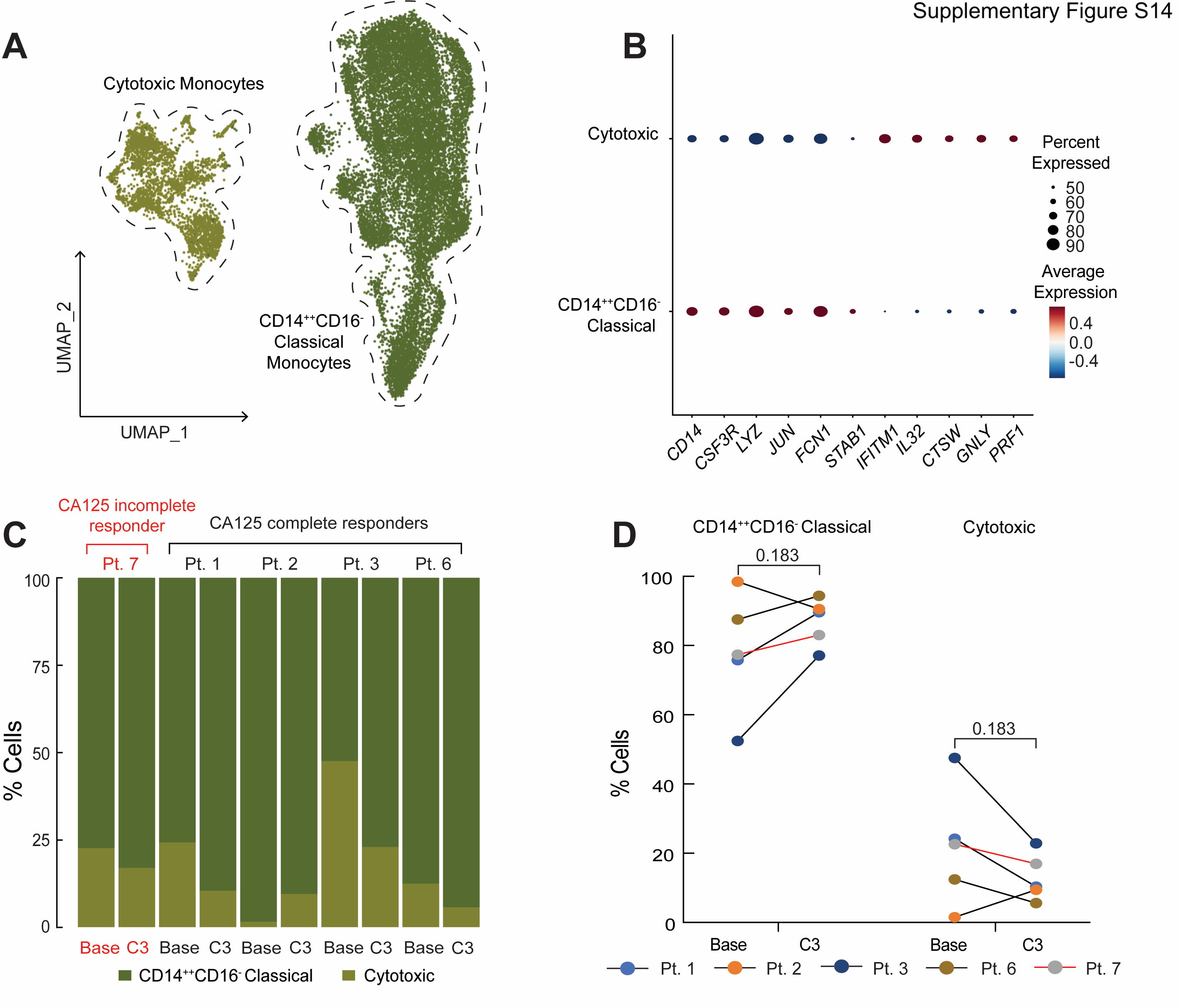
The samples processed for single-cell RNA sequencing (RNAseq) were obtained from the PBMC samples isolated after Ficoll density gradient centrifugation. The viable cells were washed and resuspended in PBS with 0.04% BSA at a cell concentration of 1,000 cells/μL. Seventeen thousand viable cells were loaded onto a 10x Genomics Chromium instrument (10x Genomics) according to the manufacturer's recommendations. Single-cell RNAseq libraries were generated either using the Chromium Next Single-Cell 5' Kit v2 (10x Genomics; Pts. 3, 6, and 7) or the Chromium Single-Cell 5' Library and Gel Bead Kit (10x Genomics; Pts. 1 and 2). Matched single-cell TCR libraries were prepared using the Chromium Single-Cell Human TCR Amplification Kit (10x Genomics). Quality controls (QC) for amplified cDNA libraries, TCR-sequencing libraries, and final RNAseq libraries were performed using the Bioanalyzer High-Sensitivity DNA Kit (Agilent). The sequencing libraries for single-cell RNAseq and single-cell TCR sequencing were diluted to 4 nmol/L and pooled using a volume ratio of 4:1. The pooled libraries of patients 3, 6, and 7 were sequenced on the Illumina NovaSeq S4 platform (sequencing parameters: Read 1 of 28 bp, Read 2 of 90 bp, Index 1 of 10 bp, and Index 2 of 10 bp), and for patients 1 and 2, on the NovaSeq SP platform (sequencing parameters: Read 1: 26 bp, Read 2: 91 bp, Index 1: 8 bp). Sequencing data were demultiplexed and aligned to GRCh38 using the cell ranger version 5.0.0 pipeline (10x Genomics). Single-cell analysis methods were described in detail previously (21). In brief, QC was performed to exclude cells with (i) fewer than 500 or more than 15,000 UMIs, (ii) fewer than 300 genes, (iii) fewer than 0.8 UMIs per gene, and (iv) mitochondrial ratio <0.2 (number of UMIs assigned to mitochondrial genes over total number of UMIs per cell). All cells passing QC were integrated and clustered using Seurat v3 (22). Before downstream analysis, two clusters of potential doublets were removed on the basis of cell marker expression. Differential expression (DE) analysis was performed between time points (pre-chemotherapy and the third cycle of chemotherapy) and clusters using the default Wilcoxon rank-sum test implementation in Seurat. DE analysis was performed on the combined patient dataset as well as on a per-patient basis, to avoid bias in DE calling. T-cell states were predicted using Seurat reference datasets that applied unsupervised weighted-nearest neighbor analysis and incorporate RNA sequencing and CITE sequencing of 211,000 human PBMCs, with large cell-surface protein marker panels (23). PBMCs of the above Seurat reference datasets were also used as one of the healthy donor controls. In addition, we downloaded pre-processed single-cell RNAseq data (count matrix) from 10x Genomics of PBMCs from a healthy donor (5k_pbmc_v3; libraries were generated with Chromium Single-Cell 3' Reagent Kits v3.1; single-cell dataset was processed by Cell Ranger 3.0.2), and a 25-year-old healthy female donor (Parent_NGSC3_DI_PBMC; libraries were generated with Chromium Single Cell 3' Reagent Kits v3.1; single-cell dataset was processed by Cell Ranger 4.0.0) as healthy donor controls. Dendritic cells (DC) and monocytes were subclustered and subpopulations were identified on the basis of marker genes identified by Villani and colleagues (24). Briefly, subclusters were defined by signature genes: CD14++CD16– classical monocytes (e.g., CD14, CSF3R, FCN1, LYZ, and STAB1), cytotoxic monocytes (e.g., IFITM1, IL32, PRF1, GNLY, and CTSW), CLEC9A DCs (e.g., BATF3 and CLEC9A), pDCs (e.g., GZMB and SERPINF1), inflammatory CD1C DCs (e.g., GZMB, SERPINF1, S100A8, and CD14), and non-inflammatory CD1C DCs (e.g., CD1C, CLEC10A).
Flow cytometry
Live lymphocytes and monocytes were gated by forward and side scatter. T cells were identified by anti–CD3-FITC (Biolegend HIT3a, all patients), CD8-APC (BD RPA-T8; Pts 1 and 2), and CD4-BV650 (BD L200; Pts 1 and 2). Monocytes were identified by anti–CD14-APC/Cy7 (BioLegend M5E2; pts 1 and 2) or CD14-APC (BD M5E2; Pts 3, 6 and 7). A healthy PBMC donor was used as control for all flow cytometry experiments.
Data availability
The single cell RNAseq and TCRseq data generated in this study are publicly available in the database of Genotypes and Phenotypes (dbGaP) at phs002862.v1.p1.
The bulk TCR sequencing data generated in this study are publicly available in the Gene Expression Omnibus (GEO) at GSE206339 (hyperlink: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE206339).
Other data analyzed in this study were obtained from Gene Expression Omnibus (GEO) at GSE164378 (hyperlink: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164378) and from 10x Genomics for samples 5k_pbmc_v3 (hyperlink: https://www.10xgenomics.com/resources/datasets/5-k-peripheral-blood-mononuclear-cells-pbm-cs-from-a-healthy-donor-v-3-chemistry-3-1-standard-3-0-2) and Parent_NGSC3_DI_PBMC (hyperlink: https://www.10xgenomics.com/resources/datasets/pbm-cs-from-a-healthy-donor-whole-transcriptome-analysis-3-1-standard-4-0-0).
Results
Patient characteristics and response to therapy
Ten patients with stage IIIC (n = 9) or IV (n = 1) high-grade serous ovarian cancer treated with NACT were included in this study (Table 1). Serial blood samples were collected at 4 time points: before initiation of chemotherapy (baseline sample), after the third cycle (C3 sample), after the sixth cycle (C6 sample), and approximately 2 months after completion of chemotherapy (post-treatment sample). All patients received standard-of-care carboplatin and paclitaxel chemotherapy IV every 3 weeks (Supplementary Fig. S1); dexamethasone premedication was administered to all patients with each chemotherapy cycle. Eight of 10 patients received 6 cycles of chemotherapy, whereas one subject (Pt. 2) received 5 cycles of chemotherapy and another (Pt. 8) received 7 cycles of chemotherapy. Treatment with recombinant G-CSF (either filgrastim or pegfilgrastim), was administered in 8 patients (all patients except Pts. 3 and 5) to prevent chemotherapy-induced neutropenia (Supplementary Fig. S1). Nine patients (all except Pt. 8) underwent interval surgery; in all 9, an optimal cytoreductive effect was achieved, defined as no residual tumor implant greater than 1 cm in size (25). In 2 patients (Pts. 1 and 7), cytoreduction to no gross residual disease was achieved.
Table 1.
Baseline clinical and pathologic characteristics for 10 subjects with EOC.
| Patient ID | Age (y) | Stage | Histology | Grade | Interval surgical effect | CA125 response per GCIG criteria after 3 cycles | CA125 response per GCIG criteria after 6 cycles |
|---|---|---|---|---|---|---|---|
| Pt. 1 | 56 | IIIC | Serous | High | Optimal | Complete | Complete |
| Pt. 2 | 85 | IIIC | Serous | High | No gross residual disease | Complete | Complete |
| Pt. 3 | 55 | IV | Serous | High | Optimal | Complete | Complete |
| Pt. 4 | 59 | IIIC | Serous | High | Optimal | Complete | Complete |
| Pt. 5 | 68 | IIIC | Serous | High | Optimal | Complete | Complete |
| Pt. 6 | 52 | IIIC | Serous | High | Optimal | Complete | Complete |
| Pt. 7 | 69 | IIIC | Serous | High | No gross residual disease | Incomplete | Incomplete |
| Pt. 8 | 89 | IIIC | Serous | High | No surgery | Incomplete | Incomplete |
| Pt. 9 | 62 | IIIC | Serous | High | Optimal | Incomplete | Complete |
| Pt.10 | 75 | IIIC | Serous | High | Optimal | Incomplete | Incomplete |
All 10 patients had an elevated baseline CA125, and all had a decrease in their CA125 by at least 50% after 3 cycles of chemotherapy. However, 6 patients had complete normalization of their CA125 levels after 3 cycles of chemotherapy, and these levels remained normal following interval surgery and completion of 6 cycles of chemotherapy; these patients were defined as CA125 complete responders per the Gynecologic Cancer InterGroup (GCIG) criteria, reflective of excellent response to chemotherapy (Fig. 1). The remaining 4 patients (Pts. 7, 8, 9, and 10) did not achieve normalization of their CA125 levels after 3 cycles of chemotherapy and were thereafter referred to as CA125 incomplete responders. Of note, for patients 7, 8, and 10, CA125 levels remained elevated even after completion of 6 cycles of chemotherapy, reflecting an insufficient response to chemotherapy (Fig. 1). For patient 9, CA125 levels decreased after the third cycle of chemotherapy but normalized at the 6th cycle of chemotherapy, suggesting a beneficial effect of the combination of tumor debulking surgery and adjuvant therapy (Fig. 1 and Table. 1).
Figure 1.

Time course of CA125 levels for the patient cohort (n = 10). Filled dots represent CA125 levels for each individual patient at the indicated sample collections. Inset, 10 patients after C3 are indicated. Black lines, CA125 complete responders; red lines, CA125 incomplete responders.
Improved peripheral T-cell function following chemotherapy
To test whether chemotherapy exposure affected T-cell function, we performed IFNγ ELISpot assays across serial timepoints to evaluate T-cell responses against common viral antigens. As FluA and CEF are common viral antigens, we chose them as representative antigens to test T-cell functions through the course of NACT treatment. Samples were available for 7 of 10 patients (Pts. 1, 2, 4, 6, 7, 8, and 10). CA125 complete responders (Pts. 1, 2, 4, and 6) exhibited increases in T-cell responses to either FluA peptide or the CEF peptide pool at C3 compared with baseline [median increase by 1.6-fold (range, 0.2–3.2) from C3 relative to baseline; Fig. 2A and B; Supplementary Fig. S2A and S3). CA125 incomplete responders (Pts. 7, 8, and 10) also exhibited increases in T-cell response to FluA or CEF during chemotherapy treatment, but these were delayed and were observed only after the C3 sample compared with responding patients (Fig. 2C; Supplementary Fig. S2B and S3), likely reflecting the contribution of surgery in these patients. We also compared the ELISpot response with viral antigens between these 7 patients and 10 healthy adult volunteers. We observed that the percentage of positive ELISpot response to CEF increased after chemotherapy and surgery in patients with ovarian cancer, achieving a level similar to that of the healthy donors, which supported our previous finding that, after chemotherapy and surgery, T-cell function recovered in response to viral antigens (Supplementary Fig. S4). Overall, these data suggested that T-cell function recovered in response to viral antigens after chemotherapy in CA125 complete responders, and also improved after chemotherapy and debulking surgery in CA125 incomplete responders.
Figure 2.

Standard-of-care first-line platinum-based chemotherapy increases T-cell responses to viral antigens following chemotherapy. A, IFNγ ELISpot responses to influenza (FluA) and CEF peptide pools over the course of chemotherapy in patient 6 (CA125 complete responder). OKT CD3 (anti-CD3ε antibody) served as a positive control. B and C, IFNγ ELISpot responses to FluA and CEF peptide pools over the course of chemotherapy in CA125 complete responders (Pts. 1, 2, 4, and 6; B) and CA125 incomplete responders (Pts. 7, 8, and 10; C). Spot-forming cell (SFC) per 106 PBMCs were background (OVA)-subtracted with n = 3 biologically independent samples. All data points represent mean ± standard error of the mean (s.e.m.). A repeated-measures regression model was used for generating P values; *, P ≤ 0.05.
Evaluation of T-cell dynamics following chemotherapy
To gain insight into the mechanisms underlying increased T-cell responses following chemotherapy, we examined longitudinal changes in the T-cell repertoire over four collected time points spanning the course of chemotherapy through bulk TCR-sequencing analysis. Eight (Pts. 1, 2, 4, 5, 6, 7, 8, and 10) of 10 patients had available samples for evaluation. The numbers of UMIs per sample undergoing bulk TCR sequencing were heterogenous (median 55,170 UMIs, range 3,680–147,180 UMIs; Supplementary Fig. S5A). The diversity of T-cell repertoires throughout chemotherapy treatment was calculated using the Normalized Shannon Index, which takes into account the richness and evenness of T-cell clones while correcting for the number of observations per sample (in Materials and Methods). These calculations revealed that the overall T-cell repertoire diversity remained stable after chemotherapy and there were no obvious differences in the overall peripheral blood T-cell repertoire between healthy donors and patients with ovarian cancer, albeit increased diversity of T-cell repertoire was observed over the course of chemotherapy in 3 patients (Pts. 1, 2, and 4; Supplementary Fig. S5B). Even at the level of individual circulating T-cell clonotypes over time, only a small percentage of T-cell clonotypes (median 0.029%, range, 0.007%–0.35%) changed after chemotherapy (Fig. 3A–C). We further validated this observation with samples from 5 additional patients (Pt. A to Pt. E; Supplementary Table S1) that also demonstrated overall TCR repertoire stability following surgery and chemotherapy (Supplementary Fig. S5C). The minimal longitudinal changes we observed in these patients were in range with natural variability of serial TCR sampling from healthy donors (Supplementary Fig. S5B). Notably, however, these dynamic clonotypes were more frequent among CA125 complete responders, where we observed more TCR clonotypes that expanded from baseline to C3 (Fig. 3D and E; Table 2). That said, when focusing on the dynamics of the most expanded clonotypes (n = 100) per patient, which we assumed would be enriched for antigen-experienced T-cell clones (Supplementary Fig. S5D), no clear patterns of change were detected. Altogether, the results of bulk TCR sequencing demonstrated that the T-cell repertoires were overall stable following chemotherapy.
Figure 3.

TCR clonotype dynamics over the course of chemotherapy and interval surgery (n = 8). A and B, Scatter plots of individual clonotype dynamics between pre-chemotherapy (baseline) and the third cycle of chemotherapy (C3). T-cell clonotypes were defined on the basis of CDR3 amino acid sequences from the TCR β chain. Clonotypes of the five CA125 complete responders (A) and three CA125 incomplete responders (B) were combined, respectively. C–E, The percentage of the overall changed clonotypes (C), and the subsets of novel and expanded (D), and disappeared and contracted (E) clonotypes between each of two consecutive timepoints during treatment in CA125 complete responders (n = 5) and CA125 incomplete responders (n = 3). Statistical analyses between patient groups and between treatment time points were performed using two-sided Wilcoxon rank-sum tests without adjustment for multiple comparisons.
Table 2.
Summary statistics of the changed clonotypes throughout chemotherapy.
| Changed clonotypes | Treatment timeline | CA125 response | Median | 90% Median confidence interval |
|---|---|---|---|---|
| Novel and expanded clonotypes | Baseline to C3 | CA125 complete responders | 0.026% | 0.012%–0.16% |
| CA125 incomplete responders | 0.0025% | 0%–0.011% | ||
| Disappeared and contracted clonotypes | C3 to C6 | CA125 complete responders | 0.058% | 0.0088%–0.099% |
| CA125 incomplete responders | 0.0063% | 0.0034%–0.0082% | ||
| C6 to Post | CA125 complete responders | 0.014% | 0.0049%–0.022% | |
| CA125 incomplete responders | 0.0016% | 0%–0.0044% |
Chemotherapy leads to increased HLA class II expression on monocytes
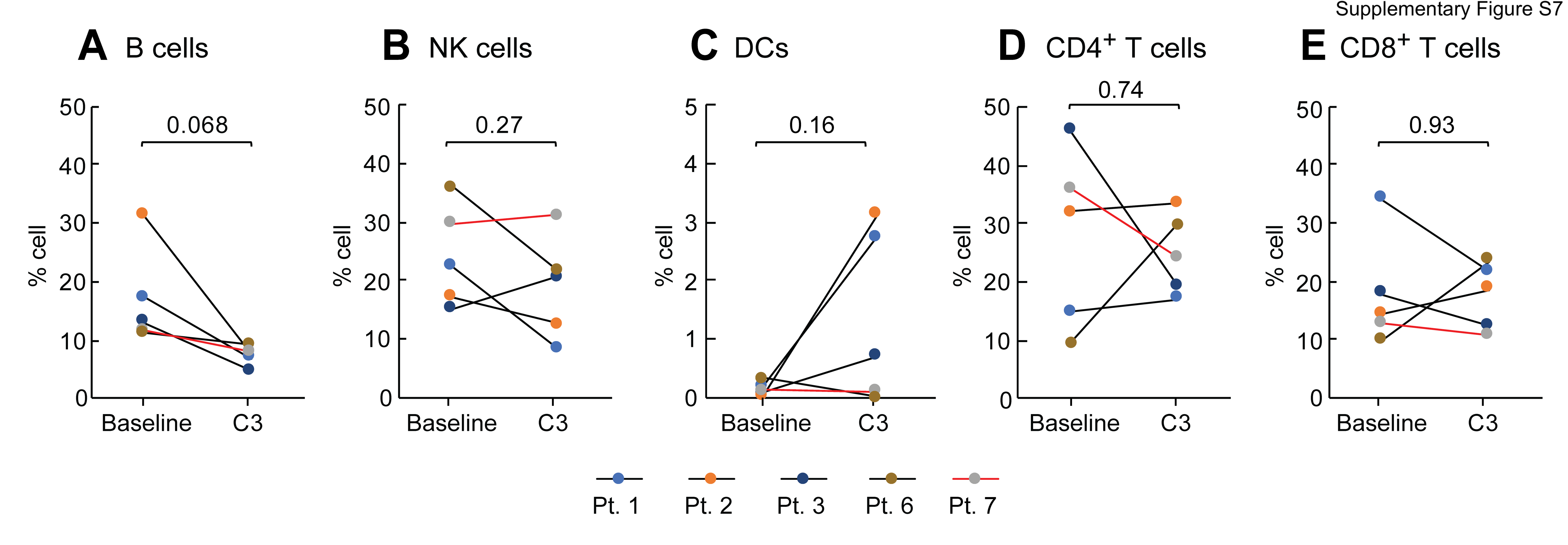
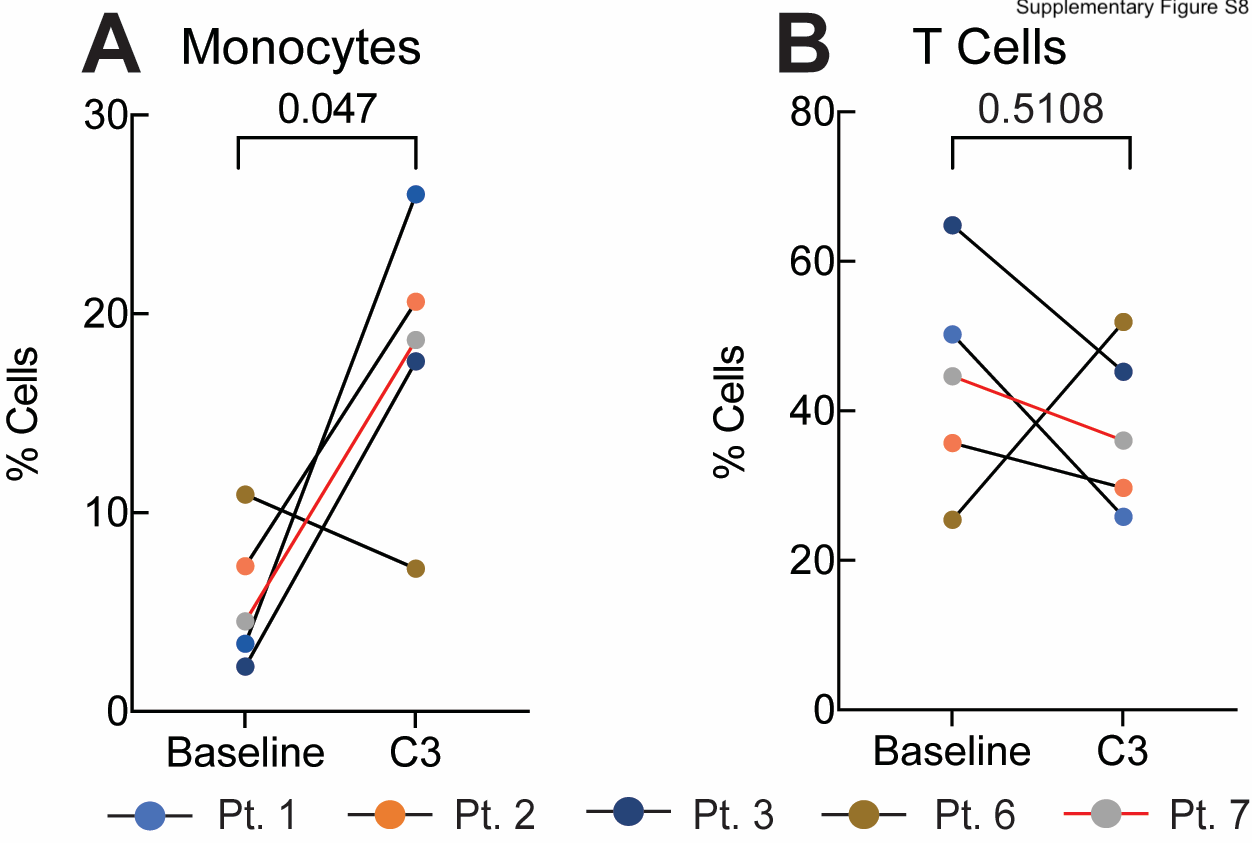
To further characterize the impact of platinum-based chemotherapy on peripheral immune cell composition, we performed single-cell RNA sequencing and paired single-cell TCR sequencing on PBMC samples of 4 CA125 complete responders (Pts. 1, 2, 3, and 6) and 1 CA125 incomplete responder (Pt. 7) collected at baseline and C3 (Fig. 4A); unfortunately, there was not adequate sample to perform these analyses on the other patients from our cohort. Immune cell populations were identified by marker gene expression (Supplementary Fig. S6) and were composed of B cells, monocytes, DCs, CD4+ T cells, CD8+ T cells, and natural killer (NK) cells (Fig. 4B). To normalize the variations in the cell counts between patient samples and quantify the phenotypic changes of immune cells after chemotherapy, the relative percentage of cells of each immune cell population was calculated (Fig. 4C). All 5 patients demonstrated a decline in B-cell numbers (P = 0.068; Fig. 4C; Supplementary Fig. S7) whereas no alternations were observed in NK and DC populations (Fig. 4C; Supplementary Fig. S7). Consistent with the TCR bulk–sequencing studies, the frequency of T cells remained stable in 3 of 5 patients (Fig. 4C; Supplementary Fig. S7). Stability in T-cell frequency after chemotherapy was also observed by flow cytometry (Supplementary Fig. S8B). We next turned to our single-cell TCR-sequencing data. We could match only four clonotypes to annotated TCRs (refs. 19, 20; data not shown), precluding meaningful analysis of antigen specificity. Similar to bulk analyses, we found that overall repertoire diversity remained stable after chemotherapy in our single-cell analysis (data not shown). To link the TCR clonotypes with their corresponding phenotypes for each individual T-cell, we identified T-cell subclusters and mapped TCR clonotypes of patients 1, 2, 3, 6, and 7 to each of the CD8+ and CD4+ T-cell states at the single-cell level (Supplementary Fig. S9; Supplementary Fig. S10A and S10E). The evaluation of clonotype dynamics by patient and by T-cell subcluster indicated subset stability in CD8+ and CD4+ T-cell subpopulations after chemotherapy except for a trend toward increasing representation of central memory CD8+ (P = 0.077) and regulatory CD4+ T clonotypes (P = 0.053) among total clonotypes after C3 (Supplementary Fig. S10 and S11).
Figure 4.

Dynamics of peripheral blood immune cell populations following chemotherapy. A, A summary of sample collection and single-cell analysis. B and C, UMAP (B) and the relative percentage (C) of immune cell populations in the integrated PBMC samples from patients 1, 2, 3, 6, and 7. No statistically significant changes were detected between pre-chemotherapy (Base) and the third cycle of chemotherapy (C3) in each immune cell population using a two-sided one-sample t test.
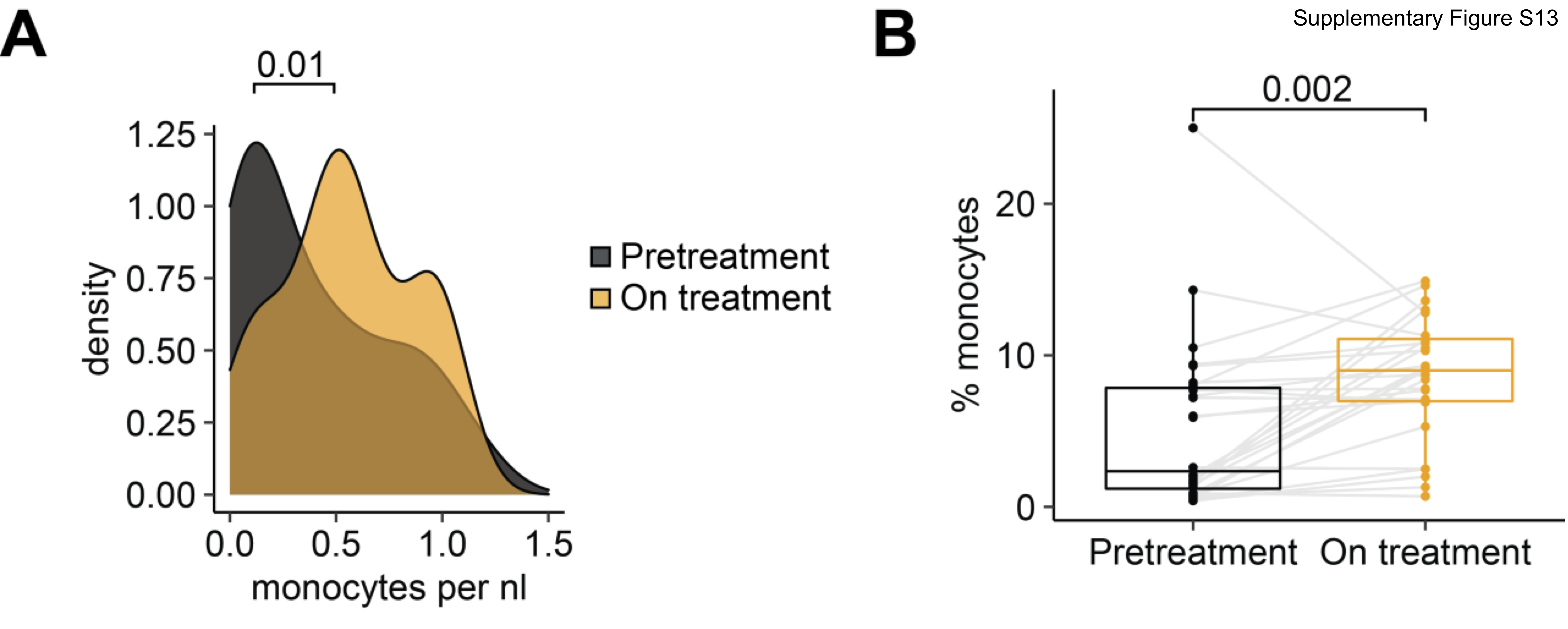
We next turned our attention to the monocyte population and noted that 4 of 5 patients exhibited a trend toward a higher monocyte frequency after chemotherapy (P = 0.061; Fig. 5A), which was consistent with increases in absolute monocytes in complete blood count measurements in our initial patient cohort [baseline (median = 0.08 K/μL), C3 (median = 0.16 K/μL, P = 0.48), C6 (median = 0.46 K/μL, P = 0.004); Supplementary Fig. S12], as well as an independent cohort of 27 patients [baseline (median = 0.26 K/μL) vs. C3 (median = 0.55 K/μL), P = 0.01; Supplementary Fig. S13]. Importantly, increased monocyte frequencies after the third cycle of chemotherapy were also seen by flow cytometric analysis of CD14+ cells in matched PBMC samples from the same 5 patients analyzed by RNAseq (Pts. 1, 2, 3, 6, and 7; Supplementary Fig. S8A). This increased frequency of monocytes was independent of (peg)filgrastim administration as only patient 3 (of the 5 patients studied), had Neupogen administration before C3 (Supplementary Fig. S1). To confirm whether chemotherapy impacted the size of the monocyte population, we also evaluated the composition of immune cells in 3 healthy donors from reference datasets (ref. 23; in Materials and Methods). The percentage of circulating monocytes in healthy donors was more comparable with patient values at C3 than at baseline, at least for 4 of 5 patients with EOC (Pts. 1, 2, 3, and 7), supporting the notion of monocyte number recovery following chemotherapy (Fig. 4C). We next asked whether increased monocyte frequency was driven by specific monocyte subpopulations. Subclustering of monocytes revealed two populations, which corresponded to CD14++CD16– classical and cytotoxic monocytes based on prior characterization of PBMCs by Villani and colleagues (ref. 24; Supplementary Fig. S14A and S14B). However, it did not appear that either monocyte population was specifically increased after chemotherapy (Supplementary Fig. S14C and S14D). We also subclustered DCs, identifying four subpopulations (CLEC9A DCs, pDCs, non-inflammatory and inflammatory DCs; ref. 24); however, we observed no clear patterns in DC subpopulation representation before or after chemotherapy, though the limited sample size and our inability to perform orthogonal analyses of DCs across multiple datasets preclude definitive conclusions regarding DCs (Supplementary Fig. S16). Overall, these analyses suggest that monocyte frequency increases after neoadjuvant chemotherapy, and that this increase is not driven by a specific monocyte subpopulation.
Figure 5.

Differential gene expression analysis in monocytes after chemotherapy. A, Quantification of the percentage of monocytes pre-chemotherapy (baseline) and after the third cycle of chemotherapy (C3). A P value was calculated using a two-sided one-sample t test. Black lines represent CA125 complete responders and the red line represents the CA125 incomplete responder. B, Differentially expressed genes in 5 patients (Pts. 1, 2, 3, 6, and 7) in the combined dataset between baseline and C3. Red dots represent genes with statistically significant change in gene expression (FDR-corrected P < 0.01 and log2 fold change > 0.5). Positive values indicate upregulated genes after C3. C, Heatmap of the average expression of the 8 HLA genes labeled in B at baseline and C3. Patients labeled in black were CA125 complete responders and the patient labeled in red was a CA125 incomplete responder. D, Enrichment score of the identified upregulated genes at C3 overlapped with the Gene Ontology Biological Process (GOBP) antigen processing and presentation pathway gene set.
Differential gene expression analysis of combined monocyte cells revealed upregulation of transcription factors such as JUN and LYZ as well as HLA class II genes after chemotherapy (Fig. 5B and C). We asked whether the upregulation of HLA class II genes was driven by a particular monocyte subset. We observed higher average HLA class II gene expression in the CD14++CD16– classical monocyte population compared with the cytotoxic monocyte population (Supplementary Fig. S15A), but both populations exhibited increased HLA class II expression at C3 compared with baseline (Supplementary Fig. S15B). Thus, the CD14++CD16– classical monocyte population is likely the major source of upregulated HLA class II within the monocyte cluster, but all monocytes appear to increase HLA class II expression after chemotherapy. Interestingly, gene set enrichment analysis of upregulated genes in the overall monocyte population at C3 showed a signature of increased antigen processing and presentation after chemotherapy (Fig. 5D; Supplementary Fig. S17); this signature was also significantly increased in both classical (P = 1.25 × 10–5) and cytotoxic (P = 0.00045) monocyte subsets. No transition from M1-like to M2-like signatures was observed in the peripheral monocytes in our patient cohort. Taken together, chemotherapy appears to increase antigen-processing and presentation programs in monocytes but did not alter the composition of circulating T cells.
Discussion
Previous studies of the immune effects of carboplatin and paclitaxel chemotherapy in EOC have demonstrated that these agents induce a more immune-inflamed phenotype within the ovarian tumor microenvironment with increased CD8+ and CD4+ tumor-infiltrating lymphocytes (TIL), induction of an IFNγ signature, increased mature DCs, and increased expression of PD-L1 in tumor and immune cells (5–9). No effect of chemotherapy on NK cells has been observed (26). Regarding immunosuppressive cells, a decreased number of CD4+CD25+FOXP3+ regulatory T cells has been reported after carboplatin/paclitaxel chemotherapy. In contrast, the number of tumor-associated macrophages (the most abundant infiltrating immune cells in the ovarian tumor microenvironment) and the ratio of their immunosuppressive M2 versus the antitumor/cytotoxic M1 phenotype does not appear to be affected by chemotherapy (9, 26). However, although understanding local tissue-based immune responses is critical for elucidating direct tumor–immune cell interactions, peripheral immune responses are increasingly recognized as occupying an important role in anticancer immunity. Furthermore, ovarian cancer exhibits a high degree of heterogeneity in its local immune microenvironments, whereby distinct tumor lesions within the same patient may exhibit dramatically different immune phenotypes ranging from the one extreme of immune activation to the other extreme of complete immune exclusion (27). Therefore, focusing on a specific immune microenvironment of an isolated tumor lesion cannot adequately capture the global immunological consequences of systemic chemotherapy.
To address these issues, we evaluated the peripheral immune effects of chemotherapy in EOC by interrogating serial blood samples from patients with advanced disease treated with NACT. We systematically characterized the evolution of peripheral immune cell function and composition across the course of EOC therapy. We report that, despite using potent cytotoxic agents such as carboplatin and paclitaxel, and despite the use of dexamethasone with every chemotherapy cycle (refs. 21, 28, 29; as per standard-of-care), we observed increased T-cell responses against viral antigens after administration of chemotherapy. Notably, the increase in T-cell responses paralleled the decrease in CA125 levels suggesting that reductions in tumor volume may alleviate tumor-associated immunosuppression leading to less inhibition of immune responses. In this regard, in CA125 complete responders, T-cell responses increased promptly, after only 3 cycles of chemotherapy, consistent with their excellent response to chemotherapy. Conversely, in CA125 incomplete responders, increased T-cell responses against viral antigens occurred later (i.e., after interval surgery and additional chemotherapy cycles), suggesting that further reduction in tumor volume was necessary to restore T-cell responsiveness in these patients. Taken together, our results support the notion that reduction in tumor volume either via chemotherapy alone (in CA125 complete responders) or via a combination of surgery and chemotherapy (in CA125 incomplete responders) correlates with the restoration of T-cell responsiveness. Similar observations of restored T-cell responsiveness after cytotoxic chemotherapy have been reported in alternative tumor settings such as breast cancer (30). However, we acknowledge that although ELISpot responses to common viral antigens support the notion that reductions in tumor volume via surgery and chemotherapy restores T-cell responsiveness, the same mechanism may not apply to exhausted tumor antigen-specific T cells in ovarian cancer.
Consistent with the increase in T-cell responses, single-cell analysis revealed an increased number of CD8+ memory TCR clonotypes, again potentially reflecting the reinvigoration of suppressed effector and memory T-cell populations due to reduced tumor burden after chemotherapy. Previous studies have shown that cross-reactivity to similar epitopes derived from other microbial genomes may elicit effector and memory T-cell populations against viral antigens that individuals may not have previously encountered (31). Thus, the result of increased T-cell responses to influenza and CEF peptide pools may not be solely due to viral infections but may also reflect cross-reactivity with epitopes from other microbes, thereby explaining the upward trend in memory T-cell populations in the single-cell RNAseq analysis. Alternatively, this may reflect increased homeostatic proliferation of memory T cells with respect to other T-cell populations, after cytotoxic chemotherapy. Consistent with our finding of increased memory T cells, and especially central memory CD8+ T-cell clonotypes, after the third cycle of chemotherapy, others have also described faster recovery of CD8+ T cells compared with CD4+ and B-cell populations, and an increased memory component in repopulating cells after chemotherapy (32, 33).
Another important finding of our study was the increased frequency of monocytes with concomitant elevation of HLA class II expression and the potential for enhanced antigen presentation, after chemotherapy. Previous studies have demonstrated that chemotherapy‐induced immunogenic cell death (ICD) and release of tumor-associated antigens may promote phagocytic uptake of cancer cells, increase antigen cross-presentation and facilitate infiltration of activated cytotoxic T cells in the tumor microenvironment (34). In this regard, several studies have demonstrated that platinum analogs and anti-tubulin agents can induce ICD, independently of their cytotoxic effects (35, 36). Of note, carboplatin also induces DNA double-strand breaks (DSB) that, especially in the setting of the homologous recombination DNA repair deficiencies that are prevalent in high-grade serous ovarian cancer, may activate the innate immune system via the STING pathway, leading to a type I interferon response and robust immunostimulatory effects on antigen-presenting and effector cells (37, 38).
Single-cell sequencing technologies have been successfully applied in various malignancies to address questions related to tumor subtypes, evolution, and response to systemic therapy (21, 28, 39, 40). A single-cell landscape of patients with ovarian cancer, previously reported by Izar and colleagues (40), demonstrated intra- and interpatient variations of tumor and immune cells in the ascites ecosystem. The same investigators also showed that despite intrapatient heterogeneity, the expression of HLA class II in subsets of malignant cells were consistent across multiple patients and were associated with increased TILs, improved prognosis, and treatment response (40). We similarly observed heterogeneous patterns of circulating immune cells, again with an upward trend in HLA class II and antigen presentation gene expression in circulating monocytes after chemotherapy. Thus, interconnections between malignant and non-malignant compartments, as well as circulating immune cells, may occur during tumor–host co-evolution and administration of systemic therapy. Nonetheless, it is important to underscore that variations between TILs and circulating immune cells may also exist. For example, unlike previous studies of the immune effects of carboplatin/paclitaxel chemotherapy discussed above, we observed neither an increase in CD8+ T cells nor an increase in PD-L1/PD-1 expression in circulating T cells. These observations highlight differences between immune cells residing in the tumor microenvironment versus the systemic circulation.
We acknowledge certain limitations of this study. The number of patients was small, as this study was hypothesis generating, focused on a comprehensive evaluation of peripheral immune responses across four different time points during the course of therapy. Samples were collected over a period of more than 6 months for each patient and, despite our best efforts, missing or insufficient samples did not permit completion of all planned analyses at all-time points for all patients. These limitations notwithstanding, our results provide clinically meaningful insights that may be relevant for immunotherapy in this disease. Specifically, ovarian cancer remains one of the few malignancies where immune checkpoint inhibitors (ICI) exhibit only modest activity as monotherapy and currently have no FDA-approved indication (41). Our results suggest that use of cytotoxic chemotherapy is unlikely to be the cause of the modest response of ovarian cancer to ICIs; on the contrary, the immune effects of carboplatin/paclitaxel chemotherapy observed in our study (increased T-cell responses to viral antigens, increased monocytes with concomitant elevation of HLA class II expression and antigen presentation genes) suggest that chemotherapy may actually prime ovarian tumors to respond to immunotherapy. Thus, although ICI monotherapy has not been shown to be widely effective in ovarian cancer (because other mechanisms of immunosuppression may play a role in ovarian cancer), our data suggest rather that novel combinatorial strategies involving ICIs may be more effective. Another implication of our findings is that use of cytotoxic chemotherapy, including use of steroids as premedication, should not preclude application of therapeutic vaccine approaches in this setting. On the basis of our findings, T-cell responses improve after administration of chemotherapy as tumor burden decreases, with the restoration of T-cell responses in all patients 2 months after completion of chemotherapy thereby providing guidance on the optimal timing of vaccine administration. Finally, our study highlights that monitoring of peripheral immune cell responses is feasible and may correlate with the efficacy of antitumor therapy.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the patients and their families for their generous participation in this research study. We thank Dr. Nicoletta Cieri for flow cytometry advice and Swati Narayan for help with collating clinical records and data. This work was funded by the Dana-Farber/Harvard Cancer Center Specialized Program of Research Excellence (SPORE) in Ovarian Cancer (NIH/NCI 2P50CA240243–01A1; to P.A. Konstantinopoulos, U.A. Matulonis, and C.J. Wu). In addition, S. Li is supported by the NCI Research Specialist Award (R50CA251956), L. Penter is a Scholar of the American Society of Hematology, and M. Sellars is supported by an NIH training grant (T32CA9172–46).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
S. Li reports grants from NCI during the conduct of the study. J.F. Liu reports personal fees from AstraZeneca, Clovis, Eisai, Genentech/Roche, GSK, Regeneron Pharmaceuticals, and EpsilaBio outside the submitted work. E.H. Stover reports grants from National Cancer Institute during the conduct of the study, as well as a patent for U.S. 16/499,393 pending to Dana-Farber Cancer Institute. K.J. Livak reports grants from NIH/NCI during the conduct of the study, as well as stock ownership in Fluidigm Corporation. U.A. Matulonis reports personal fees from Merck, AstraZeneca, Immunogen, Trillium, Agenus, Blueprint Medicines, GlaxoSmithKline, Novartis, Boehringer Ingelheim, 2X Oncology, Synphogen, Advaxis, and Alkermes outside the submitted work. C.J. Wu reports other support from BioNTech and Pharmacyclics outside the submitted work. D.B. Keskin reports equity in Affimed N.V., Armata Pharmaceuticals, Breakbio, BioMarin Pharmaceutical, Bristol Myers Squibb, Celldex Therapeutics, Editas Medicine, Exelixis, Gilead Sciences, Immunitybio, ImmunoGen, IMV, Lexicon Pharmaceuticals, Moderna, Neoleukin Therapeutics, and Regeneron Pharmaceuticals. D.B. Keskin also reports that BeiGene, a Chinese biotech company, supports unrelated research at TIGL. P.A. Konstantinopoulos reports grants from Eli Lilly; grants and personal fees from Bayer, GSK, and AstraZeneca; and personal fees from Kadmon, Mersana, Artios, Repare, and BMS outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
M. Liu: Conceptualization, data curation, formal analysis, writing–original draft. N. Tayob: Formal analysis, writing–review and editing. L. Penter: Supervision, visualization, methodology, writing–review and editing. M. Sellars: Conceptualization, resources, formal analysis, investigation, writing–review and editing. A. Tarren: Formal analysis, methodology. V. Chea: Formal analysis, writing–review and editing. I. Carulli: Formal analysis, writing–review and editing. T. Huang: Formal analysis, writing–review and editing. S. Li: Formal analysis, methodology, writing–review and editing. S.-C. Cheng: Data curation, writing–review and editing. P. Le: Data curation, formal analysis, methodology, writing–review and editing. L. Frackiewicz: Data curation, writing–review and editing. J. Fasse: Data curation, writing–review and editing. C. Qi: Data curation, writing–review and editing. J.F. Liu: Resources, validation, writing–review and editing. E.H. Stover: Resources, validation, writing–review and editing. J. Curtis: Resources, writing–review and editing. K.J. Livak: Formal analysis, writing–review and editing. D. Neuberg: Formal analysis, writing–review and editing. G. Zhang: Formal analysis, writing–review and editing. U.A. Matulonis: Resources, project administration, writing–review and editing. C.J. Wu: Conceptualization, resources, formal analysis, writing–original draft, project administration, writing–review and editing. D.B. Keskin: Conceptualization, data curation, formal analysis, supervision, writing–original draft, project administration, writing–review and editing. P.A. Konstantinopoulos: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, writing–original draft, project administration, writing–review and editing.
References
- 1. Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers 2016;2:16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Konstantinopoulos PA, Awtrey CS. Management of ovarian cancer: a 75-year-old woman who has completed treatment. JAMA 2012;307:1420–9. [DOI] [PubMed] [Google Scholar]
- 3. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov 2015;5:1137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Konstantinopoulos PA, Norquist B, Lacchetti C, Armstrong D, Grisham RN, Goodfellow PJ, et al. Germline and somatic tumor testing in epithelial ovarian cancer: ASCO guideline. J Clin Oncol 2020;38:1222–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Böhm S, Montfort A, Pearce OM, Topping J, Chakravarty P, Everitt GL, et al. Neoadjuvant chemotherapy modulates the immune microenvironment in metastases of tubo-ovarian high-grade serous carcinoma. Clin Cancer Res 2016;22:3025–36. [DOI] [PubMed] [Google Scholar]
- 6. Khairallah AS, Genestie C, Auguste A, Leary A. Impact of neoadjuvant chemotherapy on the immune microenvironment in advanced epithelial ovarian cancer: prognostic and therapeutic implications. Int J Cancer 2018;143:8–15. [DOI] [PubMed] [Google Scholar]
- 7. Lo CS, Sanii S, Kroeger DR, Milne K, Talhouk A, Chiu DS, et al. Neoadjuvant chemotherapy of ovarian cancer results in three patterns of tumor-infiltrating lymphocyte response with distinct implications for immunotherapy. Clin Cancer Res 2017;23:925–34. [DOI] [PubMed] [Google Scholar]
- 8. Mesnage SJL, Auguste A, Genestie C, Dunant A, Pain E, Drusch F, et al. Neoadjuvant chemotherapy (NACT) increases immune infiltration and programmed death-ligand 1 (PD-L1) expression in epithelial ovarian cancer (EOC). Ann Oncol 2017;28:651–7. [DOI] [PubMed] [Google Scholar]
- 9. Polcher M, Braun M, Friedrichs N, Rudlowski C, Bercht E, Fimmers R, et al. Foxp3(+) cell infiltration and granzyme B(+)/Foxp3(+) cell ratio are associated with outcome in neoadjuvant chemotherapy-treated ovarian carcinoma. Cancer Immunol Immunother 2010;59:909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- 11. Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, et al. Anti–PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med 2016;8:370ra180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer 2021;21:345–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keskin DB, Reinhold BB, Zhang GL, Ivanov AR, Karger BL, Reinherz EL. Physical detection of influenza A epitopes identifies a stealth subset on human lung epithelium evading natural CD8 immunity. Proc Natl Acad Sci U S A 2015;112:2151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reche PA, Keskin DB, Hussey RE, Ancuta P, Gabuzda D, Reinherz EL. Elicitation from virus-naive individuals of cytotoxic T lymphocytes directed against conserved HIV-1 epitopes. Med Immunol 2006;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oliveira G, Ruggiero E, Stanghellini MT, Cieri N, D'Agostino M, Fronza R, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T-cell immunological memory. Sci Transl Med 2015;7:317ra198. [DOI] [PubMed] [Google Scholar]
- 16. Hu Z, Anandappa AJ, Sun J, Kim J, Leet DE, Bozym DJ, et al. A cloning and expression system to probe T-cell receptor specificity and assess functional avidity to neoantigens. Blood 2018;132:1911–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li S, Sun J, Allesoe R, Datta K, Bao Y, Oliveira G, et al. RNase H-dependent PCR-enabled T-cell receptor sequencing for highly specific and efficient targeted sequencing of T-cell receptor mRNA for single-cell and repertoire analysis. Nat Protoc 2019;14:2571–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rempala GA, Seweryn M. Methods for diversity and overlap analysis in T-cell receptor populations. J Math Biol 2013;67:1339–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bagaev DV, Vroomans RMA, Samir J, Stervbo U, Rius C, Dolton G, et al. VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res 2020;48:D1057–D62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tickotsky N, Sagiv T, Prilusky J, Shifrut E, Friedman N. McPAS-TCR: a manually curated catalogue of pathology-associated T-cell receptor sequences. Bioinformatics 2017;33:2924–9. [DOI] [PubMed] [Google Scholar]
- 21. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T-cell responses in phase Ib glioblastoma trial. Nature 2019;565:234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data .. Cell 2021;184:3573–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017;356:4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cannistra SA. Cancer of the ovary. N Engl J Med 2004;351:2519–29. [DOI] [PubMed] [Google Scholar]
- 26. Yaniz E, Genestie C, Klein C, Salviat F, Ray-Coquard IL, Joly F, et al. Impact of chemotherapy alone or in combination with an anti-angiogenic on the immune tumor microenvironment (TME) of ovarian cancer: data from the randomized CHIVA trial (a GINECO–GINEGEPS study). J Clin Oncol 2020;38:6011. [Google Scholar]
- 27. Jimenez-Sanchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 2017;170:927–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu Z, Leet DE, Allesoe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory T-cell responses and epitope spreading in patients with melanoma. Nat Med 2021;27:515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iorgulescu JB, Gokhale PC, Speranza MC, Eschle BK, Poitras MJ, Wilkens MK, et al. Concurrent dexamethasone limits the clinical benefit of immune checkpoint blockade in glioblastoma. Clin Cancer Res 2021;27:276–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bernal-Estevez D, Sanchez R, Tejada RE, Parra-Lopez C. Chemotherapy and radiation therapy elicits tumor specific T-cell responses in a breast cancer patient. BMC Cancer 2016;16:591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity 2013;38:373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verma R, Foster RE, Horgan K, Mounsey K, Nixon H, Smalle N, et al. Lymphocyte depletion and repopulation after chemotherapy for primary breast cancer. Breast Cancer Res 2016;18:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fagnoni FF, Lozza L, Zibera C, Zambelli A, Ponchio L, Gibelli N, et al. T-cell dynamics after high-dose chemotherapy in adults: elucidation of the elusive CD8+ subset reveals multiple homeostatic T-cell compartments with distinct implications for immune competence. Immunology 2002;106:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hannani D, Sistigu A, Kepp O, Galluzzi L, Kroemer G, Zitvogel L. Prerequisites for the antitumor vaccine-like effect of chemotherapy and radiotherapy. Cancer J 2011;17:351–8. [DOI] [PubMed] [Google Scholar]
- 35. Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer 2018;118:312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rebe C, Demontoux L, Pilot T, Ghiringhelli F. Platinum derivatives effects on anticancer immune response. Biomolecules 2019;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by S-phase–specific DNA damage in breast cancer. J Natl Cancer Inst 2017;109:djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, et al. Consensus guidelines for the definition, detection, and interpretation of immunogenic cell death. J Immunother Cancer 2020;8:e000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep 2014;6:514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Izar B, Tirosh I, Stover EH, Wakiro I, Cuoco MS, Alter I, et al. A single-cell landscape of high-grade serous ovarian cancer. Nat Med 2020;26:1271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Konstantinopoulos PA, Cannistra SA. Immune checkpoint inhibitors in ovarian cancer: can we bridge the gap between IMagynation and reality? J Clin Oncol 2021;17:1833–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The single cell RNAseq and TCRseq data generated in this study are publicly available in the database of Genotypes and Phenotypes (dbGaP) at phs002862.v1.p1.
The bulk TCR sequencing data generated in this study are publicly available in the Gene Expression Omnibus (GEO) at GSE206339 (hyperlink: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE206339).
Other data analyzed in this study were obtained from Gene Expression Omnibus (GEO) at GSE164378 (hyperlink: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164378) and from 10x Genomics for samples 5k_pbmc_v3 (hyperlink: https://www.10xgenomics.com/resources/datasets/5-k-peripheral-blood-mononuclear-cells-pbm-cs-from-a-healthy-donor-v-3-chemistry-3-1-standard-3-0-2) and Parent_NGSC3_DI_PBMC (hyperlink: https://www.10xgenomics.com/resources/datasets/pbm-cs-from-a-healthy-donor-whole-transcriptome-analysis-3-1-standard-4-0-0).