

Abstract
The main protease (Mpro) plays a crucial role in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication and is highly conserved, rendering it one of the most attractive therapeutic targets for SARS-CoV-2 inhibition. Currently, although two drug candidates targeting SARS-CoV-2 Mpro designed by Pfizer are under clinical trials, no SARS-CoV-2 medication is approved due to the long period of drug development. Here, we collect a comprehensive list of 817 available SARS-CoV-2 and SARS-CoV Mpro inhibitors from the literature or databases and analyze their molecular mechanisms of action. The structure–activity relationships (SARs) among each series of inhibitors are discussed. Additionally, we broadly examine available antiviral activity, ADMET (absorption, distribution, metabolism, excretion, and toxicity), and animal tests of these inhibitors. We comment on their druggability or drawbacks that prevent them from becoming drugs. This Perspective sheds light on the future development of Mpro inhibitors for SARS-CoV-2 and future coronavirus diseases.
Graphical Abstract
1. INTRODUCTION
Developing effective drugs or therapies against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an urgent task for scientific and pharmaceutical communities. SARS-CoV-2 has an unprecedented high infection and prevalence rate, as well as a long incubation period.1 Up to October 5, 2021, 236 million SARS-CoV-2 cases had already brought 4.8 million deaths worldwide. This reminds us that although 21 distinct SARS-CoV-2 vaccines have already been approved for full or emergency use globally,2 the battle against SARS-CoV-2 is still far from the end because of various vaccine escape mutations on the spike protein.3 In comparison, mutations on enzyme active sites are rare.4 Thus, to finally control SARS-CoV-2 and return to normalcy, we must develop effective medications against the virus.
However, there is no approved SARS-CoV-2 medication due to the inherently long period of drug discovery. Pfizer has released two drug candidates PF-07321332 and PF-07304814 inhibiting the SARS-CoV-2 main protease (Mpro), which are now in clinical trials.5,6 Notably, PF-07321332 is now in phase 3 clinical trials and appears to be on its way to becoming the first Mpro drug to treat SARS-CoV-2. Traditionally, developing a new drug can take about 10–15 years,7 from initial design to entering the marketplace. Considering the urgency of the current pandemic, the drug discovery for SARS-CoV-2 must be expedited.
SARS-CoV-2 is a β-coronavirus belonging to the Coronaviridae family. β-Coronaviruses seriously threaten human health. During the first two decades of the 21st century alone, β-coronaviruses have triggered three major outbreaks of deadly pneumonia: SARS-CoV (2002), Middle East respiratory syndrome coronavirus (MERS-CoV) (2012), and SARS-CoV-2 (2019).8 SARS-CoV-2’s infection rate is even higher than SARS-CoV and MERS-CoV.9,10
A great number of drug targets of SARS-CoV-2 have been identified.11 Among them, Mpro, also called 3-chymotrypsin-like protease (3CLpro), is an attractive target for small-molecule inhibitors.12–20
First, Mpro is an indispensable enzyme for viral replication and transcription. Coronaviruses possess the largest known RNA genomes with a length of about 30 kb.21 Their genomes consist of multiple open-reading frames (ORFs). Among them, two overlapping ORFs (ORF1a and ORF1ab) are translated into two large polyproteins, pp1a and pp1ab, via a −1 translation frameshift mechanism. Then, Mpro and papain-like protease (PLpro)22 cleave pp1a and pp1ab into 16 mature nonstructural proteins (NSPs).23 Mpro formed from NSP5 cleaves the two polyproteins at 11 recognition sites and creates NSP4 to NSP10 and NSP12 to NSP16 (NSP11 is the N terminal end of NSP12), while PLpro cleaves the other 3 sites to generate NSP1 to NSP3. Notably, NSP4 to NSP16 cleaved by Mpro contain many essential viral proteins, especially the RNA-dependent RNA polymerase (NSP12), RNA binding proteins (NSP9), helicase (NSP13), exoribonuclease (NSP14), and methyltransferase (NSP16).24,25 Therefore, effectively blocking Mpro could stop SARS-CoV-2 replication in human bodies and cure the disease.
Second, according to the data from the global initiative on sharing all influenza data (GISAID), SARS-COV-2 Mpro is highly conserved. The mutation rate on its binding domain is lower than 0.001 (see section 3.2). Thus, mutations will not broadly impact the efficacy of SARS-CoV-2 Mpro inhibitors.
Last but not least, the Mpro enzymes of SARS-CoV-2 and SARS-CoV share a very high 2D sequence identity of 96.1% and a very low 3D RMSD of 0.42 Å (see Figure 1). Their binding-site sequence identity is even as high as 100%.26 Therefore, previous SARS-CoV Mpro inhibitors are still effective against SARS-CoV-2 Mpro, providing a useful resource for developing SARS-CoV-2 Mpro drugs. For instance, PF-00835231, a SARS-COV-2 Mpro inhibitor currently in clinical trials, was originally designed by Pfizer during the 2002–2003 outbreak of SARS-CoV to target SARS-CoV Mpro.6
Figure 1.
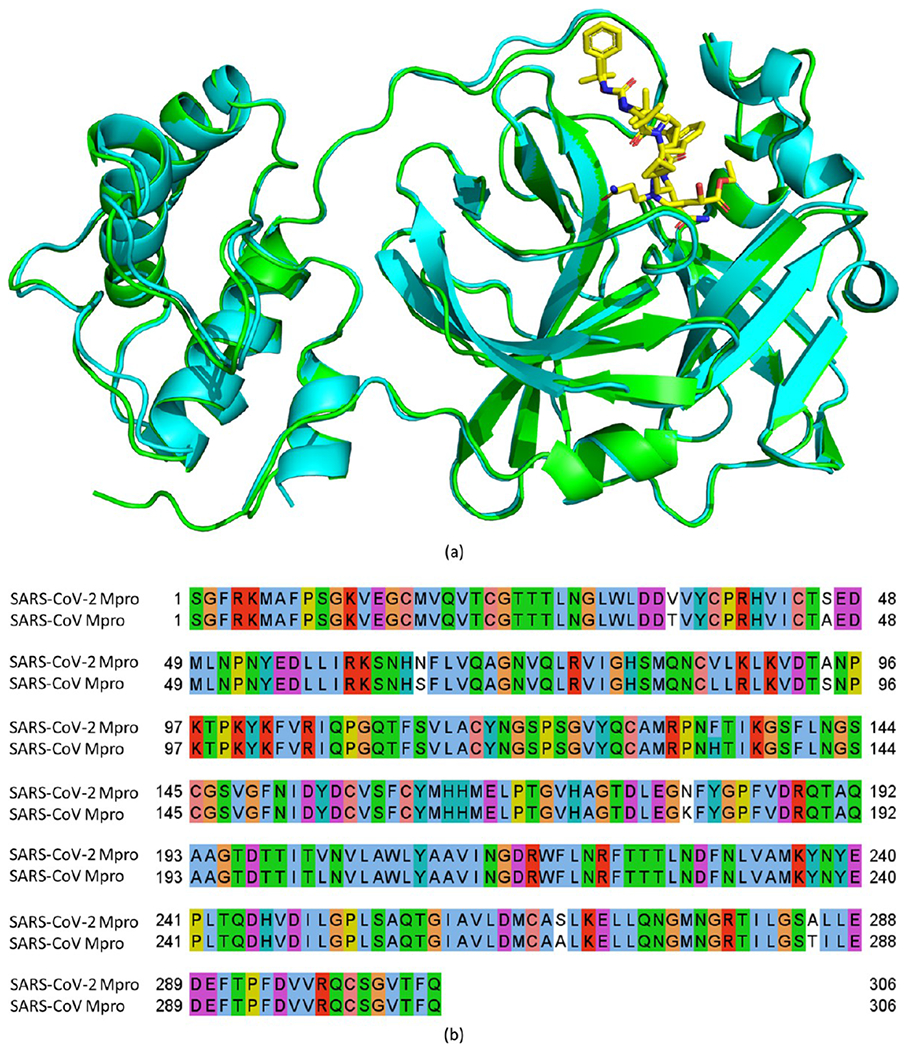
3D conformation alignment (a) and 2D sequence alignment (b) of SARS-CoV-2 and SARS-CoV main proteases (Mpro). The sequences and structures are from the Protein Data Bank (PDB) ID 7C6S (SARS-CoV-2 Mpro, cyan in part a) and 2A5I (SARS-CoV Mpro, green in part a).
The active site of SARS-CoV-2 and SARS-CoV Mpro consists of Cys145 and His41, which form a catalytic dyad. Cys145 is the common nucleophile in the proteolytic process.17,27 Figure 2 depicts the five-step process of Mpro hydrolyzing a substrate. The first step is the deprotonation of the Cys145-thiol, in which the proton of Cys145-thiol is transferred to His41. In the second step, the resulting anionic sulfur nucleophilically attacks the substrate carbonyl carbon to form a C–S bond. Then, in the third step, the peptide substrate is protonated and cleaved, and the product with an amino terminus is released, leaving the His41 in its deprotonated form. Next, in the fourth step, the resulting thioester is hydrolyzed to release a carboxylic acid. In the last step, the free enzyme is formed again.
Figure 2.
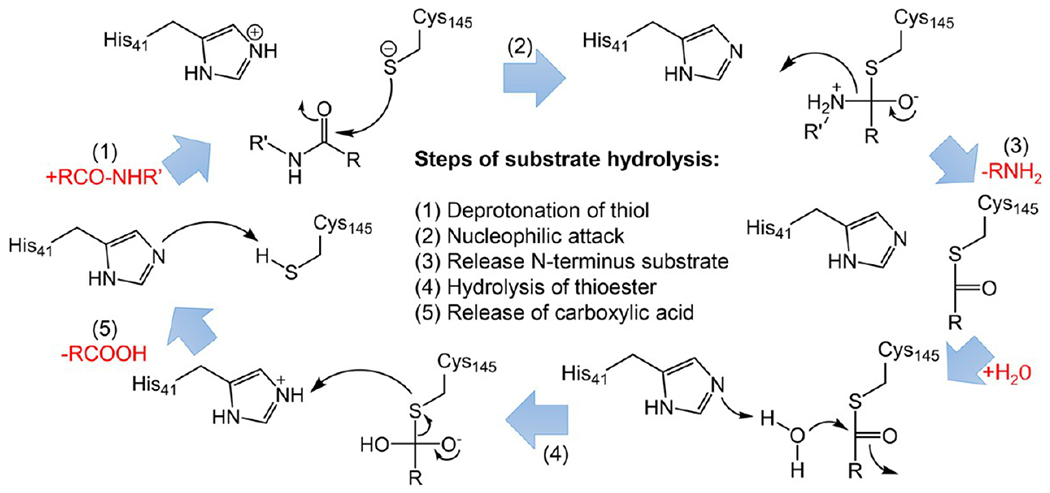
Overall scheme of SARS-CoV/SARS-CoV-2 Mpro catalytic mechanism by Cys145 and His41 at the active site.
The subsite nomenclature is a popular representation of a proteolytic enzyme and its interactions with a peptide substrate or inhibitor (Figure 3). From N-terminus to C-terminus, the amino acids in a substrate are numbered as P3, P2, P1, P1′, P2′, etc. The amide bond between P1 and P1′ represents the scissile bond, where peptide substrates are hydrolyzed.17 The protease active sites are correspondingly numbered as S3, S2, S1 S1′, and S2′. This work adopts this subsite nomenclature.
Figure 3.
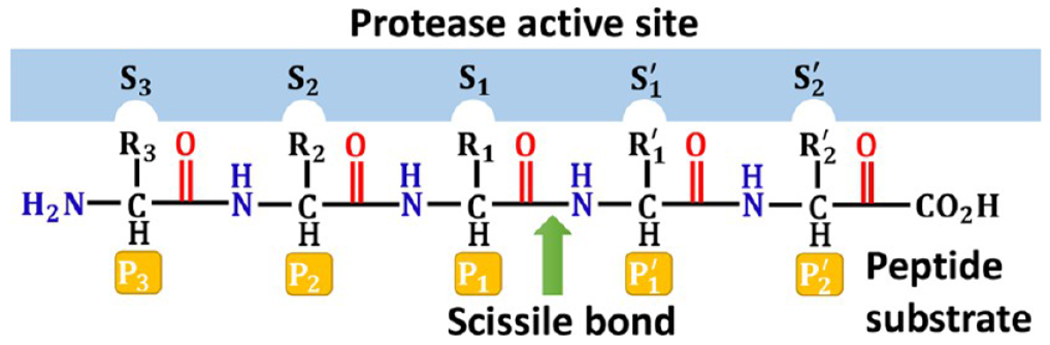
Subsite nomenclature for proteolytic enzymes. Amino acid residues to the left of the polypeptide scissile amide bond are numbered sequentially, beginning with P1 and increasing toward the N-terminus. Amino acid residues to the right of the scissile bond are numbered sequentially, beginning with P1′ and increasing toward the C-terminus. Complementary regions of the protease active site employ the corresponding S numbering.
Notably, for covalent inhibitors of SARS-CoV-2 and SARS-CoV Mpro, the kinetic scheme of covalent inhibition is illustrated as follows:
where E, I, EI, and E–I represent enzyme, inhibitor, enzyme–inhibitor complex, and covalently bonded enzyme and inhibitor, respectively. At first, the inhibitor binds to the protease noncovalently. Then, a nucleophilic attack by Cys145 triggers the protease to form a stable covalent bond with the inhibitor.28,29 Therefore, the rate of the interaction is determined by both the equilibrium-binding constant ki, (designated as k1/k2) and the inactivation rate constant for covalent bond formation k3. For a covalent inhibitor, the reported binding affinity refers to the one from the noncovalent binding in the first step.
Additionally, the existing crystal structures of SARS-CoV-2 Mpro and inhibitor complexes reveal many inhibitor binding sites. However, the major inhibitory site is at the enzyme catalytic center.30
Motivated by the pressing need for effective SARS-CoV-2 medications and the data in machine learning-based drug repositioning and generation for Mpro inhibitors,30–32 we collected 817 available SARS-CoV-2 and SARS-CoV Mpro inhibitors from the literature or databases in this Perspective. The inhibitors with enzyme inhibitory activities in sub-micromolar range are highlighted. We classify these inhibitors into different categories based on their binding mechanisms and illustrate their covalent or noncovalent binding interactions with Mpro. In each category, the structure–activity relationship (SAR) of the inhibitors and the potential path to improve potency are analyzed. Since the antiviral activities of the inhibitors are also critical, we analyze their antiviral potentials and the relationships with enzymatic activities. Moreover, we analyze their ADMET (absorption, distribution, metabolism, excretion, and toxicity), pharmacokinetics (PK), draggability data, and in vivo results, when available. More importantly, we comment on their druggable potential and possible issues preventing them from becoming drugs. Finally, we forecast the Mpro mutation impact on inhibitor efficacy as well. The data collection and perspectives in this work will provide a starting point to screen and discover drug candidates against SARS-CoV-2 Mpro.
2. SARS-COV-2 AND SARS-COV MPRO INHIBITORS WITH INHIBITORY POTENCY IN SUB-MICROMOLAR RANGE
2.1. Peptidomimetic Covalent Inhibitors.
2.1.1. Ketone-Based Covalent Inhibitors.
So far, the SARS-CoV-2 and SARS-CoV Mpro inhibitors with the strongest enzyme inhibitory activities are among the ketone-based covalent inhibitors developed by Hoffman et al. from Pfizer,26 especially the P2-modified hydroxymethylketone (HMK) inhibitors.
2.1.1.1. P2-Modified Hydroxymethylketone (HMK) Inhibitors.
A series of P2-modified HMK inhibitors developed by Hoffman et al.26 show strong inhibition of SARS-CoV-2 and SARS-CoV Mpro (Figure 4). HMKs are reversible cysteine protease inhibitors.
Figure 4.
P2-modified hydroxymethylketone (HMK) covalent inhibitors.
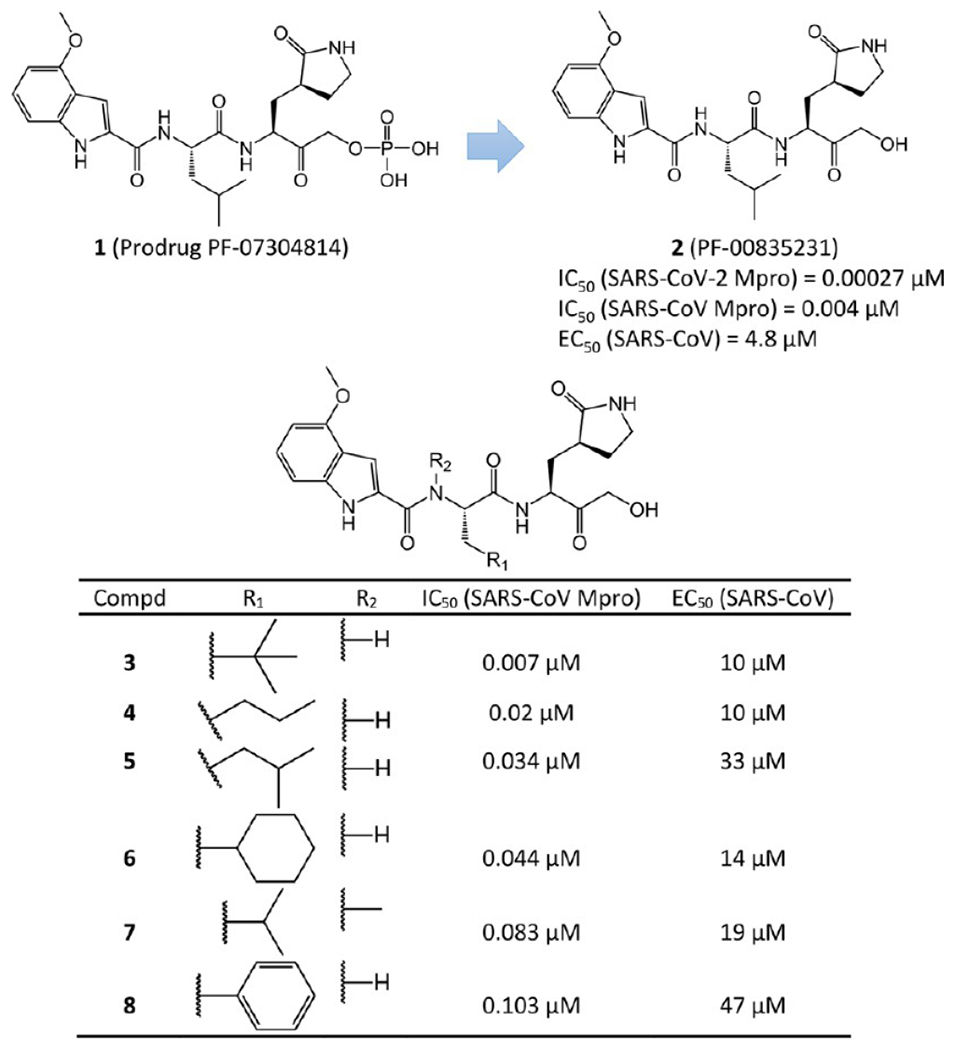
Notably, among them, 2 (PF-00835231) is the most potent SARS-CoV-2 and SARS-CoV Mpro inhibitor in enzymatic assays so far. Actually, PF-00835231 is the active metabolite of its prodrug PF-07304814 (1). PF-07304814 contains a phosphate group to improve the solubility of the compound. After entering into tissues, the phosphate group is cleaved by alkaline phosphatase enzymes and active antiviral PF-00835231 is released. Pfizer chemists originally designed it to target SARS-CoV Mpro during the 2002–2003 outbreak of SARS-CoV. At the end of the 2003 pandemic, its clinical advancement was suspended. Recently, the prodrug PF-07304814 completed a phase 1 clinical trial to treat SARS-CoV-2. PF-00835231 demonstrates efficacy against multiple strains of SARS-CoV-2 as a single agent and even demonstrates additive/synergistic activity in combination with remdesivir. Its drawbacks include its route of administration (intravenously) and its relatively high effective dose.6
PF-00835231 has an IC50 of 0.004 μM against SARS-CoV Mpro. A recent survey reported even higher efficacy against SARS-CoV-2 Mpro with an IC50 of 0.00027 μM.26 However, its antiviral activity toward SARS-CoV is much lower, with an EC50 of 4.8 μM. Other in vitro and in vivo assays indicated that PF-00835231 possesses high metabolic stability in liver microsomes (half-life time = 107 min), acceptable solubility (4.6 mg/mL), and low clearance (20.6 (mL/min)/kg in monkeys).
The crystal structures of 2 bound to the SARS-CoV Mpro and SARS-CoV-2 Mpro were solved at 1.47 and 1.26 Å resolutions (PDB ID 6XHL and 6XHM, respectively).26 As expected, since the ligand-binding domains of SARS-CoV Mpro and SARS-CoV-2 Mpro are highly conserved, their interactions with 2 are also very similar. Figure 5 shows the covalent adduct of 2 with SARS-CoV-2 Mpro. The electrophilic carbonyl C atom of 2 forms a covalent bond with the S atom of the Mpro active-site Cys145, which leads to a tetrahedral hemithioketal adduct (The C–S bond is 1.86 Å). Bridged by a water molecule, hydrogen bonds are formed between the carbinol hydroxyl of 2 and the amide NH group of Gly143, as well as the backbone NH group of Cys145. Another essential hydrogen bond is between the primary alcohol moiety of the compound and catalytic His41. Additionally, the lactam carbonyl of 2 forms a strong hydrogen bond (2.72 Å) with the side chain of His163. The NH and C2-carbonyl of the indole in the compound interact with the backbone carbonyl and NH of Glu166 via β-sheet-like hydrogen bonds. The NH group of its P2 Leu accepts a hydrogen bond (3.01 Å) from the side chain of Gln189. Another NH group of 2 also has a strong hydrogen bond (2.89 Å) with the backbone carbonyl of His164.
Figure 5.
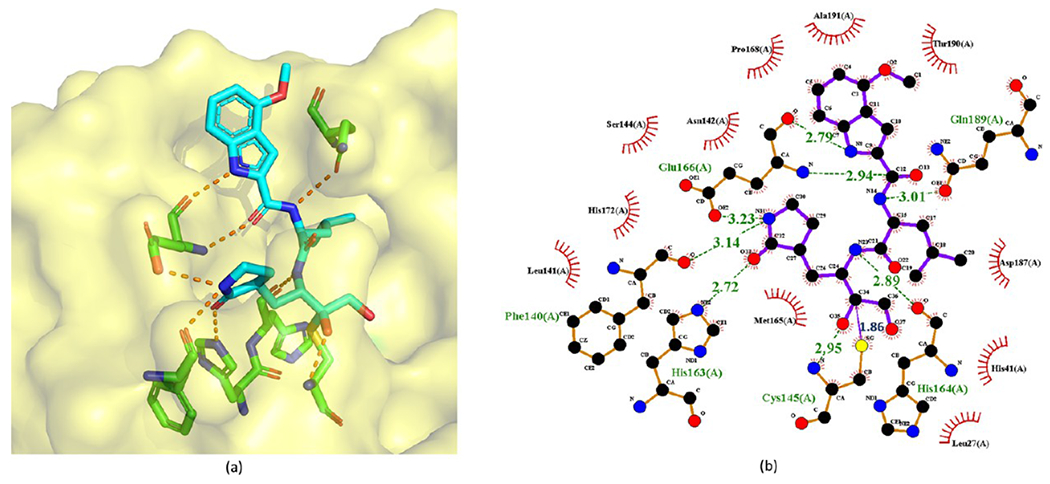
Cocrystal structure of the covalent adduct of 2 bound to SARS-CoV-2 Mpro (PDB ID 6XHM) (a) and its corresponding 2D interaction diagram (b).
Hoffman et al. also analyzed the structure–activity relationship (SAR) of these inhibitors. Notably, as revealed by Figure 4, 7 and 2 only differ by a CH3 group at the P2 site. However, the binding affinity of 7 is largely attenuated (IC50 from 0.004 μM to 0.083 μM). The explanation is, first, in the crystal structure in Figure 5, the NH group at the P2 site of 2 has a hydrogen bond with the side-chain amide of Gln189, but the CH3 substitution in 7 prevents the formation of this hydrogen bond. Second, this CH3 substitution deforms the 4-methoxy indole cap and, thus, perturbs the whole Mpro–ligand hydrogen bond network present in 2. Other potency reduction from 3 to 6 and 8 suggests a preference of smaller groups at the P2 moiety (the R1 group in Figure 4). This is probably because the S2 pocket of Mpro more easily accommodates smaller groups as seen from the crystal structure in Figure 5. Larger groups could lead to spatial contradiction and thus loosen the binding.
Figure 7.
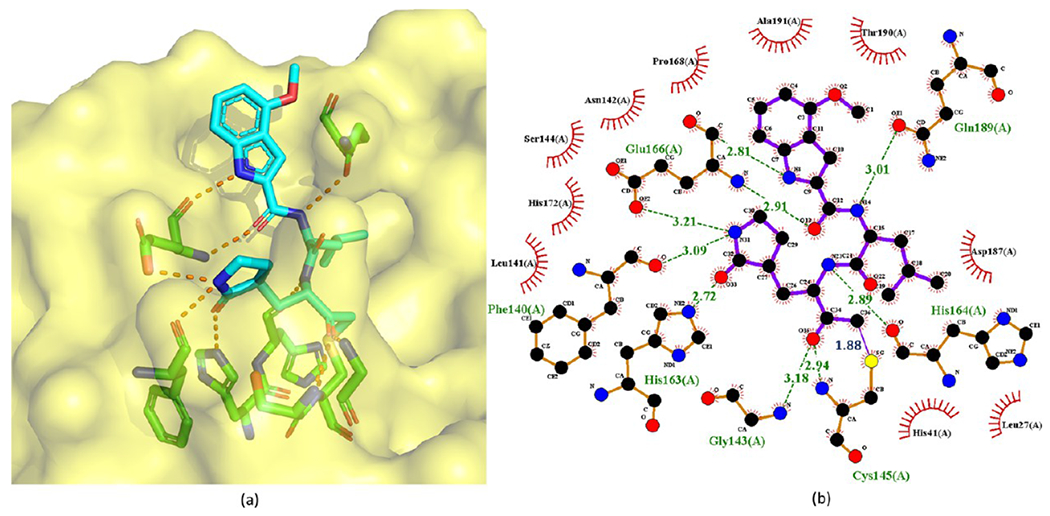
Cocrystal structure of the covalent adduct of 9 bound to SARS-CoV Mpro (PDB ID 6XHN) (a) and its corresponding 2D interaction diagram (b).
The antiviral activities of 2–8 reported by Hoffman et al.26 in terms of EC50 are shown in Figure 4 as well. It turns out that 2 has the lowest IC50 as well as the lowest EC50. These data also indicate that the antiviral EC50/enzyme IC50 ratios of 2–8 are high due to poor cell permeability. These HMK inhibitors exhibit very low permeability and high levels of efflux beyond the sensitivity of the Caco-2 in vitro assay. Currently, efforts to reduce efflux by active transporters, such as P-glycoprotein, are being attempted to decrease high EC50/IC50 ratios. An analysis of the physicochemical properties of 2 suggests that increasing log P, reducing polar surface area, and reducing the number of hydrogen bond donors and acceptors are feasible strategies to improve its cellular permeability and reduce efflux.26
2.1.1.2. Acyloxymethylketone Inhibitors.
Substituting the hydroxyl group of a HMK inhibitor with an acyloxy group results in an acyloxymethylketone inhibitor. Since the nucleophilic attack of a cysteine protease detaches the acyloxy group of an acyloxymethylketone inhibitor, acyloxymethylketone inhibitors are irreversible. As depicted in Figure 6, the acyloxymethylketone inhibitors reported by Hoffman et al.26 exhibit strong enzymatic potency against SARS-CoV Mpro. Especially, 9 possesses the lowest IC50 of 0.017 μM against SARS-CoV. The crystal structure (PDB ID 6XHN) of 9 in complex with SARS-CoV Mpro is depicted in Figure 7.
Figure 6.
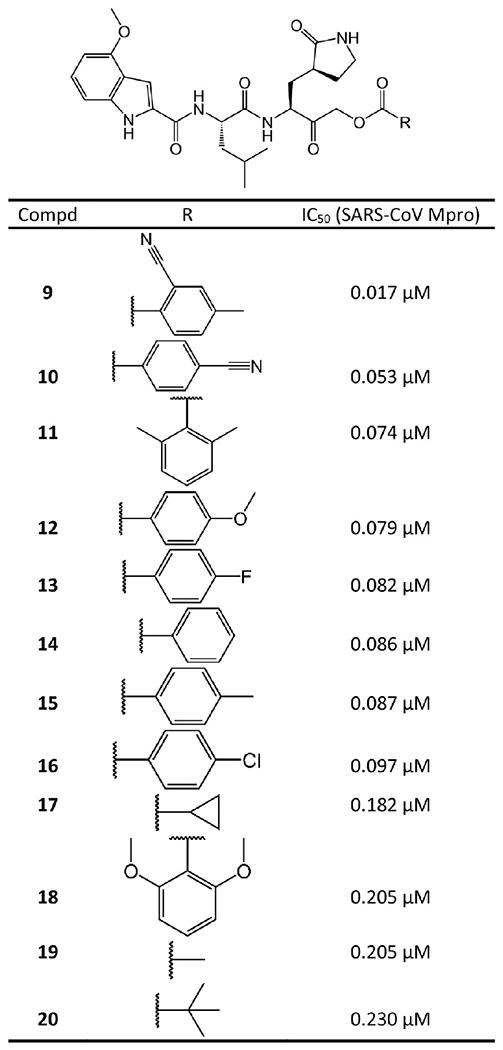
Acyloxymethylketone covalent inhibitors against SARS-CoV Mpro.
In stark contrast to HMK inhibitors, here 9 is bound to the S atom of Mpro Cys145 via an irreversible covalent bond (1.88 Å C–S bond length) at its α-methylene. The inactivation of cysteine proteases by these acyloxymethylketones can proceed via two possible mechanisms. One possibility is the direct displacement of the cyanobenzoate group by the cysteine to form the covalent thioether adduct. The second possibility involves the formation of the hemithioketal, followed by a three-membered sulfonium intermediate that rearranges to form the thioether adduct.33 The cyanobenzoate moiety serves as the leaving group. As shown in Figure 6, increasing electron density of the leaving group decreases enzymatic activity. Other critical interactions include the hydrogen bonds of the ketone carbonyl inside the oxyanion hole with the backbone NH groups of Gly143 and Cys145.
2.1.1.3. P3- and P2-Modified HMK and Alkoxymethylketone Inhibitors.
The P3- and P2-modified HMK and alkoxymethylketone inhibitors also from Hoffman et al.26 are illustrated in Figure 8.
Figure 8.
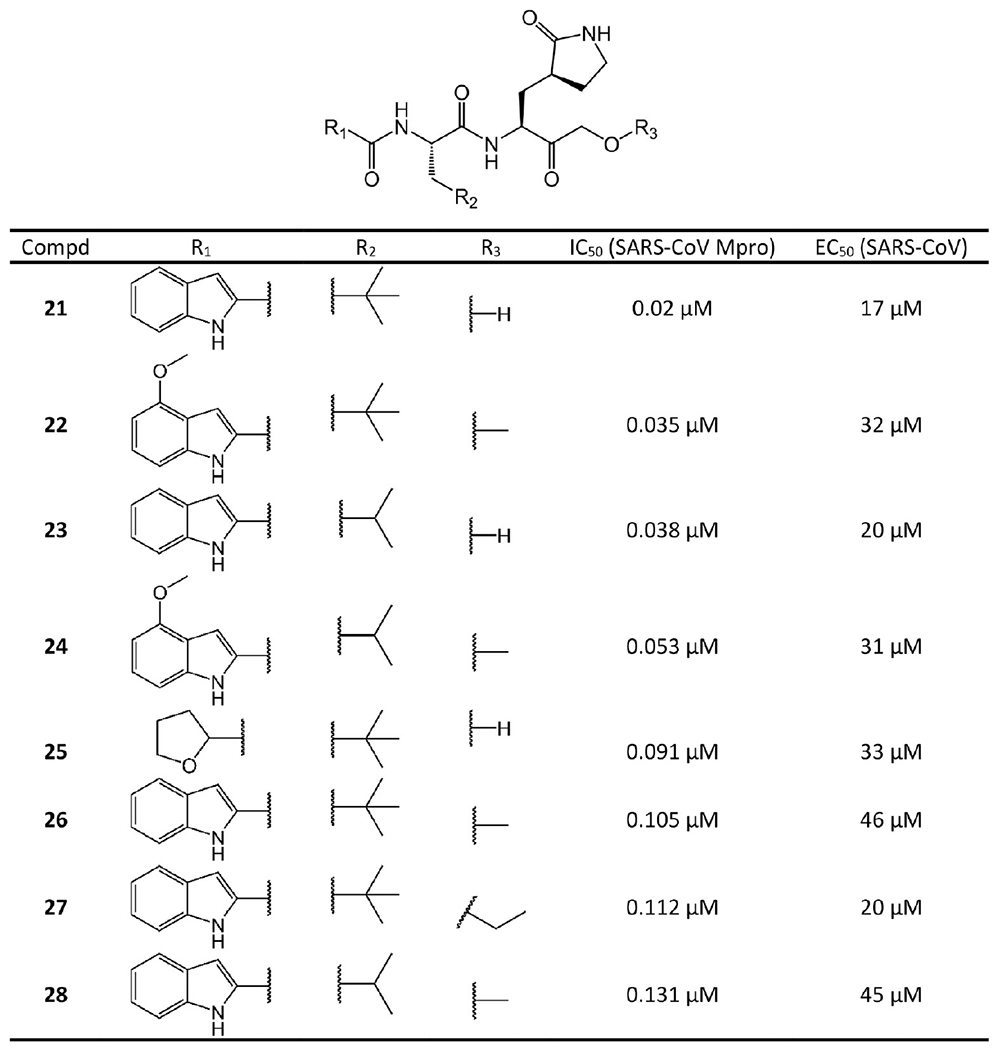
P3- and P2-modified HMK and alkoxymethylketone covalent inhibitors.
Considering the poor leaving group ability of the alkoxy moiety, it is likely that the attack of Cys145 is on the carbonyl C atom as seen with the HMK inhibitors. The comparison between the compounds in Figures 8 and 4 suggests that, first, a noteworthy reduction in enzymatic and antiviral potency can be found for each hydroxymethylketone derivative and its corresponding ether counterpart, such seen with 22 vs 3, 26 vs 21, 24 vs 2, and 28 vs 23. Second, removing the methoxy group from the indole while maintaining the two optimal P2 residues (Leu and β-tert-butyl-Ala) generally leads to slightly weaker potency in both the enzymatic and antiviral assays, such as seen with 21 vs 3, 26 vs 22, 28 vs 24, and 23 vs 2. Overall, the findings reveal that the 4-methoxy group in the indole does not play a very significant role in the P3 cap other than improving the solubility characteristics of the inhibitors.
2.1.1.4. α-Ketoamide Inhibitors.
Zhang et al. designed and synthesized several peptidomimetic α-ketoamides as broad-spectrum inhibitors against β-coronavirus, α-coronavirus, and enterovirus Mpro enzymes.34,35 The ones with both high Mpro enzymatic activity and high antiviral activity against SARS-CoV and SARS-CoV-2 are depicted in Figure 9.
Figure 9.
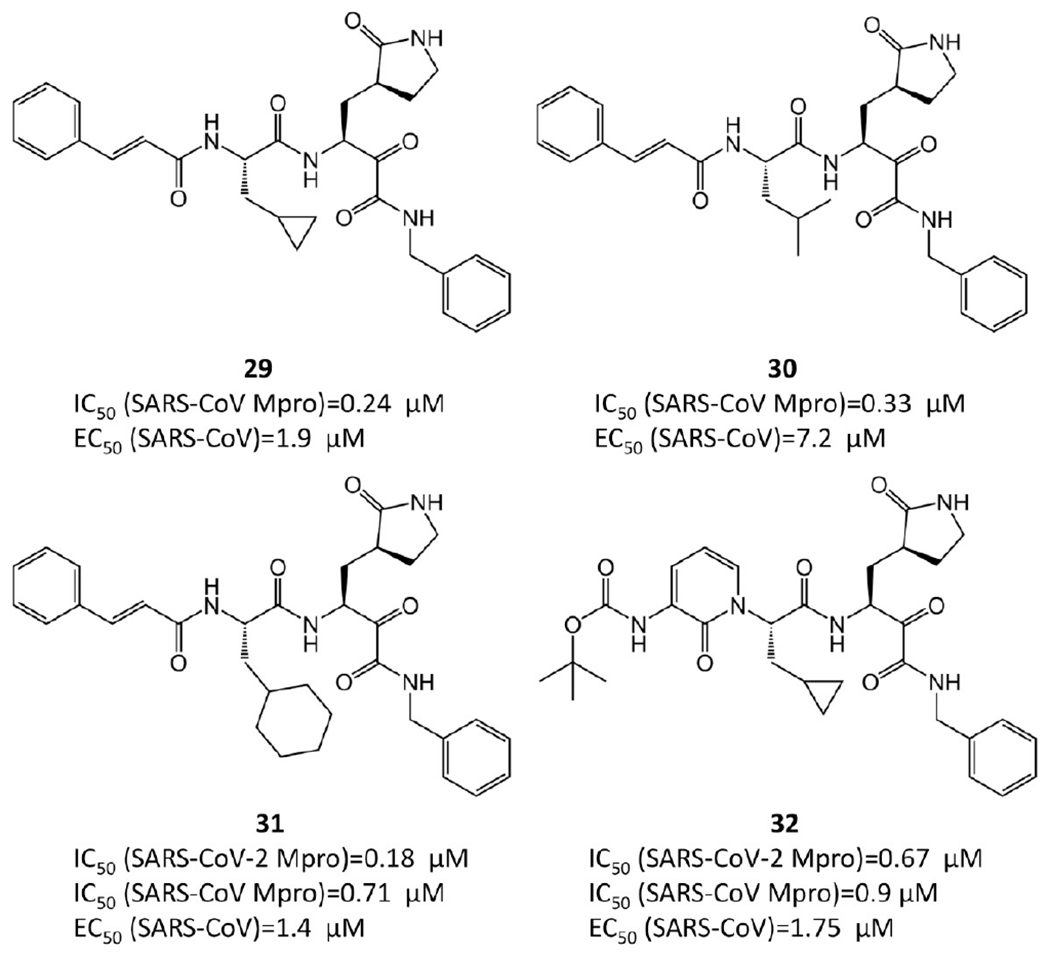
α-Ketoamide covalent inhibitors.
Zhang et al.35 performed the SAR analysis. Comparing 29, 30, and 31, which differ only in terms of the nature of the P2 groups, suggests that the binding affinities depend on the size of the P2 substituent. Unlike the enterovirus Mpro, the S2 pocket of SARS-CoV-2 Mpro displays substantial plasticity and can adapt to the shape of a smaller cyclopropane moiety. The smaller P2 group leads to lower IC50 and consequently higher efficacy. To lengthen the half-life time in plasma and prevent cellular proteases from accessing and cleaving the bond, the P3–P2 amide bond is hidden inside a pyridone ring. In addition, to increase the solubility in plasma and to reduce the binding to plasma proteins, the hydrophobic cinnamoyl moiety is replaced by a Boc-substituted amino-pyridone.
The crystal structures of 29 and 32 bound to SARS-CoV or SARS-CoV-2 Mpro are available with PDB ID 5N5O and 6Y2G, respectively (see Figure 10). These crystal structures reveal that the nucleophilic attack of Mpro Cys145 onto the α-keto group of an α-ketoamide inhibitor results in a hemithioketal. The hydroxyl group of this hemithioketal is stabilized by a hydrogen bond with His41. In addition, the carbonyl O atom of the amide accepts a hydrogen bond from the main-chain amide of Cys145. The amide O atom of 29 also forms a hydrogen bond with the main-chain amide of Gly143. This is a great merit of the α-ketoamides. Their warhead could generate multiple hydrogen-bonding interactions with the catalytic center of the target proteases, which may add an advantage over other warheads such as aldehydes36 or Michael acceptors.37
Figure 10.
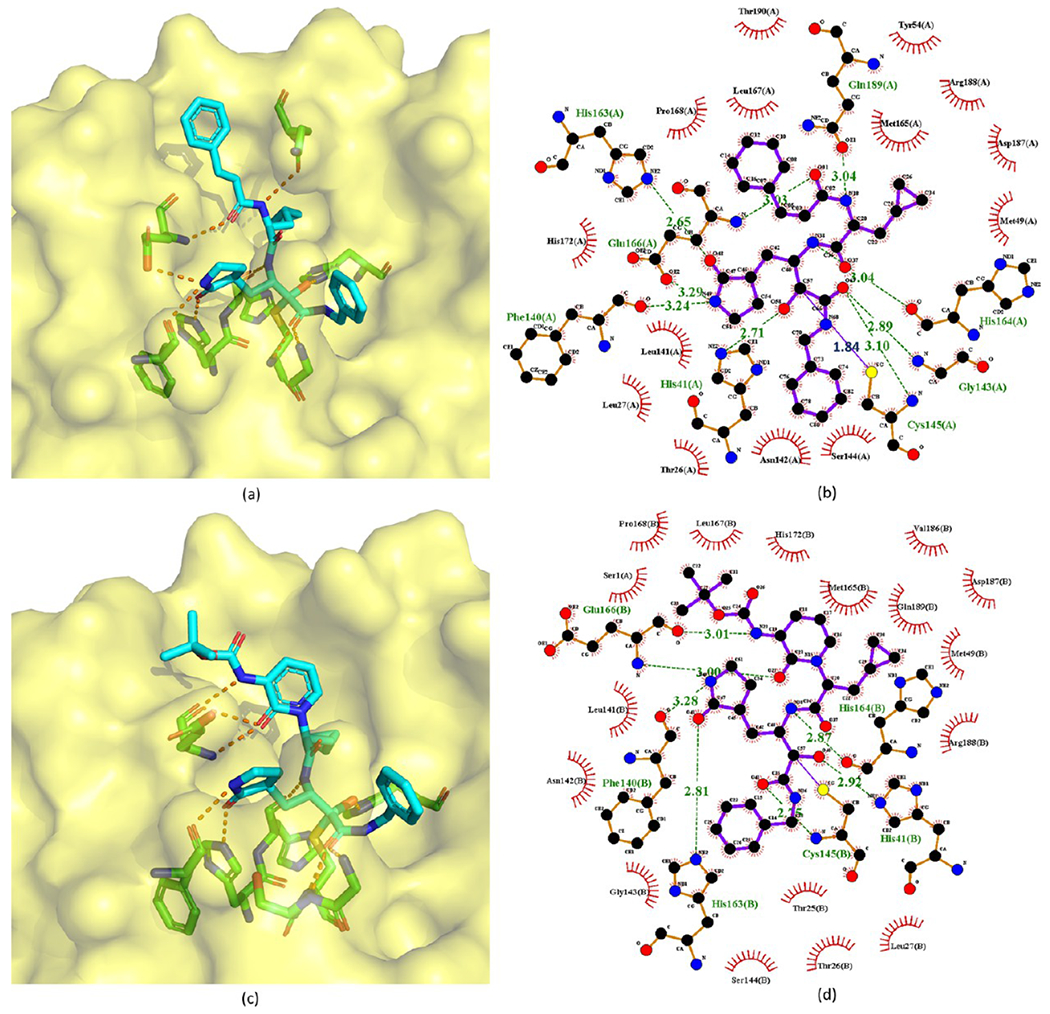
Cocrystal structures of the covalent adducts of 29 (a, b) and 32 (c, d) bound to SARS-CoV or SARS-CoV-2 Mpro (PDB ID 5N5O and 6Y2G).
To determine the ADMET properties of 32, mice were administered 32 subcutaneously at 20 mg/kg. The mean residence time is 2.7 h, and the plasma half-life is 1.8 h. Especially, its lung tissue level is promising. After 4 h, 32 still has a concentration at approximately 13 ng/g in lung tissue, which is quite beneficial since COVID-19 infects lungs. Mice do not show any adverse effects after inhalation, which suggests that 32 could be administrated directly to the lungs.35
2.1.1.5. Benzothiazole-Containing Ketone Inhibitors.
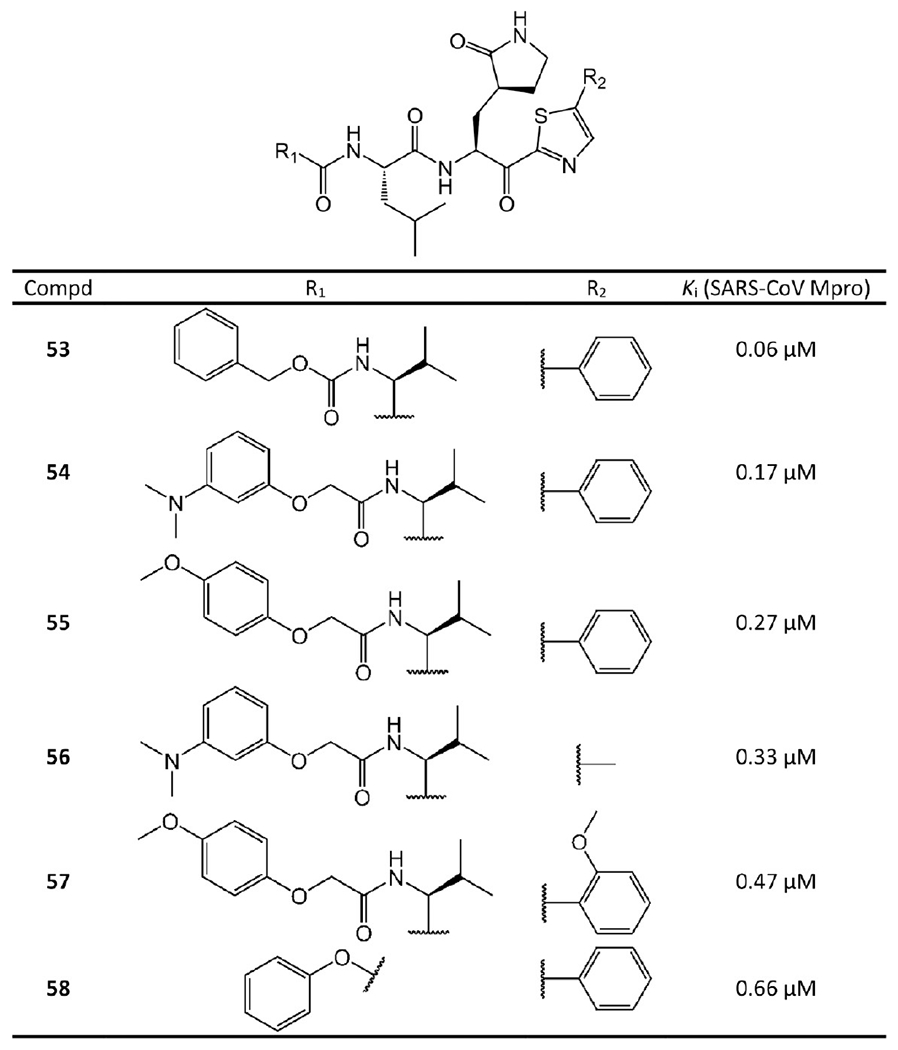
Hayashi et al.38–40 developed tens of benzothiazole-containing ketone inhibitors against SARS-CoV Mpro. Compounds 33–52 with enzyme inhibitory potency in sub-micromolar range are shown in Figures 11 and 12.
Figure 11.
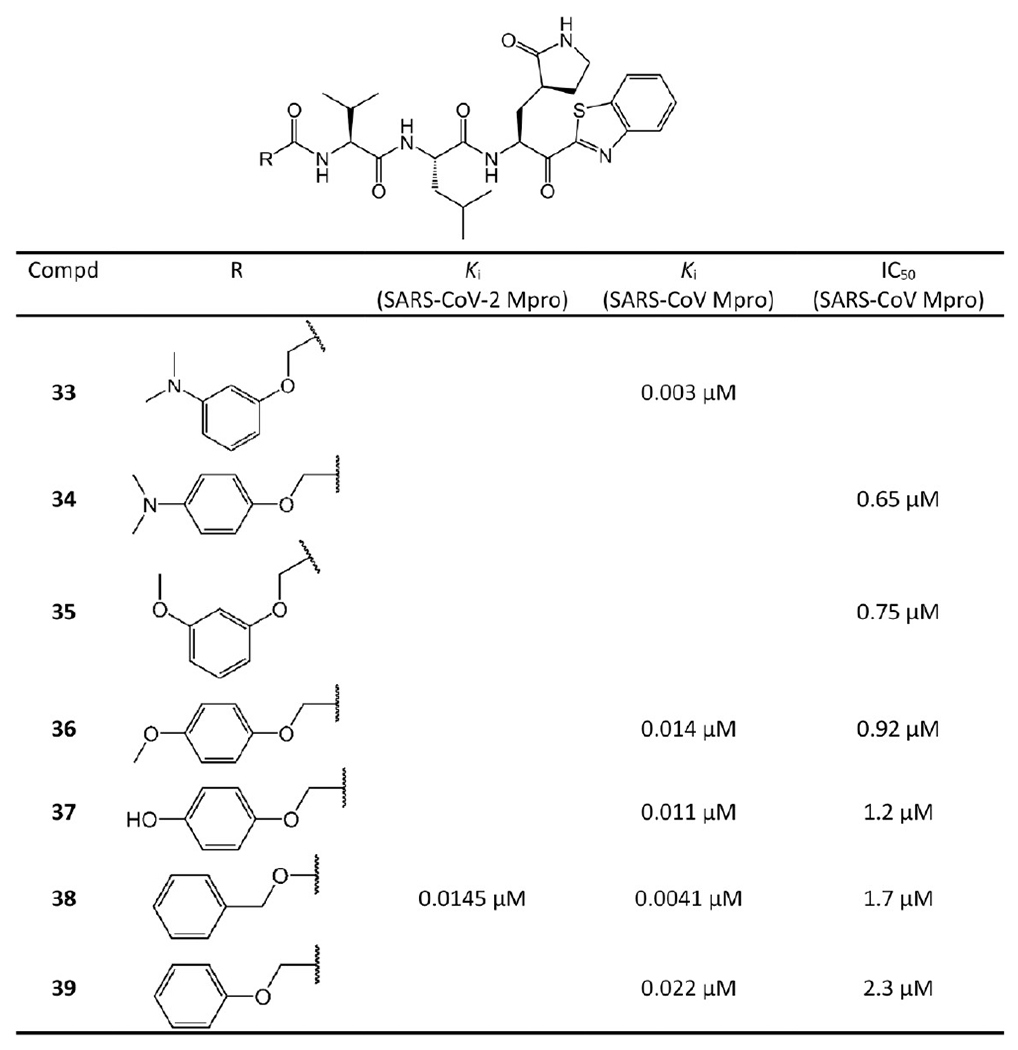
Benzothiazole ketone-containing inhibitors 33–39.
Figure 12.
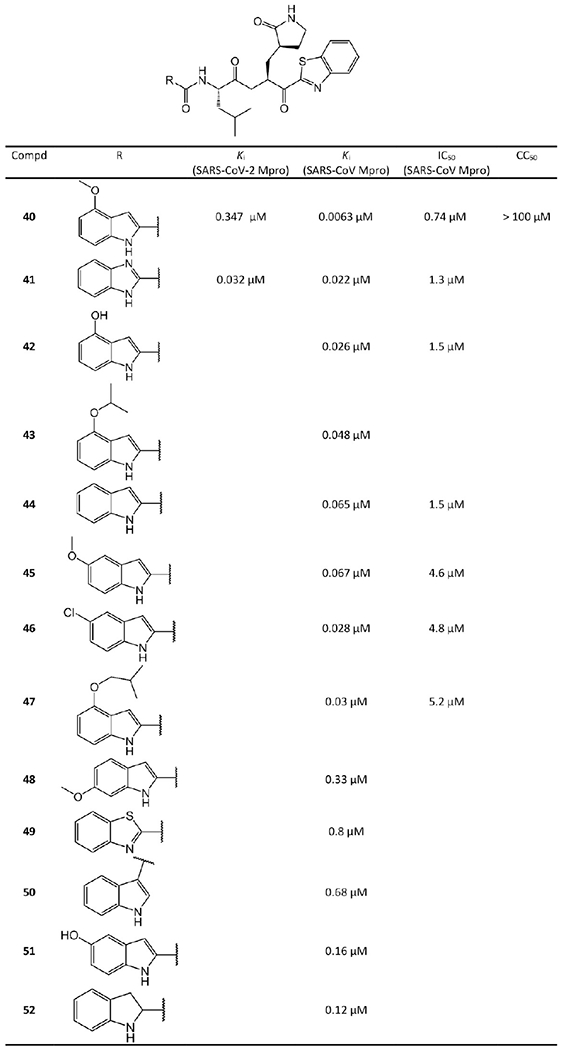
Benzothiazole ketone-containing inhibitors 40–52.
Hayashi et al. explored the SAR of this class of compounds.38 As a starting point for the compounds in Figure 11, 39 exhibits promising inhibition with a Ki value of 0.022 μM, which suggests that the benzothiazole unit is a suitable chemical warhead group for occupying the S1′-site. Thus, 39 was advanced as a lead compound for further development of 33–38. The phenyl group of the P4-moiety of 39 was functionalized with various electron-donating and -withdrawing groups. The inhibitors that contain electron-donating substituents such as methoxy, hydroxyl, or N,N′-dimethylamino at the o-, p- or m-positions were found to be more active than 39. Especially, the m-methoxy and p-N,N′ dimethylamino substituted analogs were found to strongly inhibit SARS-CoV Mpro. Notably, the m-N,N′-dimethylamino derivative 33 has a Ki of 0.003 μM and is the most potent inhibitor of this series. These results confirm that an electron-donating substituent on the phenyl ring of the P4-moiety typically increases the activity of the compounds. In the case of the 4-N,N′-dimethylamino phenoxy acetyl group, Hayashi et al.’s docking studies suggested a different folding conformation allowing a new hydrophobic interaction with Ala191 at the P4 position.
Figure 39.
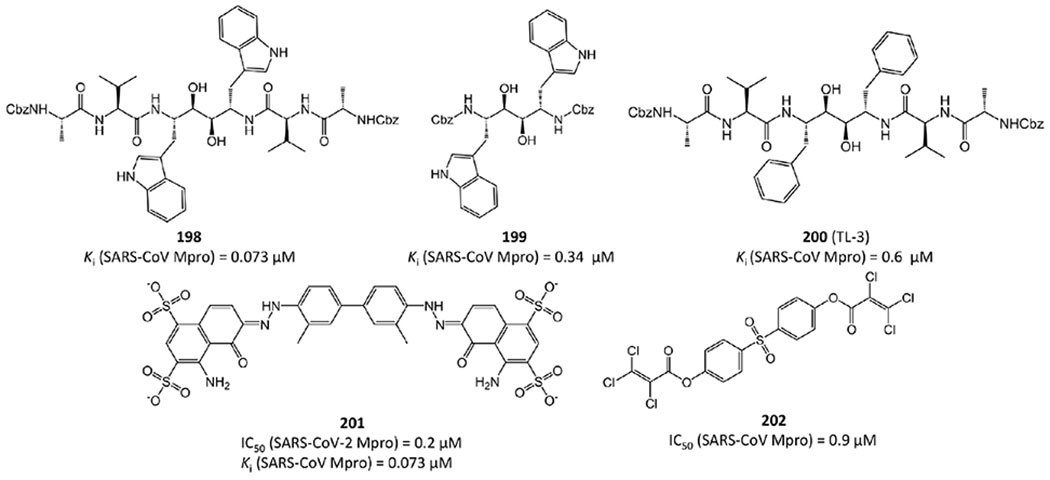
Symmetric peptides and molecules.
In Figure 12, Hayashi et al.39 selected 44 as a lead compound for further optimization. First, they substituted the 5-position of the indole unit in 44 with 5-methoxy, 5-chloro, and 5-hydroxy, which creates 45, 46, and 51, respectively. Among them, the 5-chloro substituent 46 yielded a better Ki than the lead 44. Next, a methoxy was introduced into the 6-position of the indole unit of 44, which forms 48. However, this regioisomer (48) displayed significantly lower activity. Third, the P3 indole unit of 44 was replaced by benzimidazole, benzothiazole, benzofuran, and indoline scaffolds. The benzimidazole 41 exhibits 3 times the potency of 44. However, the replacement of the indole with an indoline (52) or benzothiazole (49) provided less potent analogues. Fourth, Hayashi et al. also attempted placing a carbonyl substitution at the 3-position of the indole of 50, which leads to a 10-times potency decrease compared to the 2-substituted indole. Molecular docking suggested that this substitution may prevent critical hydrogen-bond interactions with Mpro.39
Notably, the methoxy substitution at the 4-position of the indole unit (40) exhibits excellent inhibitory activity with 10- and 55-fold increase compared to the 5-methoxy (45) and 6-methoxy (48) derivatives, respectively. This finding reveals that the methoxy at the 4-position of the indole unit plays a vital role in the inhibitor–Mpro interaction. Konno et al.’s recent report41 suggested that 40 (YH-53) has drug development potential against SARS-CoV-2, which will be discussed in section 2.1.1.7. The activity of the 4-methoxy group at the indole unit of 40 was further verified by substitutions with 4-isopropoxyl (43), 4-isobutyloxyl (47), and 4-hydroxyl (42) moieties. The decrease in activities of these analogues compared to 40 strongly suggests that the methoxy group at the 4-position is superior to the isopropoxy, isobutyloxy, or hydroxyl groups at the 4-position.
Konno et al.41 also determined the crystal structure of 40 bound to SARS-CoV-2 Mpro, which is shown in Figure 13. In this crystal structure, 40 exhibits an extended conformation. The Cys145 of Mpro forms a tetrahedral hemithioketal bond with the carbonyl C atom at the P1 position of 40. The pyrrolidin-2-one group of 40 is completely buried in the S1 pocket of Mpro. The carbonyl and amine groups of the pyrrolidine-2-one group accept hydrogen bonds from the side chains of His163 and Glu166, respectively. Additionally, the N and S atoms of the benzothiazole form a hydrogen bond network with water and His41 in the active site, respectively. More interestingly, the binding of 40 shifts a loop region (residues 188–194) of Mpro toward the inhibitor by approximately 2.5 Å. Thus, Thr190, Gln189, and Glu166 are close to the 4-methoxy-indole group at the P3 position of 40, which is important for the enhanced inhibitory activity. The side chain carbonyl of Gln189 also forms a hydrogen bond with the main chain amide group at the P2 position.
Figure 13.
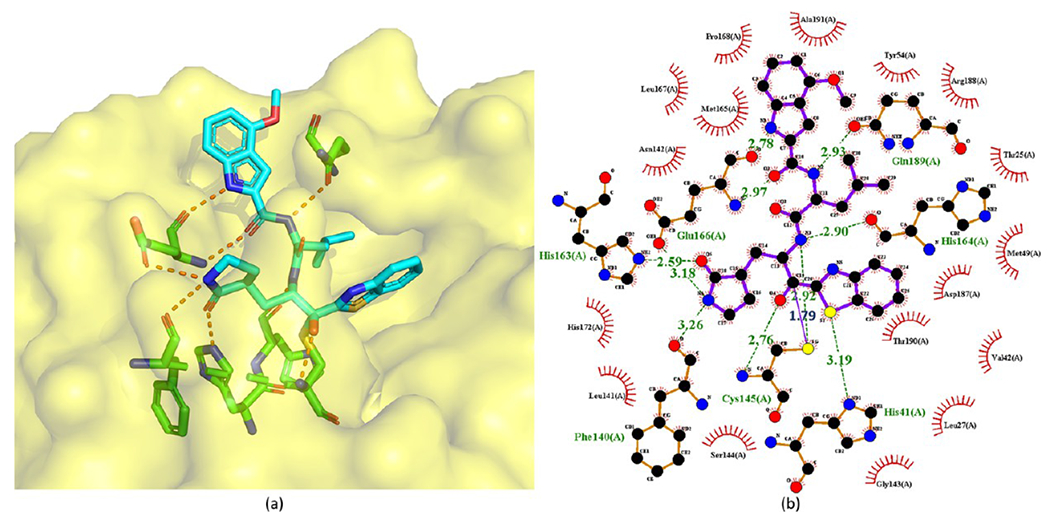
Cocrystal structures of the covalent adducts of 40 bound to SARS-CoV-2 Mpro (PDB ID 7E18) (a) and its corresponding 2D interaction diagram (b).
2.1.1.6. Thiazole-Containing Ketone Inhibitors.
Konno et al.38 and Thanigaimalai et al.40 also replaced the benzothiazole moiety at P1′ by 5-substituted thiazoles (Figure 14). However, 5-substituted thiazole replacements generally compromise inhibitory potency. The crystal structure of benzothiazole-containing ketone inhibitor 40 with SARS-CoV-2 Mpro (PDB ID 7E18, see Figure 13) reveals that the substituent in the 5-position of the thiazole at P1′ may sterically interact with the S1 pocket of Mpro thereby reducing the binding affinity of the thiazole-containing ketone inhibitors with Mpro. This comparison suggests that the benzothiazole unit is a more suitable substituent on the ketone warhead group for the P1′ moiety.
Figure 14.
Thiazole ketone-containing inhibitors from Konno et al.38 and Thanigaimalai et al.40
2.1.1.7. Perspectives on SARS-CoV-2 Ketone-Based Inhibitors.
One of the most promising ketone-based inhibitors is PF-00835231 (2). Currently its phase 1 clinical trial has been completed (https://clinicaltrials.gov/ct2/show/NCT04627532?term=PF-00835231&draw=2&rank=1). It is potent in blocking SARS-CoV-2 Mpro. Importantly, the in vitro and in vivo studies in rats indicate that both PF-00835231 and prodrug PF-07304814 exhibit no organ toxicity and minimal side effects. In addition, their work found that these compounds do not block the hERG channel.42
Another ketone-based inhibitor with drug development potential is 40 (YH-53), which has a different warhead from 2. Positives include its stability in plasma and no obvious toxicity, mutagenicity, or other side effects with regards to hERG and CYP activity. However, even though in vitro assays suggested promising ADMET properties, some irregular absorption was detected by in vivo pharmacokinetic studies.41 Nonetheless, 40 could provide a valuable lead compound for further optimization.
2.1.2. The Nitrile-Containing Drug Candidate from Pfizer.
One of the most advanced drug candidates targeting SARS-CoV-2 Mpro currently is nitrile-containing inhibitor PF-07321332 (59 in Figure 15), which was developed by Pfizer during this pandemic and disclosed at the American Chemical Society Spring 2021 meeting. PF-07321332 is now in phase 3 clinical trials (https://clinicaltrials.gov/ct2/show/NCT04960202?term=PF-07321332&draw=2&rank=3). It is the first orally administered SARS-CoV-2 inhibitor. According to a recent report from Pfizer,43 its enzymatic activity and cellular antiviral activity against SARS-CoV-2 are both high with Ki = 0.00311 μM and EC50 = 0.0745 μM. Especially, its EC50 is the lowest among all the current Mpro inhibitors, suggesting an optimal cell permeability. In mouse models, PF-07321332 can protect lung tissue well from being damaged by virus replication.43
Figure 15.
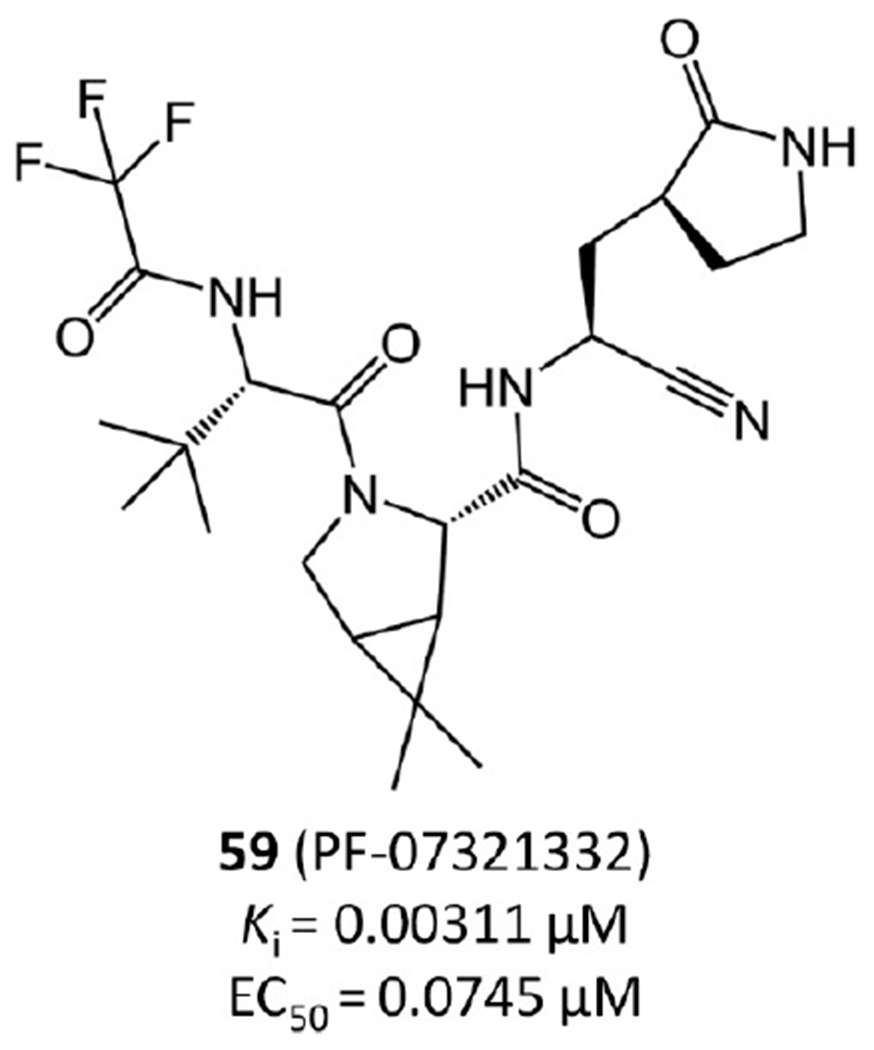
Nitrile-containing drug candidate from Pfizer (PF-07321332).
More importantly, PF-07321332 demonstrates low toxicity and side effects and does not block the hERG channel. Its ADMET properties are promising as well. For example, its plasma clearance is moderate with elimination half-lives of 5.1 and 0.79 h in rats and monkeys, respectively.
According to Pfizer’s recent report,43 the nitrile of PF-07321332 forms a covalent bond with SARS-CoV-2 Mpro in the crystal structure. However, this crystal structure has not been released to the public yet.
2.1.2.1. Perspectives on PF-07321332.
PF-07321332 is on its way to becoming the first Mpro drug to treat SARS-CoV-2. It may reach the market by the end of 2021.44 One of its major advantages is that it could be taken orally as a pill or capsule and, thus, be administrated outside of hospitals.5
2.1.3. Aldehyde-Based Inhibitors.
2.1.3.1. Bicycloproline-Containing Aldehyde Inhibitors.
Derived from approved protease inhibitor telaprevir or boceprevir, 32 bicycloproline-containing SARS-CoV-2 Mpro inhibitors were prepared by Qiao et al.45 They incorporated an aldehyde in the P1 position as the warhead to form a covalent bond with Cys145 of SARS-CoV-2 Mpro. Additionally, they adopted a bicycloproline moiety from either boceprevir or telaprevir for P2 and used various hydrophobic aryl subgroups for P3 to improve the potency and pharmacokinetic properties. As a result, all their compounds show potent SARS-CoV-2 Mpro inhibition in the FRET assay with IC50 values below 1 μM.
Compounds 60–91 in Figures 16 and 17 are the bicycloproline-containing inhibitors derived from telaprevir and boceprevir, respectively. Among them, 60, which is derived from telaprevir, has the highest Mpro binding affinity with IC50 = 0.0076 μM. To illustrate the detailed binding mode of these compounds with SARS-CoV-2 Mpro, Qiao et al.45 determined the structure of 60 in complex with SARS-CoV-2 Mpro (PDB ID 7D3I) as shown in Figure 18.
Figure 16.
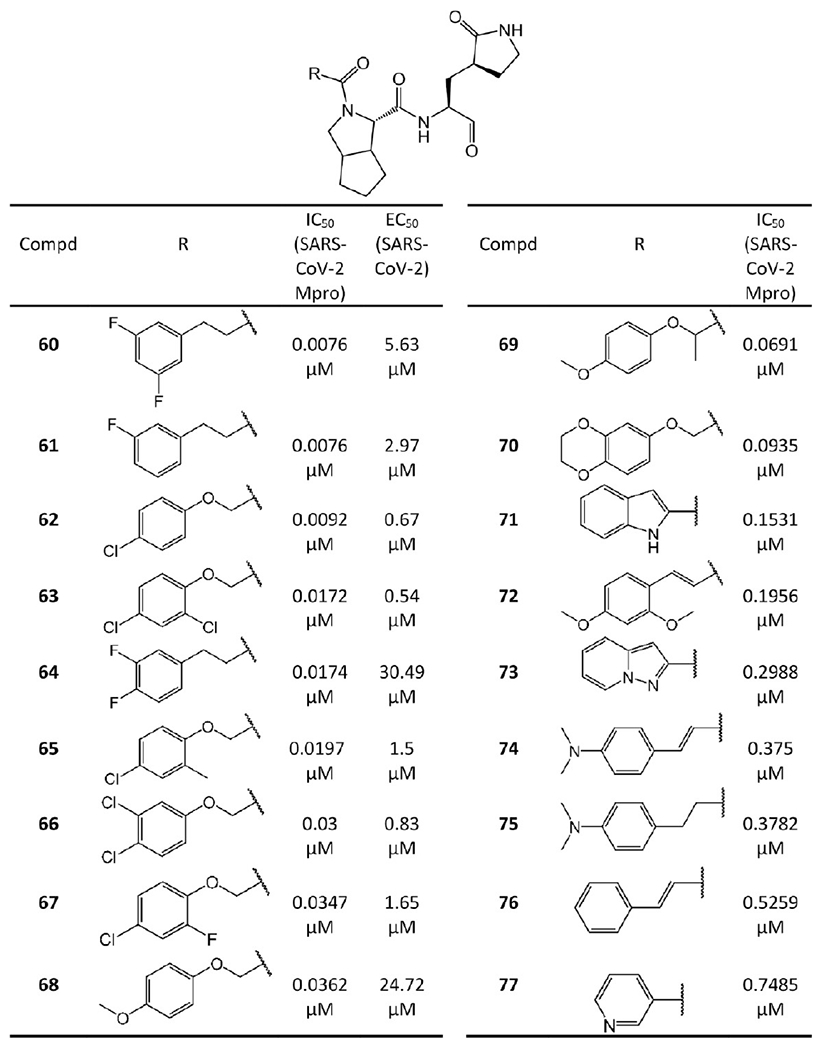
Bicycloproline-containing SARS-CoV-2 Mpro inhibitors derived from telaprevir.
Figure 17.
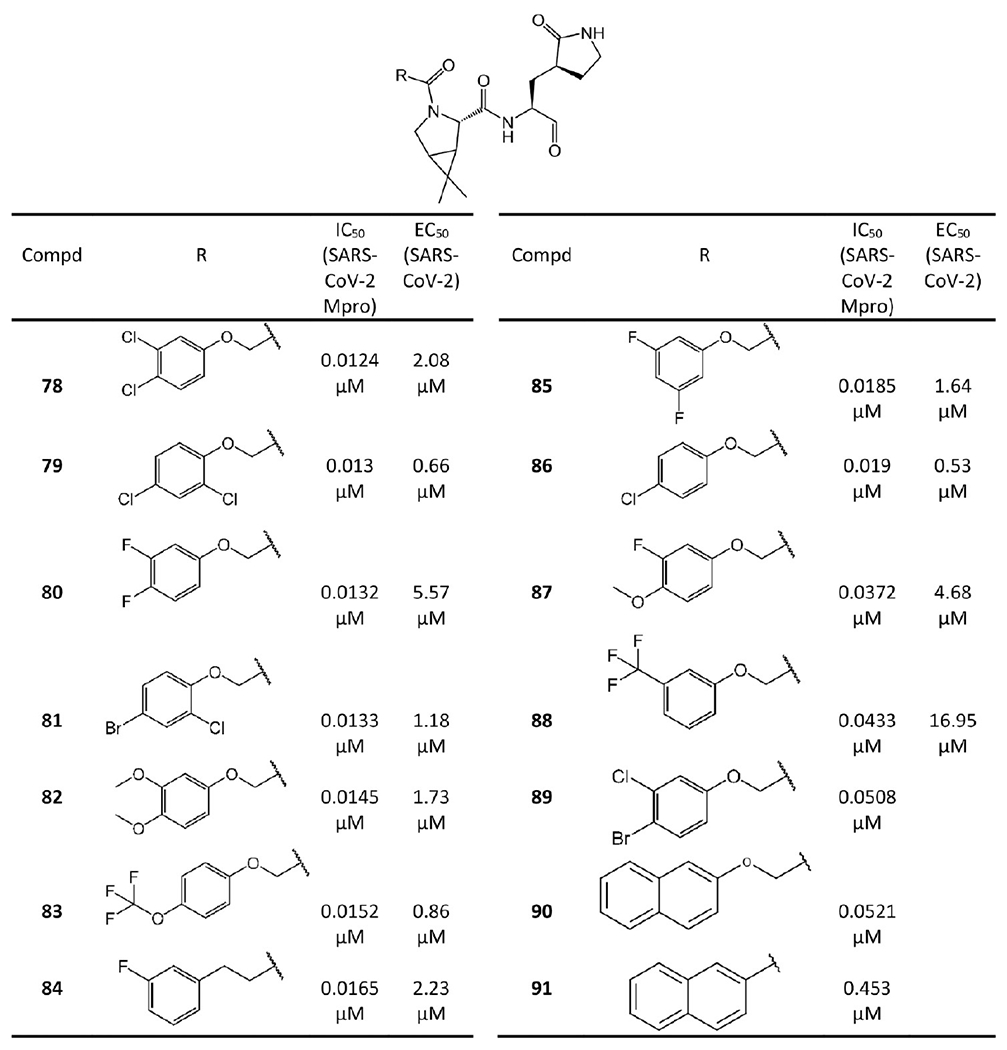
Bicycloproline-containing SARS-CoV-2 Mpro inhibitors derived from boceprevir.
Figure 18.
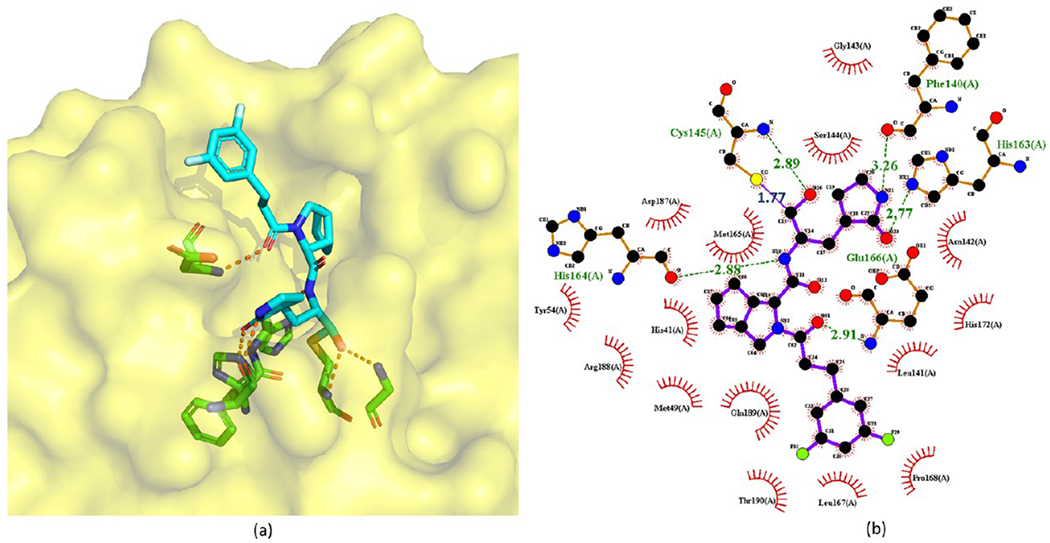
Crystal structure of 60 covalently bound to SARS-CoV-2 Mpro (PDB ID 7D3I) (a) and its corresponding 2D interaction diagram (b).
In this crystal structure, the carbonyl C atom of the aldehyde warhead of 60 reacts with the S atom of Cys145 to form a 1.77 Å covalent bond. Moreover, the O atom of the hemithioketal interacts with the main-chain amide of Cys145 by a hydrogen bond of 2.89 Å. The P1 γ-lactam ring of 60 resides deep in the S1 pocket. The O and N atoms of the lactam form hydrogen bonds with the side chain of His163 (2.77 Å) and the main chain of Phe140 (3.26 Å), respectively. The main-chain amide of P1 also accepts a 2.88 Å hydrogen bond from the backbone O atom of His164. Due to the conformational constraints inherent from the structure of proline,46 the rigid P2 bicycloproline of 60 is restrained in a trans-exo conformation with limited NCα bond rotation. As a result, the bicycloproline group stays close to the hydrophobic S2 pocket and forms hydrophobic interactions with His41, Met49, Met165, Asp187, Arg188, and Gln189 of Mpro. The backbone carbonyl O atom of P3 of 60 forms a 2.91 Å hydrogen bond with the backbone amide of Glu166. The 1-ethyl-3,5-difluorobenzene moiety of P3 occupies the S4 site with an extended conformation and forms hydrophobic interactions with Leu167, Pro168, and Gln189.45 Additionally, one F atom on the tail of 60 could form dipole–dipole interactions with the Cα atom of Pro168.
The SAR between different bicycloproline-containing inhibitors suggests the two different bicycloproline moieties at P2 do not largely impact their binding affinities to SARS-CoV-2 Mpro, such as 84 (IC50 = 0.0165 μM) vs 61 (IC50 = 0.0076 μM), 86 (IC50 = 0.019 μM) vs 62 (IC50 = 0.0092 μM), and 79 (IC50 = 0.013 μM) vs 63 (IC50 = 0.0172 μM). Moreover, the compounds with F or Cl atoms on the tail can be more active than others because these halogen atoms could form dipole–dipole interactions with Mpro such as with the Cα atom of Pro168.
In addition to the enzymatic activity, the cellular antiviral activity of 60–68 and 78–88 was also tested in cell protection assays,45 which are shown in Figures 16 and 17 in terms of the EC50. Among them, six compounds, namely, 86 (EC50 = 0.53 μM), 63 (EC50 = 0.54 μM), 79 (EC50 = 0.66 μM), 62 (EC50 = 0.67 μM), 66 (EC50 = 0.83 μM), and 83 (EC50 = 0.86 μM), exhibit sub-micromolar or low micromolar EC50 values. Notably, probably due to the poor cell membrane permeability caused by the relatively low lipophilic nature of the groups in the P3 position,47 some compounds such as 64 and 68 lack activity in the cell protection assays despite potent enzymatic inhibition.
Compounds 63 and 83 exhibit promising pharmacokinetics properties with oral bioavailability of 11.2% and 14.6% in the experiments in Sprague–Dawley rats. In light of that, Qiao et al.45 further evaluated their toxicity and antiviral activity in vivo.
Determination of tolerance in rats was evaluated at 100 and 200 mg/kg twice daily for 7 consecutive days. No noticeable toxicity was found for 63 and 83. Moreover, the in vivo antiviral activity of the compounds was assayed in a human angiotensin-converting enzyme 2 transgenic mouse model. Compounds 63 (50 mg/kg once daily) and 83 (50 mg/kg twice daily or 50 mg/kg once daily) were administered starting at 1 h prior to virus inoculation and lasting until 5 days postinfection (5 dpi). During this period, no abnormal behavior or body-weight loss was observed in the mice. More importantly, at 5 dpi, the viral RNA loads in the lung tissues of treatment groups were almost undetectable. This result suggests that 63 and 83 could efficiently inhibit SARS-CoV-2 replication and ameliorate SARS-CoV-2 induced lung lesions in vivo.
2.1.3.2. Other Aldehyde-Based Inhibitors.
In some designs,48–53 an aldehyde is selected as the warhead in the P1 moiety to form a covalent bond with Cys145 of SARS-CoV-2 and SARS-CoV Mpro (see Figure 19). In addition, most of these inhibitors are functionalized with an (S)-γ-lactam ring that occupies the S1 site of Mpro, which was already suggested by Zhang et al.34 As expected, many of them show potent IC50 or Ki values against Mpro, especially 92 and 93 with IC50 values of 0.04 μM and 0.053 μM, respectively.
Figure 19.
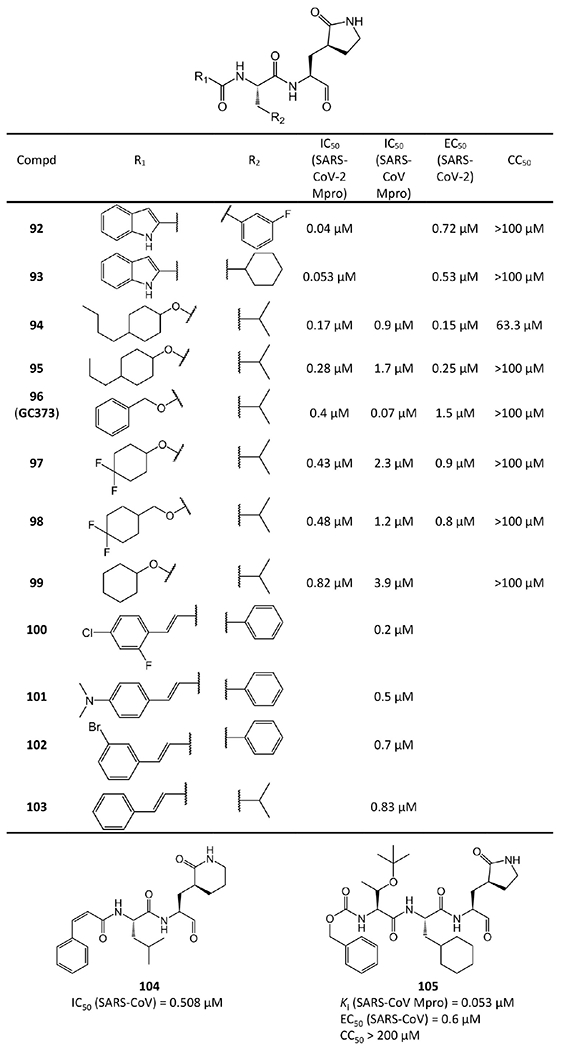
Other aldehyde-based inhibitors.
Some of these aldehyde-based inhibitors have available crystal structures with SARS-CoV-2 or SARS-CoV Mpro in PDB, such as 92 (PDB ID 6M0K), 93 (PDB ID 6LZE),48 96 (GC373, PDB ID 6WTK),50 104 (PDB ID 6LO0),52 and 105 (PDB ID 2GX4).53
As an example, the crystal structure of 92 bound to SARS-CoV-2 Mpro is chosen to depict the binding mode (Figure 20a,b). In this crystal structure, the C atom of the aldehyde group of 92 forms a standard 1.81 Å C–S covalent bond with Cys145 of SARS-CoV-2 Mpro, and the conformation is stabilized by the 2.83 Å hydrogen bond between the O atom of the hemithioketal group and the backbone of Cys145 in the S1′ site. The (S)-β-lactam ring of 92 at P1 fits well into the S1 site. The O atom of the (S)-β-lactam ring accepts a 2.71 Å hydrogen bond from the side chain of His163. The main chain of Phe140 and the side chain of Glu166 also stabilize the (S)-β-lactam ring via a 3.26 Å hydrogen bond and a 2.96 Å hydrogen bond with its NH group, respectively. Additionally, the amide bonds in the main chain of 92 form two hydrogen bonds with the main chains of His164 (3.29 Å) and Glu166 (2.81 Å). The 3-fluorophenyl moiety at P2 of 92 inserts deeply into the S2 site of Mpro, stacking with the imidazole ring of His41. The side chains of Met49, Met165, Val186, Asp187, and Arg188 are near the 3-fluorophenyl group, leading to extensive hydrophobic interactions. Just like 4 and 9, the indole group at the P3 position of 92, which is exposed to solvent, is stabilized by a 2.57 Å hydrogen bond with Glu66. Moreover, the side chains of Pro168 and Gln189 have hydrophobic interactions with the indole group of 92. Compound 95 shares with 92 a similar inhibitor binding mode (Figure 20c,d). The binding mode of GC373 (96) is as the same as that of GC376 (110) and will be illustrated in section 2.1.3.3.
Figure 20.
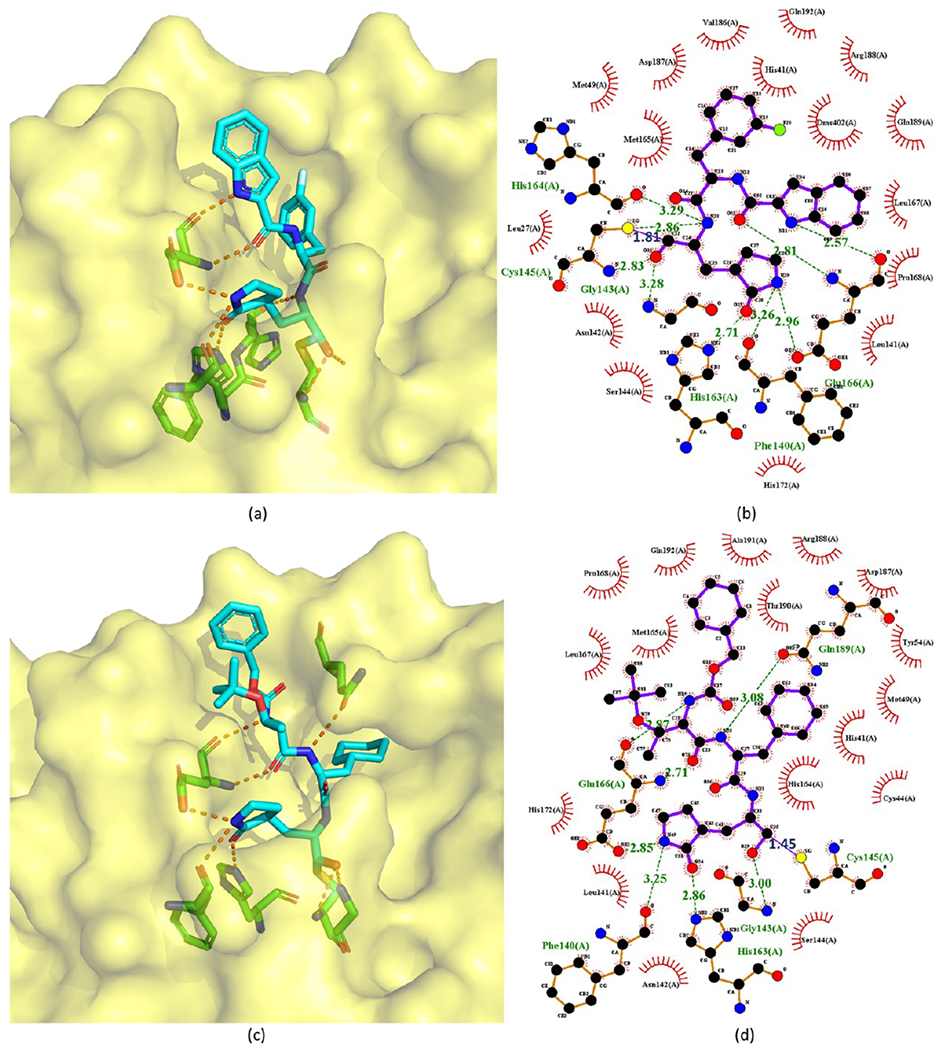
Cocrystal structures of the covalent adducts of 92 (a, b) and 105 (c, d) bound to SARS-CoV or SARS-CoV-2 Mpro (PDB ID 6M0K and 6LO0).
Comparing the enzymatic activities of these aldehyde-based inhibitors reveals that 92 and 93 are the most potent. This is because the NH group of their indole can form a hydrogen bond with the backbone carbonyl of Glu166, just like 4 and 9. This confirms that indole is an optimal design for the P3 position of Mpro inhibitors. Another finding from the comparison is the reason why 94 and 95 are the second most potent compounds. The hydrophobic tails in their P3 sites could form hydrophobic interactions with the hydrophobic amino acids in the Mpro S3 pocket, such as Met165 and Leu167.
In addition to enzymatic activity, the cellular antiviral activity of 92–98 and 105 against SARS-CoV-2 or SARS-CoV was also assayed in terms of the EC50. All of them have strong antiviral activity with EC50 values ≤1.5 μM (see Figure 19). To further examine the antiviral activity within a large dynamic range, quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed on cultures treated with 92, 93, GC373 (96), and its prodrug GC376 (110), which revealed good antiviral effect against SARS-CoV-2.48,50 Particularly, GC373 and GC376 were reported to decrease viral titers by 3-log values.50
The toxicity of 92–99 and prodrug GC376 (110) was assayed in Huh-7, CRFK, or L929 cells. Except for 94, which has a CC50 = 63.3 μM,49 none of these compounds cause obvious cytotoxicity. The CC50 values of 92, 93, and 95–99 are higher than 100 μM,48,49 and the CC50 values of 105, GC373, and GC376 are over 200 μM.50,53 More importantly, no obvious in vivo toxicity of 92 and 93 was observed during the animal tests on Sprague–Dawley rats and beagle dogs.48
Since no obvious in vivo toxicity was observed, the pharmacokinetics properties of 92 and 93 were further assessed by Dai et al.48 Compound 92 exhibits long T1/2 values (Sprague–Dawley rat, 7.6 h; beagle dog, 5.5 h), low clearance rates (rat, 4.01 (mL/min)/kg; dog, 5.8 (mL/min)/kg), and high AUC values (rat, 41 500 h·ng/mL; dog, 14900 h·ng/mL). Its potent enzymatic and antiviral activities as well as satisfying pharmacokinetic properties indicate that 92 warrants further optimization toward potential drug candidates.
2.1.3.3. Water-Soluble Aldehyde Bisulfite Adducts.
The aldehyde bisulfite adducts are prodrugs of the aldehyde-based inhibitors introduced in the last sections created to improve water solubility.50 Aldehyde bisulfites can have improved pharmacokinetic properties compared to their aldehyde counterparts, but they are typically rapidly hydrolyzed and converted to the corresponding aldehydes.54
Rathnayake et al.,49 Vuong et al.,50 and Ma et al.55 reported a series of aldehyde bisulfite adducts (see Figure 21). Notably, the crystal structure of GC376 (110) with SARS-CoV-2 Mpro is available with PDB ID 6WTJ (see Figure 22). GC376 is the prodrug of GC373 (96 in Figure 19). GC376 eliminates its bisulfite group and turns into GC373 before binding to Mpro, then the aldehyde warhead of GC373 forms a covalent bond with Cys145 of Mpro.50,55 For other interactions, GC373 mimics the peptide substrate cleaved by the protease.35 The glutamine surrogate γ-lactam ring of GC373 derived from the P1 glutamine side chain always occupies the S1 site. This ring forms hydrogen bonds with the His163 and Glu166 side chains and the Phe140 main chain. An amide bond attaches the γ-lactam side chain to the isobutyl moiety of the hydrophobic S2 site formed by His41, Met49, and Met169. A carbonate bond in GC373, which accepts hydrogen bonds from the main chain of Glu166 and the side chain of Gln189, attaches the P2 isobutyl group to a phenylmethyl ester that interacts with the aliphatic S4 site.55
Figure 21.
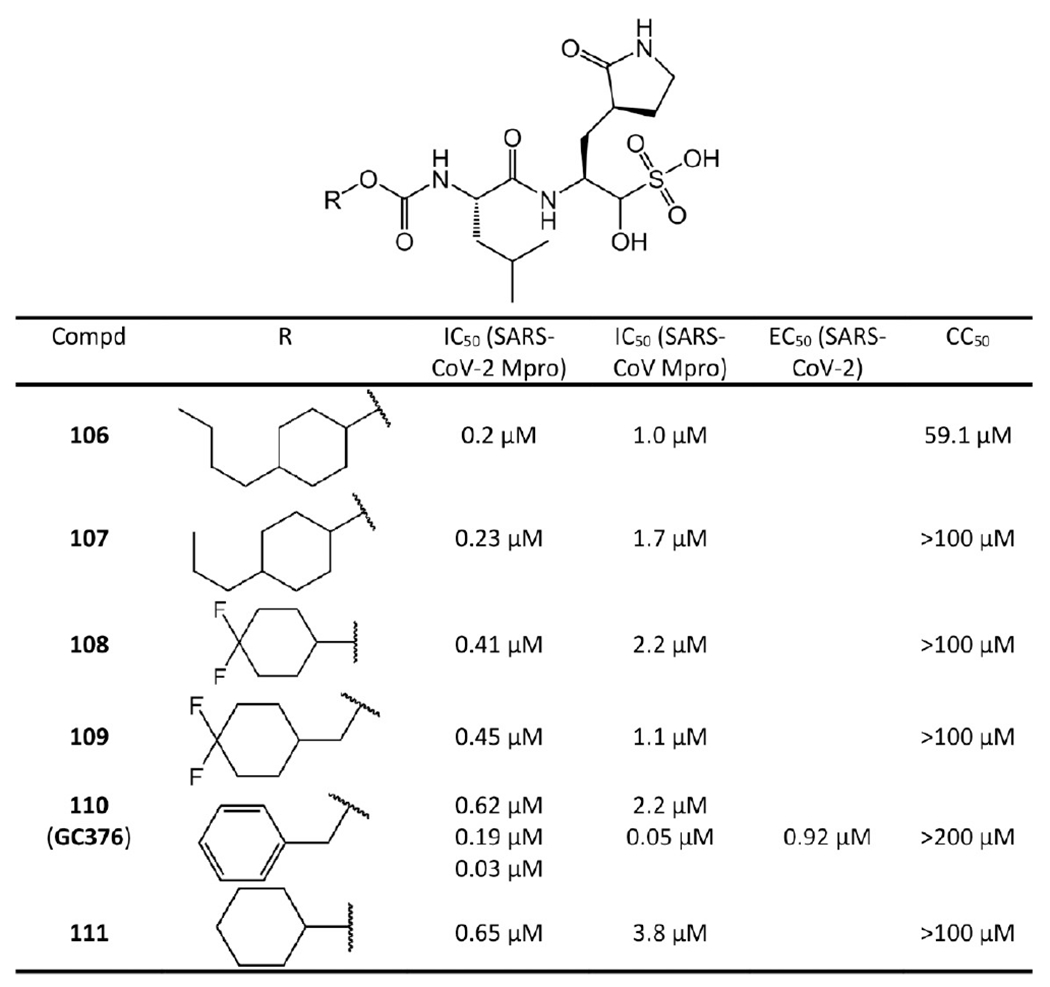
Covalent inhibitors with aldehyde bisulfite warhead. The IC50 values of 106–109 and 111 were reported by Rathnayake et al.49 For 110 (GC376), the IC50 values of 0.62 μM and 2.2 μM for SARS-CoV-2 and SARS-CoV Mpro were given by Rathnayake et al.49 The IC50 values 0.19 μM and 0.05 μM to SARS-CoV-2 and SARS-CoV Mpro were reported by Vuong et al.50 The IC50 value 0.03 μM against SARS-CoV-2 Mpro was given by Ma et al.55
Figure 22.
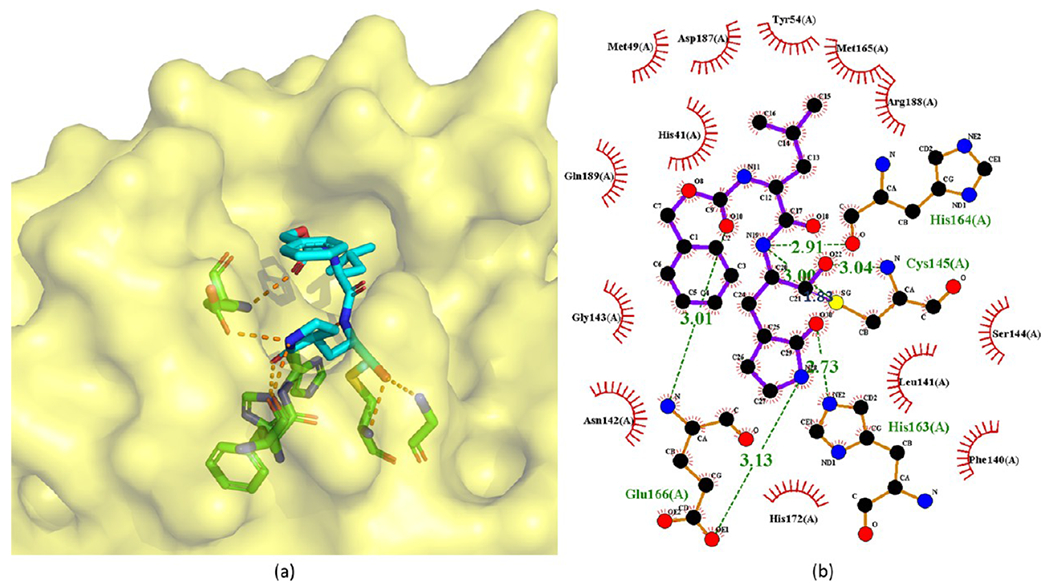
Cocrystal structures of the covalent adduct of 110 (GC376) bound to SARS-CoV-2 Mpro (PDB ID 6WTJ) (a) and its corresponding 2D interaction diagram (b).
GC376 (110) is an investigational veterinary drug to cure feline infectious peritonitis (FIP).56–58 Its target is the viral Mpro. Researchers also found that GC376 is a broad-spectrum coronavirus protease inhibitor, which is active toward the Mpro enzymes of multiple coronaviruses, including FIPV, MERS-CoV, and norovirus.56,57 In light of its broad-spectrum activity, preclinical studies have been started to examine its potential in treating COVID-19.59 Different groups have reported inconsistent but promising potency.
In the FRET enzyme assays conducted by Rathnayake et al.,49 the IC50 values of GC376 are 0.62 μM and 2.2 μM against SARS-CoV-2 Mpro and SARS-CoV Mpro, respectively. By the same technique, Vuong et al.50 achieved IC50 values of 0.19 μM and 0.05 μM against SARS-CoV-2 Mpro and SARS-CoV Mpro, respectively. While Ma et al.55 reported an IC50 value of 0.03 μM against SARS-CoV-2 Mpro. It was also reported that GC376 and its variants could increase survival of mice infected with SARS-CoV-2.60
For aldehyde bisulfite adducts such as GC376, it is their corresponding aldehydes that interact with Mpro and their binding affinities are almost identical to those of their corresponding aldehydes. For example, as shown in Figure 21, the SARS-CoV-2 Mpro IC50 values of 106, 107, 108, 109, and 111 are 0.2 μM, 0.23 μM, 0.41 μM, 0.45 μM, and 0.65 μM, respectively, while the IC50 values of their corresponding aldehydes 94, 95, 97, 98, and 99 are 0.17 μM, 0.28 μM, 0.43 μM, 0.48 μM, and 0.82 μM, respectively. Just like their corresponding aldehydes, the high enzymatic activity of 106 and 107 is also probably due to the hydrophobic tails in their P3 positions forming hydrophobic interactions with the hydrophobic amino acids in the Mpro S3 pocket.
Similar to their corresponding aldehydes, the cytotoxicity of these bisulfite adducts is also weak,49,50 except for 106, which exhibits a CC50 of 59.1 μM. All the other bisulfite adducts have CC50 values over 100 μM and GC376 (110) even has a CC50 larger than 200 μM.
2.1.3.4. Drawbacks of the Aldehyde and Aldehyde Bisulfite Inhibitors.
Aldehyde bisulfite GC373 has shown promising antiviral activity in vivo. The bisulfite prodrugs may have improved chemical stability and pharmacokinetic properties, which make them often superior drug candidates over the free aldehydes. However, considering the highly reactive nature of the electrophilic warhead, both the aldehydes and the aldehyde bisulfite prodrugs may suffer from substrate promiscuity. Reactive aldehyde species are often considered toxic, due to their formation of various Schiff bases and DNA adducts, and are known to also induce oxidative stress and inflammation.61,62 For example, GC373 and its prodrug GC376 are active against proteins cathepsin B and L with IC50 values in the micromolar range.63 Therefore, whether these types of warheads offer an advantage over ketone-based covalent inhibitors, including HMK inhibitors, still needs to be investigated carefully.
2.1.4. Michael Acceptor-Based Inhibitors.
Several Michael acceptor-based inhibitors active against SARS-CoV-2 Mpro or SARS-CoV Mpro were reported53,64–67 and are depicted in Figure 23.
Figure 23.
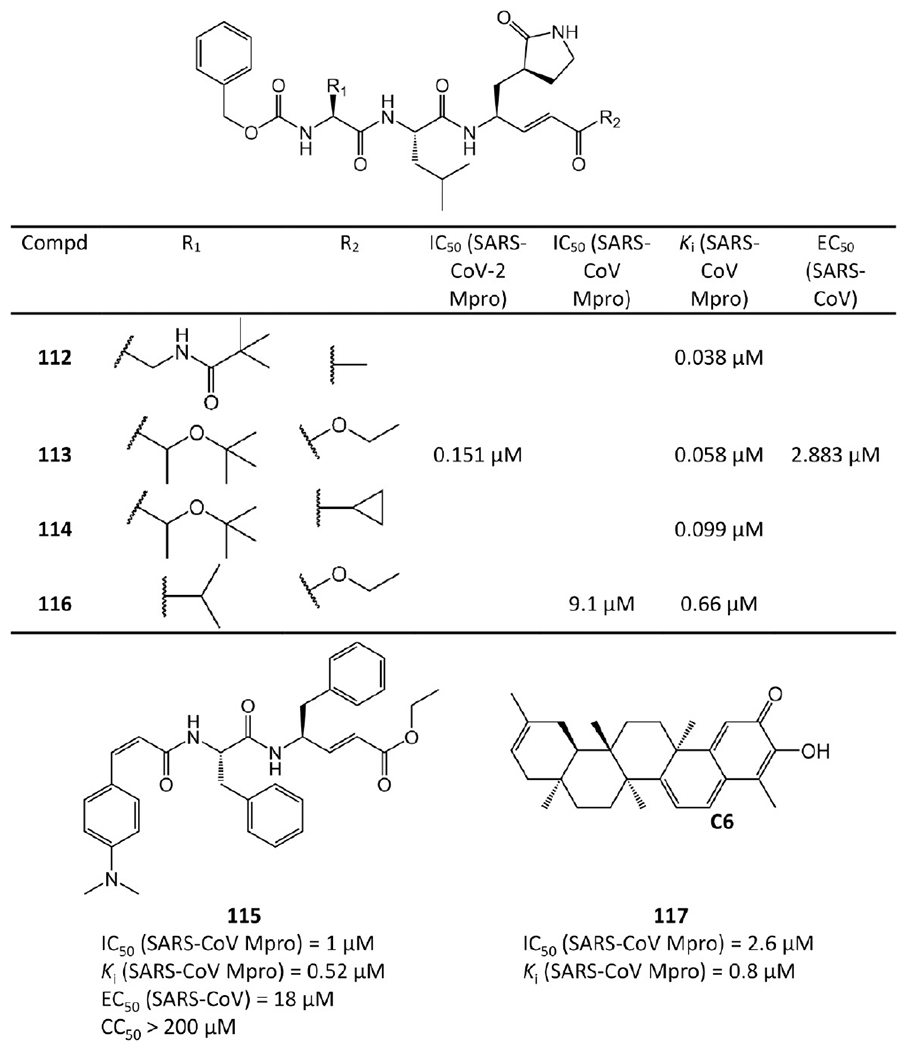
Michael acceptor-based inhibitors.
As a Michael acceptor, each of 112–116 contains an α,β-unsaturated ester with the double bond at the P1′ position to form a covalent bond with Cys145 of Mpro28 (see Figure 24b,d).
Figure 24.
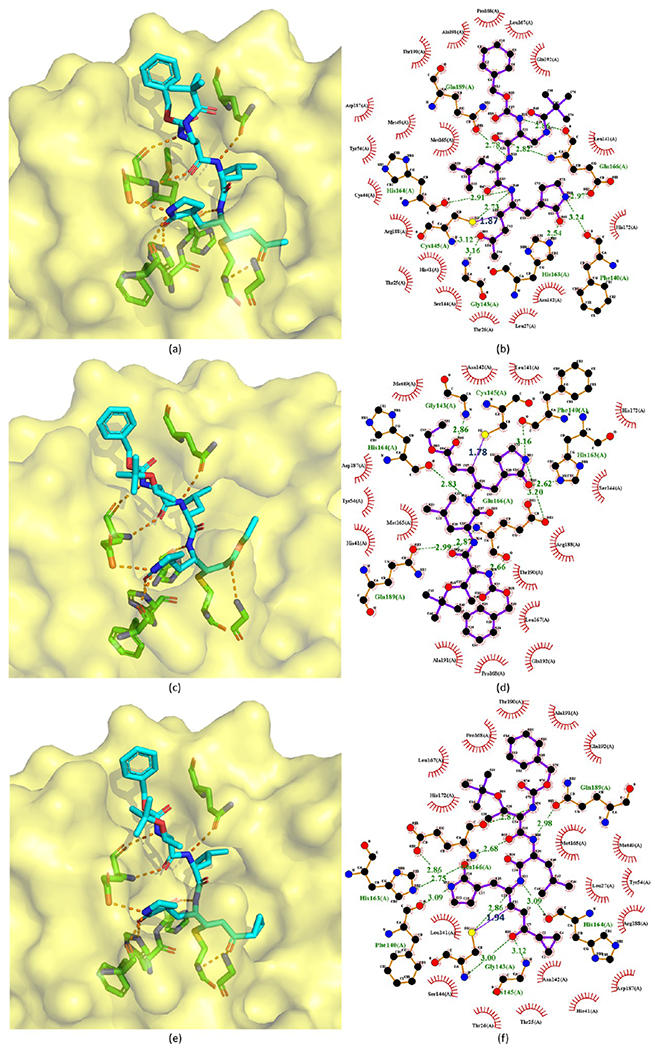
Cocrystal structures of the covalent adducts of 112 (a, b), 113 (c, d), and 114 (e, f) bound to SARS-CoV Mpro or SARS-CoV-2 Mpro (PDB ID 2ZU4, 7JT7, and 2ZU5).
However, not all Michael acceptor-based inhibitors are peptides. Ryu et al.67 extracted four quinone-methide triterpenoid derivatives from Tripterygium regelii (a woody vine). Among them, 117 has sub-micromolar potency. According to Sreeramulu et al.’s study,68 the nucleophilic group of Mpro reacts with the quinone-methide of 117 or other derivatives through a Michael addition, resulting in the formation of an adduct at C6 (see 117 in Figure 23).
The crystal structures of 113 bound to SARS-CoV-2 Mpro and 112, 113, and 114 bound to SARS-CoV Mpro are available with PDB ID 7JT7, 2ZU4, and 2ZU5, respectively,64,65 which are depicted in Figure 24. Their binding modes are almost identical. Mpro Cys145 attacks the Cβ atom of the α,β-unsaturated ketone at the P1′ position to form a covalent C–S bond (1.87 Å, 1.78 Å, and 1.94 Å, respectively), and hydrogen bonds are formed between the ketone O atom and the NH groups of Gly143 and Cys145 in the S1′ pocket. In the S1 site, the carbonyl O atom and the five-membered lactam ring’s N atom at the P1 position accept hydrogen bonds from His163 and Glu166. The P2 side chain of each compound is inside a hydrophobic S2 pocket, while the P3 group is toward the bulk solvent. The benzoxy group at the P4 position is in the S4 pocket with its phenyl ring parallel to a flat surface near Ala191. The peptide NH groups of residues P1, P2, and P3 also have four hydrogen bonds with the backbone carbonyl groups of His164 and Glu166 and the side chain of Gln189. Moreover, the carbonyl group of P3 interacts with the NH group of Glu166.
The SAR analysis of these analogues (113–116) indicates that the introduction of a lipophilic tert-butyl group at the P2 site increases binding affinity by over 10-fold. The possible explanation could be found in the crystal structure of 113 binding to Mpro (see Figure 24d). The tert-butyl group of the compound turns to the P4 site and forms hydrophobic interactions with the phenyl ring in the benzoxy group of the compound itself. Moreover, the phenyl ring of the compound interacts with Mpro and faces the P3 site directing the methylene group in a small corner pocket near Ala191.
The antiviral activity and cytotoxicity of 113 and 115 were assayed. The EC50 of 113 against SARS-CoV-2 is 2.883 μM, and the EC50 of 115 against SARS-CoV is 0.18 μM. Compound 115 also has a CC50 value over 200 μM, making 115 a promising Mpro inhibitor for further studies.
2.1.4.1. Perspectives on Michael Acceptor-Based Mpro Inhibitors.
Although with a risk of presumed indiscriminate reactivity, the covalent modification of targets using an irreversible hetero-Michael addition with a cysteine residue has gained some recent validation with the FDA approval of multiple Michael-acceptor-based drugs.69 Developing Michael acceptors could be a possible promising direction for discovering SARS-CoV-2 drug candidates. However, careful evaluations of safety and side effects are required.
2.1.5. Calpain Inhibitors.
Ma et al.55 found that calpain inhibitor XII (118) and calpain inhibitor II (119) are also potent against SARS-CoV-2 Mpro (Figure 25). A biphasic enzymatic progression curve of the inhibitors suggests a slow covalent binding. Notably, calpain inhibitors II and XII are dual inhibitors targeting both SARS-CoV-2 Mpro and human cathepsin L. Cathepsin L plays an important role in SARS-CoV-2 viral entry by activating the viral spike protein in the endosome or lysosome.70,71 Studies have proven that cathepsin L inhibitors can substantially weaken virus entry.72
Figure 25.
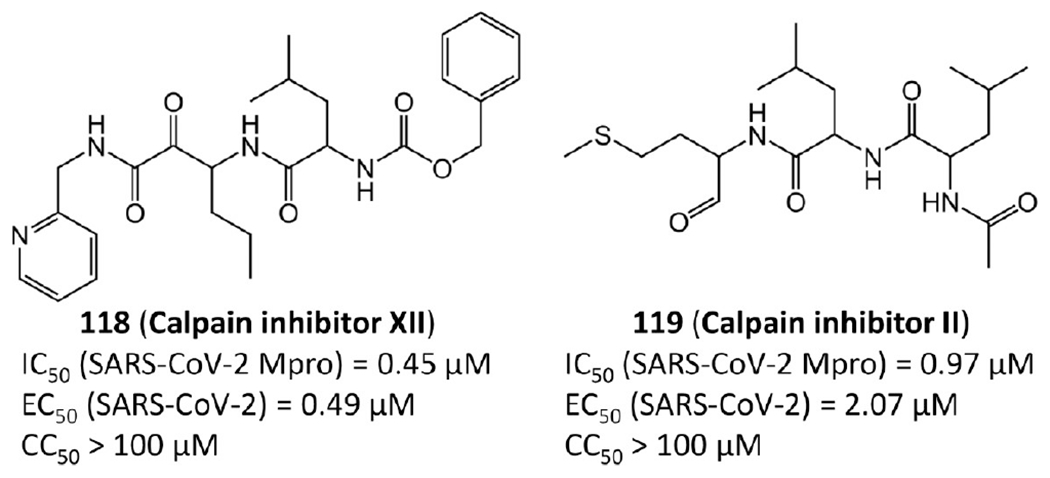
Calpain inhibitors.
The crystal structures of calpain inhibitors XII (118) and II (119) with SARS-CoV-2 Mpro are available (PDB ID 6XFN and 6XA4),73 as shown in Figure 26. These two compounds adopt an unusual binding mode compared with traditional Mpro inhibitors. Specifically, calpain inhibitors XII and II possess norvaline and hydrophobic methionine side chains at the P1 position, respectively, which challenges the previous experience that a hydrophilic glutamine mimetic is necessary at the P1 position and suggests that S1 pocket of Mpro can accommodate both hydrophilic and hydrophobic substitutions. This new finding paves the way to design dual inhibitors targeting both SARS Mpro and human cathepsin L.
Figure 26.
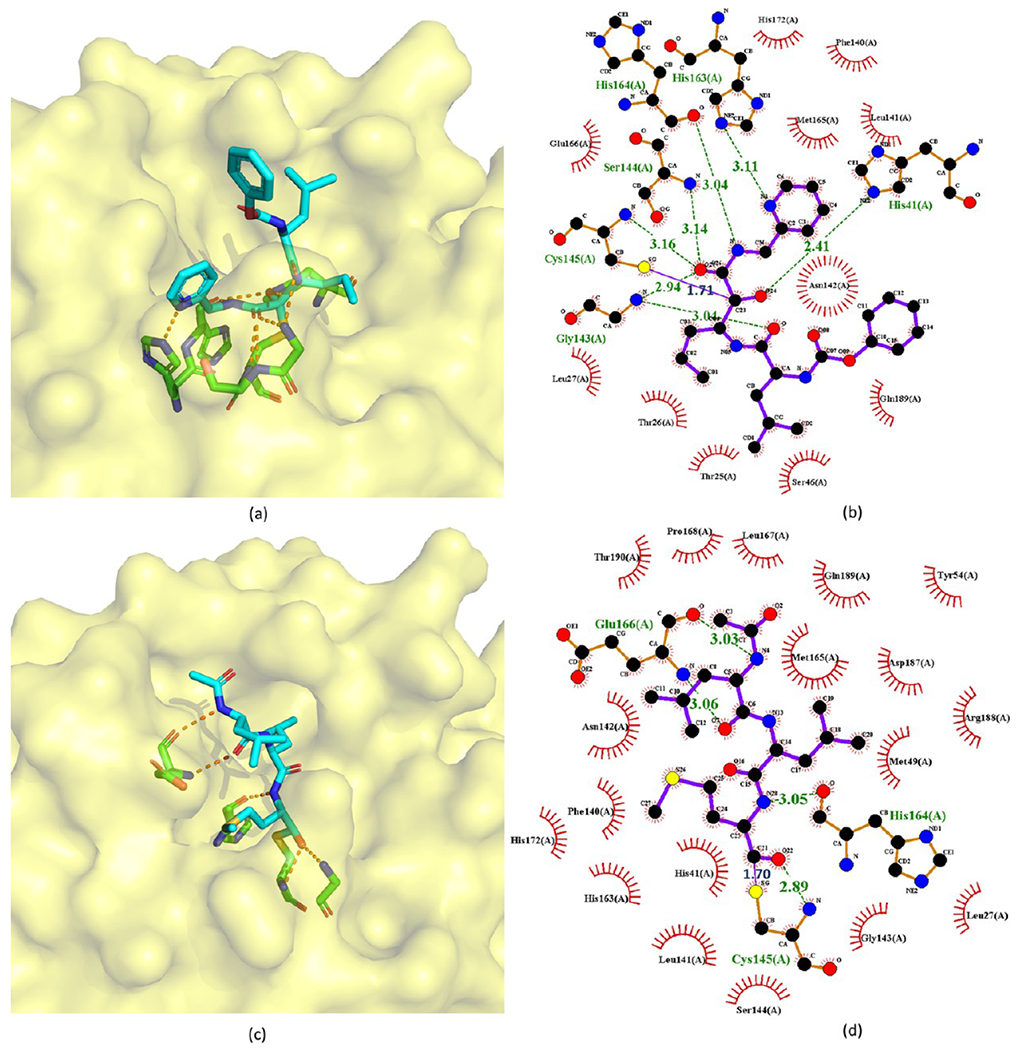
Crystal structures of the covalent adducts of 118 (a, b) and 119 (c, d) bound to SARS-CoV-2 Mpro (PDB ID 6XFN and 6XA4).
In the crystal structure of calpain inhibitor XII with Mpro, the length of the covalent bond between one ketone group of the inhibitor and Cys145 is 1.71 Å. The P1′ pyridine is positioned in the S1 site, while the P1 norvaline takes the S1′ site. The P2 leucine is toward the solvent near residues 45–51, and the terminal carboxybenzyl group facing the S1 site forces Asn142 upward and forms a water-mediated hydrogen bond with Glu166. The hydroxyl group establishes a strong hydrogen bond (2.41 Å) with the catalytic His41. The ketoamide O atom of the inhibitor forms a hydrogen bond with one residue in the oxyanion hole (2.94, 3.14, and 3.16 Å to the backbone amino of Gly143, Ser144, and Cys145, respectively), while the N atom accepts a hydrogen bond (3.04 Å) from the main chain carbonyl of His164 (Figure 26a). In the crystal structure of calpain inhibitor II with MPro, the length of the covalent bond between the aldehyde group and Cys145 is 1.70 Å. Like other peptidomimetic aldehyde inhibitors, the thiohemiacetal of calpain inhibitor II occupies the oxyanion hole consisting of the backbone amide groups of Gly143, Ser144, and Cys145. The inhibitor extends the length of the substrate-binding channel with its side chains placed in their respective recognition pockets. The P2 leucine side chain of the inhibitor forms hydrophobic interactions in the S2 pocket, while the P3 leucine occupies the solvent-accessible S3 position. Multiple hydrogen bonds form between the amide backbone of the inhibitor and the main chain of His164 and Glu166.
The antiviral activity and cytotoxicity of the two calpain inhibitors are also available. The EC50 values of calpain inhibitors XII and II against SARS-CoV-2 are 0.49 μM and 2.07 μM, respectively. Additionally, their CC50 values are both over 100 μM, representing low cytotoxicity.55
2.1.5.1. Perspectives on Calpain Inhibitors.
The calpain inhibitors represent an interesting class of inhibitors considering their potential dual role as Mpro and cathepsin L inhibitors. Whereas many peptidic Mpro inhibitors place a pyrrolidinone moiety in the P1 position to form a hydrogen bond interaction with His163, the pyridine moiety of calpain inhibitor XII was shown to be a suitable replacement. A decrease in activity is seen with calpain inhibitor II, in which this side chain is replaced with a methionine, which does not appear to be involved with a specific binding interaction according to the crystal structure. As seen with many peptide-aldehydes, the possible promiscuity of a reactive aldehyde functionality in inhibitor II may render the ketoamide XII the better lead agent in this series for further evaluation.
2.2. Non-peptidomimetic Covalent Inhibitors.
Current existing non-peptidomimetic covalent inhibitors were developed by Ghosh et al.,74 Wu et al.,75 Zhang et al.,76 and Niu et al.77 Interestingly, all these inhibitors belong to the class of esters: 3-chloropyridine, 3-chloropyridyl, or benzotriazole esters (see Figures 27 and 28).
Figure 27.
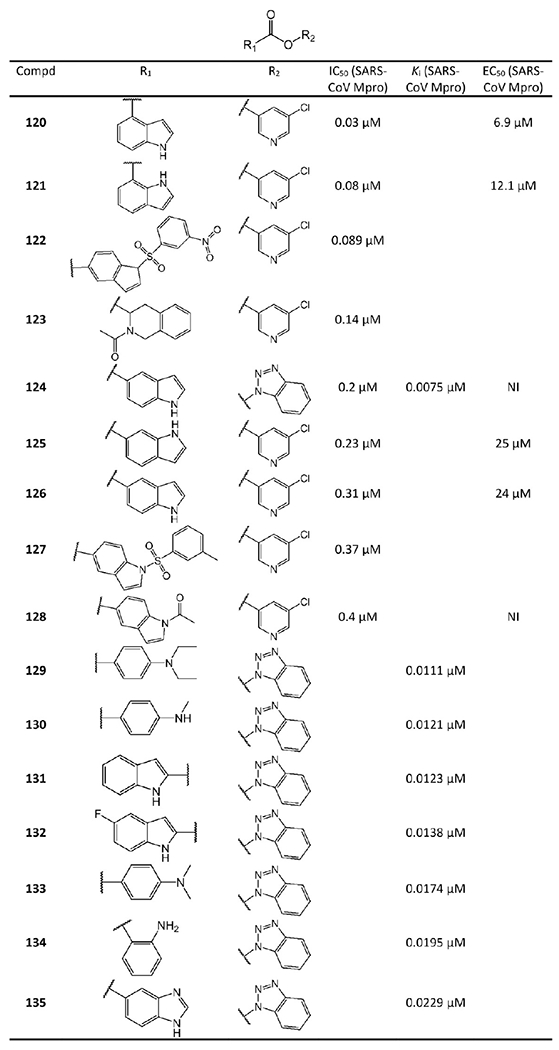
5-Chloropyridine ester and benzotriazole ester derived non-peptidomimetic covalent inhibitors from Ghosh et al.74 and Wu et al.75 NI represents no inhibition.
Figure 28.
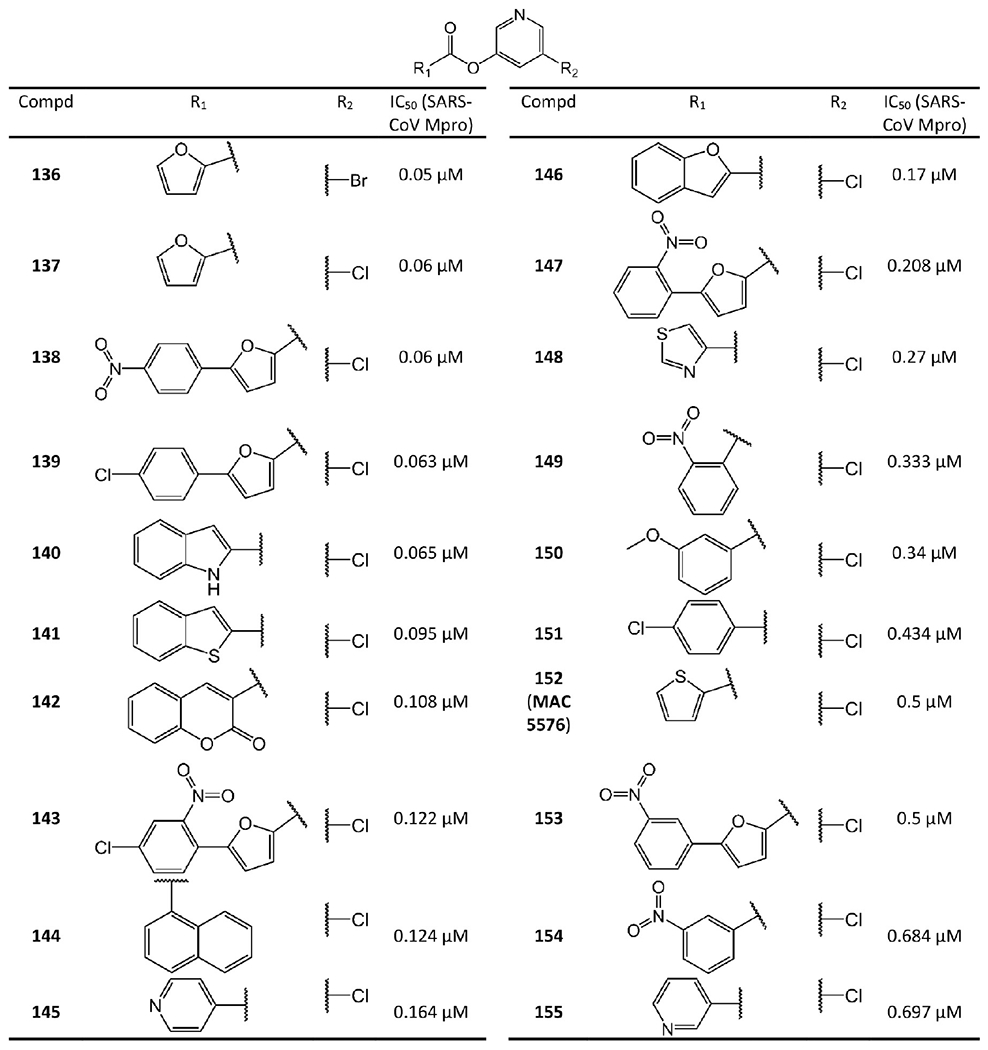
5-Chloropyridyl or 5-chloropyridine ester derived non-peptidomimetic covalent inhibitors from Zhang et al.76 and Niu et al.77
Based on their mass spectrometry experiments, Wu et al.75 proposed an inhibition mechanism of these ester-derived inhibitors, which is depicted in Figure 29. Upon binding to Mpro, the ester is attacked by the nucleophilic cysteine (Cys145). The acyl group of the ester-derived inhibitor subsequently acylates the thiol of Cys145 and generates an inactive acylated Mpro. This mechanism is further confirmed by a recent crystal structure of MAC-5576 (152) bound to SARS-CoV-2 Mpro (see Figure 30).65 In this crystal structure, the activated group of MAC-5576 acylates Cys145.
Figure 29.
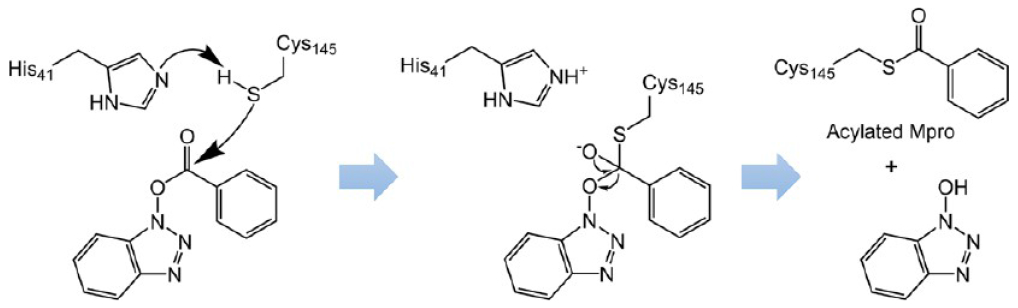
Proposed mechanism of the SARS-CoV Mpro inhibition by acylation with ester-based inhibitors.
Figure 30.
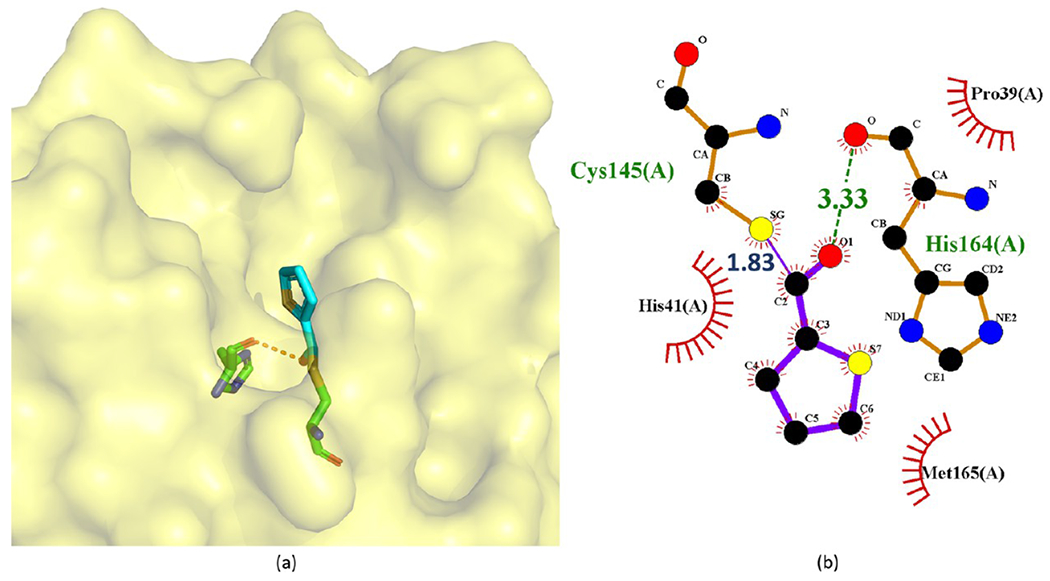
Crystal structure of the covalent complex of 152 (MAC-5576) with SARS-CoV-2 Mpro (PDB ID 7JT0) through the acylation of Cys145 (a) and its corresponding 2D interaction diagram (b).
Compounds 124 and 129–135 in Figure 27 are a series of benzotriazole esters synthesized by Wu et al. Their Ki values against SARS-CoV Mpro surveyed by the authors are also included in Figure 27. Especially 124 exhibits a Ki value as low as 0.0075 μM.75 Additionally, Wu et al.’s cell-based assays at 100 μM suggested that these esters are not toxic to Vero E6 cells.78 Notably, benzotriazole esters are well-known for their lability toward various nucleophiles, but the authors found that the compounds are relatively stable over 24 h in pH 5.0–8.0 at room temperature.75
Ghosh et al.74 also focused on 124 and its derivatives. Consistent with the reported low Ki value,78 124 has a potent IC50 value of 0.2 μM against SARS-CoV Mpro. However, as a drawback, the cell-based experiments suggested that 124 does not exhibit any SARS-CoV antiviral activity (see Figure 27, NI = no inhibition). Thus, Ghosh et al. developed more 3-chloropyridine esters derived from 124, namely, 120–123 and 125–128 in Figure 27. Replacing the benzotriazole unit with a 3-chloropyridine unit, 126 shows comparable enzymatic inhibitory potency (IC50 = 0.31 μM) with that of 124. More importantly, 126 has antiviral activity with an EC50 of 24 μM. With the indole N atom acetylated, the resulting 128 remains potent against SARS-CoV Mpro (IC50 = 0.4 μM) but without antiviral activity. Compound 127 contains a tosylated indole and has a IC50 = 0.37 μM against SARS-CoV Mpro. Interestingly, its nitrobenzenesulfonamide analog 122 indicates a quite improved IC50 value of 0.089 μM. The tetrahydroisoquinoline derivative 123 shows an IC50 value of 0.14 μM.
Additional studies suggested that the carboxylic positions on the benzene ring of indole are critical for inhibitory potency against Mpro. Accordingly, carboxylate substitutions on indole rings at the 5-, 6-, 4-, and 7-positions result in chloropyridine esters 126, 125, 120, and 121, respectively. Of this series, inhibitor 120, which contains a carboxylate at the 4-position, is the most potent inhibitor so far among all the existing non-peptidomimetic Mpro inhibitors with an IC50 value of 0.03 μM, a 10-fold potency enhancement over 126, which contains a carboxylate at the fifth position. Compound 121 is substituted with a carboxylate at the 7-position and exhibits an IC50 value of 0.08 μM as well. Encouragingly, 120 and 121 show high antiviral activity with EC50 values of 6.9 μM and 12.1 μM, respectively.
MAC-5576 (152 in Figure 28) is another non-peptidomimetic inhibitor widely investigated. Its IC50 against SARS-CoV Mpro is 0.5 μM.79 A recent study found that it is even more potent against SARS-CoV-2 Mpro with an IC50 of 0.081 μM. However, it does not block SARS-CoV-2 viral replication in cellular assays.65 With MAC-5576 as the lead compound, Zhang et al.76 and Niu et al.77 developed tens of 5-chloropyridyl ester based inhibitors. Compounds 136, 137, 139–141, 146, 148, and 150 were synthesized by Zhang et al.76 Among them, compound 136 has an IC50 of 0.05 μM against SARS-CoV Mpro. Through electrospray mass spectrometry experiments, they also suggested an inhibition mechanism identical to that from Wu et al.75 as shown in Figure 29. Compounds 138, 139, 142–145, 147, 149, 151, and 153–155 are 5-chloropyridyl ester-based inhibitors developed by Niu et al.77 Interestingly, these two research groups both reported that 138 has an IC50 value of 0.63 μM. However, the antiviral activity of these compounds was not reported.
The SAR studies by Zhang et al.76 and Niu et al.77 of the compounds in Figure 28 indicate, besides a pyridinyl ring, a second aromatic ring (furan or thiophene) is also a key component for inhibiting SARS-CoV-2 and SARS-CoV Mpro potently.76 More importantly, the positions of the electron-withdrawing substituents on the terminal aromatic rings dramatically impact inhibitory potency. Consistent with the mechanism shown in Figure 29, stronger electron-withdrawing R-groups, through either resonance or inductive effects, render a more electrophilic carbonyl of the ester warhead, resulting in stronger reactivity toward Cys145. For example, the difference in the positions of the N atom of the pyridine moiety relative to the carbonyl group in 145 and 155 causes a 4-fold disparity in their IC50 values. Compound 138 has a nitro group at the para position relative to the furan group. Changing the nitro group of 138 to either the ortho (147) or the meta (153) position leads to a 3-fold or 8-fold increase in the IC50 values, respectively. By contrast, 149, 151, and 154 bear a chloro- or a nitro-substituent on the benzene ring directly connected to the central ester group at the para, ortho, and meta positions relative to the ester bond, respectively. The position of the substituents in these three compounds has less impact on inhibiting SARS-CoV Mpro, although the meta-substituted 154 is a markedly weaker inhibitor than either 149 or 151. Unlike the ketone and aldehyde-based inhibitors, these activated esters suffer from the elimination of a relatively large leaving group that may exhibit undesirable activities itself.
Very recently, Sun et al.80 developed a series of ebselen and ebsulfur derivatives inhibiting SARS-CoV-2 Mpro as shown in Figures 31 and 32. The kinetic progression curves of these isothiazolone-type covalent modifiers showed a biphasic character, indicating that the inhibition follows pseudo-first-order rate kinetics and implying ebselen, ebsulfur, and their derivatives covalently bind to Mpro. Moreover, their assays also showed that these compounds irreversibly inhibit Mpro and do not have significant activity recovery. Ebselen and ebsulfur are the most potent among these compounds with IC50 values of 0.074 μM and 0.11 μM, respectively. Their Ki values are 0.031 μM and 0.078 μM, respectively. Other experiments confirmed that ebselen potently inhibit SARS-CoV-2 with an IC50 of 0.67 μM.81,82 Additionally, Jin et al.81 reported that ebselen has antiviral activity against SARS-CoV-2 with an EC50 of 4.67 μM. The cytotoxicity of ebselen is also extremely low (the oral median lethal dose in rats is >4600 mg/kg).83 Remarkably, the clinical evaluation of ebselen was recently initiated.84
Figure 31.
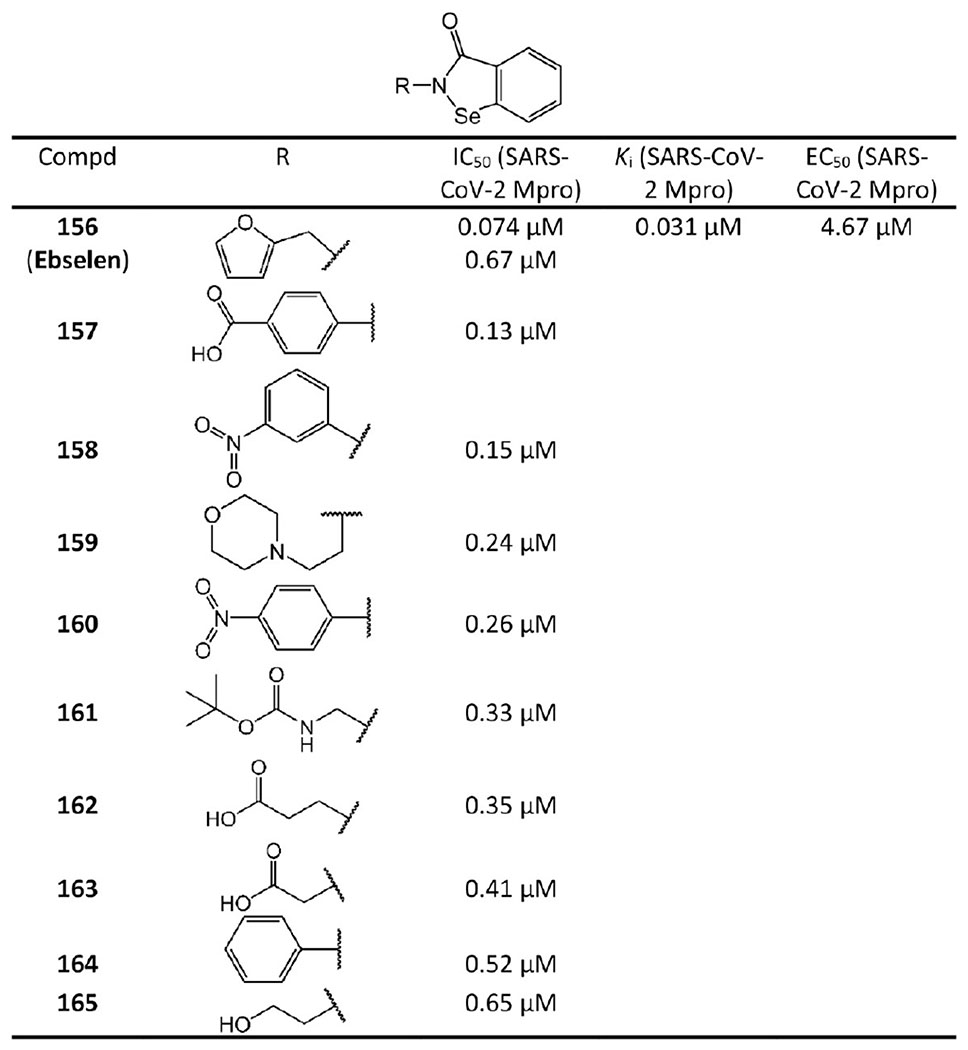
Ebselen derived non-peptidomimetic covalent inhibitors.
Figure 32.
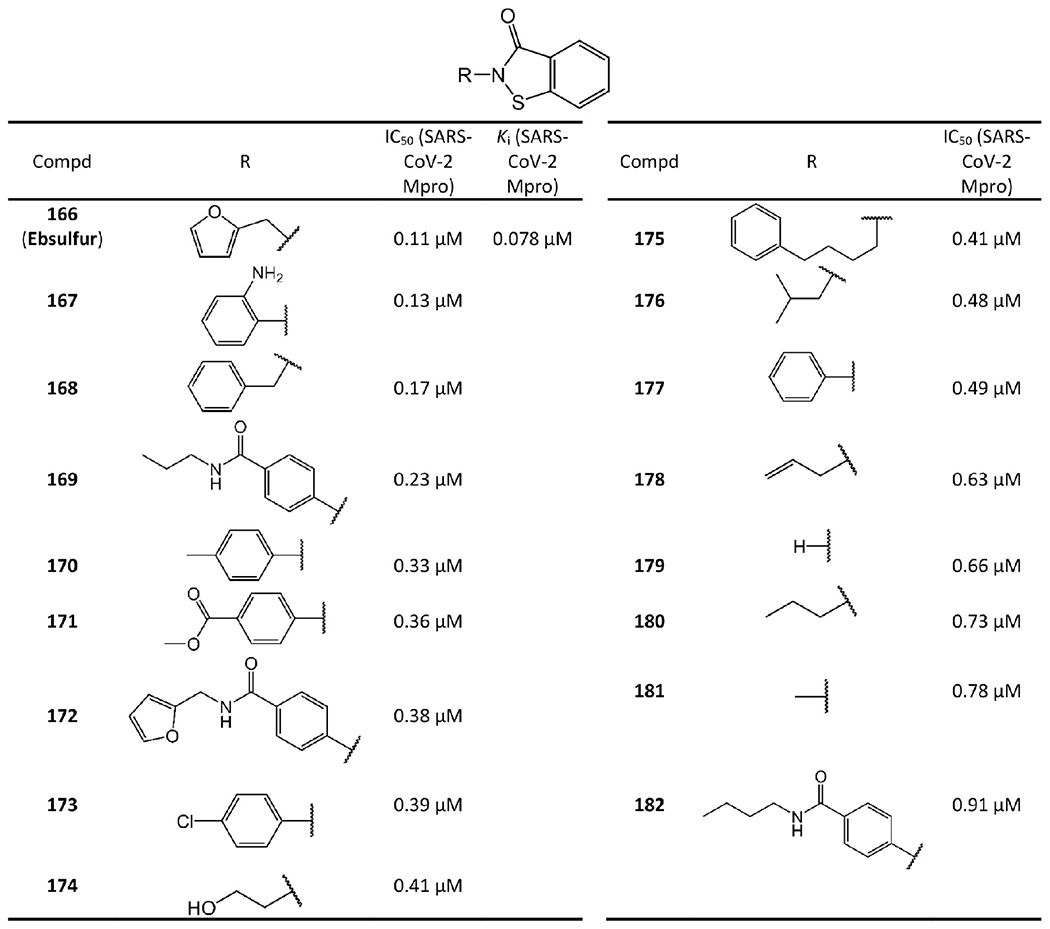
Ebsulfur derived non-peptidomimetic covalent inhibitors.
2.2.1. Drawbacks of the Current Non-peptidomimetic Covalent Inhibitors.
It should be noted that the current non-peptidomimetic covalent inhibitors lack specificity. The current non-peptidomimetic covalent inhibitors all contain isothiazolones. Isothiazolones have been labeled as some of the worst offenders as pan-assay interference compounds (PAINS) due to their high promiscuity.85 Recent assays found that ebselen and some other non-peptidomimetic inhibitors interact with a panel of cysteine proteases.82 Therefore, these leads should be carefully scrutinized to ensure that off-target effects do not cause serious consequences and that the proposed mechanism is responsible for the efficacy observed.
2.3. Noncovalent Mpro Inhibitors.
Since drugs acting through covalent modifications to the target may be associated with off-target liability and consequent potentially toxic effects,12 research efforts have also been devoted to discovering noncovalent Mpro inhibitors.
2.3.1. Aryl Boronic Acid Derivatives.
Some aryl boronic acid derivatives were found to be potent against SARS-CoV Mpro. Compound 183 (Figure 33), bearing anilide linkages, is the best performing compound among aryl boronic acid derivatives (Ki = 0.04 μM).86
Figure 33.
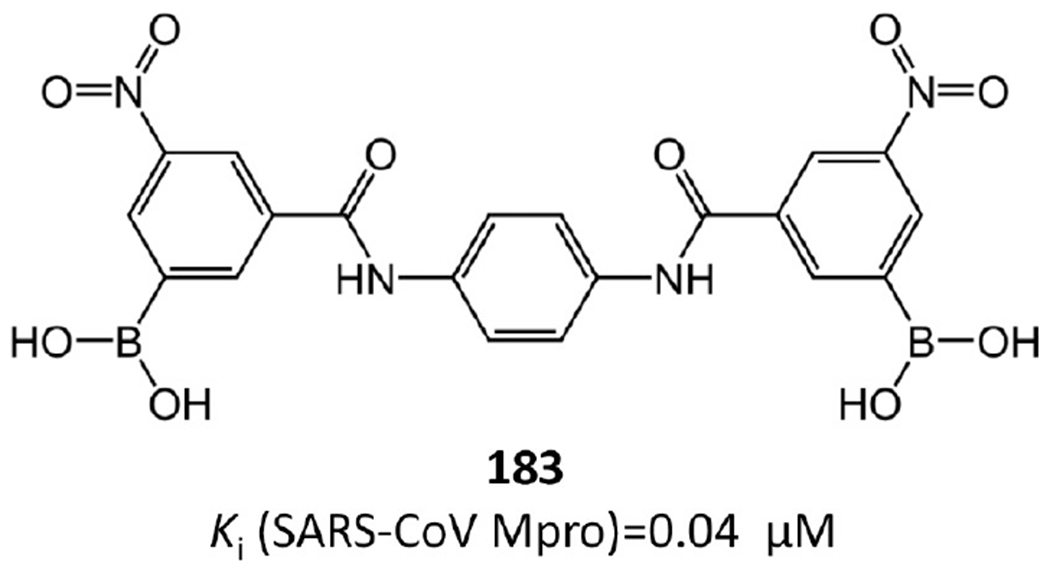
Most potent noncovalent Mpro inhibitor among aryl boronic acid derivatives.
2.3.2. Isatin Derivatives.
Lai et al.87,88 and Juang et al.89 focused on developing N-substituted isatin derivatives as noncovalent SARS-CoV-2 and SARS-CoV Mpro inhibitors (see Figure 34).
Figure 34.
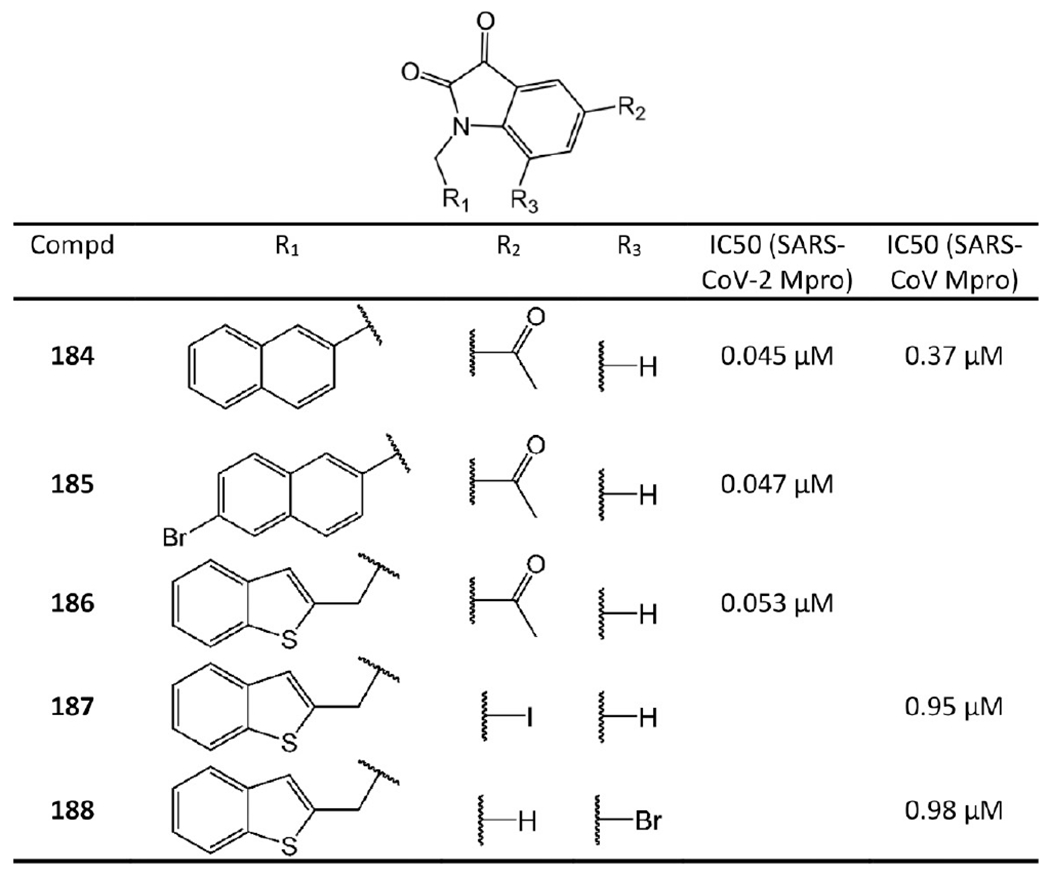
N-Substituted isatin derivatives as noncovalent SARS-CoV-2 and SARS-CoV Mpro inhibitors.
Compounds 184–186 are highly potent isatin inhibitors against SARS-CoV-2 Mpro synthesized by Liu et al.87 For instance, 184 has IC50 of 0.045 μM against SARS-CoV-2 Mpro. Lai et al.88 revealed that 184 inhibits SARS-CoV Mpro with IC50 = 0.37 μM as well. Juang et al.89 also synthesized a series of N-substituted isatin derivatives. Compounds 187 and 188 were found to have promising IC50 values against SARS-CoV.
The extensive SAR investigation by Liu et al.87 provided some suggestion concerning their binding requirements. First, due to space limitation in the Mpro active site, substituents at R1 and R2 must be small. Second, hydrophobic groups at the R1 position are necessary to ensure inhibitory effect. Finally, the carboxamide group at the R2 position is essential for high potency (see Figure 34).
2.3.3. Noncovalent Inhibitors Containing Benzotriazole.
Turlington et al.90 developed a series of noncovalent Mpro inhibitors containing benzotriazole based on their screening against the NIH molecular libraries sample collection. The potent ones are shown in Figure 35.
Figure 35.
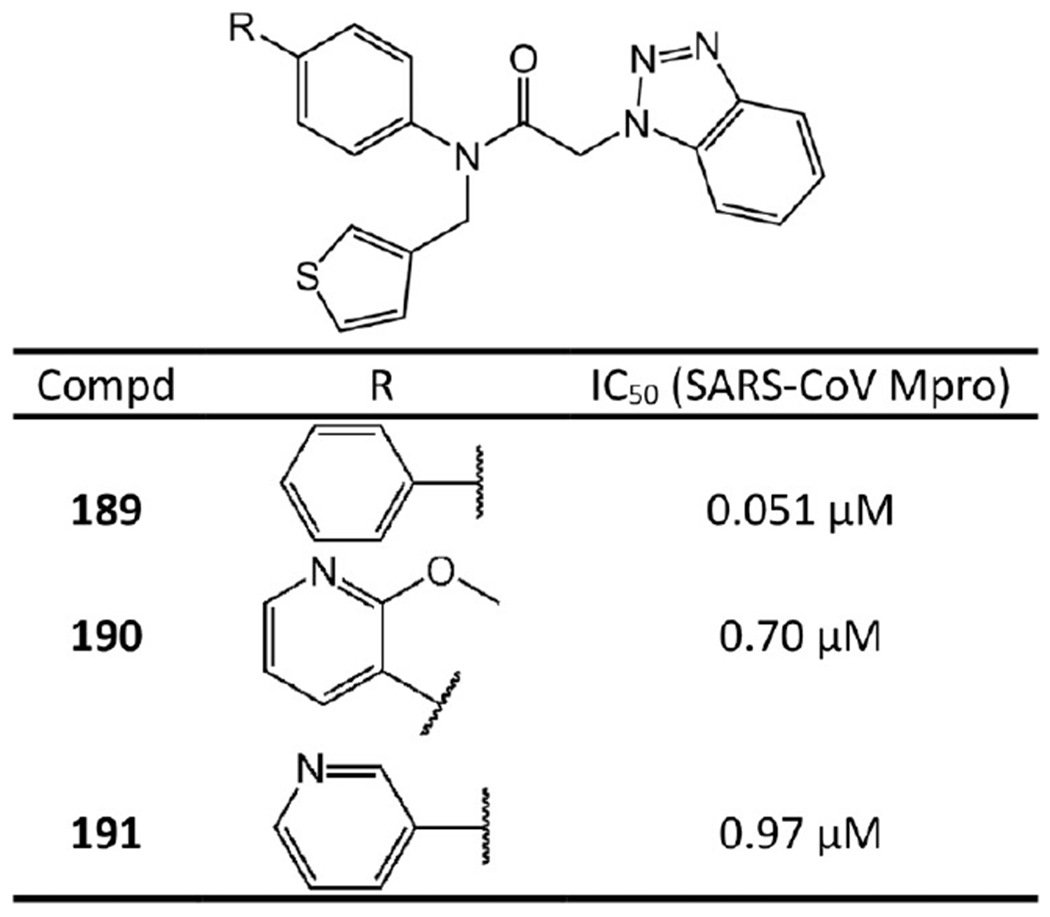
Noncovalent Mpro inhibitors containing benzotriazole.
Notably, based on the crystal structure of SID24808289 with SARS-CoV Mpro (PDB ID 4MDS), Turlington et al.’s SAR study90 suggested that a 3-pyridyl heterocycle (see 190 and 191) has potential to engage a side-chain interaction with the hydroxyl group of Thr24 or Thr25 of Mpro. As a result, 190 and 191 inhibit SARS-CoV Mpro with IC50 values of 0.70 μM and 0.97 μM, respectively. Moreover, Turlington et al.90 unexpectedly found that the parent simple phenyl biaryl (see 189) increases activity by over 700-fold relative to 190 and 191 and represents the first sub-100 nM inhibitor in the series (IC50 = 0.051 μM). This is probably because some unexpected hydrophobic interactions are formed. For example, our docking study suggested that the ring of the diaryl could form hydrophobic interactions with Ala46, Ile43, and Val42 of Mpro.
2.3.4. Anilide Based Inhibitors.
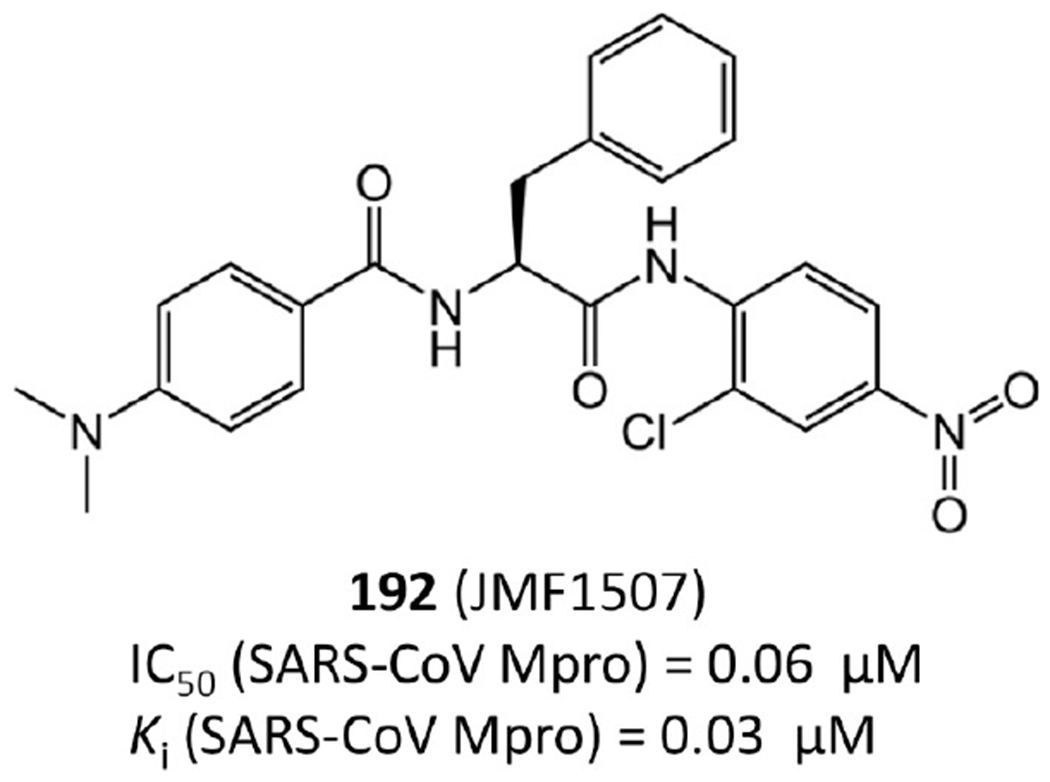
Among anilide based inhibitors, 192 (JMF1507) in Figure 36 displays the best potency with an IC50 of 0.06 μM against SARS-CoV Mpro. It is a competitive noncovalent inhibitor.91
Figure 36.
Most potent anilide based inhibitor.
2.3.5. Noncovalent Aldehyde Peptide Inhibitors.
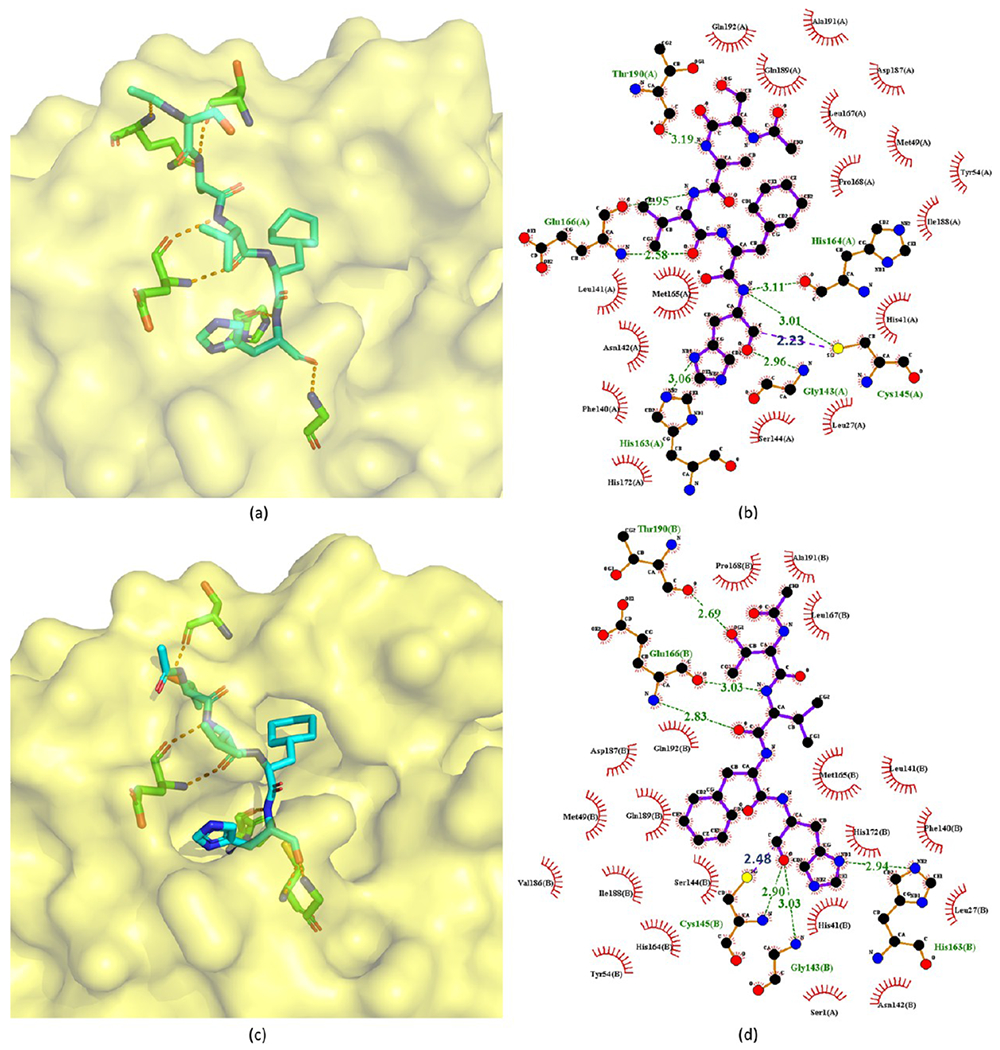
In addition to covalent aldehyde inhibitors described in section 2.1.3, there are also some noncovalent aldehyde peptide inhibitors reported by Akaji et al.92 Covalent bonds are not formed with these inhibitors because the aldehyde groups are too far from the catalytic site Cys145 of SARS-CoV Mpro in the crystal structures. For example, in the crystal structure of 193 with SARS-CoVMpro (PDB ID 3AVZ, Figure 38a,b), the C atom of the aldehyde group is 2.23 Å from the S atom of Cys145, while for 194 (PDB ID 3ATW, Figure 38c,d), this distance is 2.48 Å (see Figure 38). Considering that the length of a regular C–S bond is around 1.8 Å, their distances are not close enough to form strong covalent interactions.
Figure 38.
Crystal structures of 193 (a, b) and 194 (c, d) bound to SARS-CoV Mpro (PDB ID 3AVZ and 3ATW).
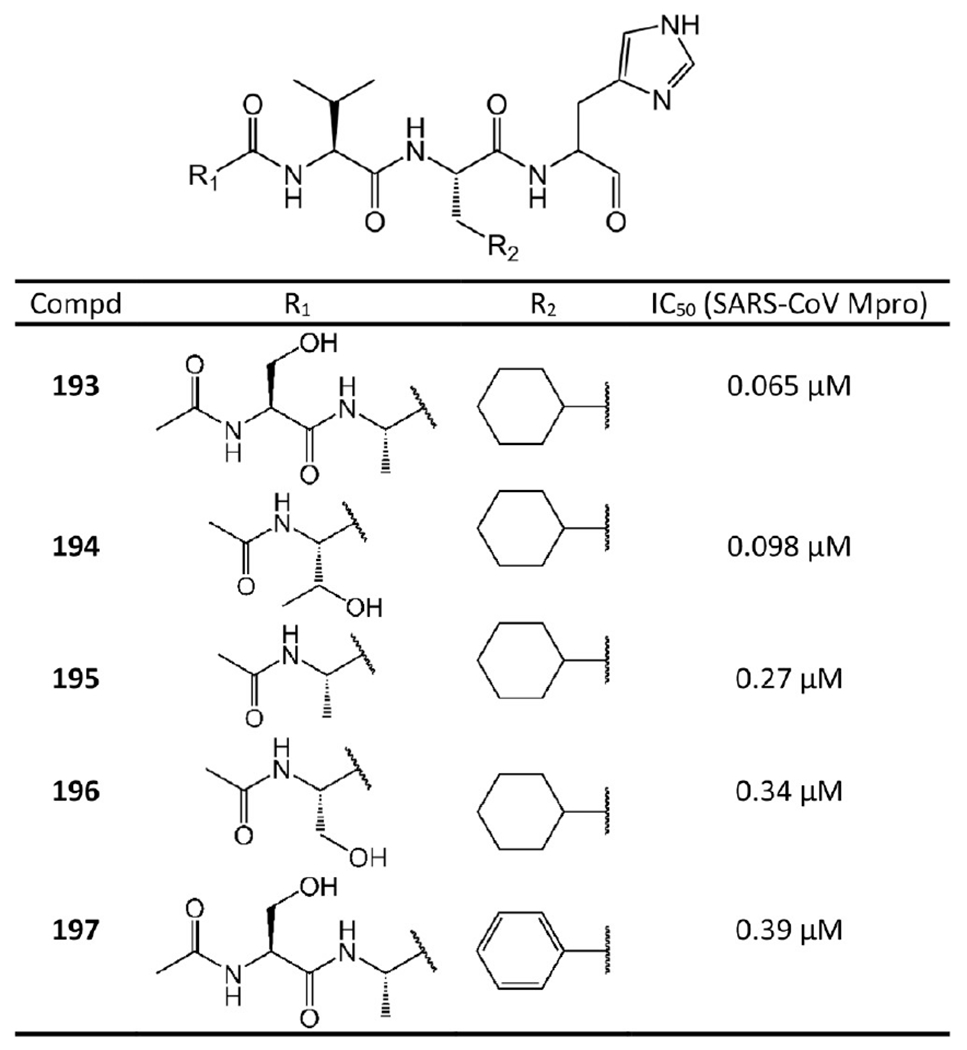
Some of these noncovalent aldehyde inhibitors are still potent against SARS-CoV Mpro (Figure 37). Akaji et al.92 designed these noncovalent aldehyde inhibitors via SAR analysis. First, they found that a bulky group at the side chain of the P2 site could fit into the S2 pocket more tightly than other groups, which leads to 193 and 197 with IC50 values of 0.065 μM and 0.39 μM, respectively. Notably, the comparison between 193 and 197 suggests that the substitution with a cyclohexyl group is more effective than that with a planar aromatic group. This is probably because a cyclohexyl group is more adjustable to more strongly interact with the S2 pocket. Second, the outwardly directed P5 site could be removed to lower the molecular weight of the inhibitors since no interactions of the corresponding side chains to the protease are detected. This strategy resulted in 195, whose activity is a bit better than that of 197. Third, to attach the P5 site-deleted inhibitors to the active-site cleft more tightly, a secondary alcohol moiety was introduced into the side chain of the P4 site to form more hydrogen bonds with Mpro, which results in 194. Compound 194 has almost the same activity (IC50 = 0.098 μM) as 193 (IC50 = 0.065 μM), while the molecular weight is reduced (534 vs 591).
Figure 37.
Noncovalent aldehyde inhibitors.
The potent inhibition of these compounds against Mpro is due to their tight hydrogen-bond interactions with Mpro.92 As shown in the crystal structures of 193 and 194 with SARS-CoV Mpro (see Figure 38), the imidazole N atom at the P1 site accepts a hydrogen bond from Mpro His163. As a result, the other side of the imidazole inserts into the S1 pocket formed by the side chains of Phe140, Leu141, and Glu166. The cyclohexyl group at the P2 site occupies most of the sizable S2 pocket consisting of His41, Met49, Met165, and Asp187. The amide-bound N and carbonyl O atoms of the P3 site have hydrogen bonds with the main chain of Glu166. The amide-bound N atom of the P4 site also accepts another hydrogen bond from Thr190 of Mpro. In the structure of 193 with Mpro, an additional hydrogen bond (3.19 Å) can also be found between the side-chain O atom at the P4 site and Thr190. Notably, along with a secondary alcohol moiety introduced at the P4 site, a strong hydrogen bond (2.69 Å) is formed between this alcohol moiety and Thr190 of Mpro, which agrees with the SAR analysis by Akaji et al.92
2.3.5.1. Drawbacks of Noncovalent Aldehyde Peptide Inhibitors.
Ample evidence suggests that the aldehyde in these agents does not form a stable covalent bond with the protease. Considering the reactive and promiscuous nature of aldehydes in general, it may be advantageous to replace this liability. Compared to the nonpeptidic and noncovalent inhibitors, these peptide-based inhibitors may also be limited by their in vivo instability and poor membrane permeability.
2.3.6. Symmetric Peptides and Molecules.
TL-3 (200 in Figure 39) is a C-2 symmetric peptide-based dimer that was identified as a human immunodeficiency virus (HIV) protease inhibitor. Wu et al.78 found that it is also active against SARS-CoV Mpro with a Ki value of 0.6 μM.
With TL-3 as the lead structure, Shao et al.93 developed several symmetric peptide-based SARS-CoV Mpro inhibitors. Among them, 198 and 199 in Figure 39 have stronger efficacy than TL-3 with Ki values of 0.073 and 0.34 μM, respectively.
Besides symmetric peptides, other symmetric molecules were also identified as SARS-CoV-2 and SARS-CoV Mpro inhibitors. For instance, recently Coelho et al.94 have found that Evans blue, a sulfonic acid-containing dye, is highly active against SARS-CoV-2 Mpro with IC50 = 0.2 μM and Ki = 0.21 μM (201 in Figure 39). Interestingly, Evans blue has been previously reported to inhibit HIV95 and hepatitis B virus (HBV).96 In addition, symmetric inhibitor 202, as one of a series of diarylsulfonyl compounds in Figure 39, has a reported IC50 = 0.9 μM against SARS-CoV Mpro97. However, the acylated phenol 201 is likely susceptible to hydrolysis or other nucleophiles, which may compromise its cellular and in vivo efficacy.
2.3.7. Aromatic-Disulfide Based Inhibitors.
Wang et al.98 synthesized a series of novel aromatic disulfides and evaluated their enzymatic activities against SARS-CoV Mpro. Notably, disulfide compounds have the potential to react with other thiols, including Cys145, and form new -S–S- covalent bonds. However, according to their experiments,98 different aryl thiols derived from the disulfides exhibit no efficacy against SARS-CoV Mpro even at very high concentration. Also, no molecular weight change of Mpro was detected before and after the reactions. These two results suggest that the interaction of Mpro to their aromatic-disulfide based inhibitors is of a noncovalent nature.
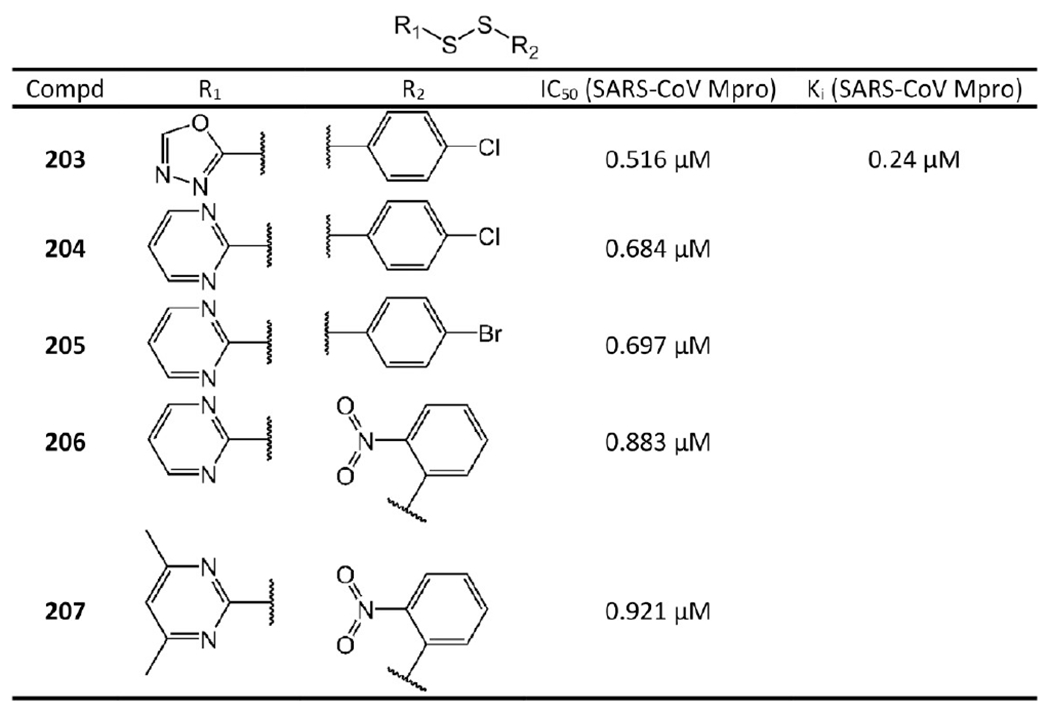
Aromatic disulfide-based inhibitors with potency in the micromolar range are illustrated in Figure 40. The best IC50 value among them is 0.516 μM (203).
Figure 40.
Aromatic-disulfide based inhibitors.
2.3.7.1. Drawbacks of Aromatic-Disulfide Based Inhibitors.
No cellular activity was reported for the aromatic-disulfide based inhibitors. It would be interesting to see the translational efficacy of these agents as multiple reductases and high levels of endogenous glutathione are known to readily clean disulfides in vivo, which may significantly affect the overall efficacy of aromatic-disulfide based inhibitors.99 These electrophilic aryl disulfide-based species are also prone to nucleophilic attack by other species, and therefore, undesirable sulfenylation events are likely to occur.
2.3.8. Other Noncovalent Mpro Inhibitors.
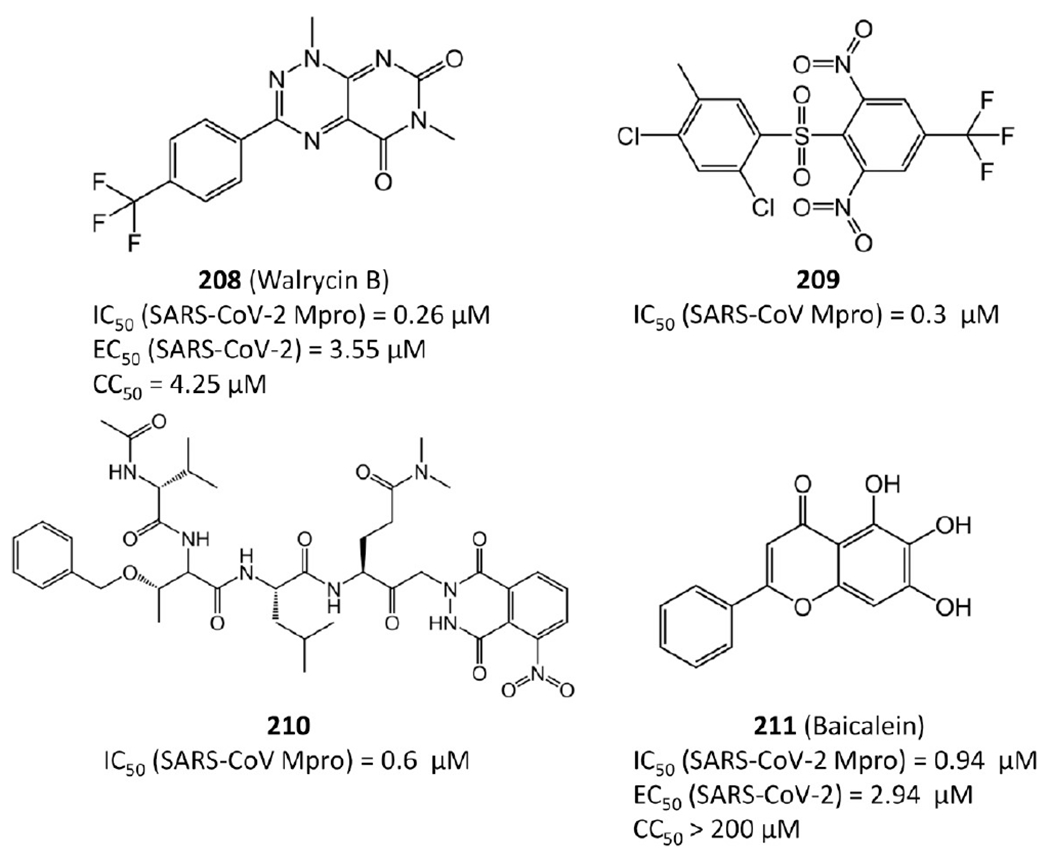
Walrycin B (208 in Figure 41) is an analog of toxoflavin. Recent repurposing work100 found that walrycin B also strongly inhibits SARS-CoV-2 Mpro with IC50 = 0.26 μM and more importantly shows potent antiviral activity against SARS-CoV-2 with EC50 = 3.55 μM. However, its CC50 of 4.25 μM suggests it may be highly cytotoxic.
Figure 41.
Other noncovalent Mpro inhibitors.
Compound 209 was identified from high-throughput screening performed by Lu et al.97 and has IC50 = 0.3 μM against SARS-CoV Mpro. Compound 210 belongs to a series of keto-glutamine analogs synthesized by Jain et al.101 and displays reversible inhibition against SARS-CoV Mpro with IC50 = 0.6 μM. The flavonoid baicalein (211) is one ingredient of traditional Chinese medicine shuanghuanglian. Su et al.102 reported that it also exhibits activity against SARS-CoV-2 Mpro with an IC50 of 0.94 μM and antiviral activity with an EC50 of 2.94 μM. However, its cytotoxicity was reported with CC50 = 86 μM in Vero cells.103
The crystal structures of 209 and 211 (baicalein) bound to SARS-CoV or SARS-CoV-2 Mpro are available with PDB ID 2GZ7 and 6M2N, respectively. In crystal structure 2GZ7 (Figure 42a,b), 209 occupies the S3–S5 pockets of SARS-CoV Mpro. The 2,4-dichloro-5-methylbenzene group sits deeply inside the hydrophobic pocket consisting of Pro39, His41, Cys145, His163, His164, Phe181, Tyr182, and Phe185. The phenyl ring of 208 has strong π–π interactions with the side chain of His41, while the dicholoro and methyl groups are close to Cys145, His164, Pro39, and Leu27. Moreover, the 1,3-dinitro-5-(trifluoromethyl) benzene group forms strong hydrogen-bond interactions with Mpro. In particular, one nitro group has a hydrogen bond with the N atom in the side chain of His41 and two hydrogen bonds with Met49 and His41 through a water molecule. The trifluoromethyl substituent generates a weak hydrogen bond with Gln192 and is adjacent to Gln192, Gln189, Leu167, and Met165. Moreover, the benzene group forms hydrophobic interactions with Met165, while the sulfone group has hydrogen-bond interactions with a water molecule.97
Figure 42.
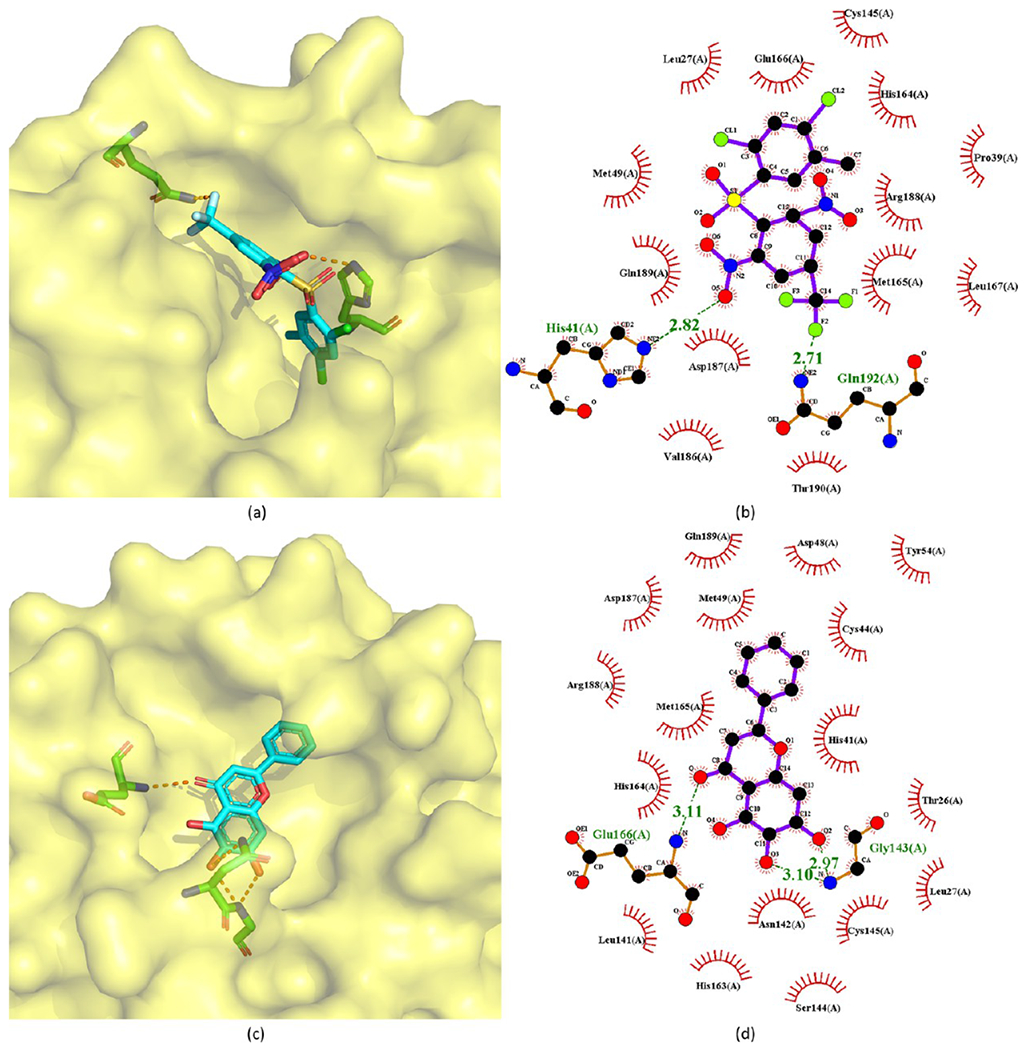
Crystal structures of 209 (a, b) and 211 (c, d) bound to SARS-CoV-2 Mpro (PDB ID 2GZ7 and 6M2N).
In the crystal structure of baicalein (211) in complex with SARS-CoV-2 Mpro, the baicalein binds to the surface of the protease between domains I and II, which is the core region of the binding site (Figure 42c,d). Three phenolic hydroxyl groups of baicalein form multiple hydrogen bonds with the main chains of Leu141 and Gly143 and the side chains of Ser144 and His163. The carbonyl group of baicalein leads to a hydrogen bond with the main chain of Glu166, while the free phenyl ring is inside the S2 subsite and forms hydrophobic interactions with Gln189, Arg188, Met49, Cys44, and His41. Notably, in addition to the hydrophobic interactions, the catalytic His41 and Cys145 also establish S–π and π–π interactions with the aromatic rings of baicalein. The side chain of Asn142 also forms NH2–π interactions with baicalein. These interactions, together with the interactions from Cys145, sandwich the phenyl ring of baicalein with three OH groups between Asn142 and Cys145. Moreover, Met165 has hydrophobic interactions with the middle ring of baicalein. As a result, baicalein blocks two catalytic residues, as well as the oxyanion loop (residues 138–145), Glu166, and the S1 and S2 subsites, thereby preventing Mpro from recognizing substrates.
2.3.8.1. Drawbacks of Walrycin B and Baicalein.
Walrycin B (208) is not only highly cytotoxic but also an analog of toxoflavin, which is a well-known offender in the arena of PAINS.85 Considering its promiscuous reactivity, the observed efficacy and how it correlates to a proposed mechanism should therefore be treated with severe caution. Baicalein (211) is also reported to have some cytotoxicity. More importantly, baicalein has promiscuity risks and is known to inhibit numerous substrates.102
2.4. Metal Conjugated SARS-CoV-2 or SARS-CoV Mpro Inhibitors.
As shown in Figure 43, 212–218 with conjugated Zn2+ or Hg2+ ions also show inhibitory potency in the sub-micromolar range against SARS-CoV-2 Mpro or SARS-CoV Mpro.94,104,105 Notably, the crystal structures of metal conjugated inhibitors bound to Mpro reveals that, after binding, the inhibitors are always dissociated and the metals covalently attach to Mpro residues such as Cys145 (see Figure 44).
Figure 43.
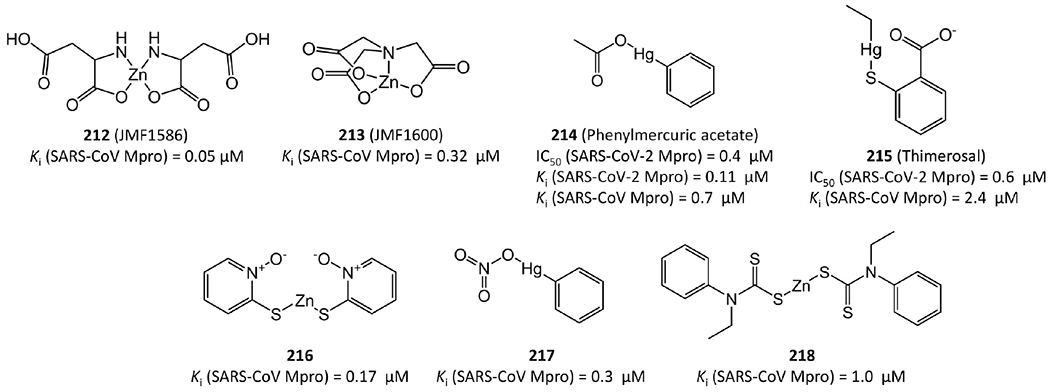
Metal conjugated SARS-CoV-2 or SARS-CoV Mpro inhibitors.
Figure 44.
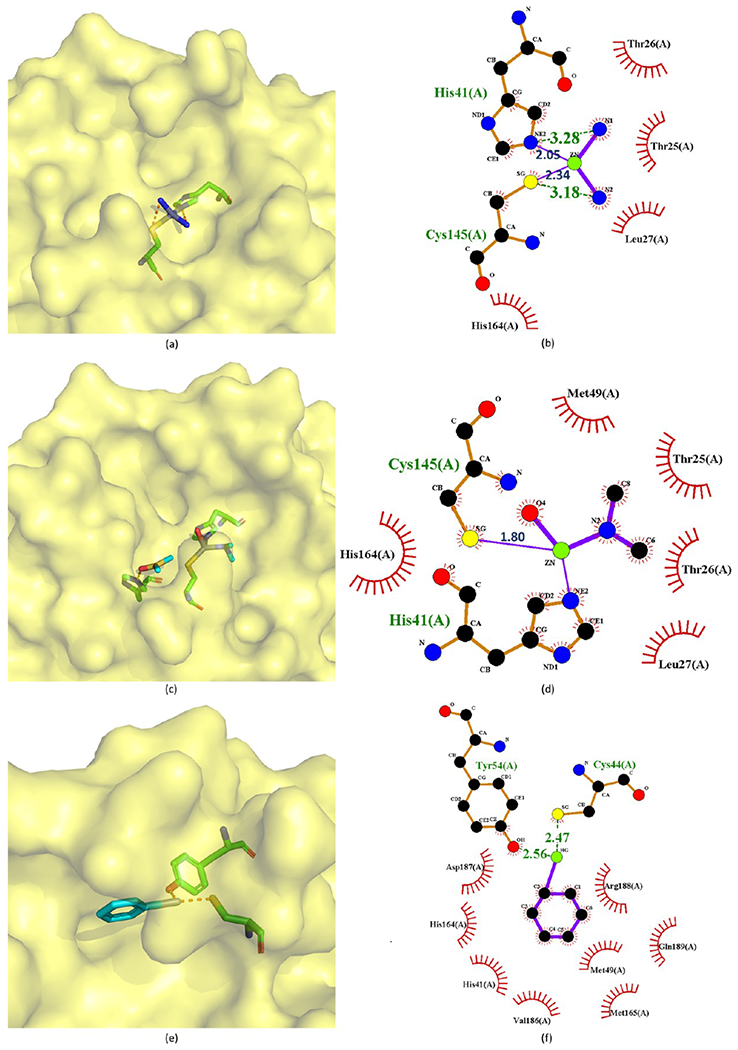
Crystal structures of 212 (a, b), 213 (c, d), and 214 (e, f) bound to SARS-CoV-2 Mpro (PDB ID 2Z9L, 2Z9K and 2Z9G).
Compounds 212–214 are good examples to depict their binding modes to Mpro. In the Mpro–JMF1586 (212) complex (Figure 44a,b), His41, Cys145, and two N atoms of the compound make up the Zn-centered tetrahedral coordination, in which two N and two O atoms chelate the Zn atom. In the Mpro–JMF1600 (213) complex (Figure 44c,d), His41, Cys145, one N atom, and a water molecule participate in the Zn coordination. The Zn atom is chelated by one N and three O atoms of JMF1600. A Zn–N bond is stronger than a Zn–O bond, which is consistent with the lower Ki value of JMF1586 than JMF1600. Both these 3D structures indicate that the metal–O bonds of JMF1586 and JMF1600 must be broken before being replaced by His41 and Cys145 to form the Zn-centered complex. In the Mpro–phenylmercuric acetate (214) complex structure (Figure 44e,f), the phenyl-bound mercury is bound to the S atom of Cys44 with a bond distance of 2.47 Å. The phenolic O atom of Tyr54 is another site to accept a 2.56 Å Hg–O bond from 214, while its acetate group is dissociated and substituted by protein residues.
2.4.1. Perspectives on Metal Conjugated Inhibitors.
Metal conjugated inhibitors provide an interesting class of lead agents and are often improperly labeled as toxic agents. However, phenylmercuric acetate and thimerosal are used as pharmaceutical excipients, and several other metal conjugates have been used as topical antimicrobial preservatives. The anticipated toxicity of metals depends on many factors, including their oxidation states.
3. DISCUSSION AND PERSPECTIVES
COVID-19 caused by SARS-CoV-2 is one of the most deadly pandemics since the new millennium. It has overwhelmed the worldwide healthcare system and devastated the global economy. Currently, there is no approved medication against the virus; thus it is imperative to discover potential therapeutic agents against SARS-CoV-2.
The main protease (Mpro) is not only critical for viral replication but also highly conserved in viral evolution. Therefore, Mpro is one of the most attractive targets for developing antiviral therapies. A large number of Mpro inhibitors have been reported since the outbreak of SARS-CoV in 2003. To provide a whole landscape of the current status of SARS-CoV-2 and SARS-CoV inhibitors, here we collect all available SARS-CoV-2 and SARS-CoV Mpro inhibitors so far from literature or databases. We highlight highly potent inhibitors, analyze their structures and functions, and illustrate their covalent or noncovalent binding interactions with Mpro. The optimization of inhibitors to improve potency is also discussed. To serve as a good starting point to design new drugs against SARS-CoV-2, we classify Mpro inhibitors into different categories based on their molecular mechanisms of action, such as small-molecule covalent inhibitors, peptidomimetic covalent inhibitors, non-covalent inhibitors, and metal conjugated inhibitors.
3.1. Drug Development Potential of Existing SARS-CoV-2 and SARS-CoV Mpro Inhibitors.
So far, more than 800 SARS-CoV-2 and SARS-CoV Mpro inhibitors are available, and this data set is provided in the Supporting Information. The classification of these inhibitors is summarized in Table 1. Considering that the binding sites of SARS-CoV-2 and SARS-CoV Mpro are almost identical, SARS-CoV Mpro inhibitors are also effective to SARS-CoV-2.
Table 1.
Classification of Current SARS-CoV-2 and SARS-CoV Mpro Inhibitors
| class | subclass | ref |
|---|---|---|
| covalent | peptidomimetic ketone | 26,34,35,38–40,106–112 |
| peptidomimetic nitrile-containing | 43 | |
| peptidomimetic aldehyde | 45,48,49,51–53 | |
| peptides with aldehyde bisulfite | 50,65,113 | |
| peptidomimetic michael acceptors | 29,64,66,67,114–116 | |
| nonpeptidomimetic covalent inhibitors | 74–77,79,80,117 | |
| noncovalent | aryl boronic acid derivative | 86 |
| istin based | 87–89,118 | |
| benzotriazole based | 90 | |
| anilide | 91 | |
| aldehyde | 92 | |
| symmetric peptide or molecule | 78,93 | |
| aromatic disulfide | 98 | |
| ketone | 119 | |
| others | 4,81,82,94,97,100–102,120–155 | |
| metal conjugated | 94,104,105 |
However, many Mpro inhibitors cannot be developed into effective drugs due to their unqualified potency, promiscuity, toxicity, side effects, or poor ADME properties. Current inhibitors with drug development potential fall mostly in the class of peptidomimetic covalent inhibitors. The most promising ones are nitrile-containing inhibitor PF-07321332 and HMK inhibitor PF-00835231. Both of them are now in clinical trials. In particular, PF-07321332 is projected to reach the market relatively soon. Aldehyde bisulfite GC376 was also reported to have druggable potential but may require further evaluation to determine if aldehyde bisulfates offer the selectivity profile needed for clinical development.
High-throughput screening efforts have identified many hits, which have resulted in many rapid reports. Unfortunately, this has also resulted in the identification of many known PAINS as possible drug leads. For example, there are several non-peptidomimetic covalent inhibitors that contain the classic isothiazolone scaffold. These isothiazolones have also been labeled as some of the worst offenders as PAINS due to their high promiscuity. Toxoflavin is another well-known offender in the arena of PAINS. A reactive aldehyde functionality could possibly render promiscuity, too. Therefore, caution should be taken in the pursuit of these agents as SARS-CoV-2 Mpro inhibitors.
3.2. Mpro Mutation Impact on Inhibitor Efficacy.
We collected 1 983 328 complete SARS-CoV-2 sequences with exact collection dates submitted to GISAID up to September 20, 2021. After applying the multiple sequence alignment (MSA) technique, we obtained the single nucleotide polymorphism (SNP) information on these 1 983 328 complete SARS-CoV-2 sequences, in which a total of 28 780 unique single mutations on the whole SARS-CoV-2 genome were identified. Among them, 1395 unique single mutations were detected on Mpro, where 906 unique single mutations were missense mutations and 489 unique single mutations were silent mutations.
Although SARS-CoV-2 Mpro admits more than ten different inhibitor binding sites, there is only one major inhibition site.30 The crystal structure of SARS-CoV-2 Mpro in complex with boceprevir is available with PBD ID 7C6S. We identify the binding domain of Mpro to be the residues located in the sphere of 10 Å radius around the center of small molecule boceprevir. Then, the number of silent mutations (Nsilent) and missense mutations (NMissense) in the binding domain can be found in Table 2. A total of 78 missense mutations were detected in the binding domain of Mpro, and we list the top 10 highest frequency missense mutations in the binding domain and nonbinding domain of Mpro in Table 3 and Table 4. It can be seen that the frequency of missense mutations in the binding domain of Mpro is much lower than that in the nonbinding domain of Mpro, indicating that the binding domain is more conserved than the nonbinding domain. The 10202→T-(L50F) mutation with the highest frequency in the binding domain of Mpro only has a ratio of 0.00049.
Table 2.
Number of the Silent Mutations (Nsilent), Missense Mutations (Nmissense), and Unique Single Mutations (Ntotal) on the Mpro, Binding Domain of Mpro, and Non-binding Domain of Mpro
| location | N silent | N missense | N total |
|---|---|---|---|
| Mpro | 489 | 906 | 1395 |
| binding domain | 59 | 78 | 137 |
| non-binding domain | 430 | 828 | 1258 |
Table 3.
Top 10 Missense Mutations on the Binding Domain of Mproa
| SNP | protein mutation | total frequency | ratio |
|---|---|---|---|
| 10202→T | L50F | 969 | 0.00049 |
| 10610→T | V186F | 749 | 0.00038 |
| 10188→T | T45I | 636 | 0.00032 |
| 10623→T | T190I | 601 | 0.00030 |
| 10201→T | M49I | 427 | 0.00022 |
| 10191→T | S46F | 416 | 0.00021 |
| 10128C→T | T25I | 93 | 0.00005 |
| 10617G→A | R188K | 77 | 0.00004 |
| 10190T→C | S46P | 52 | 0.00003 |
| 10196G→A | D48N | 51 | 0.00003 |
The frequency is the number of complete SARS-CoV-2 sequences that carry a specific mutation, and the ratio is defined by the frequency of a specific mutation over the total number of complete SARS-CoV-2 sequences.
Table 4.
Top 10 Missense Mutations in the Nonbinding Domain of Mproa
| SNP | protein mutation | total frequency | ratio |
|---|---|---|---|
| 10319C→T | L89F | 60078 | 0.03029 |
| 10323A→G | K90R | 38117 | 0.01922 |
| 10097G→A | G15S | 9695 | 0.00489 |
| 10833C→T | A260V | 5068 | 0.00256 |
| 10376C→T | P108S | 4837 | 0.00244 |
| 10667T→G | L205V | 4135 | 0.00208 |
| 10193G→A | E47K | 3947 | 0.00199 |
| 10195A→T | E47D | 3880 | 0.00196 |
| 10277C→T | L75F | 2985 | 0.00151 |
| 10533G→T | C160F | 2904 | 0.00146 |
The frequency is the number of complete SARS-CoV-2 sequences that carry a specific mutation, and the ratio is defined by the frequency of a specific mutation over the total number of complete SARS-CoV-2 sequences.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NIH grant GM126189, NSF Grants DMS-2052983, DMS-1761320, and IIS-1900473, NASA 80NSSC21M0023, Michigan Economic Development Corporation, George Mason University award PD45722, Bristol-Myers Squibb 65109, and Pfizer.
ABBREVIATIONS
- 3CLpro
3-chymotrypsin-like protease
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- AUC
area under the curve
- COVID-19
Coronavirus Disease 2019
- CRFK
Crandell–Rees feline kidney cells
- CYP
cytochrome P450
- FIP
feline infectious peritonitis
- FRET
fluorescence resonance energy transfer
- GISAID
global initiative on sharing all influenza data
- hERG
human ether-a-go-go
- HIV
human immunodeficiency virus
- HMK
hydroxymethylketone
- MERS-CoV
Middle East respiratory syndrome coronavirus
- Mpro
main protease
- NI
no inhibition
- NIH
National Institutes of Health
- NSP
nonstructural protein
- ORF
open-reading frame
- PAINS
pan-assay interference compounds
- PDB
protein data bank
- PK
pharmacokinetics
- PLpro
papain-like protease
- RT-PCR
reverse transcription polymerase chain reaction
- SAR
structure–activity relationship
- SARS-CoV
severe acute respiratory syndrome coronavirus
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SNP
single nucleotide polymorphism
Biographies
Biographies
Kaifu Gao obtained his Ph.D. degree in physical chemistry from Chinese Academy of Sciences and completed his postdoctoral studies at the University of California, San Diego, under the guidance of Prof. Michael Gilson. His Ph.D. and postdoctoral studies focused on MD simulations of protein conformational transitions and binding free energy calculations. Now, he is a research associate in Prof. Guo-Wei Wei’s group at Michigan State University. His current research concerns the application of deep learning to biological science and drug discovery, especially, automated drug-like compound generation and drug property prediction. His deep learning models have already been applied to the design and screening of SARS-CoV-2 Mpro inhibitors.
Rui Wang received her B.S. degree in mathematics at Xian Jiaotong University in 2017. Later, she joined Dr. Guo-Wei Wei’s group at Michigan State University (MSU). Currently, she is a fifth-year Ph.D. candidate in the Department of Mathematics at MSU, and she expects to finish her Ph.D. study in July 2022. Her methodological research focuses on the development of mathematical tools for the descriptive and predictive modeling of biomolecules. She has also worked on genomics analysis and mathematical modeling of infectious diseases. She is highly interested in integrating AI, mathematics, genomics, biophysics, bioinformatics, and experimental data to tackle challenges in biological sciences, human diseases, and infectious pathogens.
Jiahui Chen received his Ph.D. degree from Southern Methodist University, where his research focused on the implementation of mathematical methods for biophysics, including the Poisson–Boltzmann equation. After graduation, he joined the Department of Mathematics at Michigan State University as a research associate in Professor Guo-Wei Wei’s group. His current research focuses on topological and geometrical data analysis and machine learning algorithms with their modeling and application to biomolecules. His model studies were applied to prediction of mutation-induced binding free energy changes of the SARS-CoV-2 spike protein binding to ACE2 and antibodies.
Jetze J. Tepe is a Professor at the Department of Chemistry and Pharmacology & Toxicology at Michigan State University. He received his Ph.D. in chemistry from the University of Virginia (T.L. Macdonald) and completed his postdoctoral studies at Colorado State University (R. M. Williams). Research in his lab is focused on the synthesis of marine sponge metabolites and their unnatural drug-like analogs as antagonists or agonists of proteasome function. By development of new synthetic methodologies, drug-like derivatives of natural products are prepared and interrogated for their clinical significance in vitro, in cell culture, and in animal models, with a therapeutic focus on oncology and neurodegenerative diseases.
Faqing Huang received his Ph.D. degree in 1994 from Duke University, where he investigated the incorporation of boron into nucleic acids under the direction of Dr. Barbara Ramsay Shaw. As an NIH postdoctoral fellow under the mentorship of Dr. Michael Yarus at the University of Colorado at Boulder, he focused on the isolation of novel ribozymes that may shed light on the origin of life. He joined the faculty of the Department of Chemistry and Biochemistry at the University of Southern Mississippi in 1998. His interdisciplinary research interests include the origin and early evolution of life, RNA catalysis, chemical biology, targeted cancer therapeutics, antiviral strategies, innate immunity, novel cloning strategies, and self-amplifying mRNA based vaccines.
Guo-Wei Wei earned his Ph.D. degree from the University of British Columbia in 1996. He was awarded a fellowship from the NSERC of Canada to pursue his postdoctoral work at the University of Houston. In 1998, he joined the faculty of the National University of Singapore and was promoted to Associate Professor in 2001. In 2002, he relocated to Michigan State University, where he is an MSU Foundation Professor of Mathematics, Electrical and Computer Engineering, and Biochemistry and Molecular Biology. His current research interests include mathematical biosciences, deep learning, drug discovery, and computational geometry, topology, and graphing. He has advised over 150 research students, postdoctoral associates, and visiting scientists. Dr. Wei has served extensively on a wide variety of national and international panels, committees, and journal editorships.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00409.
Prediction of ADMET, PK, and druggability properties and summary of the 817 SARS-CoV-2 and SARS-CoV Mpro inhibitors with SMILES strings, potency information, cytotoxicity, structure availability with PDB ID, names, and references (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c00409
The authors declare no competing financial interest.
Contributor Information
Kaifu Gao, Department of Mathematics, Michigan State University, East Lansing, Michigan 48824, United States.
Rui Wang, Department of Mathematics, Michigan State University, East Lansing, Michigan 48824, United States.
Jiahui Chen, Department of Mathematics, Michigan State University, East Lansing, Michigan 48824, United States.
Jetze J. Tepe, Department of Chemistry and Pharmacology & Toxicology, Michigan State University, East Lansing, Michigan 48824, United States.
Faqing Huang, Department of Chemistry and Biochemistry, University of Southern Mississippi, Hattiesburg, Mississippi 39406, United States.
Guo-Wei Wei, Department of Mathematics, Department of Biochemistry and Molecular Biology, and Department of Electrical and Computer Engineering, Michigan State University, East Lansing, Michigan 48824, United States.
REFERENCES
- (1).Shin MD; Shukla S; Chung YH; Beiss V; Chan SK; Ortega-Rivera OA; Wirth DM; Chen A; Sack M; Pokorski JK; Steinmetz NF COVID-19 vaccine development and a potential nanomaterial path forward. Nat. Nanotechnol 2020, 15, 646–655. [DOI] [PubMed] [Google Scholar]
- (2).Zimmer C; Corum J; Wee S-L Coronavirus Vaccine Tracker. https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html, 2020.
- (3).Wang R; Chen J; Gao K; Wei G-W Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries. Genomics 2021, 113,2158–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jo S; Kim S; Yoo J; Kim M-S; Shin DH A study of 3CLpros as promising targets against SARS-CoV and SARS-CoV-2. Microorganisms 2021, 9, 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Halford B Pfizer unveils its oral SARS-CoV-2 inhibitor. https://cen.acs.org/acs-news/acs-meeting-news/Pfizer-unveils-oral-SARS-CoV/99/i13, 2020.
- (6).Halford B Pfizer’s novel COVID-19 antiviral heads to clinical trials. https://cen.acs.org/pharmaceuticals/drug-discovery/Pfizers-novel-COVID-19-antiviral/98/web/2020/09, 2020.
- (7).Lansdowne LE Exploring the Drug Development Process. https://www.technologynetworks.com/drug-discovery/articles/exploring-the-drug-development-process-331894, 2020.
- (8).Lu R; Zhao X; Li J; Niu P; Yang B; Wu H; Wang W; Song H; Huang B; Zhu N; Bi Y; Ma X; Zhan F; Wang L; Hu T; Zhou H; Hu Z; Zhou W; Zhao L; Chen J; Meng Y; Wang J; Lin Y; Yuan J; Xie Z; Ma J; Liu WJ; Wang D; Xu W; Holmes EC; Gao GF; Wu G; Chen W; Shi W; Tan W Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Walls AC; Park Y-J; Tortorici MA; Wall A; McGuire AT; Veesler D Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wrapp D; Wang N; Corbett KS; Goldsmith JA; Hsieh C-L; Abiona O; Graham BS; McLellan JS Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gao K; Wang R; Chen J; Cheng L; Frishcosy J; Huzumi Y; Qiu Y; Schluckbier T; Wei G-W Methodology-centered review of molecular modeling, simulation, and prediction of SARS-CoV-2. arXiv preprint, arXiv:2102.00971, 2021, https://arxiv.org/abs/2102.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ghosh AK; Brindisi M; Shahabi D; Chapman ME; Mesecar AD Drug development and medicinal chemistry efforts toward SARS-coronavirus and Covid-19 therapeutics. ChemMedChem 2020, 15, 907–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cannalire R; Cerchia C; Beccari AR; Di Leva FS; Summa V Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem 2020, DOI: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ullrich S; Nitsche C The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett 2020, 30, 127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Paul A; Sarkar A; Saha S; Maji A; Janah P; Kumar Maity T Synthetic and computational efforts towards the development of peptidomimetics and small-molecule SARS-CoV 3CLpro inhibitors: a review. Bioorg. Med. Chem 2021, 46, 116301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Amin SA; Banerjee S; Gayen S; Jha T Protease targeted COVID-19 drug discovery: What we have learned from the past SARS-CoV inhibitors? Eur. J. Med. Chem 2021, 215, 113294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pillaiyar T; Manickam M; Namasivayam V; Hayashi Y; Jung S-H An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem 2016, 59, 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kumar V; Jung Y-S; Liang P-H Anti-SARS coronavirus agents: a patent review (2008-present). Expert Opin. Ther. Pat 2013, 23, 1337–1348. [DOI] [PubMed] [Google Scholar]
- (19).Wang H-M; Liang P-H Pharmacophores and biological activities of severe acute respiratory syndrome viral protease inhibitors. Expert Opin. Ther. Pat 2007, 17, 533–546. [Google Scholar]
- (20).Liang P-H Characterization and inhibition of SARS-coronavirus main protease. Curr. Top. Med. Chem 2006, 6, 361–376. [DOI] [PubMed] [Google Scholar]
- (21).Wu F; Zhao S; Yu B; Chen Y-M; Wang W; Song Z-G; Hu Y; Tao Z-W; Tian J-H; Pei Y-Y; Yuan M-L; Zhang Y-L; Dai F-H; Liu Y; Wang Q-M; Zheng J-J; Xu L; Holmes EC; Zhang Y-Z A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Marra MA; Jones SJM; Astell CR; Holt RA; Brooks-Wilson A; Butterfield YSN; Khattra J; Asano JK; Barber SA; Chan SY; Cloutier A; Coughlin SM; Freeman D; Girn N; Griffith OL; Leach SR; Mayo M; Mc-Donald H; Montgomery SB; Pandoh PK; Petrescu AS; Robertson AG; Schein JE; Siddiqui A; Smailus DE; Stott JM; Yang GS; Plummer F; Andonov A; Artsob H; Bastien N; Bernard K; Booth TF; Bowness D; Czub M; Drebot M; Fernando L; Flick R; Garbutt M; Gray M; Grolla A; Jones S; Feldmann H; Meyers A; Kabani A; Li Y; Normand S; Stroher U; Tipples GA; Tyler S; Vogrig R; Ward D; Watson B; Brunham RC; Krajden M; Petric M; Skowronski DM; Upton C; Roper RL The genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. [DOI] [PubMed] [Google Scholar]
- (23).Forni D; Cagliani R; Clerici M; Sironi M Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017, 25, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen S; Chen L; Tan J; Chen J; Du L; Sun T; Shen J; Chen K; Jiang H; Shen X Severe acute respiratory syndrome coronavirus 3C-like proteinase N terminus is indispensable for proteolytic activity but not for enzyme dimerization: biochemical and thermodynamic investigation in conjunction with molecular dynamics simulations. J. Biol. Chem 2005, 280, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lee J; Worrall LJ; Vuckovic M; Rosell FI; Gentile F; Ton A-T; Caveney NA; Ban F; Cherkasov A; Paetzel M; Strynadka NCJ Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun 2020, 11 , 5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hoffman RL; Kania RS; Brothers MA; Davies JF; Ferre RA; Gajiwala KS; He M; Hogan RJ; Kozminski K; Li LY; Lockner JW; Lou J; Marra MT; Mitchell LJ; Murray BW; Nieman JA; Noell S; Planken SP; Rowe T; Ryan K; Smith GJ; Solowiej JE; Steppan CM; Taggart B Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem 2020, 63, 12725–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Anand K; Ziebuhr J; Wadhwani P; Mesters JR; Hilgenfeld R Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- (28).Matthews DA; Dragovich PS; Webber SE; Fuhrman SA; Patick AK; Zalman LS; Hendrickson TF; Love RA; Prins TJ; Marakovits JT; Zhou R; Tikhe J; Ford CE; Meador JW; Ferre RA; Brown EL; Binford SL; Brothers MA; DeLisle DM; Worland ST Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 11000–11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang H; Xie W; Xue X; Yang K; Ma J; Liang W; Zhao Q; Zhou Z; Pei D; Ziebuhr J; Hilgenfeld R; Yuen KY; Wong L; Gao G; Chen S; Chen Z; Ma D; Bartlam M; Rao Z Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nguyen DD; Gao K; Chen J; Wang R; Wei G-W Unveiling the molecular mechanism of SARS-CoV-2 main protease inhibition from 137 crystal structures using algebraic topology and deep learning. Chem. Sci 2020, 11, 12036–12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gao K; Nguyen DD; Chen J; Wang R; Wei G-W Repositioning of 8565 existing drugs for COVID-19. J. Phys. Chem. Lett 2020, 11, 5373–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gao K; Nguyen DD; Wang R; Wei G-W Machine intelligence design of 2019-nCoV drugs. bioRxiv 2020, DOI: 10.1101/2020.01.30.927889. [DOI] [Google Scholar]
- (33).Powers JC; Asgian JL; Ekici ÖD; James KE Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev 2002, 102, 4639–4750. [DOI] [PubMed] [Google Scholar]
- (34).Zhang L; Lin D; Kusov Y; Nian Y; Ma Q; Wang J; von Brunn A; Leyssen P; Lanko K; Neyts J; de Wilde A; Snijder EJ; Liu H; Hilgenfeld R α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem 2020, 63, 4562–4578. [DOI] [PubMed] [Google Scholar]
- (35).Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhu L; George S; Schmidt MF; Al-Gharabli SI; Rademann J; Hilgenfeld R Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 2011, 92, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tan J; George S; Kusov Y; Perbandt M; Anemüller S; Mesters JR; Norder H; Coutard B; Lacroix C; Leyssen P; Neyts J; Hilgenfeld R 3C protease of enterovirus 68: structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J. Virol 2013, 87, 4339–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Konno S; Thanigaimalai P; Yamamoto T; Nakada K; Kakiuchi R; Takayama K; Yamazaki Y; Yakushiji F; Akaji K; Kiso Y; Kawasaki Y; Chen S-E; Freire E; Hayashi Y Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety. Bioorg. Med. Chem 2013, 21, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Thanigaimalai P; Konno S; Yamamoto T; Koiwai Y; Taguchi A; Takayama K; Yakushiji F; Akaji K; Chen S-E; Naser-Tavakolian A; Schön A; Freire E; Hayashi Y Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem 2013, 68, 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Thanigaimalai P; Konno S; Yamamoto T; Koiwai Y; Taguchi A; Takayama K; Yakushiji F; Akaji K; Kiso Y; Kawasaki Y; Chen S-E; Naser-Tavakolian A; Schön A; Freire E; Hayashi Y Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: Structure-activity relationship study. Eur. J. Med. Chem 2013, 65, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Konno S; Kobayashi K; Senda M; Funai Y; Seki Y; Tamai I; Schäkel L; Sakata K; Pillaiyar T; Taguchi A; Taniguchi A; Gütschow M; Müller CE; Takeuchi K; Hirohama M; Kawaguchi A; Kojima M; Senda T; Shirasaka Y; Kamitani W; Hayashi Y 3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents. J. Med. Chem 2021, DOI: 10.1021/acs.jmed-chem.1c00665. [DOI] [PubMed] [Google Scholar]
- (42).Boras B; Jones RM; Anson BJ; Arenson D; Aschenbrenner L; Bakowski MA; Beutler N; Binder J; Chen E; Eng H; Hammond H; Hammond J; Haupt RE; Hoffman R; Kadar EP; Kania R; Kimoto E; Kirkpatrick MG; Lanyon L; Lendy EK; Lillis JR; Logue J; Luthra SA; Ma C; Mason SW; McGrath ME; Noell S; Obach RS; O’Brien MN; O’Connor R; Ogilvie K; Owen D; Pettersson M; Reese MR; Rogers TF; Rossulek MI; Sathish JG; Shirai N; Steppan C; Ticehurst M; Updyke LW; Weston S; Zhu Y; Wang J; Chatterjee AK; Mesecar AD; Frieman MB; Anderson AS; Allerton C Discovery of a novel inhibitor of coronavirus 3CL protease as a clinical candidate for the potential treatment of COVID-19. BioRxiv 2021, DOI: 10.1101/2020.09.12.293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Owen DR; Allerton CMN; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ; Dantonio A; Di L; Eng H; Ferre R; Gajiwala KS; Gibson SA; Greasley SE; Hurst BL; Kadar EP; Kalgutkar AS; Lee JC; Lee J; Liu W; Mason SW; Noell S; Novak JJ; Obach RS; Ogilvie K; Patel NC; Pettersson M; Rai DK; Reese MR; Sammons MF; Sathish JG; Singh RSP; Steppan CM; Stewart AE; Tuttle JB; Updyke L; Verhoest PR; Wei L; Yang Q; Zhu Y An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. medRxiv 2021, DOI: 10.1101/2021.07.28.21261232. [DOI] [PubMed] [Google Scholar]
- (44).Beer T Pfizer CEO SaysAntiviral Pill To Treat Covid Could Be Ready By The End Of The Year. https://www.forbes.com/sites/tommybeer/2021/04/27/pfizer-ceo-says-antiviral-pill-to-treat-covid-could-be-ready-by-end-of-the-year/?sh/244a1e012a0d, 2021.
- (45).Qiao J; Li Y-S; Zeng R; Liu F-L; Luo R-H; Huang C; Wang Y-F; Zhang J; Quan B; Shen C; Mao X; Liu X; Sun W; Yang W; Ni X; Wang K; Xu L; Duan Z-L; Zou Q-C; Zhang H-L; Qu W; Long Y-H-P; Li M-H; Yang R-C; Liu X; You J; Zhou Y; Yao R; Li W-P; Liu J-M; Chen P; Liu Y; Lin G-F; Yang X; Zou J; Li L; Hu Y; Lu G-W; Li W-M; Wei Y-Q; Zheng Y-T; Lei J; Yang S SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ganguly HK; Basu G Conformational landscape of substituted prolines. Biophys. Rev 2020, 12, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Raboisson P; Rognan D; Aldous D; Wermuth CG The Practice of Medicinal Chemistry; Elsevier, 2015. [Google Scholar]
- (48).Dai W; Zhang B; Jiang X-M; Su H; Li J; Zhao Y; Xie X; Jin Z; Peng J; Liu F; Li C; Li Y; Bai F; Wang H; Cheng X; Cen X; Hu S; Yang X; Wang J; Liu X; Xiao G; Jiang H; Rao Z; Zhang L-K; Xu Y; Yang H; Liu H Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Rathnayake AD; Zheng J; Kim Y; Perera KD; Mackin S; Meyerholz DK; Kashipathy MM; Battaile KP; Lovell S; Perlman S; Groutas WC; Chang K-O 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med 2020, 12, eabc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Vuong W; Khan MB; Fischer C; Arutyunova E; Lamer T; Shields J; Saffran HA; McKay RT; van Belkum MJ; Joyce MA; Young HS; Tyrrell DL; Vederas JC; Lemieux MJ Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun 2020, 11, 5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kumar V; Shin JS; Shie J-J; Ku KB; Kim C; Go YY; Huang K-F; Kim M; Liang P-H Identification and evaluation of potent Middle East respiratory syndrome coronavirus (MERS-CoV) 3CLPro inhibitors. Antiviral Res. 2017, 141, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wang H; He S; Deng W; Zhang Y; Li G; Sun J; Zhao W; Guo Y; Yin Z; Li D; Shang L Comprehensive insights into the catalytic mechanism of middle east respiratory syndrome 3C-Like protease and severe acute respiratory syndrome 3C-Like protease. ACS Catal. 2020, 10, 5871–5890. [DOI] [PubMed] [Google Scholar]
- (53).Yang S; Chen S-J; Hsu M-F; Wu J-D; Tseng C-TK; Liu Y-F; Chen H-C; Kuo C-W; Wu C-S; Chang L-W; Chen W-C; Liao S-Y; Chang T-Y; Hung H-H; Shr H-L; Liu C-Y; Huang Y-A; Chang L-Y; Hsu J-C; Peters CJ; Wang AH-J; Hsu M-C Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem 2006, 49, 4971–4980. [DOI] [PubMed] [Google Scholar]
- (54).Sharun K; Tiwari R; Dhama K Protease inhibitor GC376 for COVID-19: Lessons learned from feline infectious peritonitis. Ann. Med 2021, 61, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020,30,678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Pedersen NC; Kim Y; Liu H; Galasiti Kankanamalage AC; Eckstrand C; Groutas WC; Bannasch M; Meadows JM; Chang K-O Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg 2018,20, 378–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kim Y; Liu H; Galasiti Kankanamalage AC; Weerasekara S; Hua DH; Groutas WC; Chang K-O; Pedersen NC Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 2016, 12, e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kim Y; Lovell S; Tiew K-C; Mandadapu SR; Alliston KR; Battaile KP; Groutas WC; Chang K-O Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol 2012, 86, 11754–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Cat got your SARS-CoV-2 antiviral? https://www.genengnews.com/news/cat-got-your-sars-cov-2-antiviral/, 2020.
- (60).Dampalla CS; Zheng J; Perera KD; Wong L-YR; Meyerholz DK; Nguyen HN; Kashipathy MM; Battaile KP; Lovell S; Kim Y; Perlman S; Groutas WC; Chang K-O Postinfection treatment with a protease inhibitor increases survival of mice with a fatal SARS-CoV-2 infection. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2101555118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ahmed Laskar A; Younus H Aldehyde toxicity and metabolism: the role of aldehyde dehydrogenases in detoxification, drug resistance and carcinogenesis. Drug Metab. Rev 2019, 51, 42–64. [DOI] [PubMed] [Google Scholar]
- (62).Uchida K Role of reactive aldehyde in cardiovascular diseases. Free Radical Biol. Med 2000, 28, 1685–1696. [DOI] [PubMed] [Google Scholar]
- (63).Steuten K; Kim H; Widen JC; Babin BM; Onguka O; Lovell S; Bolgi O; Cerikan B; Neufeldt CJ; Cortese M; Muir RK; Bennett JM; Geiss-Friedlander R; Peters C; Bartenschlager R; Bogyo M Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect. Dis 2021, 7, 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Lee C-C; Kuo C-J; Ko T-P; Hsu M-F; Tsui Y-C; Chang S-C; Yang S; Chen S-J; Chen H-C; Hsu M-C; Shih S-R; Liang P-H; Wang AH-J Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem 2009, 284, 7646–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Iketani S; Forouhar F; Liu H; Hong SJ; Lin F-Y; Nair MS; Zask A; Huang Y; Xing L; Stockwell BR; Chavez A; Ho DD Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun 2021, 12, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Shie J-J; Fang J-M; Kuo T-H; Kuo C-J; Liang P-H; Huang H-J; Wu Y-T; Jan J-T; Cheng Y-SE; Wong C-H Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α, β-unsaturated esters. Bioorg. Med. Chem 2005, 13, 5240–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Ryu YB; Park S-J; Kim YM; Lee J-Y; Seo WD; Chang JS; Park KH; Rho M-C; Lee WS SARS-CoV 3CLpro inhibitory effects of quinone-methide triterpenes from Tripterygium regelii. Bioorg. Med. Chem. Lett 2010, 20, 1873–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Sreeramulu S; Gande SL; Göbel M; Schwalbe H Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem., Int. Ed 2009, 48, 5853–5855. [DOI] [PubMed] [Google Scholar]
- (69).Jackson PA; Widen JC; Harki DA; Brummond KM Covalent modifiers: A chemical perspective on the reactivity of α, β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J. Med. Chem 2017, 60, 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Shang J; Wan Y; Luo C; Ye G; Geng Q; Auerbach A; Li F Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Liu T; Luo S; Libby P; Shi G-P Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther 2020, 213, 107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Ou X; Liu Y; Lei X; Li P; Mi D; Ren L; Guo L; Guo R; Chen T; Hu J; Xiang Z; Mu Z; Chen X; Chen J; Hu K; Jin Q; Wang J; Qian Z Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun 2020, 11 , 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Sacco MD; Ma C; Lagarias P; Gao A; Townsend JA; Meng X; Dube P; Zhang X; Hu Y; Kitamura N; Hurst B; Tarbet B; Marty MT; Kolocouris A; Xiang Y; Chen Y; Wang J Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv 2020, 6, eabe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Ghosh AK; Gong G; Grum-Tokars V; Mulhearn DC; Baker SC; Coughlin M; Prabhakar BS; Sleeman K; Johnson ME; Mesecar AD Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett 2008, 18, 5684–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Wu C-Y; King K-Y; Kuo C-J; Fang J-M; Wu Y-T; Ho M-Y; Liao C-L; Shie J-J; Liang P-H; Wong C-H Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease. Chem. Biol 2006, 13, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Zhang J; Pettersson HI; Huitema C; Niu C; Yin J; James MN; Eltis LD; Vederas JC Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C-like protease based on phthalhydrazide ketones or heteroaromatic esters. J. Med. Chem 2007, 50, 1850–1864. [DOI] [PubMed] [Google Scholar]
- (77).Niu C; Yin J; Zhang J; Vederas JC; James MN Molecular docking identifies the binding of 3-chloropyridine moieties specifically to the S1 pocket of SARS-CoV Mpro. Bioorg. Med. Chem 2008, 16, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Wu C-Y; Jan J-T; Ma S-H; Kuo C-J; Juan H-F; Cheng Y-SE; Hsu H-H; Huang H-C; Wu D; Brik A; Liang F-S; Liu R-S; Fang J-M; Chen S-T; Liang P-H; Wong C-H Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 10012–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Blanchard JE; Elowe NH; Huitema C; Fortin PD; Cechetto JD; Eltis LD; Brown ED High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol 2004, 11, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Sun L-Y; Chen C; Su J; Li J-Q; Jiang Z; Gao H; Chigan J-Z; Ding H-H; Zhai L; Yang K-W Ebsulfur and Ebselen as highly potent scaffolds for the development of potential SARS-CoV-2 antivirals. Bioorg. Chem 2021, 112, 104889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [DOI] [PubMed] [Google Scholar]
- (82).Ma C; Hu Y; Townsend JA; Lagarias PI; Marty MT; Kolocouris A; Wang J Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol. Transl. Sci 2020, 3, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Renson M; Etschenberg E; Winkelmann J 2-Phenyl-1, 2-benzisoselenazol-3 (2H)-one containing pharmaceutical preparations and process for the treatment of rheumatic diseases. US Patent 4,352,799, 1982.
- (84).Haritha C; Sharun K; Jose B Ebselen, a new candidate therapeutic against SARS-CoV-2. International Journal of Surgery (London, England) 2020, 84, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Baell J; Walters MA Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481. [DOI] [PubMed] [Google Scholar]
- (86).Bacha U; Barrila J; Velazquez-Campoy A; Leavitt SA; Freire E Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry 2004, 43, 4906–4912. [DOI] [PubMed] [Google Scholar]
- (87).Liu P; Liu H; Sun Q; Liang H; Li C; Deng X; Liu Y; Lai L Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. European. Eur. J. Med. Chem 2020, 206, 112702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Zhou L; Liu Y; Zhang W; Wei P; Huang C;Pei J; Yuan Y; Lai L Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. J. Med. Chem 2006, 49, 3440–3443. [DOI] [PubMed] [Google Scholar]
- (89).Chen L-R; Wang Y-C; Lin YW; Chou S-Y; Chen S-F; Liu LT; Wu Y-T; Kuo C-J; Chen TS-S; Juang S-H Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg. Med. Chem. Lett 2005, 15, 3058–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Turlington M; Chun A; Tomar S; Eggler A; Grum-Tokars V; Jacobs J; Daniels JS; Dawson E; Saldanha A; Chase P; Baez-Santos YM; Lindsley CW; Hodder P; Mesecar AD; Stauffer SR Discovery of N-(benzo [1, 2, 3] triazol-1-yl)-N-(benzyl) acetamido) phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg. Med. Chem. Lett 2013, 23, 6172–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Shie J-J; Fang J-M; Kuo C-J; Kuo T-H; Liang P-H; Huang H-J; Yang W-B; Lin C-H; Chen J-L; Wu Y-T; Wong C-H Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease. J. Med. Chem 2005, 48, 4469–4473. [DOI] [PubMed] [Google Scholar]
- (92).Akaji K; Konno H; Mitsui H; Teruya K; Shimamoto Y; Hattori Y; Ozaki T; Kusunoki M; Sanjoh A Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem 2011, 54, 7962–7973. [DOI] [PubMed] [Google Scholar]
- (93).Shao Y-M; Yang W-B; Peng H-P; Hsu M-F; Tsai K-C; Kuo T-H; Wang AH-J; Liang P-H; Lin C-H; Yang A-S; Wong C-H Structure-based design and synthesis of highly potent SARS-CoV 3CL protease inhibitors. ChemBioChem 2007, 8, 1654–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Coelho C; Gallo G; Campos CB; Hardy L; Würtele M Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS One 2020, 15, e0240079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Clanton D; Moran R; McMahon J; Weislow O; Buckheit R Jr; Hollingshead M; Ciminale V; Felber B; Pavlakis G; Bader J Sulfonic acid dyes: inhibition of the human immunodeficiency virus and mechanism of action. J. Acquired Immune Defic. Syndr 1992, 5, 771–781. [PubMed] [Google Scholar]
- (96).Xiao Y; Liu C; Tang W; Zhang H; Chen X Evans blue inhibits hbv replication through a dual antiviral mechanism by targeting virus binding and capsid assembly. Front. Microbiol 2019, 10, 2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Lu I-L; Mahindroo N; Liang P-H; Peng Y-H; Kuo C-J; Tsai K-C; Hsieh H-P; Chao Y-S; Wu S-Y Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J. Med. Chem 2006, 49, 5154–5161. [DOI] [PubMed] [Google Scholar]
- (98).Wang L; Bao B-B; Song G-Q; Chen C; Zhang X-M; Lu W; Wang Z; Cai Y; Li S; Fu S; Song F-H; Yang H; Wang J-G Discovery of unsymmetrical aromatic disulfides as novel inhibitors of SARS-CoV main protease: chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. Eur. J. Med. Chem 2017, 137, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Zhang D; Fourie-O’Donohue A; Dragovich PS; Pillow TH; Sadowsky JD; Kozak KR; Cass RT; Liu L; Deng Y; Liu Y; Hop CE; Khojasteh SC Catalytic cleavage of disulfide bonds in small molecules and linkers of Antibody–Drug conjugates. Drug Metab. Dispos 2019, 47, 1156–1163. [DOI] [PubMed] [Google Scholar]
- (100).Zhu W; Xu M; Chen CZ; Guo H; Shen M; Hu X; Shinn P; Klumpp-Thomas C; Michael SG; Zheng W Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol. Transl. Sci 2020,3, 1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Jain RP; Pettersson HI; Zhang J; Aull KD; Fortin PD; Huitema C; Eltis LD; Parrish JC; James MNG; Wishart DS; Vederas JC Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J. Med. Chem 2004, 47, 6113–6116. [DOI] [PubMed] [Google Scholar]
- (102).Su H.-x.; Yao S; Zhao W.-f.; Li M.-j.; Liu J; Shang W.-j.; Xie H; Ke C.-q.; Hu H.-c.; Gao M.-n.; Yu K.-q.; Liu H; Shen J.-s.; Tang W; Zhang L.-k.; Xiao G.-f.; Ni L; Wang D.-w.; Zuo J.-p.; Jiang H.-l.; Bai F; Wu Y; Ye Y; Xu Y.-c. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin 2020, 41, 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Zandi K; Musall K; Oo A; Cao D; Liang B; Hassandarvish P; Lan S; Slack RL; Kirby KA; Bassit L; Amblard F; Kim B; AbuBakar S; Sarafianos SG; Schinazi RF Baicalein and baicalin inhibit SARS-CoV-2 RNA-dependent-RNA polymerase. Microorganisms 2021, 9, 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Lee C-C; Kuo C-J; Hsu M-F; Liang P-H; Fang J-M; Shie J-J; Wang AH-J Structural basis of mercury-and zinc-conjugated complexes as SARS-CoV 3C-like protease inhibitors. FEBS Lett. 2007, 581, 5454–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Hsu JT-A; Kuo C-J; Hsieh H-P; Wang Y-C; Huang K-K; Lin CP-C; Huang P-F; Chen X; Liang P-H Evaluation of metal-conjugated compounds as inhibitors of 3CL protease of SARS-CoV. FEBS Lett. 2004, 574, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Bacha U; Barrila J; Gabelli SB; Kiso Y; Mario Amzel L; Freire E Development of broad-spectrum halomethyl ketone inhibitors against coronavirus main protease 3CLpro. Chem. Biol. Drug Des 2008, 72, 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Shao Y-M; Yang W-B; Kuo T-H; Tsai K-C; Lin C-H; Yang A-S; Liang P-H; Wong C-H Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem 2008, 16, 4652–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Regnier T; Sarma D; Hidaka K; Bacha U; Freire E; Hayashi Y; Kiso Y New developments for the design, synthesis and biological evaluation of potent SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 2722–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Sydnes MO; Hayashi Y; Sharma VK; Hamada T; Bacha U; Barrila J; Freire E; Kiso Y Synthesis of glutamic acid and glutamine peptides possessing a trifluoromethyl ketone group as SARS-CoV 3CL protease inhibitors. Tetrahedron 2006, 62, 8601–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Zhang H-Z; Zhang H; Kemnitzer W; Tseng B; Cinatl J Jr.; Michaelis M; Doerr HW; Cai SX Design and synthesis of dipeptidyl glutaminyl fluoromethyl ketones as potent severe acute respiratory syndrome coronovirus (SARS-CoV) inhibitors. J. Med. Chem 2006, 49, 1198–1201. [DOI] [PubMed] [Google Scholar]
- (111).Lee T-W; Cherney MM; Huitema C; Liu J; James KE; Powers JC; Eltis LD; James MN Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol 2005, 353, 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Chuck C-P; Chen C; Ke Z; Wan DC-C; Chow H-F; Wong K-B Design, synthesis and crystallographic analysis of nitrile-based broad-spectrum peptidomimetic inhibitors for coronavirus 3C-like proteases. Eur. J. Med. Chem 2013, 59, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Fu L; Ye F; Feng Y; Yu F; Wang Q; Wu Y; Zhao C; Sun H; Huang B; Niu P; Song H; Shi Y; Li X; Tan W; Qi J; Gao GF Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun 2020, 11, 4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Kaeppler U; Stiefl N; Schiller M; Vicik R; Breuning A; Schmitz W; Rupprecht D; Schmuck C; Baumann K; Ziebuhr J; Schirmeister T A new lead for nonpeptidic active-site-directed inhibitors of the severe acute respiratory syndrome coronavirus main protease discovered by a combination of screening and docking methods. J. Med. Chem 2005, 48, 6832–6842. [DOI] [PubMed] [Google Scholar]
- (115).Ghosh AK; Xi K; Grum-Tokars V; Xu X; Ratia K; Fu W; Houser KV; Baker SC; Johnson ME; Mesecar AD Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 5876–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Ghosh AK; Xi K; Ratia K; Santarsiero BD; Fu W; Harcourt BH; Rota PA; Baker SC; Johnson ME; Mesecar AD Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem 2005, 48, 6767–6771. [DOI] [PubMed] [Google Scholar]
- (117).Breidenbach J; Lemke C; Pillaiyar T; Schäkel L; Al Hamwi G; Diett M; Gedschold R; Geiger N; Lopez V; Mirza S; Namasivayam V; Schiedel AC; Sylvester K; Thimm D; Vielmuth C; Phuong Vu L; Zyulina M; Bodem J; Gütschow M; Müller CE Targeting the main protease of SARS-CoV-2: from the establishment of high throughput screening to the design of tailored inhibitors. Angew. Chem., Int. Ed 2021, 60, 10423–10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Liu W; Zhu H-M; Niu G-J; Shi E-Z; Chen J; Sun B; Chen W-Q; Zhou H-G; Yang C Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem 2014, 22, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Jacobs J; Grum-Tokars V; Zhou Y; Turlington M; Saldanha SA; Chase P; Eggler A; Dawson ES; Baez-Santos YM; Tomar S; Mielech AM; Baker SC; Lindsley CW; Hodder P; Mesecar A; Stauffer SR Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem 2013, 56, 534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Lee H; Mittal A; Patel K; Gatuz JL; Truong L; Torres J; Mulhearn DC; Johnson ME Identification of novel drug scaffolds for inhibition of SARS-CoV 3-Chymotrypsin-like protease using virtual and high-throughput screenings. Bioorg. Med. Chem 2014, 22, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Ahn T-Y; Kuo C-J; Liu H-G; Ha D-C; Liang P-H; Jung Y-S Synthesis and evaluation of benzoquinolinone derivatives as sars-cov 3cl protease inhibitors. Bull. Korean Chem. Soc 2010, 31, 87–91. [Google Scholar]
- (122).Chen L; Chen S; Gui C; Shen J; Shen X; Jiang H Discovering severe acute respiratory syndrome coronavirus 3CL protease inhibitors: virtual screening, surface plasmon resonance, and fluorescence resonance energy transfer assays. J. Biomol. Screening 2006, 11,915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Ryu YB; Jeong HJ; Kim JH; Kim YM; Park J-Y; Kim D; Naguyen TTH; Park S-J; Chang JS; Park KH; Rho M-C; Lee WS Biflavonoids from Torreya nucifera displaying SARS-CoV 3CLpro inhibition. Bioorg. Med. Chem 2010, 18, 7940–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Li Y; Zhang J; Wang N; Zhang Y; Yang Y; Yuan Y; Jing H; Liu X; Wu S; Luo P; Zhang W; Lu D; Zeng H; Guo G; Zou Q High-throughput Screening and Experimental Identification of Potent Drugs Targeting SARS-CoV-2 Main Protease. Research square 2020, DOI: 10.21203/rs.3.rs-40014/v1. [DOI] [Google Scholar]
- (125).Park J-Y; Kim JH; Kim YM;Jeong HJ; Kim DW; Park KH; Kwon H-J; Park S-J; Lee WS; Ryu YB Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg. Med. Chem 2012, 20, 5928–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Chen L; Li J; Luo C; Liu H; Xu W; Chen G; Liew OW; Zhu W; Puah CM; Shen X; Jiang H Binding interaction of quercetin-3-β-galactoside and its synthetic derivatives with SARS-CoV 3CLpro: structure-activity relationship studies reveal salient pharmacophore features. Bioorg. Med. Chem 2006, 14, 8295–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Kuo C-J; Liu H-G; Lo Y-K; Seong C-M; Lee K-I; Jung Y-S; Liang P-H Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 2009, 583, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Martina E; Stiefl N; Degel B; Schulz F; Breuning A; Schiller M; Vicik R; Baumann K; Ziebuhr J; Schirmeister T Screening of electrophilic compounds yields an aziridinyl peptide as new active-site directed SARS-CoV main protease inhibitor. Bioorg. Med. Chem. Lett 2005, 15, 5365–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Hamill P; Hudson D; Kao RY; Chow P; Raj M; Xu H; Richer MJ; Jean F Development of a red-shifted fluorescence-based assay for SARS-coronavirus 3CL protease: identification of a novel class of anti-SARS agents from the tropical marine sponge Axinella corrugata. Biol. Chem 2006, 387, 1063–1074. [DOI] [PubMed] [Google Scholar]
- (130).Liu Y-C; Huang V; Chao T-C; Hsiao C-D; Lin A; Chang M-F; Chow L-P Screening of drugs by FRET analysis identifies inhibitors of SARS-CoV 3CL protease. Biochem. Biophys. Res. Commun 2005, 333, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Goetz D; Choe Y; Hansell E; Chen Y; McDowell M; Jonsson C; Roush W; McKerrow J; Craik C Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry 2007, 46, 8744–8752. [DOI] [PubMed] [Google Scholar]
- (132).Xue X; Yang H; Shen W; Zhao Q; Li J; Yang K; Chen C; Jin Y; Bartlam M; Rao Z Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol 2007, 366, 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Kao RY; To AP; Ng LW; Tsui WH; Lee TS; Tsoi H-W; Yuen K-Y Characterization of SARS-CoV main protease and identification of biologically active small molecule inhibitors using a continuous fluorescence-based assay. FEBS Lett. 2004, 576, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Chen C-N; Lin CP; Huang K-K; Chen W-C; Hsieh H-P; Liang P-H; Hsu JT-A Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3, 3′-digallate (TF3). Evid. Based Complementary Altern. Med 2005, 2, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Karypidou K; Ribone SR; Quevedo MA; Persoons L; Pannecouque C; Helsen C; Claessens F; Dehaen W Synthesis, biological evaluation and molecular modeling of a novel series of fused 1, 2, 3-triazoles as potential anti-coronavirus agents. Bioorg. Med. Chem. Lett 2018, 28, 3472–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Baker JD; Uhrich RL; Kraemer GC; Love JE; Kraemer BC A drug repurposing screen identifies hepatitis C antivirals as inhibitors of the SARS-CoV2 main protease. PLoS One 2021, 16, e0245962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Tripathi PK; Upadhyay S; Singh M; Raghavendhar S; Bhardwaj M; Sharma P; Patel AK Screening and evaluation of approved drugs as inhibitors of main protease of SARS-CoV-2. Int. J. Biol. Macromol 2020, 164, 2622–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Zhang J; Huitema C; Niu C; Yin J; James MN; Eltis LD; Vederas JC Aryl methylene ketones and fluorinated methylene ketones as reversible inhibitors for severe acute respiratory syndrome (SARS) 3C-like proteinase. Bioorg. Chem 2008, 36, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Chen L; Gui C; Luo X; Yang Q; Günther S; Scandella E; Drosten C; Bai D; He X; Ludewig B; Chen J; Luo H; Yang Y; Yang Y; Zou J; Thiel V; Chen K; Shen J; Shen X; Jiang H Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol 2005, 79, 7095–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Tsai K-C; Chen S-Y; Liang P-H; Lu I-L; Mahindroo N; Hsieh H-P; Chao Y-S; Liu L; Liu D; Lien W; Lin T-H; Wu S-Y Discovery of a novel family of SARS-CoV protease inhibitors by virtual screening and 3D-QSAR studies. J. Med. Chem 2006, 49, 3485–3495. [DOI] [PubMed] [Google Scholar]
- (141).Kao RY; Tsui WH; Lee TS; Tanner JA; Watt RM; Huang J-D; Hu L; Chen G; Chen Z; Zhang L; He T; Chan K-H; Tse H; To AP; Ng LW; Wong BC; Tsoi H-W; Yang D; Ho DD; Yuen K-Y Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics. Chem. Biol 2004, 11, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Ramajayam R; Tan K-P; Liu H-G; Liang P-H Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg. Med. Chem 2010, 18, 7849–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Yoshizawa S.-i.; Hattori Y; Kobayashi K; Akaji K Evaluation of an octahydroisochromene scaffold used as a novel SARS 3CL protease inhibitor. Bioorg. Med. Chem 2020, 28, 115273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Shimamoto Y; Hattori Y; Kobayashi K; Teruya K; Sanjoh A; Nakagawa A; Yamashita E; Akaji K Fused-ring structure of decahydroisoquinolin as a novel scaffold for SARS 3CL protease inhibitors. Bioorg. Med. Chem 2015, 23, 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Gurard-Levin ZA; Liu C; Jekle A; Jaisinghani R; Ren S; Vandyck K; Jochmans D; Leyssen P; Neyts J; Blatt LM; Beigelman L; Symons JA; Raboisson P; Scholle MD; Deval J Evaluation of SARS-CoV-2 3C-like protease inhibitors using self-assembled monolayer desorption ionization mass spectrometry. Antiviral Res. 2020, 182, 104924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Hanh Nguyen TT; Ryu H-J; Lee S-H; Hwang S; Breton V; Rhee JH; Kim D Virtual screening identification of novel severe acute respiratory syndrome 3C-like protease inhibitors and in vitro confirmation. Bioorg. Med. Chem. Lett 2011, 21, 3088–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Mukherjee P; Desai P; Ross L; White EL; Avery MA Structure-based virtual screening against SARS-3CLpro to identify novel non-peptidic hits. Bioorg. Med. Chem 2008, 16, 4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Wen C-C; Kuo Y-H; Jan J-T; Liang P-H; Wang S-Y; Liu H-G; Lee C-K; Chang S-T; Kuo C-J; Lee S-S; Hou C-C; Hsiao P-W; Chien S-C; Shyur L-F; Yang N-S Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem 2007,50,4087–4095. [DOI] [PubMed] [Google Scholar]
- (149).He Z; Zhao W; Niu W; Gao X; Gao X; Gong Y; Gao X Molecules inhibit the enzyme activity of 3-chymotrypsin-like cysteine protease of SARS-CoV-2 virus: the experimental and theory studies. bioRxiv 2020, DOI: 10.1101/2020.05.28.120642. [DOI] [Google Scholar]
- (150).Verschueren KH; Pumpor K; Anemüller S; Chen S; Mesters JR; Hilgenfeld R A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol 2008, 15, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Du R; Cooper L; Chen Z; Lee H; Rong L; Cui Q Discovery of Chebulagic Acid and Punicalagin as Novel Allosteric Inhibitors of SARS-CoV-2 3CLpro. Antiviral Res. 2021, 190, 105075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Rizzuti B; Grande F; Conforti F; Jimenez-Alesanco A; Ceballos-Laita L; Ortega-Alarcon D; Vega S; Reyburn HT; Abian O; Velazquez-Campoy A Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs. Biomedicines 2021, 9, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Abian O; Ortega-Alarcon D;Jimenez-Alesanco A; Ceballos-Laita L; Vega S; Reyburn HT; Rizzuti B; Velazquez-Campoy A Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol 2020, 164, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Alhakamy NA; Ahmed OA; Ibrahim TS; Aldawsari HM; Eljaaly K; Fahmy UA; Alaofi AL; Caraci F; Caruso G Evaluation of the Antiviral Activity of Sitagliptin-Glatiramer Acetate Nano-Conjugates against SARS-CoV-2 Virus. Pharmaceuticals 2021, 14, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Abdallah HM; El-Halawany AM; Sirwi A; El-Araby AM; Mohamed GA; Ibrahim SR; Koshak AE; Asfour HZ; Awan ZA; Elfaky MA Repurposing ofsome natural product isolates as SARS-COV-2 main protease inhibitors via in vitro cell free and cell-based antiviral assessments and molecular modeling approaches. Pharmaceuticals 2021, 14, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.