

Abstract
Identification of genetic alterations through next‐generation sequencing (NGS) can guide treatment decision‐making by providing information on diagnosis, therapy selection, and prognostic stratification in patients with hematological malignancies. Although the utility of NGS‐based genomic profiling assays was investigated in hematological malignancies, no assays sufficiently cover driver mutations, including recently discovered ones, as well as fusions and/or pathogenic germline variants. To address these issues, here we have devised an integrated DNA/RNA profiling assay to detect various types of somatic alterations and germline variants at once. Particularly, our assay can successfully identify copy number alterations and structural variations, including immunoglobulin heavy chain translocations, IKZF1 intragenic deletions, and rare fusions. Using this assay, we conducted a prospective study to investigate the feasibility and clinical usefulness of comprehensive genomic profiling for 452 recurrently altered genes in hematological malignancies. In total, 176 patients (with 188 specimens) were analyzed, in which at least one alteration was detected in 171 (97%) patients, with a median number of total alterations of 7 (0–55). Among them, 145 (82%), 86 (49%), and 102 (58%) patients harbored at least one clinically relevant alteration for diagnosis, treatment, and prognosis, respectively. The proportion of patients with clinically relevant alterations was the highest in acute myeloid leukemia, whereas this assay was less informative in T/natural killer‐cell lymphoma. These results suggest the clinical utility of NGS‐based genomic profiling, particularly for their diagnosis and prognostic prediction, thereby highlighting the promise of precision medicine in hematological malignancies.
Keywords: comprehensive genomic profiling, hematological malignancy, next‐generation sequencing, precision medicine, somatic alteration
We have developed an integrated DNA/RNA profiling assay for detecting germline variants and somatic alterations, including structural variations and fusions, recurrently found in hematological malignancies. We performed a prospective hospital‐based cohort study to demonstrate that our assay is feasible and useful for identifying clinically relevant alterations, particularly for diagnosis and prognostic prediction. These results suggest the clinical utility of our assay, thereby highlighting the promise of precision medicine in hematological malignancies.
1. INTRODUCTION
With the advent of next‐generation sequencing (NGS), large‐scale studies have delineated the entire landscape of genetic alterations in various types of human cancers. These efforts have broadened our knowledge of driver alterations and have revealed the molecular mechanisms of oncogenesis, bringing the promise of cancer precision medicine. 1 , 2 These discoveries have clinical implications in the diagnosis of specific subtypes defined by recurrent alterations and the prognostic prediction of patient outcome. 3 More importantly, some alterations can guide the use of molecularly targeted therapy and predict the resistance to specific inhibitors, highlighting the relevance of genomic testing in clinical oncology. 4
Hematological malignancies are diagnosed and classified using morphology, immunophenotype, and genetic alterations. A diverse array of genetic alterations underlies their pathogenesis, as exemplified by more than 250 recurrent alterations listed in the latest WHO classification. 5 In addition, reflecting their diversity and the presence of rare disease types, there have been many genetic studies in hematological malignancies, which have identified novel genetic alterations even recently. 6 , 7 , 8 Moreover, a non‐negligible proportion of patients with myeloid neoplasms, such as acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), have pathogenic germline variants in leukemia‐predisposing genes, such as RUNX1 and DDX41 6 , 9 , illuminating the potential necessity of genomic testing for germline susceptibility in hematological malignancies.
A variety of somatic alterations, including single nucleotide variants (SNVs) and insertions/deletions (indels), copy number alterations (CNAs), and structural variations (SVs), are present in hematological malignancies. 5 Among them, recurrent SVs (also called as rearrangements), such as those involving immunoglobulin heavy chain (IGH), are characteristic of specific subtypes. These include chromosomal translocations, inversions, duplications, and deletions, resulting in aberrant expression of oncogenes (activating SV), or generation of fusion transcripts (fusion‐generating SV). Several conventional methods, such as G‐banding karyotyping, FISH, and RT‐PCR, have been utilized to detect these SVs. However, these methods are designed to identify individual SVs, and the spectrum of SVs that can be evaluated by them is still limited.
Recent studies investigated the feasibility and utility of NGS‐based genomic profiling in hematological malignancies, which enables simultaneous examination of many somatic alterations at once. 10 , 11 , 12 However, its potential may be underestimated because these studies did not sufficiently cover driver genes listed in the WHO classification as well as recently discovered ones. In addition, germline controls and/or RNA panel were lacking, and several disease types, including T/natural killer‐cell non‐Hodgkin lymphoma (T/NK‐NHL), were not well investigated in these studies. Therefore, to address these issues, here we devised an integrated DNA/RNA profiling assay that can simultaneously detect various types of somatic alterations and germline variants in 452 genes recurrently altered in various hematological malignancies. Using this assay, we have performed a prospective hospital‐based cohort study to investigate the potential of NGS‐based comprehensive genomic profiling in 188 specimens of hematological malignancies.
2. MATERIALS AND METHODS
2.1. Workflow of clinical sequencing
We performed a prospective cohort study to evaluate the feasibility and clinical utility of integrated DNA/RNA profiling assay using tumor and germline specimens. The workflow is divided into four parts: (i) patient enrollment/specimen collection, (ii) library preparation, (iii) sequencing and bioinformatic analysis, and (iv) assessment of clinical relevance (Figure 1A). DNA and RNA were independently processed for library preparation and hybrid selection.
FIGURE 1.
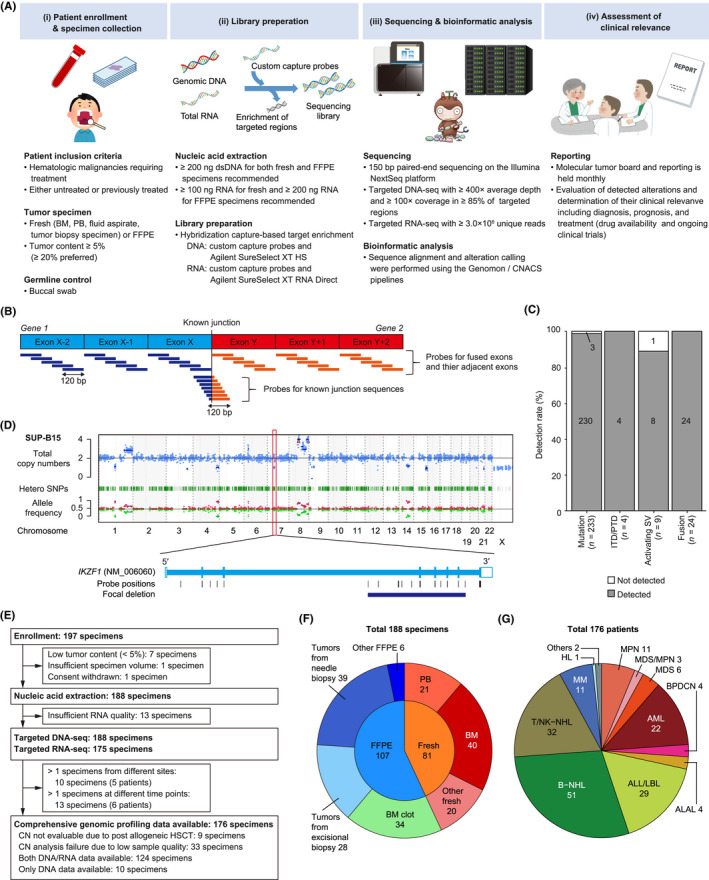
Overview of the study using comprehensive genomic profiling of patients with hematological malignancies. (A) Schema of the prospective hospital‐based cohort study. (B) Strategy for capturing fusions by targeted RNA sequencing (RNA‐seq). (C) Detection rate of known alterations using cell lines according to alteration type. (D) Representative result of copy number (CN) analysis (top) and zoom‐in view for an IKZF1 deletion (bottom) in SUP‐B15 cell line. (E) Flow diagram showing enrollment, nucleic acid extraction, and sequencing. (F) Distribution of tissues and specimen types in all 188 specimens. (G) Distribution of disease types in 176 patients. ALAL, acute leukemias of ambiguous lineage; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; BM, bone marrow; B‐NHL, B‐cell non‐Hodgkin lymphoma; BPDCN, blastic plasmacytoid dendritic cell neoplasm; DNA‐seq, DNA sequencing; FFPE, formalin‐fixed, paraffin‐embedded; HL, Hodgkin lymphoma; HSCT, hematopoietic stem cell transplantation; ITD, internal tandem duplication; LBL, lymphoblastic lymphoma; MDS, myelodysplastic syndrome; MM, multiple myeloma; MPN, myeloproliferative neoplasm; PB, peripheral blood; PTD, partial tandem duplication; SNP, single nucleotide polymorphism; SV, structural variation; T/NK‐NHL, T/natural killer‐cell non‐Hodgkin lymphoma
2.2. Patients and specimens
Patients aged 1 year or older who were diagnosed with untreated or relapsed/refractory hematological malignancies according to the WHO classification 5 and would intend to receive chemotherapy and/or hematopoietic stem cell transplantation (HSCT) were enrolled. The availability of tumor specimens with tumor content of 5% or more was required (≥20% was recommended). Fresh or formalin‐fixed paraffin‐embedded (FFPE) specimens of bone marrow (BM), peripheral blood (PB), fluid aspirate, lymph node, or tumor tissues were collected. Buccal swab was obtained as a matched germline control. The study was approved by the National Cancer Center Institutional Review Board, and all patients and/or their legal guardians (when minors were enrolled) provided written informed consent for this study.
2.3. Design of integrated DNA/RNA profiling assay
We designed a hybridization capture‐based DNA/RNA profiling assay to detect SNVs, indels, SVs, CNAs, and fusions (Tables S1–S3). These include almost all driver alterations listed in the Japanese Society of Hematology (JSH) Guideline for Genomic Testing in Hematological Malignancies (2021 release) (JSH Genome Guideline; http://www.jshem.or.jp/genomgl/) (Tables S4–S6), which covers almost all alterations listed in the WHO classification 5 and recently discovered ones in hematological malignancies. The DNA panel covered (i) entire coding sequences (CDSs) of 321 frequently mutated genes, (ii) eight non‐coding regions affected by recurrent activating SVs, including IGH translocations, GATA2/MECOM, and CD274/PDCD1LG2 truncations, (iii) immunoglobulin (IG) and T‐cell receptor (TCR) genes, (iv) HTLV‐1 provirus sequence, (v) the miR‐15a/16–1 regions, and (vi) 1296 single nucleotide polymorphism (SNP) regions to evaluate genome‐wide copy number and detect intragenic deletions and duplications, including those involving IKZF1 and KMT2A (MLL) (Table S1). The RNA panel was designed to detect (i) CDSs of 44 genes targeted by recurrent fusions, (ii) CDSs and untranslated regions of 32 genes targeted by recurrent activating SVs, and (iii) IG/TCR genes (Table S2). To increase the sensitivity to detect fusions, we have extended our previous method 13 to design probes for not only fused exons and their adjacent exons but also junction sequences of 134 well‐known fusions (Figure 1B; Table S3). The performance of our assay was assessed for (i) 233 mutations in five cell lines detected at an allele frequency of 0.05 or more by amplicon sequencing with QIAseq Human Myeloid Neoplasms Panel (Qiagen), and (ii) 35 SVs/fusions and IKZF1 intragenic deletion in 31 cell lines described in previous reports. In addition, using clinical specimens, fusions were evaluated using 13 different RT‐PCR tests and five different FISH tests, and rearrangements and deletion of 17p (del17p) using eight different FISH tests.
2.4. Library preparation and sequencing
Fresh tumor and buccal germline DNA were extracted using the QIAamp DNA Blood Mini kit (Qiagen) or the Maxwell RSC Blood DNA Kit (Promega). FFPE tumor DNA was extracted using the GeneRead DNA FFPE kit (Qiagen) and its quality was evaluated with Agilent 4200 TapeStation (Agilent Technologies). Fresh and FFPE tumor RNA were extracted using the QIAamp RNA Blood Mini Kit (Qiagen) or the Maxwell RSC simplyRNA Blood Kit (Promega) and the RNeasy FFPE Kit (Qiagen) or the Maxwell RSC RNA FFPE Kit (Promega), respectively. Library preparation for targeted DNA sequencing (DNA‐seq) was performed using a custom SureSelect library (Agilent Technologies), as previously described. 14 , 15 Library preparation for targeted RNA sequencing (RNA‐seq) was also performed using a custom SureSelect library with SureSelect XT RNA direct according to the manufacturer’s instructions. These libraries were sequenced using the NextSeq 500 (Illumina) to generate 150 bp paired‐end reads to ≥400× average depth for DNA‐seq and ≥3 million uniquely mapped reads for RNA‐seq either in‐house or at RIKEN GENESIS. Quality control results were compared using a two‐sided Welch's t‐test.
2.5. Mapping and mutation detection
For targeted DNA‐seq data, sequence alignment and mutation calling were performed using the Genomon pipeline version 2.6.2 (https://github.com/Genomon‐Project/), as previously described. 14 , 15 Sequencing reads were mapped to the custom reference genome consisting of 1000 genomes Reference Genome Sequence hs37d5 with the HTLV‐1 genome (AB513134). Putative somatic mutations with (i) EBCall 16 p < 0.01, (ii) four or more variant reads, and (iii) allele frequency ≥0.05 were adopted and filtered by excluding (i) synonymous SNVs, (ii) variants only present in unidirectional reads, and (iii) variants occurring in repetitive genomic regions. These candidate mutations were further filtered, unless they were listed 10 or more times in the COSMIC database version 70 or ASXL1 (NM_015338.5) c.1927dupG, by removing known germline variants (i) observed in NCBI dbSNP build 131, or (ii) observed at a frequency ≥0.0001 in any of the following datasets: the 1000 Genomes Project (October 2014 release), NCBI dbSNP build 131, National Heart, Lung, and Blood Institute Exome Sequencing Project 6500, the Human Genome Variation Database (version 2.00), or the Exome Aggregation Consortium r0.3.1. Finally, all detected mutations were manually checked by Integrative Genomics Viewer (IGV). 17
2.6. SV and CNA detection
SVs and CNAs were detected using the Genomon pipeline and the CNACS algorithm, respectively, as previously described. 14 , 15 Putative SVs were manually curated and further filtered by removing those (i) with fewer than five supporting reads in tumor; (ii) with allele frequency in tumor <0.02; or (iii) present in in‐house pooled normal specimens. SV breakpoints were visually inspected using IGV. Candidate focal CNAs (shorter than half a chromosome arm) were assessed in genomic regions where sequencing coverage was sufficient in unmatched control samples, and then manually reviewed. Among genes covered by the DNA panel, 374 and 193 genes (170 for deletions and 23 for amplifications) listed in the JSH Genome Guideline were analyzed for SVs and focal CNAs (Table S6).
2.7. Fusion detection
For targeted RNA‐seq data, sequence alignment and fusion calling were performed using the Genomon RNA pipeline version 2.6.2, with a slight modification of those used for poly‐A RNA‐seq. 14 , 15 Candidate fusions with 10 or more supporting reads in tumor were filtered by excluding (i) endogenous IG/TCR recombination; and (ii) those detected in in‐house pooled normal specimens. Finally, mapping errors were removed by visual inspection with IGV. As IGH/DUX4 and STIL‐TAL1 fusions are difficult to detect, these fusions were individually reviewed.
2.8. Definition and interpretation of clinically relevant alterations
Clinical evidence levels (A–D) were assigned to each genetic alteration in terms of diagnosis, treatment, and prognostic prediction for each disease subtype at the time of NGS‐based genomic profiling according to the JSH Genome Guideline (Table S6), and driver alteration was defined as those with clinical evidence in each disease subtype. Germline variants involving 59 genes responsible for (i) hematological diseases with a germline predisposition proposed in the JSH Genome Guideline, and/or (ii) hereditary cancers for which the American College of Medical Genetics and Genomics (ACMG) 18 recommends reporting of incidental and secondary findings were evaluated (Table S1). The molecular tumor board comprised of a multidisciplinary team interpreted the sequencing results, including evaluation of somatic alterations and germline variants, and determination of their clinical relevance based on the evidence level assigned for individual alterations.
3. RESULTS
3.1. Design and validation of integrated DNA/RNA profiling assay
We designed NGS‐based comprehensive genomic profiling using tumor and germline specimens. Integrated DNA/RNA analysis with elaborated panel design can provide increased breadth and improved sensitivity to detect not only fusion‐generating SVs but also activating SVs, which causes aberrant expression of oncogenes (Figure 1B, Tables S1–S3). As the excellent performance of targeted DNA‐seq for detecting SNVs and CNAs has been established (Table S7), 14 , 15 we then evaluated the analytical accuracy for fusion‐generating and activating SVs by integrated DNA/RNA analysis (Figure 1C, Table S8). We detected all 24 known fusions, three FLT3‐internal tandem duplications (ITDs), and 1 KMT2A‐partial tandem duplication (PTD), with no false positive call observed. We also detected eight of nine activating SVs, consisting of IGH translocations (including IGH/MYC, IGH/BCL2, IGH/CCND1, IGH/CRLF2, and IGH/DUX4) and GATA2/MECOM rearrangement. In addition, we confirmed successful detection of an intragenic deletion of IKZF1 19 (Figure 1D). These observations suggest acceptable sensitivity for various kinds of SVs and intragenic deletions.
3.2. Quality evaluation of NGS‐based genomic profiling
Between 2019 and 2021, 197 specimens from 185 patients were enrolled in our prospective cohort study (Figure 1A,E). Nine specimens were excluded, and the remaining 188 specimens were submitted to further analysis (Table S9). The specimens consisted of 81 fresh and 107 FFPE specimens, including tumors from excisional biopsy (n = 28) and needle biopsy (n = 39) (Figure 1F). The median average depth and 100× coverage of DNA‐seq were 1022× (range, 143–1723×) and 0.98 (0.58–0.99) in tumors, and 942× (129–1371×) and 0.97 (0.56–0.99) in normal tissues, respectively. Fresh specimens tended to show a higher depth than FFPE specimens, which included all six (4%) specimens with low coverage (<0.85), whereas no difference was observed across tissues and biopsy procedures (Figure S1A,B). Except for nine specimens collected after allogeneic HSCT, genome‐wide copy number was evaluable in 142 (79%) of 179 specimens, which contained all fresh specimens but only 64% of FFPE specimens (Figures 1E and S1C). As the amount of input RNA was too small in 13 (7%) specimens, the remaining 175 specimens were analyzed by targeted RNA‐seq (Figures 1E and S1D). The read number was higher in fresh than FFPE specimens, with a median of 7.1 (0.9–13.3) × 106 (Figure S1E). DNA integrity number was lower in FFPE specimens stored for 1 year or longer than those stored for less than 1 year (p < 0.01) (Figure S1F). The median turnaround time (TAT), defined as time from specimen receipt to molecular tumor board, was 50.5 (21–167) days.
3.3. Somatic alterations detected by NGS‐based genomic profiling
Among the 188 specimens, five patients submitted two specimens from different sites (10 specimens), and six patients submitted two or more specimens at different time points (13 specimens). Therefore, a total of 176 patients with various backgrounds were analyzed (Figure 1G, Tables 1 and S9). The integrated DNA/RNA profiling detected 1746 somatic alterations in 296 genes and at least one alteration in 171 patients (97%) (Figure 2A, Tables S10–S13). The median number of alterations per specimen was 7 (0–55), with 10 or more alterations detected in 38% of patients. The most frequently altered gene was TP53 (19%), followed by CREBBP (15%), CDKN2A/B (13%), KMT2D (13%), and NRAS (11%) (Figures 2B and S2).
TABLE 1.
Characteristics of 176 patients with hematological malignancies
| Characteristic | Number (%) |
|---|---|
| Number of patients | 176 |
| Disease type | |
| AML | 22 (13) |
| ALL/LBL | 29 (16) |
| ALAL | 4 (2) |
| BPDCN | 4 (2) |
| MPN | 11 (6) |
| MDS/MPN | 3 (2) |
| MDS | 6 (3) |
| B‐NHL | 51 (29) |
| T/NK‐NHL | 32 (18) |
| HL | 1 (1) |
| MM | 11 (6) |
| Others | 2 (1) |
| Sex | |
| Male | 111 (63) |
| Female | 65 (37) |
| Age, years | |
| 0–14 | 14 (8) |
| 15–39 | 39 (22) |
| 40–64 | 61 (35) |
| ≥65 | 62 (35) |
| Disease status | |
| Primary | 104 (59) |
| Relapsed/refractory | 72 (41) |
Abbreviations: ALAL, acute leukemias of ambiguous lineage; ALL/LBL, acute lymphoblastic leukemia/lymphoblastic lymphoma; AML, acute myeloid leukemia; B‐NHL, B‐cell non‐Hodgkin lymphoma; BPDCN, blastic plasmacytoid dendritic cell neoplasm; HL, Hodgkin lymphoma; MDS, myelodysplastic syndrome; MM, multiple myeloma; MPN, myeloproliferative neoplasm; T/NK‐NHL, T/natural killer‐cell non‐Hodgkin lymphoma.
FIGURE 2.
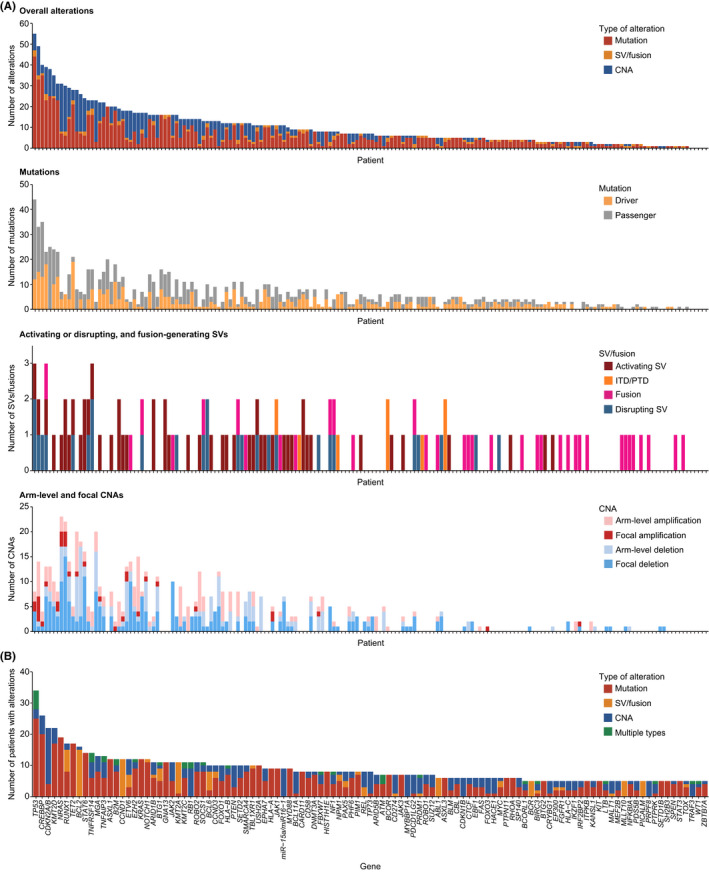
Frequencies and types of genetic alterations detected in clinical specimens from patients with hematological malignancies. (A) Number of somatic alterations detected in each patient (n = 176). (B) Number of patients with somatic alterations for each driver gene according to alteration type. Genes altered in ≥5 patients are shown. In case of >1 alteration type detected, they are considered multiple for tumor suppressor or functionally unknown genes, whereas only one major alteration type is counted for oncogenes. CNA, copy number alteration; ITD, internal tandem duplication; PTD, partial tandem duplication; SV, structural variation
Targeted DNA‐seq detected 1027 mutations (SNVs and indels), including 451 driver and 576 passenger mutations, in 238 genes, and at least one mutation was detected in 159 (90%) patients (Figures 2A; Table S10). The median number of mutations per specimen was four (0–44), including one (0–19) of driver mutations. The most frequently mutated gene was TP53 (18%), followed by CREBBP (11%), NRAS (11%), TET2 (10%), and KMT2D (10%) (Figures 2B and S2).
Combined targeted DNA‐seq and RNA‐seq identified 121 SVs, including 35 fusion‐generating, 51 activating, and 27 disrupting SVs as well as five KMT2A‐PTDs and three FLT3‐ITDs, and at least one SV was detected in 95 (54%) patients (Figure 2A, Table S11). Fusions included five PICALM‐MLLT10, five BCR‐ABL1, three RUNX1‐RUNX1T1, and five KMT2A‐related fusions (Figures 2B and S2). Among them, 21 fusions were evaluated by RT‐PCR or FISH, which were all positive. The negative and positive predictive values of our assay were all 100% for FLT3‐ITDs and fusions. Activating SVs mainly consisted of IGH translocations, such as 15 IGH/BCL2, 12 IGH/CCND1, and two IGH/BCL6 translocations, but also included two TCR translocations (Figures 2B and S2). The IGH translocations mainly occurred adjacent to DJ segments in B‐cell NHL (B‐NHL) and in the switch regions of constant genes in multiple myeloma (MM) (Figure 3A). All SV partners of BCL2 and CCND1 were IGH (Figure 3A, Table S11). Although IGH was the most common partner of MYC (50%) and BCL6 SVs (33%), multiple additional partners were identified (Figure 3B, Table S11). Other activating SVs involved REL, NOTCH1, and CD274. Among them, 23 SVs, including IGH translocations, were assessed by FISH, in which 21 were positive, but two (one IGH/MYC and one non‐IGH/MYC) were negative. When FISH was negative (n = 30), no corresponding SVs were detected by NGS, producing negative and positive predictive values of 94% and 100% for activating SVs, respectively.
FIGURE 3.
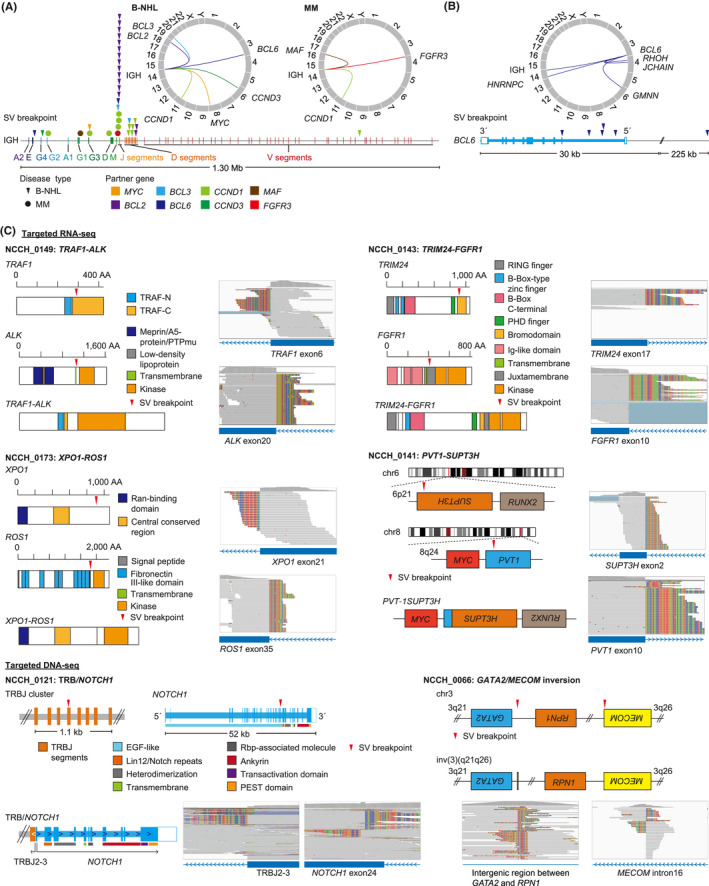
Detection of activating and fusion‐generating structural variations (SVs) in clinical specimens from patients with hematological malignancies. (A) Distribution of breakpoints and partners of immunoglobulin heavy chain (IGH) translocations in B‐cell non‐Hodgkin lymphoma (B‐NHL; triangle) and multiple myeloma (MM; circle). (B) Distribution of breakpoints and partners of BCL6 SVs. (C) Rare fusions and SVs detected in this study. Red triangles represent SV breakpoints. AA, amino acid; DNA‐seq, DNA sequencing; EGF, epidermal growth factor; Ig, immunoglobulin; PEST, proline, glutamic acid, serine, and threonine; PTP, protein tyrosine phosphatase; Rbp, Recombination signal binding protein; RING, really interesting new gene; RNA‐seq, RNA sequencing; TRAF, tumor necrosis factor receptor‐associated factor
In 134 patients in which genome‐wide copy numbers were evaluable, 322 arm‐level CNAs (163 amplifications and 159 deletions), and 277 focal CNAs (34 amplifications and 242 deletions) were identified, with a median number of CNAs per patient of three (Figure 2A, Tables S12 and S13). We identified arm‐level or focal del17p involving TP53 in 21 patients. Among them, eight were assessed by FISH, of which six were positive. Taken together, these findings suggest that our integrated DNA/RNA profiling can detect a variety of somatic alterations with adequate sensitivity.
3.4. Extensive heterogeneity of somatic alterations in our cohort
The entire cohort showed extensive heterogeneity of somatic alterations, while reflecting the genetic landscape of each disease type reported by previous genetic studies 20 , 21 , 22 , 23 , 24 (Figure S2). As confirmed in cell lines, integrated DNA/RNA profiling successfully identified one GATA2/MECOM inversion and five KMT2A‐PTDs (Figure 3C, Table S11). In addition to well‐known disease‐defining SVs and fusions, our assay detected various kinds of SVs and fusions, such as PVT1‐SUPT3H fusion and DUSP22 rearrangement characteristic of blastic plasmacytoid dendritic cell neoplasm (BPDCN) and ALK‐negative anaplastic large cell lymphoma, respectively. Moreover, we identified rare IG/TCR‐involving SVs, including IGH/BCL3 and TRB/NOTCH1, as well as kinase fusions, such as TRAF1‐ALK, TRIM24‐FGFR1, and XPO1‐ROS1, which can be a promising therapeutic target (Figure 3C).
3.5. Differences in somatic alterations across disease types
Disease‐specific frequencies revealed quite different genetic profiles and associated clinical evidence across disease types (Figures 4 and 5, Table S14). These frequencies were comparable or slightly higher than those reported by the FoundationOne Heme panel, 10 demonstrating the promising performance of our assay (Tables S15–S17). KMT2A (32%), NPM1 (23%), DNMT3A (23%), RUNX1 (23%), and TET2 (23%) were frequently altered in AML (n = 22) (Figure 4A). Among them, 95%, 91%, and 95% of patients harbored at least one genetic alteration with clinical evidence for diagnosis, treatment, and prognosis, respectively (Figure 5). In addition to those already detected by other conventional methods, integrated DNA/RNA profiling identified many subtype‐defining alterations, including four NPM1, one biallelic CEBPA, and one RUNX1 mutation, one GATA2/MECOM inversion, and one DDX41 germline variant. According to the risk stratification by the European LeukemiaNet, 25 seven (32%) patients with normal cytogenetics (n = 6) or no cytogenetics data available (n = 1) changed prognostic risk category (four from intermediate to favorable, two from intermediate to adverse, and one from not available to intermediate risk), due to the presence of four NPM1, two ASXL1, and one biallelic CEBPA mutations. Potential targetable alterations included three FLT3‐ITDs and kinase domain mutations (FLT3 inhibitor, level A) and two IDH1/2 mutations (IDH1/2 inhibitor, level D). In addition, DNMT3A mutation was associated with anthracycline resistance (level B) in five patients. 26
FIGURE 4.
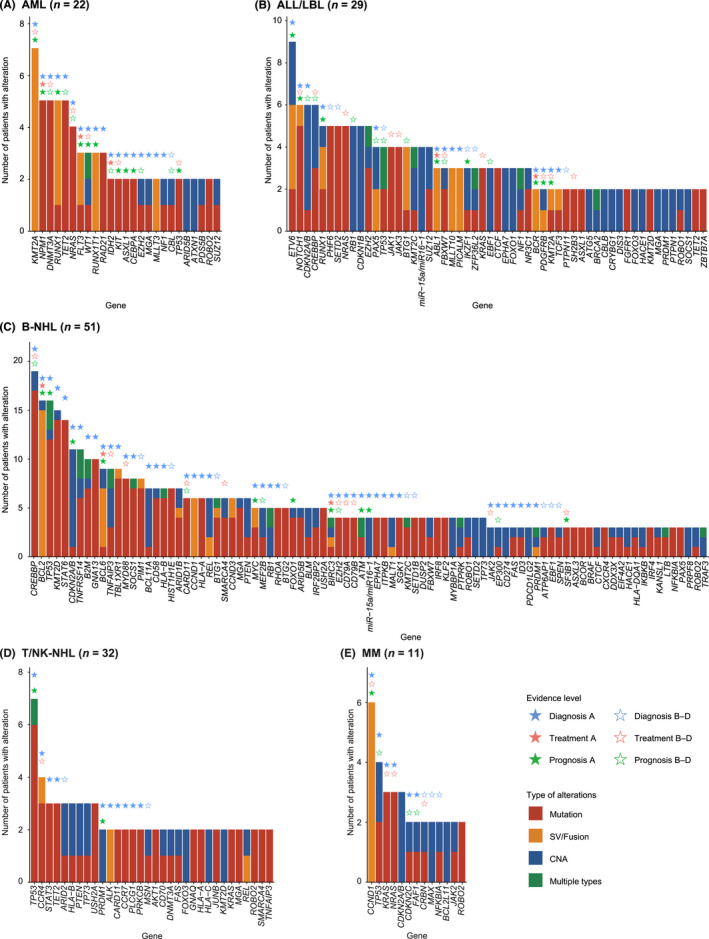
Spectrum of altered genes in clinical specimens from patients with hematological malignancies, by disease type. (A–E) Number of somatic alterations in patients diagnosed with (A) acute myeloid leukemia (AML), (B) acute lymphoblastic leukemia (ALL)/lymphoblastic lymphoma (LBL), (C) B‐cell non‐Hodgkin lymphoma (B‐NHL), (D) T/natural killer‐cell non‐Hodgkin lymphoma (T/NK‐NHL), and (E) multiple myeloma (MM). Stars represent the evidence level assigned to genes according to the Japanese Society of Hematology Genome Guideline in each disease type. In case of >1 alteration type detected, they are considered multiple for tumor suppressor or functionally unknown genes, whereas only one major alteration type is counted for oncogenes. CNA, copy number alteration; SV, structural variation
FIGURE 5.
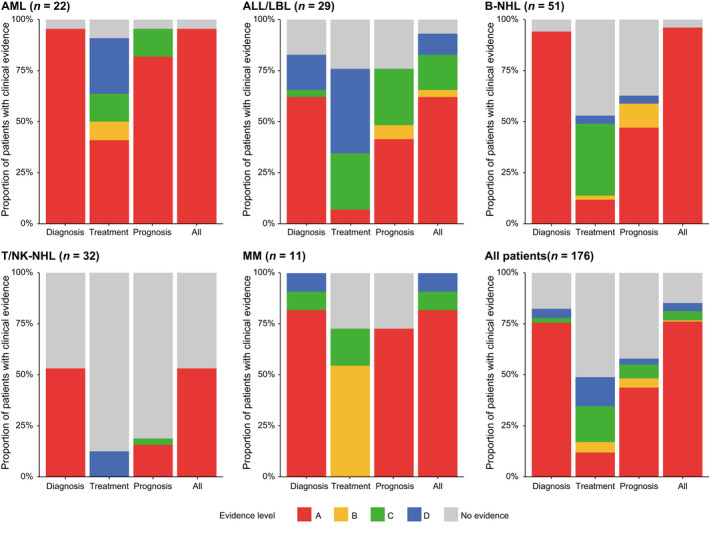
Clinical utility of comprehensive genomic profiling in hematological malignancies. Proportion of patients harboring at least one somatic alteration with indicated clinical evidence level according to the Japanese Society of Hematology Genome Guideline in acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL)/lymphoblastic lymphoma (LBL), B‐cell non‐Hodgkin lymphoma (B‐NHL), T/natural killer‐cell non‐Hodgkin lymphoma (T/NK‐NHL), and multiple myeloma (MM), and for all patients
ETV6 (31%), NOTCH1 (21%), CDKN2A/2B (21%), and CREBBP (21%) were frequently mutated in acute lymphoblastic leukemia/lymphoblastic lymphoma (ALL/LBL) (n = 29) (Figure 4B). Among them, 83%, 76%, and 76% of patients harbored at least one genetic alteration with clinical evidence for diagnosis, treatment, and prognosis, respectively (Figure 5). In addition to already detected ones, several subtype‐defining fusions were identified, including KMT2A‐MAML2, ZNF384‐EP300, ETV6‐ABL1, and ATF7IP‐PDGFRB (n = 1 for all). The patients harboring the latter two ABL‐class gene rearrangements were reclassified into Philadelphia chromosome‐like (Ph‐like) ALL. The diagnosis of one T‐ALL patient changed into myeloid/lymphoid neoplasms with eosinophilia due to the identification of ETV6‐LYN fusion (NCCH_0023), which was also the case in one mixed‐phenotype acute leukemia patient with TRIM24‐FGFR1 fusion (NCCH_0143). In addition to subtype‐defining alterations, two IKZF1 deletions and six NOTCH1 mutations were associated with worse prognosis in B‐ and T‐ALL/LBL, respectively. In addition to the aforementioned fusions, including one ABL1 fusion (ABL inhibitor, level C) and one PDGFRB fusion (PDGFRB inhibitor, level C), potential targetable alterations included those activating kinase and cytokine receptor signaling, such as one SH2B3 (JAK inhibitor, level C) and one FLT3 (FLT3 inhibitor, level C) mutations.
Frequent alterations in B‐NHL (n = 51) included CREBBP (37%), BCL2 (31%), TP53 (31%), KMT2D (29%), and STAT6 (27%) (Figure 4C). Among them, 94%, 53%, and 63% of patients harbored at least one genetic alteration with clinical evidence for diagnosis, treatment, and prognosis, respectively (Figure 5). Four patients changed diagnoses based on genome profiling: one from MALT lymphoma to follicular lymphoma (FL) due to IGH/BCL2 translocation (NCCH_0039), one from low‐grade B‐NHL to Waldenström macroglobulinemia (WM) due to MYD88 mutation (NCCH_0002), one from primary effusion lymphoma‐like lymphoma to recurrence of primary central nervous system lymphoma due to MYD88, CD79B, and PIM1 mutations (NCCH_0090), and one from high‐grade B‐cell lymphoma, not otherwise specified (HGBL, NOS) to HGBL with MYC and BCL2 and/or BCL6 rearrangement (NCCH_0180). Different alterations were associated with prognosis in different B‐NHL subtypes, according to the subtype‐specific prognostication systems, such as LymphGen classification in diffuse large B‐cell lymphoma (DLBCL), 27 the m7‐FLIPI clinicogenetic model in FL, 28 and the iwCLL guidelines in chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). 29 Due to the paucity of targeted therapy in B‐NHL, only four patients had druggable alterations, including three EZH2 mutations (EZH2 inhibitor, level C) in FL and one MYD88 mutation (BTK inhibitor, level C) in WM. In addition, four MYD88 and three CD79B mutations were reported to predict sensitivity to BTK inhibitor in DLBCL (level C).
Frequent alterations in T/NK‐NHL (n = 32) included TP53 (22%), CCR4 (13%), STAT3 (9%), TET2 (9%), and ARID2 (9%) (Figure 4D). Among them, 53%, 13%, and 19% of patients harbored at least one genetic alteration with clinical evidence for diagnosis, treatment, and prognosis, respectively (Figure 5), demonstrating a smaller proportion of patients harboring clinically relevant alterations in T/NK‐NHL. In two patients suspected of peripheral T‐cell lymphoma with follicular helper T‐cell phenotype, no somatic alterations were detected, leading to a change of diagnosis to reactive lymphoid hyperplasia (NCCH_136 and NCCH_139). These observations suggest the relevance of NGS‐based genomic profiling to determine clonality in mature T‐cell neoplasms. The only therapeutically relevant alteration was CCR4 mutations (CCR4 inhibitor, level D) in four patients. 30
CCND1 (55%), TP53 (36%), KRAS (27%), NRAS (27%), and CDKN2A/B (27%) were frequently altered in MM (n = 11) (Figure 4E). Among them, 100%, 73%, and 73% of patients harbored at least one genetic alteration with clinical evidence for diagnosis, treatment, and prognosis, respectively (Figure 5). According to International Myeloma Working Group molecular classification, 31 eight patients were classified into four subtypes, including hyperdiploidy (n = 2), IGH/CCND1 (n = 4), IGH/FGFR3 (n = 1), and IGH/MAF (n = 1). According to the Stratification for Myeloma and Risk‐Adapted Therapy (mSMART 3.0), 32 seven patients had high‐risk genetic abnormalities (such as del17q and 1q gain), including one double‐hit (IGH/MAF and TP53 mutation) and one triple‐hit (IGH/FGFR3, del17p, and 1q gain) patients. Regarding treatment, six patients had IGH/CCND1 (BCL2 inhibitor, level B) and two patients had CRBN mutations, known as resistant mutation to immunomodulatory drugs (level C).
3.6. Clinical utility in diagnosis, treatment, and prognostic prediction
In total, 145 (82%), 86 (49%), and 102 (58%) patients harbored at least one clinically relevant somatic alteration for diagnosis, treatment, and prognosis, of which 76%, 12%, and 44% were supported by level A evidence, respectively (Figure 5). Particularly, most (65%) of the relevant alterations for treatment were of potential clinical significance (evidence level C or D). Although clinically relevant alterations mainly consisted of mutations, SVs/fusions accounted for a substantial proportion of the relevant alterations for treatment (Figure S3). However, only one received targeted therapy based on the recommendation by the molecular tumor board, who had an ETV6‐ABL1 fusion and was treated with dasatinib (ABL inhibitor) as off‐label use (NCCH_0094). These differences were consistent across disease types, suggesting that comprehensive genomic profiling is informative for diagnosis followed by for prognostic prediction, whereas its utility is inadequate for therapy selection (Figure 5). Regardless of clinical purpose, patients with AML had the highest proportion of clinically relevant alterations (particularly with level A evidence), followed by those with ALL/LBL, B‐NHL, and MM, whereas their proportion was limited in T/NK‐NHL, suggesting the different degrees of utility of comprehensive genomic profiling across disease types.
3.7. Detection of cancer‐related germline variants
Germline variants causing hereditary cancers were identified in six (3%) patients, all of which were of level A evidence (Table S18). Among them, one AML patient harbored a DDX41 germline variant and one AML, one MDS, and three ALL/LBL patients harbored deleterious BRCA1 or BRCA2 germline variants associated with hereditary breast and ovarian cancers. All six patients were referred for genetic counseling, and three of them requested confirmatory testing and were validated to have the variants.
3.8. Multi‐site and multi‐timepoint analyses
Multi‐site and multi‐timepoint analyses were performed for five and six patients, respectively (Table S9). In one patient with a history of essential thrombocythemia who developed myeloid sarcoma, an MPL mutation was found in both BM and sarcoma specimens, while several alterations, including RUNX1 mutation and 7q deletion, were detected only in sarcoma specimens, suggesting that these alterations contribute to the clonal evolution (Figure S4A). In one CLL patient analyzed before and after BTK inhibitor therapy, a PLCG2 mutation, known to confer BTK inhibitor resistance (level D), was detected only after progression (Figure S4B). These results suggest that serial analysis is helpful to uncover somatic alterations underlying therapeutic resistance.
4. DISCUSSION
Through a prospective hospital‐based cohort study, we demonstrate the feasibility and clinical utility of integrated DNA/RNA profiling for hematological malignancies in the clinical setting. Our study substantiates that this assay enables comprehensive genomic profiling of clinical specimens as a single test, including the assessment of a variety of SVs and CNAs, such as IG translocations, IKZF1 intragenic deletion, and rare fusions. Particularly, comprehensive genomic profiling is clinically relevant in diagnosis and prognostic prediction, although the usefulness in guiding the use of molecularly targeted therapy is still limited. A larger proportion of patients with myeloid neoplasms can benefit from this assay, whereas it is less informative for T/NK‐NHL patients.
Consistent with previous studies, 10 , 11 , 12 targeted DNA/RNA‐seq is feasible not only for fresh but also for FFPE specimens regardless of the collection methods, except for those collected more than one year before the analysis. However, CNA assessment is still difficult for more than one‐third of FFPE specimens, suggesting that fresh specimens are recommended for certain disease types in which CNAs are clinically relevant. Although our assay successfully identified various SVs and fusions, the detection rate of MYC SVs was insufficient, probably due to the presence of non‐IG partner and widespread distribution of MYC breakpoints. 33
Comprehensive genomic profiling is most informative for diagnosis. First, our assay can identify various subtype‐defining alterations, such as fusions in AML and ALL/LBL, and IGH translocations in aggressive B‐NHL and MM. In addition, several alterations, including PVT1‐SUPT3H in BPDCN, are rare but characteristic of a specific disease type. Second, our assay can evaluate the clonality, which can help distinguishing non‐neoplastic from neoplastic diseases, particularly in patients suspected of MDS, myeloproliferative neoplasm (MPN), and peripheral T‐cell lymphoma. Third, the analysis of buccal swab specimens allows precise identification of germline variants responsible for both leukemia predisposition and other hereditary diseases, including DDX41 mutations. Such germline variants may influence transplant candidacy and donor selection, although appropriate strategies for their management and genetic counseling have not been established.
The identification of various kinds of kinase fusions provides important therapeutic information, particularly in Ph‐like ALL 34 and other kinase fusion‐mediated malignancies, as they are potential targets of molecular‐based therapies. In addition, our assay can detect resistant alterations to specific inhibitors, such as PLCG2 mutation in CLL and CRBN mutation in MM. 35 , 36 Serial sampling may provide additional therapeutically relevant information, given the mutational differences between diagnosis and relapse. 37 , 38 , 39 Although the currently targetable alterations are inadequate for hematological malignancies, comprehensive genomic profiling can serve as a platform for identifying appropriate candidate individuals for clinical trials investigating molecularly targeted therapies.
Comprehensive genomic profiling can help predict patient prognosis and determine transplant candidacy, particularly in AML and ALL/LBL. In addition, such genetic information may be useful to consider the timing of therapeutic intervention in MDS, MPN, and indolent lymphomas. As clinical evidence for the utility of genetic alterations to predict prognosis is still limited, additional clinicogenetic studies are required.
In our study, TAT was approximately 50 days, which prevents the use of the profiling result for determining induction therapy for acute leukemia. Therefore, it is essential to shorten TAT and combine with conventional methods to treat rapidly progressing malignancies. As the JSH Genome Guideline assigns clinical evidence at the gene level rather than at the position or alteration level, some clinical evidence can be assigned to alterations with uncertain significance, such as BCL2 and non‐L265P MYD88 mutations. Thus, discussion in the molecular tumor board is needed to avoid misinterpretation. As only a small number of patients were enrolled in our study, large‐scale real‐world data are warranted to validate our findings. Despite these challenges, comprehensive genomic profiling can open a new era of precision medicine in hematological malignancies.
DISCLOSURE
S.F. has received research funding from Chugai Pharmaceutical Co., Ltd. Y.O‐K., H.K., and Y.Kikukawa are employees of Otsuka Pharmaceutical Co., Ltd. Spouse of Y.O‐K. is an employee of Taiho Pharmaceutical Co., Ltd. H.M. has received research funding from Sekisui Medical Co., Ltd. T.K. has received honoraria from Eli Lilly Japan K.K. and research funding from Chugai Pharmaceutical Co., Ltd and Sysmex Corp. S.K. has received a patent royalty from Konica Minolta Inc. H.M. has received a patent royalty from Konica Minolta Inc. and research funding from Konica Minolta Inc. Y.S. has received research funding from Otsuka Pharmaceutical Co., Ltd. S.O. has received a patent royalty from Cordia Therapeutics Inc., research funding from Cordia Therapeutics Inc. and Otsuka Pharmaceutical Co., Ltd., and has accepted researchers from Cordia Therapeutics Inc. K.I. has received honoraria from Janssen Pharmaceutical K.K. and Ono Pharmaceutical Co., Ltd., and research funding from Otsuka Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., AstraZeneca K.K., AbbVie Inc., Incyte Corp., Bristol Myers Squibb K.K., Novartis Pharma K.K., Bayer Yakuhin, Ltd., Pfizer Japan Inc., Janssen Pharmaceutical K.K., Yakult Honsha Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Ono Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Beigene, Ltd., and Genmab K.K. K.K. has received research funding from Otsuka Pharmaceutical Co., Chugai Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., and Chordia Therapeutics Inc., has received scholarship endowments from Eisai Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Takeda Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Mochida Pharmaceutical Co., Ltd., JCR Pharmaceuticals Co., Ltd., and Asahi Kasei Pharma Corp., and has accepted researchers from Otsuka Pharmaceutical Co., Ltd. The other authors declare no competing financial interests. H.M. is a Deputy Editor‐in‐Chief, and T.Y., T.K., and K.K. are Associate Editors of Cancer Science.
ETHICS STATEMENT
The study was approved by the National Cancer Center Institutional Review board. All patients and/or their legal guardians (when minors were enrolled) provided written informed consent for this study.
AUTHOR CONTRIBUTIONS
S.F., K.I., and K.K. designed the clinical study. Y.O‐K., Y.Kogure, S.S., Y.S. and K.K. designed the integrated DNA/RNA profiling assay. T.K., S.K., H.M., and S.O. assisted the assay design. H.K. and Y.Kikukawa performed experiments. Y.O‐K, Y.Kogure, Y.Kikukawa, Y.S., and K.K. performed sequencing data analyses. J.K., Y.S., M.T., K.Y., and K.M. assisted experiments and data analyses. S.F., A.M‐M., H.M., M.Sugiyama, C.O., Y.I., T.F., M.Sugano, N.Y., Y.M., and K.I. provided clinical specimens. M.H. and T.Y. interpreted germline variants. S.F., Y.O‐K, Y.Kogure, and K.K. generated figures and tables and wrote the manuscript. K.I. and K.K. led the entire project. All authors participated in discussions and interpretation of the data and results.
Supporting information
Table S1
Figure S1
Figure S2
Figure S3
Figure S4
ACKNOWLEDGMENTS
This work was supported by Otsuka Pharmaceutical Co., Ltd. and National Cancer Center Research and Development Funds (30‐A‐1). Otsuka Pharmaceutical Co., Ltd. played a role in the design, analysis, and interpretation of the data and in the preparation, review, and approval of the manuscript, and as such are included in the author list. No honoraria or payments were made for authorship. We thank F. Ueki, Y. Hokama, Y. Ito, and M. Sagou for technical assistance. Nucleic acid extraction, library preparation, and sequencing were supported by RIKEN GENESIS and Fundamental Innovative Oncology Core at the National Cancer Center. The supercomputing resources were provided by the Human Genome Center, The Institute of Medical Science, The University of Tokyo. The image of sequencer is from TogoTV (©2016 DBCLS TogoTV / CC‐BY‐4.0).
Fukuhara S, Oshikawa‐Kumade Y, Kogure Y, et al. Feasibility and clinical utility of comprehensive genomic profiling of hematological malignancies. Cancer Sci. 2022;113:2763‐2777. doi: 10.1111/cas.15427
Suguru Fukuhara, Yuji Oshikawa‐Kumade, and Yasunori Kogure contributed equally.
Funding information
Otsuka Pharmaceutical Co., Ltd; National Cancer Center Research and Development Funds, Grant/Award Number: 30‐A‐1.
Contributor Information
Koji Izutsu, Email: kizutsu@ncc.go.jp.
Keisuke Kataoka, Email: kkataoka-tky@umin.ac.jp.
REFERENCES
- 1. Chang K, Creighton CJ, Davis C, et al. The Cancer Genome Atlas Pan‐Cancer analysis project. Nat Genet. 2013;45:1113‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. The ICGC/TCGA Pan‐Cancer Analysis of Whole Genomes Consortium . Pan‐cancer analysis of whole genomes. Nature. 2020;578:82‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next‐generation sequencing revolution and its impact on genomics. Cell. 2013;155:27‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McDermott U. Next‐generation sequencing and empowering personalised cancer medicine. Drug Discov Today. 2015;20:1470‐1475. [DOI] [PubMed] [Google Scholar]
- 5. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th Edition ed. International Agency for Research on Cancer; 2017. [Google Scholar]
- 6. Yasuda T, Tsuzuki S, Kawazu M, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48:569‐574. [DOI] [PubMed] [Google Scholar]
- 7. Seki M, Kimura S, Isobe T, et al. Recurrent SPI1 (PU.1) fusions in high‐risk pediatric T cell acute lymphoblastic leukemia. Nat Genet. 2017;49:1274‐1281. [DOI] [PubMed] [Google Scholar]
- 8. Kogure Y, Kameda T, Koya J, et al. Whole‐genome landscape of adult T‐cell leukemia/lymphoma. Blood. 2022;139(7):967‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21:122‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He J, Abdel‐Wahab O, Nahas MK, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004‐3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKerrell T, Moreno T, Ponstingl H, et al. Development and validation of a comprehensive genomic diagnostic tool for myeloid malignancies. Blood. 2016;128:e1‐e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yasuda T, Sanada M, Nishijima D, et al. Clinical utility of target capture‐based panel sequencing in hematological malignancies: A multicenter feasibility study. Cancer Sci. 2020;111:3367‐3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kohsaka S, Tatsuno K, Ueno T, et al. Comprehensive assay for the molecular profiling of cancer by target enrichment from formalin‐fixed paraffin‐embedded specimens. Cancer Sci. 2019;110:1464‐1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watatani Y, Sato Y, Miyoshi H, et al. Molecular heterogeneity in peripheral T‐cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia. 2019;33:2867‐2883. [DOI] [PubMed] [Google Scholar]
- 15. Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47:1304‐1315. [DOI] [PubMed] [Google Scholar]
- 16. Shiraishi Y, Sato Y, Chiba K, et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manickam K, McClain MR, Demmer LA, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence‐based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23:2029‐2037. [DOI] [PubMed] [Google Scholar]
- 19. Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. New England Journal of Medicine. 2009;360:470‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379:1416‐1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gu Z, Churchman ML, Roberts KG, et al. PAX5‐driven subtypes of B‐progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B‐cell lymphoma. N Engl J Med. 2018;378:1396‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walker BA, Mavrommatis K, Wardell CP, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guryanova OA, Shank K, Spitzer B, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med. 2016;22:1488‐1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wright GW, Huang DW, Phelan JD, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell. 2020;37(551–568):e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pastore A, Jurinovic V, Kridel R, et al. Integration of gene mutations in risk prognostication for patients receiving first‐line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population‐based registry. Lancet Oncol. 2015;16:1111‐1122. [DOI] [PubMed] [Google Scholar]
- 29. Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745‐2760. [DOI] [PubMed] [Google Scholar]
- 30. Sakamoto Y, Ishida T, Masaki A, et al. CCR4 mutations associated with superior outcome of adult T‐cell leukemia/lymphoma under mogamulizumab treatment. Blood. 2018;132:758‐761. [DOI] [PubMed] [Google Scholar]
- 31. Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia. 2009;23:2210‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mikhael JR, Dingli D, Roy V, et al. Management of newly diagnosed symptomatic multiple myeloma: Updated Mayo Stratification of Myeloma and Risk‐Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clin Proc. 2013;88:360‐376. [DOI] [PubMed] [Google Scholar]
- 33. Lieber MR. Mechanisms of human lymphoid chromosomal translocations. Nat Rev Cancer. 2016;16:387‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roberts KG, Li Y, Payne‐Turner D, et al. Targetable kinase‐activating lesions in Ph‐like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286‐2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kortum KM, Mai EK, Hanafiah NH, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood. 2016;128:1226‐1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole‐genome sequencing. Nature. 2012;481:506‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waanders E, Gu Z, Dobson SM, et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov. 2020;1:96‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Landau DA, Tausch E, Taylor‐Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figure S1
Figure S2
Figure S3
Figure S4