

Abstract
Restless legs syndrome (RLS) is a common sensorimotor disorder for which two main pathological elements are fairly well accepted: brain iron deficiency (BID) and an altered dopaminergic system. The ability to better understand the causal and consequential factors related to these two pathological elements, would hopefully lead to the development of better therapeutic strategies for treating, if not curing, this disease. The current understanding of the relationship between these two elements is that BID leads to some alterations in neurotransmitters and subsequent changes in the dopaminergic system. Therefore, rodent models based on diet-induced BID, provide a biological substrate to understand the consequences of BID on dopaminergic pathway and on alternative pathways that may be involved. In this review, we present the current research on dopaminergic changes found in RLS subjects and compare that to what is seen in the BID rodent model to provide a validation of the BID rodent model. We also demonstrate the ability of the BID model to predict changes in other neurotransmitter systems and how that has led to new treatment options. Finally, we will present arguments for the utility of recombinant inbred mouse strains that demonstrate natural variation in brain iron, to explore the genetic basis of altered brain iron homeostasis as a model to understand why in idiopathic RLS there can exist a BID despite normal peripheral iron store. This review is the first to draw on 25 years of human and basic research into the pathophysiology of RLS to provide strong supportive data as to the validity of BID model as an important translational model of the disease. As we will demonstrate here, not only does the BID model closely and accurately mimic what we see in the dopaminergic system of RLS, it is the first model to identify alternative systems from which new treatments have recently been developed.
Keywords: Restless legs syndrome, iron deficiency, rodent models, genetics, neurotransmitter systems, translational research
1. Introduction
Restless legs syndrome (RLS) is a common sensorimotor disorder. Its primary clinical component is an often hard to describe, very unpleasant, viscerally-irritating, compulsion or urge to move the legs (Earley, 2003), which would put it in the category of akathisia (Haskovec, 1902). The symptoms are triggered by rest, drowsiness or sleep. The symptoms will be immediately relieved as soon as the patient gets up and walks. It can be temporarily relieved so long as the patient is moving the legs or rubbing the legs but may restart as soon as these activities are stopped. The symptom presentation has a clear and distinct circadian pattern: being worse at night and reduced or absent in the morning (Earley, 2003). Patients with RLS are found to have rhythmic or semi-rhythmic movements of the legs during sleep, referred to as periodic leg movements of sleep (PLMS) (Earley, 2003). It has been postulated that the overlying framework of the disease is a biological bias toward maintaining alertness even in the face of severe sleepiness. RLS can be conceptualized as a biological “drive”, like hunger or thirst, whose primary purpose is to “drive” the individual to increase alertness, activity, and movement, which stops completely once the activities are started (Ferré et al., 2019). This biological drive also operates as a counter to the sleep homeostatic drive (Gamaldo et al., 2009; Saletu et al., 2002), further supporting the concept of an enhancement of the normal arousal mechanisms in this disease (Allen et al., 2009b).
Peripheral iron deficiency (ID) is known to be a contributing factor to both the worsening of the symptoms as well as a cause of the disease. Nordlander in his 1953 paper (Nordlander, 1953) reported that in patients who had an ID anemia (IDA) and concomitant RLS symptoms, treating the IDA with intravenous iron, dramatically improved the RLS symptoms. Several studies have found a strong negative correlation between peripheral iron stores as determined by serum ferritin and RLS severity: decreasing ferritin was associated with increasing RLS severity (O’Keeffe et al., 1994; Sun et al., 1998). In support of a causal relationship between peripheral ID and RLS, there have been several studies in which the prevalence of RLS is substantially greater in patients with IDA (35-45%) (Allen et al., 2013a; Aspenstroem, 1964) than that seen in the general population (7.5%) (Allen et al., 2005). Following his discovery of the effectiveness of intravenous iron in treating RLS symptoms in patients with an IDA, Nordlander went further by demonstrating that intravenous iron can also dramatically improve RLS symptoms in patients who were not ID (Nordlander, 1953). He postulated that it could be possible “that there can exist an ID in the tissues in spite of normal serum iron” (Nordlander, 1953).
It is clear from the last 25 years of research, that the tissue of interest is in fact the brain and that altered brain iron homeostasis is one of the basic pathological factors in this disease (see review: Earley et al., 2014). The first study to assess brain iron status measured cerebrospinal fluid (CSF) ferritin and transferrin levels. The study demonstrated that despite RLS and control groups showing no difference in hemoglobin and serum ferritin levels (both in the normal range), RLS subjects had significantly lower CSF ferritin and significantly elevated CSF transferrin, consistent with CNS ID (Earley et al., 2000). Four further studies utilizing CSF analysis support this initial finding of altered CNS iron in RLS (Clardy et al., 2006a; Clardy et al., 2006b; Earley et al., 2005; Mizuno et al., 2005). The second series of investigative studies utilized various magnetic resonance imaging (MRI) techniques to quantify regional brain iron. The initial study by Allen et al (Allen et al., 2001) found low brain iron concentrations in the substantia nigra (SN) of patients with RLS. Further studies (Astrakas et al., 2008; Earley et al., 2006a; Godau et al., 2008; Haba-Rubio et al., 2005; Knake et al., 2010; Lee et al., 2007; Li et al., 2016; Margariti et al., 2012; Moon et al., 2015; Rizzo et al., 2013; Stefani et al., 2019), many by independent investigators, demonstrated low iron concentrations in the SN or other brain areas. Although the majority of studies supports a relative decrease iron in several brain regions, the decrease in the SN was the most consistent finding and, at least, provides a surrogate for altered brain iron homeostasis in RLS. A distinctly different technology from that of MRI is transcranial ultrasound of the midbrain. Changes in SN echogenicity have been shown to correlate with total iron concentration in SN (Zecca et al., 2005). Utilizing this technique, two separate investigators demonstrated relatively low brain iron concentrations in the SN of RLS patients compared to controls (Godau et al., 2007; Schmidauer et al., 2005). Most subjects from the CSF, MRI and transcranial ultrasound studies cited above, were not anemic nor did they have a frank ID. The data, therefore, support the general concept of brain ID (BID) in the absence of peripheral ID. The first direct measurement of iron and its related proteins in the brain, utilized postmortem material. The initial study demonstrated a decrease in H-ferritin and increase in transferrin along with the decrease in iron staining in the SN (Connor et al., 2003). Further studies, using brain autopsy material and examining white matter, microvascular and choroid plexus, add further support to the concept of altered brain iron homeostasis that may go beyond the substantia nigra (Clardy et al., 2006b; Connor et al., 2011a; Connor et al., 2011b; Connor et al., 2004; Snyder et al., 2009).
In summary, RLS symptoms are sensitive to the concentration of peripheral iron. Systemic ID appears to be an important environmental trigger for the worsening or development of this disease. On the other hand, despite normal peripheral iron stores, RLS patients appear to have a relative BID. The findings support Nordlander’s original postulation that there can exist in the tissues an ID despite normal blood levels (Nordlander, 1953). It is clear from the data that there is a disconnect between serum-determined iron levels and CNS iron levels in patients with RLS. Cellular ID in the SN has been the most consistent finding and at least represents a viable surrogate for altered iron homeostatic mechanisms in RLS. The data support the relevance of an animal model that is based around the construct of low brain iron or BID as a translational approach towards better treatment options in this disease. The data also support the development of models to better understand the mechanisms behind variation in regional brain iron homeostasis and thus provide translational constructs that could potentially lead to an understanding of this primary pathology in RLS. As we will demonstrate here, not only does the BID model closely and accurately mimic what we see in the dopaminergic (DAergic) system of RLS, this review will also provide strong supportive data as to the validity of BID model as an important translational model of the disease.
2. Aims and Approach
The first aim of this review is to validate the diet-induced BID rodent model as a predictive model of the pathophysiology in RLS. To accomplish that aim, we reviewed all currently published research into the pathological bases of an altered DAergic system in RLS and compare that to published data on consequences of BID on the DAergic system. The second aim is to prove the utility of the BID model to predict alternative neurotransmitter systems. To accomplish that aim, we reviewed the published studies in which the BID model was used to explore alternative neurotransmitter systems on which were based therapeutic trials in RLS. The third aim was to validate a rodent model, which allows us to examine the consequences of natural variations in brain iron and explore the complex genetics behind the variations. To accomplish that aim, we reviewed the published studies that have utilized the BXD/Ty recombinant inbred (RI) strains of mice to examine variations in brain iron, its consequences, and the genetic basis for these variations.
Our approach was to review all known published papers relevant to the DAergic system in pathophysiology of RLS, which includes studies utilizing CSF analysis, brain tissue analysis, and positron emission tomography (PET) and single-photon emission computed tomography (SPECT) imaging techniques. The data collection and analysis were restricted to the basal ganglia (BG) because that is where the majority of research studies were focused and also that is where the majority of research into the consequences of BID on DAergic system focused as well. Where there were multiple studies reporting either positive or negative studies, we chose to bias the presentation towards the predominate results, whether negative or positive or at least provide a fair discussion of both data sets. If there were directional difference between studies or differences between types of investigative technique, then that was discussed. The primary focus of this paper is on BID and brain iron variability as a starting point for RLS model development and essential validation of the models utilizing current data from human studies of RLS pathophysiology. This was not meant to be a broad-based review of RLS models and therefore does not include other published models utilizing primary genetic approaches. Also, models have been used to mimics behavioral components of RLS (e.g., periodic leg movement). Although not the focused of this review, we do present, as an aside, studies that have utilized the BID or BXD models in exploring behavioral components of the disease, mostly as supportive evidence for the broader utility of the models.
3. Validating the diet-induced, BID rodent as a model of RLS.
The validation of any disease-based model should first start with its ability to predict known pathology (Salminen et al., 2020). The two main pathological elements in RLS that are fairly well accepted are BID and altered DAergic system. The current understanding of the relationship between these two is that BID leads to some alterations in neurotransmitters and subsequent change in the DAergic system (Earley et al., 2014). Thus, a validation of the BID rodent model would be how well it predicts some of the known pathological findings in RLS. The theory of an altered DAergic system in RLS is derived from the significant clinical benefits achieved with the use of levodopa and further supported by randomized clinical trials of DA agonists (Allen, 2004). As RLS is a form of akathisia and DA antagonists are known to increase the expression of akathisia, this further supports the concept of decreased DAergic activity as factor in the development of RLS (Ferré et al., 2021). An extensive review of the investigative work into understanding the DAergic pathology of RLS has been presented elsewhere (Earley et al., 2014), but will be reviewed here, in brief, in order to provide the context for comparing and thus validating the BID rodent model. The standard research approach to inducing BID in rodents, is providing rodents with an iron-insufficient diet of 4 part per million (ppm) or less of iron, compared to 35 ppm or greater iron for normal “control” diet, at day 21 of weaning for the full life of the animal. This model has been utilized extensively to explore the consequences of BID on brain maturation, neurochemical changes and cellular function. An elaboration on this basic ID-diet-at-weaning model is the introduction of an ID diet to pregnant dames or at postpartum day one. These later ID conditions are exploring the effects of in utero or neonatal ID on later brain development. Although of potential relevance to exploring the broader potential consequences of BID and thus its implication for RLS, that aspect of the BID model will not be examined here.
3.1. Changes in tyrosine hydroxylase and dopamine synthesis
Iron is an essential cofactor, along with oxygen and tetrahydrobiopterin for tyrosine hydroxylase (TH) activity, which is the rate-limiting enzyme for DA production (Cooper et al., 2008). The initial hypothesis was that BID leads to a decrease in TH activity and thus decreases in DA production (Earley et al., 2001). The data, however, proved to be just the opposite: CSF analysis found that RLS subjects had an increase in tetrahydrobiopterin, which is the primary rate determining cofactor for TH activity, and in 3-O-methyldopa (3OMD) (Allen et al., 2009a; Earley et al., 2001). 3OMD is almost exclusively seen when dihydroxyphenylalanine concentration exceeds the ability of dihydroxyphenylalanine decarboxylase to convert it DA (Earley et al., 2006b). Postmortem studies utilizing tissue from the putamen and SN showed a significant upregulation in TH and its active from, phosphorylated TH (pTH), in RLS compared to controls (Connor et al., 2009). The CSF and autopsy data suggest that there is, opposite to the prediction, an increase in DA synthesis associated with RLS (Earley et al., 2014). Sprague-Dawley rats were used to examine the effect of diet-induced BID on TH activity in the BG. The study demonstrated that BID leads to an increase in TH and pTH in line with that seen in RLS pathology (Connor et al., 2009). This finding of ID leading to an increase in TH or pTH rat brain, was also found in PC12 cells made ID with an iron chelator (Connor et al., 2009). Thus, ID shows a consistent effect of upregulation of TH and its activity, against all predictions. An explanation for this unexpected finding, is that BID is acting on the hypoxia-inducible factor (HIF) pathway (Earley et al., 2014), which is then upregulating TH (Schofield and Ratcliffe, 2004). Other studies that have explored the relation between ID, HIF and the DAergic system have found a relation between ID, increased HIF and TH upregulation (Guo et al., 2016; Nguyen et al., 2007). Back translating these animal and cell culture findings to explore the potential role of HIF in RLS, Patton et al (Patton et al., 2011) showed that RLS postmortem tissue and cortical microvasculature had increases in HIF-1α, HIF-2α and HIF-related factors, like vascular endothelial growth factor. Thus, BID model not only mimicked the findings outlined above in CSF and postmortem from RLS but also predicted the role of HIF in the disease.
3.2. Changes in extracellular dopamine and dopamine-reuptake transporter
Another approach to evaluating the DAergic system in RLS is through use of positron emission tomography (PET) and single-photon emission computed tomography (SPECT) imaging techniques. Several studies have utilized SPECT ligand [123I]-2β-carbomethoxy-3β-(4-iodophenyl) tropane, which binds to both dopamine-reuptake transporter (DAT) and the serotonin reuptake transporter. This ligand has long-data acquisition times (e.g., 22-24 hours) and likely reflects total DAT pool, not just membrane-bound DAT. Two studies that used this ligand found no change (Michaud et al., 2002; Mrowka et al., 2005), while one study in an elder RLS cohort found small increases in DAT binding (Kim et al., 2012). Studies utilizing PET or SPECT ligands with higher DAT specificity and involving short-data acquisition times most likely are assessing primarily membrane-bound DAT. Two studies involving three independent cohort of subjects that utilized these ligands, showed decreases in BG DAT concentrations (Earley et al., 2011; Lin et al., 2016), while two studies with much smaller sample sizes, found no significant change in DAT (Eisensehr et al., 2001; Tribl et al., 2002). Postmortem evaluation of tissue from the putamen and SN showed no difference in total DAT concentrations between RLS and controls (Connor et al., 2009). Given the small sample size and the fact that total cellular DAT was determined, the possibly of a relative decrease in membrane-bound DAT in RLS cannot be excluded. The three positive brain imaging studies with short-data acquisition times support a role for at least a decrease in membrane-bound DAT in RLS. In the rat, diet-induced BID was associated with decreased membrane-bound DAT and DAT mRNA as well a decrease DAT functionality in the BG (Bianco et al., 2008 ). In fact, the most consistent effect seen in rats, mice, PC12 cells and neuroblastoma cells with ID is a decrease in DAT with many of the studies suggesting a dysfunctional transporter (Bianco et al., 2008 ; Unger et al., 2014a; Wiesinger et al., 2007).
The BID rat models show an increase in extracellular DA (ecDA) (Nelson et al., 1997). There was no indication of increased storage based on the absence of any difference in the vesicular monoamine transport-2 (Bianco et al., 2008 ). Also, amphetamine-induced assessment of DA release did not indicate changes in release or intracellular DA pool that could account for the increase in ecDA (Bianco et al., 2008 ). Using other techniques, the findings implicated decreased DAT as the likely primary cause of the increase in ecDA (Bianco et al., 2008 ). The increase in ecDA in BID model was associated with an increase HVA, which is reported to reflect increase ecDA metabolism (Nelson et al., 1997). Therefore, one interpretation of the increase in CSF HVA seen in RLS is that it reflects metabolism of increased ecDA. Utilizing special PET techniques, changes in BG DA D2 receptor binding potential (D2R BP) in RLS subjects were interpreted as indicating the presence of increase ecDA in RLS (Laruelle, 2000). Given that there appears to be an increase in DA synthesis in RLS, as well as a possible decrease in DAT, the increase in ecDA may reflects increased release and/or decreased uptake. Postmortem tissue from RLS did not show any change in putamen vesicular monoamine transport-2 (though a non-significant increase in SN VMAT was found) (Connor et al., 2009). Utilizing PET-determination of BG DA D2 receptor (D2R) binding potential (BP) and its reduction following amphetamine injections, is a well establish approach to determine pre-synaptic DA release (Laruelle, 2000). Utilizing this technique in a study of D2R-BP (Earley et al., 2013), no difference in the amount of DA released was found between RLS and control subjects (data not reported). Thus, increased DA storage or release to account for the increase in ecDA in RLS, though possible, is not supported by the current data. In conclusion, RLS appears to be associated with an increase in ecDA, which may be a result of increased release or decreased uptake. The BID model again demonstrates compatible changes to those seen in RLS and potentially adds more to our understanding of the pathology. The BID rat model shows that BID affects membrane-bound to a greater degree than total DAT (Bianco et al., 2008 ; Burhans et al., 2005), which could account for the absence of total DAT changes in RLS putamen. More importantly, ID in rodent and cells significantly affects the quality of the transport, not just the quantity (Bianco et al., 2008 ; Erikson et al., 2000; Wiesinger et al., 2007). This later element cannot be assess using any current technique in postmortem tissue or humans. Therefore, any finding with postmortem DAT concentrations or with SPECT/PET assessment of DAT, can only reflect transporter quantity and not transporter functionality. Of translational relevance, studies in the BID model have shown these findings in DAT and ecDA are reversed with iron treatment (Nelson et al., 1997).
3.3. Changes in the dopamine receptors
Postmortem tissue from RLS and control subjects was used to determine the density of total D2R in the putamen. RLS patients had a significant lower level of D2R in the putamen. The density of D2R was correlated positively with RLS severity (r= 0.8, p=0.018). The majority of SPECT- and PET-based assessments of BG D2R binding potential (BP) have shown a decrease in BP (Earley et al., 2014). This decrease in D2R BP may be a result of decreased D2R affinity (Kd) or density (βmax) or a result of increased ecDA (Narendran et al., 2004). Special PET techniques have been developed to provide a crude determination of the membrane-bound D2R, Kd and βmax in the BG (Kuwabara et al., 2012). Utilizing these techniques, a decrease in D2R BP was not associated with a comparable difference in Kd and βmax in the BG (Laruelle, 2000). One interpretation of the finding is that change in D2R BP are, in part, related to increased ecDA. Another interpretation, is that methods to estimate D2R, Kd and βmax in the BG (Kuwabara et al., 2012) within the frame work of the current PET technology are not sensitive enough. Finally, even if the interpretation of the decrease in D2R BP that has been found in a majority of PET/SPECT studies, is wholly related to increased ecDA, it does not negate the changes in D2R that were found in postmortem tissues. There are notable and relevant differences between D2R assessment in postmortem tissue or by PET/SPECT techniques. For postmortem assessment, total cellular concentration was determined using antibodies that have high specificity for D2R with no interaction with the other four DA receptor subtypes. PET/SPECT ligands on the other hand, bind to both D2R and D3R that are membrane bound. Overall, the data support the concept that RLS is associated with a relative decrease in D2R in the BG. There may or may not be a quantitative or qualitative change in D2R or D3R on the membrane. Of course, neither approach is able to say whether there is a functional change in the D2R. Utilizing the BID rat model, studies have found that total D2R density in the BG is decreased with ID. Studies have demonstrated a decrease in the cAMP inhibitory response to D2R agonist, suggesting ID leads to functional change in the receptor, not just a quantitative decrease in receptors. The findings further validate the utility of the diet-induced BID rat as a model that closely mimics the underlying pathology in RLS.
3.4. Glutamatergic system in RLS
Aside from the DAergic system, the only other potential neurochemical system to be implicated in RLS is the glutamatergic system (GLUergic). In an effort to better understand one of the major paradoxes of RLS (lack of significant sleepiness despite severe sleep loss), the thalamic GLUergic system was assessed using proton magnetic resonance spectroscopy (MRS). RLS patients showed an increase in Glx, which is an indirect measurement of glutamate (Glu) and corresponds to a combination of GLU and glutamine spectral peaks (Allen et al., 2013b). This MRS measurement correlated negatively with total sleep time: increasing Glx activity was associates with decreasing sleep time (r=0.60; p=0.008). The findings were interpreted as implicating an increase in GLUergic activity in RLS (Allen et al., 2013b). Given this preliminary and suggestive finding of increased GLUergic activity in RLS, the diet-induced BID rodent model was used to explore the potential role of BID in affecting the GLUergic system. The GLUergic system that was chosen to be explored with the BID model, was the corticostriatal-GLUergic (CS-GLUergic) pathway. The selection of the system was based on several clinical aspects of RLS. The essential clinical feature of RLS is an urge to move thus implicating the motor system (Earley, 2003). The vast majority of investigative data into the pathophysiology of RLS have implicated the BG (Earley et al., 2014). Finally, transcranial magnetic stimulation has consistently found presence of a “hyperactive” motor cortex in this disease (Scalise et al., 2010; Tergau et al., 1999), which could be attributed to increase GLUergic motor pathway (Stagg et al., 2011).
The pathway was examined in diet-induced BID Sprague-Dawley male rats (Yepes et al., 2017). The CS-GLUergic pathway was stimulated at the level of the striatum using an optogenetically-based technique, with a modified microdialysis probe with an embedded optic fiber engineered to deliver light in the same brain area being sampled for GLU (Yepes et al., 2017). Stimulation at 100 Hz was effective in increasing GLU release in both BID and control rats. Stimulation at a lower frequency of 60Hz was only associated with increased GLU release in BID animals, indicating a BID-induced increased sensitivity of the CS-GLUergic terminals. As proof of concept that this finding represents an RLS-relevant finding, animals were treated with pramipexole, ropinirole and gabapentin, which are all known to markedly improve RLS symptoms (Silber et al., 2021). Optogenetically-induced release (60 Hz stimulation) of GLU from the CS-GLUergic pathway in BID animals was completely blocked by the local striatal infusion of pramipexole or gabapentin. The model was then utilized to assess DA receptor subtypes involved in the pramipexole effect. L745-870, a specific D4R antagonist, and raclopride, a D2R-D3R antagonist, completely counteracted the inhibitory effect of pramipexole on GLU release. VK4-116, a selective D3R antagonist, did not block the effects of pramipexole. These results agree with another study using the optogenetic-microdialysis technique in mice with a genetically modified D4R in which the third intracellular loop corresponding to a common human polymorphism (Bonaventura et al., 2017). This study demonstrated a significant role of D4R in the modulation of CS-GLUergic transmission (Bonaventura et al., 2017).
These results suggest that selective D4R or D2R agonists may be of better therapeutic value in RLS than the less selective DA receptor agonists used so far. This prediction awaits to be tested. There is also evidence indicating that in the CS-GLUergic terminals, D4R form complexes (heteromers) with D2R (Gonzalez et al., 2012; Sanchez-Soto et al., 2019), suggesting that D2R-D4R heteromers should be most appropriate targets (Fig. 1). However, in vitro experiments indicate that the properties of D2R-D4R heteromers depend on the D4R polymorphic variant, including their response to agonists and the constitutive activity of the D2R in the heteromer (Sánchez-Soto et al., 2019). This implies that determining the D4R polymorphic variant might be necessary to establish the efficacy of D4R agonists in RLS patients.
Figure 1.
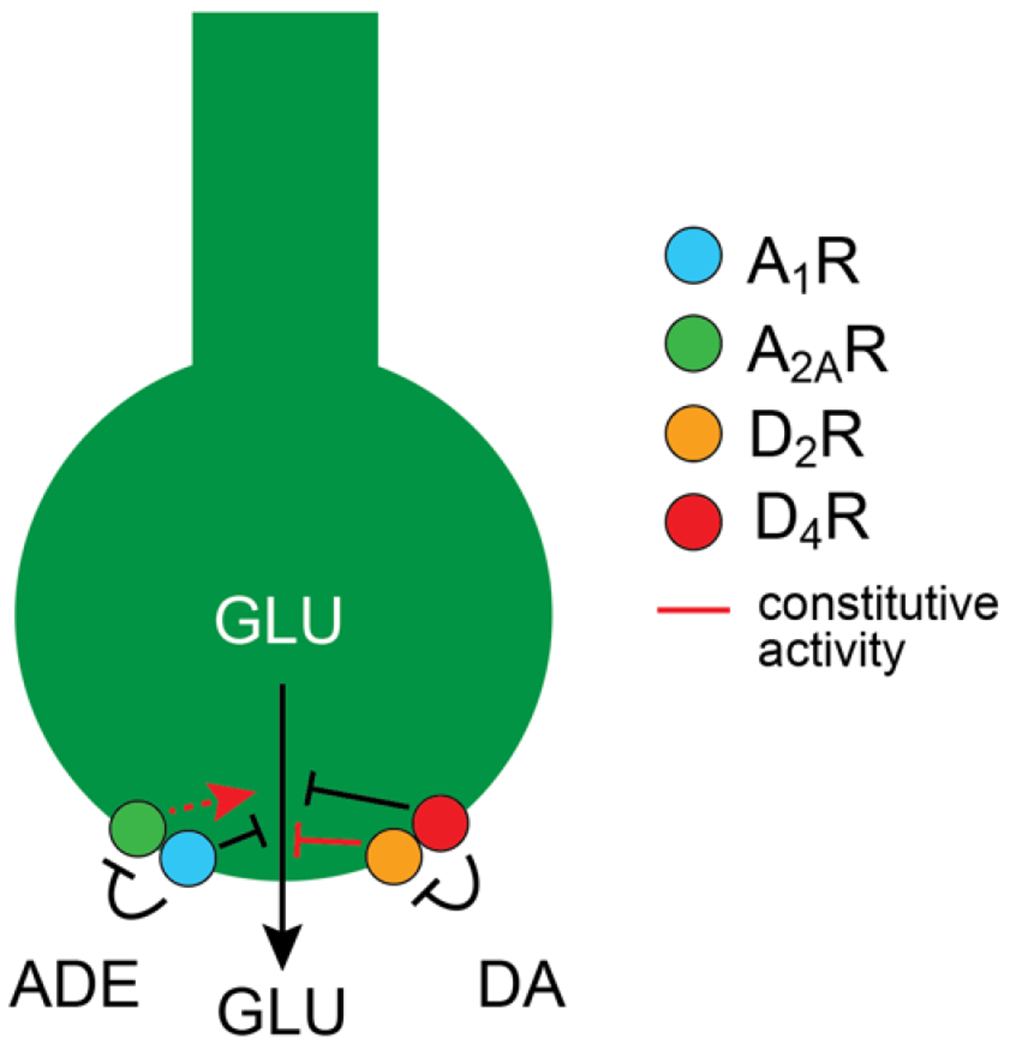
Schematic representation of a cortico-striatal glutamatergic (CS-GLUergic) terminal and its modulatory dopamine (DA) and adenosine (ADE) receptors. DA and ADE modulate glutamate (GLU) release by acting on A1R-A2AR and D2R-D4R heteromers. Both, ADE and DA exert and inhibitory modulation of the sensitivity of the CS terminal to release glutamate. A1R plays a pivotal role by mediating the effect of endogenous ADE, which has more affinity for A1R than for A2AR, and by inhibiting the constitutive activity of the A2AR. Under conditions of downregulation of A1R (such as with BID), the unleashed constitutive activity of A2AR not forming heteromers with A1R promotes and increased sensitivity of the CS-GLUergic terminal. D2R forms heteromers with D4R in the CS-GLUergic terminal and exerts a constitutive activity which decreases the sensitivity of the CS-GLUergic terminal. The ability of D4R to modulates the constitutive activity of the D2R and its response to DA in the D2R-D4R heteromer depends on the D4R polymorphic variant.
The reversal of this BID-induced hyperglutaminergic state by two medications that are pharmacologically diverse and very effective in treating RLS, is highly supportive of the ability of BID model to predict alterations in neurotransmitter systems that may underlie RLS. As a direct translational derivative of these BID-model findings, a clinical trial was undertaken to look at a clinically-available, selective AMPA receptor antagonists, perampanel, in treating RLS symptoms (Garcia-Borreguero et al., 2017). After eight weeks of treatment, perampanel significantly reduced RLS severity and, based upon polysomnogram assessment, significantly improved total sleep time and sleep efficiency. PLMS, which is a clinical signature of the disease, were also reduced by the treatment. This was the first study to develop a treatment for RLS that was based on a combination of limited human data and BID-model-derived changes in GLUergic system. Prior to this study, all other treatments were found serendipitously without clear understanding as to why they had worked. The next level of challenge for the validation of BID model is to explore the consequences of BID on other systems not yet implicated directly or indirectly in RLS and provide a potential unified theory of RLS pathology.
3.5. Translational capability of the BID model
Given the relationship between the adenosinergic (ADergic) system and DAergic systems implied by the work with Parkinson’s disease (Jenner, 2005), the earliest work into exploring alternative neurotransmitter system that might be affected by ID and might impact DAergic system, focused on the ADergic system. The potential role of adenosine in RLS has been reviewed (Ferre et al., 2017) but a brief presentation of some of those findings will be presented here. A series of four studies (Ferre et al., 2019; Gulyani et al., 2009; Quiroz et al., 2016; Quiroz et al., 2010), which included both rat and mice (C57/BL6) using ID diets of 3 ppm or 7 ppm iron, as well as recombinant inbred (RI) strains of mice (cross between C57/BL6 and DBA/2J (BXD)) with natural variation in striatal iron (Jellen et al., 2012), demonstrated the following. First, BID with 3 ppm and 7 ppm iron diet in rats and mice produce a downregulation of striatal D2R and adenosine A1 receptors (A1R). Only the 3 ppm diet produced an upregulation of adenosine A2A receptors (A2AR). Second, the BXD RI strain with the lower striatal iron concentration had a downregulation of striatal D2R and upregulation of striatal A2AR (A1R were not assessed in this study) compared to a strain with higher iron concentrations.
Analyzing the optogenetically-stimulated GLU release from CS-GLUergic neurons, the increased CS-GLUergic sensitivity previously found in BID rats, was reproduced by an A1R antagonist and counteracted by increasing extracellular adenosine by inhibiting the equilibrative nucleoside transporter (ENT)1 and ENT2 with dipyridamole (Ferre et al., 2019). Both A1R and A2AR are co-localized in CS-GLUergic terminals, where they form A1R-A2AR heteromers (Ciruela et al., 2006) (Fig. 1). The A2AR has a strong constitutive activity, which is blunted when forming heteromers with the A1R (Kofalvi et al., 2020). It has recently been demonstrated that BID in rats leads to a relative reduction of A1R versus A2AR in CS-GLUergic terminals and, therefore, to an increase in the population of A2AR not forming heteromers, which constitutive activity could be mostly responsible for the increased sensitivity of CS-GLUergic terminals (Rodrigues et al., 2022). The experiments with dipyridamole showed that an increase in the extracellular concentration of adenosine leads to a predominant activation of A1R, which is related to the higher affinity of adenosine for A1R than for A2AR. Therefore, dipyridamole or an A2AR antagonist that would also counteract the constitutive activity of A2AR (an A2AR inverse agonist) could be of potential therapeutic value. In fact, as predicted by the BID model, a randomized, placebo-controlled clinical trial with dipyridamole was recently performed and found to be effective (Garcia-Borreguero et al., 2021). Two weeks of treatment with 300 mg of dipyridamole, compared to placebo, produced significant improvement in standard subject rating scales and in polysomnographic measures of sleep quality and PLMS. These findings are an important validation of the BID model as true biological model of RLS disease.
3.6. Summary- BID rodent models as a model for RLS
A key and essential validation for any currently-based model is its ability to demonstrate compatible biological and behavioral changes that mimic or replicate changes seen in the human condition. Based on our current understanding, the primary pathophysiological characteristic of RLS is BID. Within a subgroup of this population, RLS is clearly related to a primary peripheral ID in that severe IDA creates a six-fold increase in the prevalence of disease, which is reversible with treatment. These clinical findings are the basis for the current BID rodent model. As outlined in this review, those changes in RLS that are found in the DAergic system, which include increased DAergic synthesis, increased ecDA, decreased density of DAT and D2R, are highly reproducible in BID rodent model. Limited clinical data on the role of the GLUergic system is found to be mimicked in the BID rodent model and subsequently validated by clinical therapy, which affected the GLUergic transmission. Also pertinent to its validation is the compatibility between the BID model and clinical features of the disease, BID in rats has been shown to reproduce one of the signature clinical feature of the disease, PLMS (Lai et al., 2017). Ultimately, the most important validation for any disease-based model is its ability to predict a yet unknown pathological factor for which new treatment could be developed. In this regard, BID model identified the ADergic system as a potential player in the pathophysiology of disease. The model demonstrated the value of dipyridamole, which was subsequently found in clinical trials to be of benefit in treating RLS symptoms. All current treatments for RLS were found serendipitously without clear understanding as to why they should work. The BID model not only proved effective in mimicking the known pathology in RLS, but also defined the potential role of the ADergic system and provided a new therapy. Based upon existing human pathological data in concert with the nuanced changes seen in neurotransmitter systems with BID model, a more dynamic model of RLS has been proposed (Ferré et al., 2019). More importantly, this validated BID model offers a significant opportunity to explore more nuanced changes in receptor pharmacology that could provide much more selective and specific treatment options (Ferré et al., 2021).
4. Validating the BXD recombinant mice as a model of RLS
BID rodent model provides an understanding of the consequences of BID and how that might relate to RLS. It does not, however, address the question of how RLS patients, who are otherwise not iron deficient, have BID. Also, the BID model more closely mimics the secondary form of RLS associated with severe ID rather than the more common primary form, which is not associated with peripheral ID. BXD recombinant inbred (RI) strains represent a genetic reference population that is a result of a cross between two different parental strains: C57/BL6 and DBA/2J (Ashbrook et al., 2021; Williams et al., 2001). Each RI strain (S) within the BXD/Ty RI population represents a unique composite of genes derived from either of the two parental gene pools (Fig 2). By assessing a biological element or phenotype across multiple strains, some degree of variable expression of that phenotype will be evident. Because the genetic variation of each of more than 140 extant strains is known, complex genetic interactions that are behind the phenotypic variability, can also be identified. Across a series of selected strains, studies have shown large variations in the ventral midbrain (VMB), containing the SN, and striatal iron concentrations under normal dietary iron condition as well as under ID dietary condition (Jellen et al., 2012). Studies show limited if any association between the brain iron status and peripheral iron status (Jones et al., 2003). Even within the brain, regional variance in iron concentration do not always correlate very well (Jones et al., 2003). The clear message from the BXD model, if nothing else, is that peripheral iron status based in serum values is not a reliable maker of brain iron status. This peripheral-brain iron dissociation is at the heart of both the clinical and pathological aspect of RLS and thus why the BXD model offers a more biologically compatible model of idiopathic RLS (Earley, 2009). The clinical implications are important when trying to conceptualize why iron therapy would be of value in RLS patients who are not anemic (Allen et al., 2018). The studies with BXD mice demonstrate several distinct features that support the uniqueness of the model compared the diet-induced BID rodent model.
Figure 2.
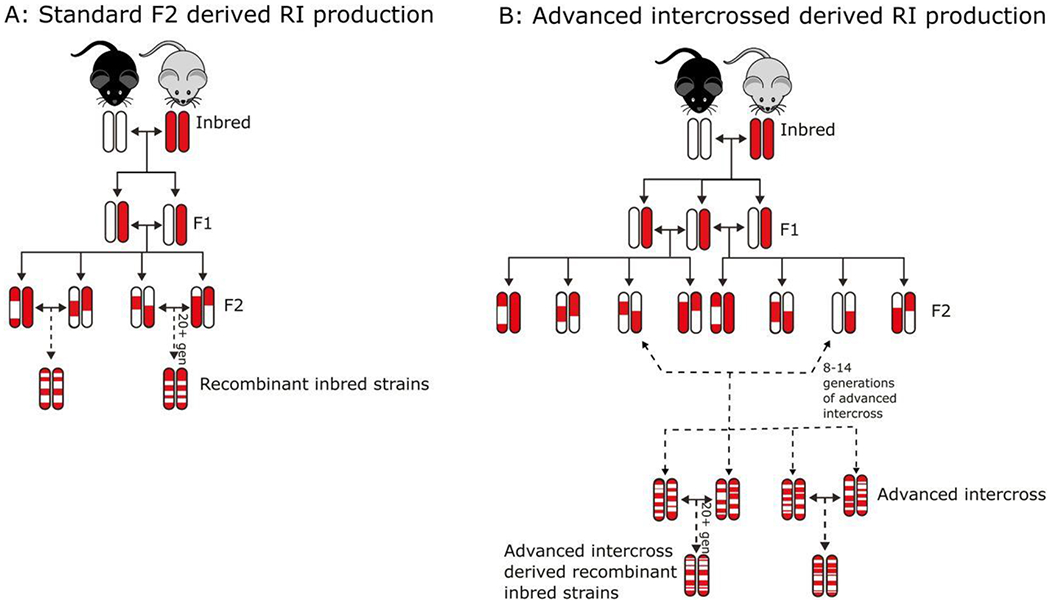
Schematic representation of derivation of the BXD recombinant inbred mouse strains. Panel A shows the derivation of the original 40 strains from an F2 intercross between C57Bl/6J (black strain) and DBA/2J (grey strain) mice. The two strains differ by more than 6 million variations in DNA and derivation from F2 maintains many genetic linkages and thus challenges gene mapping precision. Panel B helps to remedy the situation by breaking up linkages via advanced intercrosses and thus improving mapping precision. Reproduced and modified from Ashbrook and others (2021)
4.1. The BXD RI mice provide a means to understand the complex genetics behind natural or diet-induced ID variant in brain iron.
Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNP) implicating several genes that are associated with increasing risk of developing RLS (Winkelmann and Muller-Myhsok, 2008). In the Icelandic GWAS study, the RLS risk allele on btbd9 was associated with lower serum ferritin levels (Stefansson et al., 2007). In a Danish blood donor study, the allelic variant per se was associated with lower serum ferritin levels after two years of blood donation, even though ferritin levels were similar to non-allelic carriers at the beginning of blood donation (Sorensen et al., 2012). It is clear from human studies that this gene plays some role in iron homeostasis. In flies, the homolog of this gene was found to ubiquinate an important iron regular protein, iron responsive protein-2 (Freeman et al., 2012). Utilizing the BXD data on variations in VMB iron, btbd9 expression was strongly correlated with VMB iron concentrations: lower expression is associated with lower iron (Jellen et al., 2009). In looking for genes of relevance, a strong correlation was found between natural variances in VMB iron and striatal DAT function and striatal D2R (Jones et al., 2003). The strongest correlation was found between variances in VMB iron, either under normal or ID diet conditions, and glt1 expression (normal: r=0.9; ID: r=0.83) (Jellen et al., 2012). Glt1 is GLU-transporter-1, which is found primarily on astrocytes and is essential in regulating GLU metabolism and GLUergic system. In summary, the BXD model was able to demonstrate a relation between known RLS pathology and a known RLS-risk gene. The BXD model further showed its ability to provide a coherent relation between VMB iron concentrations and key elements in the DAergic and GLUergic systems, which are both important components of RLS pathology. The model has the potential to better understanding gene-gene interaction-effects on iron or any other neurotransmitter system considered pertinent to RLS.
4.2. The BXD model has the ability to explore more dynamic biological changes in iron homeostasis that may be pertinent to RLS.
In general, genes that regulate iron homeostasis for any given organ or brain region are not always the same genes that are involved in iron regulation under severe ID condition (Jellen et al., 2012; Yin et al., 2012). As simple phenotypic example of complex genetics, BXD RI strain 19 (S19) and S40 have similar hematocrits and similar VMB iron concentrations under normal iron diets (Jellen et al., 2012). With ID diet, S40 females (S40f; BXD40 males do not show the same iron management profile as the females) show minimal change in hematocrit while S19f drop by 90%. On the other hand, diet-induced ID is associated with a 60% drop in VMB iron for S40f but no change in VMB iron for S19f. In the broader perspective, the model offers an opportunity to explore genes that are involved in maintaining or countering the consequences of dietary ID. The model could be used specifically to understand why some individuals with severe ID develop RLS and while other never seem to develop RLS. Studies in humans have demonstrated natural circadian changes in serum iron regulation with the serum iron levels dropping at night (Kuhn and Brodan, 1982). In non-human primates, similar circadian changes in serum iron are found but there is a concomitant and notable increase in CSF iron and ferritin at night, again demonstrating the dissociation in iron regulation between brain and peripheral systems (Hyacinthe et al., 2015). One of the defining clinical aspects of RLS is that symptom expression follows a distinct circadian pattern being worse at night (Earley, 2003). One hypothesis is that despite normal blood iron levels, natural circadian variations in CNS iron are at fault for producing circadian changes in symptoms. Specifically, the expected nighttime rise in CNS iron levels to support an increased metabolic activity during sleep, is inadequate in RLS (Earley et al., 2014). This is, in part, supported by the finding of decrease in the late night/early morning CSF ferritin in RLS relative to controls, a difference that does not exist in sample collected 12 hours later (Earley et al., 2005). BXD strains have been used to identify mouse strains that have this relative decrease in brain iron during the inactive period (Unger et al., 2014b). Other areas of interest include assessing for variability in iron export out of the CNS in small membrane vesicle call exosomes. This can be done across time, in different strains and dietary iron status to better understand why patient with RLS show increased export of ferritin from the CNS when in fact the brain appears to have ID (Chawla et al., 2019).
4.3. The BXD model can be used to develop a single-strain prototype of RLS disease.
An equally important value of the BXD model is its ability to provide a single strain within the broader cohort of mice, whose iron homeostatic mechanism best mimics that seen in RLS (or best mimic non-RLS population) that can be used to explore more subtle changes in neurotransmitter systems, and to develop a behavioral model. Both of which could provide a source for developing new potential therapies. Two strains were selected from the larger BXD cohort to explore the effects of natural variations in brain iron on neurotransmitter systems (Gulyani et al., 2009). BXD S40f under normal dietary conditions had relatively lower striatal iron (12 μg/g tissue) compared to S1f (22 μg/g tissue). Despite the fact that the range of striatal iron levels is in the normal range for both strains -- obviously on the higher or low side of that range --, S40f showed a decrease in D2R and an increase in A2AR density in the striatum compared to S1f concentrations. This study showed that subtle natural changes in iron are clearly relevant as they can make dynamic changes in neurotransmitter systems. These natural difference in striatal iron concentration produced a similar effect to that seen with diet-induced BID model, but without concern for the physiological consequences of anemia. In developing a potential behavioral prototype, a single-strain selection would need to have a broader composite of RLS pathology. The composite that would be considered appropriate would include a mouse strain with medium to low VMB under normal diets, a drop in VMB iron but not in hemoglobin with ID diet, a natural and significant decrement in VMB brain iron during the inactive period and expressing relatively lower striatal D2R compared to a strain with higher striatal iron. Based on these distinctive properties that appear relevant to RLS iron pathology, S40f was considered to be a potential single-strain model of RLS (Earley et al., 2020). The S40f mouse along with 7 other strains, several of which had distinctly opposite biological factors to that seen in RLS, were then assessed for an RLS phenotype: increased activity at the end of the active period. S40f showed a distinct and significant increase in the home cage activity in the last two hours of the active period. A finding which was only seen with one of the other seven strains. Across all eight strains, this 2-hours period of activity was correlated with diurnal changes in VMB iron (Earley et al., 2020). A subsequent study found that this activity in S40f mice worsened with diet-induce ID and improved with levodopa, a highly effective treatment for RLS, and with quinpirole, a D2/3R agonist (Allen et al., 2020).
4.4. Summary - Utility of BXD mouse model of RLS
The primary distinguishing element of the BXD mouse model from that of the diet-induced BID rodent model, is the ability of this model to more definitively represent the idiopathic form of RLS with regards to BID. Variations in VMB iron were strongly correlated with expression of an important RLS-risk gene, btbd9, which in human also appears to play a role in iron regulation. Natural variations in striatal iron concentrations between two different BXD strains showed the ability of natural iron variations to produce significant changes in D2R that were in line with what has been seen in human pathology and in line with that seen with diet-induced BID model. The BXD model is capable of identifying strains that show natural diurnal variations in brain iron compatible with that reported for diurnal changes in CSF iron concentrations in RLS and to show the strong dissociation between peripheral and brain iron status. Finally, based solely on the pathologically-pertinent changes in brain iron, a single strain was identified that demonstrated a circadian-dependent behavioral phenotype of RLS that was aggravated with ID and blocked with treatment known to be effective in RLS. Thus, giving support for the value of this model in the broader context of iron pathology and behavior.
5. Limitations
As the primary reference point in this review, starts with human data, our understanding of the pathological bases of the DAergic system in RLS is limited and thus limiting the nature of the comparison between the more in-depth neuropharmacology seen in the animals compared to the limited data from autopsy and imaging. Also, the data collection and analysis were restricted to the BG because there is where the majority of RLS human research studies were focused and also there is where the majority of research into the consequences of BID on DAergic system focused as well. This review, therefore, does not include alternative DAergic pathways in this analysis. There are, at least, theoretical concerns that DAergic abnormalities in RLS may reside in hypothalamus or in the spinal cord (Ondo et al., 2007). In fact, this was the basis of the autopsy analysis of the number of TH positive cells in the lateral hypothalamus in RLS versus controls (Earley et al., 2009). This is the only human-RLS study of the DAergic system outside of the BG and it showed no difference. Also, there are no comparative data from BID rodents. By providing support in this review for the validity of the BID rodent model of RLS based on essentially BG data, we hope that this model could provide the basis for extending research to other brain regions like the hypothalamus or the spinal cord.
Idiopathic RLS, by definition, does not include patients with anemia. The diet-induced BID model produces a systemic ID with anemia and a concomitant BID. So, the question is how valid are the neurophysiological findings in the BID model when, in fact, an important physiological state (i.e., anemia) does not exist in the idiopathic form of RLS. This has often been a stated point of reservation about accepting the BID model as a valid representation of the disease. First, we have provided a comparison between the human data in which anemia is not present and the data from BID rodents where anemia is clearly present. At least from the point of view of changes in the BG DAergic system, we see highly comparable changes between the disease and the BID model. Second, data generated by the BID model predicted changes in the ADergic and GLUergic systems on which clinical trials with new potential treatments in idiopathic RLS were based. Finally, we demonstrated that utilizing two different BXD RI mouse strains with natural differences in VMB iron without anemia, changes in the D2R and A2aR are similar to that seen in BID model. These findings would seem to provide proof of the BID model’s ability to be representative of idiopathic RLS. However, utilizing a differential, iron-deficient diet (4 ppm versus 7 ppm), the study demonstrated that degrees of ID may produce “dose-dependent” differential effects and that 3-4 ppm iron diet at weaning may represent the more severe state of BID. Thus, the overall consequences of 3-4 ppm diet on brain neurotransmitter system may be more demonstrative of changes in other non-DAergic neurotransmitter systems in subjects whose RLS is triggered by ID anemia.
The rodent data are fairly consistent across the multitude of studies and, in fact, often further supported by in vitro cellular studies. The biggest problem in the data analysis was looking across the human studies. A much more thorough analysis of all the human-RLS studies of the DAergic system has been reviewed elsewhere (Earley et al., 2014). There were significant variations across studies in sample size, methods and technology and treatment status of the population studied, just to name a few variables. Where there were multiple studies reporting either positive or negative studies, we chose to bias the present towards the predominate results, whether negative or positive or at least provide a fair discussion of both data sets. There was, however, bias in our data presentation towards positive studies because of the statistical reality of all the human-RLS studies: none of the studies that were negative had sufficient power to exclude the possibility that a true difference could exist. If there were directional difference between studies or differences between types of the investigative technique, then that was discussed. However, unlike our prior review where we analyzed the studies in order to determine the potential underlying the pathological basis of the DAergic system in RLS (Earley et al., 2014), this review starts with just presenting what is the common or acceptable data from human-RLS studies and seeing how the animal data compare to them.
The complex genetics behind the BXD RI mouse strains are ultimately based upon genes originating from just two different parental strains: C57/BL6 and DBA/2J. If we accept that many phenotypes are a result of complex genetic interactions then the complex background genetics becomes a determinant of outcomes. So, we have accepted that these two parental strains are a limited representation of potentially broader set of complex genetics that could be found with crosses between other strains. The finding of a strong correlation between expression of the btbd9 gene (which has been identified as an RLS risk gene and involved in iron regulation) and ventral midbrain iron, is strong supporting evidence for the value of the BXD model in helping us to understanding the complex genetics behind altered iron homeostasis in RLS. Any definitive genetic findings with the BXD RI mouse can, however, be evaluated by utilizing other parental RI crosses.
6. Conclusion and Future Remarks
Altered brain iron homeostatic mechanisms are a major contributing factor to the underlying pathology of the RLS. The extensive research into brain iron status in RLS clearly supports a construct that involves BID as a model of RLS. The significant increase in prevalence of RLS under severe peripheral ID conditions, supports the construct of a BID model based on diet-induced ID. The BID rodent model has shown nearly homologous changes in DAergic system to that seen in RLS, has supported and extended the limited observation of GLUergic changes in RLS and, finally, has predicted changes in ADergic system that produced a novel treatment option for RLS. The data presented in this review provides a strong validation of the model’s predictive utility, and thus its potential for further development of novel treatments with higher specificity for disease-relevant changes in neurotransmitter systems. The BXD model, on the other hand, provides two distinct elements for exploring RLS pathology and treatment. First, it has the ability to model the peripheral-brain iron dissociation and thus a model to understand the potential genetic or epigenetic factors involved in altered brain iron homeostatic mechanisms. An understanding of why RLS has relatively lower brain iron, may allow for more direct “curative” treatment for this disease. At the current time, IV iron therapy is the only treatment that has been demonstrated to “cure” this disease but that occurs in only about 35-40% of patients (Allen et al., 2018). Why? This model has the potential to address that question. Secondly, the BXD model has the ability to model the idiopathic form of RLS with natural and diurnal variations in brain iron under normal dietary conditions that mimic the clinical situation. Natural brain-differences in iron were significant enough to produce change in DAergic compatible with that seen in RLS pathology and in the BID model. The model thus can be used to explore how iron is imported, utilized and exported from the brain and what are the consequences of natural variations on neurotransmitter system and how compatible are those finding to that seen in BID model.
Acknowledgements
SF is supported by the intramural funds of the National Institute on Drug Abuse.
Abbreviations:
- 3OMD
3-O-methyldopa
- A1R
adenosine A1 receptor
- A2AR
adenosine A2A receptor
- ADergic
adenosinergic
- βmax
receptor density
- BID
brain iron deficiency
- BP
binding potential
- BXD
recombinant inbred mouse strains from parental cross of C57/BL6 and DBA/2J
- CSF
cerebrospinal fluid
- CS-GLUergic
corticostriatal-glutaminergic
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- D4R
dopamine D4 receptor
- DAergic
dopaminergic
- DA
dopamine
- DAT
dopamine re-uptake transporter
- ecDA
extracellular DA
- ENT
equilibrative nucleoside transporter
- GLUergic
glutamatergic system
- GWAS
Genome-wide association study
- HIF
hypoxia-inducible factor
- ID
iron deficiency
- IDA
iron deficiency anemia
- Kd
dissociation constant, receptor affinity
- MRI
magnetic resonance imaging
- MRS
proton magnetic resonance spectroscopy
- PET
positron emission tomography
- PLMS
periodic leg movements of sleep
- ppm
parts per million
- pTH
phosphorylated tyrosine hydroxylase
- RI
recombinant inbred
- RLS
restless legs syndrome
- S19
strain 19
- S1
strain 1
- S40
strain 40
- SN
substantia nigra
- SNP
single nucleotide polymorphism
- SPECT
single-photon emission computed tomography
- TH
tyrosine hydroxylase
- VDCC
voltage-dependent calcium channels
- VMB
ventral midbrain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest. The authors declare no competing interests.
References
- Allen R, 2004. Dopamine and iron in the pathophysiology of restless legs syndrome (RLS). Sleep Medicine 5, 385–391. [DOI] [PubMed] [Google Scholar]
- Allen RP, Auerbach S, Bahrain H, Auerbach M, Earley CJ, 2013a. The prevalence and impact of restless legs syndrome on patients with iron deficiency anemia. Am J Hematol 88, 261–264. [DOI] [PubMed] [Google Scholar]
- Allen RP, Barker PB, Horska A, Earley CJ, 2013b. Thalamic glutamate/glutamine in restless legs syndrome: Increased and related to disturbed sleep. Neurology 80, 2028–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RP, Barker PB, Wehrl F, Song HK, Earley CJ, 2001. MRI measurement of brain iron in patients with restless legs syndrome. Neurology 56, 263–265. [DOI] [PubMed] [Google Scholar]
- Allen RP, Connor JR, Hyland K, Earley CJ, 2009a. Abnormally increased CSF 3-Ortho-methyldopa (3-OMD) in untreated restless legs syndrome (RLS) patients indicates more severe disease and possibly abnormally increased dopamine synthesis. Sleep Med 10, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RP, Earley CJ, Jones BC, Unger EL, 2020. Iron-deficiency and dopaminergic treatment effects on RLS-Like behaviors of an animal model with the brain iron deficiency pattern of the restless legs syndrome. Sleep Medicine 71, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RP, Kosinski M, Hill-Zabala CE, Calloway MO, 2009b. Psychometric evaluation and tests of validity of the Medical Outcomes Study 12-item Sleep Scale (MOS sleep). Sleep Medicine 10, 531–539. [DOI] [PubMed] [Google Scholar]
- Allen RP, Picchietti DL, Auerbach M, Cho YW, Connor JR, Earley CJ, Garcia-Borreguero D, Kotagal S, Manconi M, Ondo W, Ulfberg J, Winkelman JW, 2018. Evidence-based and consensus clinical practice guidelines for the iron treatment of restless legs syndrome/Willis-Ekbom disease in adults and children: an IRLSSG task force report. Sleep Medicine 41, 27–44. [DOI] [PubMed] [Google Scholar]
- Allen RP, Walters AS, Montplaisir J, Hening W, Myers A, Bell TJ, Ferini-Strambi L, 2005. Restless legs syndrome prevalence and impact: REST general population study. Arch Intern Med 165, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Ashbrook DG, Arends D, Prins P, Mulligan MK, Roy S, Williams EG, Lutz CM, Valenzuela A, Bohl CJ, Ingels JF, McCarty MS, Centeno AG, Hager R, Auwerx J, Lu L, Williams RW, 2021. A platform for experimental precision medicine: The extended BXD mouse family. Cell Systems 12, 235–247.e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspenstroem G, 1964. Pica and restless legs in iron deficiency. Sven Läkartidningen 61, 1174–1177. [PubMed] [Google Scholar]
- Astrakas LG, Konitsiotis S, Margariti P, Tsouli S, Tzarouhi L, Argyropoulou MI, 2008. T2 relaxometry and fMRI of the brain in late-onset restless legs syndrome. Neurology 71, 911–916. [DOI] [PubMed] [Google Scholar]
- Bianco LE, Wiesinger J, Earley CJ, Jones BC, Beard JL, 2008. Iron deficiency alters dopamine uptake and response to L-DOPA injection in Sprague-Dawley rats. J Neurochem 106, 205–215. [DOI] [PubMed] [Google Scholar]
- Bonaventura J, Quiroz C, Cai NS, Rubinstein M, Tanda G, Ferre S, 2017. Key role of the dopamine D4 receptor in the modulation of corticostriatal glutamatergic neurotransmission. Sci Adv 3, e1601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burhans MS, Dailey C, Beard Z, Wiesinger J, Murray-Kolb L, Jones BC, Beard JL, 2005. Iron deficiency: differential effects on monoamine transporters. Nutr Neurosci 8, 31–38. [DOI] [PubMed] [Google Scholar]
- Chawla S, Gulyani S, Allen RP, Earley CJ, Li X, Van Zijl P, Kapogiannis D, 2019. Extracellular vesicles reveal abnormalities in neuronal iron metabolism in restless legs syndrome. Sleep 42, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Casado V, Rodrigues RJ, Lujan R, Burgueno J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortes A, Canela EI, Lopez-Gimenez JF, Milligan G, Lluis C, Cunha RA, Ferre S, Franco R, 2006. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci 26, 2080–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clardy SL, Earley CJ, Allen RP, Beard JL, Connor JR, 2006a. Ferritin subunits in CSF are decreased in restless legs syndrome. J Lab Clin Med 147, 67–73. [DOI] [PubMed] [Google Scholar]
- Clardy SL, Wang X, Boyer PJ, Earley CJ, Allen RP, Connor JR, 2006b. Is ferroportin-hepcidin signaling altered in restless legs syndrome? J Neurol Sci 247, 173–179. [DOI] [PubMed] [Google Scholar]
- Connor JR, Boyer PJ, Menzies SL, Dellinger B, Allen RP, Earley CJ, 2003. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 61, 304–309. [DOI] [PubMed] [Google Scholar]
- Connor JR, Ponnuru P, Lee B-Y, Podskalny GD, Alam S, Allen RP, Earley CJ, Yang QX, 2011a. Postmortem and imaging based analyses reveal CNS decreased myelination in restless legs syndrome. Sleep Medicine 12, 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Ponnuru P, Wang X-S, Patton SM, Allen RP, Earley CJ, 2011b. Profile of altered brain iron acquisition in restless legs syndrome. Brain 134, 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Wang XS, Allen RP, Beard JL, Wiesinger JA, Felt BT, Earley CJ, 2009. Altered dopaminergic profile in the putamen and substantia nigra in restless leg syndrome. Brain 132, 2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Wang XS, Patton SM, Menzies SL, Troncoso JC, Earley CJ, Allen RP, 2004. Decreased transferrin receptor expression by neuromelanin cells in restless legs syndrome. Neurology 62, 1563–1567. [DOI] [PubMed] [Google Scholar]
- Cooper JR, Bloom FE, Roth RH, 2008. The Biochemical Basis of Neuropharmacology, 8th ed. Oxford University Press, New York. [Google Scholar]
- Earley CJ, 2003. Clinical practice. Restless legs syndrome. N Engl J Med 348, 2103–2109. [DOI] [PubMed] [Google Scholar]
- Earley CJ, 2009. Iron dysregulation in restless legs syndrome, in: Hening WA, Allen RP, Chokroverty S, Earley CJ (Eds.), Restless Legs Syndrome. Saunders Elsevier Philadelphia, pp. 61–68. [Google Scholar]
- Earley CJ, Allen RP, Connor JR, Ferrucci L, Troncoso J, 2009. The dopaminergic neurons of the A11 system in RLS autopsy brains appear normal. Sleep Med 10, 1155–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley CJ, Allen RP, Jones BC, Unger EL, 2020. Developing a behavioral model of Restless Legs Syndrome utilizing mice with natural variances in ventral midbrain iron. Sleep Medicine 71, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley CJ, Barker PB, Horska A, Allen RP, 2006a. MRI-determined regional brain iron concentrations in early- and late-onset restless legs syndrome. Sleep Med 7, 459–461. [DOI] [PubMed] [Google Scholar]
- Earley CJ, Connor J, Garcia-Borreguero D, Jenner P, Winkelman J, Zee PC, Allen R, 2014. Altered brain iron homeostasis and dopaminergic function in restless legs syndrome (Willis–Ekbom Disease). Sleep Medicine 15, 1288–1301. [DOI] [PubMed] [Google Scholar]
- Earley CJ, Connor JR, Beard JL, Clardy SL, Allen RP, 2005. Ferritin levels in the cerebrospinal fluid and restless legs syndrome: effects of different clinical phenotypes. Sleep 28, 1069–1075. [DOI] [PubMed] [Google Scholar]
- Earley CJ, Connor JR, Beard JL, Malecki EA, Epstein DK, Allen RP, 2000. Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology 54, 1698–1700. [DOI] [PubMed] [Google Scholar]
- Earley CJ, Hyland K, Allen RP, 2001. CSF dopamine, serotonin, and biopterin metabolites in patients with restless legs syndrome. Mov Disord 16, 144–149. [DOI] [PubMed] [Google Scholar]
- Earley CJ, Hyland K, Allen RP, 2006b. Circadian changes in CSF dopaminergic measures in restless legs syndrome. Sleep Med 7, 263–268. [DOI] [PubMed] [Google Scholar]
- Earley CJ, Kuwabara H, Wong DF, Gamaldo C, Salas R, Brasic J, Ravert HT, Dannals RF, Allen RP, 2011. The dopamine transporter is decreased in the striatum of subjects with restless legs syndrome. Sleep 34, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley CJ, Kuwabara H, Wong DF, Gamaldo C, Salas RE, Brasic JR, Ravert HT, Dannals RF, Allen RP, 2013. Increased synaptic dopamine in the putamen in restless legs syndrome. Sleep 36, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisensehr I, Wetter TC, Linke R, Noachtar S, von Lindeiner H, Gildehaus FJ, Trenkwalder C, Tatsch K, 2001. Normal IPT and IBZM SPECT in drug-naive and levodopa-treated idiopathic restless legs syndrome. Neurology 57, 1307–1309. [DOI] [PubMed] [Google Scholar]
- Erikson KM, Jones BC, Beard JL, 2000. Iron deficiency alters dopamine transporter functioning in rat striatum. J Nutr 130, 2831–2837. [DOI] [PubMed] [Google Scholar]
- Ferré S, García-Borreguero D, Allen RP, Earley CJ, 2019. New Insights into the Neurobiology of Restless Legs Syndrome. The Neuroscientist 25, 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Guitart X, Quiroz C, Rea W, García-Malo C, Garcia-Borreguero D, Allen RP, Earley CJ, 2021. Akathisia and Restless Legs Syndrome: Solving the Dopaminergic Paradox. Sleep Medicine Clinics 16, 249–267. [DOI] [PubMed] [Google Scholar]
- Ferre S, Quiroz C, Guitart X, Rea W, Seyedian A, Moreno E, Casado-Anguera V, Diaz-Rios M, Casado V, Clemens S, Allen RP, Earley CJ, Garcia-Borreguero D, 2017. Pivotal Role of Adenosine Neurotransmission in Restless Legs Syndrome. Front Neurosci 11, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre S, Quiroz C, Rea W, Guitart X, Garcia-Borreguero D, 2019. Adenosine mechanisms and hypersensitive corticostriatal terminals in restless legs syndrome. Rationale for the use of inhibitors of adenosine transport. Advances in pharmacology 84, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman A, Pranski E, Miller RD, Radmard S, Bernhard D, Jinnah HA, Betarbet R, Rye David B., Sanyal S, 2012. Sleep Fragmentation and Motor Restlessness in a Drosophila Model of Restless Legs Syndrome. Current Biology 22, 1142–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamaldo C, Benbrook AR, Allen RP, Oguntimein O, Earley CJ, 2009. Evaluating daytime alertness in individuals with Restless Legs Syndrome (RLS) compared to sleep restricted controls. Sleep Medicine 10, 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Borreguero D, Cano I, Granizo JJ, 2017. Treatment of restless legs syndrome with the selective AMPA receptor antagonist perampanel. Sleep Medicine 34, 105–108. [DOI] [PubMed] [Google Scholar]
- Garcia-Borreguero D, Garcia-Malo C, Granizo JJ, Ferre S, 2021. A Randomized, Placebo-Controlled Crossover Study with Dipyridamole for Restless Legs Syndrome. Mov Disord 36, 2387–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godau J, Klose U, Di Santo A, Schweitzer K, Berg D, 2008. Multiregional brain iron deficiency in restless legs syndrome. Mov Disord 23, 1184–1187. [DOI] [PubMed] [Google Scholar]
- Godau J, Schweitzer KJ, Liepelt I, Gerloff C, Berg D, 2007. Substantia nigra hypoechogenicity: definition and findings in restless legs syndrome. Mov Disord 22, 187–192. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Rangel-Barajas C, Peper M, Lorenzo R, Moreno E, Ciruela F, Borycz J, Ortiz J, Lluis C, Franco R, McCormick PJ, Volkow ND, Rubinstein M, Floran B, Ferre S, 2012. Dopamine D4 receptor, but not the ADHD-associated D4.7 variant, forms functional heteromers with the dopamine D2S receptor in the brain. Mol Psychiatry 17, 650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyani S, Earley CJ, Camandola S, Maudsley S, Ferré S, Mughal MR, Martin B, Cheng A, Gleichmann M, Jones BC, Allen RP, Mattson MP, 2009. Diminished iron concentrations increase adenosine A2A receptor levels in mouse striatum and cultured human neuroblastoma cells. Experimental Neurology 215, 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Hao LJ, Yang ZH, Chai R, Zhang S, Gu Y, Gao HL, Zhong ML, Wang T, Li JY, Wang ZY, 2016. Deferoxamine-mediated up-regulation of HIF-1alpha prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp Neurol 280, 13–23. [DOI] [PubMed] [Google Scholar]
- Haba-Rubio J, Staner L, Petiau C, Erb G, Schunck T, Macher JP, 2005. Restless legs syndrome and low brain iron levels in patients with haemochromatosis. J Neurol Neurosurg Psychiatry 76, 1009–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskovec L, 1902. Akathisia. Arch Bohemes Med Clin 3, 193–200. [Google Scholar]
- Hyacinthe C, De Deurwaerdere P, Thiollier T, Li Q, Bezard E, Ghorayeb I, 2015. Blood withdrawal affects iron store dynamics in primates with consequences on monoaminergic system function. Neuroscience 290, 621–635. [DOI] [PubMed] [Google Scholar]
- Jellen LC, Beard JL, Jones BC, 2009. Systems genetics analysis of iron regulation in the brain. Biochimie 91, 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellen LC, Unger EL, Lu L, Williams R, Rousseau S, Wang X, Earley CJ, Allen RP, Miles M, Jones BC, 2012. Systems genetic analysis of the effects of iron deficiency in mouse brain. Neurogenetics 13, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P, 2005. Istradefylline, a novel adenosine A2A receptor antagonist, for the treatment of Parkinson’s disease. Expert Opin Investig Drugs 14, 729–738. [DOI] [PubMed] [Google Scholar]
- Jones BC, Reed CL, Hitzemann R, Wiesinger JA, McCarhy KA, Buwen JP, Beard JL, 2003. Quantitative genetic analysis of ventral midbrain and liver iron in BXD recombinant inbred mice. Nutr Neurosci 6, 369–377. [DOI] [PubMed] [Google Scholar]
- Kim KW, Jhoo JH, Lee SB, Lee SD, Kim TH, Kim SE, Kim YK, Yoon IY, 2012. Increased striatal dopamine transporter density in moderately severe old restless legs syndrome patients. Eur J Neurol 19, 1213–1218. [DOI] [PubMed] [Google Scholar]
- Knake S, Heverhagen JT, Menzler K, Keil B, Oertel WH, Stiasny-Kolster K, 2010. Normal regional brain iron concentration in restless legs syndrome measured by MRI. Nature and Science of Sleep 2, 19–22. [PMC free article] [PubMed] [Google Scholar]
- Kofalvi A, Moreno E, Cordomi A, Cai NS, Fernandez-Duenas V, Ferreira SG, Guixa-Gonzalez R, Sanchez-Soto M, Yano H, Casado-Anguera V, Cunha RA, Sebastiao AM, Ciruela F, Pardo L, Casado V, Ferre S, 2020. Control of glutamate release by complexes of adenosine and cannabinoid receptors. BMC biology 18, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn E, Brodan V, 1982. Changes in the circadian rhythm of serum iron induced by a 5-day sleep deprivation. Eur J Appl Physiol Occup Physiol 49, 215–222. [DOI] [PubMed] [Google Scholar]
- Kuwabara H, McCaul ME, Wand GS, Earley CJ, Allen RP, Weerts EM, Dannals RF, Wong DF, 2012. Dissociative changes in Bmax and KD of dopamine D2/D3 receptors with aging observed in functional subdivisions of striatum: A revisit with an improved data analysis method. Journal of Nuclear Medicine 53, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Cheng YH, Hsieh KC, Nguyen D, Chew KT, Ramanathan L, Siegel JM, 2017. Motor hyperactivity of the iron-deficient rat - an animal model of restless legs syndrome. Mov Disord 32, 1687–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M, 2000. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab 20, 423–451. [DOI] [PubMed] [Google Scholar]
- Lee B-Y, Farace E, Vesek J, Connor J, Yang Q, 2007. In vivo measurement of iron deficiency in restless legs syndrome(RLS) with voxel-based R2 relaxometry. Proc Intl Soc Mag Reson Med 15, 2170. [Google Scholar]
- Li X, Allen RP, Earley CJ, Liu H, Cruz TE, Edden RAE, Barker PB, van Zijl PCM, 2016. Brain iron deficiency in idiopathic restless legs syndrome measured by quantitative magnetic susceptibility at 7 tesla. Sleep Medicine 22, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Fan YM, Lin GY, Yang FC, Cheng CA, Lu KC, Lin JC, Lee JT, 2016. 99mTc-TRODAT-1 SPECT as a Potential Neuroimaging Biomarker in Patients With Restless Legs Syndrome. Clin Nucl Med 41, e14–17. [DOI] [PubMed] [Google Scholar]
- Margariti PN, Astrakas LG, Tsouli SG, Hadjigeorgiou GM, Konitsiotis S, Argyropoulou MI, 2012. Investigation of unmedicated early onset restless legs syndrome by voxel-based morphometry, T2 relaxometry, and functional MR imaging during the night-time hours. AJNR Am J Neuroradiol 33, 667–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud M, Soucy JP, Chabli A, Lavigne G, Montplaisir J, 2002. SPECT imaging of striatal pre- and postsynaptic dopaminergic status in restless legs syndrome with periodic leg movements in sleep. J Neurol 249, 164–170. [DOI] [PubMed] [Google Scholar]
- Mizuno S, Mihara T, Miyaoka T, Inagaki T, Horiguchi J, 2005. CSF iron, ferritin and transferrin levels in restless legs syndrome. J Sleep Res 14, 43–47. [DOI] [PubMed] [Google Scholar]
- Moon HJ, Chang Y, Lee YS, Song H, Chang HW, Ku J, Allen RP, Earley CJ, Cho YW, 2015. A comparison of MRI tissue relaxometry and ROI methods used to determine regional brain iron concentrations in restless legs syndrome. Med Devices (Auckl) 8, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrowka M, Jobges M, Berding G, Schimke N, Shing M, Odin P, 2005. Computerized movement analysis and beta-CIT-SPECT in patients with restless legs syndrome. J Neural Transm 112, 693–701. [DOI] [PubMed] [Google Scholar]
- Narendran R, Hwang DR, Slifstein M, Talbot PS, Erritzoe D, Huang Y, Cooper TB, Martinez D, Kegeles LS, Abi-Dargham A, Laruelle M, 2004. In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (−)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]-raclopride. Synapse 52, 188–208. [DOI] [PubMed] [Google Scholar]
- Nelson C, Erikson K, Pinero DJ, Beard JL, 1997. In vivo dopamine metabolism is altered in iron-deficient anemic rats. J Nutr 127, 2282–2288. [DOI] [PubMed] [Google Scholar]
- Nguyen MV, Pouvreau S, El Hajjaji FZ, Denavit-Saubie M, Pequignot JM, 2007. Desferrioxamine enhances hypoxic ventilatory response and induces tyrosine hydroxylase gene expression in the rat brainstem in vivo. J Neurosci Res 85, 1119–1125. [DOI] [PubMed] [Google Scholar]
- Nordlander NB, 1953. Therapy in restless legs. Acta Med Scand 145, 453–457. [PubMed] [Google Scholar]
- O’Keeffe ST, Gavin K, Lavan JN, 1994. Iron status and restless legs syndrome in the elderly. Age and ageing 23, 200–203. [DOI] [PubMed] [Google Scholar]
- Ondo WG, Zhao HR, Le WD, 2007. Animal models of restless legs syndrome. Sleep Medicine 8, 344–348. [DOI] [PubMed] [Google Scholar]
- Patton SM, Ponnuru P, Snyder AM, Podskalny GD, Connor JR, 2011. Hypoxia-inducible factor pathway activation in restless legs syndrome patients. Eur J Neurol 18, 1329–1335. [DOI] [PubMed] [Google Scholar]
- Quiroz C, Gulyani S, Ruiqian W, Bonaventura J, Cutler R, Pearson V, Allen RP, Earley CJ, Mattson MP, Ferré S, 2016. Adenosine receptors as markers of brain iron deficiency: Implications for Restless Legs Syndrome. Neuropharmacology 111, 160–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz C, Pearson V, Gulyani S, Allen R, Earley C, Ferre S, 2010. Up-regulation of striatal adenosine A(2A) receptors with iron deficiency in rats: effects on locomotion and cortico-striatal neurotransmission. Exp Neurol 224, 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo G, Manners D, Testa C, Tonon C, Vetrugno R, Marconi S, Plazzi G, Pizza F, Provini F, Malucelli E, Gramegna LL, Lodi R, 2013. Low brain iron content in idiopathic restless legs syndrome patients detected by phase imaging. Mov Disord 28, 1886–1890. [DOI] [PubMed] [Google Scholar]
- Rodrigues MS, Ferreira SG, Quiroz C, Earley CJ, Garcia-Borreguero D, Cunha RA, Ciruela F, Kofalvi A, Ferre S, 2022. Brain Iron Deficiency Changes the Stoichiometry of Adenosine Receptor Subtypes in Cortico-Striatal Terminals: Implications for Restless Legs Syndrome. Molecules 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saletu B, Anderer P, Saletu M, Hauer C, Lindeck-Pozza L, Saletu-Zyhlarz G, 2002. EEG mapping, psychometric, and polysomnographic studies in restless legs syndrome (RLS) and periodic limb movement disorder (PLMD) patients as compared with normal controls. Sleep Med 3 Suppl, S35–42. [DOI] [PubMed] [Google Scholar]
- Salminen AV, Silvani A, Allen RP, Clemens S, Garcia-Borreguero D, Ghorayeb I, Ferré S, Li Y, Ondo W, Picchietti DL, Rye D, Siegel JM, Winkelman JW, Manconi M, 2020. Consensus Guidelines on Rodent Models of Restless Legs Syndrome. Mov Disord. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Soto M, Yano H, Cai NS, Casado-Anguera V, Moreno E, Casado V, Ferre S, 2019. Revisiting the Functional Role of Dopamine D4 Receptor Gene Polymorphisms: Heteromerization-Dependent Gain of Function of the D4.7 Receptor Variant. Mol Neurobiol 56, 4778–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalise A, Pittaro-Cadore I, Janes F, Marinig R, Gigli GL, 2010. Changes of cortical excitability after dopaminergic treatment in restless legs syndrome. Sleep Medicine 11, 75–81. [DOI] [PubMed] [Google Scholar]
- Schmidauer C, Sojer M, Seppi K, Stockner H, Hogl B, Biedermann B, Brandauer E, Peralta CM, Wenning GK, Poewe W, 2005. Transcranial ultrasound shows nigral hypoechogenicity in restless legs syndrome. Ann Neurol 58, 630–634. [DOI] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ, 2004. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5, 343–354. [DOI] [PubMed] [Google Scholar]
- Silber MH, Buchfuhrer MJ, Earley CJ, Koo BB, Manconi M, Winkelman JW, Scientific, Medical Advisory Board of the Restless Legs Syndrome, F., 2021. The Management of Restless Legs Syndrome: An Updated Algorithm. Mayo Clin Proc 96, 1921–1937. [DOI] [PubMed] [Google Scholar]
- Snyder AM, Wang X, Patton SM, Arosio P, Levi S, Earley CJ, Allen RP, Connor JR, 2009. Mitochondrial ferritin in the substantia nigra in restless legs syndrome. J Neuropathol Exp Neurol 68, 1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen E, Grau K, Berg T, Simonsen AC, Magnussen K, Erikstrup C, Hansen MB, Ullum H, 2012. A genetic risk factor for low serum ferritin levels in Danish blood donors. Transfusion 52, 2585–2589. [DOI] [PubMed] [Google Scholar]
- Stagg CJ, Bestmann S, Constantinescu AO, Moreno Moreno L, Allman C, Mekle R, Woolrich M, Near J, Johansen-Berg H, Rothwell JC, 2011. Relationship between physiological measures of excitability and levels of glutamate and GABA in the human motor cortex. The Journal of Physiology 589, 5845–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani A, Mitterling T, Heidbreder A, Steiger R, Kremser C, Frauscher B, Gizewski ER, Poewe W, Högl B, Scherfler C, 2019. Multimodal Magnetic Resonance Imaging reveals alterations of sensorimotor circuits in restless legs syndrome. Sleep 42. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Rye DB, Hicks A, Petursson H, Ingason A, Thorgeirsson TE, Palsson S, Sigmundsson T, Sigurdsson AP, Eiriksdottir I, Soebech E, Bliwise D, Beck JM, Rosen A, Waddy S, Trotti LM, Iranzo A, Thambisetty M, Hardarson GA, Kristjansson K, Gudmundsson LJ, Thorsteinsdottir U, Kong A, Gulcher JR, Gudbjartsson D, Stefansson K, 2007. A genetic risk factor for periodic limb movements in sleep. N Engl J Med 357, 639–647. [DOI] [PubMed] [Google Scholar]
- Sun ER, Chen CA, Ho G, Earley CJ, Allen RP, 1998. Iron and the restless legs syndrome. Sleep 21, 371–377. [PubMed] [Google Scholar]
- Tergau F, Wischer S, Paulus W, 1999. Motor system excitability in patients with restless legs syndrome. Neurology 52, 1060–1063. [DOI] [PubMed] [Google Scholar]
- Tribl GG, Asenbaum S, Klosch G, Mayer K, Bonelli RM, Auff E, Zeitlhofer J, Happe S, 2002. Normal IPT and IBZM SPECT in drug naive and levodopa-treated idiopathic restless legs syndrome (letter to editor). Neurology 59, 649–650. [DOI] [PubMed] [Google Scholar]
- Unger EL, Bianco LE, Jones BC, Allen RP, Earley CJ, 2014a. Low brain iron effects and reversibility on striatal dopamine dynamics. Experimental Neurology 261, 462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger EL, Jones BC, Bianco LE, Allen RP, Earley CJ, 2014b. Diurnal variations in brain iron concentrations in BXD RI mice. Neuroscience 263, 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesinger JA, Buwen JP, Cifelli CJ, Unger EL, Jones BC, Beard JL, 2007. Down-regulation of dopamine transporter by iron chelation in vitro is mediated by altered trafficking, not synthesis. J Neurochem 100, 167–179. [DOI] [PubMed] [Google Scholar]
- Williams RW, Gu J, Qi S, Lu L, 2001. The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol 2, RESEARCH0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelmann J, Muller-Myhsok B, 2008. Genetics of restless legs syndrome: a burning urge to move. Neurology 70, 664–665. [DOI] [PubMed] [Google Scholar]
- Yepes G, Guitart X, Rea W, Newman AH, Allen RP, Earley CJ, Quiroz C, Ferre S, 2017. Targeting hypersensitive corticostriatal terminals in restless legs syndrome. Ann Neurol 82, 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Unger EL, Jellen LC, Earley CJ, Allen RP, Tomaszewicz A, Fleet JC, Jones BC, 2012. Systems genetic analysis of multivariate response to iron deficiency in mice. Am J Physiol Regul Integr Comp Physiol 302, R1282–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Berg D, Arzberger T, Ruprecht P, Rausch WD, Musicco M, Tampellini D, Riederer P, Gerlach M, Becker G, 2005. In vivo detection of iron and neuromelanin by transcranial sonography: A new approach for early detection of substantia nigra damage. Mov Disord 20, 1278–1285. [DOI] [PubMed] [Google Scholar]