

Abstract
Plate readers are commonly used to measure cell growth and fluorescence, yet the utility and reproducibility of plate reader data is limited by the fact that it is typically reported in arbitrary or relative units. We have previously established a robust serial dilution protocol for calibration of plate reader measurements of absorbance to estimated bacterial cell count and for green fluorescence from proteins expressed in bacterial cells to molecules of equivalent fluorescein. We now extend these protocols to calibration of red fluorescence to the sulforhodamine-101 fluorescent dye and blue fluorescence to Cascade Blue. Evaluating calibration efficacy via an interlaboratory study, we find that these calibrants do indeed provide comparable precision to the prior calibrants and that they enable effective cross-laboratory comparison of measurements of red and blue fluorescence from proteins expressed in bacterial cells.
Keywords: calibration, units, fluorescence, cell count
1. Introduction
Plate readers are one of the most commonly used instruments for collecting data from cell cultures. Absorbance (optical density) is commonly used for estimating the concentration of cells in a liquid suspension, typically at a wavelength of 600 nm (OD600). Likewise, expression of fluorescent reporters is commonly used for quantifying gene expression levels and fluorescent dyes for quantifying a wide range of other biological properties. With plate readers, such measurements can be collected from large numbers of samples simultaneously with minimal disruption, low cost and a high degree of automation.
Unfortunately, both the utility and reproducibility of plate reader data have generally been limited by the fact that the measurement units are not directly linked to the biological properties that are being quantified. Absorbance values depend not just on the density of cells but also on the length of the light path through the sample, the arrangement of cells within that sample and the geometry of the plate reader optics. Fluorescence measurements even less reproducible, as they are typically collected as relative fluorescent units, with values dependent upon instrument components and settings including excitation light intensity, emission light collection using photomultiplier devices and bandpass windows, optical configuration and other machine settings.
Many studies have attempted to make fluorescence measurements more reproducible via normalization to a biological sample that has been cultured in parallel with the experimental samples (e.g. (9, 12, 13)). Such normalization approaches produce markedly less precise measurements than normalization to an independent calibrant (5), likely due to the ill-defined potential variability of the biological samples used for normalization. Recent work, however, has identified low-cost calibrants that can be used to produce precise estimates of bacterial cell-count from 600 nm absorbance and of molecules equivalent of fluorescein (MEFL) from green fluorescence (3–5).
We now extend these protocols to calibration of red and blue fluorescence, specifically using the dyes sulforhodamine-101 as a calibrant for red fluorescence and Cascade Blue as a calibrant for blue fluorescence. We evaluated the efficacy of the extended protocol for calibration of red and blue fluorescence via an interlaboratory study, finding that these calibrants do indeed provide comparable precision and that they enable effective cross-laboratory comparison of measurements of red and blue cellular fluorescence.
2. Methods: criteria for data selection
Data from each participating laboratory were included in the analysis only if they met a set of minimal data quality criteria for calibrant measurements, specifically that values are non-negative and generally decrease with increasing dilution. Of the nine participating laboratories, eight passed quality control, while one was excluded due to insufficient calibrant data.
Culturing data was excluded if the measurements were implausibly high, indicating a likely issue with instrument settings, protocol execution or data handling. In particular, we set a threshold for exclusion of culture data if the highest value of the media blank was more than the highest non-saturated value for OD600 or more than 10% of the highest non-saturated value for fluorescence. No data sets had such range issues for OD600, while for fluorescence one dataset was excluded for both red and green fluorescence and three different datasets were excluded for blue fluorescence.
Individual cell samples without sufficient growth were also removed from analysis, i.e. any individual sample with less than an estimated 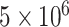 cells after 6 h of growth, a value well-separated from both the high (‘growing’) or lower (‘not growing’) clusters found in the data. Likewise, the analysis omitted any replicate set with a mean cell count that was either less than
cells after 6 h of growth, a value well-separated from both the high (‘growing’) or lower (‘not growing’) clusters found in the data. Likewise, the analysis omitted any replicate set with a mean cell count that was either less than 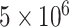 cells or less than the media blank mean plus 2 standard deviations, and any replicate set with only a single replicate. All told, of the 146 replicate sets from the eight labs passing quality control, 20 were excluded for failure to grow. Constructs in the pOpen_v4 backbone had a higher failure rate (16 of 40 replicate sets) than the pOpen_v3 backbone (4 of 106 replicate sets).
cells or less than the media blank mean plus 2 standard deviations, and any replicate set with only a single replicate. All told, of the 146 replicate sets from the eight labs passing quality control, 20 were excluded for failure to grow. Constructs in the pOpen_v4 backbone had a higher failure rate (16 of 40 replicate sets) than the pOpen_v3 backbone (4 of 106 replicate sets).
3. Methods: unit scaling factor computation
Unit scaling factors are computed using the same methods as presented in (4), substituting alternative reference values as appropriate.
The scaling factor, S, for relating molecules of fluorescent calibrant (or number of silica particles) to arbitrary fluorescent (or absorbance) units is computed as one parameter of a fit to a model of the dilution series measurement that includes a term for systematic pipetting error.
If we ignore pipetting error, then the model for serial dilution has an initial population of calibrant p0 that is diluted n times by a factor of α at each dilution. So, the expected population of calibrant for the ith dilution level is:
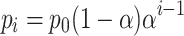 |
(1) |
In the case of the specific protocols used here, α = 0.5; and 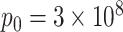 for silica particles,
for silica particles, 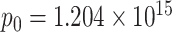 molecules for sulforhodamine-101, and
molecules for sulforhodamine-101, and 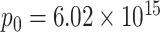 molecules for fluorescein and Cascade Blue.
molecules for fluorescein and Cascade Blue.
The model that includes systematic pipetting error modifies the intended dilution factor α with the addition of an unknown bias β, such that the expected biased population bi for the ith dilution level is:
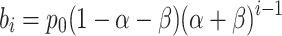 |
(2) |
We then simultaneously determine β and the scaling factor, S, using a least-squares fit of the data to the form:
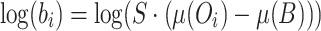 |
(3) |
where 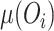 is the mean of the observed values for the ith dilution level and
is the mean of the observed values for the ith dilution level and 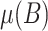 is the mean observed value for the blanks; and where we use only data over the longest sequence of dilution levels for which
is the mean observed value for the blanks; and where we use only data over the longest sequence of dilution levels for which 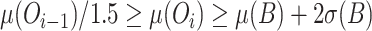 , where
, where 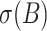 is the standard deviation of the blanks. Note that results are not particularly sensitive to the tolerance constant of 1.5 (used also in (4) and (3)), as the absolute maximum pipetting error β found for any sequence is just over 10%. Note also that a potential alternative model substitutive instrument non-linearity for pipetting bias can be found in (6).
is the standard deviation of the blanks. Note that results are not particularly sensitive to the tolerance constant of 1.5 (used also in (4) and (3)), as the absolute maximum pipetting error β found for any sequence is just over 10%. Note also that a potential alternative model substitutive instrument non-linearity for pipetting bias can be found in (6).
The OD600 and fluorescence a.u. data from Escherichia coli samples are converted into calibrated units by subtracting the mean blank media values for OD600 and fluorescence a.u., then multiplying by the corresponding scaling factors for the corresponding fluorescent calibrant and OD600.
We note that in (8), it was found that the overlap between red fluorescent proteins and OD600 can cause overestimates of cell population that lead to fluorescence per cell underestimates of up to 10%. This bias will be present in the data presented here, due to the use of OD600, but the effect magnitude is small compared to total observed variation and should in any case be consistent across laboratories.
4. Methods: statistics and reproducibility
As reproducibility is the main subject of this study, see the Results section above for its full presentation. In addition to the discussion of statistical analyses in the Results section, we note the following details of statistical analyses:
Coefficient of variation is computed per its definition, as the ratio of the standard deviation to the mean.
Fluorescence values are analyzed in terms of geometric mean and geometric standard deviation, rather than the more typical arithmetic statistics, due to the typical log-normal distribution of gene expression (2).
Data analysis was performed with Matlab.
5. Extension of serial dilution protocol to new colors
The serial dilution calibration protocol we use was first developed for calibrating GFP measurements using a fluorescein calibrant (5), then adapted for calibrating cell count measurements to a silica particle calibrant (4). In both cases, the concept is simple: beginning with a stock of calibrant at a defined concentration, execute a 2-fold serial dilution series to produce a range of known concentrations. A conversion factor can then be computed by fitting the observed readings against the expected concentrations in the dilution, and further tuned by accounting for pipetting error as well.
The starting concentration and length of the dilution series are chosen to cover the full anticipated measurement range of a typical fluorescence plate reader, from above the highest expected biological value to near-zero in the background diluent. Likewise, excitation and emission wavelengths are chosen based on the spectrum of the calibrant and target class of fluorescent protein, preferably matching a common flow cytometry channel where possible. With the protocol established in (4), fluorescein was used with 10 µmol/l as the starting concentration and diluted 10 times with PBS as the diluent. Then, fluorescence was measured with excitation at 488 nm (or closest applicable bandpass) and an emission filter of 530 nm/30 nm (i.e. a 30 nm bandpass filter centered at 530 nm, spanning the range of 515 to 545 nm). Silica particles are used with 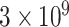 microspheres per ml as the starting concentration and are diluted 10 times with water as the diluent. Then absorbance was measured at 600 nm.
microspheres per ml as the starting concentration and are diluted 10 times with water as the diluent. Then absorbance was measured at 600 nm.
The fluorescein and silica particle calibrants were selected using the following criteria:
Good spectral match for biological measurement target: Spectral match does not need to be perfect, but if it is not a good approximation, then calibration cannot apply.
Readily soluble in PBS or water: Materials that require other solvents, such as DMSO, increase handling difficulty.
Widely available at low cost: The materials cost for fluorescein and silica particles is well under $1 US per protocol execution.
Stable for room temperature shipping: Using materials that do not require a cold-chain also increases accessibility, as well as allowing preparation of low-cost kits.
Matches a flow cytometry calibrant: Equivalence between flow cytometry and plate reader data have been demonstrated in (4) and (3). Thus, while flow cytometry was not used in this study, choosing fluorescent calibrants that match an established flow cytometry calibrant can enhance value by enabling data fusion between the two modalities.
To extend these protocols to new calibrants for red fluorescence and blue fluorescence, we needed to identify dyes that met as many of these criteria as possible.
For red fluorescence, we first considered Nile Red and PE-Texas Red, which are good spectral matches for many red fluorescent proteins and which, like fluorescein, match well-established flow cytometry channels in SpheroTech flow cytometry calibration beads (11). Unfortunately, Nile Red proved difficult to dissolve and PE-Texas Red was too expensive for our requirements. Given these drawbacks, we shifted to sulforhodamine 101, which is similar both in molecular structure and spectral properties to PE-Texas Red, but which could be obtained at a much lower expense. Unfortunately, however, this does not match a current flow cytometry calibrant. From preliminary experimentation with sulforhodamine 101, we selected 2 µmol/l as the starting concentration and dilution 10 times with a PBS diluent. Measurement was set for excitation at 561 nm with an emission filter of 610 nm/20 nm, a common flow cytometry red fluorescence measurement channel.
For blue fluorescence, we first considered Pacific Blue and Coumarin 30, which are good spectral matches for many blue and cyan fluorescence protein and which also match well-established flow cytometry channels. Here we found much the same challenge as with red fluorescence: Coumarin 30 was difficult to dissolve and Pacific Blue was too expensive, as well as presenting some stability problems. Given these drawbacks, we shifted to Cascade Blue, which could be obtained at a reasonable expense and is soluble in water. From preliminary experimentation with Cascade Blue, we selected 10 µmol/l as the starting concentration and dilution 10 times with a water diluent. Measurement was set for excitation at 405 nm with an emission filter of 450 nm/50 nm, a common flow cytometry blue fluorescence measurement channel.
To facilitate the preparation of the reference solutions across labs we have determined the extinction coefficients of the calibrants, finding for fluorescein 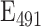 =68.029 M−1 cm−1; Cascade Blue
=68.029 M−1 cm−1; Cascade Blue 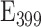 =28.902 M−1 cm−1; and sulforhodamine 101
=28.902 M−1 cm−1; and sulforhodamine 101 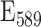 = 98.856 M−1 cm−1 (Supplementary 4 - Measurement of Extinction Coefficients). Protocols users can therefore determine the concentration of their reference solutions from a simple spectrophotometric measurement without requiring more painstaking analytical techniques.
= 98.856 M−1 cm−1 (Supplementary 4 - Measurement of Extinction Coefficients). Protocols users can therefore determine the concentration of their reference solutions from a simple spectrophotometric measurement without requiring more painstaking analytical techniques.
6. Experiment design
To test our selections for red and blue calibrants, we organized an interlaboratory study. The protocol for this study was closely based on that used in (4) for dilution series calibration and cell culturing (omitting only the alternative cell count protocols that were tested in that study). Briefly, each laboratory was provided with two 96-well plates containing a set of plasmid constructs (see below). Laboratories were instructed to transform each construct into E. coli DH5-alpha cells and select two of the resulting colonies for each construct. Each colony was grown in liquid culture overnight, then diluted and grown (in duplicate) for an additional 6 h. Absorbance (OD600) and fluorescence were then measured for each culture in a plate reader, i.e., each condition should have a replicate set containing a total of four replicates.
Laboratories were also instructed to prepare and measure a dilution series for each calibrant as described above. To accommodate the two additional calibrants, we modified the dilution series calibration protocol from (4) to reduce each calibrant from four replicates to two replicates, i.e., each condition should have a replicate set containing two replicates. This reduction allows calibration with the same total amount of effort and resources, at a tradeoff of a relatively small increase in risk given the level of reliability and precision found for the serial dilution protocol in the prior study. The complete protocol as provided to participating laboratories is provided in Supplementary 1-Protocol.
To maximize comparability with prior experiments, five of the same constitutive GFP constructs were used as in (4): the positive GFP control for the study (I20270, which is constitutive expression of GFP with promoter J23151) along with constitutive GFP expressed with a strong promoter (J23100), medium promoters (J23106, J23116) and a weak promoter (J23117). Due to constraints in fabrication versus the experimental timeline, however, we needed to switch to a different plasmid backbone, for which we selected the FreeGenes (https://stanford.freegenes.org/) pOpen_v3 backbone, which uses the same origin of replication as the iGEM pSB1C3 used in the prior study. Likewise, fabrication constraints dictated the use of a new negative control containing no sequence for expressing any fluorescent protein.
In addition to GFP, we needed to have red and blue fluorescent proteins, all to be expressed constitutively for simplicity. Here, we used FPbase (10) to select proteins that were strong, stable, with spectral properties that overlapped reasonably well with the selected calibrants, and, where possible, without active patent restrictions. For this purpose, we selected the well-established mRFP1 and mCherry for red. For blue calibration, we considered both blue and cyan proteins: EBFP, mTagBFP2, and mmilCFP for blue, and ECFP, mCerulean3, and meffCFP for cyan. Specifically, for sequences for GFP, mRFP1 and mCherry we used iGEM registry parts with many previous reports of success in E. coli (respectively BBa_E0040, BBa_E1010 and BBa_J06504); for the other fluorescent proteins we used amino acid sequences from FPbase codon-optimized for E. coli using the IDT codon optimization tool (https://www.idtdna.com/CodonOpt). We then included these eight fluorescent proteins, plus GFP, in constitutive constructs driven either by the strong J23101 or medium J23106, in a pOpen_v3 backbone. These constructs, plus the negative control and five other constitutive GFP constructs, made a total of 24 genetic constructs to be evaluated in the culturing protocol.
Unfortunately, issues encountered during the design and fabrication process led to two deviations from this experimental design. First, the J23106 promoter was fabricated with two base-pairs switched (except for the J23106 GFP combination, which was produced in two forms, one correct and one with the switched base-pairs); we designate this accidental alternate promoter pNew in our presentation of results below. Second, many of the strong promoter strains failed to properly fabricate in the pOpen_v3 backbone and had to be switched into the alternative pOpen_v4 backbone, which is designed to be an easier cloning target with a lower copy count. These latter strains were also not able to be delivered until after some participating laboratories had completed data collection, and thus were only tested in approximately half of the participating laboratories. Given time constraints and the fact that the primary focus of the study was the calibrants and not the cells, however, a decision was made to proceed: strains that fail to express a fluorescent protein should still produce equivalent autofluorescence and background fluorescence readings in every lab, and thus there is value in comparing measurements from cells that do not significantly express a fluorescent protein, as well as from those that do. Genetic construct designs, including the pOpen_v3 and pOpen_v4 backbones, and the construct plate information that was provided to participating laboratories are provided in Supplementary 2-Designs.
7. Results
Nine participating laboratories collected data following the experiment design given above, of which eight passed a calibrant quality control assessment ensuring that values were non-negative and decrease with dilution. Similarly, quality control for cell culture data excluded channels with an implausibly high fluorescent background and individual cell samples without significant growth. Additional detail on criteria for data selection is provided in section 2.
We first analyzed the data collected from calibrants, then used the unit conversions from this analysis in order to analyze cell culture data. To be effective, the dilution series needs to provide a predictable sequence of values across a large linear range of measurement. Not all measurement values are linear, however: at the high end measurements can become compressed or overflow due to sensor saturation, while at the low end the measurements can become indistinguishable from background noise. Figure 1 shows the number of dilutions in the linear range identified for each dilution series (section 3). In general, the linear range is wide, ranging from a mean of 7.1 steps (140-fold range) for silica particles to a mean of 9.3 steps (660-fold) for fluorescein. The new sulforhodamine and Cascade Blue calibrants exhibit similar linear ranges to the established calibrants, with a mean of 7.8 steps (220-fold) and 8.9 steps (430-fold), respectively. In only three cases did the identified linear range include less than five dilution levels, two apparently due to instrument settings resulting in a high background and one apparently due to inconsistency in pipetting (as indicated by mean ratios between dilution levels ranging from 1.12 to 3.25).
Figure 1.
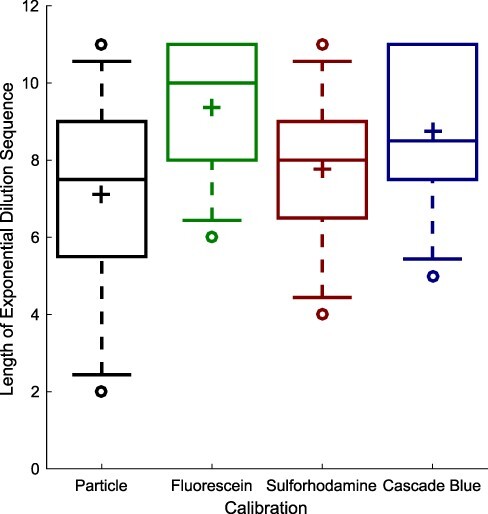
Serial dilution of sulforhodamine 101 and Cascade Blue calibrants is able to produce dilution sequences with an exponential decrease in value across a similarly large range of measurement as for fluorescein and silica particles. Each box summarizes data from the eight labs passing quality control criteria: plus indicates geometric mean, center line indicates median, top and bottom edges indicate 25th and 75th percentiles, and whiskers extend from 9–91%.
Figure 2 shows the coefficients of variation for the individual replicates sets for each calibrant at each dilution level in the linear range in each lab, which offer one view of the precision with which an individual can construct the dilution series. These are quite low, ranging from a geometric mean of 0.019 for silica particles to a geometric mean of 0.046 for sulforhodamine 101, meaning that in general one can reasonably require replicates to be within a few percent of one another. Again, there is no notable difference between the precision offered by the established calibrants versus the new red and blue calibrants.
Figure 2.
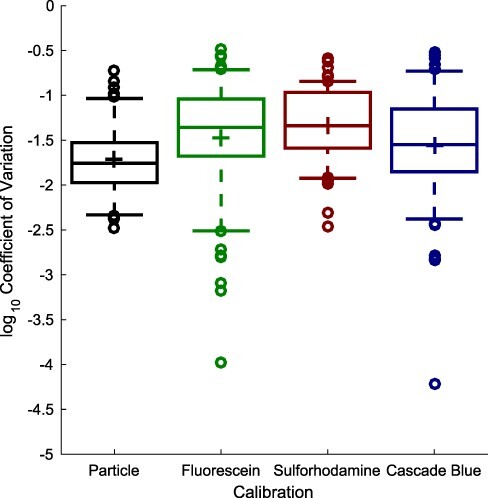
Coefficient of variation for calibrant replicate sets (two replicates each) shows sulforhodamine 101 and Cascade Blue replicate sets have similar levels of consistency that to fluorescein and silica particles. In each box, plus indicates geometric mean, center line indicates median, top and bottom edges indicate 25th and 75th percentiles, and whiskers extend from 9–91%.
The actual unit conversion between measurements and calibrated values is computed by fitting the linear-range measurements against a dilution model incorporating a term for systematic pipetting error, as presented in (4) and reviewed in section 3. Figure 3 shows that the calibrant curves take the expected form, with the mean values of each pair of replicates forming generally smooth exponential curves across the identified linear range. Figure 4(a) shows that there do not appear to be any systematic pipetting issues associated with either of the new calibrants. All four calibrants have a mean absolute pipetting error less than 2%, and the distributions show no notable difference between the established calibrants versus the new red and blue calibrants. Likewise, the residuals shown in Figure 4(b) indicate that the empirical data also matches well to the model for all four calibrants. All together, this analysis of the four calibrants indicates that sulforhodamine 101 and Cascade Blue can be used to construct serial dilutions as robust and precise as those constructed with silica particles or fluorescein.
Figure 3.
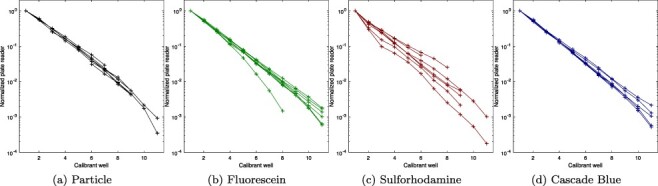
Geometric mean for calibrant replicate sets (two replicates each), normalized to dilution level, shows tightly clustered exponential decrease for most calibrants in most labs.
Figure 4.
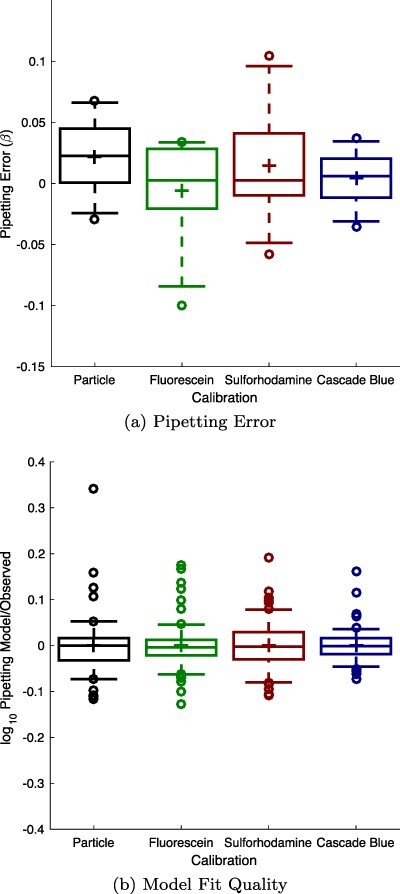
Model fitting for sulforhodamine 101 and Cascade Blue finds no notable difference in either the pipetting error observed (a) or the fit quality of the resulting model, shown from the plot of the residual between the fitted response and the experimental data (b). In each box, plus indicates geometric mean, center line indicates median, top and bottom edges indicate 25th and 75th percentiles, and whiskers extend from 9–91%.
We now turn to using these calibrant curves to compare measurements across laboratories. Figure 5(a) shows the results for the calibrated cell count for each strain at each laboratory that was able to successfully culture it (section 2). Unfortunately, due to the problems in construct fabrication noted above, the nine pOpen_v4 constructs, comprising most of the strong promoter strains, were only able to be tested in five laboratories. These constructs also encountered growth problems for unknown reasons: only 24 replicate sets successfully cultured and passed quality control for these nine strains, and one strain (pOpen_v4 J23101 EBFP) did not have a single successful replicate set. Note that we still include all of the strains, including failed strains, both here and in the analysis of fluorescence below, because the calibration that is the primary focus of the study helped us to assess that failure and to compare the measured values with biological expectations.
Figure 5.
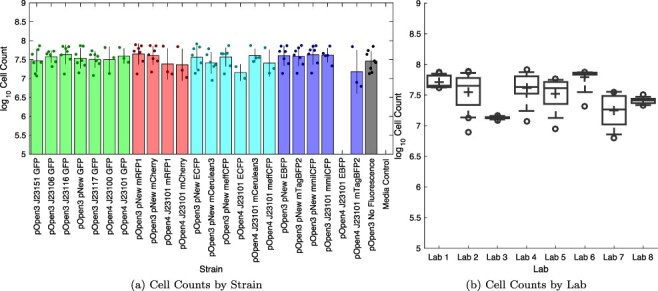
Samples showing estimate cell count for all replicate sets above 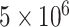 cell minimum growth threshold (section 2), sorted by strain (a) or by laboratory (b). Bars show mean and ±1 standard deviation for each calibrant; dots show values for individual replicate sets. In each box, plus indicates geometric mean, center line indicates median, top and bottom edges indicate 25th and 75th percentiles, and whiskers extend from 9–91%. The value for each replicate set is computed as the geometric mean over 2–4 replicates, depending on exclusions, with per-set exclusion information found in Supplementary 3-Data Digests.
cell minimum growth threshold (section 2), sorted by strain (a) or by laboratory (b). Bars show mean and ±1 standard deviation for each calibrant; dots show values for individual replicate sets. In each box, plus indicates geometric mean, center line indicates median, top and bottom edges indicate 25th and 75th percentiles, and whiskers extend from 9–91%. The value for each replicate set is computed as the geometric mean over 2–4 replicates, depending on exclusions, with per-set exclusion information found in Supplementary 3-Data Digests.
Amongst strains with at least two labs producing successful replicate sets, the geometric mean of geometric standard deviations in estimated cell count is 1.92-fold (note that (4) did not evaluate consistency of cell count, only its derivative value of fluorescence per cell). There is also a relatively large amount of strain-to-strain variation in cell growth within some individual labs (Figure 5(b)), with the geometric mean of geometric standards deviations in estimates within a lab being 1.48-fold. This indicates that a significant portion of the variation for each strain can be attributed to variation in culturing, as opposed to variation in calibration. For example, the visibly lower cell count values for the pOpen_v4 J23101 mTagBFP2 construct come from Lab 2 and Lab 7, which have the highest degree of variation amongst all of the laboratories. Differences in agitation and aeration may play a significant role in such variation, and would be an appropriate topic for future study.
Figure 6 shows the calibrated fluorescence per cell for each strain with at least two valid replicate sets for all three fluorescent colors, in units of Molecules of Equivalent Sulforhodamine 101 (MESR), MEFL, and Molecules of Equivalent Cascade Blue (MECB). Note the increased data drop-outs due to range issues with fluorescence measurements (section 2): in one dataset, calibration and culturing were apparently collected with inconsistent instrument settings for the red and green fluorescence, and three other datasets had their blue fluorescence data excluded due to high media background. Unfortunately, the confluence of these issues and the construct fabrication issues means that fewer than expected of the constructs show significant fluorescence: the accidentally constructed pNew promoter does not appear to function, and not all J23101 strains have two valid replicate sets.
Figure 6.
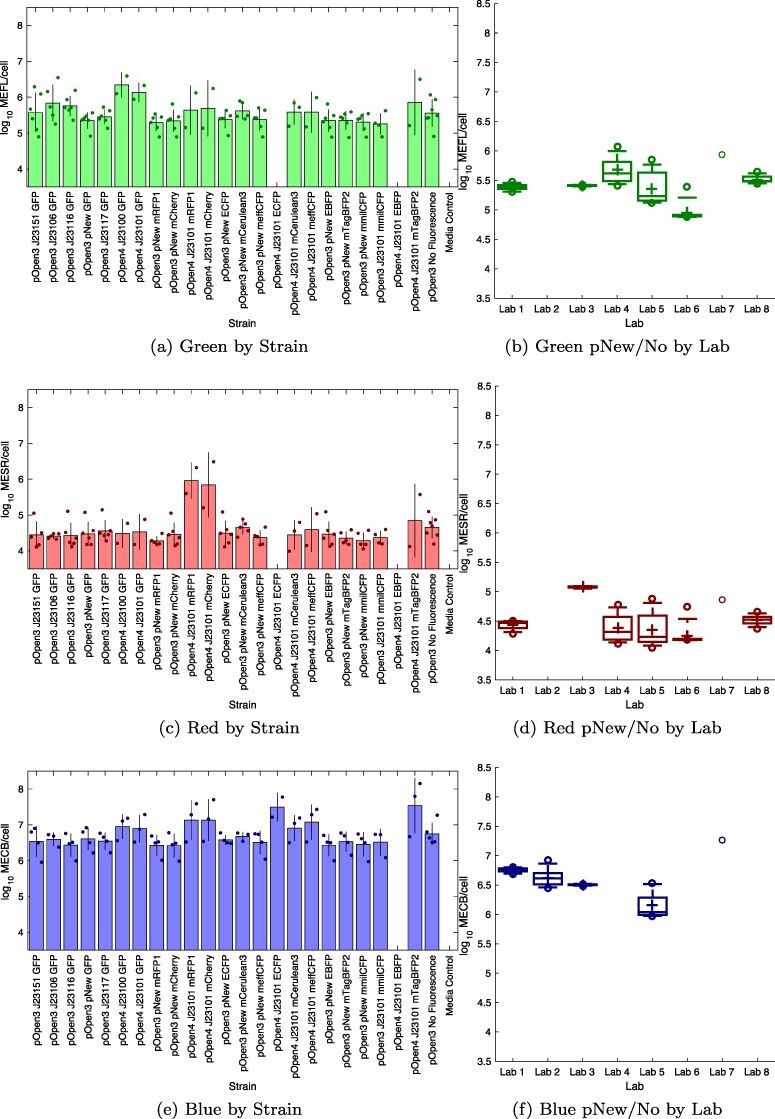
Fluorescence per cell for each strain with at least two valid replicate sets; blank columns indicate strains without sufficient valid sets: (a) four of the six constitutive GFP strains express green fluorescence above background, (c) J23101 mRFP1 and J23101 mCherry strains show strong red fluorescence, (e) J23101 ECFP and J23101 mTagBFP2 strains appear to express blue fluorescence above background, while several other strains are ambiguous. Bars show mean and ±1 standard deviation for each calibrant; dots show values for individual replicate sets; blank columns indicate strains with <2 valid. Fluorescence per cell for each laboratory for all non-fluorescent and pNew strains with at least two valid replicate sets is shown for (b) green, (d) red and (e) blue fluorescence. In each box, plus indicates geometric mean, center line indicates median, top and bottom edges indicate 25th and 75th percentiles, and whiskers extend from 9–91%. Blank columns indicate laboratories without sufficient valid sets. The value for each replicate set is computed as the geometric mean over 2–4 replicates, depending on exclusions, with per-set exclusion information found in Supplementary 3-Data Digests.
In subsequent discussion, we will consider significant difference to be two standard deviations of separation from the non-fluorescent control: specifically, for condition X, the ratio of geometric mean X over geometric mean non-fluorescent greater than twice the geometric mean of the standard deviations of X and the non-fluorescent control. Due to challenges with high background and small numbers of data points, very few conditions meet this test for significance. We thus do not emphasize significance of distinction from background, since we do not believe this is a strong conclusion to be taken away from this study, i.e., any protein that meets the significance test is likely good to use, but proteins that do not meet the significance test may not be disqualified.
For the five strong constitutive GFP strains, four are measured as expressing green fluorescence with a mean above that of the mean for non-fluorescent cells, though due to the high background only pOpen_v4 J23100 GFP meets our test for significance. The observed values for pOpen_v3 J23106 GFP, pOpen_v3 J23116 GFP, pOpen_v4 J23100 GFP and pOpen_v4 J23101 GFP are within error of those established in (4), while pOpen_v3 J23151 GFP (i.e. I20270) is not distinguishable from the higher-than-expected background. The pOpen_v3 J23117 GFP construct is not distinguishable from background (as expected given its established value of approximately 103.5 MEFL/cell in (4) and (3)), as are all non-GFP constructs.
For red fluorescent proteins, both of the pOpen_v4 J23101 strains have strong protein expression at an MESR/cell level quite similar to the pOpen_v4 J23101 GFP MEFL/cell level: 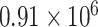 MESR for mRFP1,
MESR for mRFP1, 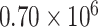 MESR for mCherry and
MESR for mCherry and 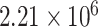 MEFL for GFP. The high standard deviation of J23101 mCherry, however, means that only J23101 mRFP1 meets our test for significance. While MESR and MEFL cannot be directly compared without additional analysis of spectra or dual constructs, the similarity of these numbers is nonetheless encouraging, since the calibrants were chosen with the intention of making as close a spectral match as feasible. All non-RFP constructs are not distinguishable from background, as expected.
MEFL for GFP. The high standard deviation of J23101 mCherry, however, means that only J23101 mRFP1 meets our test for significance. While MESR and MEFL cannot be directly compared without additional analysis of spectra or dual constructs, the similarity of these numbers is nonetheless encouraging, since the calibrants were chosen with the intention of making as close a spectral match as feasible. All non-RFP constructs are not distinguishable from background, as expected.
For blue and cyan fluorescent proteins, the pOpen_v4 J23101 strains for mTagBFP2 and ECFP have measured means well above background (though the higher standard deviation of mTagBFP2 means that only ECFP meets our significance test), while those for meffCFP and mCerulean3 are weaker but still possibly above background. The pOpen_v4 J23101 mmilCFP strain is not distinguishable from background, however, and the pOpen_v4 J23101 EBFP strain did not grow well in any participating laboratory. Several of the pOpen_v4 strains with other fluorescent proteins also appear to be potentially distinguishable from background, which should not be the case: the means of pOpen_v4 J23100 GFP, pOpen_v4 J23101 GFP, pOpen_v4 J23101 mRFP1 and pOpen_v4 J23101 mCherry are noticeably higher from those of other non-blue constructs, which is of potential concern even the actual data in the study does not meet our threshold for significance. Looking at raw data (Supplementary 3-Data Digests) it appears this may be an artifact of the high background in this channel for those particular data sets. Both this high background fluorescence and the expression levels measured for high blue fluorescent protein expression also bring into question the relationship between molecules of Cascade Blue and blue fluorescent proteins. The extinction coefficient for Cascade Blue is notably lower than that for the other fluorescent calibrants (Supplementary 4-Measurement of Extinction Coefficients), which hints that the actual ratio of dye molecules to protein molecules may be significantly higher in this range than for red and green.
Finally, we can evaluate how well the calibrants allow comparison of fluorescence measurements between different labs. Across all strains with at least two valid replicate sets (section 2), the geometric mean of geometric standards deviations is 2.37-fold for red fluorescence, 2.44-fold for green fluorescence, and 2.27-fold for blue fluorescence. As with cell growth, there is also a relatively large amount of strain-to-strain variation in the fluorescence measured from the cells without a functional promoter (non-fluorescent control and nine pNew strains) within some individual labs (Figure 6(b), 6(d), 6(f)). The geometric mean of geometric standards deviations in estimates within a labs being 1.34-fold for green, 1.40-fold for red and 1.27-fold for blue. Overall, this is a smaller portion of variation than with cell growth, but still significant. Moreover, the fact that some labs have much lower variation than others and that all labs have similar degrees of variation across colors. As with cell growth, this too suggests that a significant portion of the variation for each strain can be attributed to variation in culturing, as opposed to variation in calibration.
Although the level of variation between data sets is not quite as good as in (4), a slight reduction in precision is consistent with the fact that (4) also filtered data based on expected control values, while in this study we do not attempt such filtering because there are few datasets and no pre-established values to compare against for red or blue fluorescence. More importantly for the present study, however, the distributions show no notable difference between the established calibrants versus the new red and blue calibrants.
8. Discussion
We have extended the prior serial dilution protocols for calibration of plate reader measurements of green fluorescence and cell count to be applicable to red and blue fluorescence, using calibrants sulforhodamine 101 and Cascade Blue, respectively. By means of an interlaboratory study, we have shown that these new calibrants appear to provide a similar level of robustness and precision in constructing serial dilutions. We have also shown that these dilutions can be used to compare fluorescence measurements between laboratories, a key requirement for reproducible red and blue fluorescence measurements.
There are some important limitations of the current study, however. Notably, the MECB/cell values for blue fluorescence are much higher than expected, even given the difference in extinction coefficient, and suggests that there is need to better understand the relationship between Cascade Blue and blue fluorescent proteins (e.g. via purification of fluorescent protein as proposed in (7)). The challenges with background level also suggest ways that the protocol might be improved in the future, notably by adding a procedure for adjusting the fluorescence range to be measured by the plate reader and by switching from LB to some other culture medium with lower background levels of fluorescence. The protocol might also be improved by taking into account specific properties of the microplates and the potential for well-to-well variation in the operation of a plate reader, per (1) That said, we believe that it is at least clear that the serial dilution calibration protocol can be effectively applied for other fluorescent colors, even if additional study and adjustment of the protocol and interpretation may provide further improvements.
We note also that some may be reluctant to introduce calibration protocols, due to the fact that adding these protocols does add some complexity and with it the potential for errors in collection or interpretation of calibration data. We would argue, however, that such potential costs are more than offset by the opportunity to identify issues in the biological side of a protocol, which are common, often costly, and much more difficult to debug without the aid of reference points enabled by calibration. Anomalies discovered via calibration can also be used as a decision point for initiating more in-depth and costly debugging procedures, such as sequence verification (which was not part of this study).
Given this success, a useful future activity would be to extend the serial dilution calibration protocol to additional calibrants for other classes of fluorescent molecules (e.g. far-red/IR or larger separation between excitation and emission). Similarly, the protocol could be extended to other classes of cells grown in suspension, such as yeast, mammalian cells, plant protoplasts, by matching these larger cell types with appropriately sized silica particles. In both cases, the mechanisms involved are no different than the current cases, and thus the extension is anticipated to involve no more than a straight-forward change in the selected calibrant. Calibration of luminescence would be desirable as well, but will be more complex due to the mechanisms for producing luminescence. The methods should also be extensible beyond 96-well plates to other high-throughput labware, such as 384-well plates or microfluidic systems. Finally, a more detailed computation of the relationship between spectra may allow a mapping from equivalent calibrant molecules to an estimate of the actual molecules of interest.
Supplementary Material
Acknowledgement
We thank FreeGenes and Twist for supplying the DNA constructs for this experiment. The following additional people also contributed useful comments or feedback that supported the development of this project: Keoni Gandall, Rene Inckemann, Rahmi Lale, Dennis Mishler, Gonzalo Vidal Pena.
Contributor Information
Jacob Beal, Intelligent Software and Systems, Raytheon BBN Technologies, 10 Moulton Street, Cambridge 02138, MA, USA.
Cheryl A Telmer, Department of Biological Sciences, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh 15213, PA, USA.
Alejandro Vignoni, Synthetic Biology and Biosystems Control Group, Instituto de Automatica e Informatica Industrial, Universitat Politecnica de Valencia, Camino de Vera s/n, Valencia 46022, Spain.
Yadira Boada, Synthetic Biology and Biosystems Control Group, Instituto de Automatica e Informatica Industrial, Universitat Politecnica de Valencia, Camino de Vera s/n, Valencia 46022, Spain.
Geoff S Baldwin, Department of Life Sciences, Imperial College London, South Kensington Campus, Exhibition Road, London SW7 2AZ, UK.
Liam Hallett, Department of Life Sciences, Imperial College London, South Kensington Campus, Exhibition Road, London SW7 2AZ, UK.
Taeyang Lee, Department of Life Sciences, Imperial College London, South Kensington Campus, Exhibition Road, London SW7 2AZ, UK.
Vinoo Selvarajah, iGEM Foundation, 45 Prospect Street, Cambridge 02139, MA, USA.
Sonja Billerbeck, Molecular Microbiology, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, Groningen 9747 AG, The Netherlands.
Bradley Brown, School of Engineering, Newcastle University, Devonshire Building, Devonshire Terrace, NE1 7RU Newcastle Upon Tyne, UK.
Guo-nan Cai, School of Life Sciences, Fudan University, 220 Handan Road, Shanghai 200433, China.
Liang Cai, School of Life Sciences, Fudan University, 220 Handan Road, Shanghai 200433, China.
Edward Eisenstein, Institute of Bioscience and Biotechnology Research, Fischell Department of Bioengineering, University of Maryland, 9600 Gudelsky Drive, Rockville 20850, MD, USA.
Daisuke Kiga, School of Advanced Science and Engineering, Waseda University, 2-2 Wakamatsu Cho, Totsukamachi, Shinjuku City 169-8050, Tokyo, Japan.
David Ross, Material Measurement Laboratory, National Institute of Standards and Technology, 100 Bureau Dr., Gaithersburg 20899, MD, USA.
Nina Alperovich, Material Measurement Laboratory, National Institute of Standards and Technology, 100 Bureau Dr., Gaithersburg 20899, MD, USA.
Noah Sprent, Department of Chemical Engineering, Imperial College London, South Kensington Campus, Exhibition Road, London SW7 2AZ, UK.
Jaclyn Thompson, Chemical Engineering, Worcester Polytechnic Institute, 100 Institute Road, Worcester 01609-2280, MA, USA.
Eric M Young, Chemical Engineering, Worcester Polytechnic Institute, 100 Institute Road, Worcester 01609-2280, MA, USA.
Drew Endy, Bioengineering, Stanford University, 443 Via Ortega, Stanford 94305, CA, USA.
Traci Haddock-Angelli, iGEM Foundation, 45 Prospect Street, Cambridge 02139, MA, USA.
Supplementary Data
Supplementary Data are available at SYNBIO Online.
Material availability
All constructs used in this manuscript can be obtained under the unilateral OpenMTA from https://stanford.freegenes.org/.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Funding
This work was supported in part by the following funding sources: J.B. was supported by Air Force Research Laboratory (AFRL) and DARPA contract FA8750-17-C-0184. N.S. was supported by funding from the BBSRC under award BB/M011178/1. G.B., L.H., and T.L. were supported by the EPSRC under award EP/R034915/1 and EP/S022856/1. G.C. and L.C. were supported by funds from YF Capital and the National Top Talent Undergraduate Training Program, China. A.V. and Y.B. were funded by Grant MINECO/AEI, EU DPI2017-82 896-C2-1-R and MCIN/AEI/10.13039/501 100 011 033 grant number PID2020-117271RB-C21. Y.B. was supported by Secretaría de Educación Superior, Ciencia, Tecnología e Innovación-Ecuador (Scholarship Convocatoria Abierta 2011). D.K. was supported by JST, CREST Grant Number JPMJCR21N4, Japan. E.Y. was supported by the National Science Foundation under Grant No. 1 939 860. J.T. was supported by the Office of the Director of National Intelligence (ODNI), Intelligence Advanced Research Projects Activity (IARPA) under Finding Engineering Linked Indicators (FELIX) program contract N66001-18-C-4507.
This document does not contain technology or technical data controlled under either the U.S. International Traffic in Arms Regulations or the U.S. Export Administration Regulations. Views, opinions, and/or findings expressed are those of the author(s) and should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. Government.
Author contributions statement
Conceptualization and Methodology: J.B., C.A.T., A.V., G.B., V.S. Project administration: J.B., C.A.T., V.S., T.H.-A. Resources (DNA): D.E. Investigation: S.B., B.B., L.C., E.E., D.K., D.R., N.A., N.S., J.T., E.Y., A.V., Y.B., G.B., L.H., T.L. Data Curation and Formal Analysis: J.B. Writing: original draft: J.B., review and editing: all authors.
Conflict of interest statement.
The authors declare that they have no conflict of interest.
References
- 1. Auld D.S., Coassin P.A., Coussens N.P., Hensley P., Klumpp-Thomas C., Michael S., Sitta Sittampalam G., Joseph Trask O., Wagner B.K. and Weidner J.R.. et al. (2020) Microplate selection and recommended practices in high-throughput screening and quantitative biology. Assay Guidance Manual [Internet].
- 2. Beal J. (2017) Biochemical complexity drives log-normal variation in genetic expression. Eng. Biol., 1, 55–60.doi: 10.1049/enb.2017.0004. [DOI] [Google Scholar]
- 3. Beal J., Baldwin G.S., Farny N.G., Gershater M., Haddock-Angelli T., Buckley-Taylor R., Dwijayanti A., Kiga D., Lizarazo M. and Marken J.. et al. (2021) Comparative analysis of three studies measuring fluorescence from engineered bacterial genetic constructs. PloS One, 16, e0252263.doi: 10.1371/journal.pone.0252263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beal J., Farny N.G., Haddock-Angelli T., Selvarajah V., Baldwin G.S., Buckley-Taylor R., Gershater M., Kiga D., Marken J. and Sanchania V.. et al. (2020) Robust estimation of bacterial cell count from optical density. Commun. Biol., 3, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beal J., Haddock-Angelli T., Baldwin G., Gershater M., Dwijayanti A., Storch M., Kim M., Lizarazo M., Rettberg R. and the iGEM Interlab Study Contributors (2018) Quantification of bacterial fluorescence using independent calibrants. PloS One, 13, e0199432.doi: 10.1371/journal.pone.0199432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boada Y., Vignoni A., Alarcon-Ruiz I., Andreu-Vilarroig C., Monfort-Llorens R., Requena A. and Picó J. (2019) Characterization of gene circuit parts based on multiobjective optimization by using standard calibrated measurements. ChemBioChem, 20, 2653–2665.doi: 10.1002/cbic.201900272. [DOI] [PubMed] [Google Scholar]
- 7. Csibra E. and Stan G.-B. (2021) FPCountR: Absolute quantification of fluorescent proteins for synthetic biology. bioRxiv. doi: 10.1101/2021.12.06.471413. [DOI]
- 8. Hecht A., Endy D., Salit M. and Munson M.S. (2016) When wavelengths collide: bias in cell abundance measurements due to expressed fluorescent proteins. ACS Synth. Biol., 5, 1024–1027. [DOI] [PubMed] [Google Scholar]
- 9. Kelly J.R., Rubin A.J., Davis J.H., Ajo-Franklin C.M., Cumbers J., Czar M.J., Mora K., Glieberman A.L., Monie D.D. and Endy D. (2009) Measuring the activity of biobrick promoters using an in vivo reference standard. J. Biol. Eng., 3 4.doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lambert T.J. (2019) FPbase: a community-editable fluorescent protein database. Nat. Methods, 16, 277–278.doi: 10.1038/s41592-019-0352-8. [DOI] [PubMed] [Google Scholar]
- 11. SpheroTech (2001) Measuring molecules of equivalent fluorescein (mefl), pe (mepe) and rpe-cy5 (mepcy) using sphero rainbow calibration particles. Technical Report SpheroTechnical Notes: STN-9, Rev C 071398, SpheroTech, October.
- 12. Stanton B.C., Nielsen A.A., Tamsir A., Clancy K., Peterson T. and Voigt C. (2014) Genomic mining of prokaryotic repressors for orthogonal logic gates. Nat. Chem. Biol., 10, 99–105.doi: 10.1038/nchembio.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yordanov B., Dalchau N., Grant P.K., Pedersen M., Emmott S., Haseloff J. and Phillips A. (2014) A computational method for automated characterization of genetic components. ACS Synth. Biol., 3 , 578–588.doi: 10.1021/sb400152n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.