

Abstract
New treatments, particularly second‐line options, are needed to improve outcomes for patients with recurrent/metastatic cervical cancer (r/mCC). Tisotumab vedotin (TV) is an antibody–drug conjugate directed to tissue factor, a transmembrane protein commonly expressed in cancer cells, to deliver cytotoxic monomethyl auristatin E. This single‐arm, open‐label phase 1/2 trial evaluated the consistency of safety and efficacy outcomes of TV in Japanese patients with r/mCC to bridge the current findings with those reported in previous trials in non‐Japanese patients in the United States and Europe. In part 1 (dose escalation; N = 6), patients with advanced solid tumors received TV 1.5 or 2.0 mg/kg once every 3 weeks to determine the maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D). Part 2 (dose expansion; N = 17) evaluated the RP2D in r/mCC patients with 1–2 prior lines of therapy. In part 1, no dose‐limiting toxicities were observed, the MTD was not reached, and TV 2.0 mg/kg was established as the RP2D. In part 2, the most common treatment‐emergent adverse events were anemia (58.8%), nausea (58.8%), alopecia (47.1%), epistaxis (47.1%), and diarrhea (35.3%); adverse events of special interest were bleeding (76.5%), ocular events (35.3%), and peripheral neuropathy (17.6%), and were mostly grade 1/2. In part 2, confirmed objective response rate was 29.4%, median duration of response was 7.1 months, and median time to response was 1.2 months. In Japanese patients with r/mCC, TV demonstrated a manageable and tolerable safety, pharmacokinetics, and efficacy profile consistent with that observed in non‐Japanese patients.
Keywords: female, recurrence, thromboplastin, tisotumab vedotin, uterine cervical neoplasms
There is a need for second‐line treatments for patients with recurrent/metastatic cervical cancer (r/mCC). Tisotumab vedotin (TV), an antibody–drug conjugate, has been approved for treatment of r/mCC following a large phase 2 study in a non‐Japanese population. The current study extends the feasibility of this regimen for use in Japanese patients with r/mCC.

Abbreviations
- ADA
antidrug antibody
- ADC
antibody‐drug conjugate
- ADCC
antibody‐dependent cellular cytotoxicity
- ADCP
antibody‐dependent cellular phagocytosis
- AE
adverse event
- AESI
adverse event of special interest
- AUC
area under the curve
- CI
confidence interval
- CR
complete response
- %CV
percent coefficient of variation
- DLT
dose‐limiting toxicity
- DOR
duration of response
- FDA
US Food and Drug Administration
- MMAE
monomethyl auristatin E
- MTD
maximum tolerated dose
- ORR
objective response rate
- OS
overall survival
- PFS
progression‐free survival
- PR
partial response
- Q3W
every 3 weeks
- RECIST v1.1
Response Evaluation Criteria in Solid Tumors, version 1.1
- r/mCC
recurrent/metastatic cervical cancer
- RP2D
recommended phase 2 dose
- SAE
serious adverse event
- SD
stable disease
- TF
tissue factor
- TEAE
treatment‐emergent adverse event
- TTR
time to response
- TV
tisotumab vedotin
1. INTRODUCTION
Cervical cancer is both the fourth most prevalent and the fourth deadliest cancer globally, with more than 500,000 new cases diagnosed each year that account for 7.5% of cancer‐related deaths in women. 1 , 2 In Japan, an estimated 34,000 new patients were diagnosed with cervical cancer in 2016; contrary to the global trend, recent studies suggest that the incidence of cervical cancer has been increasing in Japan over the past 2 decades. 3 , 4 , 5 , 6 Until recently, the preferred first‐line systemic options for the treatment of recurrent/metastatic cervical cancer (r/mCC) were a taxane (eg, paclitaxel, topotecan), a platinum‐containing agent (eg, cisplatin, carboplatin), and bevacizumab. 7 , 8 , 9 Pembrolizumab was recently approved by the US Food and Drug Administration (FDA) as a single agent or in combination with chemotherapy, with or without bevacizumab, for patients with persistent, recurrent, or metastatic cervical cancer whose tumors express programmed death–ligand 1 (as expressed by a combined positive score ≥ 1). 10 , 11 , 12 However, there remains a need to establish effective second‐line treatments for patients who progress on first‐line treatment or who are unable to tolerate first‐line treatment. Accordingly, second‐line treatments for r/mCC are being explored in Japan and globally.
Tisotumab vedotin (TV) is an antibody–drug conjugate (ADC) in clinical development for the treatment of several solid tumors. 13 , 14 , 15 , 16 The antibody is directed to tissue factor (TF), a transmembrane protein whose primary role is to initiate the coagulation cascade. TF has also been shown to play a role in the tumor growth, angiogenesis, and metastasis of cancer, 17 and is highly prevalent in cervical cancer, including squamous and adenocarcinoma histological subtypes. 18 , 19 The antibody moiety of TV is conjugated to monomethyl auristatin E (MMAE) via a valine citrulline linker, which is proteolytically cleaved and released following internalization of TV into cancer cells expressing TF. 15 , 16 MMAE is a microtubule disruptor and kills actively dividing cancer cells that have internalized TV. 15 TV has antitumor activity on multiple tumor types and kills target cells by direct cytotoxicity, bystander cytotoxicity, antibody‐dependent cellular cytotoxicity (ADCC), antibody‐dependent cellular phagocytosis (ADCP), and immunogenic cell death. 13
Previously, a phase 1/2 trial (innovaTV 201; NCT02001623) of TV monotherapy in US and European patients reported a confirmed objective response rate (ORR) of 22% (95% CI, 12–35) as assessed by independent review in the cohort of patients with previously treated r/mCC (N = 55). 20 More recently, a pivotal phase 2, single‐arm, open‐label study (innovaTV 204; NCT03438396) evaluating TV monotherapy in a larger cohort of women with r/mCC (N = 101) in the United States and Europe reported a confirmed ORR as assessed by independent review of 24% (95% CI, 16–33), with a median duration of response (DOR) of 8.3 months. 21 The median progression‐free survival (PFS) and overall survival (OS) in this study were 4.2 and 12.1 months, respectively. The most common treatment‐related adverse events (AEs) were alopecia, epistaxis, nausea, conjunctivitis, fatigue, and dry eye, and most (65%) were mild to moderate (grade 1–2) in severity. It was this pivotal innovaTV 204 study that led to accelerated approval of the use of TV monotherapy in adult patients with previously treated r/mCC with disease progression on or after chemotherapy by the US FDA. 22 TV monotherapy has been shown to provide clinically meaningful and durable antitumor activity with a manageable safety profile in women with previously treated r/mCC.
Here, we performed a single‐arm phase 1/2 trial (innovaTV 206) to evaluate the safety, pharmacokinetics, and efficacy of TV in Japanese patients with solid tumors (part 1) and r/mCC (part 2).
2. PATIENTS AND METHODS
2.1. Study population and design
The innovaTV 206 trial (GCT1015‐06, NCT03913741) was a single‐arm, open‐label phase 1/2 trial designed to evaluate the safety, efficacy, pharmacokinetics, and pharmacodynamics of intravenous TV in Japanese patients with advanced solid malignancies. The trial was conducted in compliance with the protocol ICH Good Clinical Practice E6 (R2) and applicable regulatory requirements.
The study consisted of two parts: Part 1, which enrolled patients with solid tumors in sequential dose escalation cohorts, sought to determine the maximum tolerated dose (MTD) and establish the recommended phase 2 dose (RP2D). Part 2 was an expansion phase designed to assess the safety and antitumor activity of TV in Japanese patients with r/mCC at the RP2D of TV determined in part 1.
Patients enrolled in part 1 had locally advanced or metastatic solid tumors with disease progression on standard therapy or were intolerant of or not eligible for standard therapy. Eligibility criteria for part 2 included presence of extrapelvic r/mCC, including squamous cell, adenocarcinoma, or adenosquamous histological subtypes; disease progression during or following treatment with platinum doublet or paclitaxel and nogitecan plus bevacizumab (where eligible); ≤2 prior lines of therapy in the recurrent/metastatic setting; and not being a candidate for curative treatment. In parts 1 and 2, additional eligibility criteria included the presence of measurable disease according to Response Evaluation Criteria in Solid Tumors, version 1.1 (RECIST v1.1); age ≥ 20 years; Eastern Cooperative Oncology Group performance status of 0 or 1; acceptable renal function, liver function, hematological status, and coagulation status; not being of childbearing potential or willing to use ≥1 adequate form of contraception; being able to provide a fresh or archival biopsy; and being able to provide written signed informed consent.
Exclusion criteria for parts 1 and 2 included the presence of coagulation defects or active major bleeding; presence of ocular surface disease; presence of other previous or current malignancy; active brain metastases; presence of peripheral neuropathy ≥ grade 2; previous treatment with an MMAE‐containing drug or recent chemotherapy, radiation, surgery, or live vaccine; or presence of active infection.
2.2. Study procedures
For parts 1 and 2, patients were given one 30‐min intravenous infusion of TV every 3 weeks (Q3W) until disease progression or unacceptable toxicity. Thus, each cycle was 21 days in duration. For part 1, dose escalation was performed according to a 3 + 3 design. Initially, three patients were given TV 1.5 mg/kg intravenously and monitored carefully for dose‐limiting toxicities (DLTs) during the first cycle. Following the absence of DLTs, three additional patients were given TV 2.0 mg/kg and similarly monitored. The MTD was considered to be the dose below the lowest dose that caused a DLT in at least one‐third of patients. Patients were followed for ≥2 cycles before the RP2D was defined. For part 2, patients were given TV 2.0 mg/kg Q3W, which was determined to be the RP2D.
AEs were graded according to the National Cancer Institute's Common Terminology Criteria for Adverse Events (version 5.0) and monitored throughout treatment and for 30 days after treatment ended.
Treatment to prevent ocular AEs was mandated for all study patients and consisted of the use of preservative‐free lubricating eye drops throughout the study, steroid eye drops (first dose was given before start of infusion and for the first 3 days of each treatment cycle), a local ocular vasoconstrictor before TV infusion, and eye‐cooling pads during infusion. Laboratory tests for hematology, biochemistry, and coagulation assessments were performed within 5 days before the first cycle and within 24 hours before TV infusion for each cycle beginning with cycle 2. For pharmacokinetics assessments, blood sampling was performed before infusion; at the end of infusion; and at 2, 5, 12, 24 (day 2), 72 (day 4), 168 (day 8), and 336 (day 15) hours after infusion for cycles 1 and 2; before infusion for cycle 3 and for every fourth cycle beginning with cycle 7; at the end of treatment; and at follow‐up visits. These samples were collected during cycles 1 and 2 to characterize the pharmacokinetics profile for TV (ADC), and free MMAE, and to compare the pharmacokinetics of these analytes in this Japanese population with Western populations in other trials. Plasma concentrations of ADC were determined using a validated enzyme‐linked immunosorbent assay. 20 Plasma concentrations of MMAE were determined using a validated liquid chromatography method with tandem mass spectrometric detection. 20
Computed tomography was the preferred modality for the acquisition of tumor images. Contrast‐enhanced magnetic resonance imaging was used when computed tomography with iodinated contrast is contraindicated or when mandated by local practice. Tumor imaging assessments were performed during the screening period (≤28 days before cycle 1 day 1), every 6 weeks beginning with the first dose until 30 weeks, and every 12 weeks thereafter. Tumor response was determined based on RECIST v1.1 criteria. Objective response (ie, complete response [CR] or partial response [PR]) was confirmed by a repeat imaging assessment ≥4 weeks after the first observed indication of a response. In the case of stable disease (SD), the measurement must have been performed at least 5 weeks after the first dose of study treatment to meet the criterion for SD.
For antidrug antibody (ADA) assessments, sampling was performed before infusion for cycles 1–3, at every fourth cycle beginning with cycle 7, at the end of treatment, and at follow‐up visits. Serum samples were screened for antibodies binding to TV, and the titer of confirmed positive samples was determined.
Biopsies provided at screening were retrospectively analyzed for TF expression (on the cell surface or membrane) using an analytically validated immunohistochemistry assay. A TF histology score (H‐score) was calculated to combine both expression and intensity of staining as described previously. 20 Tumor cells with any proportion of TF expression were considered positive.
2.3. Endpoints
The primary endpoints were assessment of AEs (with laboratory tests, electrocardiogram, and vital signs), DLTs, ADAs, and pharmacokinetics of TV and free MMAE. The secondary endpoints were confirmed ORR, DOR, and time to response (TTR) based on RECIST v1.1 and were determined by the investigator in part 1 and by independent review in part 2. Exploratory endpoints included OS, PFS (based on investigator review for part 1 and independent review for part 2), TF expression in pretreatment tumor biopsies (membrane H‐score) and circulating TF, and changes in other serum proteins.
2.4. Sample size considerations
For part 1, the sample size was based on the standard 3 + 3 design. For part 2, the sample size was based on the following considerations: (1) the confirmed ORR in patients with previously treated r/mCC in innovaTV 201, which enrolled US and European patients, was 26% (based on data cutoff of May 16, 2018); and (2) the antitumor activity of TV was expected to be similar in Japanese and non‐Japanese patients. To demonstrate consistency between the ORR in Japanese patients and the ORR in non‐Japanese patients, the ORRJapan/ORRNon‐Japan had to be >0.5, according to method 1 in the Pharmaceuticals and Medical Devices Agency's Basic Principles on Global Clinical Trials. 23 Assuming that the ORR in non‐Japanese patients will be approximately 25%, this implies that ≥2 confirmed responders must be observed among 15 patients in part 2 of this trial. With 15 patients enrolled in part 2, there is 92% probability of observing ≥2 confirmed responders under the assumption of a true confirmed ORR of 25%.
2.5. Statistical analyses
All analyses were based on patients who received ≥1 dose of TV. Safety assessments included AEs, DLTs, and laboratory assessments and were summarized by dose for each trial part. Pharmacokinetics parameters and ADA assessments were summarized by descriptive statistics by trial part and dose. Confirmed and confirmed/unconfirmed ORRs, by dose and trial part, were estimated with exact 95% CIs calculated by the Clopper‐Pearson method. DOR, TTR, PFS, and OS were analyzed using the Kaplan‐Meier method, and medians were estimated with 95% CIs, where possible.
3. RESULTS
3.1. Study population and demographics
At data cutoff (August 14, 2020), the median follow‐up for patients was 14.0 months (range, 2–17) in part 1 and 6.3 months (range, 1–12) in part 2. All patients had discontinued study treatment by the data cutoff and four and nine patients in part 1 and 2, respectively, were in survival follow‐up.
Baseline characteristics of patients enrolled in part 1 are shown in Table 1. Six patients with various types of solid tumors were enrolled. Three patients (median age, 38.0 years) were given TV 1.5 mg/kg. No DLTs were observed at this dose level, so three additional patients (median age, 56.0 years) were given TV 2.0 mg/kg. For patients receiving TV 2.0 mg/kg in part 1, the median number of cycles was six and the median duration of exposure was 4.6 months (Table S1). No DLTs were observed at the TV 2.0 mg/kg dose level; therefore, the MTD was not reached and the RP2D for part 2 was established as TV 2.0 mg/kg Q3W.
TABLE 1.
Baseline characteristics and type of cancer at time of diagnoses for patients in the dose escalation cohort (Part 1)
| Parameters | TV 1.5 mg/kg (N = 3) | TV 2.0 mg/kg (N = 3) |
|---|---|---|
| Age, years, median (range) | 38 (31–71) | 56 (42–70) |
| Male sex, n (%) | 3 (100) | 1 (33.3) |
| ECOG performance status at baseline, n (%) | ||
| 0 | 2 (66.7) | 1 (33.3) |
| 1 | 1 (33.3) | 2 (66.7) |
| Type of cancer at time of diagnosis, n (%) | ||
| Alveolar soft part sarcoma | 1 (33.3) | 0 |
| Cervical squamous cell carcinoma | 0 | 1 (33.3) |
| Esophagus squamous cell carcinoma | 1 (33.3) | 0 |
| Large cell neuroendocrine cancer | 1 (33.3) | 0 |
| Ovarian granulosa cell tumor | 0 | 1 (33.3) |
| Sigmoid colon adenocarcinoma | 0 | 1 (33.3) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; TV, tisotumab vedotin.
Baseline demographics and disease characteristics of the 17 patients with r/mCC enrolled in part 2 are shown in Table 2. Most patients were aged <50 years (58.8%), and similar proportions had adenocarcinoma (47.1%) or squamous cell carcinoma (52.9%), one (47.1%) or two (52.9%) previous lines of therapy, and first‐line bevacizumab in combination with chemotherapy doublet treatment (47.1%) or not (52.9%). Patients in part 2 received a median of five cycles, and the median duration of exposure was 3.7 months (Table S2). This exposure to TV was consistent with the previous innovaTV 204 study. 21
TABLE 2.
Baseline demographics and disease characteristics of patients in the dose expansion cohort (part 2)
| Parameters | TV 2.0 mg/kg (N = 17) |
|---|---|
| Age (years) | |
| Mean (standard deviation) | 48.1 (11.3) |
| Median (range) | 47.0 (33.0–66.0) |
| Age, years (%) | |
| <50 | 10 (58.8) |
| ≥50 to ≤65 | 5 (29.4) |
| >65 | 2 (11.8) |
| Weight (kg) | |
| Mean (standard deviation) | 53.5 (11.7) |
| Median (range) | 53.0 (30.0–72.0) |
| Histology type, n (%) | |
| Squamous cell carcinoma | 9 (52.9) |
| Adenocarcinoma | 8 (47.1) |
| ECOG performance status at baseline, n (%) | |
| 0 | 9 (52.9) |
| 1 | 8 (47.1) |
| Metastatic disease at screening, n (%) | |
| Yes | 15 (88.2) |
| No | 2 (11.8) |
| Recurrent disease at screening, n (%) | |
| Yes | 13 (76.5) |
| No | 4 (23.5) |
| Prior lines of systemic therapy in the recurrent or metastatic setting, n (%) | |
| 1 line | 8 (47.1) |
| 2 lines | 9 (52.9) |
| Bevacizumab in combination with chemotherapy doublet as first‐line systemic regimen, n (%) | |
| Yes | 8 (47.1) |
| No | 9 (52.9) |
| Response to last systemic regimen, n (%) | |
| Yes | 4 (23.5) |
| No | 11 (64.7) |
| Not known | 2 (11.8) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; TV, tisotumab vedotin.
3.2. Safety
All patients in part 1 experienced ≥1 treatment‐emergent AE (TEAE) (Table 3). Two patients, one at each dose level, experienced ≥1 grade 3 or greater TEAE. No TEAEs leading to treatment discontinuation and no TEAEs associated with death were observed. The most common TEAEs in part 1 included nausea (83.3%) and epistaxis (50%). AEs of special interest (AESIs) included bleeding (66.7%) at TV 1.5 mg/kg dose and ocular events (66.7%), bleeding (66.7%), and peripheral neuropathy (33.3%) at TV 2.0 mg/kg dose. All AESIs were grade 1/2. The safety profile was consistent with that of the previous innovaTV 204 study. 21
TABLE 3.
TEAEs in patients in the dose escalation (part 1) and dose expansion (part 2) cohorts
| Patient with any grade TEAE, n (%) | Part 1 | Part 2 | |
|---|---|---|---|
| TV 1.5 mg/kg | TV 2.0 mg/kg | TV 2.0 mg/kg | |
| (N = 3) | (N = 3) | (N = 17) | |
| TEAE | 3 (100) | 3 (100) | 17 (100.0) |
| Related to TV | 3 (100) | 3 (100) | 17 (100.0) |
| Grade ≥3 TEAE | 1 (33.3) | 1 (33.3) | 14 (82.4) |
| Related to TV | 0 | 1 (33.3) | 9 (52.9) |
| TESAE | 0 | 1 (33.3) | 8 (47.1) |
| Related to TV | 0 | 0 | 4 (23.5) |
| Fatal TEAE | 0 | 0 | 0 |
| Related to TV | 0 | 0 | 0 |
| Dose‐limiting toxicities | 0 | 0 | NA |
| TEAE leading to treatment interruption | 1 (33.3) | 1 (33.3) | 2 (11.8) |
| TEAE leading to dose reduction | 0 | 1 (33.3) | 3 (17.6) |
| TEAE leading to drug withdrawal | 0 | 0 | 1 (5.9) |
| Preferred term, TEAEs in ≥2 patients in any arm a | |||
| Abdominal pain, upper | 0 | 0 | 4 (23.5) |
| Alanine aminotransferase increased | 0 | 1 (33.3) | 3 (17.6) |
| Alopecia | 0 | 2 (66.7) | 8 (47.1) |
| Anemia | 0 | 2 (66.7) | 10 (58.8) |
| Anxiety | 0 | 0 | 2 (11.8) |
| Aspartate aminotransferase increased | 1 (33.3) | 1 (33.3) | 3 (17.6) |
| Back pain | 0 | 0 | 2 (11.8) |
| Blood alkaline phosphatase increased | 0 | 0 | 2 (11.8) |
| Conjunctivitis | 0 | 1 (33.3) | 3 (17.6) |
| Constipation | 2 (66.7) | 0 | 0 |
| Decreased appetite | 1 (33.3) | 1 (33.3) | 2 (11.8) |
| Diarrhea | 1 (33.3) | 0 | 6 (35.3) |
| Epistaxis | 2 (66.7) | 1 (33.3) | 8 (47.1) |
| γ‐Glutamyltransferase increased | 0 | 1 (33.3) | 2 (11.8) |
| Genital hemorrhage | 0 | 0 | 2 (11.8) |
| Insomnia | 1 (33.3) | 0 | 2 (11.8) |
| Lower gastrointestinal hemorrhage | 0 | 0 | 3 (17.6) |
| Malaise | 0 | 0 | 2 (11.8) |
| Myalgia | 0 | 0 | 2 (11.8) |
| Nausea | 3 (100.0) | 2 (66.7) | 10 (58.8) |
| Neutrophil count decreased | 0 | 2 (66.7) | 3 (17.6) |
| Peripheral edema | 0 | 0 | 2 (11.8) |
| Peripheral sensory neuropathy | 0 | 1 (33.3) | 3 (17.6) |
| Pyrexia | 1 (33.3) | 1 (33.3) | 3 (17.6) |
| Rash | 0 | 1 (33.3) | 2 (11.8) |
| Stomatitis | 0 | 0 | 2 (11.8) |
| Tumor hemorrhage | 0 | 0 | 2 (11.8) |
| Vomiting | 1 (33.3) | 0 | 3 (17.6) |
| White blood cell count decreased | 0 | 2 (66.7) | 4 (23.5) |
Abbreviations: NA, not applicable; TEAE, treatment‐emergent adverse event; TESAE, treatment‐emergent serious adverse event; TV, tisotumab vedotin.
TEAEs experienced by ≥2 patients in either part 1 (ie, both dose levels [N = 6]) or part 2.
All patients in part 2 experienced ≥1 TEAE (Table 3). The most common TEAEs in part 2 were anemia (58.8%), nausea (58.8%), alopecia (47.1%), epistaxis (47.1%), and diarrhea (35.3%). Fourteen patients (82.4%; 52.9% were related to TV) experienced ≥ grade 3 TEAEs, with the most common being anemia (35.3%), tumor hemorrhage (11.8%), and leukopenia (11.8%). Eight patients (47.1%; 23.5% were related to TV) experienced serious TEAEs (grade 2/3) (Table S3). No patients experienced grade 4/5 SAEs. One TEAE (lower gastrointestinal hemorrhage) led to treatment discontinuation. No TEAEs were associated with death. AESIs included grade 1–3 bleeding events (76.5% all grades; 17.6% grade 3), grade 1/2 ocular events (35.3%), and grade 1 peripheral neuropathy (17.6%) (Table 4). Of the grade 3 bleeding events, one patient (5.9%) had a lower gastrointestinal hemorrhage and two patients (11.8%) had tumor hemorrhage. No patients experienced grade 4/5 AESIs.
TABLE 4.
Adverse events of special interest in the dose expansion cohort (part 2)
| Preferred term | N = 17 | |||
|---|---|---|---|---|
| Grade 1 | Grade 2 | Grade 3 | Any grade | |
| Patients with ≥1 ocular TEAE | 3 (17.6) | 3 (17.6) | 0 | 6 (35.3) |
| Conjunctivitis | 1 (5.9) | 2 (11.8) | 0 | 3 (17.6) |
| Conjunctivitis allergic | 0 | 1 (5.9) | 0 | 1 (5.9) |
| Scleritis | 0 | 1 (5.9) | 0 | 1 (5.9) |
| Hordeolum | 1 (5.9) | 0 | 0 | 1 (5.9) |
| Vision blurred | 1 (5.9) | 0 | 0 | 1 (5.9) |
| Patients with ≥1 peripheral neuropathy TEAE | 3 (17.6) | 0 | 0 | 3 (17.6) |
| Peripheral sensory neuropathy | 3 (17.6) | 0 | 0 | 3 (17.6) |
| Patients with ≥1 bleeding TEAE | 8 (47.1) | 2 (11.8) | 3 (17.6) | 13 (76.5) |
| Epistaxis | 8 (47.1) | 0 | 0 | 8 (47.1) |
| Lower gastrointestinal hemorrhage | 2 (11.8) | 0 | 1 (5.9) | 3 (17.6) |
| Genital hemorrhage | 1 (5.9) | 1 (5.9) | 0 | 2 (11.8) |
| Tumor hemorrhage | 0 | 0 | 2 (11.8) | 2 (11.8) |
| Anal hemorrhage | 0 | 1 (5.9) | 0 | 1 (5.9) |
| Hematochezia | 1 (5.9) | 0 | 0 | 1 (5.9) |
| Vaginal hemorrhage | 1 (5.9) | 0 | 0 | 1 (5.9) |
Abbreviation: TEAE, treatment‐emergent adverse event.
3.3. Pharmacokinetics assessments
Pharmacokinetics data for part 1 and part 2 are shown in Figure 1, Table S4, and Table S5. For both parts, the maximum concentration of TV was reached shortly after the end of infusion. For part 2, the geometric mean for cycle 1 maximum concentration for TV and MMAE was 28.6 μg/ml (percent coefficient of variation [%CV], 32.5) and 5.3 ng/ml (%CV, 59.8), respectively. No appreciable accumulation was observed with repeated dosing for TV and free MMAE; the geometric means of the area under the curve from time zero to infinity (AUC0➔∞) for TV in part 2 was 49.2 day μg/ml in cycle 1 and 48.3 day μg/ml in cycle 2, and the corresponding geometric means for free plasma MMAE was 42.0 day ng/ml and 36.0 day ng/ml, respectively (Table S5). Exposures in Japanese patients enrolled in parts 1 and 2 were consistent with exposure in non‐Japanese patients.
FIGURE 1.
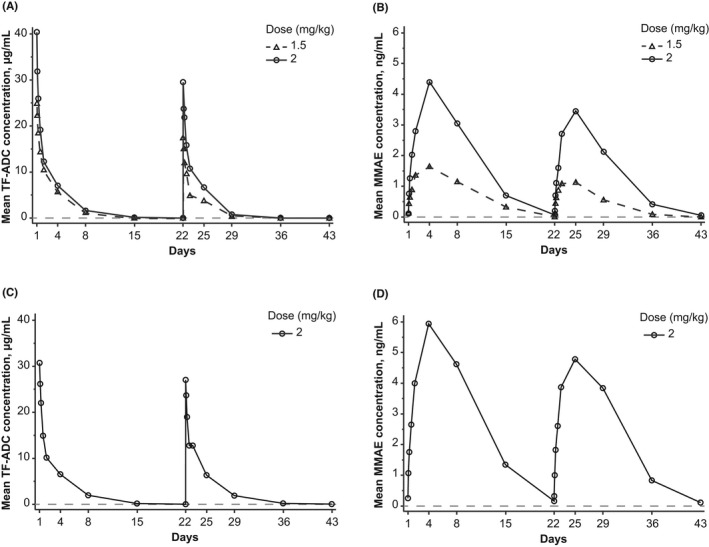
Pharmacokinetic profile of the TV ADC and free MMAE. Plasma concentrations of the ADC (A, C) and free MMAE (B, D) in plasma from patients in the dose escalation cohort (part 1; A, B) and the dose expansion cohort (part 2; C, D) for cycles 1 and 2. The gray dashed line represents the lower limit of quantitation. ADC, antibody–drug conjugate; MMAE, monomethyl auristatin E; TF‐ADC, tissue factor to antibody–drug conjugate; TV, tisotumab vedotin
3.4. Efficacy
Of the 17 patients enrolled in part 2, the confirmed best overall response of CR, PR, SD, PD, and not evaluable occurred in zero, five (29.4%), seven (41.2%), three (17.6%), and two (11.8%) patients, respectively, as assessed by the independent review. The confirmed ORR (CR + PR) was 29.4% (5/17 patients; 95% CI, 10.3–56.0), and the confirmed disease control rate (CR + PR + SD) was 70.6% (12/17 patients; 95% CI, 44.0–89.7). The maximum percentage change in sum of diameters of target lesions for the 15 treated patients with available postbaseline scans is shown in Figure 2.
FIGURE 2.
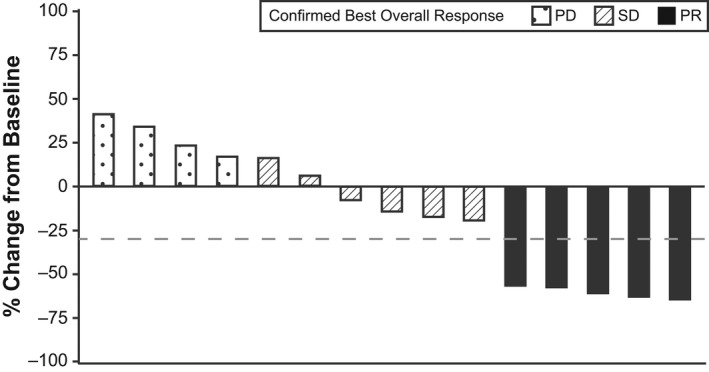
Maximum percentage change in target lesions in patients in dose expansion phase (part 2). The best overall response was evaluated by the independent review. Data from 15 patients are shown. Two patients were not evaluable because they did not undergo postbaseline scans because of withdrawal (one death and one patient decision). The dashed line represents a 30% decrease. PD, progressive disease; PR, partial response; SD, stable disease
As assessed by the independent review, the median TTR was 1.2 months (range, 1.1–2.7 months), and the median DOR was 7.1 months (range, 3.1 months to not reached). The TTR and DOR for the five patients who achieved a PR is shown in Figure 3. The median PFS was 3.1 months (95% CI, 1.2–7.1), and Kaplan–Meier estimation showed that the percentage of patients with PFS ≥6 months was 26.1% (95% CI, 7.2–50.2). The median OS was 11.4 months (95% CI, 6.2‐not reached). Kaplan–Meier estimates showed that the percentages of patients with an OS ≥6 months and ≥12 months were 81.6% (95% CI, 53.0–93.7) and 25.7% (95% CI, 1.6–63.9), respectively.
FIGURE 3.
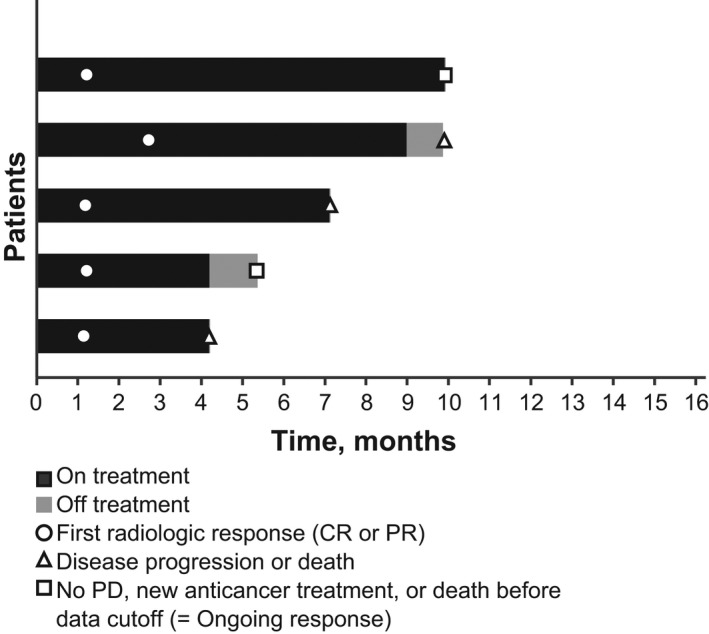
Time to response (TTR) and duration of response (DOR) in the five patients in dose expansion phase (part 2) who achieved a confirmed response. Response was assessed by independent review. One patient had one prior line of therapy in the recurrent or metastatic setting, and the others had two prior lines. All patients with two prior lines had received bevacizumab in combination with a chemotherapy doublet as first‐line systemic treatment. CR, complete response; PD, progressive disease; PR, partial response
3.5. ADA
No patients were positive for treatment‐emergent ADAs in part 1. One patient (5.9%) in part 2 became positive for treatment‐emergent ADAs. This was consistent with low rates of immunogenicity (~5.4%) observed in the innovaTV 204 study. 21
3.6. TF expression
All 17 patients provided evaluable biopsies, and membrane TF expression (≥1%) was confirmed for all patients (median overall membrane H‐score, 65.0 [range, 1.0–235.0]). Response to TV was observed regardless of the level of TF expression on tumor cell membrane (Figure 4); however, the analysis of association between TF expression and response was limited by the small sample size.
FIGURE 4.
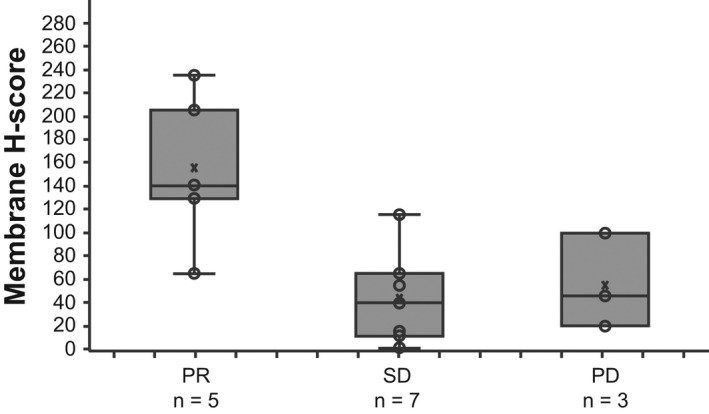
Tumor membrane H‐score for tissue factor at baseline among patients in dose‐expansion phase (part 2) by best confirmed overall response. The lines within the boxes represent the median, whiskers extend to the most extreme observation within 1.5 times the interquartile range from the nearest quartile, X's represent the mean, and circles represent individual tumor samples. PR, partial response; PD, progressive disease; SD, stable disease
4. DISCUSSION
Tisotumab vedotin monotherapy showed a manageable and tolerable safety profile at both dose levels (1.5 mg/kg and 2 mg/kg Q3W) in Japanese patients. The efficacy/safety results in the r/mCC cohort enrolled in part 2 are consistent with those reported in the pivotal innovaTV 204 study, 21 suggesting that the overall benefit‐to‐risk ratio in Japanese patients with r/mCC is relatively comparable with that in non‐Japanese patients.
The most common TEAEs were anemia, nausea, alopecia, and epistaxis, all of which were grade 1/2. AESIs, including ocular events, peripheral neuropathy, and bleeding, were generally mild to moderate in nature and were effectively managed with dose modification and supportive care. AESIs reported in part 2 of the study (r/mCC patients) included bleeding in 76.5% (all grade 1–3), ocular events in 35.3% (all grade 1/2), and peripheral neuropathy in 17.6% (all grade 1) of patients; the AESI profile was overall consistent with what was reported in non‐Japanese patients participating in innovaTV 204. 21
In part 2, TV demonstrated clinically meaningful efficacy in Japanese patients with r/mCC. TV efficacy in the current study was consistent with that in non‐Japanese patients observed in innovaTV 204. 21 The ORR of 29.4% (95% CI, 10.3–56.0) as assessed by independent review in part 2 compares favorably to the corresponding confirmed ORR reported in the innovaTV 204 study (24%) 21 and the innovaTV 201 study (22%). 20 The confirmed ORR is promising, given that only 23.5% of patients in part 2 of this study had a documented response to their previous therapy. The median DOR was 7.1 months in part 2 of the current study, 6.0 months in innovaTV 201, 20 and 8.3 months in innovaTV 204 21 ; all were determined by independent review, suggesting comparable durability of clinical responses in Japanese and non‐Japanese populations with r/mCC.
The pharmacokinetics profile and rates of immunogenicity were also consistent with those in non‐Japanese patients. In the current study, no patients in part 1 were positive for treatment‐emergent ADAs, and one patient (5.9%) in part 2 became positive for treatment‐emergent ADAs. These data are consistent with the low rate of immunogenicity (~5.4%) observed in the innovaTV 204 study. 21
Tissue factor expression was assessed in biopsy specimens from patients before TV treatment. All 17 evaluable biopsy specimens showed TF expression on the cell membrane of tumor cells. Taken together with other studies in which comparable distribution of TF expression was observed among different response groups, 20 , 21 the totality of data suggests that a response to TV occurs regardless of membrane TF expression‐levels in r/mCC. The data potentially reflect that the binding of TV may be sufficient to initiate target cell killing regardless of surface TF expression levels due to its multimodal mechanisms of action, which include direct and bystander cytotoxicity of actively dividing tumor cells, ADCC, ADCP, and immunogenic cell death. 10 , 11 , 21
Limitations of this open‐label phase 2 study include that it had only one treatment group, making it difficult to fully assess the effect of therapy on patient survival given the absence of a control group. Moreover, comparisons with historical studies assessing chemotherapy are limited by the differences in the study populations, especially because the historical comparator studies did not enroll patients who received the current first‐line standard of care with bevacizumab, and also because of differences in study conduct and procedures (ie, use of RECIST criteria and confirmation of objective response). Further, conclusions about the relationship between TF expression and response in Japanese patients could not be reached because of the small sample size.
In conclusion, results from this study were consistent with data from trials enrolling US and European women in that TV was similarly tolerable and showed comparable efficacy in Japanese and Western women. These results support the continued development of TV as a viable second‐line systemic option for Japanese patients with r/mCC with disease progression during or after chemotherapy.
DISCLOSURE
Kan Yonemori reports lecture fees from Eisai, Taiho, Pfizer, Eli Lilly, AstraZeneca, Takeda, Fujifilm, and Chugai; an advisory/consultancy role with Takeda, Eisai, Novartis, Chugai, Ono, AstraZeneca, Genmab, and Haihe Biopharma; and grants for clinical trials from Merck Sharp & Dohme, Daiichi‐Sankyo, AstraZeneca, Taiho, Haihe Biopharma, Pfizer, Novartis, Takeda, Chugai, Ono, Sanofi, Seagen, Eisai, Eli Lilly, Genmab, Boehringer Ingelheim, Kyowa Kirin, and Nippon Kayaku. Chaitali Passey reports advisory/consultancy remuneration, and profit from shares, from Genmab US Inc. Jeppe Klint Buchbjerg, Jeffrey R. Harris, Camilla Mondrup Andreassen, and Ibrahima Soumaoro are employees of Genmab. Leonardo Nicacio is an employee of Seagen. Kazuhiro Takehara reports lecture fees from Takeda Pharmaceutical Company. Keiichi Fujiwara reports lecture fees and research funds from Genmab KK. Takashi Iwata reports research funds from Tella Co. and MSD Co. Yasutoshi Kuboki reports research funds from Genmab. Kosei Hasegawa, Hidenori Kato, Yasuyuki Hirashima, and Hisamori Kato have no conflicts of interest to declare.
ETHICS STATEMENT
All clinical studies were performed in accordance with Good Clinical Practice Guidelines from the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use and the principles of the Declaration of Helsinki. Protocols were approved by appropriate institutional review boards.
CONSENT
Written informed consent was provided by all participants.
REGISTRY AND THE REGISTRATION OF THE STUDY/TRIAL
Clinical trial number NCT03913741 (innovaTV 206/GCT‐1015‐06); JapicCTI‐194639.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank the patients and their families and caregivers for participating in this study and all site personnel. This study was funded and sponsored by Genmab A/S (Copenhagen, Denmark) and Seagen Inc. (Bothell, USA). Tisotumab vedotin (TV) is being codeveloped by Genmab and Seagen Inc. Medical writing assistance in the development of the manuscript was provided by Emma Bone, PhD, of ApotheCom and was funded by Genmab A/S.
Yonemori K, Kuboki Y, Hasegawa K, et al. Tisotumab vedotin in Japanese patients with recurrent/metastatic cervical cancer: Results from the innovaTV 206 study. Cancer Sci. 2022;113:2788‐2797. doi: 10.1111/cas.15443
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. doi: 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 2. World Health Organization . Cancer Fact Sheets: Cervical Cancer.
- 3. Yamagami W, Nagase S, Takahashi F, et al. Clinical statistics of gynecologic cancers in Japan. J Gynecol Oncol. 2017;28(2):e32. doi: 10.3802/jgo.2017.28.e32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Noguchi T, Zaitsu M, Oki I, et al. Recent increasing incidence of early‐stage cervical cancers of the squamous cell carcinoma subtype among young women. Int J Environ Res Public Health. 2020;17(20):7401. doi: 10.3390/ijerph17207401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Utada M, Chernyavskiy P, Lee WJ, et al. Increasing risk of uterine cervical cancer among young Japanese women: comparison of incidence trends in Japan, South Korea and Japanese‐Americans between 1985 and 2012. Int J Cancer. 2019;144(9):2144‐2152. doi: 10.1002/ijc.32014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. National Cancer Center Cancer Information Service . Cancer type statistics: cervix. Accessed March 2022. https://ganjoho.jp/reg_stat/statistics/stat/cancer/17_cervix_uteri.html
- 7. Kitagawa R, Katsumata N, Shibata T, et al. Paclitaxel plus carboplatin versus paclitaxel plus cisplatin in metastatic or recurrent cervical cancer: the open‐label randomized phase III trial JCOG0505. J Clin Oncol. 2015;33(19):2129‐2135. doi: 10.1200/JCO.2014.58.4391 [DOI] [PubMed] [Google Scholar]
- 8. Tewari KS, Sill MW, Long HJ 3rd, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370(8):734‐743. doi: 10.1056/NEJMoa1309748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ebina Y, Mikami M, Nagase S, et al. Japan Society of Gynecologic Oncology guidelines 2017 for the treatment of uterine cervical cancer. Int J Clin Oncol. 2019;24(1):1‐19. doi: 10.1007/s10147-018-1351-y [DOI] [PubMed] [Google Scholar]
- 10. US Food and Drug Administration . Keytruda [package insert]. Merck Sharp Dohme. Accessed May 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125514Orig1s054lbl.pdf [Google Scholar]
- 11. Colombo N, Dubot C, Lorusso D, et al. Pembrolizumab for persistent, recurrent, or metastatic cervical cancer. N Engl J Med. 2021;385(20):1856‐1867. doi: 10.1056/NEJMoa2112435 [DOI] [PubMed] [Google Scholar]
- 12. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) . Cervical Cancer. Accessed May 2022. https://www.nccn.org/patients/guidelines/content/PDF/cervical‐patient‐guideline.pdf.
- 13. Safety and efficacy of tisotumab vedotin monotherapy & in combination with other cancer agents in subjects with cervical cancer. Accessed March 2022. https://clinicaltrials.gov/ct2/show/NCT03786081
- 14. Efficacy and safety study of tisotumab vedotin for patients with solid tumors (innovaTV 207). Accessed March 2022. https://clinicaltrials.gov/ct2/show/NCT03485209
- 15. Breij EC, de Goeij BE, Verploegen S, et al. An antibody‐drug conjugate that targets tissue factor exhibits potent therapeutic activity against a broad range of solid tumors. Cancer Res. 2013;74(4):1214‐1226. doi: 10.1158/0008-5472.CAN-13-2440 [DOI] [PubMed] [Google Scholar]
- 16. de Goeij BE, Satijn D, Freitag CM, et al. High turnover of tissue factor enables efficient intracellular delivery of antibody‐drug conjugates. Mol Cancer Ther. 2015;14(5):1130‐1140. doi: 10.1158/1535-7163.MCT-14-0798 [DOI] [PubMed] [Google Scholar]
- 17. Versteeg HH, Spek CA, Peppelenbosch MP, Richel DJ. Tissue factor and cancer metastasis: the role of intracellular and extracellular signaling pathways. Mol Med. 2004;10(1–6):6‐11. doi: 10.2119/2003-00047.versteeg [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao X, Cheng C, Gou J, et al. Expression of tissue factor in human cervical carcinoma tissue. Exp Ther Med. 2018;16(5):4075‐4081. doi: 10.3892/etm.2018.6723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cocco E, Varughese J, Buza N, et al. Expression of tissue factor in adenocarcinoma and squamous cell carcinoma of the uterine cervix: implications for immunotherapy with hI‐con1, a factor VII‐IgGFc chimeric protein targeting tissue factor. BMC Cancer. 2011;11:263. doi: 10.1186/1471-2407-11-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hong DS, Concin N, Vergote I, et al. Tisotumab vedotin in previously treated recurrent or metastatic cervical cancer. Clin Cancer Res. 2020;26(6):1220‐1228. doi: 10.1158/1078-0432.CCR-19-2962 [DOI] [PubMed] [Google Scholar]
- 21. Coleman RL, Lorusso D, Gennigens C, et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG‐3023/ENGOT‐cx6): a multicentre, open‐label, single‐arm, phase 2 study. Lancet Oncol. 2021;22(5):609‐619. doi: 10.1016/S1470-2045(21)00056-5 [DOI] [PubMed] [Google Scholar]
- 22. US Food and Drug Administration Tivdak™ (tisotumab verdotin‐tftv) [package insert]. Seagen Inc. Updated September 2021. Accessed October 6, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761208Orig1s000lbledt.pdf [Google Scholar]
- 23. Pharmaceuticals and Medical Devices Agency (PMDA) Basic principles on Global Clinical Trials. Accessed July 12, 2021. https://www.pmda.go.jp/files/000153265.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1