

Abstract
Obesity is a global epidemic and a major predisposing factor for cancer. Increasing evidence shows that obesity-associated stress is a key driver of cancer risk and progression. Previous work has identified the phase-separation organelles, stress granules (SGs), as mutant KRAS-dependent mediators of stress adaptation. However, the dependence of tumorigenesis on these organelles is unknown. Here, we establish a causal link between SGs and pancreatic ductal adenocarcinoma (PDAC). Importantly, we uncover that dependence on SGs is drastically heightened in obesity-associated PDAC. Furthermore, we identify a previously unknown regulator and component of SGs, namely the serine/arginine protein kinase 2 (SRPK2), as a specific determinant of SG formation in obesity-associated PDAC. We show that SRPK2-mediated SG formation in obesity-associated PDAC is driven by hyperactivation of the IGF1/PI3K/mTOR/S6K1 pathway, and that S6K1 inhibition selectively attenuates SGs and impairs obesity-associated PDAC development.
INTRODUCTION
Cancer cells share a set of properties that are collectively referred to as the stress phenotype of cancer, which reflects the prominent levels of various cellular stresses that are present in cancer tissues (1). As cellular stress is detrimental to survival, tumorigenesis is interlocked with the capacity of cancer cells to activate stress adaptive mechanisms such as macropinocytosis, autophagy, and unfolded protein response (UPR) (2–4). Each of these stress adaptive mechanisms is specific to the type of stress and promotes tumorigenesis by enhancing the fitness of cancer cells as well as resistance to therapies. In contrast to stress-specific adaptive mechanisms, SGs are non-membranous organelles that assemble in response to several tumor-associated stresses including oxidative stress, hypoxia, ER-stress, nutrient deprivation, and osmotic stress (5–10). SGs are biomolecular condensates that form by liquid-liquid phase separation (LLPS) and compartmentalize hundreds of proteins and thousands of mRNA molecules (11–17). While much remains to be discovered on the biological pathways impacted by SGs, they are critical to cell survival under stress with particular relevance to pancreatic ductal adenocarcinoma (PDAC) cells (18–20). PDAC is the prototypical KRAS-driven cancer and an aggressive disease that is continually increasing in incidence; the 5-year survival rate for PDAC is only 10% and therapeutic options are severely deficient (21). Mutant KRAS was shown to upregulate the capacity of PDAC cells to form SGs leading to enhanced resistance to several stress stimuli and chemotherapeutic agents (18). Direct evidence linking SGs to tumorigenesis, however, is lacking.
In considering the relevance of SGs to tumorigenesis, it is important to note the ample epidemiological evidence and studies in mouse models that show that pre-existing stress and inflammatory conditions promote cancer occurrence and development (22–24). Among these conditions, obesity affects ~2/3 of adults in the US and ~50% worldwide, and is a growing global epidemic (25–28). Obesity doubles the incidence risk and mortality for pancreatic cancer, and even higher relative risks and mortality rates have been observed for prevalent cancers such as colon, breast, liver, kidney, stomach, and uterine cancer (29,30). The precise mechanisms though which obesity promotes cancer occurrence and progression are unknown. However, obesity is a strong inducer of ER-, oxidative-, genotoxic-, and biomechanical-stress, and evidence points to obesity-associated cellular stress as a critical intermediary in obesity-associated cancer (31–34). Here we aimed to ascertain that SGs, as a pan-stress adaptive mechanism, would be required in PDAC development, and that the pre-existing stress of obesity would dictate a higher selective pressure for SG-mediated stress adaptation, and a higher reliance of obesity-associated PDAC on SGs.
RESULTS
SGs promote pancreatic tumorigenesis.
Recent work has shown that SG formation is determined by the collective interactions of ~36 proteins and their associated mRNAs, and that G3BP proteins are a central node of this network (14). To assess the role of SGs in PDAC growth, we utilized PDAC cell lines derived from the KPC (LSL-KrasG12D/+;LSL-Trp53R172H/+; Pdx-1-Cre) genetically engineered mouse model (GEMM) of KRAS-driven murine PDAC (mPDAC) (35–38). KPC cells were engineered to stably express one of two independent doxycycline (Dox)-inducible small hairpin (sh) RNAs that target G3bp1, or a control non-targeting (NT) shRNA (Figure 1A–B). Immunofluorescence microscopy for two SG markers, Pumilio and endogenous eukaryotic initiation factor 4G (Eif4g), revealed that specific knockdown of G3bp1 attenuated SG formation in KPC cells exposed to oxidative stress via treatment with sodium arsenate (SA) (Figure 1A). As expected, SGs are not present in the absence of stress stimuli in cell culture conditions. Furthermore, oxidative stress induces the translocation of Eif4G and Pumilio to SGs but has no impact on their expression levels. Quantification of the SG index by computing the cell area occupied by SGs (labeled by Pumilio or Eif4g) as a fraction of total cell area, showed that knockdown of G3bp1 attenuated SG formation by 80% for G3bp1 sh #1, and by ~60% for G3bp1 sh # 2 (Figure 1C) (18). Attenuation of the SG index reflects a diminished percentage of SG positive cells and average size of SGs (Figure S1A–B). As cancer cells within a tumor are exposed to diverse types of stress and it has been suggested that the key nucleator molecules driving SG formation may change accordingly, we assessed whether G3bp1 knockdown impacted SG formation under ER stress and hypoxia (Figure 1D) (15,39,40). Knockdown of G3bp1 attenuated SG formation in KPC cells exposed to ER stress and hypoxia by ~60% and ~95%, respectively (Figure 1D). Consistent with a role for G3bp1 in stress adaptation, knockdown of G3bp1 had no impact on cell proliferation under normal growth conditions, but impaired cell survival under oxidative stress (Figure 1E and S1C–D). Altogether these results show that knockdown of G3bp1 attenuates SG formation in PDAC cells under several prevalent tumor-associated stresses.
Figure 1. G3BP1 knockdown impairs SG formation and pancreatic cancer growth.
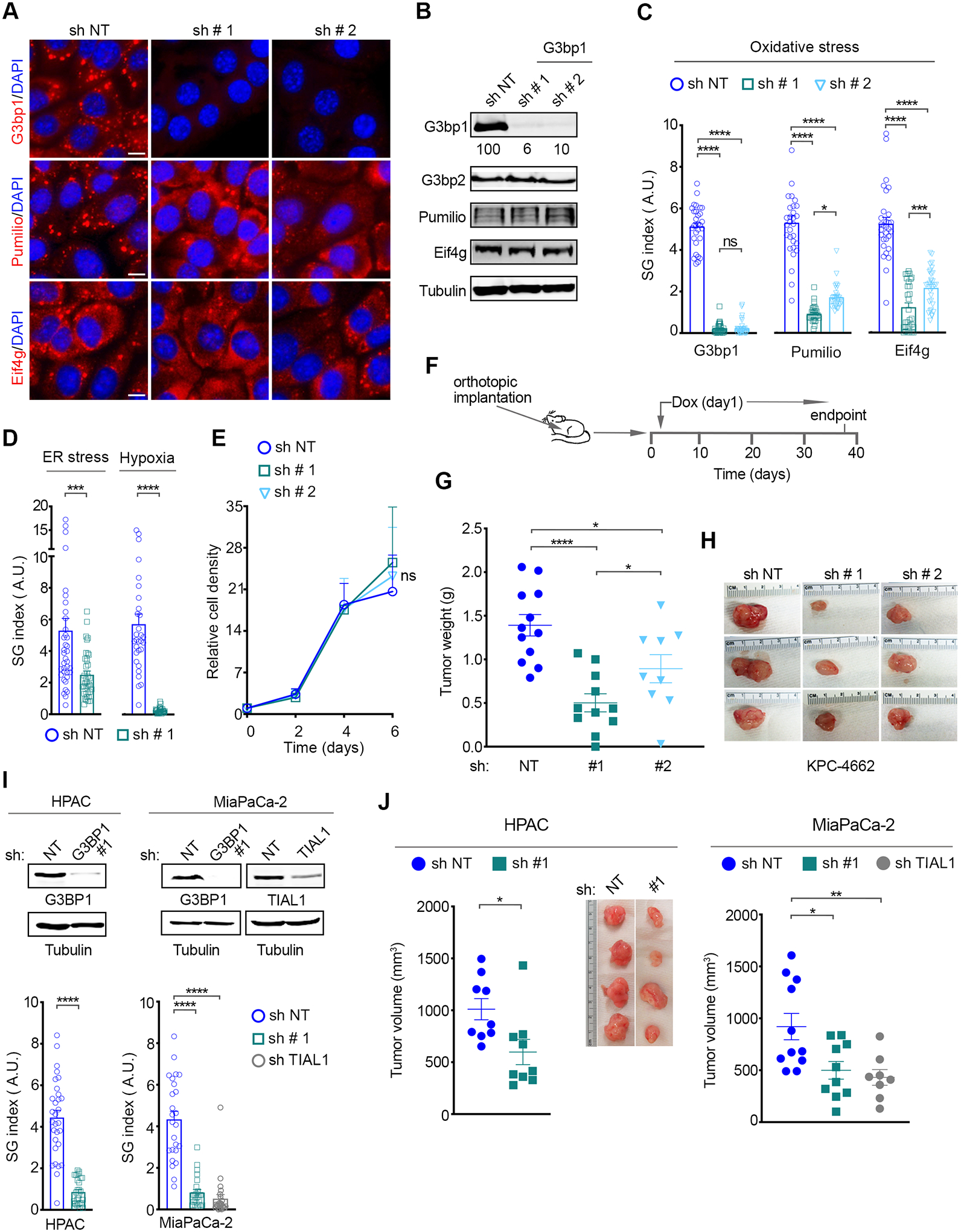
(A) Representative immunostaining of the SG markers G3bp1, Pumilio, Eif4g and DAPI in KPC-4662 expressing a Dox-induced non-targeting shRNA (sh NT) and two shRNAs for G3bp1 (sh # 1, sh # 2) and subjected to oxidative stress. Oxidative stress was induced via treatment with 200 μM sodium arsenate (SA) for 1hr. Scale bar, 10 μm.
(B) Western Blot (WB) of lysates from KPC-4662 cells expressing the indicated shRNAs as in A.
(C) Quantification of SG index as detected by G3bp1, Pumilio, and Eif4g, in KPC-4662 cells as in A. Data from a representative experiment are shown in arbitrary units (a.u.) for each marker and as mean +/− SEM for 30 non-overlapping fields of view (FOV) imaged at 20x. The individual values for each FOV are also shown. ****p<0.0001 by unpaired t-test. See also Figure S1A.
(D) Quantification of SG index in the indicated KPC-4662 cells subjected to ER stress (Thapsigargin, 25 μM, 6 hr) or hypoxia (0.5% O2, 24 hr) as detected by Eif4G and Pumilio, respectively. Measurements from a representative experiment as in C are shown. ***p<0.001, ****p<0.0001 by unpaired t-test.
(E) Relative cell density of KPC-4662 cells expressing the indicated shRNAs measured using a Syto60 stain. Data is mean +/− SD, n=3. ns = non-significant by unpaired t-test.
(F) Schematic of the experimental approach used to assess the impact of G3bp1 knockdown on mPDAC growth. KPC-4662 cells were implanted orthotopically in the pancreas. shRNA expression was induced 24 hr after implantation through Dox administration via drinking water which was changed every 3 days.
(G-H) mPDAC tumor growth at experimental endpoint on day 37 post orthotopic implantation of KPC-4662 cells expressing the indicated shRNAs as in F. (G) Mean mPDAC tumor weight at endpoint +/− SEM and individual tumor weights for each mouse. (H) Representative orthotopic tumors. sh NT n=12, sh # 1 G3bp1 n=11, sh # 2 G3bp1 n=11. *p<0.05, **** p<0.0001 by Mann Whitney t-test.
(I) Top Panel-WB of lysates from human PDAC cell lines MiaPaCa-2 and HPAC expressing the indicated Dox-inducible shRNAs. Bottom panel-Quantification of SG index as detected by eIF4G in cells under oxidative stress (SA, 100 μM, 1 hr). Measurements from a representative experiment as in C are shown. ****p<0.0001 by unpaired t-test. See also Figure S1D–E.
(J) Xenografts of human PDAC cell lines expressing the indicated shRNAs. HPAC and MiaPaCa-2 cells were subcutaneously implanted in athymic nude mice. shRNA expression was induced as in F. Data is endpoint mean tumor volume +/− SEM. The individual tumor volumes for each mouse are shown. HPAC sh NT n=9, sh # 1 G3BP1 n=9; MiaPaCa-2 sh NT n=10, sh #1 G3BP1 n=10, TIAL1 sh n=8. *p<0.05, ** p<0.01 by Mann Whitney t-test.
To assess the contribution of SGs to PDAC development, we initially implanted KPC cells harboring NT- or G3bp1 shRNAs into the pancreata of syngeneic (C57BL/6) mice, and induced shRNA expression via Dox administration through drinking water, starting at 24 hours post-implantation (Figure 1F). As previously reported, tumors arising from orthotopic implantation of KPC-4662 cells recapitulate the histopathological complexity and dense, desmoplastic microenvironment of human disease (Figure S1E) (36–38). G3bp1 knockdown led to a ~50% reduction in mPDAC growth (60% and 40% reduction for mG3bp1 sh # 1 and sh # 2, respectively) compared to control (sh NT) (Figure 1G–H). Knockdown of G3BP1 also led to robust inhibition of SG formation in human PDAC (hPDAC) cell lines (by 80% and 90% in HPAC and MiaPaCa-2 cells respectively) (Figure 1I and S1F–G) and, as seen with KPC cells, had no impact on cell proliferation under normal growth conditions (Figure S1H). Consistent with our observations in mPDAC, G3BP1 knockdown in xenografts of hPDAC cells in athymic nu/nu mice, reduced tumor growth by ~50% (Figure 1J).
To establish that the impact of G3BP1 knockdown on tumor growth is due to its function as a SG nucleator, we assessed whether targeting another SG nucleator, the T-Cell-Restricted Intracellular Antigen Like 1 (TIAL1) protein, would phenocopy the effect of G3BP1 knockdown (14,41). Dox treatment of MiaPaCa-2 cells harboring a TIAL1 shRNA downregulated TIAL1 levels and inhibited SG formation under oxidative stress comparable to G3BP1 knockdown (Figure 1I and S1F–G). TIAL1 knockdown had no impact on cell proliferation under normal growth conditions (Figure S1I). TIAL1 suppression in-vivo however, impaired tumor growth by ~50% (Figure 1J). These data show that individual downregulation of two otherwise functionally distinct SG nucleator molecules has a similar impact on hPDAC growth. As such, these results support the idea that the impact of G3BP1 and TIAL1 knockdown on tumor growth is mediated by SGs.
Previous work has shown that the ability of G3BP1 to regulate SG formation is determined by its capacity to LLPS with RNA, which is controlled by the dimerization of G3BP1 via its NTF2-like (NTF2L) domain and the interplay between its three intrinsically disordered regions (IDRs) (14). To causally link SG formation with PDAC growth we initially took advantage of a G3bp1 mutant where the NTF2L domain has been deleted (dNTF2L-G3bp1) and which has been shown to be deficient in SG formation (14,42). As such, G3bp1-knockdown KPC cells were engineered to express shRNA-resistant GFP-G3bp1 WT or GFP-dNTF2L-G3bp1 to comparable levels to endogenous G3bp1 (Figure 2A). Expression of G3bp1 WT or GFP-dNTF2L-G3bp1 in G3bp1-knockdown cells had no impact on proliferation under normal growth conditions (Figure S2A). As expected, the formation of SGs under oxidative stress conditions was fully rescued by expression of shRNA resistant GFP-G3bp1 WT, but not GFP-dNTF2L-G3bp1 (Figure 2B and S2B–D). Furthermore, GFP-G3bp1 WT expression rescued the growth of G3bp1 knockdown mPDAC tumors, whereas expression of GFP-dNTF2L-G3bp1 failed to do so (Figure 2C–D and S2E). Given that the NTF2L domain is critical to SG formation, these results show that the rescue of tumor growth by expression of GFP-G3bp1 WT is determined by its SG-nucleating capacity.
Figure 2. SGs promote pancreatic tumorigenesis.
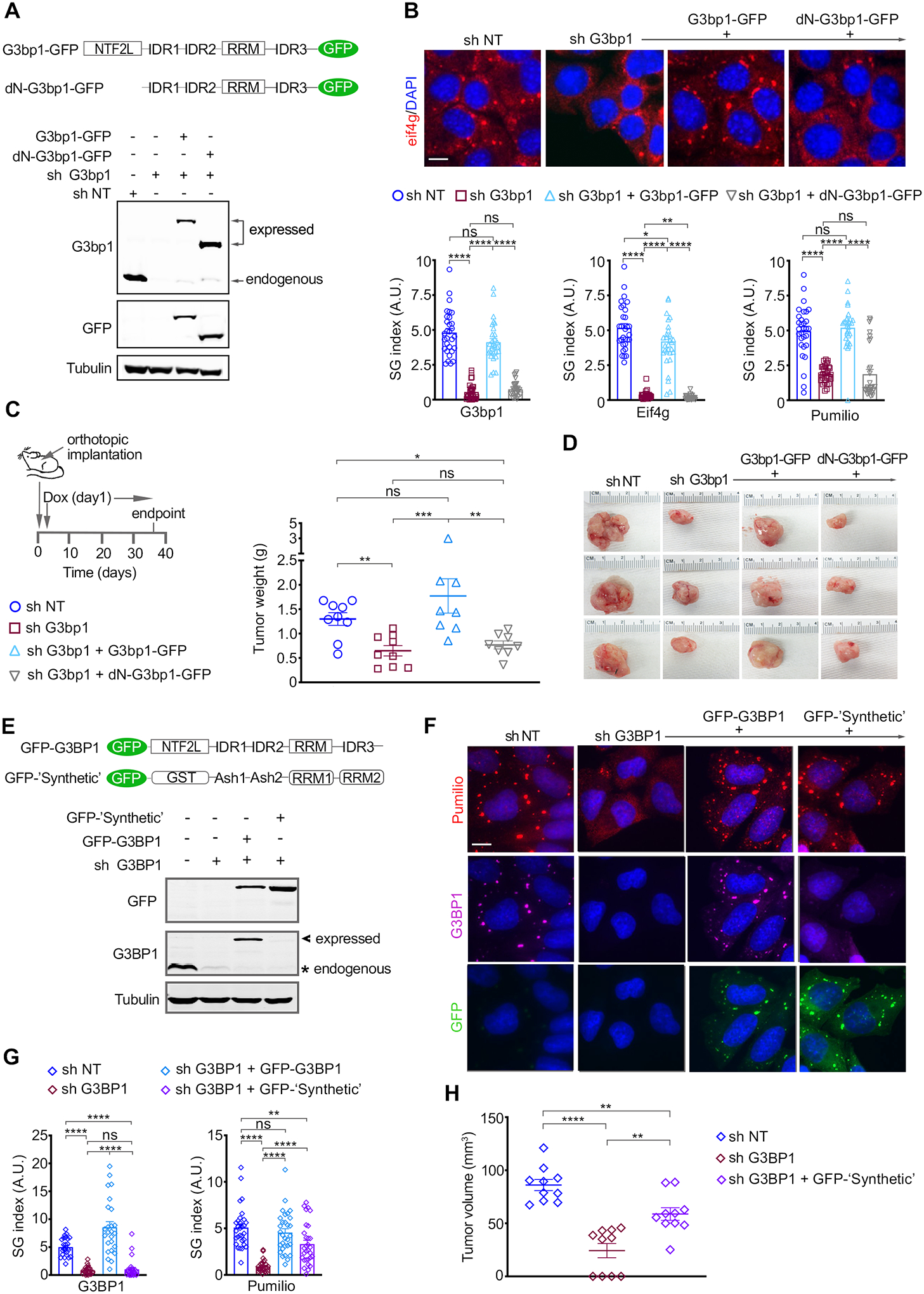
(A) Schematic of G3bp1 expression constructs and WB of lysates of KPC-4662 cells expressing the indicated Dox-inducible shRNA and shRNA-resistant G3bp1 constructs.
(B) Top Panel-Representative immunostaining of Eif4g and DAPI in KPC-4662 cells expressing the indicated constructs as in A and stressed as in 1A. Scale bar, 10 μm. Bottom Panel-Quantification of SG index as detected by G3bp1, Pumilio, and Eif4g. Measurements as in 1C are shown. *p<0.05, ** p<0.01, ****p<0.0001 by unpaired t-test. See also Figure S2A–C.
(C) Left Panel-Schematic of the experimental approach used to assess the impact of SG inhibition on mPDAC growth. KPC-4662 cells were implanted and induced to express the indicated shRNA and shRNA-resistant constructs as in 1F. Right Panel-Mean mPDAC tumor weight at endpoint +/− SEM and individual tumor weights. sh NT n=9, sh # 3 G3bp1 n=9, sh # 3 G3bp1+ G3bp1-GFP n=8, sh # 3 G3bp1+ dN-G3bp1-GFP n=8. *p<0.05, ** p<0.01, *** p<0.001 by Mann Whitney t-test.
(D) Representative mPDAC tumors from mice implanted with the KPC-4662 cell lines in C.
(E) Schematic of expression constructs and WB of lysates of MiaPaCa-2 cells expressing the indicated Dox-inducible shRNAs, and shRNA-resistant G3BP1 and ‘synthetic’ constructs.
(F) Representative immunostaining of G3BP1 and Pumilio in MiaPaCa-2 cells expressing the indicated constructs and stressed as in 1I. Scale bar, 10 μm.
(G) Quantification of SG index as detected by G3BP1 and Pumilio in MiaPaca-2 cells in F. Measurements as in 1C are shown. **p<0.01, ****p<0.0001 by unpaired t-test. See also Figure S2.
(H) Xenografts of MiaPaCa-2 cells expressing the indicated shRNAs and ‘synthetic’ construct. Cell implantation and induction of shRNA expression was performed as in 1J. Data are endpoint mean tumor volume +/− SEM and the individual tumor volumes for each mouse. MiaPaCa-2 sh NT n=10, sh # 2 G3BP1 n=10, sh # 2 G3BP1+ GFP- ‘Synthetic’ n=10. **p<0.01, ****p<0.0001 by Mann Whitney t-test.
To conclusively determine that the impact of G3BP1 knockdown on PDAC growth is due to the inhibition of SG formation, we utilized a ‘synthetic’ construct comprised of domains heterologous to the domains of G3BP1 that are involved in SG formation, and was previously shown to rescue SG formation in G3BP1/G3BP2 knockout cells (Figure 2E) (14). Expression of GFP- ‘synthetic’ in hPDAC cells where G3BP1 was knocked down had no impact on cell proliferation under normal growth conditions but led to a ~3x increase in SGs when cells were under oxidative stress (Figure S2F–G). However, at the attainable expression levels, GFP- ‘synthetic’ was weaker (by 40%) than shRNA-resistant GFP-G3BP1 WT, which rescued SG formation to levels comparable to sh NT expressing cells (Figure 2F–G and S2G). Nonetheless, in agreement with its capacity to rescue SG formation, GFP- ‘synthetic’ rescued the growth deficiency of G3BP1 knockdown tumors by ~2.5x (Figure 2H). Altogether these results causally link SGs to PDAC development.
SGs are upregulated in Obesity-associated PDAC.
Previous work reporting that mutant KRAS upregulated SG formation showed that SG inhibition led to higher cell death in mutant KRAS vs. KRAS wild-type cells under oxidative stress, hence indicating that the levels of SGs can correlate with their requirement for survival (18). To determine the dependence of obesity-associated PDAC on SGs, we initially assessed SG levels in syngeneic orthotopic tumors in mice fed a high-fat chow to model diet-induced obesity (DIO) (Figure 3A). For comparison, we utilized standard weight mice (ST) that were fed standard chow (Figure 3A). Consistent with previous studies, DIO promoted mPDAC growth (Figure 3B and Figure S3A–B) (43–45). As such, tumors in three orthotopic mPDAC models (KPC-4662, KPC-6560, and ES-149) in DIO mice were ~ 3x larger than their counterparts in ST mice (Figure 3B and Figure S3A–B) (36,37,46). In agreement with previous studies, assessment of cell death and proliferation by quantification of the fraction of tumor area positive (+) for cleaved caspase-3 (CC3) and Ki-67, respectively, indicated a ~2.5x decrease in cell death and a ~5x increase in proliferation in DIO vs. ST tumors (Figure S3C–D) (47). Quantification of the SG index revealed a ~5–6x increase in SG levels in DIO vs. ST tumors for KPC-4662, KPC-6560, and ES-149 orthotopic models (Figure 3C–D and Figure S3E). Analysis of tumors of comparable size in the DIO and ST cohorts (Figure S3F) also revealed a ~5x difference in SG levels, indicating that enhanced SG levels in obesity-associated PDAC are linked to obesity and not tumor size. Of note, no SGs were detected in normal pancreata from DIO and ST mice (Figure S3G). In addition, quantification of tumor SGs reflects predominantly cancer cell SGs as stroma cells form relatively, significantly less (1/10th – 1/20th) SGs (Figure S3H).
Figure 3. SGs are upregulated in obesity-associated pancreatic cancer.
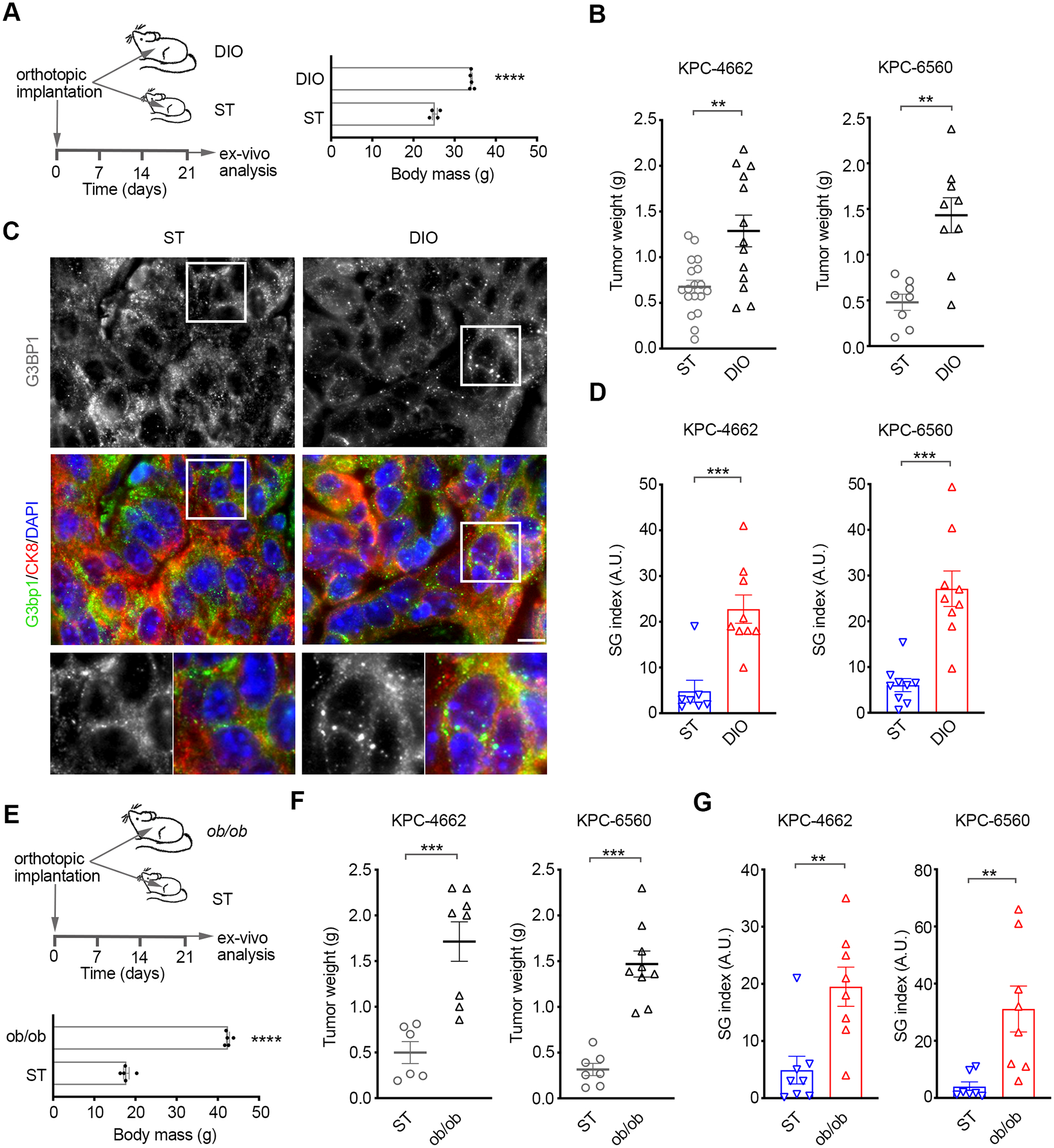
(A) Left Panel-Schematic of the experimental approach used to assess the effect of diet induced obesity (DIO) on mPDAC growth and SG levels. Right Panel-Body weight of age-matched (11–13 weeks) mice fed a standard diet (ST) and high-fat diet (DIO) at implantation of KPC cells. ****p<0.0001 by unpaired t-test.
(B) mPDAC tumor growth at experimental endpoint on day 21 post orthotopic implantation of KPC-4662 and KPC-6560 cells in mice as in A. Data are mean tumor weight at endpoint +/− SEM and individual tumor weights for each mouse. KPC-4662 ST n=18, KPC-4662 DIO n=13, KPC-6560 ST n=8, KPC-6560 DIO n=9, ** p<0.01 by Mann Whitney t-test.
C) Representative immunostaining of G3bp1 and DAPI in KPC-4662 orthotopic tumors in B. Scale bar, 10 μm. Lower panels are 2x zoom-in of boxed regions.
(D) Quantification of SG index in sections from tumors in B as detected by G3bp1. Data are mean SG index +/− SEM and individual values for each tumor. The SG index for each tumor section is the average of 30 non-overlapping FOV imaged at 40x. *** p<0.001 by Mann Whitney t-test.
(E) Schematic of the experimental approach used to assess mPDAC growth and SG levels in ob/ob mice. Body weight of age-matched (7 weeks) of ST and ob/ob mice at implantation. ****p<0.0001 by unpaired t-test.
(F) mPDAC tumor growth at experimental endpoint on day 21 post orthotopic implantation of KPC-4662 and KPC-6560 cells in ST and ob/ob mice. Data are mean tumor weight at endpoint +/− SEM and individual tumor weights for each mouse. KPC-4662 ST n=6, KPC-4662 ob/ob n=8, KPC-6560 ST n=7, KPC-6560 ob/ob n=9, *** p<0.001 by Mann Whitney t-test.
(G) Quantification of SG index in sections from tumors in F as detected by G3bp1. Data was derived and shown as in D. ** p<0.01 by Mann Whitney t-test.
To determine whether the heightened levels of SGs in mPDAC tumors in DIO vs. ST mice were truly a consequence of obesity and not solely a high fat diet, we next utilized the leptin-deficient (ob/ob) genetic model of obesity (Figure 3E and Figure S3I–J). Tumors arising from KPC cells implanted in the pancreata of ob/ob mice vs. age-matched wild-type mice on standard chow were ~3–4x larger and showed a ~5x increase in SG levels (Figure 3F–G and Figure S3J–K). The same difference in SG levels was observed when tumors of comparable size in the ob/ob and ST cohorts were analyzed (Figure S3L). These results are consistent with a model whereby the stress phenotype of obesity-associated PDAC dictates a heightened SG formation capacity.
Dependence of obesity-associated PDAC on SGs.
To assess the dependence of obesity-associated PDAC on SGs we transduced KPC-4662 cells harboring NT- or G3bp1 shRNAs (Figure 1A–B) with a firefly luciferase lentiviral construct to track the development of mPDAC orthotopic tumors in real time by bioluminescent imaging. Western blotting confirmed equal levels of luciferase expression in the established cell lines, and IVIS imaging upon implantation confirmed equal luciferase activity among all cohorts (Figure S4A). As expected, control (sh NT) tumors in DIO mice showed an earlier onset, whereby tumor burden at the earliest measurement on Day 7 in DIO mice was comparable to day 35 in ST mice (Figure 4A–C and Figure S4B). In addition, growth of control DIO tumors proceeded at an accelerated rate compared to control ST tumors and control DIO tumors were significantly larger at endpoint (Figure 4B–C). Inhibition of SGs in tumors in ST mice had no initial impact on tumor burden which became significantly smaller from control tumors only from Day 38 onwards; from this point, SG-inhibited tumors in ST mice progressed with a slower growth rate compared to control ST tumors and showed a ~50% decrease in bioluminescence at endpoint (60% and 40% for G3bp1 sh#1 and sh#2 respectively). The 50% decrease in bioluminescence is consistent with our findings with tumor weight measurements (Figure 4B–C and Figure 1H). Distinct from their ST counterparts, SG-inhibited DIO tumors showed a drastic difference in tumor burden compared to control DIO or ST tumors throughout the duration of the study (Figure 4B–C and Figure S4B). In DIO mice, SG-inhibited tumors showed a significantly smaller tumor burden compared to control from Day 7 (Figure 4C and Figure S4B). This difference expanded as control tumors grew exponentially whereas SG inhibited tumors showed little growth. SG inhibition had no impact on the body weight of ST or DIO mice (Figure S4C). Confirming that the bioluminescence readings accurately reflect tumor size in the DIO model, tumor weight measurements and bioluminescence reads on Day 35 show a similar ~ 5-fold difference between control and SG-inhibited tumors (Figure 4B and Figure S4D). At endpoint measurement on Day 42, SG-inhibited G3bp1 sh#1 and sh#2 tumors in DIO mice were ~ 1/14th (93% smaller) and ~1/7th (87% smaller), respectively, of control tumors (Figure 4B–C). A higher dependence of obesity-associated PDAC on SGs was also observed in a second (KPC-6560) orthotopic model (Figure S4E–G). Knockdown of G3bp1 in KPC-6560 cells inhibited SG formation to a similar extent as in KPC-4662 cells and had no impact on cell proliferation under normal growth conditions (Figure S4E–F). The tumor weight of SG-inhibited KPC-6560 tumors in DIO mice was ~70% smaller than control tumors on Day 21; at this point, consistent with the KPC-4662 model, no difference in tumor weight is observed between SG-inhibited and control tumor in ST mice (Figure S4G–H). Inhibition of SGs also had a heightened impact on established KPC-4662 tumors in DIO vs. ST mice (Figure S4I–J and Supplemental Methods). Dox administration in ST and DIO mice harboring similar size tumors led to a ~ 7x inhibition of the growth of G3bp1 sh vs. NT sh expressing tumors in DIO mice but only a ~2x inhibition in ST mice. In addition, orthotopic mPDAC tumors in the ob/ob model of obesity showed a dependence on SGs comparable to DIO (Figure 4D–E and Figure S4K). SG-inhibited tumors in ob/ob mice were smaller than control starting at the first measurement on Day 6 and throughout the duration of the study with a 91% reduction at endpoint on Day 25 (Figure 4D–E). Of note, tumor burden data past day 42 for DIO, day 45 for ST mice, and Day 25 for ob/ob mice were excluded from the analysis as mice in control conditions were euthanized due to disease progression. Altogether, these data show that orthotopic PDAC onset, progression, and maintenance in obese mice is significantly impaired by SG inhibition, and that the growth and maintenance of obesity-associated PDAC has a higher dependence on SGs.
Figure 4. Dependence of obesity-associated pancreatic cancer on SGs.
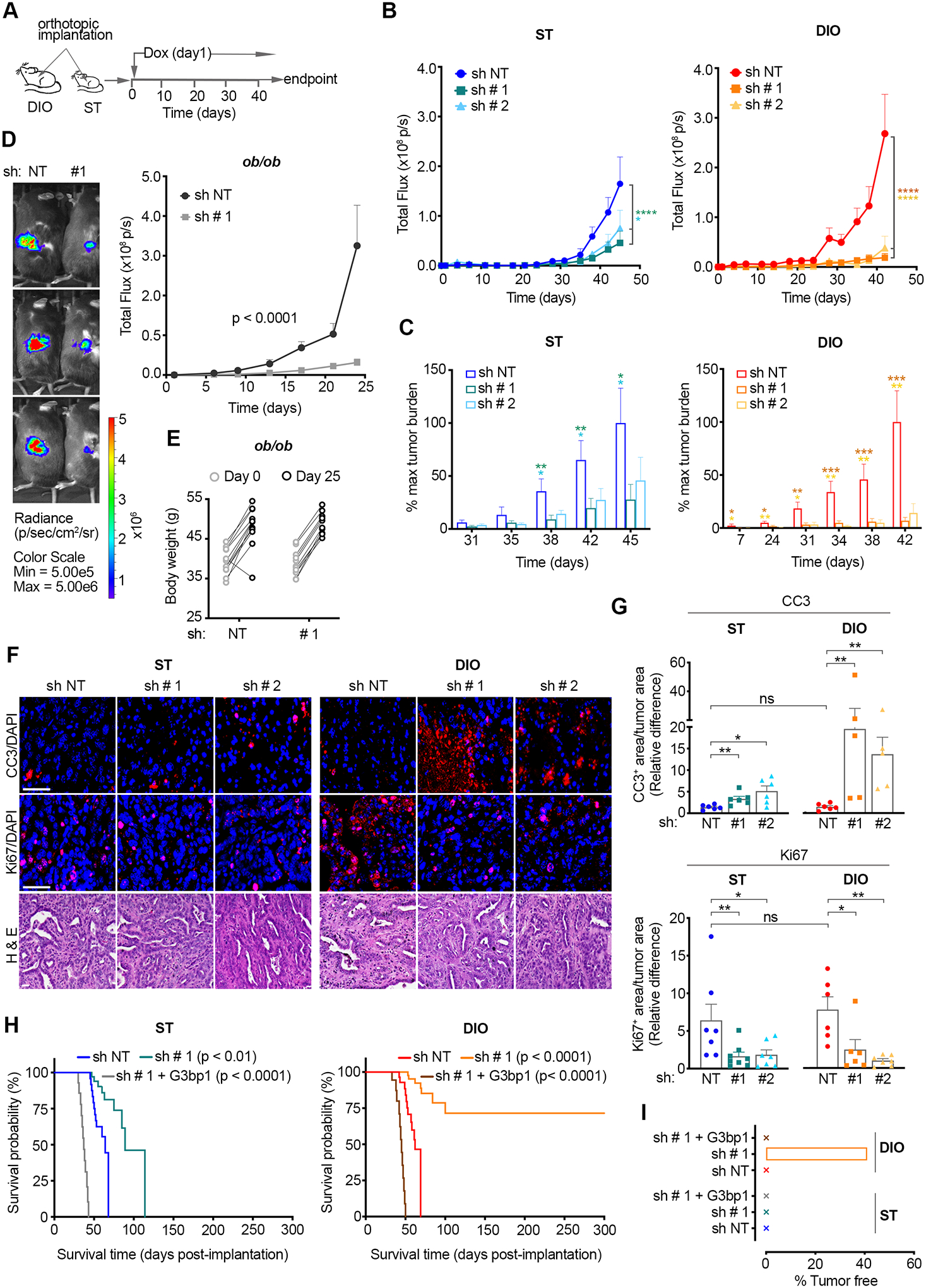
(A) Schematic of the experimental approach used to assess the impact of SG inhibition on mPDAC growth in DIO and ST immunocompetent mice. Luciferase expressing KPC-4662 cells were implanted orthotopically and expression of the indicated shRNAs was induced as in 1F.
(B) Quantification of tumor bioluminescent signal (total flux p per second) over time post orthotopic implantation of KPC-4662 cells in ST and DIO mice as in A. Data are mean signal +/− SEM of individual tumors in each group; ST (sh NT n=12, sh # 1 G3bp1 n=13, sh # 2 G3bp1 n=13), DIO (sh NT n=12, sh # 1 G3bp1 n=14, sh # 2 G3bp1 n=9). *p<0.05, ****p<0.0001 by two-way ANOVA.
(C) Quantification of tumor growth over time as percent of mean bioluminescent signal at endpoint in B. Data are mean +/− SEM of individual mice in each group as in B. *p<0.05, **p<0.01, ***p<0.001 by Mann Whitney t-test.
(D) Left Panel - Representative bioluminescent images. Images taken on day 24 post orthotopic implantation of the indicated KPC-4662 cells in ob/ob immunocompetent mice are shown. The scale indicates radiance expressed as p/sec/cm2/sr. Right Panel - Quantification of bioluminescent signal over time post orthotopic implantation of KPC-4662 cells expressing the indicated shRNAs. Data are presented as mean +/− SEM of individual mice in each group. ob/ob (sh NT n=12, sh # 1 G3bp1 n=12). ****p<0.0001 by two-way ANOVA.
(E) Body weight of ob/ob mice on day 0 and 25 post orthotopic implantation of KPC-4662 cell lines expressing the indicated shRNAs as in D.
(F) Representative immunostaining of cleaved caspase 3 (CC3), Ki67, DAPI, and H&E in sections from KPC-4662 orthotopic tumors in A-C. Scale bar, 50 μm.
(G) Quantification of CC3 or Ki67 positive (+) areas in tumors in A-C. Data are mean caspase 3 area, or Ki67 area, over total tumor area +/− SEM of individual tumors in mice in each group of A-C. The values for each tumor represent the average of non-overlapping FOV imaged at 20x and covering ~50% of each section. * p<0.05, ** p<0.01 by Mann Whitney t-test.
(H) Kaplan-Meier survival curves for ST and DIO mice implanted with KPC-4662 cells and induced to express the indicated shRNA and shRNA-resistant constructs. ST (sh NT n=12, sh # 1 G3bp1 n=9, sh # 1 G3bp1 + G3bp1 WT n=12), DIO (sh NT n=12, sh # 1 G3bp1 n=12, sh # 1 G3bp1 + G3bp1 WT n=12). p-values by log-rank (Mantel-Cox) t-test are shown.
(I) Percent tumor-free mice as in F on day 300.
We next evaluated whether the differential impact of SG inhibition on the growth of obesity-associated vs. ST mPDAC was due to cell death and/or proliferation. SG inhibition led to higher cell death in DIO mice compared to ST mice (Figure 4F–G). As such, CC3+ fractions for G3bp1 sh#1 and sh#2 tumors were respectively ~17x and 8x higher than control (sh NT) tumors in DIO mice, but only ~2x and ~4x higher than control tumors in ST mice (Figure 4F–G). No differences in CC3+ fractions were observed in control tumors in DIO vs. ST. In contrast to cell death, the impact of SG inhibition on proliferation in DIO and ST tumors was similar; G3bp1 sh#1 and sh#2 reduced Ki-67 levels by ~3x and ~7x, respectively, in DIO mice, and by ~4x in ST mice (Figure 4F–G). These results show that SGs can contribute to both cancer cell proliferation and survival in vivo. However, the specific enhancement of tumor cell death that was observed when SGs were inhibited in the DIO setting indicates that obesity-associated PDAC have a higher dependence on SGs for survival.
Following the initial 45-day period, mice were continually monitored for a total of 300 days to determine their rate of survival (Figure 4H–I). Despite accelerated tumor growth, the median survival between DIO and ST mice bearing control tumors was comparable, 62 and 64 days, respectively (Figure 4H). The median survival of ST mice bearing G3bp1-knockdown tumors relative to control was extended by ~1.5x to 89 days. G3bp1-knockdown in tumors in DIO mice on the other hand led to significantly longer survival (Figure 4H). Expression of an shRNA resistant GFP-G3bp1 WT in G3bp1 knockdown cells rescued SG formation and countered the impact of SG inhibition on survival similarly in DIO and ST mice, leading to a median survival of 45 and 37 days, respectively (Figure 4H and Figure S4L–M). Notably, ~40% of DIO mice with SG-inhibited tumors showed no detectable tumors by bioluminescence or gross examination of pancreata and surrounding tissues at endpoint on Day 300 (Figure 4I). Altogether, these data demonstrate that the development and progression of obesity-associated PDAC is highly dependent on SGs.
IGF1 mediates SG upregulation in obesity-associated PDAC
The heightened dependence of obesity-associated PDAC on SGs and enhanced SG levels suggests that obesity may modulate cellular signaling to promote SG formation in cancer cells under stress. The pathobiology of obesity is intricate and multisystemic including major signaling alterations due to secreted factors in the bloodstream from adipose tissue, and perturbed metabolic pathways and tissue homeostasis mechanisms (48). To determine the mechanism(s) through which obesity enhances SG levels in PDAC, we initially assessed how secreted factors with altered circulating levels in obese subjects, namely Interferon gamma (IFNγ), Tumor Necrosis Factor alpha (TNFα), Insulin-like Growth Factor 1 (IGF1), Interleukin-1beta,−6,−10, −4, −13 (IL1β, IL6, IL8, IL10, IL4, IL13), Plasminogen Activator Inhibitor (PAI), Leptin, and Adiponectin, affect the capacity of PDAC cells to form SGs (49–57). Evaluation of SGs in hPDAC cells treated with each of the obesity-associated secreted factors for 2 hours prior to oxidative stress revealed that IGF1 enhanced SG-formation by ~4.5x (Figure 5A–B). Of note, oxidative stress had no impact on IGF1 levels (Figure S5A). A modest upregulation of SGs was observed with IL6, IFNγ, IL10, and IL13 (~1.25x for IL6, IFNγ, IL10 and ~1.14x for IL13) whereas TNFα, Adiponectin, and IL1β led to a slight reduction (~0.75x for TNFα, Adiponectin and ~0.9x for IL1β) (Figure 5A). IL8 and Leptin had no effect. A previous study linked obesity-associated PDAC growth to a crosstalk between locally produced, obesity-driven cholecystokinin (CCK) and pancreatic epithelium (45). While the concentration at which CCK can accumulate locally is unknown, stimulation of hPDAC cells with the reported concentration of CCK in circulation in obesity-associated mPDAC or 3x above, had no impact on SG formation (Figure S5B and Supplemental Methods). The capacity of IGF1 to potently promote SG formation was maintained throughout a panel of mouse and human PDAC cell lines under oxidative and ER stress (Figure 5C–D and S5C). As such, these data identify IGF1 as the top obesity-associated secreted factor that promotes SG formation in PDAC cells.
Figure 5. Obesity-associated IGF1 and hyperactivation of IGF1R mediates SG upregulation in obesity-associated pancreatic cancer.
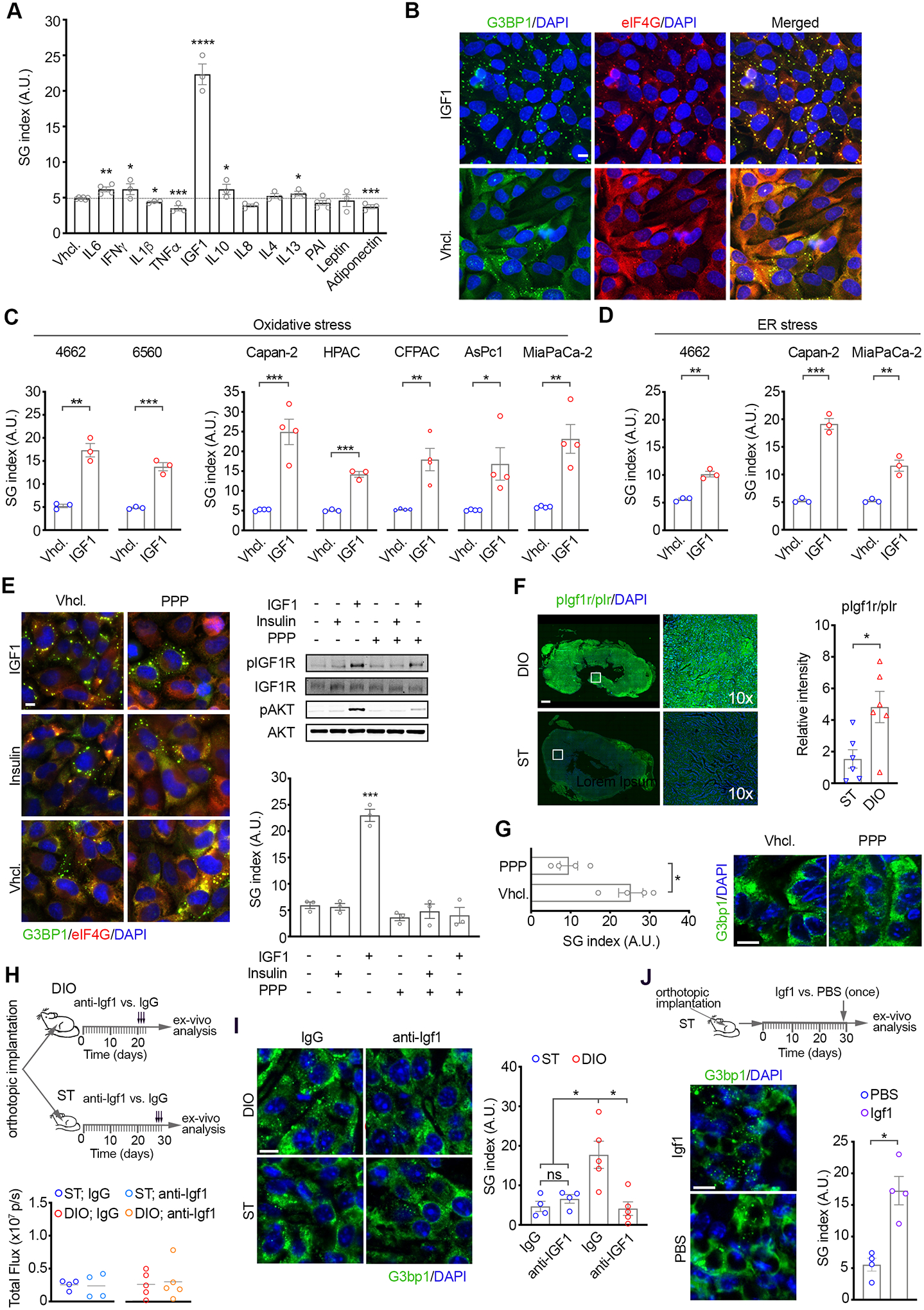
A) Quantification of SG index as detected by G3BP1 in MiaPaCa-2 cells treated with the indicated obesity-associated factors for 2 hr followed by oxidative stress (SA, 100 μM, 1 hr). Data are mean SG index +/− SEM, n=3, and individual values for each experiment. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by unpaired t-test.
(B) Representative immunostaining of G3BP1, eIF4g and DAPI in MiaPaCa-2 cells pre-treated with IGF1 (100 ng/ml, 2 hr) and stressed as in A. Scale bar, 10 μm.
(C) Quantification of SG index as detected by G3BP1 in a panel of mouse (KPC-4662, KPC-6560) and human (Capan-2, HPAC, MiaPaCa-2, CFPAC, and AsPc-1) PDAC cells pretreated with murine or human IGF1 (100 ng/ml, 2 hr) accordingly, and stressed as in A. See also S5.
(D) Quantification of SG index as detected by G3BP1 in cells treated with IGF1 as in B and subjected to ER stress (Thapsigargin).
(C-D) Data are mean SG index +/− SEM, n=3, and individual values for each experiment. *p<0.05, **p<0.01, ***p<0.001 by unpaired t-test.
(E) Left Panel-Representative immunostaining of G3BP1, eIF4G, and DAPI in MiaPaCa-2 cells stressed in the presence of IGF1 (100 ng/ml, 2 hr), Insulin (5 ng/ml, 2 hr), or vehicle and Picropodophyllin (PPP, 0.5 μM). Scale bar, 10 μm. Right Panel- WB of lysates from MiaPaCa-2 cells treated as indicated. Quantification of SG index as detected by G3BP1 in MiaPaCa-2 cells treated as in the left panel from at least 3 independent experiments. Data are mean SG index +/− SEM, ****p<0.0001 by unpaired t-test.
(F) Representative immunostaining of pIGF1R/pIr and DAPI in tissue sections from KPC-4662 tumors in ST and DIO mice as in 3B. Scale bar, 50 μm. Right panels are 10x-zoom in of boxed regions. Graph shows mean immunofluorescence intensity +/− SEM and values for each tumor. The value for each tumor is the average of non-overlapping FOV covering ~50% of each section imaged at 20x. *** p<0.001 by Mann Whitney t-test.
(G) Left Panel-SG index in KPC-4662 tumors in DIO mice as in 3A-C. Two weeks post implantation mice were treated with 20 mg/kg PPP every 12 hr by i.p. injection for a total of 48 hr. Quantification of SG index in tumor sections was performed as in 3D. Data are mean SG index +/− SEM, and individual values for each tumor, *** p<0.05 by Mann Whitney t-test. Right Panel- Representative immunostaining of G3bp1 and DAPI in sections from tumors treated as indicated. Scale bar, 10 μm.
(H) Top Panel - Schematic of the experimental approach used to assess the impact of an Igf1-neutralizing antibody and IgG control on SGs in mPDAC in DIO and ST immunocompetent mice. Luciferase expressing KPC-4662 cells were implanted orthotopically. On ~day 20 and 27 post implantation in DIO and ST mice respectively, 0.1 μg/g of anti-Igf1 or IgG was administered every 24 hr by i.p. injection for a total of 72 hr. Bottom Panel - Bioluminescence measurements of orthotopic implants of KPC-4662 cells in DIO and ST mice on day of first anti-Igf1 or IgG administration showing equivalent tumor size.
(I) Representative immunostaining of G3BP1 and DAPI and quantification of SG index in KPC-4662 tumor sections from DIO and ST mice in H. Scale bar, 10 μm.
(J) Top Panel - Schematic of the experimental approach used to assess the impact of Igf1 on SGs in mPDAC in ST immunocompetent mice. Luciferase expressing KPC-4662 cells were implanted orthotopically in the pancreata of ST mice. Recombinant Igf1 was administered by i.p. injection (once) at 100 ng/mouse. Bottom Panel - Representative immunostaining of G3BP1 and DAPI and quantification of SG index in KPC-4662 tumor sections from ST mice treated as indicated. Scale bar, 10 μm.
(I-J) Data are mean SG index +/− SEM, and individual values for each tumor, *p<0.05 by Mann Whitney t-test.
IGF1 binds specifically to the IGF1 receptor (IGF1R) leading to phosphorylation and activation of the tyrosine kinase activity of the receptor and its downstream effector pathways. The IGF1R inhibitor picropodophyllin (PPP), which specifically inhibits phosphorylation of tyrosine Y1136 in the activation loop of the IGF1R kinase domain, impaired IGF1-stimulated phosphorylation of IGF1R and downstream activation of AKT in hPDAC cells and diminished SGs to levels equivalent to control vehicle-treated cells (Figure 5E and Figure S5D) (58). Moreover, SG inhibition by PPP is specific to the IGF1-treated condition as in the absence of IGF1, PPP had no impact on SG levels. These results suggest that upregulation of SGs in obesity-associated PDAC is mediated by a distinct IGF1/IGF1R-driven pathway. In principle however, circulating Insulin, which is also increased in obesity, can stimulate IGF1R and, consequently, upregulate SGs (59–61). Nonetheless, treatment of hPDAC cells with Insulin, at concentrations reported in the pancreas of obese mice and in circulation in obese patients, had no impact on IGF1R phosphorylation, activation of downstream signaling, or SG levels (Figure 5E and Figure S5D) (59,60). These findings suggest that activation of IGF1R in obesity-associated PDAC is preferentially driven by elevated IGF1 vs. Insulin.
Consistent with reported heightened levels of IGF1 and Insulin in obesity, quantification of phospho-Igf1r (pIgf1r)/phospho-Insulin receptor (pIr) in DIO and ST tumor tissues revealed a ~2.5x increase in pIgf1r/p-Ir in DIO vs. ST tumor tissues (Figure 5F and Figure S5E). A similar stimulation was observed when mPDAC cells were treated with Igf1 (Figure S5F). To discern whether SG upregulation in obesity-associated PDAC is mediated by Igf1r activation we next administered the specific Igf1r inhibitor PPP (every 12 hr for 48 hr) or vehicle to DIO mice with established orthotopic tumors and evaluated SG levels (Figure 5G and Figure S5G). PPP treatment resulted in a ~2.5x reduction in SG levels in DIO tumors thus demonstrating that SG upregulation in obesity-associated PDAC is driven by Igf1r activation. To determine whether Igf1r-mediated upregulation of SGs in obesity-associated PDAC is driven by Igf1, we administered an Igf1-neutralizing antibody or IgG control (once per day for 72 h) to DIO and ST mice with orthotopic PDAC tumors of similar size (Figure 5H, Figure S5H, and Supplemental Methods) (62). Confirming target engagement, tumors from DIO mice treated with the Igf1-neutralizing antibody showed impaired phosphorylation of the downstream effector molecule S6 kinase 1 (S6k1) compared to tumors from IgG control-treated DIO mice (Figure S5I). Moreover, Igf1-neutralizing antibody treatment reduced SGs (~5x reduction) in DIO tumors to levels comparable to control-treated ST tumors (Figure 5I). Treatment of ST mice with the Igf1-neutralizing antibody had no impact on tumor SG levels. These data demonstrate that neutralizing Igf1 inhibits SG formation in DIO tumors specifically and indicate that SG upregulation in obesity-associated PDAC is driven by elevated Igf1. Based on these observations we next tested whether administration of Igf1 at levels found in obesity was sufficient to stimulate SG formation in PDAC tumors in ST mice (Figure 5J, Figure S5J, and Supplemental Methods). Confirming target engagement, Igf1 treatment enhanced S6k1 phosphorylation in ST tumors (Figure S5K). Notably, a single administration of Igf1 enhanced SG levels by ~3x (Figure 5J). Altogether these results indicate that SG upregulation in obesity-associated PDAC is driven by IGF1/IGF1R activation.
IGF1 promotes SG formation by modulating S6K1-mediated partitioning of SRPK2 to SGs and activation.
Previous work reported that mutant KRAS regulation of SG formation in hPDAC cells is driven by 15-deoxy-12,14-prostaglandin J2 (15-d-PGJ2) via KRAS/Mitogen Activated Protein Kinase (MAPK) regulation of cyclooxygenase 2 and 15-hydroxyprostaglandin dehydrogenase (HPGD) (18). Assessment of the interstitial fluid of DIO and ST tumors of comparable size revealed similar levels of 15-d-PGJ2 (Figure S5L and Supplemental Methods). In addition, no changes in the transcript levels of Cox1, Cox2, and Hpgd were observed between ST and DIO tumors (Figure S5L and Supplemental Methods). These observations support a model whereby Igf1 promotes SGs independent of prostaglandins (Figure S5M).
Given that the phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT) and mitogen-activated protein kinase (MAPK) are the two major pathways downstream of IGF1/IGF1R activation we assessed the impact of PI3K and MEK inhibition on SG levels in IGF1- and vehicle-treated cells (Figure 6A–B and S6A) (63). Consistent with previous work that mutant KRAS regulation of SG formation in hPDAC cells is MAPK-dependent, the protein kinase mitogen activated kinase 1 (MEK1) inhibitor PD98059 impaired SG formation in the absence of IGF1 by ~60% and had a modest impact (25% inhibition) in SG levels in IGF1-treated cells (Figure 6A–B) (18). On the other hand, the PI3K inhibitor LY294002, as well as inhibition of the downstream PI3K/AKT effector mTOR with rapamycin, attenuated SG formation in IGF1-treated cells but had no impact in the absence of IGF1 (Figure 6A–B). Of note, LY294002 and rapamycin diminished SGs in IGF1-treated cells to levels equivalent to vehicle-treated cells. Inhibitor treatments at the indicated doses and duration had no impact on cell death or cell cycle (Figure S6B–C) These results show that IGF1 promotes SG formation via a MEK-independent but PI3K/AKT/mTOR-dependent mechanism.
Figure 6. IGF1 promotes SG formation in PDAC cells by modulating S6K1-mediated partitioning of SRPK2 to SGs and activation.
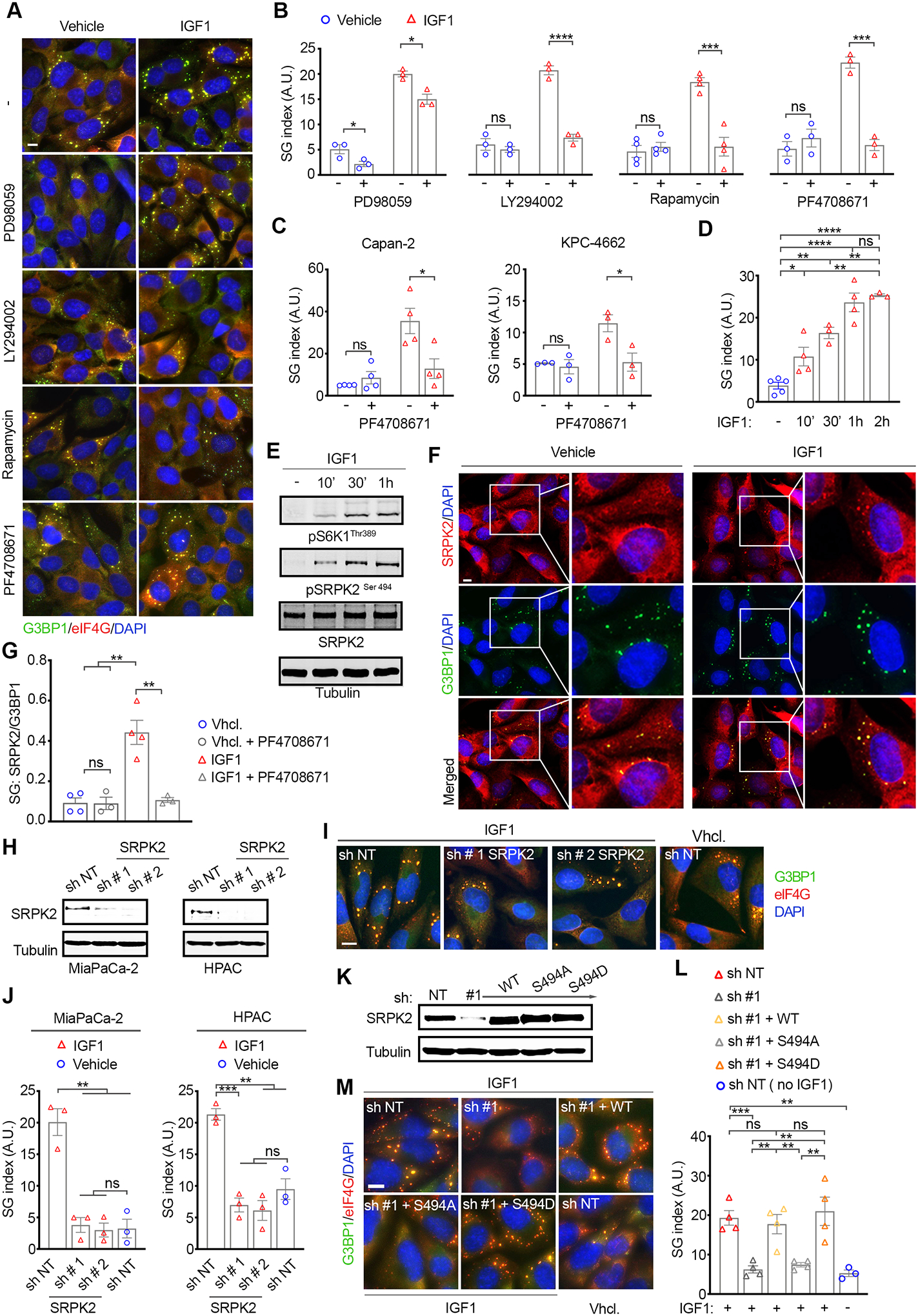
(A) Representative immunostaining of G3BP1, eIF4G, and DAPI in MiaPaCa-2 cells stressed in the presence or absence of IGF1 as in 5A-C, and the MEK (PD98059) AKT, (LY294002), mTOR (Rapamycin), and S6K1 (PF4708671) inhibitors. Scale bar, 10 μm. See also S7A.
(B) Quantification of SG index as detected by G3BP1 in MiaPaCa-2 cells in A.
(C) Quantification of impact of S6K1 inhibition on SG index in mouse (KPC-4662) and human (Capan-2) cells treated as in A.
(D) Quantification of SG index in MiaPaCa-2 cells pre-treated with IGF1 for the indicated time points and followed by oxidative stress (SA, 100 μM, 1 hr.)
(B-D) Data are mean SG index +/− SEM, n=3, *p<0.05, ***p<0.001, ***p<0.0001 by unpaired t-test.
(E) WB of MiaPaCa-2 cells treated with IGF1 as in D.
(F) Representative immunostaining of SRPK2, G3BP1, and DAPI in MiaPaCa-2 cells treated as in A. Scale bar, 10 μm. Right panels for vehicle and IGF1 treated are 2x-zoom in of boxed regions.
(G) Quantification of the partitioning of SRPK2 to SGs relative to G3BP1. Graph shows the ratio of SRPK2-SG area over G3BP1-SG area. Data are mean +/− SEM, n=3, and values for each experiment. At least 300 SG positive cells per condition were analyzed in each experiment. **p<0.01 by unpaired t-test.
(H) WB of lysates from MiaPaCa-2 and HPAC cells expressing the indicated shRNAs.
(I) Representative immunostaining of G3BP1 and DAPI in MiaPaCa-2 cells expressing the indicated shRNAs and treated as in A. Scale bar, 10 μm.
(J) Quantification of SG index as detected by G3BP1 in MiaPaCa-2 cells in I. Data are mean +/− SEM, n=3, and values for each experiment. **p<0.01, ***p<0.001 by unpaired t-test.
(K) WB of lysates from MiaPaCa-2 induced to express the indicated shRNAs and shRNA resistant constructs.
(L) Quantification of SG index as detected by G3BP1 in MiaPaCa-2 cells in K. Data are mean +/− SEM, n=4, and values for each experiment. **p<0.01, ***p<0.001, ns = non-significant by unpaired t-test.
(M) Representative images and quantification of SG index as detected by G3BP1 in MiaPaCa-2 cells expressing the indicated shRNAs and constructs. Data are mean +/− SEM, n=4, and values for each experiment. **p<0.01, ***p<0.001, ns = non-significant by unpaired t-test.
A previous study reported that the PI3K/AKT/mTOR effector molecules, S6K1 and S6K2 mediated SG formation in Hela cells under oxidative stress by regulating the phosphorylation of eIF2α via an unknown mechanism, and eIF2α-independent mechanism(s) (64). Given these findings we sought to determine the contribution of S6K1/ S6K2-signaling to IGF1-mediated SG formation in hPDAC cells. Treatment with the S6K1 specific inhibitor PF4708671 blocked IGF1-mediated SG formation in hPDAC cells but had no impact on SG formation in the absence of IGF1 (Figure 6A–C). In addition, IGF1 treatment had no impact on eIF2α phosphorylation in the presence or absence of oxidative stress, thus indicating that IGF1 promotes SG formation in hPDAC cells via an eIF2α-independent mechanism (Figure S6D). No changes were observed in the expression levels of the SG nucleators G3BP1 and TIAL, or the KRAS mediators of SG formation, namely COX2 and HPGD (Figure S6D) (18). Altogether these results show that IGF1 promotes SG formation through a S6K1-dependent but MEK and eIF2α-independent mechanism.
Since mutant KRAS-mediated SG formation is MEK-dependent, the observation that IGF1 promotes SG formation independent of MEK suggests that IGF1-driven SG formation is independent of mutant KRAS. To test this idea, hPDAC cells expressing an shRNA targeting KRAS or control shRNA were subjected to oxidative stress and assessed for SG formation (Figure S6E and Supplemental Methods). As expected, KRAS knockdown impaired SG formation in the absence of IGF1 (18). KRAS knockdown had no impact on SG formation in the presence of IGF1 however, demonstrating that IGF1 stimulates SGs independent of mutant KRAS. Given that SGs function to enhance cellular stress resistance we tested whether IGF1 stimulation would also restore the fitness of KRAS knockdown cells concomitant with SG upregulation (Figure S6F and Supplemental Methods). As expected, in the absence of IGF1, KRAS knockdown lowered stress resistance and led to a ~3x increase in cell death under lethal oxidative stress conditions. In contrast, IGF1 treatment fully abrogated cell death in KRAS knockdown cells to levels comparable to control IGF1 treated cells. Of note, IGF1 treatment enhanced the resistance of control cells by impairing cell death by ~40%. Altogether these results indicate that IGF1 drives SG formation independent of mutant KRAS and can counter the impact of KRAS inhibition on SG formation and cellular fitness.
To decipher the molecular mediator(s) through which IGF1/S6K1 promote SG formation, we initially assessed if temporal dynamics of IGF1 stimulation impacted SG levels (Figure 6D). Quantification of SG levels under oxidative stress following a time-course (10 min, 30 min, 1hr and 2 hr pre-treatment with IGF1) revealed a ~2x enhancement with as short as 10 min pre-treatment with IGF1; SG levels increased over time and plateaued at ~4x over control at the 1 hr time point-onwards, mirroring the dynamics of S6K1 phosphorylation (Figure 6E). The evidence that a short treatment with IGF1 and S6K1 activation promote SGs formation point to IGF1/S6K1-driven posttranslational modifications of an effector(s) as a mediator of this process. S6K1 regulates several effector molecules with roles in cellular processes including cytoskeletal organization, mRNA splicing, inflammation, apoptosis, and metabolism (65). In addition, S6K1 also stimulates protein translation via the elongation factor 2 kinase (eEF2K), eukaryotic initiation factor 3 (eIF3), and ribosomal protein 6 (S6), which as such would counteract SG formation (66). Focusing on the former group of effectors therefore, we identified the SRSF2 Protein kinase 2 (SRPK2) as distinct in that several of its binding partners are components of SGs (13,67–70). For this reason, we next investigated the localization of SRPK2 in IGF1- and vehicle-treated hPDAC cells under oxidative stress (Figure 6F and Figure S6G). Notably, SRPK2 was detected in SGs in both IGF1-and vehicle-treated cells. However, the partitioning of SRPK2 to SGs was significantly higher in IGF1-treated cells (Figure 6F and Figure S6G). As such, quantification of the relative fraction of SRPK2 in G3BP1-SGs showed a ~4x enrichment in IGF1-treated cells (Figure 6G). No changes were observed in SRPK2 levels with IGF1 stimulation whereas phosphorylation of SRPK2 followed the same temporal dynamics as IGF1-induced S6K1 phosphorylation (Figure 6E). To investigate if IGF1/S6K1-induced phosphorylation of SRPK2 impacted the partitioning of SRPK2 to SGs, we assessed the effect of the S6K1 inhibitor PF4708671 on the localization of SRPK2 (Figure 6G and Figure S6H). PF4708671 impaired SRPK2 phosphorylation and attenuated the partitioning of SRPK2 into SGs in IGF1-treated cells to levels comparable to those in the absence of IGF1. These results identify SRPK2 as a novel component of SGs and show that IGF1 promotes the partitioning of SRPK2 into SGs via S6K1.
The IGF1/S6K1-dependent partitioning of SRPK2 into SGs suggest that the capacity of IGF1 to promote SG formation may be mediated by SRPK2 phosphorylation. To evaluate this idea, we initially utilized Dox-inducible lentiviral shRNAs to target SRPK2 in hPDAC cells (Figure 6H). Knockdown of SRPK2 attenuated SG formation in hPDAC cells stimulated with IGF1 to levels comparable to unstimulated control (Figure 6H–J and Figure S6I). Expression of an shRNA resistant wild-type SRPK2 (SRPK2 WT) in SRPK2 knockdown cells or of a phosphomimetic SRPK2 mutant where the S6K1 phosphorylation amino acid Ser 494 was mutated to aspartate (SRPK2 S494D) fully rescued IGF1-driven SG formation, whereas expression of a phosphodeficient SRPK2 where Ser 494 was mutated to an alanine (SRPK2 S494A) failed to do so (Figure 6K–M and Figure S6J) (69). In addition, PF4708671 treatment attenuated SG formation in SRPK2 knockdown cells expressing shRNA resistant SRPK2 WT but had no effect when shRNA resistant, phosphomimetic SRPK2 S494D was expressed (Figure S6K). Altogether, these results identify S6K1-phosphorylated SRPK2 as an essential mediator of IGF1-stimulated SG formation in hPDAC cells.
Selective dependence of obesity-associated PDAC on S6K1 for SG formation and tumor growth.
Our results indicate that SG upregulation in obesity-associated PDAC is driven by IGF1/IGF1R activation and that IGF1 promotes SG formation specifically through S6K1/SRPK2 (Figure S7A). Along with the heightened dependence of obesity-associated PDAC on SGs, these data suggest that SG formation and growth of obesity-associated PDAC may be selectively dependent on S6K1 activity. To evaluate this idea, KPC-4662 and KPC-6560 cells expressing luciferase were orthotopically implanted in the pancreata of syngeneic DIO and ST mice; 24 hr post-implantation (Day1) mice were treated with the S6K1 inhibitor PF4708671 or vehicle control via a once-a-day intraperitoneal injection for the duration of the study (Figure 7A). Bioluminescence measurements on Day 1 demonstrate equal cell number implantations in all cohorts (Figure S7B). As expected, S6K1 is hyperactivated in DIO vs. ST tumors (Figure S7C). Confirming target engagement, treatment of DIO mice with PF4708671 led to diminished phosphorylation of the S6 protein in orthotopic tumors (Figure 7B). PF4708671 treatment attenuated SG levels in DIO tumors by ~80% but had no effect on SG levels in tumors in ST mice, establishing that SG formation in obesity-associated PDAC is specifically dependent on S6K1 activity (Figure 7C and Figure S7D). Consistent with the role of S6K1 as a mediator of SG formation specific to IGF1-stimulation and the heightened dependence of obesity-associated PDAC on SGs, PF4708671 treatment significantly impaired mPDAC growth in DIO mice but had no effect on mPDAC growth in ST mice (Figure 7D–E and Figure S7E–H). Of note, PF4708671 treatment had no impact on the body weight of DIO or ST animals (Figure S7E–F). KPC-4662 and KPC-6560 tumors in PF4708671-treated DIO mice diverged in growth rate from their vehicle treated counterparts starting on Day 25, and at endpoint were respectively ~1/5th and ~1/8th (82% and 88% smaller) of tumors in vehicle-treated DIO mice (Figure 7E and Figure S7G). In contrast, the growth rate of tumors in PF4708671- and vehicle-treated ST mice was indistinguishable for the duration of the study.
Figure 7. Selective dependence of obesity-associated PDAC on S6K1 for SG formation and tumor growth.
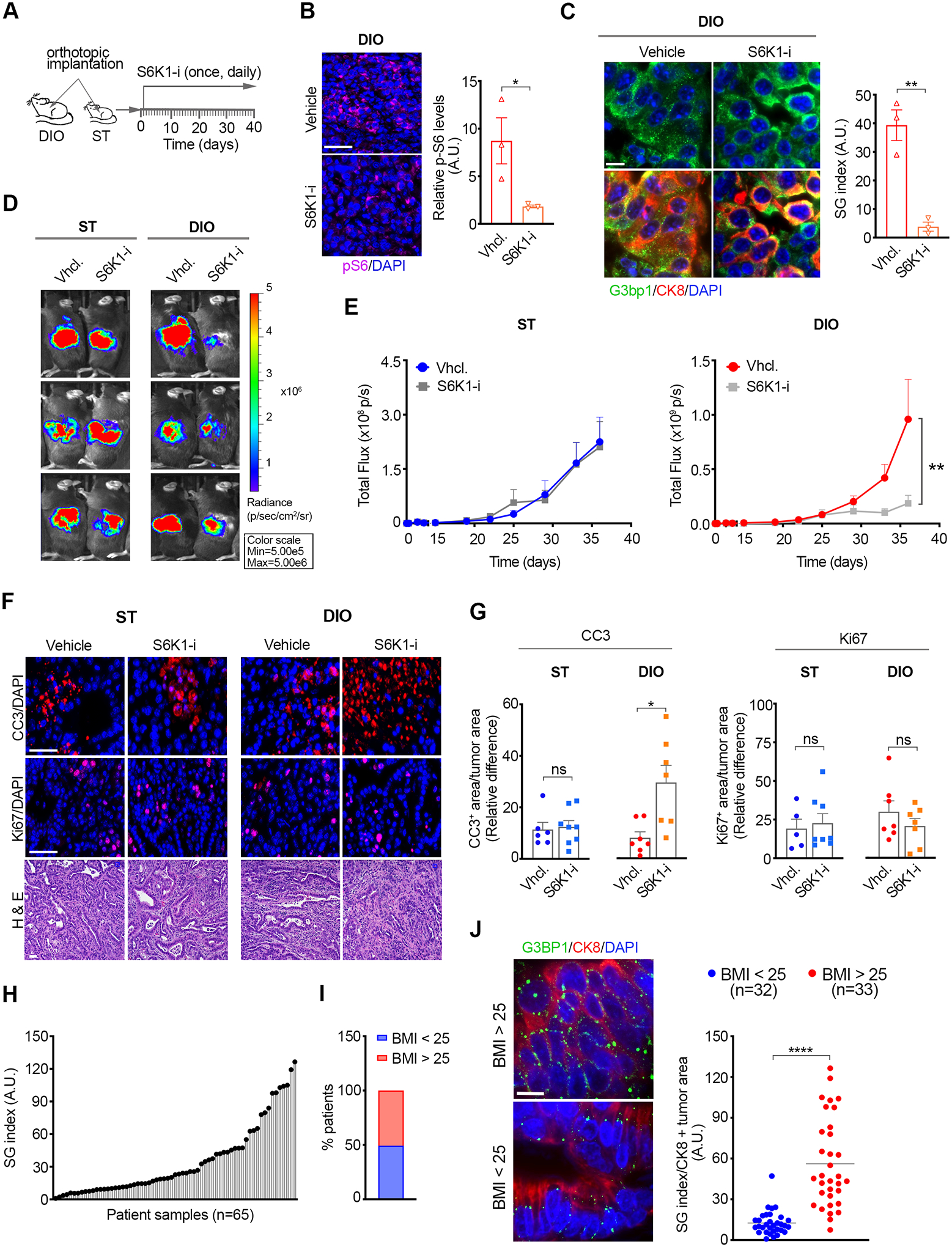
(A) Schematic of the experimental approach used to assess the impact of PF4708671 on mPDAC growth in ST and DIO mice. Vehicle or PF4708671 (50 mg/kg) were administered by i.p. injection once per day starting on Day 1 post-implantation of KPC-4662 and KPC-6560 cells expressing luciferase.
(B) Representative immunostaining of pS6 and DAPI in KPC-4662 tumor sections from DIO mice treated with PF4708671 for 3 days. Graph shows pS6 mean immunofluorescence intensity +/− SEM and values for each tumor. The intensity value for each tumor is the average of non-overlapping FOV covering ~50% of each section imaged at 20x. Scale bar, 50 μm. * p<0.05 by Mann Whitney t-test.
(C) Representative images and quantification of SG index in sections from tumors in B as detected by G3bp1. Data are mean SG index +/− SEM and individual values for each tumor. The SG index for each tumor section is the average of 30 non-overlapping FOV imaged at 40x. ** p<0.01 by Mann Whitney t-test. Scale bar, 10 μm.
(D) Representative bioluminescent images of ST and DIO mice implanted with KPC-4662 cells and treated as in A. The scale indicates radiance expressed as p/sec/cm2/sr.
(E) Quantification of tumor bioluminescent signal over time post orthotopic implantation of KPC-4662 cells in ST and DIO mice treated with vehicle or PF4708671 as in A. Data are mean signal +/− SEM of individual tumors in each group; ST (vehicle n=9, PF4708671 n=9), DIO (vehicle n=13, PF4708671 n=12). **p<0.01 by two-way ANOVA.
(F) Representative immunostaining of CC3, Ki67, and DAPI, H&E in sections from orthotopic tumors in E. Scale bar, 50 μm.
(G) Quantification of CC3 or Ki67 positive areas in tumors in E. Data was derived and quantified as in 4F-G. * p<0.05 by Mann Whitney t-test.
(H) Distribution of SG index in PDAC patient samples (n=65)
(I) Patient distribution according to Body Mass Index (BMI)
(J) Representative immunostaining of G3BP1, CK8, and DAPI, and SG index in tumor sections of PDAC patients as in H, segregated by BMI. Data are mean SG index of CK8 positive (+) tumor areas +/− SEM, and individual values for each tumor. The SG index for each tumor is the average of 2 sections, 15 non-overlapping FOV for each section, imaged at 40x.**** p<0.0001 by Mann Whitney t-test. Scale bar, 10 μm.
SG abrogation by G3BP1 knockdown impacted cell death and proliferation in both ST and DIO mPDAC but resulted in greater cell death in DIO tumors (Figure 4). Inhibition of S6K1 does not impact the basal levels of SGs as S6K1 specifically augments SG levels in obesity-associated PDAC. Therefore, S6K1-inhibited PDAC tumors would still retain the basal levels/function of SGs and treatment with PF4708671 would be expected to selectively impact cell death in DIO tumors. Consistent with this model, PF4708671-treatment enhanced cell death specifically in DIO tumors but had no impact on proliferation in DIO or ST tumors (Figure 7F–G). Altogether, these observations identify IGF1/S6K1 as a specific pathway mediating SG formation in obesity-associated PDAC and a potential target for the treatment of this disease.
To confirm the relevance of our findings to human PDAC, we obtained tumor samples of 65 patients and examined the SG index in the epithelial compartments as marked by cytokeratin 8 (CK8) (Figure 7H). SGs were consistently observed across all patient samples and their prevalence was heterogeneous (Figure 7H). Approximately 50% of the patients had a body mass index (BMI) > 25 and as such were qualified as overweight/obese (Figure 7I). Notably, SG levels differed significantly between the different BMI cohorts (Figure 7J). As such, we found an ~5-fold increase in SG levels in tumor samples from patients with BMI > 25 compared to those with BMI < 25 (Figure 7J). Altogether, these results establish SG upregulation as a key feature and unique vulnerability of obesity-associated PDAC.
DISCUSSION
Biomolecular condensates that form through LLPS are increasingly recognized as important means of organizing and compartmentalizing cellular signaling (71). However, the understanding of their physiological relevance is lacking. SGs are a prime example of such biomolecular condensates that are found in vivo in tissue samples from patients and mouse models of pancreatic cancer and sarcomas, and have been implicated in the stress adaptation of cancer cells and resistance to therapy (18,72,73). Recent work has identified that the shift in saturation concentration that enables phase separation, and consequently SG formation, is determined by the collective interactions of ~36 proteins and their associated mRNAs. Notably, G3BP1 and G3BP2 are a central node of this network (14). In agreement with this model, we show that G3BP1 knockdown significantly diminishes SG formation in PDAC cells. Importantly, we show that G3BP1 knockdown impairs PDAC growth. Three lines of evidence indicate that SGs specifically, and not an alternative function of G3BP1, are required for tumorigenesis. First, targeting another node in the core SG network, namely TIAL1, phenocopies the effect of G3BP1 knockdown on PDAC growth. Second, deletion of the dimerization domain of G3BP1 which results in impaired SG formation, also impairs PDAC growth. Third, a ‘synthetic’ protein consisting of dimerization-, central IDR-, and tandem RNA binding-domains that are heterologous to the domains of G3BP1 necessary for SG formation, can substitute for G3BP1 in SG formation and PDAC tumorigenesis to a similar extent, despite harboring completely different primary sequences. Collectively, these results establish a pathophysiological function of SGs as critical mediators of pancreatic tumorigenesis. Adding to these findings, our results provide insight into the specific functions of SGs under physiological stress stimuli. In vitro evidence has linked SGs to stress adaptation primarily through the role SGs have in blocking cell death. Our results show that in the setting of tumorigenic stress, SGs contribute to both tumor proliferation and evasion of cell death. Whether proliferation is regulated directly by SGs, or indirectly as a corollary of stress adaptation, remains to be determined.
We predicted that stress acts as a selective pressure in tumorigenesis and that tumors that arise in pre-existing stress conditions like obesity would face a greater selective pressure and have a higher dependence on SGs. In support of this idea, our results show that SG inhibition has a heightened impact on obesity-associated PDAC: the relative decrease in tumor growth is several-fold larger compared to standard weight, survival is prolonged, and 40% of the mice are tumor free at endpoint (300 days post-implantation) whereas all standard weight mice succumb to disease. These findings provide important insight on the pathophysiological role of SGs and inform potential chemo-preventive strategies. Given the heightened dependence of obesity-associated PDAC, approaches targeting SG formation may succeed at blunting the occurrence risk imparted by obesity. In addition, these approaches may be relevant to the myriad of tumors associated with obesity, several of which are continually increasing in occurrence. Moreover, the findings that the heightened dependence of established obesity-associated PDAC on SGs for tumor maintenance indicates that targeting SGs may also prove a beneficial therapeutic option for the treatment of established tumors. On this note, the observation that the heightened dependence of obesity-associated PDAC on SGs is accompanied by augmented SG levels provide further evidence that the level of SG dependence correlates with the cellular capacity to form SGs and that SG levels may serve as biomarkers dictating optimized outcome.
Our results establish a specific pathway, namely IGF1/IGF1R/S6K1/SRPK2, through which obesity promotes SG formation in PDAC. While IGF1 and Insulin downstream signaling overlap significantly and circulating levels of both IGF1 and Insulin are elevated in obesity, we show that IGF1 is the driver of IGF1R and downstream effector activation. This may be due to higher levels of IGF1 compared to circulating and pancreatic levels of Insulin in obesity. In addition, ~40–90% IGF1R in tissues is present in complex with IR and these heterodimers have a higher affinity for IGF1 than for Insulin (74–76). SRPK2 is a recently identified substrate of S6K1 with roles in mRNA stability, splicing, and lipid metabolism (69). Our results add to these roles by identifying SRPK2 as an IGF1/S6K1 dependent constituent of SGs, and a determinant of IGF1-driven SG formation. The mechanisms through which SRPK2 mediates SG formation are yet to be characterized. The nodes of the core SG network are known to contribute to SG formation to varying degrees and it is possible that S6K1-stimulated SRPK2 may impact their contribution; this is consistent with the predicted model whereby SG assembly is subject to regulation by positive and negative cooperativity of extrinsic factors with the core network interactions (14). In support of this possibility, SRPK2 is known to interact with SG constituents such SRSF proteins and Tau in a phosphorylation-dependent manner (67,70). SRSF3 is a component of the core SG network and therefore may represent a means through which SRPK2 alters the core interaction network to promote SG assembly. Another possibility is that as a constituent of SGs, SRPK2 itself may contribute to the core SG network in IGF-treated cells. Clearly, further investigation will be required to decipher the mechanisms through which SRPK2 can mediate SG assembly. Nonetheless, the IGF1/S6K1 dependent localization of SRPK2 to SGs highlights that the composition of SGs is context dependent. Previous studies have shown that the composition of SGs can vary with stress. The varying composition, therefore, extends not only to different stress stimuli, but also to macromolecular stimuli.
In addition to SGs, tumors engage several stress-specific adaptive mechanisms. As such, it will be important to determine whether the heightened dependence of obesity-associated PDAC on SGs indicates a broad characteristic of these tumors and extends to other stress adaptive mechanisms. Furthermore, it will be critical to determine whether any of these dependencies extends to the many obesity-associated cancers. On this note, the findings that IGF1 stimulates SG formation even under conditions of mutant KRAS inhibition support a model whereby obesity-associated SG upregulation is independent of the tumor mutational status. Together with a better understanding of signaling alteration in obesity and other cancer-predisposing pathological conditions, as they pertain to SG formation and stress adaptation, such studies can have important implications for the development of new and optimized therapeutic approaches.
METHODS
Mice
All animal work was approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. Standard C57BL/6 (ST) (RRID:IMSR_JAX:000664), diet-induced obese C57BL/6 (DIO) (RRID:IMSR_JAX:380050), and Lepob (ob/ob) (RRID:IMSR_JAX:000632) mice were obtained from Jackson Laboratory. CrTac:NCr-Foxn1nu (Nude) (RRID:IMSR_TAC:ncrnu) mice were obtained from Taconic Farms. 11–13 weeks old ST and DIO and 5–7 weeks old ST and Ob/Ob mice were used for orthotopic tumor models and associated controls. 6–8 weeks old nude mice were used for xenograft tumor models and associated controls.
Cells
Lentiviral transductions of all cancer cell lines were approved by the Thomas Jefferson University Institutional Biosafety Committee. The human pancreatic cancer cell lines, Mia-PaCa-2 (AddexBio Cat# C0018002/59, RRID:CVCL_0428), HPAC (ATCC Cat# CRL-2119, RRID:CVCL_3517), CFPAC (ATCC Cat# CRL-1918, RRID:CVCL_1119), ASPC1 (ATCC Cat# CRL-1918, RRID:CVCL_1119) and Capan-2 (ATCC Cat# HTB-80, RRID:CVCL_0026) were obtained and originally authenticated by short tandem repeat (STR) from American Type Culture Collection (ATCC). MiaPaCa-2, HPAC and CFPAC cells were cultured in Dulbecco’s Modified Eagles Medium (DMEM; Corning, 10-017-CV) supplemented with 10% FBS (Corning, 35-011-CV), 1% penicillin/streptomycin (Gibco, 15140122) and 1% HEPES Buffer 1M solution (Fisher, BP299-100). Capan-2 were cultured in McCoy’s 5A medium (GIBCO, 16600082) and ASPC1 were cultured RPMI 1640 medium (Fisher scientific, 11-875-093) supplemented as above. Mouse pancreatic cancer cell lines, KPC-4662 and KPC-6560 were isolated from the LSL-KrasG12D/+;LSL-Trp53R172H/+; Pdx-1-Cre genetically engineered mouse model of KRAS-driven murine PDAC and were a kind gift by Dr. Robert H. Vonderheide, University of Pennsylvania (36)) and cultured in DMEM supplemented as above. All cells were cultured at 37°C with 5% CO2 and routinely tested for mycoplasma contamination every 4 months after the first thaw by Mycoplasma PCR detection kit (ABM, cat.no. G238). All cells were frozen at low passage and were no longer used after 20 passages after first thaw.
Constructs
The following constructs were purchased from Dharmacon: SMARTvector inducible mCMV/TurboRFP, G3BP1 shRNA (V3SH11252-226176431; V3IHSMCR_6114081), TIAL1 shRNA (V3SH11252-227445578; V3IHSMCR_7383228), SRPK2 shRNA#1 (V3SH11252-225843065), SRPK2 shRNA#2 (V3SH11252-226279721); pTRIPZ inducible/TurboRFP, G3BP1 shRNA#1 (RHS4696-200750396; V3THS_329105); SMARTvector inducible hCMV/TurboGFP, non-targeting shRNA (VSC11707) used in human cells lines, non-targeting shRNA (VSC11651) used in mouse cell lines, G3bp1 shRNA#1 (V3SM11253-231226612; V3IMMMCG_11164262), G3bp1 shRNA#2 (V3SM11253-231617629; V3IMMMCG_11555279), G3bp1 shRNA (V3SM11253-234970000; V3IMMMCG_14907650). pTRIPZ inducible/TurboRFP, KRAS shRNA (Open Biosystems V2THS-275818). The luciferase expression plasmid pLenti-PGK-Blast-V5-LUC (w528-1) (RRID:Addgene_19166) was a gift from Eric Campeau & Paul Kaufman (Addgene plasmid #19166; http://n2t.net/addgene:19166;RRID:Addgene_191166, RRID:Addgene_19166). pLenti-CMV-Blast-DEST (706-1) (RRID:Addgene_17451) was a gift from Eric Campeau & Paul Kaufman (Addgene plasmid #17451; http://n2t.net/addgene:17451;RRID:Addgene_17451, RRID:Addgene_17451). The doxycycline inducible pCW57.1 plasmid was a gift from David Root (Addgene plasmid # 41393; http://n2t.net/addgene:41393; RRID: Addgene_41393). Mouse G3bp1-turboGFP in pCMV-6-AC was obtained from Origene (MG207441). BP-LR cloning was used to insert G3bp1-tGFP into pcW57.1. The following primers were used to add attB1 sites to generate the full-length G3bp1-GFP and the N-terminus deletion dN-G3bp1-GFP constructs cloned into pcW57.1: G3bp1-GFP Forward: 5’ GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GAG ATC TGC CGC GAT CGC C 3’; dN-G3bp1-GFP Forward: 5’ GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GAT GTT TGT CAC AGA GCC TCA AGA GGA AT 3’; GFP-Reverse: 5’ GGG GAC CAC TTT GTA CAA GAA AGC TGG GTC AGG CAC TGG GGA GGG GTC ACA GG 3’. The following primers were used to generate a full length G3bp1-GFP resistant to G3bp1 shRNA # 1 via QuickChange mutagenesis (QuickChange II Site-Directed Mutagenesis Kit, Agilent, cat.no.200523): P1 Forward: 5’ GAC ACC AGA GGT CGT CCC CGA TGA TTC TGG AAC TTT CTA TG 3’; P1 Reverse: 5’ CAT AGA AAG TTC CAG AAT CAT CGG GGA CGA CCT CTG GTG TC 3’; P2 Forward: 5’ GAC ACC AGA GGT CGT CCC CGA CGA CTC CGG AAC TTT CTA TG 3’; P2 Reverse : 5’ CAT AGA AAG TTC CGG AGT CGT CGG GGA CGA CCT CTG GTG TC 3’. Plasmids containing EGFP-G3BP1-WT and EGFP-GST-2xAsh1-IDR-hnRNPA1-tandem RRM (‘synthetic’) in pEGFP-C3 (RRID:Addgene_53755) were kindly obtained from Dr. Paul Taylor. BP-LR cloning was used to insert EGFP-G3BP1-WT and GFP- ‘synthetic’ into pLenti-CMV-Blast-DEST (RRID:Addgene_17451). The following primers were used to add attB1 sites for BP-LR cloning: Synthetic Forward: 5’ GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC CAC CAT GGT GAG CAA G 3’; Synthetic Reverse: 5’ GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT CAG TTA TCT AGA TCC GGT GGA TCC 3’; EGFP-G3BP1-WT Forward: 5’ GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GCT ACC GGT CGC CAC C 3’; EGFP-G3BP1-WT Reverse: 5’ GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT CAG TTA TCT AGA TCC GGT GGA TCC 3’.
Human pDONR223-SRPK2 was a gift from William Hahn & David Root (Addgene plasmid # 23766; http://n2t.net/addgene:23766; RRID:Addgene_23766) and human SRPK2-MycD in pCMV-6-AC was obtained from Origene (Cat #RC205134). The following primers were used to generate point mutations in SRPK2-MycD-pCMV-6-AC via QuickChange mutagenesis: S494D forward: 5’ CAT GAC AGA AGC AGA ACG GTT GAT GCC TCC AGT ACT GGG GA 3’; S494D reverse: 5’ TCC CCA GTA CTG GAG GCA TCA ACC GTT CTG CTT CTG TCA TG 3’; S494A forward: 5’ GAC AGA AGC AGA ACG GTT GCC GCC TCC AGT ACT GG 3’; S494A reverse: 5’ CCA GTA CTG GAG GCG GCA ACC GTT CTG CTT CTG TC 3’. The following primers were used to add attB1 sites to these SRPK2 mutants: SRPK2 forward: 5’ GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GCG AGG AGA TCT GCC GCC GCG ATC G 3’ reverse: 5’ GGG GAC CAC TTT GTA CAA GAA AGC TGG GTC GGA AAC AGC TAT GAC CGC G 3’. BP-LR cloning was used to insert Wildtype SRPK2, SRPK2 S494D, or SRPK2 S494A into pLenti-CMV-BLAST-DEST.
Lentivirus Production
HEK293T (RRID:CVCL_HA71) cells were transduced using the calcium phosphate transfection method. Briefly, 500 μl of CaCl2 2.5 M, 7 μg pMD2G (RRID:Addgene_12259) (Gift from Didier Trono, Addgene plasmid # 12259; http://n2t.net/addgene:12259; RRID:Addgene_12259), 13 μg psPAX2 (RRID:Addgene_12260) (Gift from Didier Trono, Addgene plasmid # 12260; http://n2t.net/addgene:12260; RRID:Addgene_12260), and 20 μg of construct vector was added dropwise to 500 μl of HEPES Buffered Saline (HEBS) 2x while vortexing. After 25–40min the DNA CaPO4 coprecipitate was transferred dropwise onto HEK293T (RRID:CVCL_HA71) cells. Media was harvested 24 hours and 48 hours after transfection and passed through 0.45 μm filters. Finally, viruses were concentrated by spinning in 100 KD cutoff Amicon Filter tubes for 30 minutes at 4°C at 4,000 RPM.
Viral Transduction
40,000 cells of interest were seeded in serum free media and transduced with the lentiviral particles containing the indicated constructs. Multiplicity of infection (MOI) for KPC cells was 5; all other cell lines, MOI=10. Following transduction, KPC cells were selected with puromycin 5 μg/ml for 3 days and the top 20% cells based on their GFP expression after a 16 hour induction with 1μg/ml doxycycline (Sigma-Aldrich, D9891) were sorted by flow cytometry with a BD FACSAria II Cell Sorter. For tumor growth luminescence tracking by IVIS, sorted KPC-4662 and KPC-6560 cells harboring the indicated constructs were additionally transduced with lentiviral particles containing the luciferase expression construct and selected with Blasticidin (10 μg/ml) for 3 days. For KPC-4662 cells expressing G3bp1-GFP or dN-G3bp1-GFP resistant to shRNA, cells were transduced with lentivirus particle containing G3bp1-GFP or dN-G3bp1-GFP expression constructs cells and selected with puromycin (5 μg/ml) for 2 days. Subsequently, cells were transduced with lentivirus particles containing G3bp1 shRNA # 3, which targets the 3’ untranslated region (UTR) of G3bp1, or a NT shRNA construct, and selected with puromycin (5 μg/ml) for an additional 3 days.
MiaPaCa-2 and HPAC cells transduced with lentiviral particles containing the indicated shRNA constructs were selected with puromycin 5 ug/ml for 3 days. For MiaPaCa-2 cells expressing shRNA resistant GFP-G3BP1 or GFP-synthetic, cells harboring G3BP1 shRNA #2, which targets the 3’ untranslated region (UTR) of G3BP1, were transduced with lentiviral particles containing the GFP-G3BP1 or GFP-synthetic expression construct and selected for 3 days with blasticidin (10 μg/ml). Cells were subjected to sorting by flow cytometry with a BD FACSAria II Cell Sorter for the top 20% GFP expressing cells. G3BP1 knock down, expression of G3BP1-GFP, dN-G3bp1-GFP, GFP-G3BP1, GFP- ‘synthetic’, and luciferase were confirmed by western blot after 3–5 days of doxycycline induction at 1 μg/ml.
Stress Induction
Cells were plated on coverslips and stressed when at 70% confluent. Where indicated, cells were treated with doxycycline (1μg/ml) for 3–5 days to induce shRNA or protein expression, prior to stress. Oxidative stress was induced with Sodium Arsenate (SA) for 1 hour at 200 μM for KPC-4662 and KPC-6560 and 100 μM for human PDAC cell lines; ER stress was induced by treatment with Thapsigargin (Tocris, cat.no. 1138) for 6 hours at 25 μM for KPCs 4662 and Capan-2, and 16 μM for MiaPaCa-2. Hypoxia was induced using the Celldiscoverer 7 Automated Microscope incubation chamber system (Zeiss) set up at 0.5% O2 and 5% CO2 for 12 hours for MiaPaCa-2, or 24 hours for KPC 4662 cells.
Cell Treatments
For treatment with obesity associated factors cells were cultured as described above, starved for 16 hours in serum-free DMEM, and then treated for 2 hours with vehicle control (PBS), 100 ng/ml IGF-1 (Human IGF-I, PeproTech, cat. no. 100-11; murine IGF1, PeproTech, cat. no. 250-19), 50 ng/ml PAI (PeproTech, cat. no. 140-04), Leptin (PeproTech, cat. no. 300-27); 10pg/ml recombinant IFNγ (PeproTech, cat. no. 300-02), 5 ng/mL or 15 ng/mL Cholecystokinin (CCK) (Thermo Fisher Scientific, J66669.EXD), 5 ng/ml Insulin (EMD Millipore, cat.no. 407709), IL1β (PeproTech, cat. no. 200-01B), IL6 (PeproTech, cat. no. 200-06), IL8 (PeproTech, cat. no. 200-08), IL10 (PeproTech, cat. no. 200-10), IL4 (PeproTech, cat. no. 200-04), IL13 (PeproTech, cat. no. 200-13), Adiponectin (PeproTech, cat. no. 450-24), TNFα (PeproTech, cat. no. 300-01A), according to reported serum levels in obese subjects. Subsequently, stress was induced as described above.
For inhibitor treatments cells were starved for 16 hours in serum-free DMEM or McCoy’s 5A. Cells were then treated with recombinant IGF-1 or vehicle control, in combination with each of the following inhibitors for 1 hour: the IGF1R inhibitor Picropodophyllin (PPP) (EMD Millipore, 407247) at 0.5 μM, the PI3K inhibitor LY294002 (Cayman, cat. no. 70920) at 0.2 μM, the mTOR inhibitor Rapamycin (Selleck Chemicals, cat. no. S1039) at 2 nM, the MEK inhibitor PD98059 (Selleck Chemicals, cat. no. S1177) at 0.7 μM, the S6K1-inhibitor (PF-4708671) (Selleck Chemical, cat. no. S2163, or MedChem, cat.no. HY-15773) at 200 nM for Capan-2 cells and 20 nM for all other cells. Treatment concentrations were determined empirically and based on EC50 values. Finally, stress was induced for 1 hour with 100 μM SA for human PDAC cells or 200 μM for KPC cells.
Immunofluorescence
All cells were grown on coverslips, fixed in 4% paraformaldehyde, immunostained, and mounted in ProlongGold (Invitrogen, P36934). Immunostaining was performed using 3 well-described markers of stress granules: G3BP1 (1:2000 Bethyl Cat# A302-033A, RRID:AB_1576539), eIF4G (1:500 Cell Signaling Technology Cat# 2498S, RRID:AB_2096025 or 1:500 Santa Cruz Biotechnology Cat# sc-133155, RRID:AB_2095748) and Pumilio (1:500 Abcam Cat# ab10361, RRID:AB_297098). Additionally, cells were stained for SRPK2 (1:200 Proteintech Cat# 25417-1-AP, RRID:AB_11205747), phospho-IGF1R (1:200, Abcam Cat# ab39398, RRID:AB_731544) and Cleaved Caspase 3 (CC3) (1:500e Cell Signaling Technology Cat# 9661, RRID:AB_2341188)
Tissue Immunohistochemistry and Immunofluorescence
All tissues samples were rinsed in PBS and fixed in 10% formalin for 24 hours. All tissues samples where then placed in cassettes and stored at 4°C in 70% ethanol until embedded in paraffin, sectioned (5-μm), and mounted on glass slides. Slides of tissue sections were de-paraffinized in xylene and rehydrated through a reversed ethanol gradient. Then sections were subjected to heat-mediated antigen retrieval with citrate buffer (0.01M sodium citrate/0.05% Tween-20 pH 6.0). Tissue samples were blocked for 1 hour at room temperature (RT) in a 10 mM Tris-HCl, 0.1M MgCl2, 0.05% Tween-20, 1% BSA, 10% solution. Slides were stained with primary antibody overnight at 4°C in a humified chamber. Primary antibodies were used as follows: phospho-IGF1R (1:200, Abcam Cat# ab39398, RRID:AB_731544), CK8 (1:200, DSHB Cat# TROMA-I, RRID:AB_531826), G3BP1 (1:400, Bethyl Cat# A302-033A, RRID:AB_1576539), phospho-S6K1 (1:100; Aviva Systems Biology Cat# OAAF07416, RRID:AB_2630782), phospho-S6 (1:800; Cell Signaling Technology Cat# 5364S, RRID:AB_10694233), cleaved caspase 3 (1:200; Cell Signaling Technology Cat# 9664S, RRID:AB_2070042), Ki67 (1:200; Proteintech Cat# 27309-1-AP, RRID:AB_2756525). Slides were washed in TBST (0.1% Tween-20, TBS) and incubated with the appropriate secondary antibodies for 1 hour at RT. After washing in TBST, sections were stained with DAPI for 5 minutes at RT and coverslips were mounted with ProlongGold (Invitrogen, P36934).
Imaging
Imaging was performed with a Celldiscoverer 7 Automated Microscope system (Zeiss) using ZEISS Plan-APOCHROMAT 20× / 0.95 Autocorr Objective, a ZEISS Plan-APOCHROMAT 20× / 0.95 Autocorr Objective with a 2x tubelens. Images were acquired with an Axiocam 506 mono camera and the LSM 900 with Airyscan 2 imaging software. For SG index quantification in cells in culture, a z-stack of 12 serial optical sections every 0.49 μm at 20x, or of 25 serial optical sections every 0.2 μm at 40x or 20x were captured in 15–30 non-overlapping fields randomly positioned in grid format to span the coverslip. For SG quantification in tissues, a z-stack of 25 serial optical sections were captured every 0.2 μm at 40x, in 30 random non-overlapping fields with similar % epithelial composition. For tissue immunofluorescence (mean intensity and relative tumor area) a Z-stack of 6 serial optical sections were captured every 1 μm for each field of view. Non-overlapping fields of view were randomly positioned to span the tissue section in a grid like fashion imaging ~50% of each tissue section. For SRPK2 localization to SGs imaging was performed with an Axio Observer 7 microscope (ZEISS) with a Plan-Apochromat 63x/1.40 Oil M27 Oil objective and an Axiocam 702 camera.
Imaging Quantification
Quantification of the SG index was performed as previously described (18). Quantification of the percentage of cells positive for SGs was based on a manual count of a minimum of 200 cells per experimental condition, in a minimum of 5 random fields of view, for each for at least 3 independent experiments. Quantification of protein levels in tissue sections, was done by measuring the fluorescence intensity using Image J (https://imagej.nih.gov/ij, RRID:SCR_003070). Z-stacks from tissue sections were projected onto one maximum intensity projection image per field of view. Subsequently, the mean intensities of each field of view were averaged and background was subtracted to give the mean intensity of each tissue section. For the quantification of tumor area positive for Ki67 and cleaved caspase 3 (CC3) maximum projection images were thresholded to black-and-white, and subsequently used to determine the tumor area positive for the protein of interest or the total tumor area per field of view. Batch processing was utilized to apply set thresholds to all images from the same experiment. The relative Ki67 or CC3 positive area per tumor was computed as fraction of the total tumor area for each field of view and then averaged across all fields. The same method was applied to determine the cell area positive for cleaved caspase 3 (CC3), in a minimum of 8 random fields of views in each condition and then averaged across all fields.
Cell Death Measurement by Live Imaging
KPC cells were induced with doxycycline (1 μg/ml) 3 days prior seeding in 24-well plate. Doxycycline was maintained in the media for the duration of the experiments. Oxidative stress was induced by 300 μM SA for 6 hr. Imaging was performed using a Celldiscoverer 7 Automated Microscope system (Zeiss) with a ZEISS Plan-APOCHROMAT 20× / 0.95 Autocorr Objective. A z-stack of 7 serial optical brightfield sections were captured every 1μm, in a minimum of 5 random fields, hourly. The percentage of dead cells was derived by manual count based on morphology and blebbing and is the mean of 5 random fields of view for each condition.
Western Blotting
Cells were lysed in 2x Laemli sample buffer. All protein samples were denaturized by heating at 95°C for 5 minutes. SDS-PAGE electrophoresis was then performed, transferred to a nitrocellulose membrane (Odyssey® Nitrocellulose Membranes; Licor), blocked with TBS-BSA 3% (w/v) at RT for 20 minutes, incubated with primary antibodies overnight at 4°C, followed by 1 hour with secondary antibodies coupled with an Alexa Fluor dye 680 or 800. Fluorescence detection and quantification of immunoblot bands was achieved using the LI-COR imager and software (Li-COR Biosciences). The following antibodies were used: G3BP2 (1:000, Thermo Fisher Scientific Cat# A302-033A, RRID:AB_1576539), G3BP1 (1:20000, Bethyl Laboratories Cat# A302-033A, RRID:AB_1576539); Pumilio (1:5000, Abcam Cat# ab10361, RRID:AB_297098); eIF4G (1:1000 Cell Signaling Technology Cat# 2498S, RRID:AB_2096025); Alpha Tubulin (1:5000, Sigma Aldrich Cat# T6199, RRID:AB_477583); eIF2 alpha (1:1000 Cell Signaling Technology Cat# 9722S, RRID:AB_2230924); phospho-eIF2 alpha (1:1000 Cell Signaling Technology Cat# 3398P, RRID:AB_2096481); Cox2 (1:500 Abcam Cat# ab15191, RRID:AB_2085144); TIAL1 (1:1000 Abcam Cat# ab169547, RRID:AB_2910194), turbo GFP (1:2000, Origene Technologies Cat# TA150041, RRID:AB_2622256), GFP (1:1000, Cell Signaling Technology Cat# 2555S, RRID:AB_10692764), HPGD (1:250 Abcam Cat# ab187161, RRID:AB_2861359), 1:1000 for: phospho-IGF1R (Abcam Cat# ab39398, RRID:AB_731544), IGF1R (Cell Signaling Technology Cat# 3027S, RRID:AB_2122378), phospho-AKT (Cell Signaling Technology Cat# 4060S, RRID:AB_2315049), AKT (Abcam Cat# ab8805, RRID:AB_306791), phospho-ERK1/2 (Cell Signaling Technology Cat# 4370S, RRID:AB_2315112), ERK1/2 (Cell Signaling Technology Cat# 9107S, RRID:AB_10695739), phospho-S6K1thr389 (Aviva Systems Biology Cat# OAAF07416, RRID:AB_2630782), SRPK2 (ProteinTech Cat# 25417-1-AP, RRID:AB_2880068), phospho-SRPK2ser494 (EMD, Millipore Cat# 07-1817, RRID:AB_11205747), IGF1 (1:500, R&D Systems Cat# AF-291-NA, RRID:AB_2122119).
Cell Proliferation
Cells were induced with doxycycline (1μg/ml) and doxycycline was replenished every 48hr for the duration of the experiment. For growth curves, 5000 cells were plated in 24-well plate and fixed in 4% PFA at day 0, 2, 4 and 6. All samples were stained with SYTO® 60 red fluorescent nucleic acid stain (5 mM stock solution solution) (Fisher Scientific, S11342) at 1/5000 in 0.1% Triton/PBS for 1 hour. Results were imaged, and relative cell density was quantified with a Licor system.
Orthotopic Implantations
DIO mice were fed with rodent high fat (60% kcal from fat) chow (Research Diets, D12492i) during the entire experiment. Standard mice received standard chow. As female mice are not susceptible to diet-induced obesity, ST and DIO mice for the comparative studies were male of 11–13 weeks of age. OB/Ob mice were male and female of 5–7 weeks of age at the time of orthotopic implantation for IVIS experiments; ST mice for the comparative studies were age-matched. Mice were weighed before cell implantation and at endpoint. For IVIS and survival experiments mice were also weighed twice a week through the entire duration of experiments. For all orthotopic implantations, 50000 cells resuspended in a 1:1 solution of DMEM and Matrigel at a final volume of 50 μl were injected in the tail of the pancreas. Prior to each surgery mice were injected with Buprenex at 0.08 mg/kg (Buprenorphine Hydrochloride, injection 0.3 mg/ml; NDC 42023-179-05). For each surgery mice were anesthetized with inhalation of ~3% v/v vaporized isoflurane. Mice implanted with inducible cell lines received doxycycline in their water starting at 24 hours post implantation at a final concentration of 200 μg/ml; doxycycline-water was changed every 3 days. At end point tumors were weighed, measured, and pictured.
Xenografts
Human PDAC cells lines (1M/flank; MiaPaCa2 and HPAC) were resuspended in a 1:1 solution of PBS and Matrigel in a final volume of 100 μl and were implanted subcutaneously in both left and right flanks of 6–8 weeks old male and female Nude mice. Tumor volume was determined using electronic calipers to measure length (l), width (w), and the formula (w2 × l)/2. Experiments were terminated when tumor volume in control mice reached ~ 2000 mm3.
IVIS
For bioluminescence measurements animals were anesthetized with inhalation of ~3% v/v vaporized isoflurane. All mice were imaged with a positive control (orthotopically implanted with an NT shRNA cell line). To detect orthotropic pancreatic tumors, mice were injected with XenoLight D-Luciferin - K+ Salt Bioluminescent Substrate (15 mg/ml stock) (D-luciferin potassium salt, Gold Biotech) by intraperitoneal (i.p.) injection at a final concentration of 50 mg/kg. The bioluminescence images were acquired 8 minutes after IP substrate administration using the IVIS Lumina XR® system. Measurements were set up as follows: exposure times ranged from 5 seconds to 1 minute (auto settings); binning on medium; f/stop at 1. The gray scale photograph and pseudo-color luminescent images were superimposed for identification of the location of any bioluminescent signal of the labeled cells. Optical images were displayed and analyzed with IVIS Living Image software packages. Regions of interest (ROI) were drawn to assess relative signal intensity emitted. Bioluminescent signal was expressed as photon intensity, in units of photons/second (p/s) within the ROI.
Survival Studies
After KPC cells implantation mice were tracked everyday post-surgery until death. Survival studies were performed using humane endpoints including a 20% loss in body weight, loss of locomotor activity, or any sign of distress. Mice surviving over 300 days post KPC cell implantations were euthanized.
In vivo treatments
Experimental treatments with PPP were performed by i.p. injections (20 mg/kg/12 hours) of the compound in 10 μl volume of DMSO: vegetable oil, 10:1 (v/v). For experimental treatments with the S6K1 inhibitor, PF-4708671 (MedChem, HY-15773) was dissolved in 10% DMSO (Thermo Fisher Scientific, D1391) first and further diluted in 30% PEG400 (Selleck Chemical, S6705), 0.5% Tween 80 (Selleck Chemical, S6702) and 5% propylene glycol (Sigma-Aldrich, 528072), to achieve a final DMSO concentration of 1%. Vehicle was made of 10% DMSO diluted in 30% PEG400, 0.5% Tween 80 and 5% propylene glycol. Mice were then injected at a final concentration of 50 mg/kg daily for 35 (KPC-6560) or 40 (KPC-4662) days and tumor development was assessed with IVIS Lumina XR® system.
To assess target engagement of PF-4708671, DIO mice with established tumors (day 21 post implantation) or ST mice with established tumors (day 27 post implantation) were injected for 3 days with vehicle or PF-4708671 50 mg/kg. Subsequently mice were euthanized, tumors excised, and tissue were processed and stained for phosphorylated S6 as described above.
Mouse Igf1 antibody (R&D Systems Cat# AF791, RRID:AB_2248752) and IgG control (R&D Systems Cat# AB-108-C, RRID:AB_354267) were resuspended in PBS and administered to mice at a final concentration of 0.1 μg/g by i.p. injections. DIO and ST mice were injected daily for 3 days, starting at day 20 and 27 post KPC-4662 implantation, respectively. Recombinant Mouse Igf-I/Igf-1 Protein (R&D systems, Cat# 791-MG) was resuspended in PBS and administered to ST mice at 100 ng/mouse by i.p. injection; PBS was used as control. Mice were injected once on ~day 29 post KPC-4662 implantation. Subsequently mice were euthanized, tumors excised, and tissue were processed and stained for pS6 and SG index as described above.
Human Tissue Micro Array
Human tissue samples of n = 65 patients with histologically confirmed pancreatic ductal adenocarcinoma were provided by the tissue bank of the University Medical Center Mainz in accordance with the regulations of the tissue biobank and the approval of the ethics committee of University Medical Center Mainz (2019–14390; Landesärztekammer RLP). All patients were therapy naïve and underwent primary surgical resection. Of this cohort, a tissue micro array was created. To overcome heterogeneity, 4 array spots of each tumor sample were generated (2 center, 2 periphery; diameter: 1 mm). Clinical and pathological data, including the BMI of the patients were obtained. Tissue microarrays were immuonstained with anti-CK5, anti-G3BP1, and DAPI and 50% of each tumor-center spot was imaged at 40x. Imaging and analysis was performed blinded. SGs index in CK8 positive cells was derived as described above.
Quantification and Statistical Analysis
Statistical analysis was performed with GraphPad 8 Prism Software (GraphPad Prism, RRID:SCR_002798). Statistical significance was determined with the Mann–Whitney’s two-tailed, unpaired t test for non-parametric values and the student’s two-tailed unpaired t-test for parametric values. For tumor growth tracking with IVIS, data were analyzed by 2-way ANOVA (Time/Total Flux). A p value <0.05 was considered significant.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
We show that stress adaptation via the phase-separation organelles stress granules (SGs) mediates PDAC development. Moreover, pre-existing stress conditions such as obesity are a driving force behind tumor SG-dependence, and enhanced SG levels are key determinants and a chemo-preventive target for obesity-associated PDAC.
ACKNOWLEDGMENTS
This study was supported by an NIH/NCI R37CA160495 grant and a V-Scholar Award to EG and grants of the German Research Foundation (Ga 1818/2-3, SFB1292 TPQ1) to M.M.G. The Thomas Jefferson University Flow Cytometry Core, Translational Research/Pathology Core, and Animal Core are shared facilities partially supported by the National Cancer Center Support Grant (P30 CA056036) to the Sidney Kimmel Cancer Center at Jefferson.
Footnotes
Conflict of Interest Statement: The authors declare no potential conflicts of interest.
Data and Materials Availability
Requests for data and reagents may be directed to, and will be fulfilled by the corresponding author, Dr. Elda Grabocka (elda.grabocka@jefferson.edu).
REFERENCES
- 1.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009;136(5):823–37 doi 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanzo R, Bartkova J, Merchut-Maya JM, Hall A, Bouchal J, Dyrskjot L, et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ 2020;27(3):1134–53 doi 10.1038/s41418-019-0403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497(7451):633–7 doi 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer 2004;4(12):966–77 doi 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 5.Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol 1999;147(7):1431–42 doi 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 2015;4 doi 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P 3rd, Good SK, et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol 2011;31(5):1098–108 doi 10.1128/MCB.01279-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol 2008;28(11):3729–41 doi 10.1128/MCB.02284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goulet I, Boisvenue S, Mokas S, Mazroui R, Cote J. TDRD3, a novel Tudor domain-containing protein, localizes to cytoplasmic stress granules. Hum Mol Genet 2008;17(19):3055–74 doi 10.1093/hmg/ddn203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amen T, Kaganovich D. Stress granules inhibit fatty acid oxidation by modulating mitochondrial permeability. Cell Rep 2021;35(11):109237 doi 10.1016/j.celrep.2021.109237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin Y, Protter DS, Rosen MK, Parker R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell 2015;60(2):208–19 doi 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015;163(1):123–33 doi 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016;164(3):487–98 doi 10.1016/j.cell.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang P, Mathieu C, Kolaitis RM, Zhang P, Messing J, Yurtsever U, et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 2020;181(2):325–45 e28 doi 10.1016/j.cell.2020.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018;172(3):590–604 e13 doi 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R. The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol Cell 2017;68(4):808–20 e5 doi 10.1016/j.molcel.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Namkoong S, Ho A, Woo YM, Kwak H, Lee JH. Systematic Characterization of Stress-Induced RNA Granulation. Mol Cell 2018;70(1):175–87 e8 doi 10.1016/j.molcel.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grabocka E, Bar-Sagi D. Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell 2016;167(7):1803–13 e12 doi 10.1016/j.cell.2016.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol 2008;10(11):1324–32 doi 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- 20.Thedieck K, Holzwarth B, Prentzell MT, Boehlke C, Klasener K, Ruf S, et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013;154(4):859–74 doi 10.1016/j.cell.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 21.Park W, Chawla A, O’Reilly EM. Pancreatic Cancer: A Review. JAMA 2021;326(9):851–62 doi 10.1001/jama.2021.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019;51(1):27–41 doi 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010;31(1):37–49 doi 10.1093/carcin/bgp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zitvogel L, Pietrocola F, Kroemer G. Nutrition, inflammation and cancer. Nat Immunol 2017;18(8):843–50 doi 10.1038/ni.3754. [DOI] [PubMed] [Google Scholar]
- 25.Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD, Ogden CL. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA 2016;315(21):2284–91 doi 10.1001/jama.2016.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogden CL, Carroll MD, Lawman HG, Fryar CD, Kruszon-Moran D, Kit BK, et al. Trends in Obesity Prevalence Among Children and Adolescents in the United States, 1988–1994 Through 2013–2014. JAMA 2016;315(21):2292–9 doi 10.1001/jama.2016.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collaborators GBDO, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N Engl J Med 2017;377(1):13–27 doi 10.1056/NEJMoa1614362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roser HRaM. Obesity. Published online at OurWorldInDataorg2017.
- 29.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003;348(17):1625–38 doi 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 30.Petrelli F, Cortellini A, Indini A, Tomasello G, Ghidini M, Nigro O, et al. Association of Obesity With Survival Outcomes in Patients With Cancer: A Systematic Review and Meta-analysis. JAMA Netw Open 2021;4(3):e213520 doi 10.1001/jamanetworkopen.2021.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep 2012;2:799 doi 10.1038/srep00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K, et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med 2015;7(301):301ra130 doi 10.1126/scitranslmed.3010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004;114(12):1752–61 doi 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bousquenaud M, Fico F, Solinas G, Ruegg C, Santamaria-Martinez A. Obesity promotes the expansion of metastasis-initiating cells in breast cancer. Breast Cancer Res 2018;20(1):104 doi 10.1186/s13058-018-1029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7(5):469–83 doi 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 36.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012;21(6):822–35 doi 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 2016;1(14) doi 10.1172/jci.insight.88328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res 2015;75(14):2800–10 doi 10.1158/0008-5472.CAN-14-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchan JR, Yoon JH, Parker R. Stress-specific composition, assembly and kinetics of stress granules in Saccharomyces cerevisiae. J Cell Sci 2011;124(Pt 2):228–39 doi 10.1242/jcs.078444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aulas A, Fay MM, Lyons SM, Achorn CA, Kedersha N, Anderson P, et al. Stress-specific differences in assembly and composition of stress granules and related foci. J Cell Sci 2017;130(5):927–37 doi 10.1242/jcs.199240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bley N, Lederer M, Pfalz B, Reinke C, Fuchs T, Glass M, et al. Stress granules are dispensable for mRNA stabilization during cellular stress. Nucleic Acids Res 2015;43(4):e26 doi 10.1093/nar/gku1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, et al. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol 2003;160(6):823–31 doi 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov 2016;6(8):852–69 doi 10.1158/2159-8290.CD-15-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zyromski NJ, Mathur A, Pitt HA, Wade TE, Wang S, Nakshatri P, et al. Obesity potentiates the growth and dissemination of pancreatic cancer. Surgery 2009;146(2):258–63 doi 10.1016/j.surg.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 45.Chung KM, Singh J, Lawres L, Dorans KJ, Garcia C, Burkhardt DB, et al. Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma. Cell 2020;181(4):832–47 e18 doi 10.1016/j.cell.2020.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Recouvreux MV, Moldenhauer MR, Galenkamp KMO, Jung M, James B, Zhang Y, et al. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J Exp Med 2020;217(9) doi 10.1084/jem.20200388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, et al. PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin Cancer Res 2016;22(12):2993–3004 doi 10.1158/1078-0432.CCR-15-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer 2011;11(12):886–95 doi 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- 49.Roytblat L, Rachinsky M, Fisher A, Greemberg L, Shapira Y, Douvdevani A, et al. Raised interleukin-6 levels in obese patients. Obes Res 2000;8(9):673–5 doi 10.1038/oby.2000.86. [DOI] [PubMed] [Google Scholar]
- 50.Park HS, Park JY, Yu R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res Clin Pract 2005;69(1):29–35 doi 10.1016/j.diabres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 51.Olszanecka-Glinianowicz M, Zahorska-Markiewicz B, Janowska J, Zurakowski A. Serum concentrations of nitric oxide, tumor necrosis factor (TNF)-alpha and TNF soluble receptors in women with overweight and obesity. Metabolism 2004;53(10):1268–73 doi 10.1016/j.metabol.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 52.Pardina E, Ferrer R, Baena-Fustegueras JA, Rivero J, Lecube A, Fort JM, et al. Only C-reactive protein, but not TNF-alpha or IL6, reflects the improvement in inflammation after bariatric surgery. Obes Surg 2012;22(1):131–9 doi 10.1007/s11695-011-0546-3. [DOI] [PubMed] [Google Scholar]
- 53.Azizian M, Mahdipour E, Mirhafez SR, Shoeibi S, Nematy M, Esmaily H, et al. Cytokine profiles in overweight and obese subjects and normal weight individuals matched for age and gender. Ann Clin Biochem 2016;53(6):663–8 doi 10.1177/0004563216629997. [DOI] [PubMed] [Google Scholar]
- 54.Caroleo M, Carbone EA, Greco M, Corigliano DM, Arcidiacono B, Fazia G, et al. Brain-Behavior-Immune Interaction: Serum Cytokines and Growth Factors in Patients with Eating Disorders at Extremes of the Body Mass Index (BMI) Spectrum. Nutrients 2019;11(9) doi 10.3390/nu11091995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Somodi S, Seres I, Lorincz H, Harangi M, Fulop P, Paragh G. Plasminogen Activator Inhibitor-1 Level Correlates with Lipoprotein Subfractions in Obese Nondiabetic Subjects. Int J Endocrinol 2018;2018:9596054 doi 10.1155/2018/9596054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aleidi SM, Shayeb E, Bzour J, Abu-Rish EY, Hudaib M, Al Alawi S, et al. Serum level of insulin-like growth factor-I in type 2 diabetic patients: impact of obesity. Horm Mol Biol Clin Investig 2019;39(1) doi 10.1515/hmbci-2019-0015. [DOI] [PubMed] [Google Scholar]
- 57.Garneau L, Parsons SA, Smith SR, Mulvihill EE, Sparks LM, Aguer C. Plasma Myokine Concentrations After Acute Exercise in Non-obese and Obese Sedentary Women. Front Physiol 2020;11:18 doi 10.3389/fphys.2020.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Girnita A, Girnita L, del Prete F, Bartolazzi A, Larsson O, Axelson M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res 2004;64(1):236–42 doi 10.1158/0008-5472.can-03-2522. [DOI] [PubMed] [Google Scholar]
- 59.Imai Y, Patel HR, Hawkins EJ, Doliba NM, Matschinsky FM, Ahima RS. Insulin secretion is increased in pancreatic islets of neuropeptide Y-deficient mice. Endocrinology 2007;148(12):5716–23 doi 10.1210/en.2007-0404. [DOI] [PubMed] [Google Scholar]
- 60.He M, Su H, Gao W, Johansson SM, Liu Q, Wu X, et al. Reversal of obesity and insulin resistance by a non-peptidic glucagon-like peptide-1 receptor agonist in diet-induced obese mice. PLoS One 2010;5(12):e14205 doi 10.1371/journal.pone.0014205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stern JH, Smith GI, Chen S, Unger RH, Klein S, Scherer PE. Obesity dysregulates fasting-induced changes in glucagon secretion. J Endocrinol 2019;243(2):149–60 doi 10.1530/JOE-19-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nackiewicz D, Dan M, Speck M, Chow SZ, Chen YC, Pospisilik JA, et al. Islet Macrophages Shift to a Reparative State following Pancreatic Beta-Cell Death and Are a Major Source of Islet Insulin-like Growth Factor-1. iScience 2020;23(1):100775 doi 10.1016/j.isci.2019.100775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hakuno F, Takahashi SI. IGF1 receptor signaling pathways. J Mol Endocrinol 2018;61(1):T69–T86 doi 10.1530/JME-17-0311. [DOI] [PubMed] [Google Scholar]
- 64.Sfakianos AP, Mellor LE, Pang YF, Kritsiligkou P, Needs H, Abou-Hamdan H, et al. The mTOR-S6 kinase pathway promotes stress granule assembly. Cell Death Differ 2018;25(10):1766–80 doi 10.1038/s41418-018-0076-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tavares MR, Pavan IC, Amaral CL, Meneguello L, Luchessi AD, Simabuco FM. The S6K protein family in health and disease. Life Sci 2015;131:1–10 doi 10.1016/j.lfs.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 66.Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell 2009;36(6):932–41 doi 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Long Y, Sou WH, Yung KWY, Liu H, Wan SWC, Li Q, et al. Distinct mechanisms govern the phosphorylation of different SR protein splicing factors. J Biol Chem 2019;294(4):1312–27 doi 10.1074/jbc.RA118.003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jang SW, Liu X, Fu H, Rees H, Yepes M, Levey A, et al. Interaction of Akt-phosphorylated SRPK2 with 14-3-3 mediates cell cycle and cell death in neurons. J Biol Chem 2009;284(36):24512–25 doi 10.1074/jbc.M109.026237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 2017;171(7):1545–58 e18 doi 10.1016/j.cell.2017.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong Y, Chan CB, Kwon IS, Li X, Song M, Lee HP, et al. SRPK2 phosphorylates tau and mediates the cognitive defects in Alzheimer’s disease. J Neurosci 2012;32(48):17262–72 doi 10.1523/JNEUROSCI.3300-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lyon AS, Peeples WB, Rosen MK. A framework for understanding the functions of biomolecular condensates across scales. Nat Rev Mol Cell Biol 2021;22(3):215–35 doi 10.1038/s41580-020-00303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Somasekharan SP, El-Naggar A, Leprivier G, Cheng H, Hajee S, Grunewald TG, et al. YB-1 regulates stress granule formation and tumor progression by translationally activating G3BP1. J Cell Biol 2015;208(7):913–29 doi 10.1083/jcb.201411047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhan Y, Wang H, Ning Y, Zheng H, Liu S, Yang Y, et al. Understanding the roles of stress granule during chemotherapy for patients with malignant tumors. Am J Cancer Res 2020;10(8):2226–41. [PMC free article] [PubMed] [Google Scholar]
- 74.Bailyes EM, Nave BT, Soos MA, Orr SR, Hayward AC, Siddle K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem J 1997;327 (Pt 1):209–15 doi 10.1042/bj3270209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Federici M, Porzio O, Zucaro L, Fusco A, Borboni P, Lauro D, et al. Distribution of insulin/insulin-like growth factor-I hybrid receptors in human tissues. Mol Cell Endocrinol 1997;129(2):121–6 doi 10.1016/s0303-7207(97)04050-1. [DOI] [PubMed] [Google Scholar]
- 76.Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 2014;6(1) doi 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Requests for data and reagents may be directed to, and will be fulfilled by the corresponding author, Dr. Elda Grabocka (elda.grabocka@jefferson.edu).