

Abstract
Background:
Unfolded protein response (UPR) is a multifaceted signaling cascade that alleviates protein misfolding. Although well studied in nucleated cells, UPR in absence of transcriptional regulation has not been described. Intricately associated with cardiovascular diseases, platelets, despite being anucleate, respond rapidly to stressors in blood. We investigate the UPR in anucleate platelets and explore its role, if any, on platelet physiology and function.
Methods:
Human and mouse platelets were studied using a combination of ex vivo and in vivo experiments. Platelet lineage specific knockout mice were generated independently for each of the three UPR pathways, protein kinase RNA (PKR)-like ER kinase (PERK), X-binding protein (XBP1) and activating transcription factor 6 (ATF6). Diabetes mellitus (DM) patients were prospectively recruited and platelets were evaluated for activation of UPR under chronic pathophysiological disease conditions.
Results:
Tunicamycin induced the IRE1α-XBP1 pathway in human and mouse platelets, while oxidative stress predominantly activated the PERK pathway. PERK deletion significantly increased platelet aggregation and apoptosis, and phosphorylation of PLCγ2, PLCβ3, and p38 MAPK. Deficiency of XBP1 increased platelet aggregation, with higher PLCβ3 and PKCδ activation. ATF6 deletion mediated a relatively modest effect on platelet phenotype with increased PKA. Platelets from DM patients exhibited a positive correlation between disease severity, platelet activation and protein aggregation, with only IRE1α-XBP1 activation. Moreover, IRE1α inhibition increased platelet aggregation, while clinically approved chemical chaperone, 4-PBA reduced the platelet hyperactivation.
Conclusion:
We show for the first time, that UPR activation occurs in platelets and can be independent of genomic regulation, with selective induction being specific to the source and severity of stress. Each UPR pathway plays a key role and can differentially modulate the platelet activation pathways and phenotype. Targeting the specific arms of UPR may provide a new anti-platelet strategy to mitigate thrombotic risk in DM and other cardiovascular diseases.
Keywords: Platelets, Unfolded protein response, anucleate, diabetes, protein misfolding, XBP, PERK, ATF6
Keywords: Basic Science Research, Cell Signaling/Signal Transduction, Platelets, Translational Studies, Vascular Biology
Graphical Abstract
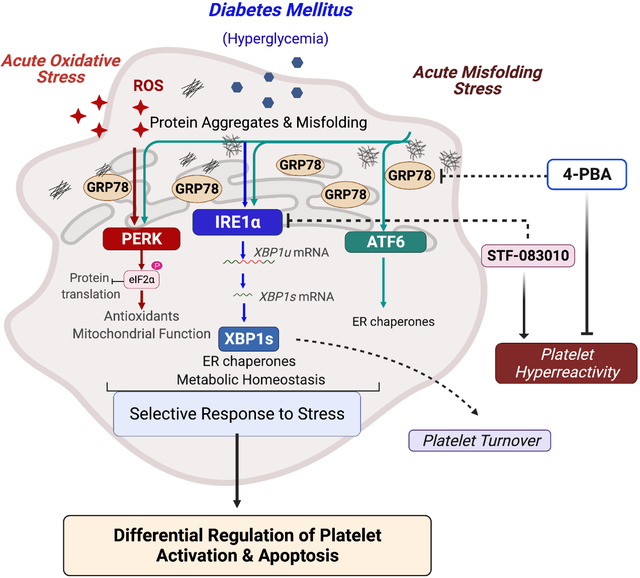
Introduction
Several stressors, including hypoxia, oxidative stress and nutrient stress, can affect the protein folding capacity of the endoplasmic reticulum (ER), leading to the accumulation of misfolded proteins (ER stress) 1. To alleviate the misfolded protein load and ensure protein folding fidelity, the unfolded protein response (UPR) is activated, which includes multifaceted signaling pathways to reprogram gene transcription, mRNA translation, and protein expression. Glucose regulated protein (GRP78), an ER chaperone, forms the master regulator of the UPR and regulates activation of transmembrane ER stress sensors protein kinase RNA (PKR)-like ER kinase (PERK), inositol-requiring enzyme-1alpha (IRE1α) and activating transcription factor 6 (ATF6), which work in a concerted manner in nucleated cells to restore protein folding homeostasis 2.
Platelets, while anucleate, contain a variety of cellular organelles (including the dense tubular system, DTS), granules, and mitochondria. Platelets, short lived and small (2–5 μm), result from the fractionation of megakaryocytes, and ‘inherit’ the cytoplasm 3. Although platelets lack genomic DNA, they contain a heterogenous array of functional coding and non-coding RNAs, spliceosome components and translational machinery 4, 5. They are thus capable of highly efficient de novo protein synthesis 4 and upon activation, can rapidly synthesize proteins and respond efficiently to perturbations through activation of diverse signaling cascades. Indeed, platelets possess the machinery for several stress induced signaling pathways including apoptosis, autophagy, ubiquitin proteasome system and mitophagy 6–9; the UPR however remains largely unexplored in these anucleate cells.
Beyond hemostasis and thrombosis 10, platelets are involved in pathophysiological conditions including cardiovascular diseases (CVD), neurodegenerative diseases and cancer 11, 12. Diabetes mellitus Type II (DM) affects millions globally and CVD forms the leading cause of morbidity and mortality in DM patients. Such patients present a higher risk for thrombotic events and several factors including hyperglycemia, impaired insulin signaling, and oxidative stress, contribute to the platelet hyperreactivity 7, 13. Here, we set out to explore (1) whether the multifaceted UPR can be launched in platelets despite the lack of transcriptional regulation, (2) whether the UPR plays a role in platelet function in normal physiology (3) whether chronic pathophysiological (diabetes) stress affects the platelet UPR and 4) its therapeutic relevance in platelets (if any).
Methods
Data Availability.
The data supporting the findings of the study are available upon reasonable request from the corresponding authors.
A detailed description of materials and methods is available in the Online Supplemental Material.
Results
UPR induction by tunicamycin selectively activates IRE1α in platelets
The UPR has not been previously described or characterized in platelets. A robust expression of GRP78, IRE1α and spliced XBP1 (Fig S1A) was observed in resting platelets; PERK and ATF6 were minimally detected (Fig S1B). To investigate whether UPR plays a role in platelets, we treated platelets with a classic inducer of ER stress, tunicamycin (Tm), an N-linked glycosylation inhibitor. Platelet ER (DTS) exhibits rapid ultrastructural changes in response to platelet agonists and is involved in platelet activation 19. Following Tm treatment, the ER was visibly distended into a more elongated vesicular form (Figure 1A). Similar ultrastructural changes, following ER stress, have been shown in yeast, possibly due to the need for an increased ER surface area for translational reprogramming in response to misfolded proteins 20. Next, Proteostat® dye, a fluorescent molecular rotor dye, was used to estimate the relative percentage of protein aggregation in human platelets. Significantly higher protein aggregation was observed in Tm treated platelets as compared to control human (HC) platelets (Figure 1B). Activation of platelets with thrombin or ADP did not increase protein aggregation (Figure S1C). Tm increased ER chaperone expression including GRP78 (Figure 1C–D, Figure S1D), GRP94 and PDI. Incubation with tunicamycin increased surface P-selectin levels (released by the alpha granules upon platelet activation) (Figure S1 E). Immunostaining, as visualized by confocal microscopy, further confirmed the increase in GRP78 and P-selectin (Figure 1E). Surprisingly, Tm selectively induced the IRE1α pathway; PERK and ATF6α remained unaffected (Figure 1F–G). This was consistent, even with higher Tm concentrations or longer treatments with Tm (Figure S1 F–G). Activation of IRE1α causes the cytoplasmic mRNA splicing of X-binding protein (XBP1) and higher XBP1s protein expression (Figure 1G–H). We thus find, that, unlike most in vitro and in vivo studies in nucleated cells, which demonstrate the induction of ER stress within days of Tm treatment 21, anucleate platelets appear to quickly mount the ER stress response (within hours); thus demonstrating extreme sensitivity to changes in proteostasis.
Figure 1: Tunicamycin selectively activates IRE1α in platelets.

A-H. Freshly isolated human platelets were treated with Tm (5 μg/ml) or vehicle (saline) for 3 h at 37°C. A. Transmission electron microscopy (TEM) images of vehicle and Tm treated platelets, acquired on FEI Tecnai Biotwin (LaB6, 80 kV). Arrows show distended ER within treated platelets. Scale bar, 500nm. n=3 independent experiments for Veh and Tm. B. Percentage of protein aggregates in platelets measured using a rotor dye, ProteoStat® in n=8/group. Values calculated based on standard curve generated using lysozyme IgG standards and controls provided by manufacturer and expressed as percentage. C-D. Representative immunoblots and respective quantitation for expression of ER chaperones following Tm treatment (3 × 108 platelets/ml). n=6/group. E. Confocal microscopy images showing localization of GRP78 (red) and P-selectin (blue) on platelets stained with CD41 (green). Scale bar, 10 μm. Images acquired on Perkin Elmer 5-Laser Spinning Disk Confocal Microscope (equipped with Nikon Ti-E Eclipse inverted microscope), objective 100X (oil). Platelet counts adjusted between samples prior to staining. n=3/group. F-G. Immunoblotting and respective quantitation for key UPR proteins in platelets. For phosphorylated proteins, quantification represents ratio of phosphorylated to total protein, normalized to the loading control, GAPDH. H. Levels of spliced and unspliced XBP1 mRNA measured by qPCR in platelet RNA. For Fig I-H, n=6/group. See also Supplemental Figure S1. I-N. Wild type (WT) mice injected with 1 μg/ml Tm, i.p or vehicle (150 mM dextrose), n=15 per group, 8 males/7 females. Platelets isolated 24 h post injection. I. Soluble P-selectin measured in plasma from WT and Tm injected mice (n=14/group). J. TMRE assay for mitochondrial apoptosis in mice platelets (n=5/group), with or without addition of calcium. K. Protein aggregate levels in mice platelets (n=15/ group). L. Intraplatelet (green, CD41) expression of GRP78 (red) as seen by confocal microscopy in mice platelets. Scale bar, 10 μm. Images acquired on Perkin Elmer 5-Laser Spinning Disk Confocal Microscope with 100× oil immersion lens, n=3/group. M-N. Representative images for UPR protein expression by immunoblotting and respective quantification from n=4/group. Values expressed as mean ± SEM and the p value corresponds to Student t test (B, I, K) or Mann-Whitney test (D, G, H, N). See also Supplemental Figure S1–S2.
Examining the response in vivo, we injected mice with Tm 21 and observed higher surface and soluble sP-selectin levels (Figure S2A–B, Figure 1I), loss of membrane mitochondrial potential (Figure 1J, Figure S2C), and higher apoptosis (caspase-3 and Bax) (Figure S2D–E). Platelets from Tm-injected mice had higher intraplatelet protein aggregates (Figure 1K), along with increased GRP78 expression (Figure 1L–N, Figure S2F). Concurrent with human platelets, we observed a selective increase in only IRE1α and spliced XBP1 (Figure 1M–N). In contrast, heart and liver tissues isolated from same mice exhibited simultaneous activation of all three arms of the UPR (Figure S2G).
Oxidative stress selectively activates only PERK while DTT stress-induced protein misfolding activates all three UPR arms in platelets
We next investigated whether other stressors could selectively activate different arms of UPR. Oxidative stress (OS), through the disruption of ER redox homeostasis, inhibits protein folding; thereby inducing ER stress and UPR 22. We applied hydrogen peroxide, H2O2 (oxidative stressor) onto freshly isolated human platelets 14. Accompanied with higher platelet apoptosis (Figure S3A), transmission electron microscopy (TEM) revealed altered platelet morphology, with presence of protein aggregate like fibrils and degranulation (Figure 2A). OS mediated a significant increase in protein carbonylation (Figure S3B) and intraplatelet protein aggregation (Figure 2B). Surprisingly, while GRP78 expression increased (Figure 2C–D, Figure S3C,E), IRE1α levels and spliced XBP1 remained unaffected, at protein (Figure 2C, Figure S3C) and mRNA (Figure S3D) levels. Interestingly, OS mediated a distinct increase in PERK expression (Figure 2E–F) along with increase in downstream effectors including phosphorylated eIF2α and ATF4 (Figure 2E–F) and was consistent even with higher H2O2 concentrations and longer incubation times (Figure S3F–G). Expression of cleaved (activated) ATF6α also increased (Figure 2E–F). This provided further evidence of pathway selectivity of the platelet UPR. Although more extensive studies are needed to detail the roles of ATF4 and ATF6 in absence of a nucleus, the rapid selective induction of UPR components within platelets is likely an effective strategy when dealing with specific stressors.
Figure 2: Acute oxidative stress activates only PERK pathway while stress-induced protein misfolding activates all three arms of the UPR in platelets.

A-G. Freshly isolated human platelets were treated with H2O2 (50 μM) or vehicle (saline) for 30 min at 37°C. A. TEM images of vehicle and H2O2 treated platelets acquired on FEI Tecnai Biotwin (LaB6, 80 kV). Arrows show protein aggregate fibrils and microparticle release. Scale bar, 1 μm. B. Protein aggregation in platelets. n=5/group. C. Representative immunoblots for expression of GRP78, IRE1α and sXBP1 following H2O2 treatment. D. GRP78 (red) levels in individual platelets (green) as seen by microscopy. Scale bar, 10 μm. Images acquired on Perkin Elmer 5-Laser Spinning Disk Confocal Microscope (equipped with Nikon Ti-E Eclipse inverted microscope), 100× oil immersion lens. Platelet counts adjusted between samples prior to staining. TEM and immunostaining micrographs representative of n=3 independent experiments. E-F. Expression and quantification of PERK pathway proteins and ATF6. n=6/group. See also Supplemental Figure S3. G-J. Freshly isolated human platelets were treated with DTT (2 mM) or vehicle (saline) for 30 min at 37°C. G. TEM images of the vehicle and DTT treated platelets acquired on FEI Tecnai T12 (LaB6, 120 kV). Arrows indicate presence of protein aggregates. Zoom in shows magnified view of protein fibril aggregates. Scale bar, 1 μm. Higher magnification of HC platelets shown in Online Data Supplemental Figure S3. TEM micrographs representative of n=3/group. H. Protein aggregation in DTT-treated platelets. n=6/control group and n=9/DTT treated group. I. Representative immunoblots for expression of UPR proteins following DTT treatment; n= 6/group. J. Confocal Microscopy images showing co-localization of IRE1α (magenta) and PERK (cyan) on the platelets stained with CD41 (green). Scale bar, 10 μm. Zoom in depicts levels of IRE1α and PERK in individual platelets. n= 3 independent experiments/group. For phosphorylated proteins, quantification represents ratio of phosphorylated to total protein, normalized to GAPDH. Values expressed as mean ± SEM and precise p value corresponds to Mann-Whitney test.
Treatment of human platelets with dithiothreitol (DTT; disulfide bond disruption and protein misfolding) led to a significant increase in protein aggregates/fibril like structures, by TEM (Figure 2G, higher magnification in Figure S3H) and biochemically (Figure 2H), along with elevated mitochondrial depolarization and apoptosis (Figure S3I–J). Such visible direct evidence of protein aggregates in platelets has not been reported previously. Protein misfolding led to simultaneous upregulation of IRE1α (Figure 2I, FigureS3 K), XBP1 splicing (Figure 2I, Figure S3K–L), PERK and ATF6α (Figure 2I, Figure S3K), confirmed further by the distinct colocalization of IRE1α and PERK (Figure 2J, Figure S3M). Our results, for the first time, demonstrate the presence of a robust UPR in the anucleate platelet; this selective UPR likely equips circulating platelets with a finely tuned, rapid, highly sensitive response to stress
Platelet-specific deletion of UPR genes differentially modulates platelet phenotype
Next, we wanted to investigate the pathophysiological significance of UPR in platelets in vivo. Since global knockout mice for most UPR genes are embryonically lethal 23, we utilized the platelet factor 4-Cre recombinase (Pf4-Cre) megakaryocyte lineage specific transgenic mice18, to generate platelet specific knockout mice (Figure 3A).
Figure 3: Platelet-specific deletion of UPR genes differentially modulates platelet physiology.
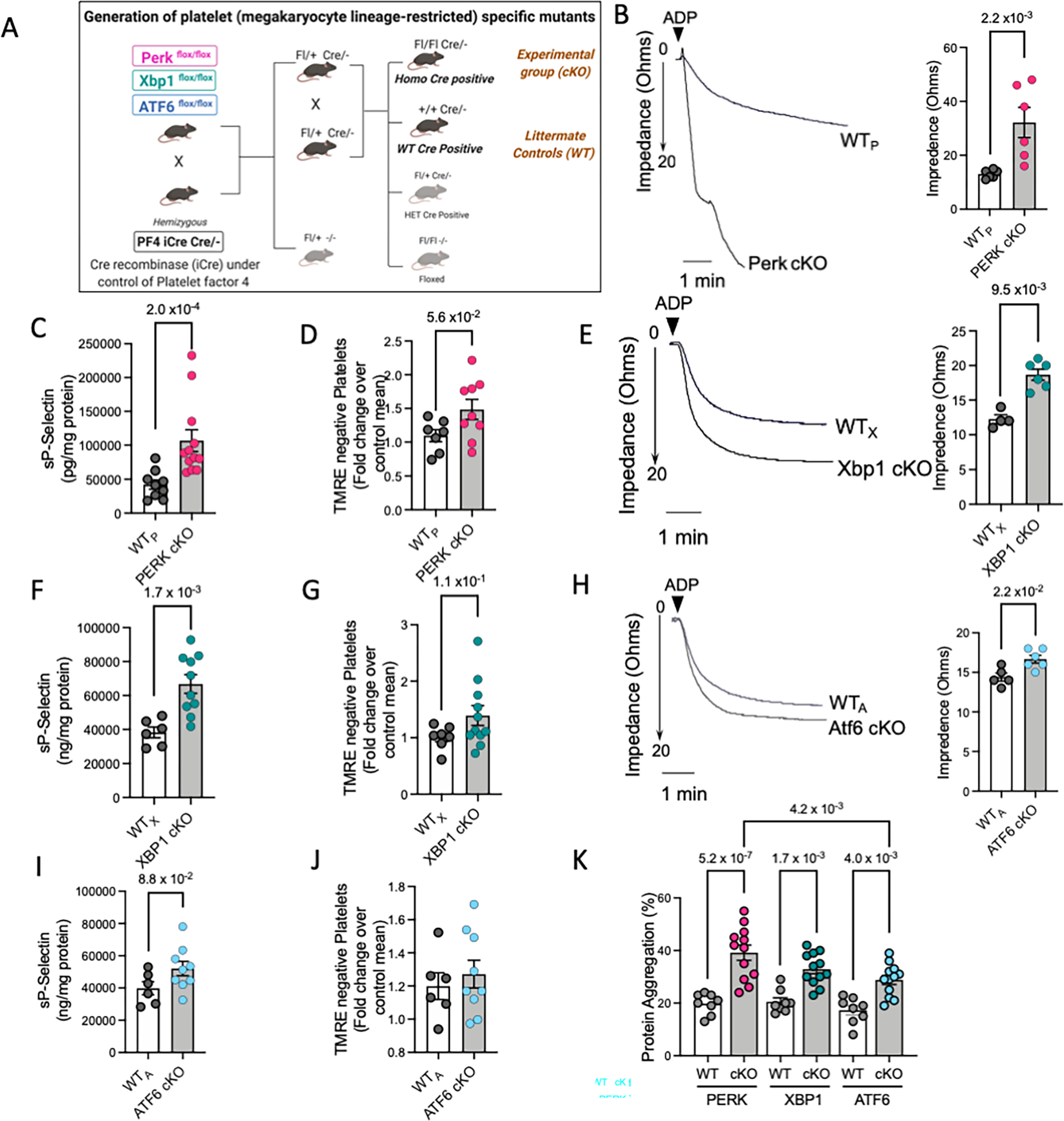
A. Platelet-specific knockout mice were generated by crossing respective floxed mice with Pf4-iCre transgenic mice, as depicted in schematic. Freshly drawn blood and isolated platelets from each KO mice strain and their respective littermate controls (n=16 mice per group per strain, 8 males and 8 females) were used for experiments. Results from genotyping are shown in Supplemental Figure S4. Panel B-D represents results from the PERK conditional KO mice. Panel E-G depicts results from XBP1 cKO mice. Panel H-J shows results from ATF6 cKO mice. B, E, H. Representative platelet aggregation tracings in response to ADP (20μM) for each strain and quantitation shown taking 20 ohms (maximal impedance) as the 100% response. Aggregation response to collagen and CLEC-2 stimulatory antibody shown in Supplemental Figure S5. Aggregation curves reflect results from n=6 WTP and n=6 PERK cKO, n=4 WTX and n, =6 XBP1 cKO, n=5 WTA and n=6 ATF6 cKO mice respectively. C, F, I. Levels of soluble P-selectin in platelet-specific KO mice. Results shown are from n=9 WTP and n=16 PERK cKO, n=6 WTX and n=10 XBP1 cKO, n=6 WTA and n=9 ATF6 cKO mice respectively. D, G, J. Quantification of TMRE assay in the platelets following deletion of specific UPR genes. Graphs reflect results from n=7 WTP and n=9 PERK cKO, n=7 WTX and n=9 XBP1 cKO, n=6 WTA and n=9 ATF6 cKO mice respectively. Representative flow cytometry scatter plots provided in Supplemental Figure S6. K. Intraplatelet protein aggregation levels in platelet-specific KO mice. Results from n=8 WTP and n=12 PERK cKO, n=8 WTX and n=12 XBP1 cKO, n=8 WTA and n=12 ATF6 cKO mice respectively. Values expressed as Mean ± SEM. Exact p values correspond to Mann Whitney test (B-C, E-F, I-J), Student t test (D, G) or one way ANOVA, with Tukey multiple comparisons (K). See also Supplemental Figure S4–S8.
Mice with the platelet-specific PERK deletion (PERK cKO) demonstrated normal litter frequencies and survival (Figure S4A); both male and female PERK cKO mice had lower body weights (Figure S4B) and platelet counts (Figure S4C), compared to WT littermate (WTP) controls. Agonist-induced platelet aggregation in whole blood forms the standard assay for measurement of prothrombotic tendency. PERK deficient platelets had significantly higher platelet aggregation in response to agonists like ADP (Figure 3B), collagen (Figure S5 A, C) and CLEC-2 (C-type lectin-like receptor 2) stimulatory antibody (Figure S5B, D). A marked elevation was also observed in P-selectin (Figure 3C) and mitochondrial depolarization (Figure 3D, Figure S6A). Additionally, a modest reduction occurred in the percentage of reticulated platelets (immature platelets) (Figure S6B). Platelets lacking PERK had elevated protein aggregates (Figure 3K) and a higher expression of ER chaperones, GRP78 and GRP94 (Figure S7 A–C).
PERK cKO platelets also displayed higher IRE1α and spliced XBP1 expression (Figure S7 A, D–E), even at basal conditions, likely as a compensatory mechanism to cope with physiological stressors. Levels of phosphorylated eIF2α, which occurs downstream of PERK, were significantly lowered (Figure S7 A,G). Interestingly, the UPR-mediated increase in PERK expression also regulates mitochondrial integrity and function in other cells 24. This likely explains the marked increase in mitochondrial dysfunction, and subsequent platelet activation and aggregation observed in PERK cKO mice. This is consistent with our results from the ex vivo studies showing the selective increase in PERK and not IRE1α, following acute oxidative stress. These results further highlight the presence of a highly sensitive yet selective stress response in platelets, despite the lack of transcriptional machinery.
We next targeted the IRE1α-XBP1 pathway to identify its effect on platelet function. Mice with platelet-specific deletion of XBP1 (XBP1 cKO, Figure S4D), had normal survival, body weight, and platelet counts (Figure S4E–F). ADP and collagen induced platelet aggregation was higher in the XBP1 cKO than the WTX (Figure 3E, collagen, Figure S5 A, C), along with increased platelet activation (Figure 3F). In contrast to PERK cKO, the XBP1 deficient platelets had a modest increase in apoptosis (Figure 3G, Figure S6C). The percentage of reticulated platelets was significantly higher (Figure S6D), suggesting a potential role for XBP1 in the regulation of platelet production from megakaryocytes or platelet lifespan in circulation. Although not significant, the expression of GRP78, GRP94 and PERK was elevated in the XBP1 cKO mice platelets (Figure S7), further supporting a distinct role for XBP1 as compared to PERK in platelets.
Finally, to identify the role of ATF6, we generated platelet-specific knockouts (ATF6 cKO, Figure S4G). Body weight and platelet counts in the ATF6 cKO mice did not differ significantly from littermate controls (WTA) (Figure S4H–I). Unlike the PERK and XBP1 cKO mice, ATF6 cKO mice showed higher platelet aggregation in response to only ADP (Figure 3H) but not collagen and CLEC-2 stimulatory antibody (Figure S5) and platelet activation (Figure 3I) was distinctly lower. No significant difference was observed in apoptosis markers (Figure 3J, Figure S6E) and reticulated platelet levels in the ATF6 cKO (Figure S6F). Albeit still significant, the increase in protein aggregates was relatively lower in ATF6 cKO mice (Figure 3K). Concurrent with the overall relatively lower consequence of ATF6 on platelet phenotype, there were no significant changes in the expression of the ER chaperones and UPR pathway proteins (PERK, IRE1α, XBP1s) (Figure S7). Consistent with the differential induction of UPR observed in human ex vivo studies, platelet-specific ablation of specific UPR components causes distinct changes in the platelet phenotype in vivo. Even at normal physiological conditions, PERK and XBP1 deficient platelets displayed a higher prothrombotic tendency than the ATF6 cKO and WT mice.
We next wanted to investigate whether UPR pathways play a selective and differential role in thrombus formation in vivo. We used the ferric chloride-induced carotid artery injury model which employed the topical application of FeCl3 to induce oxidative stress arterial damage, and subsequent thrombus formation. Post injury, carotid arteries from cKO mice were stained to visualize the thrombus composition. Different components of the thrombus display different colors in Carstairs staining (bright red for fibrin, gray-blue to navy blue for platelets, and bright blue for collagen). Platelet-fibrin rich thrombus area was highest in the PERK cKO and XBP1 cKO mice as compared to both wild type littermate controls and ATF6 cKO mice (Figure S8 A–B). This in vivo evidence is consistent with the distinct platelet aggregation response, and increase in activation biomarkers observed in platelets from the cKO mice ex vivo as well. Indeed, platelets from PERK cKO mice, which display the most pronounced increase in aggregation in response to all three agonists, also show the maximal increase in platelet rich thrombus formation following injury. Taken together, it is evident that each arm of the UPR pathway plays a key protective role in platelets; indeed, deletion of the UPR pathways promotes platelet hyperactivation and prothrombotic phenotype in mice.
Platelet-specific deletion of UPR genes selectively affects activation signaling
Based on increased platelet aggregation and thrombus formation associated with the PERK cKO and XBP1 cKO, we now needed to address how the key UPR response pathways intersect with the platelet activation signaling cascades. We also wanted to explore whether the distinct differences in the role of PERK, XBP1 or ATF6 on the observed platelet phenotype, arises from their differential regulation of the complex network of platelet activation responses. Using our platelet specific KO mice, we thus set out to investigate the effect of deletion of these UPR components on some of the known signaling pathways involved in platelet activation.
Early events of receptor mediated signaling, followed by the convergence and amplification of the signaling networks, leads to intraplatelet cytoskeletal re-arrangements, eventually causing platelet aggregation. There are two major classes of activation receptors in platelets – G-protein coupled receptors (GPCRs) and immunoreceptor tyrosine-based activation motif (ITAM) receptors 25. Phospholipase C (PLC) in platelets plays a key role in transducing surface receptor activation stimuli into a intracellular signaling cascade events. The GPCR pathway primarily activates PLC-β2/3 26, while ITAM pathway has been known to activate PLCγ2 27. Both PLCγ2 and PLCβ2/3 are involved in earlier events of αIIbβ3 inside -out signaling, forming a major determinant of platelet activation 25, 28, 29. PLCγ2 phosphorylation increased significantly in PERK cKO but not XBP1 cKO and the ATF6 cKO platelets (Figure 4A). PLCβ2/3 expression and phosphorylation increased significantly in the platelets from PERK cKO and XBP1 cKO mice (Figure 4B).
Figure 4: Platelet-specific deletion of UPR genes selectively affects specific platelet activation pathways.
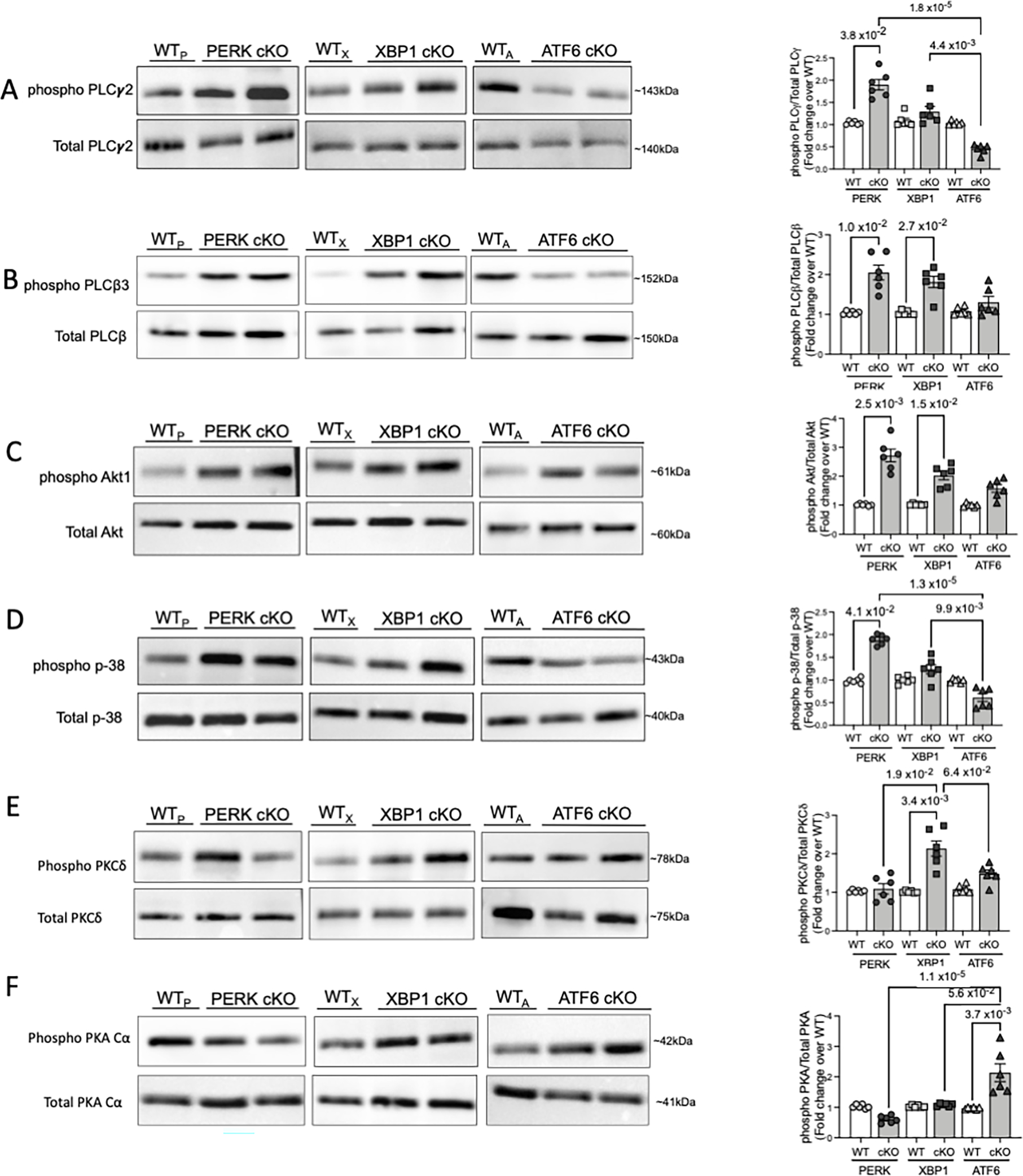
Platelets (3 × 108 per mL) from each of the mouse strains were lysed using RIPA buffer, resolved on SDS PAGE and immunoblotted for respective proteins. A total of n=10 mice (n=4 males, n=6 females) per strain and respective littermate controls used for platelet activation signaling responses. Immunoblotting and respective quantitation for A. phospho- PLCγ2 and total PLCγ2. B. phospho PLCβ3 and Total PLCβ3. C. phospho-Akt1 and Total Akt. D. phospho-p38 and Total p-38. E. phospho-PKCδ and Total PKCδ. F. phospho-PKA Cα and total PKA Cα. Quantification of immunoblotting reflects the results from n=6 WTP and n=6 PERK cKO, n=6 WTX and n=6 XBP1 cKO, n=6 WTA and n=6 ATF6 cKO mice respectively. Data presented as mean ± SEM. Precise p value corresponds to Kruskal-Wallis test with Dunn’s multiple comparisons.
Platelets are known to express all isoforms of Akt, which are involved in signal amplification of initial receptor signals, leading to robust platelet activation responses 30, 31. Deletion of UPR genes led to upregulation of Akt1 in all our three cKO mice (Figure 4C). This is not surprising, as the precise mechanism by which Akt modulates platelet activation response (positive or negative) is multifactorial and is still not completely understood. Mitogen Activated Protein Kinases (MAPKs) are a family of serine/threonine kinases present in all nucleated cells and p38 MAPK regulates platelet aggregation and apoptosis 32. Mice deficient in PERK show higher phospho-p38 levels, as compared to the other UPR cKO mice (Figure 4D). While p38 phosphorylation in nucleated cells can be modulated by the PERK/ATF4/e-IF2α pathway 33, 34, the p38-RTN4-Bcl-xl pathway was recently shown to regulate ER physiology in platelet activation 35. We find, that, consistent with the higher aggregation in response to ADP, collagen as well as CLEC-2, PERK cKO mouse platelets show the most pronounced increase in phosphorylation and activation of downstream signaling effectors. Our results provide new insights into PERK as an integral regulator of platelet physiological responses via modulation of PLC and p38 activation pathways.
Another signaling mediator, Protein Kinase C δ (PKCδ) regulates platelet activation response via calcium independent activation and aggregation 36, 37 and plays a complex role with opposing signaling functions36. Phospho-PKCδ expression increased significantly in the XBP1 cKO and ATF6 cKO platelets, but not in PERK cKO mice (Figure 4E). PKCδ has been linked with ER stress induced apoptosis, primarily via the IRE1α/JNK pathway and has been associated with atherosclerotic plaque stability 38. Our results highlight a possible mechanism for the elevated platelet aggregation and activation observed in the XBP1 cKO mice. Cyclic AMP (cAMP) and cAMP-dependent protein kinase (PKA) form the negative regulators of platelet activation 39. Platelets deficient in PERK and XBP1 displayed no significant PKA activation; a marked increase in phospho-PKA was observed in ATF6 cKO platelets (Figure 4F). PKA serves as regulator of apoptosis and determines platelet life span and survival 40. PKA activation likely mediates the least increase in platelet aggregation and no changes in apoptosis, as observed with the ATF6 ckO platelets.
These results are consistent with our observations on the modulation of platelet aggregation phenotype in response to ADP, Collagen and CLEC-2. ADP signals through the GPCRs (Gq), while collagen and CLEC-2 mediate their effects through the ITAM (GpVI) pathway25, 41. These synergistic platelet activation pathways and their complex downstream events, lead to cytoskeletal rearrangements within the platelets, ultimately causing aggregation. Platelets, following deletion of the individual UPR components, particularly PERK and XBP1, appear to be primed for enhanced hyperreactivity, as evident in effects on key signaling mediators of GPCR and ITAM pathways. ATF6 cKO platelets display no change in aggregation due to collagen and CLEC-2; this may be mediated by the higher PKA expression, which acts via a negative feedback mechanism. Thus, platelet specific ablation of individual UPR components, PERK and XBP1 differentially increases platelet aggregation (in response to agonists), activation and promotes a pro thrombotic phenotype in vivo, likely via enhanced activation of selective platelet signaling pathways.
Platelet-specific deletion of UPR genes differentially affects the response to ex vivo stressors
We next wanted to explore whether ablation of platelet UPR genes modifies the stress response in platelets using oxidative stress as an acute stressor 14. Deficiency of PERK and XBP1 leads to increased sensitivity to oxidative stress 42, 43. This was evident with maximum increase in protein aggregation levels in PERK and XBP1 deficient platelets (Figure 5A). Elevated mitochondrial depolarization following H2O2 treatment was most pronounced in PERK cKO platelets (Figure 5B–C). OS induced apoptosis was highest in the PERK cKO platelets, followed closely by the XBP1 cKO (Figure 5D). Interestingly, PERK expression increased in both XBP1 and ATF6 deficient platelets; PERK deficient platelets displayed higher IRE1α and XBP1, likely as a compensatory mechanism in absence of PERK (Figure 5E, Figure S9). Our results demonstrate that both PERK and XBP1 form a key component of the pro-survival mechanism in platelets as well as their ability to respond to external stress stimuli.
Figure 5: Platelet-specific deletion of UPR genes differentially affects the response to ex vivo stressors.
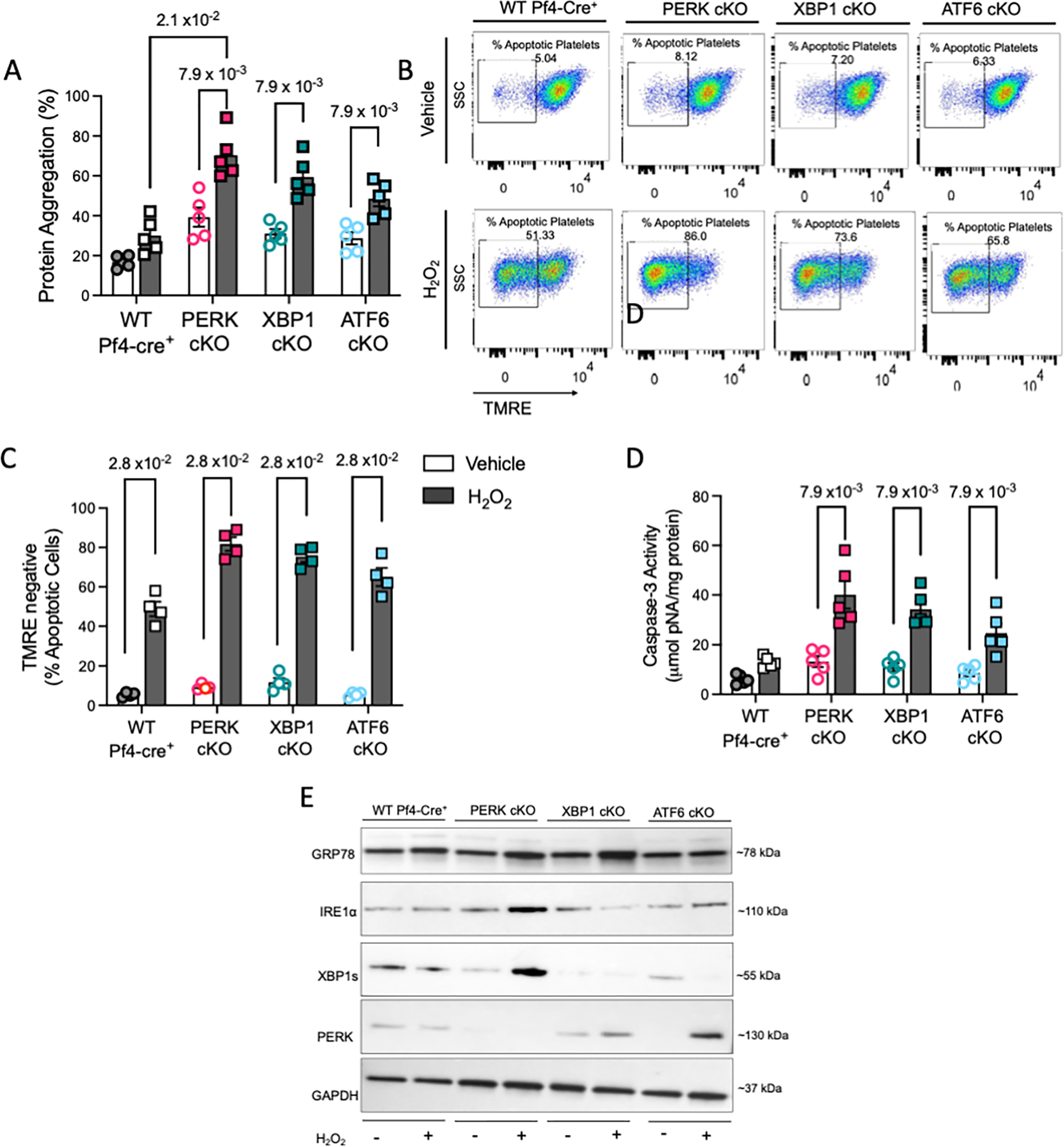
A-F. Freshly isolated platelets (3 × 108 platelets per mL, n=3 samples pooled to obtain the appropriate platelet counts) from each KO strain and littermate controls, incubated for 30 min with 50 μM H2O2 or vehicle. Blood drawn from a total of n=12 mice/ strain. For PERK and XBP1 strain, n=7 male/ n=5 female mice were used. For ATF6, n=6 male/ n=6 female mice were used. A. Protein aggregation in platelets following acute stress (n=5 pooled samples). B-C. Representative plots and quantification for TMRE assay each mouse strain (n=4 pooled samples). D. Intraplatelet Caspase-3 activity following H2O2 treatment (n=4 pooled samples). E. Representative immunoblotting for UPR protein expression in KO mice platelets. Quantitation shown in Supplemental Figure S9. Data presented as mean ± SEM. The precise p value corresponds to Mann Whitney test for comparison between vehicle and H2O2 treated groups; or Kruskal-Wallis test with Dunn’s multiple comparisons between treated strains. See also Supplemental Figure S9.
Diabetes mellitus activates IRE1α in platelets
Hyperglycemia and hyperlipidemia, observed in diabetes mellitus (DM), leads to ER stress and UPR44, 45. Previously, our lab has shown the role of stress-induced signaling in platelets from DM patients 7, 46. We now explored whether the chronic stress, associated with Type II DM, activates the UPR in platelets (Supplementary Table 1). Consistent with our previous study 7, DM platelets displayed higher activation and apoptosis (Figure 6A, S10A). TEM revealed a more pronounced and clearly distended ER in the DM platelets compared to HC (Figure 6B, Figure S10B). Consistent with previous studies in yeast 20, increased ER surface area and membrane expansion likely allows for more efficient clearance and resolution of misfolded protein accumulation and subsequent stress under the hyperglycemic conditions 45. Presence of intraplatelet protein aggregation was significantly increased in DM platelets (Figure 6C). DM platelets showed a marked upregulation of IRE1α and spliced XBP1 protein (Figure 6D–E); however, PERK (and downstream effectors) and ATF6α expression did not change remarkably (Figure 6D–E). Platelet gene expression array for UPR, confirmed upregulation of genes associated predominantly with the IRE1α pathway (Figure 6F), the increase being more pronounced in patients with more severe DM (hemoglobin A1c (HbA1c) > 8.5, a prognostic marker for diabetes). This is likely due to higher persistent hyperglycemic stress faced by not only the platelets, but also megakaryocytes. Levels of selected genes were validated by qPCR experiments (Figure S11 A–F). Colocalization immunostaining studies further demonstrated a higher number of DM platelets, positive for both GRP78 and IRE1α vs HC platelets (Figure 6G, Fig S11G). Interestingly, levels of protein aggregates correlated strongly with the platelet activation marker (sP-selectin), (r=0.690, p=1.1 × 10−11; Figure 6H) and HbA1c levels (r=0.710, p=1.3 × 10−12; Figure 6I). These results from our prospectively recruited DM patients highlight the clinical and translational relevance of platelet UPR.
Figure 6: Diabetes mellitus selectively activates IRE1α in platelets.
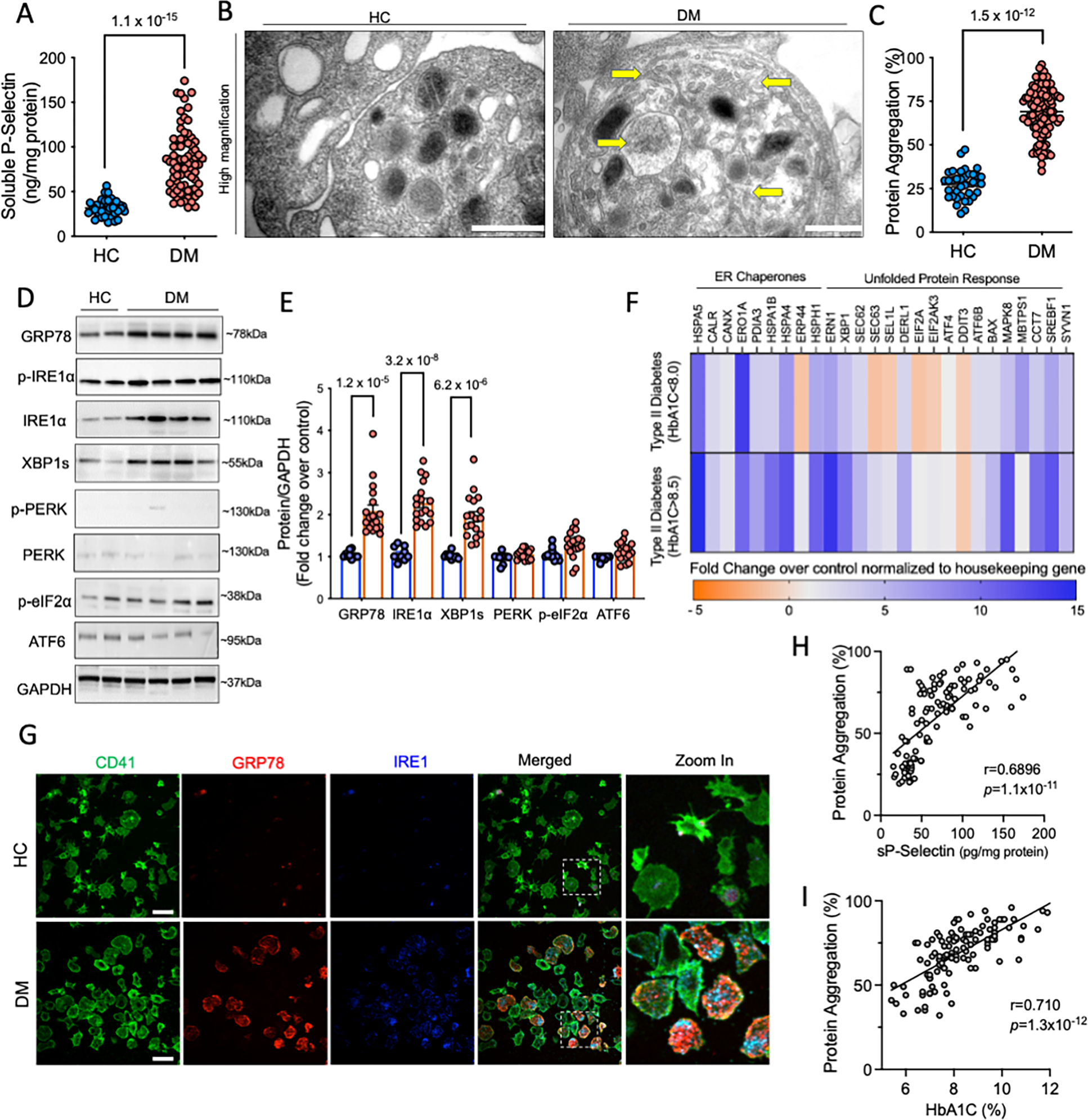
Freshly isolated platelets from DM patients (n=102) were compared to healthy age matched controls(HC), processed simultaneously. A. Levels of soluble P-selectin in DM (n=75) and controls (n=38). B. Representative TEM from DM and HC platelets, acquired on FEI Tecnai Biotwin (LaB6, 80 kV). Arrows highlight presence of protein aggregates and ER expansion within DM platelets. Scale bar, 500 nm. Data representative of n=3 independent experiments. Lower magnification shown in Supplemental Figure S10. C. Protein aggregation in DM (n=102) and HC platelets (n=40) measured using the Proteostat Dye. D-E. Platelets (3 × 108 per mL) were used for immunoblotting. Representative immunoblots and quantification for expression of UPR pathway proteins in HC (n=10) and DM (n=18) samples. F. Heat map shows differential expression of key ER stress/UPR genes measured and quantified using the RT2 Profiler Human Unfolded Protein Response PCR array. Heat map represents fold change in patients with less (HbA1c<8.5) and more (HbA1c>8.5) severe diabetes. Results normalized to housekeeping genes and expressed over HC. n=6 each, for HC and DM platelets. Validation by qPCR for UPR genes shown in Supplemental Figure S11. G. Confocal microscopy images from DM and control platelets (green, CD41) stained for IRE1α (blue) with GRP78 (red). Zoom in as shown by dotted line shows expression in individual platelets. Images acquired on Perkin Elmer 5-Laser Spinning Disk Confocal Microscope (equipped with Nikon Ti-E Eclipse inverted microscope), 100X (oil) objective. Microscopy images representative of n=3 independent experiments. Platelet counts adjusted between samples prior to staining. Scale bar, 10 μm H. Pearson’s correlation between protein aggregates and P-selectin. I. Pearson’s Correlation between protein aggregates and HbA1c (%), an indicator of the severity of diabetes. Values expressed as mean ± SEM. Precise p value corresponds to Mann Whitney test (A) and Student’s t test (C, E). See also Supplemental Data Table 1 and Supplemental Figure S10–12
IRE1α-XBP1 pathway has been shown to regulate glucose biosynthesis and mitochondrial respiration 47–50 and we hypothesize that its selective activation in platelets is mediated by glucose levels and overall metabolic homeostasis in diabetes. Ex vivo hyperglycemia (Figure S12A–C) led to a trend (not significant) towards increase in protein aggregates in human platelets (Figure S12A) along with significantly higher IRE1α and XBP1s expression; PERK remained unchanged (Figure S12B–C). To further exclude effects of comorbidities and medications, we used the recognized streptozotocin (STZ) mouse model of diabetes. STZ platelets had elevated protein aggregation and consistent with human studies, only IRE1α and XBP1s but not PERK was increased (Figure S12E–F). In contrast, all three UPR pathways were activated in heart tissues from the same mice (Figure S12G). Taken together, the disruption of glucose homeostasis in human and mouse platelets, both acute (ex vivo) and chronic (DM), selectively induces the IRE1α-XBP1 pathway.
Chemical chaperone 4-PBA and IRE1α inhibition differentially modulates platelet reactivity in diabetes mellitus.
Growing recognition of UPR in human diseases has led to an emerging interest in therapeutic targeting of UPR components. Pharmacological chaperones are low molecular mass compounds that stabilize protein conformation and buffer the misfolded protein aggregates 51. Sodium 4-phenylbutyrate (PBA) is the most well studied chemical chaperone and is clinically approved for urea cycle disorders. Previously, PBA has been shown to improve insulin sensitivity, glucose homeostasis and leptin resistance in diabetes mellitus 52, 53. With the significant increase in protein misfolding observed in our patient cohort, we were interested in investigating the effect of PBA on platelet phenotype. Ex vivo administration of PBA reduced platelet aggregation in both HC and DM platelets, with the decline being significant in DM platelets (Figure 7A–B). Both ADP and Collagen induced aggregation was substantially decreased following treatment with PBA in the control samples (Figure S13A). Further, PBA considerably reduced platelet apoptosis (Figure 7C) and protein aggregates levels (Figure 7D) in both the HC and DM platelet samples, ex vivo. These results demonstrate the ability of PBA to potentially mitigate platelet hyperactivation in diabetes mellitus.
Figure 7: Chemical chaperone 4-PBA and IRE1α inhibition differentially modulates platelet activation in diabetes mellitus.
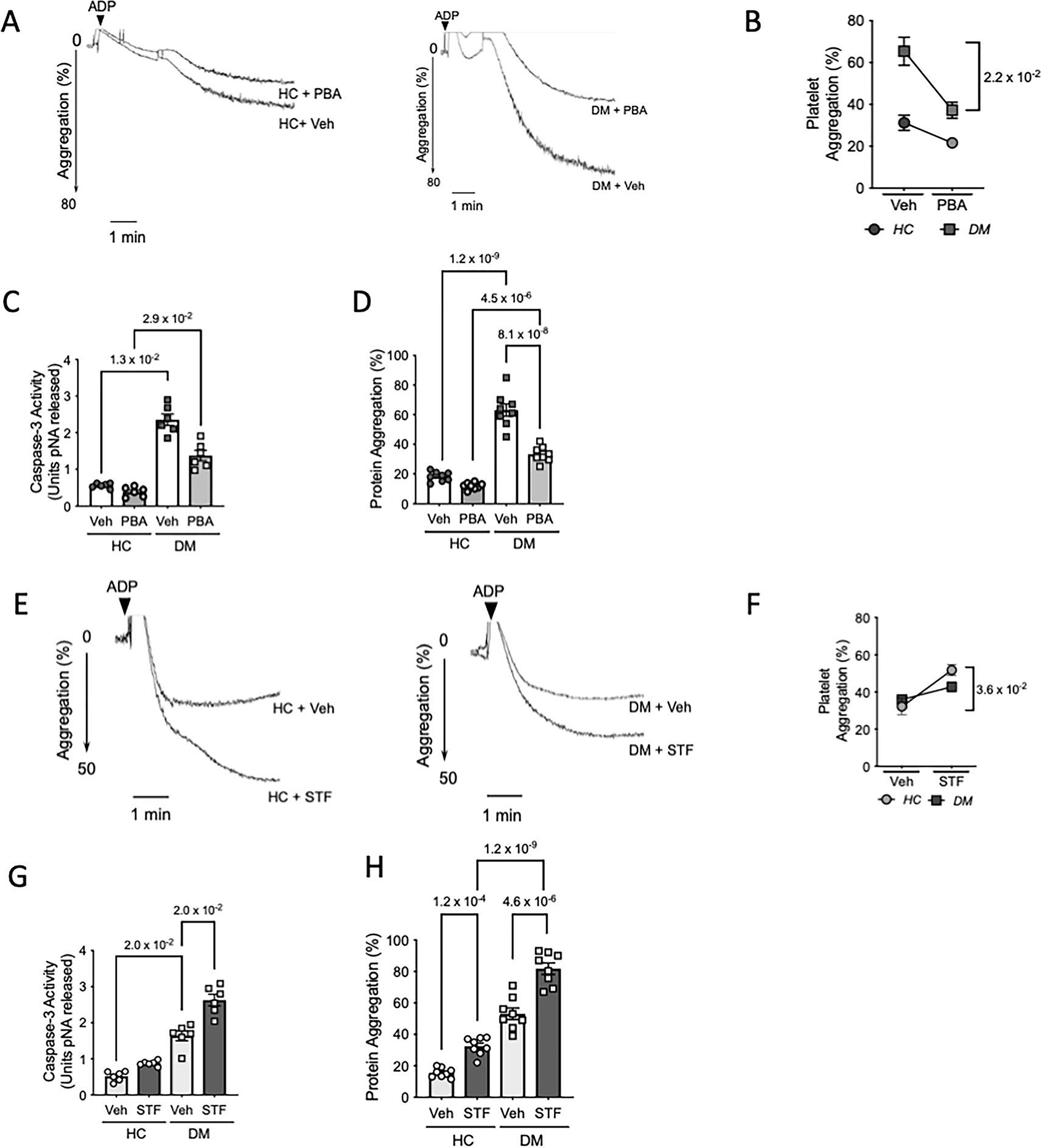
Healthy control (HC) and diabetes mellitus (DM) patients were randomly selected for ex vivo interventions to study effect of UPR modulation on platelet phenotype. A-D. Freshly isolated platelet rich plasma (PRP) from either HC or DM patients (n=8 per group) incubated with 4- phenylbutyric acid (PBA, 5mM) or vehicle (PBS) for 3 hr at 37°C. A-B. Representative platelet aggregation tracings using ADP (2.5μM) as agonist. Quantitation shows percentage light transmittance. n=5 samples per group used for aggregation studies. p-value shown between DM veh vs DM + PBA corresponds to Wilcoxon test between pre- and post PBA treated samples. C. Caspase-3 activity (n=6 per group). D. Protein aggregation levels in HC and DM samples (n=8 per group) treated with Veh or PBA were measured in the lysates. E-H. Freshly isolated PRP from HC and DM patients (n=8 per group) was incubated with STF-083010 (STF, 30μM) or vehicle. E-F. Representative platelet aggregation tracings from HC and DM platelet rich plasma (n=5 per group) treated with STF and respective quantitation. Exact p-value shown between HC veh vs HC + STF corresponds to Wilcoxon test between pre- and post STF treated samples. G. Caspase -3 activity following incubation of HC and DM samples (n=6/group) with STF. H. Intraplatelet protein aggregates in HC and DM samples (n=8 per group). Exact p value corresponds to Wilcoxon test between pre- and post-treatment groups (B, F), Kruskal-Wallis test with Dunn’s multiple comparisons (C, G) or one way ANOVA with Tukey multiple comparisons (D-H).
With the observed selective activation of the IRE1α-XBP1 pathway in DM platelets, we next investigated whether pharmacological targeting of this pathway affects the prothrombotic phenotype observed in DM. STF-080310 is a novel IRE1α inhibitor which specifically inhibits the RNase activity of IRE1α and thus can block XBP1 splicing and decrease XBP1s levels 54. Ex vivo treatment with STF led to higher platelet aggregation (Figure 7E–F, Figure S13B), caspase-3 activity (Figure 7G) and protein aggregates (Figure 7H) in both HC and DM platelets. In DM platelets, this selective upregulation of the IRE1α-XBP1 likely plays a protective role and limits platelet hyperactivation. Consistent with XBP1 cKO mice results, this further confirms the role and translational significance of XBP1 pathway in platelet pathophysiology. Although more in depth studies are needed to completely elucidate the effect of PBA in ameliorating platelet hyper-reactivity, our results, statistically significant even in a relatively small cohort, suggest its therapeutic value as an anti-platelet agent. As shown comprehensively using a combination of ex vivo, in vivo and human studies, the understanding of the finely tuned UPR in platelets could pave the way for the development of an attractive new milieu for developing safer anti-platelet therapies.
Discussion
The UPR is a ubiquitous response to alleviate the load of misfolded proteins in nucleated cells; however, whether it can occur in absence of nucleus and transcriptional regulation has not been described. Here, we report, for the first time, that platelets contain a functional highly selective UPR machinery; the presence of mRNA transcripts indicates that all components for this complex process are prepackaged in the platelet. Our platelet specific knockout models for specific UPR genes, further exemplify the differential role played by each pathway in the functional platelet phenotype. This is particularly important in DM wherein the platelet mediated atherothrombosis forms a major cause of morbidity and mortality. Results from our DM cohort (and the STZ mice) reveal a significant, yet selective, activation of IRE1α following a robust increase in the intraplatelet protein aggregate accumulation. Targeting the protein misfolding, using the chemical chaperone PBA, was beneficial in diminishing platelet hyperactivation in DM platelets, and could be developed as a potential new therapeutic strategy.
ER stress and UPR has been identified in the platelet precursor cells, the megakaryocytes and the role of ER stress induced apoptosis described in platelet formation and thrombopoiesis55, 56. Inhibition of eIF2α phosphorylation, inhibits pro-platelet formation from Meg-01 cell lines, in vitro56. Although, a few limited studies have suggested the presence of UPR proteins (mainly ER chaperones) 57, the key signaling components of this multifaceted response are not yet described in platelets. In our human (ex vivo and translational) and mouse studies, inhibition of N-linked glycosylation (Tm) and hyperglycemia both led to an increase in the IRE1α-XBP1 but not the PERK pathway. XBP1 modulates the N-linked glycome 58 and with N-linked glycosylation forming a crucial posttranslational modification for several platelet proteins 59, the role of XBP1 in platelets likely extends beyond the UPR. Indeed, selective activation of XBP1 is involved in anti-tumor immunity and host defense 48, 60, 61 and more studies are needed to elucidate its role in platelets. Metabolic dysregulation associated with diabetes has been linked to vascular dysfunction and DM platelets display exaggerated aggregation and prothrombotic risk 62. The selective activation of the IRE1α-XBP1 in platelets from diabetic patients is likely mediated by glucose levels and metabolic homeostasis. While the XBP1-deficient mice platelets displayed increased activation and apoptosis and a higher population of immature platelets, the inhibition of IRE1α-XBP1 by STF worsened platelet apoptosis in both HC and DM platelets. Thus, along with upregulating the translation of cytoprotective proteins, the selective activation of the IRE1α-XBP1 pathway likely contributes to the maintenance of platelet integrity, function, and lifespan.
PBA is an clinically approved orally bioavailable chemical chaperone, shown to improve glucose homeostasis 52, 53. In patients with insulin resistance, treatment with PBA improved glucose tolerance 63. We demonstrate the effect of PBA on platelets for the first time in reducing platelet aggregation, thereby decreasing the associated prothrombotic risk. With its low toxicity, stability and ease of oral supplementation, PBA is under consideration for use in several diseases including thalassemia, neurodegenerative diseases and cancers 64. Although more extensive studies are needed to investigate the effect of other chemical chaperones on platelets, we show that PBA is beneficial in alleviating platelet hyperactivation and could be potentially developed as a clinical therapy in vascular diseases.
PERK pathway plays a key role in the hypoxia tolerance of tumor cells 65–67 with an increase in hypoxia-induced cell death in PERK mutant cell lines 65. Induction of OS preferentially activated PERK pathway in human platelets; this could be an acute stress response providing the signal for upregulation of antioxidant proteins and reduction of intraplatelet ROS levels. PERK deficient platelets showed a distinct procoagulant phenotype and enhanced sensitivity to oxidative stress. Since ROS is a signaling molecule in platelets and promotes platelet pro-coagulant phenotype 68, higher PERK and eIF2α phosphorylation could drive both pro-survival and apoptotic decisions. Platelet mitochondria regulate the pro-thrombotic function via redox signaling and initiation of apoptosis 69 and PERK can modulate mitochondrial function 24. PERK, thus, may be involved in the protection of mitochondria against stress, regulation of platelet function and translation of selective proteins.
Genetic manipulation of UPR in murine models provide insights into its role in normal physiology and disease conditions 23. Although few individual studies have investigated the tissue-specific deletion of all three UPR sensors, Gass et al demonstrated that the deletion of XBP1, but not PERK or ATF6, impaired B cell differentiation 70. Our mouse studies establish, for the first time, that ablation of these genes differentially affects platelet aggregation, activation and apoptosis responses as well as thrombus formation, in vivo. Mechanistically, enhanced activation of PLCγ2 and p38 MAPKs in PERK cKO mice, likely forms the basis of increased aggregation in absence of PERK; suggesting the role of PERK in regulating platelet prothrombotic propensity. XBP1 deletion led to higher aggregation and activation likely via increase in PKC δ. Conversely, deletion of ATF6 cKO led to higher phosphorylation of PKA, the negative regulator of platelet activation. Our results, thus, highlight a critical role for UPR proteins in modulating these intricately linked complex pathways in platelets; further, it would be interesting to explore the regulation of these ubiquitous signaling pathways in nucleated cells by UPR components. Platelet functional responses encompasses complex mechanisms to maintain platelets in their resting state, modulate the activation pathways by platelet agonists and thrombus formation as well as negative feedback mechanisms 25. As we observed the regulation of selective platelet activation pathways by individual UPR molecules, it would be interesting to examine the effect on UPR when studying platelet activation pathways. Our study, provides novel directions and forms the basis of future more detailed and focused studies for each key signaling molecule and how it may be regulated by activation of a selective yet robust UPR. Although the deletion of PERK and XBP1 increases the propensity for platelet rich thrombus formation in the semi-occlusive carotid artery injury model used, our study did not measure the occlusion time. Future studies involving other murine thrombosis models such as the photochemical injury model, mechanical injury model 71 could provide more detailed insights into the role of platelet UPR during in vivo thrombus formation.
Previously, using siRNA mediated knockdown or chemical inhibitors of specific UPR arms, it has been shown that inhibition of one UPR branch did not necessarily lead to compensatory activation of other branches of UPR72, 73. In platelets from our UPR cKO mice, however, both under physiological baseline conditions and OS, we observed a compensatory increase in the arms of UPR. With UPR being an integral stress response, it is likely that platelets deficient in one of the UPR branches, would modulate the compensatory upregulation of other UPR arms. Moreover, since UPR is critical for ER stress induced apoptosis and subsequent platelet formation from megakaryocytes55, 56, the compensatory role of each of the arms is highly relevant. This is also reflected in the selective stress-specific regulation of individual UPR arms in platelets, which has been observed in few studies with nucleated cells74. More detailed studies using megakaryocytes from cKO mice would provide new insights into the compensatory mechanisms within the platelet UPR.
While we show the effect of PBA and STF on platelet function in DM, it would be important to study the impact, if any, of other modulators of UPR pathways, including pharmacological chaperones on platelet phenotype, especially in atherothrombosis. IRE1α, upon activation, can also induce degradation of mRNAs through a process termed as RIID (Regulated IRE1α-Dependent Decay)75. While RIDD and its mechanism is still being explored in other nucleated cells, platelet RNA decay is a growing field of interest76 and it would be exciting to use IRE1α cKO mice to investigate RIDD in platelets. With XBP1 being crucial for platelet production and lifespan, it would be interesting to evaluate its role in thrombocytopenia and bleeding disorders. The apoptosis-like phase, which, forms a key step in megakaryocyte maturation and platelet formation, has been previously shown to be induced by ER stress56. It is likely that lineage specific deletion of UPR genes could affect thrombopoiesis and megakaryocyte maturation, as is also evident by the decreased platelet counts in the PERK cKO mice. Further, UPR could potentially affect the downstream signaling and consequences in the bone marrow niche, especially under physiological conditions and hypoxia.
Thus, the diversity of the signaling responses observed, demonstrates for the first time, a non-redundant, specific role for the UPR sensors in platelets. Our results emphasize the distinct role of platelet UPR under various stimuli and in different microenvironments and how these may independently or in a concerted manner modulate the platelet activation signaling. Indeed, this specificity could be closely linked to the ability of platelets to sense and respond rapidly to multiple kinds of blood stressors. More in-depth studies are needed to investigate the relevance of selective UPR in context of other diseases including cancers, cardiovascular and neurodegenerative diseases, with a known platelet etiology. A highly specific platelet UPR highlights the significance of therapeutic targeting of selective arms and provides novel avenues for development of anti-platelet therapy. Beyond platelet function and physiology, our results also have significance in understanding the transcription independent regulation of UPR and reveal a new dimension of UPR in absence of a nucleus.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is known?
The unfolded protein response (UPR) is well described in nucleated cells under diverse conditions of stress.
Platelet hyper-reactivity is associated with increased thrombotic events and cardiovascular diseases including diabetes mellitus (DM)
ER stress induced apoptosis has been shown to be involved in megakaryocyte maturation and thrombopoiesis
What new information does this article contribute?
Circulating platelets contain the UPR pathway components, each of which can be activated selectively in absence of a nucleus.
Platelet UPR components PERK, XBP1 and ATF6 regulate platelet function via distinct underlying platelet activation signalling pathways.
Protein aggregate levels in platelets from Type II DM patients correlate significantly with the disease severity, and the IRE1a-XBP-1 UPR axis is selectively activated in platelets from these DM patients.
UPR modulators including chemical chaperones could be developed as potential targeted novel anti-platelet therapies in cardiovascular diseases.
The diversity of the platelet responses, demonstrate for the first time, a non-redundant, yet defined role for the UPR sensors in platelets. Our results emphasize the distinct role of platelet UPR under various stimuli and in different microenvironments and how these may, independently or in a concerted manner, modulate the platelet activation signaling. Diabetes Mellitus Type II (DM) affects millions globally and potentiates a risk for thrombotic events as well as cardiovascular diseases. Platelet hyperreactivity is a known phenomenon in DM. Understanding platelet UPR pathways and its effect on platelet reactivity highlights the significance of therapeutic targeting of selective UPR arms, especially in DM. Our results thus also provide novel avenues for development of anti-platelet therapy. Beyond platelet function and physiology, our results are relevant in understanding the transcription independent regulation of UPR and thus reveal a new dimension of UPR in the absence of a nucleus.
Acknowledgements
We are grateful to Dr. Laurie H. Glimcher (President, Dana Farber Cancer Institute) for the kind gift of Xbp1flox/flox mice and Prof Xinran Liu (Director, Yale Biological EM Facility) for his helpful advice. We thank Dr. Hongyu Zhou (Ira V. Hiscock Professor of Biostatistics, Yale School of Public Health) for his inputs in the statistical analysis. We also thank Life Science Editors (https://www.lifescienceeditors.com/) for editing assistance.
Source of Funding
These studies have been supported by grants from NIH-NHLBI, RO1-HL122815, RO1-HL150515, and RO1-HL115247.
Non-standard Abbreviations and Acronyms
- ADP
Adenosine diphosphate
- ATF6
Activating Transcription Factor 6
- cKO
conditional knockout
- CLEC-2
C-type lectin-like receptor 2
- CVD
Cardiovascular Diseases
- DM
Diabetes Mellitus
- DTS
Dense Tubular System
- DTT
Dithiothreitol
- eIF2
eukaryotic Initiation Factor 2
- ER
Endoplasmic Reticulum
- GPCR
G-protein coupled receptors
- GRP78
Glucose Regulated Protein 78
- GRP94
Glucose Regulated Protein 94
- HC
Healthy Control
- IRE1α
Inositol-Requiring Enzyme-1alpha
- ITAM
immunoreceptor tyrosine-based activation motif
- OS
Oxidative Stress
- PBA
Sodium 4-phenylbutyrate
- PDI
Protein Disulfide Isomerase
- PERK
Protein kinase RNA (PKR)-like ER kinase
- Pf4
Platelet factor 4
- PKA
Protein Kinase A
- PKC
Protein Kinase C
- PLC
Phospholipase C
- RIID
Regulated IRE1-Dependent Decay
- ROS
Reactive Oxygen Species
- RTN4
Reticulon-4
- SEM
Standard Error of Mean
- STZ
Streptozotocin
- TEM
Transmission Electron Microscopy
- Tm
Tunicamycin
- UPR
Unfolded Protein Response
- XBP1
X Binding Protein
Footnotes
References
- 1.Mori K Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–4. [DOI] [PubMed] [Google Scholar]
- 2.Walter P and Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. [DOI] [PubMed] [Google Scholar]
- 3.Wagner DD and Burger PC. Platelets in Inflammation and Thrombosis. 2003;23:2131–2137. [DOI] [PubMed] [Google Scholar]
- 4.Weyrich AS, Schwertz H, Kraiss LW and Zimmerman GA. Protein synthesis by platelets: historical and new perspectives. J Thromb Haemost. 2009;7:241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowley JW, Schwertz H and Weyrich AS. Platelet mRNA: the meaning behind the message. Curr Opin Hematol. 2012;19:385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leytin V Apoptosis in the anucleate platelet. Blood Reviews. 2012;26:51–63. [DOI] [PubMed] [Google Scholar]
- 7.Lee SH, Du J, Stitham J, Atteya G, Lee S, Xiang YZ, Wang DD, Jin Y, Leslie KL, Spollett G, Srivastava A, Mannam P, Ostriker A, Martin KA, Tang WH and Hwa J. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. Embo Molecular Medicine. 2016;8:779–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colberg L, Cammann C, Greinacher A and Seifert U. Structure and function of the ubiquitin-proteasome system in platelets. J Thromb Haemost. 2020;18:771–780. [DOI] [PubMed] [Google Scholar]
- 9.Ouseph MM, Huang Y, Banerjee M, Joshi S, MacDonald L, Zhong Y, Liu H, Li X, Xiang B, Zhang G, Komatsu M, Yue Z, Li Z, Storrie B, Whiteheart SW and Wang QJ. Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood. 2015;126:1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leslie M Cell biology. Beyond clotting: the powers of platelets. Science. 2010;328:562–4. [DOI] [PubMed] [Google Scholar]
- 11.Koupenova M, Clancy L, Corkrey HA and Freedman JE. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ Res. 2018;122:337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregg D and Goldschmidt-Clermont PJ. Cardiology patient page. Platelets and cardiovascular disease. Circulation. 2003;108:e88–90. [DOI] [PubMed] [Google Scholar]
- 13.Colwell JA and Nesto RW. The platelet in diabetes: focus on prevention of ischemic events. Diabetes Care. 2003;26:2181–8. [DOI] [PubMed] [Google Scholar]
- 14.Jain K, Tyagi T, Patell K, Xie Y, Kadado AJ, Lee SH, Yarovinsky T, Du J, Hwang J, Martin KA, Testani J, Ionescu CN and Hwa J. Age associated non-linear regulation of redox homeostasis in the anucleate platelet: Implications for CVD risk patients. EBioMedicine. 2019;44:28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K and Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, Mathis D and Hotamisligil GS. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci Transl Med. 2013;5:211ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee AH, Scapa EF, Cohen DE and Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tiedt R, Schomber T, Hao-Shen H and Skoda RC. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109:1503–6. [DOI] [PubMed] [Google Scholar]
- 19.Ebbeling L, Robertson C, McNicol A and Gerrard JM. Rapid ultrastructural changes in the dense tubular system following platelet activation. Blood. 1992;80:718–23. [PubMed] [Google Scholar]
- 20.Schuck S, Prinz WA, Thorn KS, Voss C and Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. 2009;187:525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdullahi A, Stanojcic M, Parousis A, Patsouris D and Jeschke MG. Modeling Acute ER Stress in Vivo and in Vitro. Shock. 2017;47:506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malhotra JD and Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–93. [DOI] [PubMed] [Google Scholar]
- 23.Bommiasamy H and Popko B. Animal models in the study of the unfolded protein response. Methods Enzymol. 2011;491:91–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MC and Wiseman RL. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018;22:2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Estevez B and Du X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology (Bethesda). 2017;32:162–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Offermanns S, Toombs CF, Hu YH and Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389:183–6. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki-Inoue K, Inoue O, Frampton J and Watson SP. Murine GPVI stimulates weak integrin activation in PLCgamma2−/− platelets: involvement of PLCgamma1 and PI3-kinase. Blood. 2003;102:1367–73. [DOI] [PubMed] [Google Scholar]
- 28.Wonerow P, Pearce AC, Vaux DJ and Watson SP. A critical role for phospholipase Cgamma2 in alphaIIbbeta3-mediated platelet spreading. J Biol Chem. 2003;278:37520–9. [DOI] [PubMed] [Google Scholar]
- 29.Lee SB, Rao AK, Lee KH, Yang X, Bae YS and Rhee SG. Decreased expression of phospholipase C-beta 2 isozyme in human platelets with impaired function. Blood. 1996;88:1684–91. [PubMed] [Google Scholar]
- 30.Woulfe DS. Akt signaling in platelets and thrombosis. Expert Rev Hematol. 2010;3:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, De S, Damron DS, Chen WS, Hay N and Byzova TV. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Zhang G, Feil R, Han J and Du X. Sequential activation of p38 and ERK pathways by cGMP-dependent protein kinase leading to activation of the platelet integrin alphaIIb beta3. Blood. 2006;107:965–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang Q, Li F, Shi K, Wu P, An J, Yang Y and Xu C. Involvement of p38 in signal switching from autophagy to apoptosis via the PERK/eIF2alpha/ATF4 axis in selenite-treated NB4 cells. Cell Death Dis. 2014;5:e1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lumley EC, Osborn AR, Scott JE, Scholl AG, Mercado V, McMahan YT, Coffman ZG and Brewster JL. Moderate endoplasmic reticulum stress activates a PERK and p38-dependent apoptosis. Cell Stress Chaperones. 2017;22:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Babur O, Ngo ATP, Rigg RA, Pang J, Rub ZT, Buchanan AE, Mitrugno A, David LL, McCarty OJT, Demir E and Aslan JE. Platelet procoagulant phenotype is modulated by a p38-MK2 axis that regulates RTN4/Nogo proximal to the endoplasmic reticulum: utility of pathway analysis. Am J Physiol Cell Physiol. 2018;314:C603–C615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chari R, Getz T, Nagy B Jr., Bhavaraju K, Mao Y, Bynagari YS, Murugappan S, Nakayama K and Kunapuli SP. Protein kinase C[delta] differentially regulates platelet functional responses. Arterioscler Thromb Vasc Biol. 2009;29:699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kostyak JC, Mauri B, Patel A, Dangelmaier C, Reddy H and Kunapuli SP. Phosphorylation of protein kinase Cdelta positively regulates thromboxane generation in platelets. J Biol Chem. 2021:100720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larroque-Cardoso P, Swiader A, Ingueneau C, Negre-Salvayre A, Elbaz M, Reyland ME, Salvayre R and Vindis C. Role of protein kinase C delta in ER stress and apoptosis induced by oxidized LDL in human vascular smooth muscle cells. Cell Death Dis. 2013;4:e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raslan Z and Naseem KM. The control of blood platelets by cAMP signalling. Biochem Soc Trans. 2014;42:289–94. [DOI] [PubMed] [Google Scholar]
- 40.Zhao L, Liu J, He C, Yan R, Zhou K, Cui Q, Meng X, Li X, Zhang Y, Nie Y, Zhang Y, Hu R, Liu Y, Zhao L, Chen M, Xiao W, Tian J, Zhao Y, Cao L, Zhou L, Lin A, Ruan C and Dai K. Protein kinase A determines platelet life span and survival by regulating apoptosis. J Clin Invest. 2017;127:4338–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bye AP, Unsworth AJ and Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost. 2016;14:918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, Cavener D and Diehl JA. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29:3881–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Adachi M, Zhao S, Hareyama M, Koong AC, Luo D, Rando TA, Imai K and Shinomura Y. Preventing oxidative stress: a new role for XBP1. Cell Death Differ. 2009;16:847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Back SH and Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang WH, Stitham J, Jin Y, Liu R, Lee SH, Du J, Atteya G, Gleim S, Spollett G, Martin K and Hwa J. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation. 2014;129:1598–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Harg JM, van Heest JC, Bangel FN, Patiwael S, van Weering JR and Scheper W. The UPR reduces glucose metabolism via IRE1 signaling. Biochim Biophys Acta Mol Cell Res. 2017;1864:655–665. [DOI] [PubMed] [Google Scholar]
- 48.Wu R, Zhang QH, Lu YJ, Ren K and Yi GH. Involvement of the IRE1α-XBP1 pathway and XBP1s-dependent transcriptional reprogramming in metabolic diseases. DNA Cell Biol. 2015;34:6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitchell T, Johnson MS, Ouyang X, Chacko BK, Mitra K, Lei X, Gai Y, Moore DR, Barnes S, Zhang J, Koizumi A, Ramanadham S and Darley-Usmar VM. Dysfunctional mitochondrial bioenergetics and oxidative stress in Akita(+/Ins2)-derived beta-cells. Am J Physiol Endocrinol Metab. 2013;305:E585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vannuvel K, Van Steenbrugge M, Demazy C, Ninane N, Fattaccioli A, Fransolet M, Renard P, Raes M and Arnould T. Effects of a Sublethal and Transient Stress of the Endoplasmic Reticulum on the Mitochondrial Population. J Cell Physiol. 2016;231:1913–31. [DOI] [PubMed] [Google Scholar]
- 51.Cortez L and Sim V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion. 2014;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ and Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr. and Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. [DOI] [PubMed] [Google Scholar]
- 54.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M and Koong AC. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morishima N and Nakanishi K. Proplatelet formation in megakaryocytes is associated with endoplasmic reticulum stress. Genes Cells. 2016;21:798–806. [DOI] [PubMed] [Google Scholar]
- 56.Lopez JJ, Palazzo A, Chaabane C, Albarran L, Polidano E, Lebozec K, Dally S, Nurden P, Enouf J, Debili N and Bobe R. Crucial role for endoplasmic reticulum stress during megakaryocyte maturation. Arterioscler Thromb Vasc Biol. 2013;33:2750–8. [DOI] [PubMed] [Google Scholar]
- 57.Hernández Vera R, Vilahur G, Ferrer-Lorente R, Peña E and Badimon L. Platelets derived from the bone marrow of diabetic animals show dysregulated endoplasmic reticulum stress proteins that contribute to increased thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:2141–8. [DOI] [PubMed] [Google Scholar]
- 58.Wong MY, Chen K, Antonopoulos A, Kasper BT, Dewal MB, Taylor RJ, Whittaker CA, Hein PP, Dell A, Genereux JC, Haslam SM, Mahal LK and Shoulders MD. XBP1s activation can globally remodel N-glycan structure distribution patterns. Proc Natl Acad Sci U S A. 2018;115:E10089–E10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mammadova-Bach E, Jaeken J, Gudermann T and Braun A. Platelets and Defective N-Glycosylation. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo-Garcia JR and Glimcher LH. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell. 2015;161:1527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinon F, Chen X, Lee AH and Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vinik AI, Erbas T, Park TS, Nolan R and Pittenger GL. Platelet dysfunction in type 2 diabetes. Diabetes Care. 2001;24:1476–85. [DOI] [PubMed] [Google Scholar]
- 63.Xiao C, Giacca A and Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes. 2011;60:918–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iannitti T and Palmieri B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D. 2011;11:227–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG and Koumenis C. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fels DR and Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther. 2006;5:723–8. [DOI] [PubMed] [Google Scholar]
- 67.Pereira ER, Frudd K, Awad W and Hendershot LM. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF). J Biol Chem. 2014;289:3352–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qiao J, Arthur JF, Gardiner EE, Andrews RK, Zeng L and Xu K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018;14:126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Melchinger H, Jain K, Tyagi T and Hwa J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front Cardiovasc Med. 2019;6:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gass JN, Gifford NM and Brewer JW. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J Biol Chem. 2002;277:49047–54. [DOI] [PubMed] [Google Scholar]
- 71.Westrick RJ, Winn ME and Eitzman DT. Murine models of vascular thrombosis (Eitzman series). Arterioscler Thromb Vasc Biol. 2007;27:2079–93. [DOI] [PubMed] [Google Scholar]
- 72.Chang TK, Lawrence DA, Lu M, Tan J, Harnoss JM, Marsters SA, Liu P, Sandoval W, Martin SE and Ashkenazi A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol Cell. 2018;71:629–636 e5. [DOI] [PubMed] [Google Scholar]
- 73.Duran-Aniotz C, Cornejo VH, Espinoza S, Ardiles AO, Medinas DB, Salazar C, Foley A, Gajardo I, Thielen P, Iwawaki T, Scheper W, Soto C, Palacios AG, Hoozemans JJM and Hetz C. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017;134:489–506. [DOI] [PubMed] [Google Scholar]
- 74.Pierre N, Barbe C, Gilson H, Deldicque L, Raymackers JM and Francaux M. Activation of ER stress by hydrogen peroxide in C2C12 myotubes. Biochem Biophys Res Commun. 2014;450:459–63. [DOI] [PubMed] [Google Scholar]
- 75.Maurel M, Chevet E, Tavernier J and Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci. 2014;39:245–54. [DOI] [PubMed] [Google Scholar]
- 76.Mills EW, Green R and Ingolia NT. Slowed decay of mRNAs enhances platelet specific translation. Blood. 2017;129:e38–e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leytin V, Allen DJ, Mykhaylov S, Mis L, Lyubimov EV, Garvey B and Freedman J. Pathologic high shear stress induces apoptosis events in human platelets. Biochemical and Biophysical Research Communications. 2004;320:303–310. [DOI] [PubMed] [Google Scholar]
- 78.Landry P, Plante I, Ouellet DL, Perron MP, Rousseau G and Provost P. Existence of a microRNA pathway in anucleate platelets. Nature Structural and Molecular Biology. 2009;16:961–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yoon SB, Park YH, Choi SA, Yang HJ, Jeong PS, Cha JJ, Lee S, Lee SH, Lee JH, Sim BW, Koo BS, Park SJ, Lee Y, Kim YH, Hong JJ, Kim JS, Jin YB, Huh JW, Lee SR, Song BS and Kim SU. Real-time PCR quantification of spliced X-box binding protein 1 (XBP1) using a universal primer method. PLoS One. 2019;14:e0219978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Severin S, Pollitt AY, Navarro-Nunez L, Nash CA, Mourao-Sa D, Eble JA, Senis YA and Watson SP. Syk-dependent phosphorylation of CLEC-2: a novel mechanism of hem-immunoreceptor tyrosine-based activation motif signaling. J Biol Chem. 2011;286:4107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jarvis GE, Atkinson BT, Snell DC and Watson SP. Distinct roles of GPVI and integrin alpha(2)beta(1) in platelet shape change and aggregation induced by different collagens. Br J Pharmacol. 2002;137:107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Angenieux C, Maitre B, Eckly A, Lanza F, Gachet C and de la Salle H. Time-Dependent Decay of mRNA and Ribosomal RNA during Platelet Aging and Its Correlation with Translation Activity. PLoS One. 2016;11:e0148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heidt T, Deininger F, Peter K, Goldschmidt J, Pethe A, Hagemeyer CE, Neudorfer I, Zirlik A, Weber WA, Bode C, Meyer PT, Behe M and von Zur Muhlen C. Activated platelets in carotid artery thrombosis in mice can be selectively targeted with a radiolabeled single-chain antibody. PLoS One. 2011;6:e18446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carstairs KC. The identification of platelets and platelet antigens in histological sections. J Pathol Bacteriol. 1965;90:225–31. [DOI] [PubMed] [Google Scholar]
- 85.Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif). 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 86.Tang WH, Stitham J, Gleim S, Di Febbo C, Porreca E, Fava C, Tacconelli S, Capone M, Evangelista V, Levantesi G, Wen L, Martin K, Minuz P, Rade J, Patrignani P and Hwa J. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. Journal of Clinical Investigation. 2011;121:4462–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of the study are available upon reasonable request from the corresponding authors.
A detailed description of materials and methods is available in the Online Supplemental Material.