

Abstract
Background:
The use of immune checkpoint inhibitors combined with vascular endothelial growth factor (VEGF)-targeted therapy as second-line treatment for metastatic clear cell renal cancer (mRCC) has not been evaluated prospectively.
Objective:
To evaluate the efficacy and safety of atezolizumab + bevacizumab following disease progression on atezolizumab or sunitinib monotherapy in patients with mRCC.
Design, setting, and participants:
IMmotion150 was a multicenter, randomized, open-label, phase 2 study of patients with untreated mRCC. Patients randomized to the atezolizumab or sunitinib arm who had investigator-assessed progression as per RECIST 1.1 could be treated with second-line atezolizumab + bevacizumab.
Intervention:
Patients received atezolizumab 1200 mg intravenously (IV) plus bevacizumab 15 mg/kg IV every 3 wk following disease progression on either atezolizumab or sunitinib monotherapy.
Outcome measurements and statistical analysis:
The secondary endpoints analyzed during the second-line part of IMmotion150 included objective response rate (ORR), progression-free survival (PFS), and safety. PFS was examined using Kaplan-Meier methods.
Results and limitations:
Fifty-nine patients in the atezolizumab arm and 78 in the sunitinib arm were eligible, and 103 initiated second-line atezolizumab + bevacizumab (atezolizumab arm, n = 44; sunitinib arm, n = 59). ORR (95% confidence interval [CI]) was 27% (19–37%). The median PFS (95% CI) from the start of second line was 8.7 (5.6–13.7) mo. The median event follow-up duration was 19.4 (12.9–21.9) mo among the 25 patients without a PFS event. Eighty-six (83%) patients had treatment-related adverse events; 31 of 103 (30%) had grade 3/4 events. Limitations were the small sample size and selection for progressors.
Conclusions:
The atezolizumab + bevacizumab combination had activity and was tolerable in patients with progression on atezolizumab or sunitinib. Further studies are needed to investigate sequencing strategies in mRCC.
Patient summary:
Patients with advanced kidney cancer whose disease had worsened during treatment with atezolizumab or sunitinib began second-line treatment with atezolizumab + bevacizumab. Tumors shrank in more than one-quarter of patients treated with this combination, and side effects were manageable.
Keywords: Atezolizumab, Bevacizumab, Cancer immunotherapy, Metastatic, Renal cell carcinoma, Second line, Sunitinib, Vascular endothelial growth factor inhibitor
1. Introduction
Both immune checkpoint inhibitor (ICI) and vascular endothelial growth factor (VEGF)-targeted therapy are first-line standard-of-care treatments for metastatic clear cell renal cancer (mRCC) [1]. The observation of enhanced T-cell infiltration by VEGF-targeted therapies [2–5] provides a strong rationale for combining a VEGF-targeted therapy with an ICI to treat patients with mRCC.
Atezolizumab is an engineered, humanized, immunoglobulin G1 monoclonal antibody that binds selectively to programmed death-ligand 1 (PD-L1) and blocks the interactions of PD-L1 with programmed death-1 (PD-1) in the tumor microenvironment, which can reinvigorate suppressed tumor-specific cytotoxic T cells [2,3]. Atezolizumab led to durable responses in patients with treatment-naive and VEGF inhibitor–resistant mRCC [6,7]. Bevacizumab is a VEGF-targeted therapy that has also shown single-agent efficacy in patients with mRCC [8–10]. The combination of atezolizumab + bevacizumab demonstrated a progression-free survival (PFS) advantage over sunitinib monotherapy in patients with treatment-naïve PD-L1–positive mRCC in both the phase 2 IMmotion150 and phase 3 IMmotion151 studies [7,11].
IMmotion150 (NCT01984242) was a randomized phase 2 trial to evaluate atezolizumab, sunitinib, and atezolizumab + bevacizumab as first-line treatments in patients with untreated mRCC. The coprimary endpoints of PFS in the intention-to-treat (ITT) and PD-L1+ populations have been published previously [7]. In a subsequent phase of the study, patients who experienced disease progression on atezolizumab or sunitinib monotherapy could begin treatment with atezolizumab + bevacizumab. To our knowledge, it is the first prospective study investigating the clinical activity and safety of an anti-VEGF and immunotherapy combination as second-line treatment after an ICI or VEGF targeted therapy. Here, we describe the efficacy, safety, and biomarker correlates of atezolizumab + bevacizumab following progression on either atezolizumab (immunotherapy) or sunitinib (VEGF-targeted therapy) in patients with mRCC.
2. Patients and methods
2.1. Study design and participants
The study design of this global, multicenter, open-label, phase 2 study of first-line atezolizumab, sunitinib, and atezolizumab + bevacizumab has been described previously [7]. In a subsequent portion of the study, patients receiving atezolizumab or sunitinib monotherapy could begin treatment with atezolizumab + bevacizumab after experiencing disease progression, as assessed by the investigator according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1).
Inclusion in this phase was restricted to patients who participated in the first-line phase of IMmotion150. They had to have measurable metastatic clear cell or sarcomatoid RCC that had not been treated previously with systemic therapy. Patient characteristics have been described previously [7]. Additional requirements included Karnofsky performance status of ≥70 at progression and a postprogression biopsy prior to atezolizumab + bevacizumab treatment, if clinically feasible.
2.2. Interventions
Patients had previously been randomly assigned (1:1:1) to (1) atezolizumab 1200 mg intravenously (IV) plus bevacizumab 15 mg/kg IV every 3 wk, (2) atezolizumab 1200 mg IV every 3 wk, or (3) sunitinib 50 mg/d for 4 wk followed by a 2-wk rest period. Those in either monotherapy arm could initiate atezolizumab + bevacizumab (1200 mg intravenously every 3 wk) upon disease progression (Fig. 1). No interval was specified before patients could receive combination therapy; however, no interim therapy was allowed between progression and subsequent combination therapy. Combination treatment following atezolizumab therapy was not allowed in the European centers due to local health authority feedback that patients who progressed should not receive a second investigational therapy on the trial.
Fig. 1–
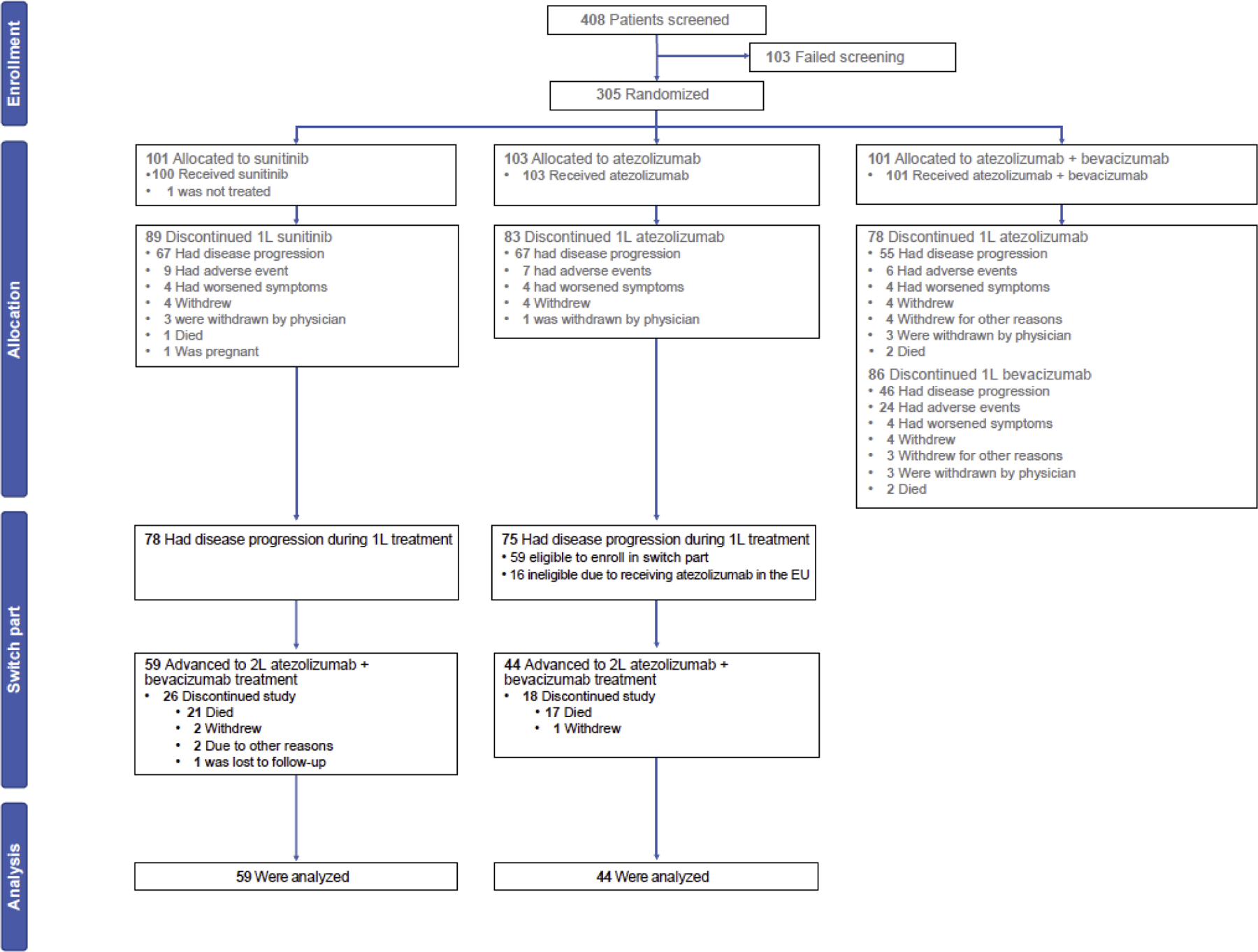
CONSORT diagram of patient disposition for the IMmotion150 trial. Patients who entered the first-line (1L) part are shown in allocation. 2L = second line.
2.3. Outcomes
Efficacy outcomes during the second-line part of IMmotion150 were secondary endpoints, including objective response rate (ORR), PFS, and duration of response (DOR; based on investigator assessment as per RECIST 1.1). Patients who initiated atezolizumab + bevacizumab underwent a new baseline tumor assessment ≤28 d before starting second-line treatment, and tumor assessments were made every 12 wk thereafter. Safety endpoints included incidence, nature, and severity of all adverse events (AEs), including grade ≥3 laboratory toxicities as per the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Exploratory endpoints included PD-L1 status in tumor tissue collected at baseline and progression, and gene expression levels (T-effector [Teff] and myeloid inflammation [Myeloid] signatures) at baseline [7].
2.4. Statistical analysis
PFS from the time of starting second-line treatment to progression or death was assessed in all patients who began second-line atezolizumab + bevacizumab. Subgroups were analyzed according to the originally allocated treatment (atezolizumab or sunitinib), and by PD-L1 status at baseline and prior response during first-line treatment. The Kaplan-Meier method was used to estimate the probability of PFS and to estimate median PFS. The Brookmeyer-Crowley methodology was used to construct the 95% confidence interval (CI) for median PFS. ORR during the second-line part was summarized among patients who had a tumor assessment at the time of starting second-line treatment and at least one second-line visit, with 95% CI constructed using the Clopper-Pearson method. DOR was analyzed using a similar method as for PFS. All analyses were descriptive, and no formal hypothesis testing was conducted. Safety was summarized for all patients who initiated second-line atezolizumab + bevacizumab.
2.5. Biomarker analysis
At baseline, tissue specimens collected within 12 mo of initiation of study treatment or from a recent tumor biopsy were submitted. Subsequent biopsies, preferably of a progressing metastatic lesion, were obtained when medically safe at the time of radiographic progression for patients receiving second-line atezolizumab + bevacizumab. Tumor specimens were tested for PD-L1 expression on tumor-infiltrating immune cells (ICs) by a central laboratory using the VENTANA SP142 immunohistochemistry (IHC) assay (Ventana), and gene expression analyses were generated using TruSeq RNA Access technology (Illumina) [7]. Specimens were scored as IC0, IC1, IC2, or IC3 if <1%, 1–<5%, 5–<10%, or ≥10% of immune cells per tumor area were PD-L1 positive, respectively. PD-L1 positivity was defined as ≥1% of ICs expressing PD-L1. Gene expression analyses of baseline tumor samples were performed as previously described [7] and focused on angiogenesis, Teff, and Myeloid signatures.
3. Results
3.1. Patients
The data cutoff for analysis in the second-line part was April 19, 2017. Overall, 75 patients in the first-line atezolizumab arm and 78 in the first-line sunitinib arm had disease progression. Sixteen of the patients in the atezolizumab arm were being treated in EU countries and were thus ineligible to proceed (Fig. 1). Of these patients, 103 entered the second-line part and received atezolizumab + bevacizumab: 75% (44/59) from the atezolizumab arm and 76% (59/78) from the sunitinib arm.
Baseline characteristics of patients in the second-line part are shown in Table 1 and were similar to those of the overall ITT population from the first-line treatment phase [7]. Among the eligible patients who did not advance to the second-line part, there was no consistent pattern of difference from the patients who did. Of the patients who advanced, 61% had tumors that were PD-L1 positive at baseline, 9% had responded to first-line atezolizumab, and 24% had responded to first-line sunitinib (Supplementary Table 1). The median PFS in patients who advanced to second-line treatment was 4.8 mo (95% CI, 2.8–5.5 mo) with first-line atezolizumab and 5.7 mo (95% CI, 5.4–8.1 mo) with first-line sunitinib (Supplementary Table 1).
Table 1–
Patient baseline characteristics
| Characteristic | All second line n = 103) |
Second line after atezolizumab (n = 44) |
Second line after sunitinib (n = 59) |
|---|---|---|---|
| Age (yr), median (IQR) | 61 (52–67) | 61 (53–67) | 61 (52–67) |
| Male, n (%) | 83 (81) | 35 (80) | 48 (81) |
| KPS ≥80, n (%) | 98 (97) a | 41 (98) | 57 (97) |
| Predominant clear cell histology, n (%) | 96 (93) | 40 (91) | 56 (95) |
| Sarcomatoid component, n (%) | 11 (11) | 5 (11) | 6 (10) |
| MSKCC risk category, n (%) | |||
| Favorable (0) | 22 (21) | 12 (27) | 10 (17) |
| Intermediate (1 or 2) | 78 (76) | 30 (68) | 48 (81) |
| Poor (≥3) | 3 (3) | 2 (5) | 1 (2) |
| ≥1% of IC expressing PD-L1, n (%) b | 63 (61) | 26 (59) | 37 (63) |
| Geographic region, n (%) | |||
| USA | 86 (83) | 44 (100) | 42 (71) |
| European Union | 17 (17) | 0 c | 17 (29) |
IC = tumor-infiltrating immune cell; IQR = interquartile range; KPS = Karnofsky performance status; MSKCC = Memorial Sloan Kettering Cancer Center; PD-L1 = programmed death-ligand 1.
Clinical cutoff date: April 19, 2017.
n = 101.
Denominators include patients who were not evaluable.
Advancing to second-line atezolizumab + bevacizumab from atezolizumab monotherapy was not allowed in European centers.
3.2. Efficacy
In 100 of 103 patients evaluable for response (44 from the atezolizumab arm and 56 from the sunitinib arm), the ORR was 27% (27/100; 95% CI, 19–37%). The ORRs were 25% (11/44; 95% CI, 13–40%) in patients who previously received atezolizumab and 29% (16/56; 95% CI, 17–42%) in patients who previously received sunitinib (Table 2). The ORR in patients who responded to first-line treatment was 44% (7/16; 95% CI, 20– 70%), and it was 24% (20/84; 95% CI, 15–34%) in nonresponders (Table 2). Responses to atezolizumab + bevacizumab occurred in nonresponders to both prior atezolizumab (n = 9; 23% [95% CI, 11–39%]) and sunitinib (n = 11; 25% [95% CI, 13– 40%]). Forty patients experienced either progressive disease (PD; n = 21) or stable disease (SD; n = 19) as the best response to first-line atezolizumab, and subsequently four patients (19%) with initial PD on atezolizumab achieved a partial response (PR) on atezolizumab + bevacizumab. In patients with PD-L1–positive tumors at baseline, the response rate to second-line atezolizumab + bevacizumab was 28% (95% CI, 17–41%; Table 2); it was 22% (95% CI, 10–38%) in patients with PD-L1–negative tumors (Supplementary Table 2).
Table 2–
Confirmed investigator-assessed responses (RECIST 1.1) in patients who advanced to second-line atezolizumab + bevacizumab
| Response | All second line a,b (n = 100) | |
|---|---|---|
| Response during second-line treatment | ||
| ORR, n (%) [95% CI] | 27 (27) [19–37] | |
| CR (%) | 1 | |
| PR (%) | 26 | |
| SD (%) | 37 | |
| PD (%) | 32 |
|
| After atezolizumab b
(n = 44) |
After sunitinib a (n = 56) | |
|
|
||
| ORR by previous first-line treatment | ||
| ORR, n (%) [95% CI] | 11 (25) [13–40] | 16 (29) [17–42] |
|
|
||
| Responder (n = 16) |
Nonresponder (n = 84) |
|
|
|
||
| ORR by response to previous first-line treatment | ||
| ORR, n (%) [95% CI] | 7 (44) [20–70] | 20 (24) [15–34] |
|
|
||
| All second line (n = 60) |
||
| ORR in PD-L1–positive patients at study baseline c | ||
| ORR, n (%) [95% CI] | 17 (28) [17–41] | |
CI = confidence interval; CR = complete response; ORR = objective response rate; PD = progressive disease; PD-L1 = programmed death-ligand 1; PR = partial response; RECIST = Response Evaluation Criteria in Solid Tumors; SD = stable disease.
Three patients in the postsunitinib arm without postbaseline tumor assessment were not included in the analysis.
One patient in the postatezolizumab arm had non-CR/non-PD as the best overall response.
Based on archival tissue and/or biopsy.
No clear trend in response could be identified based on Memorial Sloan Kettering Cancer Center risk score. For patients with favorable (n = 22), intermediate (n = 75), and poor (n = 3) risk, ORRs were 18% (95% CI, 5–40%; n = 4), 31% (95% CI, 21–42%; n = 23), and 0% (95% CI, 0–71%), respectively. For patients whose tumors had a sarcomatoid component (n = 11), ORR was 9% (95% CI, 0–41%; n = 1), and for those without a sarcomatoid component (n = 88), ORR was 28% (95% CI, 19–39%; n = 25).
The estimated median DOR in all patients who responded after advancing to second-line was 18.3 mo (95% CI, 11.6 mo–not estimable). DOR was immature because 14 of 27 patients (52%) continued to respond as of the last tumor assessment at data cutoff (five of 11 after atezolizumab and nine of 16 after sunitinib).
The median PFS in second line for all 103 patients was 8.7 mo (95% CI, 5.6–13.7 mo; Fig. 2). Twenty-five patients did not have a PFS event as of the data cutoff. The median event follow-up duration among these patients was 19.4 mo (95% CI, 12.9–21.9 mo). The respective median PFS was 11.1 mo (95% CI, 5.1–17.0 mo) and 7.7 mo (95% CI, 4.5–11.2 mo) in patients who previously received atezolizumab and sunitinib (Supplementary Fig. 1A). In patients with PD-L1–positive tumors at baseline, the median PFS with second-line atezolizumab + bevacizumab was 6.0 mo (95% CI, 4.2–13.7 mo), and in patients with PD-L1–negative tumors at baseline, the median PFS was 9.8 mo (95% CI, 6.0–14.3 mo; Supplementary Fig. 1B).
Fig. 2–
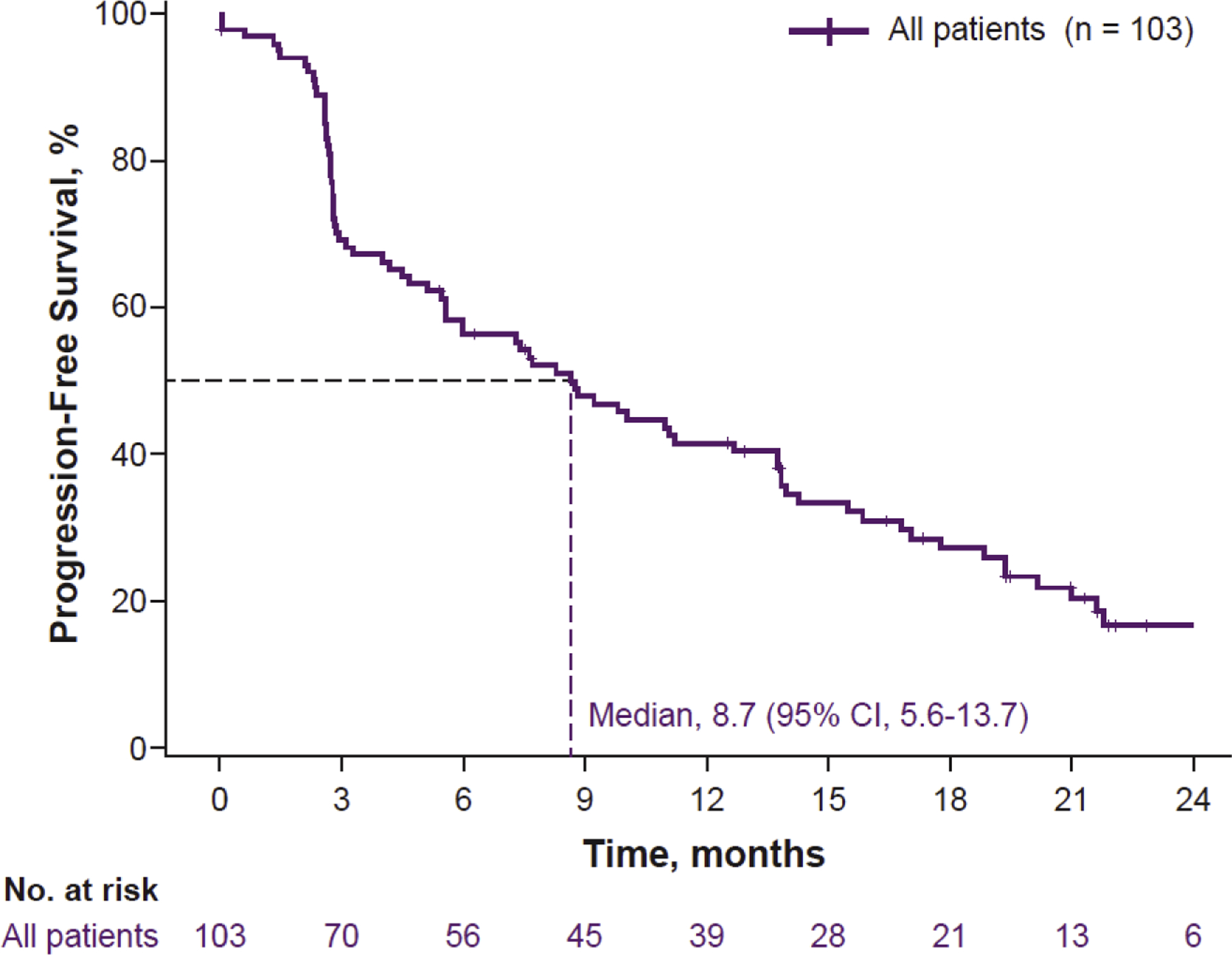
Kaplan-Meier estimate of investigator-assessed progression-free survival (PFS) outcomes in all patients who advanced to second-line treatment. CI = confidence interval.
3.3. Safety
The median treatment durations during the second-line part were 8.3 mo with atezolizumab and 6.9 mo with bevacizumab. Of 103 patients, 86 (83%) experienced a treatment-related AE of any grade, and 31 (30%) experienced a related grade 3/4 event, with no grade 5 AEs (Table 3). Of 103patients, 12 (12%) had an AE that led to withdrawal of any treatment component. Discontinuation of both atezolizumab and bevacizumab due to AEs occurred in 6% (six of 103) of patients (Table 3). The most common treatment-related grade 3/4 AEs were proteinuria and hypertension, and no unexpected AEs were identified (Table 3). The most common AEs of special interest were rash, pruritus, and hypothyroidism, and all were of grade 1/2, with none leading to discontinuation (Supplementary Table 3).
Table 3–
Safety summary in all patients who advanced to second-line treatment
| Adverse event, no. (%) | All second line (n = 103) | |
|---|---|---|
| Patients with ≥1 AEs | ||
| Treatment-related AEs | 86 (83) | |
| Treatment-related grade 3/4 AEs | 31 (30) | |
| Treatment-related grade 5 AEs | 0 | |
| Treatment-related AEs leading to withdrawal | 9 (9) | |
| AE leading to treatment discontinuation | 12 (12) | |
| AE leading to discontinuation of atezolizumab | 1 (1) | |
| AE leading to discontinuation of bevacizumab | 5 (5) | |
| AE leading to discontinuation of atezolizumab + bevacizumab | 6 (6) | |
|
Any grade |
Grade 3/4 |
|
| Treatment-related AEs occurring in ≥10% of patients | ||
| Fatigue | 36 (35) | 2 (2) |
| Proteinuria | 33 (32) | 13 (13) |
| Hypertension | 21 (20) | 10 (10) |
| Diarrhea | 20 (19) | 1 (1) |
| Nausea | 15 (15) | 0 |
| Arthralgia | 14 (14) | 0 |
| Epistaxis | 13 (13) | 0 |
| Hypothyroidism | 12 (12) | 0 |
AE = adverse event; IQR = interquartile range; 1L = first line. Clinical cutoff date: April 19, 2017.
Median treatment duration: atezolizumab + bevacizumab 1L (atezolizumab, 11.8 mo; bevacizumab, 10.4 mo); all second line (atezolizumab, 8.3 [IQR, 2.3–17.5] mo; bevacizumab, 6.9 [IQR, 2.1–15.4] mo).
3.4. Exploratory biomarker studies
Thirty patients had a tumor sample taken at progression before second-line treatment, and 29 patients had paired baseline and at-progression tumor samples with evaluable IHC status. Of these, 38% were PD-L1 positive at progression compared with 59% in baseline samples (Supplementary Fig. 2A). Two of three responders to atezolizumab + bevacizumab after first-line sunitinib showed an increase in PD-L1 expression between baseline and progression, and no clear pattern of modulation or association with response was seen after advancing from atezolizumab monotherapy (Supplementary Fig. 3A and 3B). Based on PD-L1 status at progression, the ORR of atezolizumab + bevacizumab was 42% of 12 patients with PD-L1–positive tumors and 22% of 18 patients with PD-L1–negative tumors (Supplementary Table 2). The median PFS was 10.9 mo (95% CI, 3.3–19.4 mo) in patients with PD-L1–positive tumors and 5.8 mo (95% CI, 2.8 mo–not estimable) in those with PD-L1–negative tumors (Supplementary Fig. 3C).
Exploratory gene expression evaluation focused on associating Teff and Myeloid gene expression in baseline tumors with clinical outcome in second-line treatment. Objective responses to second-line atezolizumab + bevacizumab were observed in patients with both TeffHigh (ORR, 30%; 12/40) and TeffLow (ORR, 25%; 12/48) tumors, and responses were observed irrespective of prior treatment. Complete response, PR, or SD with atezolizumab + bevacizumab was also observed after first-line atezolizumab monotherapy in TeffHigh MyeloidHigh tumors, although the number of patients in this subgroup was small (n = 9; Supplementary Fig. 2B). This observation supported previous findings that, in patients with this gene signature, first-line atezolizumab + bevacizumab had improved outcomes compared with atezolizumab monotherapy [7].
4. Discussion
To our knowledge, IMmotion150 is the first prospective sequential study to evaluate the efficacy and safety of an anti-VEGF + ICI combination after progression on single-agent ICI or VEGF-targeted therapy. As patients underwent new baseline tumor measurements prior to advancing from first-line therapy, a full assessment of PFS and response to second-line atezolizumab + bevacizumab was possible. Response rates were 27% overall and were similar after either atezolizumab or sunitinib. While these data suggest that patients who progress on ICI or VEGF-targeted therapy may have developed resistance that can prevent responses even when therapies are combined, these also show that the combination was active in some patients regardless of prior treatment.
Among the patients in IMmotion150 who advanced to second-line combination therapy, the response rate to first-line atezolizumab monotherapy was 9% and the median PFS was 4.8 mo [7]. Therefore, the response rate of 25% with atezolizumab + bevacizumab in patients who previously experienced progression on atezolizumab monotherapy along with the median PFS of 11.1 mo is encouraging. Interestingly, responses also occurred in nonresponders to first-line therapy, with a response rate of 23% in the nonresponders to first-line atezolizumab. These results, along with the durability of the responses observed in patients who advanced to the combination treatment, may suggest that patients progressing early on ICI monotherapy may benefit from the addition of VEGF-targeted therapy. They also address the need for data on second-line options [12] for patients with mRCC who have progressed on recently approved first-line ICI-based regimens.
AEs were consistent with the safety profiles of the individual agents and also with the safety profile observed for the combination in the front-line setting [7,11,13]. The atezolizumab + bevacizumab safety profile compared favorably with that of sunitinib in the first-line setting and was manageable.
PD-L1 expression in tissue collected at baseline correlated with improved outcomes for atezolizumab + bevacizumab versus sunitinib in previously untreated mRCC patients [11], but was not associated with a relevant enhancement of treatment benefit in the second-line part. This finding is consistent with published data, where PD-L1 expression was prognostic but not predictive for nivolumab treatment in previously treated mRCC patients [14,15].
Exploratory gene expression analyses in baseline tumors showed that, while both first-line atezolizumab and atezolizumab + bevacizumab led to similar PFS in TeffHigh MyeloidLow patients, atezolizumab + bevacizumab improved PFS versus atezolizumab monotherapy in TeffHigh MyeloidHigh patients [7]. Corresponding exploratory evaluation in patients who received second-line treatment showed that clinical benefit with atezolizumab + bevacizumab was observed in all Teff and Myeloid gene signature expression subgroups. Notably, patients with TeffHigh MyeloidHigh tumors also showed improved clinical outcomes to atezolizumab + bevacizumab after advancing from atezolizumab monotherapy. This result appears to support previous findings that the addition of bevacizumab to atezolizumab may overcome resistance to single-agent atezolizumab treatment in tumors with high Myeloid signatures [7].
Sequential collection of tissue in mRCC is challenging, and prospective data from interventional trials are limited. In this study, PD-L1 expression was lower in several progression biopsies than in baseline tissue, including in patients who achieved a best response of PR or SD before progression on atezolizumab. Both ICIs and VEGF-targeted therapies have immunomodulatory effects on the tumor microenvironment [16,17], and dynamic changes to biomarkers, including PD-L1, are expected. The decrease in PD-L1 expression in progressing tumors may provide insight into the mechanism of resistance to ICI monotherapy and should be a subject of future research.
Study limitations include the small sample size, especially in the subgroup and biomarker analyses, and the nonrandomized design of the second-line cohorts. Patients in the second-line cohort had shorter PFS than the ITT cohort (atezolizumab: 4.8 vs 5.5 mo; sunitinib: 5.7 vs 7.8 mo), indicating that patients enrolled in the second-line cohort were more likely to be nonresponders to and early progressors on first-line treatment, leading to a potential selection bias. The second-line part did not include patients who had not yet progressed on first-line therapy, those who chose not to advance to the atezolizumab + bevacizumab combination at progression, and those in EU countries who were not allowed due to regulatory restrictions. Results should be interpreted in this context and warrant confirmation in future studies.
5. Conclusions
Data on RCC treatment sequencing are needed to inform clinical practice [12,18]. IMmotion150 showed that atezolizumab + bevacizumab combination is well tolerated and efficacious in the first-[7] and second-line settings. Biomarker studies demonstrated that dynamic changes in tumor PD-L1 status can occur following first-line treatment and suggested hypothesis-generating insights into potential mechanisms of resistance to therapy. Further studies are needed to inform sequencing strategies in mRCC.
Supplementary Material
Acknowledgments:
The authors would like to acknowledge Jiaheng Qiu and Cindy (Qian) Zhu at Genentech, Inc., for contributions to data analyses. Patients treated at Memorial Sloan Kettering Cancer Center were supported in part by Memorial Sloan Kettering Cancer Center Support Grant/Core Grant (P30 CA008748). Medical writing assistance for this manuscript was provided by Christopher Lum, PhD, of Health Interactions, Inc., and funded by F. Hoffmann-La Roche, Ltd.
Funding/Support and role of the sponsor:
This study was sponsored by F. Hoffmann-La Roche Ltd. Authors were funded by National Cancer Institute grants P50 CA101942 to Drs. David F. McDermott and Toni K. Choueiri, P30 CA008748 to Dr. Robert J. Motzer, and P30 CA14599 to Dr. Walter M. Stadler. F. Hoffmann-La Roche Ltd. was involved in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial disclosures: Thomas Powles certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: All authors report funding of editorial support from F. Hoffmann-La Roche, Ltd. Thomas Powles has received research funding from AstraZeneca and Roche; and honoraria from AstraZeneca, Roche, Bristol-Myers Squibb, Pfizer, Novartis, Exelixis, and Merck Sharp & Dohme. Michael B. Atkins has received honoraria or fees for serving on advisory boards for Bristol-Meyers Squibb (BMS), Merck, Novartis, Arrowhead, Pfizer, Galactone, Werewolf, Fathom, Pneuma, and Leads; has received consulting fees from BMS, Merck, Novartis, Pfizer, Genentech/Roche, Exelixis, Eisai, Aveo, Array, AstraZeneca, Idera, Aduro, ImmunoCore, Boehringer Ingelheim, Iovance, Newlink Pharma, Surface, Alexion, Acceleron, Cota, and Amgen; has received institutional support from BMS, Merck, Pfizer, and Genentech; and owns stock options in Werewolf and Pyxis Oncology. Bernard Escudier has received grants from Aveo, BMS, and Novartis; and personal fees from BMS, Roche, Pfizer, Oncorena, Aveo, and Ipsen. Robert J. Motzer has received honoraria for consulting roles from Bristol-Myers Squibb, Roche-Genentech, Pfizer, Novartis, Exelixis, Eisai, Incyte, Eli Lilly, and Merck; and institutional support from Bristol-Myers Squibb, Roche/Genentech, Pfizer, Novartis, Exelixis, and Eisai. Brian I. Rini has received grants and honoraria from Roche/Genentech and Pfizer; has received grants to his institution and honoraria for consulting roles from Merck, Peloton, Aveo, BMS; has received grants to his institution from AstraZeneca; has received honoraria for consulting roles from Novartis, Synthorx, Compugen, Corvus, and Exelixis; and owns stock in PTC therapeutics. Richard W. Joseph has received honoraria for serving on advisory boards for BMS, Array, and Exelixis. Sumanta K. Pal has received consulting fees from Roche/Genentech, Aveo, Eisai, Pfizer, Novartis, Exelixis, Ipsen, BMS, and Astellas. Mario Sznol has received consulting fees from Genentech/Roche, BMS, AstraZeneca/MedImmune, Pfizer, Novartis, Kyowa Kirin, Seattle Genetics, Nektar, Pierre Fabre, Lilly, Merck US, Theravance, Biodesix, Vaccinex, Janssen/Johnson & Johnson, Modulate Therapies, Baxalta-Shire, Incyte, NewLink Genetics, Iovance, AgonOx, Arbutus, Celldex, Inovio, Gritsone, Molecular Partners, Innate Pharma, AbbVie, Immunocore, Genmab, Almac, Hinge, Allakos, Anaeropharma, Array, GI Innovation, Genocea, and Chugai-Roche; has received fees for serving on advisory boards for Symphogen, Adaptimmune, Omniox, Pieris, and Torque, and formerly for Lycera; and owns stock options in Torque, Amphivena, Adaptive Biotechnologies, Intensity, and Actym. John Hainsworth has received institutional support from Roche/Genentech for participation in a clinical trial. Walter M. Stadler has received consulting fees from AstraZeneca, Bayer, BMS, Caremark/CVS, Clovis, Eisai, Genentech, Merck, Pfizer, and Sotio; has received institutional support from AbbVie, AstraZeneca, Astellas/Medivation, Bayer, BMS, Boehringer Ingelheim, Calithera, Clovis, Eisai, Exelixis, Genentech/Roche, Johnson & Johnson/Janssen, Merck, Novartis, Pfizer, Seattle Genetics, Tesaro, and X4 Pharmaceuticals; has served on the speaker bureau of the following CME providers (sponsorship unknown): Applied Clinical Education, Dava Oncology, Global Academy for Medical Education, OncLive, PeerView, Vindico; and serves as an editor for Cancer and UpToDate. Thomas E. Hutson has received honoraria or fees for serving on advisory boards, has received honoraria or fees for consulting roles, has received research support, and served on the speaker bureau for Pfizer, BMS, Janssen, Astellas, EMD Serono, Aveo, Exelixis, Merck, and Novartis. Alain Ravaud has received fees and nonfinancial support from Pfizer, AstraZeneca, BMS, Ipsen, Roche, and Novartis; and institutional support from Pfizer. Sergio Bracarda has received honoraria and travel support for advisory roles from Novartis, Astellas, Janssen, Pfizer, BMS, Roche, and Ipsen; honoraria from Merck Sharp & Dohme, and travel support from Exelixis. Cristina Suárez has received grants from Roche for conduct of the study; advisory board, and speaking and travel fees from BMS and Pfizer; advisory board and speaking fees from Ipsen and Astellas; advisory board fees from Sanofi, Bayer, and Merck Sharp & Dohme; and travel expenses from Roche. Toni K. Choueiri has received institutional support, grants, fees for consulting and advisory boards, and travel and accommodations from Pfizer, Exelixis AstraZeneca, Bayer, BMS, Cerulean, Eisai, Foundation Medicine, Genentech/Roche, GlaxoSmithKline, Merck, Novartis, Peloton, Prometheus Labs, Corvus, and Ipsen; has received institutional and research support from Tracon; has received consulting and advisory board fees and travel and accommodations from Allegiant, UpToDate, NCCN, Analysis Group, Michael J. Hennessy Associates, Inc, OncLive, PER, L-path, Kidney Cancer Journal, Clinical Care Options, Platform Q, Navinata Healthcare, Harborside Press, American Society of Medical Oncology, NEJM, and Lancet Oncology; has received research and institutional support from Calithera and Takeda; and holds patents related to two pending and issued patents on biomarkers predictive of immune checkpoint response. James Reeves has received institutional support from the Sarah Cannon Research Institute, Eli Lilly, Tesaro, TG Therapeutics, Genentech, Celgene, Merck, BMS, Boston Biomedical Inc, AstraZeneca, Novocure, Calithera Biosciences, Novartis, Guardant Health, Acerta Pharma, Rhizen Pharmaceuticals, Takeda Pharmaceuticals, Onconova Therapeutics, Sanofi, and CTI Biopharma; and received speaking fees and travel support from Eisai and Janssen. Allen Cohn has received speaking fees from BMS and Ipsen. Beiying Ding and Kenji Hashimoto are employees and stockholders of Roche. Mahrukh Huseni and Ning Leng are employees and hold stock options in Roche/Genentech. Christina Schiff was previously employed and previously owned stock options in Roche/Genentech. David F. McDermott has received consulting fees from BMS, Pfizer, Merck, Novartis, Array BioPharma, Eli Lilly, EMD Serono, Jounce Therapeutics, Peloton, and Alkermes; and research grants from Prometheus Laboratories and BMS.
Data sharing:
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available at https://vivli.org/members/ourmembers/. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
References
- [1].National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology. Kidney cancer. V2.2019 2018https://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf [DOI] [PubMed] [Google Scholar]
- [2].Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy--inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res 2012;18:6580–7. [DOI] [PubMed] [Google Scholar]
- [3].Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res 2013;73:2943–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wallin JJ, Bendell JC, Funke R, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun 2016;7:12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].McDermott DF, Sosman JA, Sznol M, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase la study. J Clin Oncol 2016;34:833–42. [DOI] [PubMed] [Google Scholar]
- [7].McDermott DF, Huseni MA, Atkins MB, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018;24:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee CH, Hötker AM, Voss MH, et al. Bevacizumab monotherapy as salvage therapy for advanced clear cell renal cell carcinoma pretreated with targeted drugs. Clin Genitourin Cancer 2016;14:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hainsworth JD, Shipley DL, Reeves J Jr, Arrowsmith ER, Barnes EK, Waterhouse DM. High-dose bevacizumab in the treatment of patients with advanced clear cell renal carcinoma: a phase II trial of the Sarah Cannon Oncology Research Consortium. Clin Genitourin Cancer 2013;11:283–9.e1. [DOI] [PubMed] [Google Scholar]
- [10].Turnbull JD, Cobert J, Jaffe T, Harrison MR, George DJ, Armstrong AJ. Activity of single-agent bevacizumab in patients with metastatic renal cell carcinoma previously treated with vascular endothelial growth factor tyrosine kinase inhibitors. Clin Genitourin Cancer 2013;11:45–50. [DOI] [PubMed] [Google Scholar]
- [11].Motzer RJ, Powles T, Atkins MB, et al. IMmotion151: a randomized phase III study of atezolizumab plus bevacizumab vs sunitinib in untreated metastatic renal cell carcinoma (mRCC). J Clin Oncol 2018;36(suppl 6S):578. [Google Scholar]
- [12].Rini BI, Battle D, Figlin RA, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of advanced renal cell carcinoma (RCC). J Immunother Cancer 2019;7:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Escudier B, Motzer R, Rini B, et al. Patient-reported outcomes in IMmotion151: atezolizumab plus bevacizumab vs sunitinib in treatment-naive metastatic renal cell carcinoma. J Clin Oncol 2018;36(15 suppl):4511. [Google Scholar]
- [14].Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol 2015;33:1430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol 2018;52:117–24. [DOI] [PubMed] [Google Scholar]
- [17].Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- [18].Maia MC, Yang ES, Agarwal N, Pal SK. A step towards predicting checkpoint inhibitor response in kidney cancer. Lancet Oncol 2017;18:982–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.