

Abstract
Hereditary tyrosinemia type 1 is an inborn error of amino acid metabolism characterized by deficiency of fumarylacetoacetate hydrolase (FAH). Only limited treatment options (e.g., oral nitisinone) are available. Patients must adhere to a strict diet and face a life-long risk of complications, including liver cancer and progressive neurocognitive decline. There is a tremendous need for innovative therapies that standardize metabolite levels and promise normal development. Here, we describe an mRNA-based therapeutic approach that rescues Fah-deficient mice, a well-established tyrosinemia model. Repeated intravenous or intramuscular administration of lipid nanoparticle-formulated human FAH mRNA resulted in FAH protein synthesis in deficient mouse livers, stabilized body weight, normalized pathologic increases in metabolites after nitisinone withdrawal, and prevented early death. Dose reduction and extended injection intervals proved therapeutically effective. These results provide proof of concept for an mRNA-based therapeutic approach to treating hereditary tyrosinemia type 1 that is superior to the standard of care.
Keywords: hereditary tyrosinemia type 1, fumarylacetoacetate hydrolase, nitisinone supplementation, mRNA-based therapy, mouse model
Graphical abstract
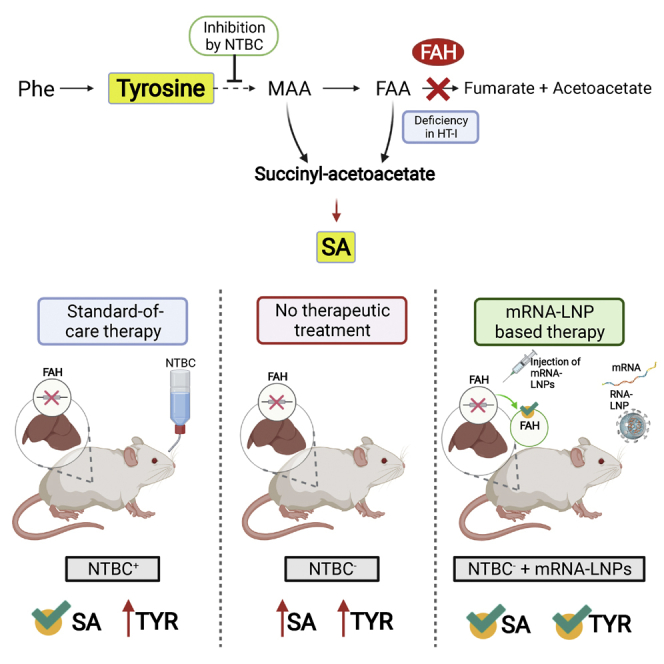
This study provides pre-clinical evidence for an mRNA-based therapy in hereditary tyrosinemia type I. Repeated intravenous injection of formulated therapeutically active mRNA saved mutant mice representing the disease model from body weight loss, early death, and accumulation of toxic metabolites. The mRNA-based therapy was superior to standard-of-care treatment.
Introduction
Hereditary tyrosinemia type 1 (HT1) is an inborn error of amino acid metabolism caused by deficiency of functional fumarylacetoacetate hydrolase (FAH).1 FAH deficiency results in accumulation of toxic and carcinogenic metabolites, such as succinylacetone (SA), tyrosine (TYR), maleylacetoacetate, and fumarylacetoacetate.1, 2, 3 In newborn screening, tyrosinemia was detected in 1 of 100,000 births.4 Acute presentation of symptoms includes liver failure, vomiting, bleeding, hypoglycemia, and tubulopathy; chronic manifestations present as hepatomegaly, cirrhosis, growth retardation, rickets, tubulopathy, and neuropathy. Tyrosinemia patients are at increased risk to develop neurologic crisis, renal failure, and early-onset hepatocellular carcinoma (HCC). Standard of care (SOC) for patients includes supplementation with nitisinone, 2-(2-nitro-4-trifluoromethyl benzoyl) cyclohexane-1,3-dione (NTBC), administered orally twice daily.3 NTBC is a strong inhibitor of 4-hydroxyphenyl-pyruvatdioxigenase, which catalyzes the second step in tyrosine degradation.5 While suppressing the accumulation of toxic tyrosine-derived metabolites, 4-hydroxyphenyl-pyruvatdioxigenase inhibition promotes accumulation of tyrosine, leading to eye symptoms, neurocognitive defects, and potential development of a condition that mimics hereditary tyrosinemia type 2.1 Consequently, a strict life-long diet low in TYR and phenylalanine remains an essential component of disease management.
NTBC supplementation significantly improves disease management, particularly when started early in life. However, some patients still develop liver cancer, while a subset of patients fail to respond to NTBC treatment; long-term risk assessment remains to be completed. Importantly, despite treatment, affected children do not develop normally and might face neurocognitive problems.1,3,6, 7, 8 It is essential for patients to adhere strictly to uninterrupted NTBC supplementation; discontinuation could result in a life-threatening neurological crisis that requires hospitalization.9 The ultimate treatment option is liver transplantation, which is complicated by shortage of donor organs, organ rejection, and side effects associated with immunosuppression.1 Therefore, there is an urgent need for alternate treatment options.
Several point mutations in patients suffering from tyrosinemia type 1 have been described, affecting the FAH gene.10 Mice bearing similar point mutations are most suitable to model human tyrosinemia type 1. Of several mouse mutants induced by N-ethyl-N-nitroso-urea that were identified, one closely mimics the chronic form of human HT1.11 Without treatment, homozygotes exhibit postnatal lethality within 24 h after birth. NTBC supplementation can normalize pathologic increases in metabolites, but not fully (e.g., serum TYR levels remain high), and rescue Fah-deficient mice from death.12
To date, experimental approaches to treat tyrosinemia type 1 involve adenovirus- and AAV-mediated gene transfer,13,14 gene delivery of naked DNA constructs,15,16 genome editing,17, 18, 19 and ex vivo therapy.20, 21, 22 The approach presented herein focuses on mRNA-based therapy, a powerful tool with tremendous potential to treat a variety of indications; e.g., infectious diseases, personalized cancer therapy, protein replacement therapy, and gene editing.23, 24, 25
FAH is expressed predominantly in liver and kidneys; consequently, hepatocytes and renal proximal tubular epithelial cells are most often affected by the absence of a functional gene.2,10 mRNA provides a safe and transient tool to express therapeutic proteins, such as FAH, in target organs. Targeting the liver to synthesize FAH could prove to be a valuable therapeutic option in treating tyrosinemia patients. Naked mRNA, however, is quickly degraded in the bloodstream after intravenous (i.v.) injection. mRNA encapsulated in lipid nanoparticles (LNPs), on the other hand, is protected from degradation and delivered mainly to the liver, the target organ for treating tyrosinemia.23,24,26 US Food and Drug Administration (FDA) approval of Onpattro (patisiran) and treatment of polyneuropathy using LNP-mediated delivery of therapeutic small interfering RNA (siRNA) to the liver facilitates LNP-mediated RNA delivery as an approach to treat a variety of diseases.27,28 Indeed, LNP-formulated therapeutic mRNA targeted to the liver is currently being investigated in pre-clinical studies as an approach to cure hemophilia B, thrombotic thrombocytopenic purpura, methylmalonic academia, ornithine transcarbamylase deficiency, Fabry disease, acute intermittent porphyria, Crigler-Najjar syndrome type 1, alpha-1 antitrypsin deficiency, hereditary spastic paraplegia type 5, and glycogen storage disease type I,24,29, 30, 31, 32, 33, 34, 35 with some of these applications already being tested in clinical trials.
In the study presented here, FAH mRNA-based therapy reversed the progression of disease in Fah-deficient mice. Repeated injections of LNP-formulated FAH mRNA via both i.v. and intramuscular (i.m.) administration routes resulted in FAH protein production in the liver, restoration of normal hepatic morphology, abrogation of pathologic increases in SA and TYR, stabilization of body weight, and prevention of early death after withdrawal of NTBC supplementation. Dosing studies showed that LNP-formulated FAH mRNA can provide therapeutic protection at low doses. Moreover, extending the intervals between injections up to 2 weeks proved therapeutically effective. Taken together, these findings suggest that LNP-formulated therapeutic FAH mRNA could provide an alternate treatment option for tyrosinemia type 1 patients. This approach would benefit children, especially, by preventing the adverse complications associated with SOC; i.e., continued NTBC supplementation and the burden of a strict, life-long adherence to a special diet.
Results
Luciferase expression in Fah-deficient mice injected with PpLuc mRNA-LNPs
Fah-deficient mice (Fah1RTyrc/RJ), a human tyrosinemia model, are liver compromised due to underlying disease. To demonstrate the ability to target protein synthesis to the livers of Fah-deficient mice, PpLuc mRNA formulated in LNPs was injected i.v. into the tail vein or i.m. into both tibialis muscles. Luciferase signals were recorded 6 h after injection. Luciferase expression was observed in predominantly the liver following i.v. injection (Figure S1). Expression was detected in both the muscles and (to a lower extent) in livers of Fah-deficient mice injected with PpLuc mRNA-LNPs i.m., demonstrating transport of PpLuc mRNA-loaded LNPs to the liver via the bloodstream and expression at a distant site consistent with results described by Pardi et al.36 Injection of the PBS control did not elicit a detectable luciferase signal.
Stable human FAH protein synthesis, and SA and TYR reduction after a single i.v. FAH mRNA-LNP administration
Untreated Fah-deficient mice die within 24 h after birth. To prevent early postnatal lethality, Fah-deficient mice are supplemented with NTBC in drinking water throughout life (pregnant and nursing females; experimental homozygotes). To induce the tyrosinemia type I disease phenotype, NTBC supplementation was withdrawn 5 days before start of treatment. Fah-deficient mice were subjected to one of the following regimens: (1) Fah-deficient mice received NTBC supplementation without interruption (NTBC+PBS+); (2) NTBC supplementation was withdrawn on day 5 prior to treatment, and PBS was injected on day 0 (NTBC−PBS+); and (3) in the experimental group, NTBC supplementation was stopped on day 5 prior to treatment, and a single dose of LNP-formulated therapeutic FAH mRNA (NTBC−RNA+) was injected i.v. on day 0 (Figure 1A). Blood and liver samples were collected on days 1, 2, and 4 after FAH mRNA-LNP injection. The therapeutic effect was shown by a significant decrease in serum SA levels detected in the experimental group at all time points. Levels were comparable with those found in mice that received continued NTBC supplementation, and, importantly, equivalent to physiologic levels found in wild-type (WT) mice (Figure 1B). Moreover, TYR levels were reduced to physiologic levels following FAH mRNA-LNP treatment, which were not achieved by NTBC supplementation (Figures 1C and S2). These findings highlight the efficacy of FAH mRNA-LNP in promoting physiologic metabolic pathways and normalizing TYR levels.
Figure 1.

A single FAH mRNA-LNP injection i.v. reduces pathologic SA and TYR serum levels in Fah-deficient mice
(A) Experiment schedule. NTBC supplementation was stopped on day 5 prior to treatment. Blood was collected on day 1 before treatment (pre-bleeding [PB]) and on termination days 1, 2, and 4 after single i.v. injection. One group of mice received NTBC supplementation (NTBC+PBS+) throughout the experiment. The second group (NTBC−PBS+) was withdrawn from NTBC supplementation on day 5 prior to treatment and injected with PBS on day 0. The experimental cohort was withdrawn from NTBC supplementation on day 5 prior to treatment and injected i.v. with a single dose of LNP-formulated therapeutic FAH mRNA (NTBC−RNA+) on day 0. (B) Absolute succinylacetone (SA) and (C) tyrosine (TYR) levels in Fah-deficient mouse serum at the times indicated (four mice/group). (D) Western blot analyses of Fah-deficient and wild-type (WT) mouse liver lysates from day 1 after single injections and normalized to β-actin loading control (representative samples; three livers/treatment group). Fah-deficient mouse livers with or without NTBC supplementation were used as controls. (E) Quantitation of FAH protein bands normalized to β-actin loading control (three livers/group). (F) Western blot analyses of liver lysates from WT mice and Fah-deficient mice on day 4 after single injections. (G) Quantitation of FAH protein bands normalized to β-actin loading control (three livers/group). Data are the means ± SEM. Significantly different from NTBC+PBS+: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed Student’s t test).
To demonstrate FAH production in the target organ, the livers of Fah-deficient and WT mice were dissected 24 h after single i.v. injections, and FAH was quantified by western blot analysis (Figure 1D). Approximately 50% of WT FAH levels were observed in livers of Fah-deficient mice injected with FAH mRNA-LNP (Figure 1E). Notably, a substantial quantity of FAH protein (∼30% of that determined in WT mice) was still detected in livers at 4 days post injection (Figures 1F and 1G). Control livers (PBS-injected, Fah-deficient mice ± NTBC) lacked detectable FAH protein. Taken together, these findings demonstrate prolonged FAH protein in livers of Fah-deficient mice following a single FAH mRNA-LNP injection and a concomitant, stable reduction in toxic serum metabolites, i.e., both SA and TYR, which was not observed in mice maintained on an NTBC diet.
Repeated i.v. FAH mRNA-LNP injection normalizes pathologic increases in serum SA and TYR levels and rescues Fah-deficient mice from body weight loss and death
To determine whether the tyrosinemia disease phenotype could be prevented for a prolonged period of time, Fah-deficient mice were injected repeatedly i.v. with FAH mRNA-LNPs. NTBC supplementation was withheld on day 5. Blood was collected on day 1 prior to injection to serve as a baseline, at 24 h post FAH mRNA-LNP injection, at periodic intervals thereafter, and at the experimental endpoint (Figure 2A). One Fah-deficient mouse control group was provided NTBC supplementation without interruption (NTBC+PBS+). A second group was denied NTBC supplementation on day 5 before repeated PBS injections (NTBC−PBS+). The experimental group, deprived of NTBC on day 5 prior to treatment, received LNP-formulated FAH mRNA i.v. every 5 days for five times (NTBC−RNA+). NTBC+PBS+ and NTBC−RNA+ Fah-deficient mice survived until the end of the 21-day study period (Figure 2B). All NTBC−PBS+ Fah-deficient mice were euthanized due to body weight loss before the end of the study. Importantly, Fah-deficient mice administered FAH mRNA-LNPs did not lose weight, nor did their weight differ significantly from the NTBC+PBS+ group, thus demonstrating treatment efficacy. Serum SA and TYR levels, normalized to pre-treatment levels, were monitored throughout the study (Figures 2C and 2D). NTBC+PBS+ Fah-deficient mice exhibited low SA levels, but consistently high TYR levels, relative to WT mice throughout the experiment. Absolute SA and TYR values are shown in Figure S3. NTBC−PBS+ mice showed substantially elevated SA and TYR levels and were euthanized prematurely. NTBC treatment did not reduce TYR levels. FAH mRNA-LNP treated Fah-deficient mice recovered quickly after two i.v. injections and exhibited low serum SA levels comparable with NTBC-treated mice. Moreover, FAH mRNA-LNP-treated Fah-deficient mice exhibited a marked decrease in TYR that reached physiologic levels, suggesting that FAH mRNA-LNP therapy might be superior to the SOC for treating HT1.
Figure 2.

Repeated FAH mRNA-LNPs injections i.v. rescue Fah-deficient mice from death
(A) Experiment schedule. NTBC supplementation was withdrawn on day 5 prior to injections. Blood was collected on day 1 before treatment (PB), at intermittent times 24 h after each injection (i.e. B1 day 6; B2 day 11; B3 day 16), and on termination day 21. NTBC+PBS+ received continuous NTBC supplementation and repeated PBS injections throughout the experiment. NTBC−PBS+ mice were stopped for NTBC supplementation on day 5 prior to treatment and injected with PBS repeatedly. NTBC−RNA+ mice were withdrawn from NTBC supplementation on day 5 prior to treatment; LNP-formulated therapeutic FAH mRNA was injected i.v. every 5 days (i.e., days 0, 5, 10, 15, and 20; repeated injection 1–5). (B) Normalized body weights of Fah-deficient mice. (C) Normalized serum SA and (D) TYR levels in Fah-deficient mice (five mice/group). (E) Western blot analyses of FAH protein in Fah-deficient and WT mouse livers, normalized to β-actin loading control (repeated injection; WT mice received single PBS injection). (F) Quantitation of FAH protein bands normalized to β-actin loading control. (G) Representative images of immunohistochemical FAH protein staining on paraffin-embedded Fah-deficient mouse liver sections after repeated i.v. injections of FAH mRNA-LNPs. (H) Alanine transaminase (ALT), (I) aspartate transaminase (AST), (J) alkaline phosphatase (AP), and (K) creatine kinase (CK) were evaluated in Fah-deficient mouse serum at termination. Data are the means ± SEM. Significantly different from the control group: ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed Student’s t test). All NTBC−PBS+Fah-deficient mice were euthanized before the scheduled end of the experiment due to weight loss.
Serum levels of glycine, alanine, proline, valine, threonine, ornithine, aspartic acid, glutamine, glutamic acid, phenylalanine, citrulline, homocitrulline, tryptophan, arginine, asparagine, and methionine were quantified in the three experimental groups (Table 1). With exceptions of leucine/isoleucine and histidine, all levels were comparable in Fah-deficient mice administered FAH mRNA-LNP and WT animals. At the end of the 21-day experiment, i.e., 24 h after the last injection, western blot analysis and quantitation of FAH protein revealed FAH protein synthesis in livers of FAH mRNA-LNP-treated Fah-deficient mice was ∼20% of endogenous FAH protein present in WT mouse livers (Figures 2E and 2F). Serum alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (AP), and creatine kinase (CK) levels, markers of liver and muscle function, were assessed. All analyte levels were increased in Fah-deficient mice injected with PBS only (i.e., NTBC−PBS+); however, levels measured in NTBC-treated and FAH mRNA-LNP-treated mice were indistinguishable and equivalent to those found in WT mice, demonstrating therapeutic efficacy of this approach (Figures 2H–2K). The localization of FAH protein production in livers of FAH mRNA-LNP-treated and control mice were determined by immunohistochemical analysis of paraffin-embedded liver samples. No FAH signal was observed in NTBC+PBS+ and NTBC−PBS+ mice. In contrast, FAH mRNA-LNP-treated animals showed a strong FAH signal localized mainly in hepatocytes, demonstrating FAH protein production in the target organ (Figure 2G). Taken together, these findings suggest that even relatively low amounts of FAH protein can rescue Fah-deficient mice from body weight loss, premature death, and pathologic increases in metabolite levels.
Table 1.
Serum amino acid levels in WT and Fah-deficient mice
| Amino acid levels in serum (μM) | Fah-deficient mice NTBC−PBS+ | Fah-deficient mice NTBC+PBS+ | Fah-deficient mice NTBC−RNA+ | WT mice NTBC−PBS+ |
|---|---|---|---|---|
| Gly | 555.6 ± 106.6∗ | 125.0 ± 22.1 | 149.7 ± 16.6 ns | 144.9 ± 8.3 |
| Ala | 2,077.1 ± 280.3∗∗ | 353.8 ± 93.0 | 455.7 ± 92.7 ns | 305.1 ± 35.1 |
| Pro | 390.9 ± 70.4∗ | 92.4 ± 20.6 | 125.5 ± 29.1 ns | 102.5 ± 14.2 |
| Val | 313.2 ± 23.7∗∗ | 146.6 ± 11.6 | 207.1 ± 23.9 ns | 126.1 ± 7.5 |
| Thr | 62.5 ± 20.5 ns | 13.3 ± 2.7 | 13.8 ± 1.4 ns | 13.1 ± 0.7 |
| Leu/Ile | 374.9 ± 22.8∗∗∗ | 191.2 ± 18.2 | 307.4 ± 51.3 ns | 147.5 ± 11.3 |
| Orn | 855.2 ± 256.8 ns | 119.9 ± 7.5 | 178.6 ± 21.7 ns | 110.4 ± 17.3 |
| Asp | 164.1 ± 51.6 ns | 20.0 ± 1.7 | 30.9 ± 4.8 ns | 15.2 ± 2.4 |
| Gln | 11,798.4 ± 1,952.2∗ | 2,933.8 ± 446.2 | 4,231.4 ±612.8 ns | 3,462.4 ± 274.0 |
| Glu | 681.5 ± 192.9∗ | 56.9 ± 5.9 | 86.5 ± 18.2 ns | 51.5 ± 6.6 |
| His | 12,177.6 ±1,062.7∗∗ | 1,117.4 ± 104.1 | 1,497.0 ±204.3 ns | 715.4 ± 125.9 |
| Phe | 376.8 ± 231.2 ns | 75.1 ± 9.5 | 116.2 ± 20.3 ns | 69.1 ± 7.5 |
| Cit | 206.7 ± 68.6 ns | 60.5 ± 7.8 | 68.7 ± 8.3 ns | 50.6 ± 2.1 |
| Hci | 0.8 ± 0.5 ns | 1.8 ± 0.7 | 1.3 ± 0.7 ns | 3.7 ± 0.5 |
| Trp | 891.8 ± 56.4∗∗∗ | 437.5 ± 50.9 | 542.3 ± 67.1 ns | 415.7 ± 28.6 |
| Arg | 15.6 ± 10.9 ns | 62.8 ± 18.9 | 83.8 ± 21.5 ns | 72.8 ± 18.7 |
| Asa | 1.5 ± 0.9 ns | 1.1 ± 0.3 | 0.9 ± 0.3 ns | 0.5 ± 0.2 |
| Met | 164.8 ± 56.8 ns | 47.3 ± 8.5 | 55.7 ± 5.4 ns | 41.2 ± 3.5 |
Fah-deficient mice were treated as follows: NTBC+, supplemented consistently with NTBC; NTBC−, NTBC withdrawn on day 5 prior to initial treatment; PBS+, injected with PBS; RNA+, injected with FAH mRNA-LNPs. Number of mice analyzed: NTBC−PBS+ n = 5 Fah-deficient mice; NTBC+PBS+ n = 5 Fah-deficient mice; NTBC−RNA+ n = 5 Fah-deficient mice; NTBC−PBS+ n = 5 WT mice. Analyses were performed on serum collected from Fah-deficient mice on day 21 according to the schedule depicted in Figure 2A. WT mice, shown for comparison purposes, were injected once i.v. with PBS then euthanized after 24 h. All NTBC−PBS+ Fah-deficient mice were euthanized before the scheduled end of the experiment due to a 20% weight loss. Gly, glycine; Ala, alanine, Pro, proline; Val, valine; Thr, threonine; Leu/Ile, leucine/isoleucine; Orn, ornithine; Asa, argininosuccinate; Gln, glutamine; Glu, glutamic acid; Phe, phenylalanine; Cit, citrulline; Hci, homocitrulline, Trp, tryptophan; Arg, arginine; Asn, asparagine; Met, methionine. Significantly different from NTBC+PBS+-treated mice: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (Student’s t test).
Repeated i.m. and i.v. injections of FAH mRNA-LNPs are equally effective in treating Fah-deficient mice
i.m. injection is a more patient-friendly route of administration than i.v. From biodistribution studies we demonstrated transport of mRNA-loaded LNPs to the liver of Fah-deficient mice via the bloodstream and expression in liver. We hypothesized that this protein expressed in the liver after i.m. injection induces therapeutic effects. To demonstrate the effectiveness of administering FAH mRNA-LNPs i.m. versus i.v., Fah-deficient mice were injected with 1 mg/kg every 5 days (Figure 3A). Fah-deficient mice injected i.m. survived until scheduled termination on day 21 without showing visual signs of health decline. Body weight was stable and indistinguishable from mice repeatedly injected i.v. (Figure 3B). Normalized SA and TYR levels dropped rapidly to near WT mouse levels and stayed low throughout the experiment (Figures 3C and 3D). At the end of the experiment, absolute SA and TYR levels were decreased substantially compared with pre-treatment values regardless of FAH mRNA-LNPs administration route (Figure S4). Serum levels of a metabolite panel were quantified. Aside from histidine, all metabolite levels in Fah-deficient mice administered FAH mRNA-LNPs either i.v. or i.m. were comparable with levels found in WT mice (Table 2). Livers of treated mice were subjected to western blot analysis to correlate FAH protein synthesis with therapeutic effects. Importantly, FAH protein was synthesized in Fah-deficient mouse livers after repeated i.m. injection (Figures 3E and 3F), demonstrating transport of FAH mRNA-loaded LNPs from distant injection sites (i.e., tibialis muscles) via the bloodstream to the liver. As expected, protein production was lower than that observed in livers of mice repeatedly injected i.v., which directly targets LNPs to the liver. Similar results were obtained by immunohistochemical evaluations. FAH production was evident in livers of both animal cohorts, but the staining was much stronger in the cohort injected i.v. (Figure 3G). The amount of FAH present in the livers of i.m.-injected mice was sufficient, however, to prevent body weight loss and death. With the exception of AST, other markers of liver and muscle function and injury (ALT, AP, and CK levels) differed insignificantly between repeated i.v. and i.m. administration (Figures 3H–3K).
Figure 3.

Repeated i.m. and i.v. injections with FAH mRNA-LNPs in Fah-deficient mice exhibit comparable therapeutic effects
(A) NTBC supplementation was withdrawn from Fah-deficient mice. Blood was collected 1 day before treatment (PB; day −1), at intermediate bleeding time points 24 h after injections (B1–B3 on days 6, 11, and 16), and on termination day (T; day 21) (Fah-deficient mice injected i.m., n = 5; Fah-deficient mice injected i.v., n = 4). (B) Normalized body weights. (C) Normalized serum SA levels. (D) Normalized serum TYR levels. (E) Western blot analyses of FAH protein in livers of Fah-deficient mice dissected on day 21; β-actin served as loading control. (F) Quantitation of FAH protein bands normalized to β-actin. FAH amount in livers of Fah-deficient mice injected repeatedly i.v. was set to 100%. (G) Representative images of immunohistochemical FAH protein staining on paraffin-embedded Fah-deficient mouse liver sections after repeated i.v. versus i.m. injections of FAH mRNA-LNPs. (H) Alanine transaminase (ALT), (I) aspartate transaminase (AST), (J) AP, and (K) CK were evaluated in Fah-deficient mouse serum at termination. Data are the means ± SEM. Significantly different from the control group: ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed Student’s t test).
Table 2.
Serum amino acid levels in Fah-deficient mice repeatedly injected i.v. or i.m. with FAH mRNA-LNPs
| Amino acid levels in serum (μM) | Fah-deficient mice NTBC−RNA+ i.m. | Fah-deficient mice NTBC−RNA+ i.v. | WT mice NTBC−PBS+ |
|---|---|---|---|
| Gly | 154.7 ± 58.1 | 96.9 ± 18.8 ns | 144.9 ± 8.3 |
| Ala | 331.1 ± 103.7 | 372.6 ± 77.9 ns | 305.1 ± 35.1 |
| Pro | 119.1 ± 34.0 | 86.9 ± 14.2 ns | 102.5 ± 14.2 |
| Val | 174.8 ± 22.0 | 129.4 ± 8.5 ns | 126.1 ± 7.5 |
| Thr | 19.8 ± 9.5 | 13.7 ± 1.8 ns | 13.1 ± 0.7 |
| Leu/Ile | 203.0 ± 19.9 | 164.7 ± 16.2 ns | 147.5 ± 11.3 |
| Orn | 193.4 ± 48.8 | 130.2 ± 20.5 ns | 110.4 ± 17.3 |
| Asp | 35.1 ± 11.1 | 21.9 ± 3.3 ns | 15.2 ± 2.4 |
| Gln | 4,624.6 ±1,934.1 | 3,745.3 ± 535.5 ns | 3,462.4 ± 274.0 |
| Glu | 89.6 ± 23.2 | 50.1 ± 7.1 ns | 51.5 ± 6.6 |
| His | 2,065.8 ± 925.3 | 1,207.0 ± 59.3 ns | 715.4 ± 125.9 |
| Phe | 56.8 ± 15.0 | 57.0 ± 9.2 ns | 69.1 ± 7.5 |
| Cit | 48.0 ± 8.4 | 48.5 ± 7.0 ns | 50.6 ± 2.1 |
| Hci | 1.3 ± 0.7 | 0.8 ± 0.3 ns | 3.7 ± 0.5 |
| Trp | 495.3 ± 55.6 | 443.1 ± 46.4 ns | 415.7 ± 28.6 |
| Arg | 112.1 ± 10.5 | 87.6 ± 26.9 ns | 72.8 ± 18.7 |
| Asa | 0.4 ± 0.3 | 0.8 ± 0.6 ns | 0.5 ± 0.2 |
| Met | 55.0 ± 19.1 | 39.3 ± 5.2 ns | 41.2 ± 3.5 |
NTBC was withheld from Fah-deficient mice on day 5 prior to repeated injection i.m. or i.v. with FAH mRNA-LNPs. Number of mice analyzed: Fah-deficient mice NTBC−RNA+ i.m. n = 5; Fah-deficient mice NTBC−RNA+ i.v. n = 4; WT mice NTBC−PBS+ n = 5. Analyses were performed on serum collected from Fah-deficient mice on day 21 according to the schedule depicted in Figure 3A. WT, shown for comparison purposes, were injected once i.v. with PBS and euthanized 24 h later. Gly, glycine; Ala, alanine; Pro, proline; Val, valine; Thr, threonine; Leu/Ile, leucine/isoleucine; Orn, ornithine; Asa, argininosuccinate; Gln, glutamine; Glu, glutamic acid; Phe, phenylalanine; Cit, citrulline; Hci, homocitrulline; Trp, tryptophan; Arg, arginine; Asn, asparagine; Met, methionine. Serum amino acid levels in mice injected repeatedly i.m. versus i.v. were not significantly different: p > 0.05 (Student’s t test).
Repeated FAH mRNA-LNP administration maintains hepatic morphology
The efficacy of mRNA-based therapy and repeated FAH mRNA-LNP injections i.v. or i.m. were evaluated by conventional histologic examination of paraffin-embedded Fah-deficient mouse liver sections. Livers of NTBC−PBS+-treated Fah-deficient mice exhibited characteristics of severe acute liver injury with single-cell and grouped necrosis, toxic-type microvesicular steatosis, regeneration phenomena with hepatocytes of different sizes, as well as a concomitant inflammatory infiltrate surrounding necrotic hepatocytes (Figure 4). Normal liver architecture, however, was preserved. Histologic examination of liver sections derived from NTBC+PBS+-treated mice revealed negligible or mild changes with minimal steatosis. Upon i.v. administration of FAH mRNA-LNPs (NTBC−RNA+), some hepatocyte swelling and minimal to mild microvesicular steatosis were apparent in Fah-deficient mice, quantified in Figure S5. Only minimal to mild steatosis was observed in the livers of mice repeatedly injected i.v. or i.m. No single-cell or grouped necrosis, fibrosis, distortion of liver architecture or foci was observed.
Figure 4.

Injected FAH mRNA-LNPs normalizes liver histopathology of Fah-deficient mice
Repeated i.v. injections were performed according to the schedule depicted in Figure 2A. (A) H&E-stained liver sections. N, hepatocyte necrosis; asterisk, binucleated cells; arrow, steatosis. (B) Single-cell and grouped necrosis were quantified according to the scaling system outlined in Figure S5. (C) H&E-stained paraffin-embedded Fah-deficient mouse liver sections after repeated FAH mRNA-LNPs injections i.v. or i.m. (D) Quantitation of single-cell (left) and grouped (right) necrosis after repeated i.v. or i.m. injections. Note: all NTBC−PBS+Fah-deficient mice were euthanized before the scheduled end of the experiment due to weight loss.
Therapeutic effects are achieved at lower doses
To determine whether doses lower than 1 mg/kg used in previous experiments would reduce serum SA and TYR concentrations to levels found in WT mice, we administered doses of 0.5 mg/kg and 0.1 mg/kg into Fah-deficient mice every 5 days (Figure 5A). Both 0.5 and 0.1 mg/kg doses administered i.v. stabilized body weights throughout life phase (Figure 5B) and reduced serum SA and TYR levels (normalized to pre-treatment values) (Figures 5C and 5D). Moreover, absolute SA and TYR values assessed at the end of the experiment reached levels found in WT mice (Figures S6A and S6B). In contrast, a clear difference between doses was observed when FAH mRNA-LNPs were administered i.m. Only animals dosed with 0.5 mg/kg FAH mRNA-LNPs survived to the scheduled end of the experiment (Figure 5E). The 0.1 mg/kg dose was not sufficient to guarantee therapeutic effects. Animals started to lose weight quickly, necessitating euthanasia when body weight loss reached 20% (Figure 5F). Both normalized and absolute SA and TYR levels increased throughout the experiment, reaching very high levels at termination (Figures 5G, 5H, S6C, and S6D). These findings correlate with western blot analyses of liver lysates derived from the same animals (Figure S6E). Highest FAH protein levels in liver were achieved when dosing 0.5 mg/kg FAH mRNA-LNPs i.v. followed by lower levels at 0.1 mg/kg i.v. On the other hand, only a small amount of FAH protein was detected in livers of mice injected with 0.5 mg/kg FAH mRNA-LNPs i.m.; liver FAH expression in mice injected i.m. with 0.1 mg/kg FAH mRNA-LNPs was below the level of detection (Figure S6F).
Figure 5.

Repeated i.v. injections of 0.1 mg/kg or 0.5 mg/kg FAH mRNA-LNPs, or 0.5 mg/kg FAH mRNA-LNPs injected i.m. rescue Fah-deficient mice from death
(A) NTBC supplementation was withdrawn from Fah-deficient mice 5 days before treatment. Blood was collected 1 day before treatment (PB; day −1), at intermediate bleeding time points 24 h after injections (B1–B3 on days 6, 11, and 16), and on day of termination (T; day 21) (n = 5 Fah-deficient mice/condition). (B) Normalized body weights of Fah-deficient mice that were repeatedly injected i.v. (C) Normalized serum SA and (D) TYR levels in Fah-deficient mice repeatedly injected i.v. with the dose of FAH mRNA-LNPs indicated. (E) Survival of Fah-deficient mice repeatedly injected i.m. with the dose of FAH mRNA-LNPs indicated (five mice/group). (F) Normalized body weights of Fah-deficient mice repeatedly injected i.m. with FAH mRNA-LNPs. (G) Normalized serum SA and (H) TYR levels in Fah-deficient mice repeatedly injected i.m. with FAH mRNA-LNPs. Data are the means ± SEM. Significantly different from mice that received a 0.1-mg/kg dose: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed Student’s t test).
Therapeutic effects are achieved over longer intervals between injections
To determine whether the therapeutic window could be extended beyond the 5-day interval (as shown above), we took into account that FAH protein was stable up to 8 days after single i.v. injection (Figures S6A and S6B), and SA and TYR levels were rescued compared with pre-treatment levels (Figures S6C–S6F). Following a single i.v. injection, western blot analysis revealed the presence of FAH protein at day 14 (Figures S7G and S7H).
A single FAH mRNA-LNP dose injected i.m. showed a very weak FAH protein signal on day 14 (Figures S7I and S7J), although SA and TYR levels were comparable with those found in the i.v. cohort (Figures S7K–S7N).
Given the capacity of a single FAH mRNA-LNP dose to exert therapeutic effects over a 14-day period, mice were injected five times with 1 mg/kg FAH mRNA-LNPs i.v. or i.m. at 1-week (Figure 6A) or 2-week (Figure 6E) intervals. Weekly injections by both routes were effective. Mice exhibited no visual signs of stress or suffering, nor changes in body weight (Figure 6B). Moreover, serum SA and TYR levels reached low, therapeutic values over time regardless of the route of administration (Figures 6C, 6D, S8A, and S8B). Fah-deficient mice injected i.v. or i.m. every 14 days survived the entire 57-day experiment. Some fluctuations in body weight and periods of weakness and recovery occurred in mice injected i.m., especially during the latter part of the experiment (Figure 6F). Mice injected i.v. showed no fluctuations in body weight, signs of stress, or health decline. Serum SA levels decreased after both i.m. and i.v. injections, reaching therapeutic values characteristic of WT animals (Figures 6G and S8C). Only mice injected i.v., however, exhibited a sustained decrease in serum TYR levels; mice injected i.m. showed marked fluctuations in TYR, which were elevated at the end of the study (Figures 6H and S8D). Western blot analyses of liver lysates revealed comparable amounts of FAH protein in the livers of mice injected repeatedly i.v. over the course of the 2-week schedule (Figures S8E and S8F) compared with livers of mice administered a single i.v. injection (Figures 1D and 1E). As little as ≤2.5% of WT FAH protein levels achieved after repeated i.m. administration on both 1- and 2-week schedules were sufficient to rescue Fah-deficient mice from premature death.
Figure 6.

Interval extension studies for repeated i.v. and i.m. injections of FAH mRNA-LNPs into Fah-deficient mice
(A) Weekly experimental protocol. NTBC supplementation was withdrawn from Fah-deficient mice for 5 days. Subsequently, five mice/group were repeatedly injected (1–5) i.v. or i.m. at 1-week intervals with 1 mg/kg FAH mRNA-LNPs; blood was collected (PB, B1–B3, and T) according to the schedule shown. (B) Normalized body weights recorded at 1-week intervals. (C) Normalized serum SA and (D) TYR levels recorded at 1-week interval. (E) Biweekly experimental protocol. NTBC supplementation was withdrawn from Fah-deficient mice for 5 days. Subsequently, five mice/group were repeatedly injected (1–5) i.v. or i.m. at 2-week intervals with 1 mg/kg FAH mRNA-LNPs; blood was collected (PB, B1–B7, and T) according to the schedule shown. (F) Normalized body weights determined at 2-week intervals. (G) Normalized serum SA and (H) TYR levels quantified at 2-week intervals. Data are the means ± SEM. Significantly different between i.v. and i.m. injected mice: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed Student’s t test).
Discussion
HT1 is an autosomal recessive disorder that affects tyrosine metabolism, resulting primarily in damage to the liver, kidneys, and peripheral nerves.1 It affects approximately 1:100,000 live births.4 Clinical manifestations of the disease, i.e., poor growth, liver failure, and hepatomegaly, typically occur within the first 2 years after birth. Indeed, disease severity correlates with the time of onset; more severe cases occur early. HT1 is caused by a deficiency in FAH activity.1 FAH is predominantly expressed in hepatocytes and renal proximal tubular epithelium, accounting for expression of the principal effects of an enzyme deficiency in the liver and kidneys.2,10 FAH is the last enzyme in the TYR catabolic pathway. Mutations leading to a deficiency in FAH activity result in elevated concentrations of TYR in the bloodstream and the accumulation of upstream metabolites; i.e., fumarylacetoacetate and maleylacetoacetate (two hepatotoxic metabolic intermediates), which are converted to succinyl acetoacetate, then SA.10
Approximately 100 mutations in the gene encoding FAH have been reported.10 These mutations include 45 missense mutations, 13 nonsense mutations, four frameshift mutations alterations, 10 deletions, and 23 splice defects. Relatively few account for the majority of HT1 cases identified clinically.37 Mutations that inhibit enzyme activity may either target the catalytic site directly or affect enzyme stability, leading to a loss of function. In the Fah-deficient mouse model used in the current study (Fah1R Tyrc/RJ), a splice mutation (Fah5961SB) induced by N-ethyl-N-nitrosourea treatment resulted in severe reduction of FAH mRNA and absence of FAH protein.11 We demonstrated absence of FAH protein in liver lysates derived from NTBC−PBS+ and NTBC+PBS+ mice by western blot. These findings enabled a clear and direct assessment of FAH mRNA LNP efficacy in reversing health decline as well as rescuing Fah-deficient mice from death.
Daily NTBC supplementation and strict adherence to a diet low in phenylalanine and tyrosine is the SOC for HT1 treatment.37 The advent of NTBC in 1992 revolutionized the prognosis of patients.8 Early NTBC supplementation can prevent liver failure, tubulopathy, and neurological crises, and decrease the risk for hepatocellular carcinoma. NTBC supplementation, however, does not guarantee normal growth or completely prevent the development of liver cancer in affected patients.38 In fact, an estimated 10% of patients are unresponsive to NTBC treatment.39 Additionally, there is considerable risk for liver cancer in patients who begin NTBC treatment late.39 Liver transplantation is the only option for such patients.37 Despite the beneficial effects of early NTBC supplementation, ophthalmologic and dermatologic problems can still occur as a consequence of increased plasma TYR concentration.8 The cost of NTBC is exorbitant, estimated at $51,493 per year to treat patients undergoing early, and $64,895 undergoing late, intervention.8,40
Currently, there is no cure or treatment for HT1 that precludes daily medication, dietary restrictions, neurotoxicity, or potential liver transplantation. Gene (FAH)-targeted therapy provides an alternate, superior approach to treating HT1.1 Predominant vectors used experimentally to treat HT1 to date are adeno-associated viral (AAV) and lentiviral (LV) vectors. AAV vectors exist primarily as episomes in host cells and, consequently, are not transferred to both daughter cells upon cell division. As such, they lose efficacy over a relatively short period of time and require repeated administration. While AAV therapies are promising, gene delivery can infrequently result in genotoxicity, and repeated dosing can induce neutralizing antibody production and potentially fatal immune responses.41, 42, 43
In contrast to AAV, LV vectors are stably integrated into the host cell genome and transferred to both daughter cells during cell division, thus alleviating the need for repeated administration.1 The livers of mice in a model of HT1 were repopulated and the metabolic function was restored in transduced hepatocytes following administration of an LV vector expressing functional FAH.44 Although not carcinogenic, a primary safety concern associated with the use of LV vectors in gene therapy remains their potential to integrate into sites that regulate oncogene expression and tumorigenesis.1
LNP-mediated transport of mRNA offers an alternate means of transfecting host cells, circumventing some of the side effects, e.g., integration and genotoxicity, associated with viral vectors.45 This approach has tremendous potential to treat a variety of indications, e.g., infectious diseases, personalized cancer therapy, protein replacement therapy, and gene editing.23,24,46 Aside from the report here, the only other study dedicated to mRNA-based HT1 therapy reported treating FAH (−/−) mice with dendrimer-based lipid nanoparticle (mDLNPs)-encapsulated FAH mRNA.47 FAH (−/−) mice denied NTBC-supplemented water were injected i.v. repeatedly with FAH mRNA in mDLNPs every 3 days with 10 μg per mouse per injection (∼0.35 mg/kg). FAH (−/−) mice repeatedly administered FAH mRNA mDLNPs survived the 30-day experimental schedule and did not lose body weight, whereas PBS- and mCherry mDLNPs-treated FAH (−/−) mice lost body weight and had to be sacrificed before the scheduled end of the experiment. Immunohistochemistry analysis demonstrated FAH expression throughout liver sections derived from FAH mDLNP-treated animals specifically.47
Here, NTBC-deprived, Fah-deficient mice injected with FAH mRNA-LNPs exhibited sustained FAH synthesis in liver. An i.v.-administered 1-mg/kg dose showed sustained therapeutic effects for at least 14 days. Subsequent studies proved that this dose could be reduced considerably while remaining within the therapeutic window. Even a 10-fold reduction in dose (i.e., 0.1 mg/kg) injected i.v. resulted in decreased serum SA and TYR levels while preserving animal health. On the other hand, only 1 mg/kg and 0.5 mg/kg doses injected i.m. showed therapeutic protection. These findings served as the basis for designing interval-finding studies. Fah-deficient mice exhibited sustained and functional expression of FAH protein in liver when injected repeatedly i.v. or i.m. with 1 mg/kg every 14 days. FAH mRNA LNP-treated mice readily survived the maximum 57-day experimental period without exhibiting signs of weight loss despite markedly lower FAH protein levels compared with WT mice. While i.v. administration produced the greatest decrease in toxic metabolites, FAH mRNA LNP administered i.m. also generated a marked decrease in metabolic markers. These findings indicate that relatively little FAH activity is required to achieve a therapeutic effect and rescue Fah-deficient mice from body weight loss and early death. Remarkably, there was no indication of liver injury even at very low enzyme levels. Serum ALT, AST, AP, and CK levels were equivalent in NTBC- and FAH mRNA-treated, Fah-deficient mice, and significantly less than quantified in sera obtained from the NTBC−PBS+ group.
The accumulation of SA is unique to an FAH deficiency; its presence is diagnostic for HT1.48,49 It is notable that serum SA levels in Fah-deficient mice administered FAH mRNA LNP were equivalent to those found in WT mice. TYR levels in the sera of FAH mRNA LNP-treated, Fah-deficient mice were significantly less than those found in mice fed an NTBC-supplemented diet. FAH mRNA LNP treatment did not significantly alter or negatively affect the levels of other metabolites relative to those found in WT animals. Taken together, these findings suggest that an FAH mRNA LNP-based approach to treating HT1 offers a distinct advantage over SOC, which includes oral NTBC supplementation. NTBC supplementation does not reduce serum TYR levels or influence the imbalance of other metabolites. Patients administered NTBC must adhere to a diet low in TYR and phenylalanine. The mRNA-based approach outlined here conveys the immense benefit of reducing not only SA but also TYR levels to an approximately normal physiological state, thus potentially preventing the accompanying neurocognitive decline frequently observed in tyrosinemia patients.
In summary, from our pre-clinical studies in a mouse model mimicking human HT1, we conclude that (1) very low amounts of FAH expression in the liver are sufficient to rescue Fah-deficient mice from body weight loss; (2) repeated i.v. injections of FAH mRNA-LNPs provide a valuable therapeutic approach; (3) repeated i.m. injections of FAH mRNA-LNPs lead to transport of FAH mRNA-loaded LNPs to the liver via the blood stream, resulting in liver FAH protein synthesis in a sufficient amount to rescue Fah-deficient mice from body weight loss; (4) our mRNA-based therapy shows superiority in TYR-lowering potential to physiological levels, compared with NTBC supplementation; (5) therapeutic efficacy is sustained upon dose lowering and interval extension. Notably, serum SA (diagnostic for HT1) and TYR levels were reduced to concentrations well below those found in NTBC-supplemented animals, suggesting that no additional dietary measures might be needed to enormously raise the life quality of an FAH mRNA LNP-treated patient. Remarkably, FAH mRNA LNP doses as low as 0.5 mg/kg administered i.m. periodically over a 20-day period sustained the body weight of Fah-deficient mice on an NTBC-free diet and drastically reduced serum concentrations of TYR as well as SA. These findings provide the potential basis for clinical application of mRNA technology to HT1 treatment.
Methods
Animal model
An Fah-deficient mouse model (Fah1R Tyrc/RJ) that mimics human HT1 was available upon cryorecovery (Jackson Laboratory, Bar Harbor, ME, USA; JAX stock number 018129; Allele synonyms Fah5981SB, Fah5961SB, Fah5981SB).11 This chemically induced (ENU) mouse mutant provides a model for severe tyrosinemia type 1. Neonatal pups without intervention die within 24 h of birth. The mutation is a G-to-A transition at the last base of exon 7 leading to the splicing of exon 6 to exon 8, resulting in a transcript that lacks exon 7. The absence of exon 7 in the transcript results in a frameshift and subsequently the introduction of a premature stop codon at amino acid position 303. Heterozygous breeding to expand the colony was initiated, switched to breeding homozygotes once available in sufficient number. All animals used experimentally resulted from mating homozygotes. To avoid early postnatal lethality of Fah-deficient mice, pregnant and nursing females were provided drinking water supplemented with 15 mg/L NTBC; homozygotes were provided water supplemented with 7.5 mg/L NTBC throughout life unless otherwise stated. Both female and male Fah-deficient mice were used at 10–12 weeks of age. All mice received unlimited access to food and water. All animal procedures were approved by the local authorities (Landesuntersuchungsamt Rhineland-Palatinate, G 19-1-080). Ethical approval was granted by the Landesuntersuchungsamt LUA, Koblenz, AK G 19-1-080.
mRNA and LNP encapsulation
An mRNA sequence that encodes human FAH (FAH mRNA) was designed and synthesized in vitro. The mRNA encoded protein was based on the human NCBI reference sequence NM_000137.4, for full-length human FAH mRNA. The mRNA construct was codon optimized to improve translation and half-life of the mRNA. Only the most GC-rich codons were used for each amino acid (as described in Thess et al.50). mRNAs were generated using non-modified nucleotides. Each mRNA contained a CleanCap (TriLink), followed by a 5′ UTR from the human hydroxysteroid 17-beta dehydrogenase 4 gene (HSD17B4), an open reading frame (ORF) encoding human FAH, a 3′ UTR from human proteasome 20S subunit beta 3 gene (PSMB3), and a template-encoded poly(A) sequence. mRNAs were further polyadenylated using A-plus poly(A) polymerase tailing kit according to manufacturer’s recommendation (Biozym). Human FAH mRNA was encapsulated in LNPs for in vivo injections. FAH mRNA was encapsulated in LNPs for in vivo injections. Formulation of mRNAs in LNPs used in this study were performed at Acuitas Therapeutics (Vancouver, Canada) as previously described (Thess et al.50; Pardi et al.36). LNPs are prepared using a self-assembly process in which an aqueous solution of mRNA at pH 4.0 is rapidly mixed with a solution of lipids dissolved in ethanol (see in Maier et al.51). LNPs used in this study contain an ionizable cationic lipid/phosphatidylcholine/cholesterol/PEG-lipid. At blood pH, LNPs exhibit a net neutral surface charge but become positively charged in acidified endosomes following ApoE-mediated endocytosis by hepatocytes in vivo, resulting in endosome disruption and release of mRNA into the cytoplasm (Thess et al.50 and references therein). Aliquots of FAH mRNA-LNPs (1 g/L) were diluted in PBS prior to injection. Physicochemical characterization determined a 93% encapsulation efficiency of FAH mRNA in LNPs, a particle diameter of 70 nm, a zeta potential of −2.08 mV, and a homogeneous size distribution (as judged by a 0.047 polydispersity index).
In vivo imaging
NTBC supplementation was stopped to induce the tyrosinemia phenotype in Fah-deficient mice. After 5 days, cohorts of mice were injected i.v. or i.m. (both tibialis muscles) with PpLuc mRNA-LNPs; Fah-deficient mice injected with PBS served as a negative control. In vivo imaging was performed 6 h after PpLuc mRNA-LNP injection. Ten minutes before image acquisition, 150 μL of luciferin substrate (20 g/L) was injected intraperitoneally (i.p.). After imaging, mice were euthanized.
Single i.v. injection of FAH mRNA-LNPs into Fah-deficient mice
To induce the disease phenotype in Fah-deficient mice, NTBC supplementation was withdrawn 5 days before treatment in all experimental cohorts. Body weight of Fah-deficient mice was determined before the start of injections, during life phase, and at termination. Blood was collected 1 day before treatment to determine pre-treatment metabolite levels, and on day of termination. Three groups of Fah-deficient mice were subjected to the following treatments: (1) one cohort of Fah-deficient mice received NTBC supplementation throughout their lives and injection of PBS (NTBC+PBS+), (2) a second group of Fah-deficient mice stopped NTBC supplementation 5 days before treatment and was injected with PBS on day 0 (NTBC−PBS+), and (3) a third group of Fah-deficient mice stopped NTBC treatment 5 days before treatment and received a single i.v. injection of LNP-formulated therapeutic FAH mRNA (NTBC−RNA+) (dose 1 mg/kg). Fah-deficient mice were terminated on designated days after a single i.v. injection (days 1, 2, and 4), and liver and serum were collected. Control groups (cohorts 1 and 2) were terminated at time point 24 h after single injection.
Repeated i.v. and i.m. injections of FAH mRNA-LNPs into Fah-deficient mice
Similar to the single injection schedule, NTBC supplementation was withdrawn 5 days prior to treatment to induce the disease phenotype. Blood was collected 1 day before treatment (pre-bleeding [PB]), at intermediate bleeding time points 24 h after injections (B1–B3), and on day of termination (T) (Figures 2A and 3A). One cohort of mice received NTBC supplementation throughout their lives, and repeated PBS injections along with the other experimental cohorts. A second group of Fah-deficient mice stopped NTBC supplementation 5 days before start of treatment and received repeated PBS injections. The third group of Fah-deficient mice stopped NTBC treatment 5 days prior treatment and received five i.v. injections of LNP-formulated therapeutic FAH mRNA every 5 days. On day of termination (day 21), final bleeding was performed, and liver and tibialis muscles were harvested for further analysis.
In a second experimental setup, Fah-deficient mice were repeatedly injected i.v. or i.m. to compare administration routes. FAH mRNA-LNPs were administered at a 20-μg dose, irrespective of the weight of Fah-deficient mice for the i.v. route, and 10 μg of FAH mRNA-LNPs were administered into both tibialis muscles to result in a total dose of 20 μg FAH mRNA-LNPs, comparable with the dose of i.v. treated Fah-deficient mice.
In dose-finding studies, lower doses of mRNA-LNPs were administered via repeated i.v. and i.m. route (0.5 mg/kg and 0.1 mg/kg) five times every 5 days (Figure 5A). For interval-finding studies, repeated i.v. and i.m. injections were applied in a 1-week versus 2-week schedule (Figures 6A–6E).
SA, TYR, and amino acid quantitation
To quantify amino acids and acylcarnitines, including SA and TYR, a 4.7-mm disk was punched out of a blank filter card (Whatman 903 paper) and placed in a 96-well filter plate. A 5-μL serum sample was transferred to the disk and dried overnight at room temperature. Samples were prepared with reagents in the MassChrom kit for analysis of amino acids and acylcarnitines (57000F, non-derivatized, Chromsystems Instrument and Chemicals, Graefelfing, Germany) using the following steps: a 150-μL dilution of the internal standards (internal standard, succinylacetone, 1:1 v/v) and 75 μL of the extraction buffer (succinylacetone) were transferred to the disk. The analytes were extracted by incubating for 30 min at 45°C on a thermoshaker (Bio-Rad Laboratories, Feldkirchen, Germany) rotating at 600 rpm. After centrifugation at 3,200 × g for 2 min in a 96-well V-bottom plate, 10 μL of the supernatant was injected into the tandem mass spectrometry (MS/MS) system via flow-injection (FIA-MS/MS). Amino acids, including succinylacetone and acylcarnitines, in serum were quantified by electrospray ionization MS/MS (ESI-MS/MS) using a Quattro TQD triple quadrupole mass spectrometer (Micromass, Manchester, UK) equipped with an electrospray ion source and a Micromass MassLynx data system. Mass transition in positive ionization mode of succinylacetone is 155 > 137, and the corresponding internal standard succinylacetone-13C5 is 160 > 142. The internal standard ensures a precise quantification.
ALT, AST, AP, and CK quantitation
Measurements from serum samples were performed using an Abbott Alinity ci-series module. Reagents and standardized protocols were used according to Abbott’s recommendations.
Western blot analysis
Livers were lysed in 1 mL of RIPA buffer supplemented with Complete Mini protease inhibitors cocktail (Roche). Lysates were diluted and mixed with loading buffer containing β-mercaptoethanol. Samples were heated for 5 min at 95°C, vortexed, centrifuged at 14,000 relative centrifugal force (rcf) at room temperature, and loaded onto gels (10% Criterion TGX Precast Midi Protein Gel, 26 wells, 12-μL samples). Gels were electrophoresed at 100 V for 90 min. Wet transfer was performed at 100 V for 45 min with Mini Trans-Blot (Electrophoretic Transfer Cell/Bio-Rad) in 1× transfer buffer. Membranes were blocked with 1× Tris-buffered saline (TBS) and 5% skim milk in H2O for 45 min at room temperature with shaking. Membranes were washed and incubated for 4 h at room temperature with primary antibodies: polyclonal rabbit anti-FAH (Abcam, Cambridge, UK) or monoclonal mouse anti-β-actin (Abcam), which served as loading control. Membranes were washed again, incubated with secondary antibodies (IRDye 800CW-conjugated goat anti-rabbit IgG; LI-COR Biosciences, Lincoln, NE, USA or IRDye 680RD-conjugated goat anti-mouse IgG, LI-COR) for 1 h at room temperature, followed by washing. An Odyssey CLX imaging system (Odyssey CLx LI-COR) was used for quantitation and analysis.
Histochemical analysis
Formalin-fixed, paraffin-embedded liver tissue was cut in 2-μm slides and stained with hematoxylin & eosin (H&E). Single-cell and grouped necrosis were evaluated microscopically (counts per 15 high power fields). Inflammation was graded from 0 to 3, steatosis was characterized as micro- or macrovesicular concerning lipid droplet size, and amount of lipid droplets was grouped from 0–3 (0, none,; 1, 1%–10%; 2, 11%–50%; 3, >50% with relation to liver parenchyma). As well, fibrosis, liver architecture and the presence of foci was described.
Statistical analysis
Two-tailed Student’s t test; p > 0.05 not significant (ns), ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Acknowledgments
The authors are grateful to Dr. Stephen H. Gregory (Providence, RI, USA) for editing this manuscript. We would like to thank Aylin Odabasi, Charlotte Kogel, Margarida Cardoso-Henriques, and Lisa Endig for their excellent experimental support, and Igor Splawski for critical proof-reading of the manuscript. LNP formulations used in this study were kindly provided by Ying Tam and Paulo Lin (Acuitas Therapeutics, Vancouver, Canada). We would also like to acknowledge Mansure Famian, Katja Hilbert, and Johannes Eckert for their important support with in vivo experiments.
Author contributions
C.W., M.L.C., S.G., F.C., and N.H. designed and planned in vivo experiments. M.L.C. performed in vivo experiments. D.F. assisted with in vivo experiments. M.L.C. and C.W. processed animal samples and tissues. C.W. and M.L.C. analyzed data and performed statistical analyses. C.W. drafted figures and the manuscript. M.L.C. helped with manuscript editing. J.H. and J.G.O. contributed to the acquisition and interpretation of amino acids and acylcarnitine acquisition data. S.W. assisted in experimental design and data analysis. B.K.S. performed histological and immunohistochemical studies. All authors performed critical revision and approved final manuscript text and figures.
Declaration of interests
C.W. and F.C. are salaried employees of CureVac AG, Tübingen, Germany. N.H. was salaried employee of CureVac AG, Tübingen, Germany at the beginning of the project; and is salaried employee of MRM Health, Zwijnaarde, Belgium. M.L.C., D.F., S.W., S.G., B.K.S., J.H., J.G.O., J.B.H., and F.Z. declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.07.006.
Supplemental information
Data availability
The data that support the findings of this study are available from the corresponding author, M.L.C., upon request. The data are not publicly available due to restrictions that could compromise proprietary information.
References
- 1.Thompson W.S., Mondal G., Vanlith C.J., Kaiser R.A., Lillegard J.B. The future of gene-targeted therapy for hereditary tyrosinemia type 1 as a lead indication among the inborn errors of metabolism. Expert Opin. Orphan Drugs. 2020;8:245–256. doi: 10.1080/21678707.2020.1791082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angileri F., Roy V., Morrow G., Scoazec J.Y., Gadot N., Orejuela D., Tanguay R.M. Molecular changes associated with chronic liver damage and neoplastic lesions in a murine model of hereditary tyrosinemia type 1. Biochim. Biophys. Acta. 2015;1852:2603–2617. doi: 10.1016/j.bbadis.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Das A.M. Clinical utility of nitisinone for the treatment of hereditary tyrosinemia type-1 (HT-1) Appl. Clin. Genet. 2017;10:43–48. doi: 10.2147/TACG.S113310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stinton C., Geppert J., Freeman K., Clarke A., Johnson S., Fraser H., Sutcliffe P., Taylor-Phillips S. Newborn screening for tyrosinemia type 1 using succinylacetone - a systematic review of test accuracy. Orphanet J. Rare Dis. 2017;12:48. doi: 10.1186/s13023-017-0599-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindstedt S., Holme E., Lock E.A., Hjalmarson O., Strandvik B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340:813–817. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- 6.Schady D.A., Roy A., Finegold M.J. Liver tumors in children with metabolic disorders. Transl. Pediatr. 2015;4:290–303. doi: 10.3978/j.issn.2224-4336.2015.10.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.García M.I., de la Parra A., Arias C., Arredondo M., Cabello J.F. Long-term cognitive functioning in individuals with tyrosinemia type 1 treated with nitisinone and protein-restricted diet. Mol. Genet. Metab. Rep. 2017;11:12–16. doi: 10.1016/j.ymgmr.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Ginkel W.G., Rodenburg I.L., Harding C.O., Hollak C.E.M., Heiner-Fokkema M.R., van Spronsen F.J. Long-term outcomes and practical considerations in the pharmacological management of tyrosinemia type 1. Paediatr. Drugs. 2019;21:413–426. doi: 10.1007/s40272-019-00364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson C., Ramachandran R., Safdar S., Murphy E., Swayne O., Katz J., Newsome P.N., Geberhiwot T. Severe neurological crisis in adult patients with tyrosinemia type 1. Ann. Clin. Transl. Neurol. 2020;7:1732–1737. doi: 10.1002/acn3.51160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macias I., Laín A., Bernardo-Seisdedos G., Gil D., Gonzalez E., Falcon-Perez J.M., Millet O. Hereditary tyrosinemia type I-associated mutations in fumarylacetoacetate hydrolase reduce the enzyme stability and increase its aggregation rate. J. Biol. Chem. 2019;294:13051–13060. doi: 10.1074/jbc.RA119.009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aponte J.L., Sega G.A., Hauser L.J., Dhar M.S., Withrow C.M., Carpenter D.A., Rinchik E.M., Culiat C.T., Johnson D.K. Point mutations in the murine fumarylacetoacetate hydrolase gene: animal models for the human genetic disorder hereditary tyrosinemia type 1. Proc. Natl. Acad. Sci. USA. 2001;98:641–645. doi: 10.1073/pnas.98.2.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grompe M., Lindstedt S., al-Dhalimy M., Kennaway N.G., Papaconstantinou J., Torres-Ramos C.A., Ou C.N., Finegold M. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat. Genet. 1995;10:453–460. doi: 10.1038/ng0895-453. [DOI] [PubMed] [Google Scholar]
- 13.Paulk N.K., Wursthorn K., Wang Z., Finegold M.J., Kay M.A., Grompe M. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 2010;51:1200–1208. doi: 10.1002/hep.23481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Junge N., Yuan Q., Vu T.H., Krooss S., Bednarski C., Balakrishnan A., Cathomen T., Manns M.P., Baumann U., Sharma A.D., Ott M. Homologous recombination mediates stable Fah gene integration and phenotypic correction in tyrosinaemia mouse-model. World J. Hepatol. 2018;10:277–286. doi: 10.4254/wjh.v10.i2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Held P.K., Olivares E.C., Aguilar C.P., Finegold M., Calos M.P., Grompe M. In vivo correction of murine hereditary tyrosinemia type I by phiC31 integrase-mediated gene delivery. Mol. Ther. 2005;11:399–408. doi: 10.1016/j.ymthe.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Montini E., Held P.K., Noll M., Morcinek N., al-Dhalimy M., Finegold M., Yant S.R., Kay M.A., Grompe M. In vivo correction of murine tyrosinemia type I by DNA-mediated transposition. Mol. Ther. 2002;6:759–769. doi: 10.1006/mthe.2002.0812. [DOI] [PubMed] [Google Scholar]
- 17.Shao Y., Wang L., Guo N., Wang S., Yang L., Li Y., Wang M., Yin S., Han H., Zeng L., et al. Cas9-nickase-mediated genome editing corrects hereditary tyrosinemia in rats. J. Biol. Chem. 2018;293:6883–6892. doi: 10.1074/jbc.RA117.000347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song C.Q., Jiang T., Richter M., Rhym L.H., Koblan L.W., Zafra M.P., Schatoff E.M., Doman J.L., Cao Y., Dow L.E., et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2020;4:125–130. doi: 10.1038/s41551-019-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneller J.L., Lee C.M., Bao G., Venditti C.P. Genome editing for inborn errors of metabolism: advancing towards the clinic. BMC Med. 2017;15:43. doi: 10.1186/s12916-017-0798-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicolas C.T., Kaiser R.A., Hickey R.D., Allen K.L., Du Z., Vanlith C.J., Guthman R.M., Amiot B., Suksanpaisan L., Han B., et al. Ex vivo cell therapy by ectopic hepatocyte transplantation treats the porcine tyrosinemia model of acute liver failure. Mol. Ther. Methods Clin. Dev. 2020;18:738–750. doi: 10.1016/j.omtm.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X., al-Dhalimy M., Lagasse E., Finegold M., Grompe M. Liver repopulation and correction of metabolic liver disease by transplanted adult mouse pancreatic cells. Am. J. Pathol. 2001;158:571–579. doi: 10.1016/S0002-9440(10)63999-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu G., Liu N., Rittelmeyer I., Sharma A.D., Sgodda M., Zaehres H., Bleidissel M., Greber B., Gentile L., Han D.W., et al. Generation of healthy mice from gene-corrected disease-specific induced pluripotent stem cells. PLoS Biol. 2011;9:e1001099. doi: 10.1371/journal.pbio.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kowalski P.S., Rudra A., Miao L., Anderson D.G. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 2019;27:710–728. doi: 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao W., Hou X., Vick O.G., Dong Y. RNA delivery biomaterials for the treatment of genetic and rare diseases. Biomaterials. 2019;217:119291. doi: 10.1016/j.biomaterials.2019.119291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kowalzik F., Schreiner D., Jensen C., Teschner D., Gehring S., Zepp F. mRNA-based vaccines. Vaccines. 2021;9:390. doi: 10.3390/vaccines9040390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witzigmann D., Kulkarni J.A., Leung J., Chen S., Cullis P.R., van der Meel R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020;159:344–363. doi: 10.1016/j.addr.2020.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akinc A., Maier M.A., Manoharan M., Fitzgerald K., Jayaraman M., Barros S., Ansell S., Du X., Hope M.J., Madden T.D., et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019;14:1084–1087. doi: 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- 28.Adams D., Gonzalez-Duarte A., O'Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L., et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 29.Roseman D.S., Khan T., Rajas F., Jun L.S., Asrani K.H., Isaacs C., Farelli J.D., Subramanian R.R. G6PC mRNA therapy positively regulates fasting blood glucose and decreases liver abnormalities in a mouse model of glycogen storage disease 1a. Mol. Ther. 2018;26:814–821. doi: 10.1016/j.ymthe.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X., Yin L., Theisen M., Zhuo J., Siddiqui S., Levy B., Presnyak V., Frassetto A., Milton J., Salerno T., et al. Systemic mRNA therapy for the treatment of Fabry disease: preclinical studies in wild-type mice, Fabry mouse model, and wild-type non-human primates. Am. J. Hum. Genet. 2019;104:625–637. doi: 10.1016/j.ajhg.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang L., Park J.S., Yin L., Laureano R., Jacquinet E., Yang J., Liang S., Frassetto A., Zhuo J., Yan X., et al. Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia. Nat. Commun. 2020;11:5339. doi: 10.1038/s41467-020-19156-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei G., Cao J., Huang P., An P., Badlani D., Vaid K.A., Zhao S., Wang D.Q.H., Zhuo J., Yin L., et al. Synthetic human ABCB4 mRNA therapy rescues severe liver disease phenotype in a BALB/c.Abcb4(-/-) mouse model of PFIC3. J. Hepatol. 2021;74:1416–1428. doi: 10.1016/j.jhep.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.An D., Schneller J.L., Frassetto A., Liang S., Zhu X., Park J.S., Theisen M., Hong S.J., Zhou J., Rajendran R., et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017;21:3548–3558. doi: 10.1016/j.celrep.2017.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Apgar J.F., Tang J.P., Singh P., Balasubramanian N., Burke J., Hodges M.R., Lasaro M.A., Lin L., Millard B.L., Moore K., et al. Quantitative systems pharmacology model of hUGT1A1-modRNA encoding for the UGT1A1 enzyme to treat Crigler-Najjar syndrome type 1. CPT Pharmacometrics Syst. Pharmacol. 2018;7:404–412. doi: 10.1002/psp4.12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hauser S., Poenisch M., Schelling Y., Höflinger P., Schuster S., Teegler A., Betten R., Gustafsson J.Å., Hübener-Schmid J., Schlake T., et al. mRNA as a novel treatment strategy for hereditary spastic paraplegia type 5. Mol. Ther. Methods Clin. Dev. 2019;15:359–370. doi: 10.1016/j.omtm.2019.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pardi N., Tuyishime S., Muramatsu H., Kariko K., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chinsky J.M., Singh R., Ficicioglu C., van Karnebeek C.D.M., Grompe M., Mitchell G., Waisbren S.E., Gucsavas-Calikoglu M., Wasserstein M.P., Coakley K., Scott C.R. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet. Med. 2017;19:1380–1395. doi: 10.1038/gim.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Spronsen F.J., Bijleveld C.M.A., van Maldegem B.T., Wijburg F.A. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro- methylbenzoyl)-1, 3-cyclohexanedione treatment. J. Pediatr. Gastroenterol. Nutr. 2005;40:90–93. doi: 10.1097/00005176-200501000-00017. [DOI] [PubMed] [Google Scholar]
- 39.Holme E., Lindstedt S. Nontransplant treatment of tyrosinemia. Clin. Liver Dis. 2000;4:805–814. doi: 10.1016/s1089-3261(05)70142-2. [DOI] [PubMed] [Google Scholar]
- 40.Simoncelli M., Samson J., Bussières J.F., Lacroix J., Dorais M., Battista R., Perreault S. Cost-consequence analysis of nitisinone for treatment of tyrosinemia type I. Can. J. Hosp. Pharm. 2015;68:210–217. [PMC free article] [PubMed] [Google Scholar]
- 41.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chandler R.J., Sands M.S., Venditti C.P. Recombinant adeno-associated viral integration and genotoxicity: insights from animal models. Hum. Gene Ther. 2017;28:314–322. doi: 10.1089/hum.2017.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mingozzi F., High K.A. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaiser R.A., Nicolas C.T., Allen K.L., Chilton J.A., Du Z., Hickey R.D., Lillegard J.B. Hepatotoxicity and toxicology of in vivo lentiviral vector administration in healthy and liver-Injury mouse models. Hum. Gene Ther. Clin. Dev. 2019;30:57–66. doi: 10.1089/humc.2018.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J., Guo Z., Tian H., Chen X. Production and clinical development of nanoparticles for gene delivery. Mol. Ther. Methods Clin. Dev. 2016;3:16023. doi: 10.1038/mtm.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlake T., Thran M., Fiedler K., Heidenreich R., Petsch B., Fotin-Mleczek M. mRNA: a novel avenue to antibody therapy? Mol. Ther. 2019;27:773–784. doi: 10.1016/j.ymthe.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng Q., Wei T., Jia Y., Farbiak L., Zhou K., Zhang S., Wei Y., Zhu H., Siegwart D.J. Dendrimer-based lipid nanoparticles deliver therapeutic FAH mRNA to normalize liver function and extend survival in a mouse model of hepatorenal tyrosinemia type I. Adv. Mater. 2018;30:e1805308. doi: 10.1002/adma.201805308. [DOI] [PubMed] [Google Scholar]
- 48.Pettit B.R., MacKenzie F., King G.S., Leonard J.V. The antenatal diagnosis and aid to the management of hereditary tyrosinaemia by use of a specific and sensitive GC-MS assay for succinylacetone. J. Inherit. Metab. Dis. 1984;7:135–136. doi: 10.1007/978-94-009-5612-4_42. [DOI] [PubMed] [Google Scholar]
- 49.Grenier A., Lescault A., Laberge C., Gagné R., Mamer O. Detection of succinylacetone and the use of its measurement in mass screening for hereditary tyrosinemia. Clin. Chim. Acta. 1982;123:93–99. doi: 10.1016/0009-8981(82)90117-6. [DOI] [PubMed] [Google Scholar]
- 50.Thess A., Grund S., Mui B.L., Hope M.J., Baumhof P., Fotin-Mleczek M., et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 2015;23:1456–1464. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maier M.A., Jayaraman M., Matsuda S., Liu J., Barros S., Querbes W., Tam Y.K., Ansell S.M., Kumar V., Qin J., et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 2013;21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, M.L.C., upon request. The data are not publicly available due to restrictions that could compromise proprietary information.