

Abstract
Immune checkpoint inhibitors (ICIs) have advanced the field of cancer immunotherapy in patients by sustaining effector immune cell activity within the tumor microenvironment. However, the approach in general is still faced with issues related to ICI response duration/resistance, treatment eligibility, and safety, which indicates a need for further refinements. As immune checkpoint upregulation is inextricably linked to cancer-induced angiogenesis, newer clinical efforts have demonstrated the feasibility of disrupting both tumor-promoting networks to mediate enhanced immune-driven protection. This review focuses on such key evidence stipulating the necessity of co-applying ICI and anti-angiogenic strategies in cancer patients, with particular interest in highlighting newer engineered antibody approaches that may provide theoretically superior multi-pronged and safe therapeutic combinations.
Keywords: anti-angiogenic strategies, immune checkpoints, immune checkpoint inhibitors, bispecific antibodies, tumor angiogenesis
GRAPHICAL ABSTRACT
Images created with Biorender.com.
Introduction
The tumor protective role of immune effector cells (such as cytotoxic CD8+ T cells) has historically been appreciated for some time, although the clinical translation of basic concepts worked out in preclinical models took decades longer to materialize from large-scale clinical trials [1]. Arguably, a larger perception of “success” surrounding cancer immunotherapy followed the appreciation that components of the tumor microenvironment (TME) instigated immunosuppression by engaging immune checkpoints (ICs) expressed by components of the immune system [2]. If the brakes (so to speak) of lymphocytes could be released (through IC inhibitors [ICIs]), anti-tumor T cell activation/proliferation could be sustained during early and late phase priming events in lymphoid tissues and the periphery, respectively, to better engage and destroy malignant cells [3]. Early clinical trials confirmed such suggestions through blocking the ICs CTLA-4 and PD-1 and eventually paved the way for the paradigm shifting immunotherapeutic strategies now commonplace in patients receiving FDA approved ICI-based regimens (as outlined in Figure 1).
Figure 1. Immune checkpoint blockade unleashes effector T cell activities in the tumor microenvironment.
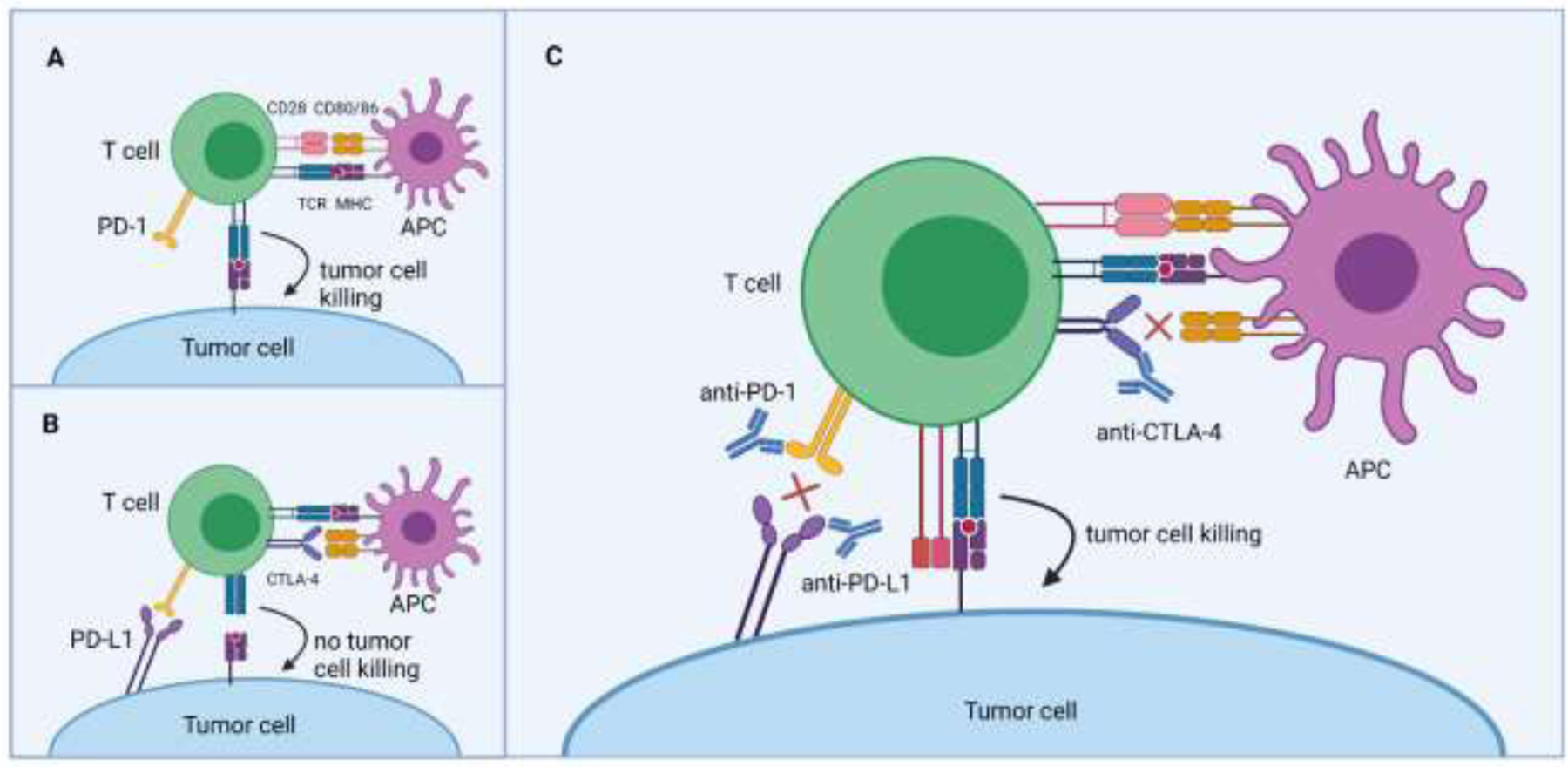
(Panel A) A naïve T cell is first activated in a lymph node by receiving stimulatory signals (e.g., upon TCR:peptide/MHC and CD28:CD80/CD86 ligations) from an antigen-presenting cell. Activated T cells that infiltrate a tumor lesion are then capable of killing cancer cells in an antigen-specific manner. (Panel B) Cognate ligands engaging immune checkpoint molecules such as CTLA-4 or PD-1 on activated T cells will prevent the proper effector functions of these cells. (Panel C) The therapeutic use of antibodies that neutralize CTLA-4, PD-1, or PD-L1 binding will sustain T cell activation in the tumor microenvironment, which improves cytotoxic responses and protection against cancer growth. Abbreviations used: TCR, T cell receptor; MHC, major histocompatibility complex. Images created with Biorender.com.
In this review we focus on the major IC classes that work to subvert immune-based responses against cancer. In particular, although ICIs have provided select patients phenomenal clinical responses, further improvements to this generalized strategy are needed to improve patient eligibility, safety, and long-term efficacy. One such promising area involves optimizing ICIs with tumor vascular-disabling agents. Angiogenesis is certainly a well-known/-established impediment to therapeutics since unrestrained blood vessel development helps establish invasive and immunosuppressive tumor lesions. Yet, cancer angiogenesis appears suited to also fuel IC-based suppression of tumor infiltrating T cells, which opens new possibilities for novel therapeutic interventions.
Immune checkpoints (ICs)
The major IC classes on which FDA-approved therapeutics have focused, thus far, are CTLA-4, PD-1, and PD-L1 [4]. Under general circumstances, these ICs serve to prevent autoimmunity and limit the extent/duration of immune responses following antigen stimulation (e.g., during a viral infection) [5]. While immune regulation is an essential and important host function, expression and engagement of ICs on tumor cells and T cells can actually prevent immune-directed clearance mechanisms by inducing an immunosuppressive state within the TME that helps cancer lesions grow and progress [6].
CTLA-4
CTLA-4 (CD152) is a cell-surface receptor that is expressed on activated T cells. Following T cell receptor interaction with its cognate peptide/MHC complex and co-stimulation through CD28, intracellularly-stored CTLA-4 traffics to the cell surface to further suppress co-stimulation [7]. CTLA-4 binds CD80 (B7–1) and CD86 (B7–2)-bearing antigen presenting cells (APCs), which interrupts CD80/CD86 interaction with T cell-derived CD28. In addition to dampening these extrinsic activation signals, CTLA-4 also exerts inhibitory effects inside the T cell, such as abrogating transcription factors such as NF-kB, NFAT, and AP-1 that help govern T cell activation, proliferation, and cytokine production [8, 9].
PD-1
PD-1 (CD279) regulates T cell activation through interaction with PD-L1 (B7-H1) or PD-L2 (B7-DC). Following ligand binding, PD-1 attenuates T cell activation by recruiting the tyrosine phosphatase SHP2, which dephosphorylates nearby effector proteins (e.g., ZAP70, CD28) to consequently reduce cytokine production and T cell proliferation [10, 11]. CD28 appears to be a preferential target of SHP2, indicating that PD-1 and CTLA-4 exhibit similar molecular mechanisms of T cell attenuation, although their functions are non-redundant based on PD-1/CTLA-4 upregulation during distinct phases of T cell activation (early phase [CTLA-4] vs. late phase [PD-1]), location of cognate ligands (many cell types [PD-L1/L2] vs. mostly APCs [CD80/86]), as well as intracellular signaling pathways that are largely distinct [12, 13]. PD-1 is also crucial for the maintenance of peripheral T cell tolerance, as evidenced by the development of a lupus-like autoimmune condition in mice following deletion of Pdcd1, which encodes PD-1 [14].
PD-L1
PD-L1 is constitutively expressed by various immune cells including T cells, dendritic cells (DCs), B cells, and macrophages along with nonhematopoietic cells such as vascular endothelial cells, epithelial cells, hepatocytes, and astrocytes [15]. PD-L1 expression can also be induced by interferons (especially IFN-γ) that arise as a result of immune activity [16]. Therefore, given the widespread expression profile of PD-L1 outside lymphoid tissues, the PD-1/PD-L1 axis plays a crucial role in systemically regulating CD8+ and Th1 CD4+ T cell function in order to maintain peripheral tolerance against self antigens and prevent autoimmune pathologies [7]. Although PD-L1 is the principal ligand for PD-1, PD-L2 is solely expressed by APCs and also binds PD-1 (albeit with higher affinity than PD-L1) [17]. During a Th2-biased immune response, however, PD-L2 is upregulated and can prominently alter T cell activity. PD-L1 is also capable of binding the CD80 costimulatory ligand, demonstrating some level of duplicative properties between the CTLA-4 and PD-1/PD-L1 pathways [18].
Ultimately IC-based mechanisms are important for maintaining normal immune cell/tissue homeostasis, but these regulatory molecules are frequently co-opted by tumors for evasion of immune system defenses involving, for example, CD8+ T cells [19]. Tumor cells generally circumvent immune detection through various means including MHC class I downregulation, mutations that confer resistance to cell death induction, and recruitment of immunosuppressive stromal cells, with each situation requiring a unique therapeutic intervention to potentially overcome [20]. For effective and efficient immune responses against cancer cells, MHC-presented peptide plays a vital role in engaging antigen-specific T cells. Progressing tumors (65–90%, depending on the type of cancer) tend to lose expression of various MHC class I molecules that allows escape from the destructive power of CD8+ T cells [21]. Spontaneous mutations arising in tumor cells may also confer survival benefits despite the prevalence of immune cells and/or cancer therapies. For example, acquired beta-tubulin mutations can disrupt microtubule stability and garner resistance to chemotherapeutic agents like paclitaxel [22, 23]. The stromal cell composition of the TME can additionally influence the immunosuppressive nature of cancer lesions. Distinct immune cell subtypes such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) are typically recruited to tumor lesions from the surrounding tissue or circulation and help fuel tumor growth and immunosuppression through the expression of pro-angiogenic molecules (e.g., vascular endothelial growth factor [VEGF]) and immunomodulatory compounds (like cytokines [e.g., IL-10, TGF-beta], reactive oxygen species, and IDO) [24]. However, a relatively more recent appreciation for tumor escape is the upregulation of ICs within the TME. CTLA-4 is often expressed by Tregs, exhausted T cells, and tumor cells [25, 26]. PD-L1 is also evident on tumor cells, immune cells (such as MDSCs and TAMs), and vascular endothelial cells [27].
Strategies to mitigate ICs
To counter IC-instigated inhibitory signals in the TME, ICIs (most notably instituted by neutralizing monoclonal antibodies) can permit continued anti-tumor responses [28], despite IC prevalence and sustained antigen exposure [29]. PD-1 blockade diminishes effector T cells binding TME-retained PD-L1/PD-L2 to instead remain functionally active. Neutralizing CTLA-4 antibodies likewise may help instigate tumor rejection by facilitating T cell priming/activation in lymphoid tissues, namely, by preventing T cell exposure to inhibitory signals from APCs [30]. But, it has also been proposed that Tregs may bear the brunt of CTLA-4 blockade, effectively reversing immunosuppression within the TME. Tregs bearing anti-CTLA-4 antibodies appear to be selectively depleted in preclinical studies though an FcγRIV-dependent mechanism involving FcγR-expressing macrophages [31, 32]. The effects of directly blocking PD-L1 are generally expected to mimic that of neutralizing PD-1 on T cells, but system-specific deviations may arise based on the use of distinct ICI antibodies (and their isotypes) [33]. For example, PD-1 inhibition promoted tumor protection in an FcγR-independent manner in a murine colon cancer model while an anti-PD-L1 antibody in the same model augmented anti-tumor responses by eliminating PD-L1-expressing myeloid cells in the TME (via activating FcγRs) [34]. Additionally, cis interactions between PD-L1 and CD80 and/or prevention of PD-L1/PD-L2 engagement could be responsible for improved therapeutic outcomes with PD-1 blockade in certain scenarios as opposed to the strategy of only targeting PD-L1 (although head-to-head comparisons have not been formally studied clinically) [35, 36]. In vitro studies also suggest PD-L1:CD80 cis-heterodimerization prevents T cell-derived CTLA-4 interaction but permits CD28 engagement of CD80, implicating the need for anti-CTLA-4 neutralization if PD-L1 will also be targeted [37].
Clinical experience: CTLA-4 blockade
Despite potential modeling variations, a pivotal moment in the history of cancer immunotherapy came with the approval of the first ICI, ipilimumab (Yervoy®), for treatment of unresectable, late-stage melanoma in 2011. Ipilimumab is a humanized IgG1 antibody that was developed against CTLA-4. In the crucial phase 3 trial, ipilimumab significantly improved both 1-year (45.6% vs. 25.3%) and 2-year (23.5% vs. 13.7%) survival rates when compared to a gp100 vaccine control (outlined in Figure 2) [38]. Aside from extending overall survival (OS), post-trial observations revealed that some patients experienced prolonged durable responses, with 5-year survival rates ranging from 13–25% depending on the trial and ipilimumab-based protocol instituted [39]. Notably, select trial participants who experienced partial tumor regressions eventually transitioned to complete responses (CRs) months to years after receiving the therapy. In some cases, CR rates increased to 17% in patients receiving ipilimumab + IL-2 [39].
Figure 2. Patient overall survival from select clinical trials leading to FDA approved immune checkpoint inhibitor-based regimens.
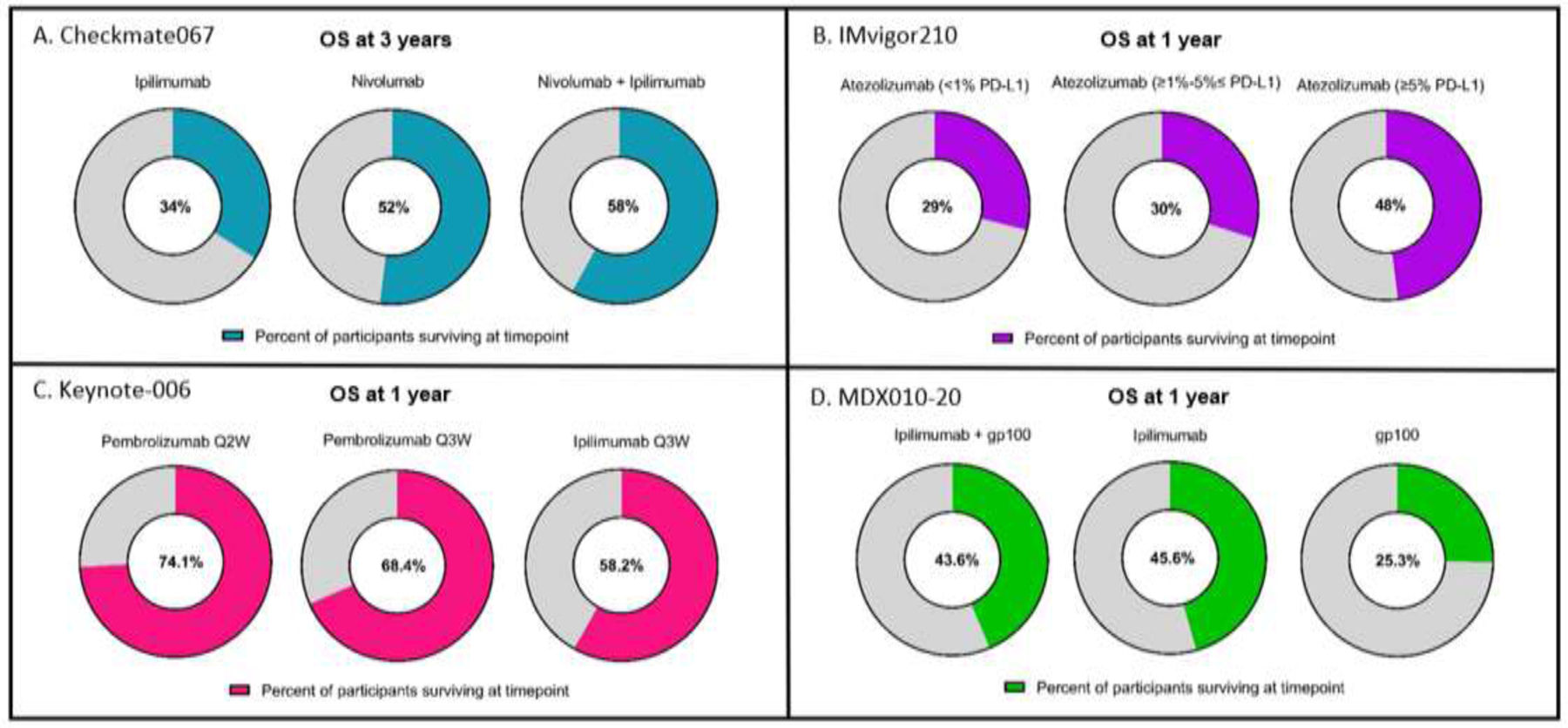
(Panel A) Checkmate067 phase 3 trial (nivolumab + ipilimumab focus) – OS was a primary endpoint. (Panel B) IMvigor210 phase 2 trial (atezolizumab focus) – OS was a secondary endpoint (Panel C) Keynote-006 phase 3 trial (pembrolizumab focus) – OS was a primary endpoint. (Panel D) MDX010–20 phase 3 trial (ipilimumab focus) – OS was a primary endpoint. Abbreviation used: OS, overall survival.
Clinical experience: PD-1 blockade
While ipilimumab is currently the only approved antibody against CTLA-4, several ICIs are in clinical use for the PD-1/PD-L1 IC pathway. PD-1 blocking antibodies include nivolumab (Opdivo®), pembrolizumab (Keytruda®), and cemiplimab (Libtayo®) (summarized in Figure 2 and compiled in Table 1) [4]. Initial FDA approval of nivolumab came after it improved objective response rates (ORRs) over investigator’s choice of chemotherapy from 10.6% to 31.7% in 120 patients (previously progressing on ipilimumab and BRAF inhibition) enrolled in the Checkmate-037 trial [40]. Pembrolizumab, like nivolumab, is a humanized IgG4 antibody against PD-1 [41]. The open-label, phase 1b KEYNOTE-001 trial enrolled 655 patients with advanced melanoma (151 of which were treatment-naïve and 496 individuals having received prior treatments) and dosed pembrolizumab accordingly until the occurrence of disease progression, intolerable adverse events (AEs), or investigator determination of withdrawal [42]. After a median exposure of 5.6 months to the drug, a 5-year follow up revealed an estimated OS rate of 34% in all patients and 41% in treatment-naïve patients and a 5-year estimated progression free survival (PFS) rate of 21% and 29% in these patient cohorts, respectively [43, 44]. Later trials (KEYNOTE-002 and KEYNOTE-006) reported similar durable response rates ranging from 30–40% [45, 46]. Such notable successes helped lead nivolumab and pembrolizumab to approvals for numerous other solid and hematological malignancies [47, 48]. Lastly, cemiplimab was first approved for advanced cutaneous squamous cell carcinoma (CSCC) in 2018 after trial participants experienced an ORR of 47% against metastatic disease [49] and 44% against locally advanced CSCC [50]. The drug has since been approved for advanced basal cell carcinoma [51] and non-small cell lung cancer (NSCLC) [52] – both events occurring in 2021.
Table 1.
Clinical trials helping lead to FDA approved ICI-based regimens
| Drug(s) | Target(s) | Trial (Clinicaltrials.gov registry) | Phase | Cancer indication(s) | Treatment group(s) | Major outcome(s) |
|---|---|---|---|---|---|---|
| Ipilimumab (Yervoy®) | CTLA-4 | MDX010–20 (NCT00094653) | 3 | Unresectable, late-stage melanoma | ipilimumab + gp100 vs. ipilimumab vs. gp100 | Median OS (months): 10 vs. 10.1 vs. 6.4 |
| Nivolumab (Opdivo®) | PD-1 | Checkmate-037 (NCT01721746) | 3 | Unresectable or metastatic melanoma with ICI relapse | nivolumab vs. investigator’s choice of chemotherapy | ORR: 31.7% vs. 10.6%; Median OS (months): 12.3 vs. 8 |
| Pembrolizumab (Keytruda®) | PD-1 | KEYNOTE-006 (NCT01866319) | 3 | Unresectable or metastatic ICI-naïve melanoma | pembrolizumab Q3W vs. pembrolizumab Q2W vs. ipilimumab Q3W | Median PFS (months): 4.1 vs. 5.5 vs. 2.8; OS (at 1 year): 68.4% vs. 74.1% vs. 58.2% |
| Cemiplimab (Libtayo®) | PD-1 | R2810-ONC-1540 (NCT02760498) | 2 | Metastatic and locally advanced CSCC | cemiplimab | ORRs: 44–47% |
| Avelumab (Bavencio®) | PD-L1 | JAVELIN Merkel 200 (NCT02155647) | 2 | Merkel cell carcinoma | avelumab | ORR: 31.8% |
| Durvalumab (Imfinzi®) | PD-L1 | PACIFIC (NCT02125461) | 3 | Advanced and unresectable NSCLC | durvalumab vs. placebo | OS (at 2 years): 66.3% vs. 55.6% |
| Atezolizumab (Tecentriq®) | PD-L1 | IMvigor210 (NCT02951767) | 2 | Locally advanced or metastatic UC that progressed on platinum-containing chemotherapy | atezolizumab in UC with 1% - <5% PD-L1 vs. atezolizumab in UC with ≥5% PD-L1 |
ORR (all subtypes): 19%; ORR (1% - <5% PD-L1): 22%; ORR (≥5% PD-L1): 27% |
| Nivolumab + Ipilimumab | CTLA-4 + PD-1 | Checkmate067 (NCT01844505) | 3 | Previously untreated and advanced melanoma | nivolumab + ipilimumab vs. nivolumab vs. ipilimumab | OS (at 3 years): 58% vs. 52% vs. 34% |
Abbreviations used: CSCC, cutaneous squamous cell carcinoma; ICI, immune checkpoint inhibitor; NSCLC, non-small cell lung cancer; ORR, objective response rate; OS, overall survival; PFS, progression free survival; Q2W, once every 2 weeks; Q3W, once every 3 weeks; UC, urothelial carcinoma
Clinical experience: PD-L1 blockade
PD-L1 inhibitors available for clinical use include avelumab (Bavencio®), durvalumab (Imfinzi®), and atezolizumab (Tecentriq®) (see Figure 2 and Table 1) [4]. Avelumab was approved in 2017 based on the JAVELIN Merkel 200 trial for treatment of metastatic chemotherapy-resistant Merkel cell carcinoma (MCC) after trial patients experienced an ORR of 31.8% [53]. Notably, avelumab is the first FDA approved treatment for metastatic MCC. Durvalumab was initially granted accelerated approval from the FDA in 2017 for patients with PD-L1+ inoperable or metastatic urothelial bladder cancer (mUC) that had progressed following treatment with a platinum-based course of therapy [54] although this was later withdrawn in 2020 following a phase 3 trial that did not meet its primary endpoints [55]. A separate phase 3 study (PACIFIC trial), however, in patients with surgically unresectable stage III NSCLC demonstrated significant improvements in patients treated with durvalumab as compared to placebo in PFS (17.2 months vs. 5.6 months), OS at 2 years (66.3% vs. 55.6%), and time to distant metastases or death (28.3 months vs. 16.2 months) prompting FDA approval in 2018 [56]. Atezolizumab received FDA approval in 2016 for the treatment of locally advanced or mUC in patients who progressed on platinum-containing chemotherapy based on the results of the IMvigor210 trial [57]. The following year, atezolizumab achieved approval for treatment of mUC in patients not eligible for chemotherapy regardless of PD-L1 tumor expression; however, in 2018, the FDA clarified that atezolizumab should only be used in patients [i] with tumor expression of PD-L1 ≥ 5% or [ii] who are not eligible for a platinum-containing chemotherapy regimen regardless of PD-L1 expression [4]. In addition to other indications (such as NSCLC, small cell lung cancer, hepatocellular carcinoma [HCC], and melanoma), atezolizumab received accelerated approval in 2019 for the treatment of metastatic triple-negative breast cancer (TNBC) harboring PD-L1 expression based on results of the phase 3 Impassion 130 study [58]. Briefly, patient cohorts receiving atezolizumab plus nab-paclitaxel demonstrated significantly increased PFS of 7.2 months compared to placebo plus nab-paclitaxel treatment of 5.5-months. Median OS also increased from 17.6 to 21.3 months in atezolizumab-treated patients overall and from 15.5 to 25.0 months in individuals with PD-L1 positive tumors.
Targetable ICs on the horizon
Although anti-CTLA-4 and anti-PD-1/PD-L1 therapies have conveyed impressive clinical results in subsets of patients, research is ongoing to identify other targetable ICs that are capable of sustaining immune-directed attacks against cancer lesions. Recent promising preclinical and early phase clinical results have led to intense interest in assessing other distinct ICs including LAG-3, TIM-3, TIGIT, and VISTA in phase 2 and 3 trials (overviewed in Table 2) [59, 60]. LAG-3 is absent on naïve CD4+ and CD8+ T cells, with expression being induced following antigen stimulation. Interestingly, LAG-3 presence can also fluctuate on other immune cells such as Tregs, NK cells, B cells, and plasmacytoid DCs [59]. The downstream effects of LAG-3 blockade in various studies have been modest [61], but co-expression of PD-1 and LAG-3 on exhausted CD8+T cells has kindled interest in combining PD-1/LAG-3 neutralizing antibodies as a potential means to overcome PD-1 blockade resistance [62–64]. Accordingly, simultaneous disruption of PD-1 and LAG-3 has demonstrated improved effects in reversing T cell exhaustion over either ICI alone in preclinical TNBC, melanoma, and colorectal cancer (CRC) models [65]. TIM-3 expression was initially recognized on CD4+ Th1 and CD8+ T cells, and is classified as an IC based on an ability to mediate T cell exhaustion through binding either Gal-9, CEACAM-1, HMGB1, or phosphatidylserine [66]. In the context of cancer, TIM-3 has been utilized as a marker for dysfunctional subsets of tumor-infiltrating CD8+ T cells that also express PD-1, with dysfunction being further identified by poor effector cytokine secretion [67, 68]. Like LAG-3, TIM-3 is frequently co-expressed with PD-1 on T cells and is observed to be upregulated following blockade of PD-1. TIM-3 is also frequently expressed by Tregs [69]. In mouse models of melanoma and CRC, TIM-3 and PD-1 neutralization resulted in an improved re-establishment of T cell functionality [68, 70–72]. TIGIT is expressed by activated CD8+ and CD4+ T cells, NK cells, Tregs, and follicular helper T cells [59] and interacts with CD155 (PVR) or CD112 (PVRL2), which are commonly present on cancer cells and APCs in the TME [66]. In mouse tumor models, TIGIT impairs T cell proliferation and function by binding CD155 on DCs, triggering an internal signaling cascade that results in reduced production of IL-12 and increased secretion of IL-10 alongside an overwhelmingly tolerogenic phenotype [73, 74]. TIGIT neutralization has demonstrated therapeutic benefits in combination with PD-1/PD-L1 blockade [75]. Additionally, preclinical studies have noted enhanced anti-tumor immune responses when TIGIT inhibition is combined with TIM-3 blockade [76]. VISTA (also known as PD-1H or B7-H5) is a recently identified coinhibitory protein of the CD28/B7 family [77]. With significant homology to both PD-1 and PD-L1, VISTA is commonly expressed on myeloid cells, naïve CD4+/CD8+ T cells, and Foxp3+ CD4+ Tregs [78]. VISTA has been shown to suppress CD4+ and CD8+ T cell proliferation, effector cytokine production, and cytotoxicity, with preclinical studies demonstrating increased anti-tumor benefits following VISTA blockade [79–82]. Yet, it currently remains mechanistically unclear how VISTA’s functions as a ligand and receptor contribute to T cell inhibition [83]. Additionally, VISTA can promote proliferation of tumor-specific CD4+ Foxp3+ Tregs, further contributing to VISTA’s T cell inhibitory functions [84]. In a study of 16 patients with metastatic melanoma initially responding to (but later relapsing from) anti-PD1 or anti-PD1/CTLA-4 therapies, VISTA+ lymphocytes significantly increased in 67% post-disease progression biopsies, potentially implicating the protein as a marker for acquired resistance to common ICIs [85]. However, in other cancer types, VISTA expression has been found to be positively correlated to survival [86, 87]. No doubt, while VISTA may be a promising immunotherapeutic target, further research is required to elucidate its complex roles across various malignancies.
Table 2.
Notable ICI antibody combinations under clinical trial evaluation
| Targets | Drugs | Phase | Cancer indication(s) | Clinicaltrials.gov registry |
|---|---|---|---|---|
| LAG-3 PD-1 |
Favezelimab Pembrolizumab |
3 | CRC | NCT05064059 |
| LAG-3 PD-1 |
Relatlimab Nivolumab |
2/3 | Melanoma | NCT03470922 |
| LAG-3 PD-1 |
LAG525 Spartalizumab |
2 | Advanced solid tumors | NCT03365791 |
| TIM-3 PD-1 |
TSR-022 Dostarlimab |
2 | Melanoma | NCT04139902 |
| TIM-3 PD-1 |
MBG453 PDR001 |
1/2 | Advanced solid tumors | NCT02608268 |
| TIM-3 PD-1 |
BMS-986258 Nivolumab |
1/2 | Advanced solid tumors | NCT03446040 |
| TIGIT PD-1 |
Vibostolimab Pembrolizumab |
3 | NSCLC | NCT04738487 |
| TIGIT PD-1 |
Domvanalimab Zimberelimab |
2 | NSCLC | NCT04262856 |
| VISTA PD-1 |
W0180 Pembrolizumab |
1 | Advanced solid tumors | NCT04564417 |
| TIM-3 PD-L1 |
LY3321367 LY3300054 |
1 | Advanced solid tumors | NCT03099109 |
| TIGIT PD-L1 |
Tiragolumab Atezolizumab |
3 | NSCLC | NCT04294810 |
| TIGIT PD-1 CTLA-4 |
BMS-986207 Nivolumab Ipilimumab |
1/2 | Advanced solid tumors | NCT02913313 |
| LAG-3 TIM-3 PD-1 |
INCAGN02385 INCAGN02390 INCMGA00012 |
1/2 | Melanoma | NCT04370704 |
Abbreviations used: ICI, immune checkpoint inhibitor; CRC, colorectal cancer; NSCLC, non-small cell lung cancer
Clinical obstacles to ICIs
Despite clinical benefits in subsets of patients, resistance to ICIs is a prevailing issue [88]. Some individuals never demonstrate an observable clinical response to ICI therapy (i.e., inherent insensitivity), while other patients exhibit acquired resistance following a period of responsiveness. Broadly, successful ICI interventions are generally proportional to tumor mutational burden (TMB) and, as a by-product, neoantigen processing/presentation that can elicit cytotoxic T lymphocyte activity [89]. For example, treatment-naive patients with high microsatellite instability (MSI-H) or mismatch repair deficiency (dMMR) CRC (i.e., leading to increased TMBs) provided pembrolizumab experienced a more than doubled PFS (16.5 months) over individuals receiving a standard-of-care chemotherapy-containing regimen (8.2 months) in the phase 3 KEYNOTE-177 trial [90]. Yet, in microsatellite stable (MSS) or mismatch repair proficient CRCs (i.e., displaying low TMB), ICIs do not typically impart clinically beneficial endpoints [91]. Such observations also apply to high TMB malignancies such as melanoma and NSCLC, where carcinogen-driven events endow immunogenicity through expression of neoantigens [92, 93]. Tumor-extrinsic mechanisms may also contribute an important role toward ICI resistance. Immunosuppressive cells populated in the TME (such as Tregs, MDSCs, and TAMs) can supply an ever-present cocktail of inhibitory cytokines and receptors that downmodulate the function of CD8+ T cells and APCs, which, in turn, discourages cytotoxic-driven immune responses against cancer cells [94–96].
In cases where patients display an initial promising ICI response following treatment, a substantial segment of individuals (nearly 80%) will not attain durable clinical responses in many cancer types such as those arising in the lung, breast, colon, and prostate [97–99]. The explicit details surrounding acquired resistance are not currently fully understood, but several mechanisms have been proposed. One major culprit involves the upregulation of other ICs to compensate for the loss of CTLA-4 or PD-1/PD-L1 following blockade [100]. An additional evasive avenue for the TME includes the expansion of cells expressing gene variants that institute loss-of-function mutations in JAK family proteins [101] and beta-2-microglobulin (B2M) [102], that ultimately confer cancer cell survival. For example, a small study of whole-exome sequencing in four patients with metastatic melanoma who, after initial tumor regression on pembrolizumab, experienced delayed relapses revealed resistance-associated mutations. One patient’s post-progression tumor biopsy included a frameshift deletion in B2M, resulting in an absence of cell surface-retained MHC class I and inability of CD8+ T cells to recognize the material [103]. Two patients’ biopsies (from the same study) post-ICI progression exhibited inactivation of JAK1 or JAK2, which detrimentally impacts IFN-γ responses that play important roles in nurturing APC function and inhibiting the progression of TME components such as tumor cells and the supportive vasculature [103]. Interestingly, other studies have found epigenetic mechanisms that may similarly institute acquired resistance to ICI treatment [101, 104]. Lastly, cell-mediated resistance mechanisms have also been implicated in resuscitating tumorigenesis following ICI treatment. In a preclinical CRC study employing in vivo imaging to determine the fate of an anti-PD1 antibody after administration, the antibody successfully bound PD1+ tumor-infiltrating CD8+ T cells but was quickly removed from the T cell surface by TAMs. This effect appeared to be driven by Fc-FcyR binding interactions and was recapitulated in vitro using nivolumab and human primary CD8+ T cells and macrophages [105].
Since CTLA-4 and PD-1/PD-L1 neutralizing antibodies impart distinct immunologic effects, ICI cocktails have gained interest as a potential route to discourage resistance and increase/extend clinical responses. Currently, ipilimumab + nivolumab is the only FDA-approved combination for cancer indications that include melanoma, renal cell carcinoma, MSI-H/dMMR metastatic CRC (mCRC), HCC, NSCLC, and malignant pleural mesothelioma [106–109]. Dual checkpoint blockade tends to improve OS rates (58% in the ipilimumab/nivolumab group, 52% in the nivolumab cohort, and 34% in the ipilimumab group in untreated melanoma patients [CheckMate067 phase 3 trial] [110]) and PFS (11.5 months for combination treatment vs. 6.9 months or 2.9 months for nivolumab and ipilimumab monotherapies, respectively, in CheckMate067) where data are available to directly compare single and combined ICI agents. Yet, additive ICI regimens may drastically increase immune-related AEs [111]. In CheckMate067, 59% of ipilimumab/nivolumab patients reported treatment-related grade 3 or 4 AEs compared to individuals treated with nivolumab (21%) or ipilimumab (28%) [110]. To counteract such issues, recent experience supports smaller doses of ipilimumab (alongside other ICIs) that may help abrogate increased toxicities while still conferring enhanced clinical benefits [112, 113]. Newer ICI combinations (as also mentioned above) are also currently being evaluated in clinical trials and may provide additional benefits to patients (see Table 2 for further details).
Intriguingly, stromal cells in the TME including endothelial cells and fibroblasts can drastically influence ICI responsiveness indirectly through various means. As an example, co-administration of tumor-associated fibroblasts expressing FAP in a murine model of MSS CRC promoted immunosuppression and resistance to anti-PD1 treatment by recruiting MDSCs via CCL2 chemotaxis [114]. Tumor stroma exposed to TGFβ (expressed by cancer cells or fibroblasts) could also be implicated in resistance to ICI blockade by antagonizing T cell infiltration and cytotoxic differentiation, which appears relevant to ICI responding/nonresponding mUC patients from the IMvigor210 trial [115, 116]. Notably, in various murine models like CRC and TNBC, TGFβ inhibition significantly increased sensitivity to IC blockade to better promote anti-tumor immunity [116]. Endothelial cells (immersed in a rich milieu of pro-angiogenic factors) also downregulate important adhesion molecules like ICAM1, VCAM1, and E-selectin to prevent circulating leukocytes extravasating into the underlying tumor bed [117]. More directly, endothelial cells can express ICs (e.g. PD-L1/L2) or soluble mediators like VEGF that upregulate ICs on infiltrating T cells [118]. So, overall, the physical and therapeutic constraints imposed by the TME must also be considered within the confines of ICI-based regimens.
Tumor angiogenesis
The TME consists of accessory cells such as immune cells, fibroblasts, and blood and lymphatic vessels that promote cancer growth and progression. Angiogenesis tends to be a crucial TME factor that is paramount to tumor survival and expansion, and, in the absence of blood vessel development, primary (avascular) tumors will usually only grow to a limited size with poor metastatic potential [119]. On the other hand, prolific tumorigenesis requires a sufficient blood vessel network that enables transport of oxygen, nutrients, and waste to and from cancer cells. However, the disordered and rapid growth of cancer cell proliferation ultimately strains vascular function; in turn, leading to intratumoral hypoxia, acidosis, and interstitial pressure, which further facilitates the release of signals that promote angiogenesis [120]. This phenomenon for vessel growth is initially managed by a balance of pro- and anti-angiogenic factors, but upon an “angiogenic switch” being tripped, such equilibrium is disrupted to favor unfettered vascular growth [121]. Ultimately, the TME’s tortuous blood vessel architecture consists of “immature” blood vessels (i.e., endothelial cells with low pericyte coverage and deficient basement membranes) that are leaky and permit the extravasation of tumor cells at distant sites [122, 123] that may eventually propagate macroscopic disease [124, 125].
Given the crucial need for angiogenesis in tumor development (and the strict control of blood vessel genesis/maintenance in healthy adult tissues), irregular characteristics of the tumor vasculature have enticed development of specific therapeutic interventions [126, 127]. Over the past decade alone, the FDA has approved a variety of anti-angiogenic cancer drugs that largely fall into three classes: monoclonal antibodies, small molecule drugs (namely, tyrosine kinase inhibitors [TKIs]), and fusion proteins (summarized in Figure 3 and detailed in Table 3). These vessel-targeting therapies work through various discrete molecular mechanisms, but the end result is usually endothelial cell destruction or normalization (at least in the short-term). Tumor-derived vessel destruction culminates in the abrogation of a necessary infrastructure for tumor tissue survival, depriving cells of needed oxygen/nutrients and expunging metabolic waste. Yet, despite these potential benefits, vessel demolition may elicit evasive immune cell maneuvering (to rearm tumor vascularity, as an example) or potentiate the emergence and dissemination of aggressive hypoxic-derived tumor cell variants [128]. Tissue normalization, on the other hand, results in a culling of “immature” endothelial branches and stabilization of “mature” vessels (i.e., those displaying increased pericyte coverage) that serves to reinstitute vessel integrity and normal aspects of blood flow and oxygenation. Vessel normalization also tends to aid the improved TME perfusion and performance of small molecule drugs/chemotherapeutics or immune interventions, underscoring the enhanced protective index of appropriately designed combinatorial strategies for patients [126, 129, 130].
Figure 3. Vascular-targeting strategies mediate tumor protection by abrogating important tumor angiogenesis pathways.
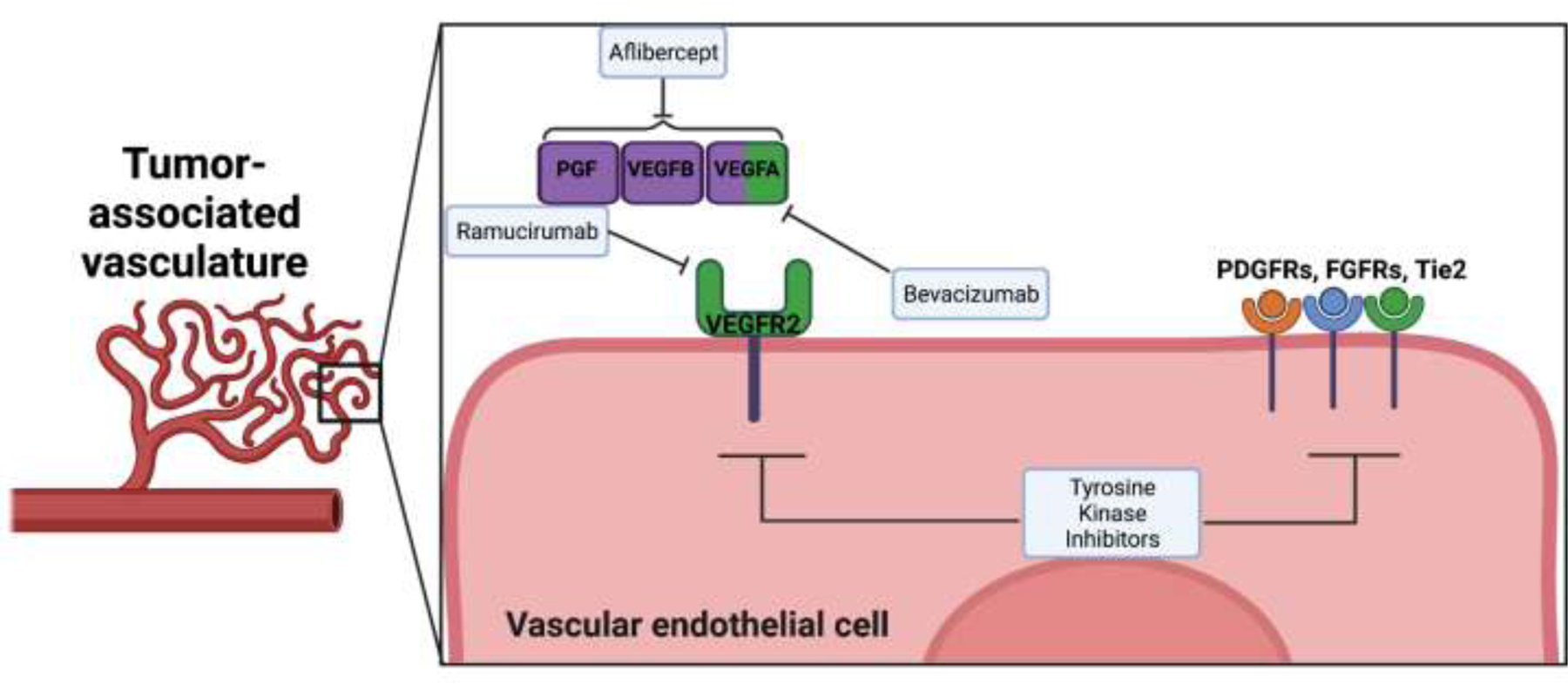
Blood vessel development/maintenance within the tumor microenvironment helps promote tumor cell survival and invasion. Yet, FDA approved monoclonal antibodies (like bevacizumab or ramucirumab), tyrosine kinase inhibitors, and fusion protein (e.g., aflibercept) therapeutics work to specifically interact with vascular components to disrupt crucial cancer angiogenesis pathways. The resulting destruction of tumor-derived vessels deprives cancer cells of a needed support system to grow and progress. Abbreviations used: FGFR, fibroblast growth factor receptors; PDGFR, platelet-derived growth factor receptors; PGF, placental growth factor; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; Images created with Biorender.com.
Table 3.
Pivotal clinical trials leading to FDA approval of select anti-angiogenic therapies for cancer
| Drug | Class (Target[s]) | Phase | Cancers indication(s) | Treatment groups | Major outcome(s) |
|---|---|---|---|---|---|
| Bevacizumab (Avastin®) | mAb (VEGF) | 3 | mCRC | bevacizumab + chemotherapy vs. placebo + chemotherapy | Median OS (months): 20.3 vs. 15.6; Median PFS (months): 10.6 vs. 6.2 |
| Ramucirumab (Cyramza®) | mAb (VEGFR2) | 3 | Gastric cancer | ramucirumab vs. placebo | Median OS (months): 5.2 vs. 3.8; Median PFS (months): 2.1 vs. 1.3 |
| Sorafenib (Nexavar®) | TKI (Multiple) |
3 | RCC | sorafenib vs. placebo | Median PFS (months): 5.5 vs. 2.8 |
| Sunitinib (Sutent®) | TKI (Multiple) |
3 | RCC | sunitinib vs. interferon alfa | Median PFS (months): 11 vs. 5; ORR: 31% vs. 6% |
| Pazopanib (Votrient®) | TKI (Multiple) |
3 | RCC | pazopanib vs. placebo | Median PFS (months): 9.2 vs. 4.2; ORR: 30% vs. 3% |
| Axitinib (Inlyta®) | TKI (Multiple) |
3 | RCC | axitinib vs. sorafenib | Median PFS (months): 6.7 vs. 4.7 |
| Regorafenib (Stivarga®) | TKI (Multiple) |
3 | mCRC | regorafenib vs. placebo | Median OS (months): 6.4 vs. 5.0; Median PFS (months): 2.0 vs. 1.7 |
| Cabozantinib (Cabometyx®) | TKI (Multiple) |
3 | RCC | cabozantinib vs. everolimus | Median PFS (months): 7.4 vs. 3.8; ORR: 21% vs. 5% |
| Lenvatinib (Lenvima®) | TKI (Multiple) |
3 | Differentiated thyroid cancer | lenvatinib vs. placebo | Median PFS (months): 18.3 vs. 3.6; ORR: 64.8% vs. 1.5% |
| Aflibercept (Zaltrap) | Fusion protein (VEGFs, PGF) | 3 | mCRC | aflibercept + chemotherapy vs. placebo + chemotherapy | Median OS (months): 13.5 vs. 12; Median PFS (months): 6.9 vs. 4.7 |
Abbreviations used: mAb, monoclonal antibody; mCRC, metastatic colorectal cancer; ORR, objective response rate; OS, overall survival; PFS, progression free survival; PGF, placental growth factor; RCC, renal cell carcinoma; TKI, tyrosine kinase inhibitor; VEGFs, vascular endothelial growth factors
Anti-angiogenic monoclonal antibodies are designed to bind vascular growth factors or cell-surface receptors that are major drivers of the pro-angiogenic cascade. Of the two antibodies approved, bevacizumab (Avastin®) was first in 2004 to demonstrate efficacy in patients with mCRC [131]. Bevacizumab acts by binding VEGF-A (an extremely potent angiogenesis inducer), which prevents the molecule from interacting with VEGFR2 on vascular endothelial cells, endothelial progenitor cells, and megakaryocytes [126]. Importantly, VEGF-A that is typically found in excess in the TME is neutralized and tumor vascularity ablated (at least in the interim) [132, 133]. Combining this effect with additional targeting strategies serves to increase tumor exposure to, for example, cytotoxic chemotherapies, presumably through a vessel normalization phenomenon [134, 135], that has demonstrated significant increases to both survival and response rates for indications like ovarian, NSCLC, renal cell carcinoma (RCC), and CRCs [136–144]. Ramucirumab (Cyramza®) is a distinct monoclonal anti-VEGFR2 antibody that directly interferes with VEGF binding, creating a similarly normalized vascular environment, wherein, traditional chemotherapies can better elicit anti-tumor effects. The FDA has approved ramucirumab for the treatment of NSCLC, mCRC, HCC, and gastric cancer [145].
The TKI class instigates tumor regressions by blocking important angiogenic-driven activation signals. Those tyrosine kinases implicated with the tumor angiogenic pathway often involve VEGF receptors (VEGFRs), platelet-derived growth factor receptors (PDGFRs), and fibroblast growth factor receptors (FGFRs). When targeted, TKIs interfere with kinase activity and downstream signaling events important for vascular proliferation, migration, and survival [146]. The TKI sorafenib (abrogating TKs such as VEGFRs, PDGFR-β, and FLT-3) was first FDA approved against advanced RCC [147]. Sorafenib’s activity against HCC also yielded an increased OS of 2.8 months (versus the placebo group) and its adoption as a first-line treatment in advanced cases [146, 148]. Sunitinib is another established TKI that is FDA approved for use against gastrointestinal stromal tumors (GIST), pancreatic neuroendocrine tumors, and metastatic RCC (mRCC) [149–151]. Sunitinib acts by interfering with TKs like VEGFRs, PDGFRs, and FLT-3 [152]. Further iteration of this class has been realized in second generation TKIs that demonstrate a more pharmacologically desirable effect (i.e., with reduced toxicities) [134]. Drugs such as regorafenib demonstrate potent anti-tumoral activity in their capacity to target kinases involved in the regulation of tumor angiogenesis (e.g., VEGFRs, FGFRs, PDGFRs, TIE2) and oncogenesis [153, 154]. Such drug development advances have yielded potent clinical activity, culminating in FDA approval for the treatment of mCRC, GIST, and HCC [155]. Uniquely, unlike the antibody class, TKI combination therapies incorporating chemotherapy typically improve PFS but at the expense of exacerbating severe AEs in patients that can lead to treatment discontinuation or death [156, 157].
The fusion protein class is currently represented by one compound – ziv-aflibercept (Zaltrap®). Ziv-aflibercept is composed of the extracellular domains of VEGFR-1 and VEGFR-2 that are fused to the Fc region of IgG1. The drug works as a decoy to immobilize/neutralize important angiogenic cues such as VEGF-A, VEGF-B, and placental growth factor, thereby, halting the pro-angiogenic effects of the VEGFR signaling pathway [158, 159]. Ziv-aflibercept was approved by the FDA in 2012 as a combination therapy with chemotherapy for the treatment of mCRC, after demonstrating a significantly improved OS and PFS in study participants [160, 161].
Co-applying anti-angiogenic and ICI strategies
In general, clinical experience tends to diverge from preclinical observations that anti-angiogenic antibodies induce tumor regressions as monotherapies. Vascular-disruptive small molecule drugs also usually only provide short-term patient benefits [162, 163]. Therefore, the potential for this class of therapeutics (at-large) is likely best realized in rationally designed combination therapies to make use of their unique effects on the TME. Since many pro-angiogenic molecules are associated with immunosuppressive effects, the administration of anti-angiogenic drugs should help sustain anti-tumor immune responses. Indeed, numerous preclinical models have demonstrated that combined anti-angiogenic and immunotherapeutic regimens resulted in normalized tumor vessels that promoted the enhanced infiltration and function of activated immune effector cells [135, 164, 165]. For example, VEGF neutralization by antibodies not only reduces tumor vascularity but also increases antigen-specific T cell infiltration and activity [166–169]. Small molecule drugs like axitinib and sunitinib also restore aberrant characteristics of the TME (e.g., diminishing hypoxia and Treg recruitment) to ultimately improve vaccine-driven T cell cytotoxicities [170, 171]. Interestingly, though anti-angiogenic interventions tend to upregulate ICs on tumor cells such as PD-L1 - presumably due to improved immune effector function within the TME (such as through IFN-γ signaling), their use in tandem with ICIs significantly reduced tumor growth/metastases and prolonged survival in cancer models related to the pancreas, breast, liver, and brain [172–177]. Consequently, given such corroborated evidence from preclinical systems, co-applied ICIs and anti-angiogenic therapies have made considerable progress in clinical trials in recent years, particularly for RCC, HCC and NSCLC (summarized in Table 4).
Table 4.
Examples of combined anti-angiogenic & ICI therapies
| Drugs (ICI/anti-angio) | Trial (Clinicaltrials.gov registry) | Phase | Cancers indication | Treatment groups | Major outcome(s) |
|---|---|---|---|---|---|
| Atezolizumab + bevacizumab | IMbrave150 (NCT03434379) | 3 | HCC | ICI/anti-angio vs. sorafenib | Median PFS (months): 6.8 vs. 4.3; Median OS (at 1-year): 67.2% vs. 54.6% |
| IMpower150 (NCT02366143) | 3 | NSCLC | ICI/anti-angio + carboplatin + paclitaxel vs. bevacizumab + carboplatin + paclitaxel | Median PFS (months): 8.3 vs. 6.8; Median OS (months): 19.2 vs. 14.7 |
|
| IMmotion151 (NCT02420821) | 3 | RCC | ICI/anti-angio vs. sunitinib | Median PFS (months): 11.2 vs. 7.7 |
|
| Avelumab + axitinib | JAVELIN Renal 101 (NCT02684006) | 3 | RCC | ICI/anti-angio vs. sunitinib | Median PFS (months): 13.8 vs. 8.4 |
| Pembrolizumab + axitinib | KEYNOTE-426 (NCT02853331) | 3 | mRCC | ICI/anti-angio vs. sunitinib | Median PFS (months): 15.1 vs. 11.1; ORR: 59.3% v. 35.7% |
Abbreviations used: HCC, hepatocellular carcinoma; ICI, immune checkpoint inhibitor; mRCC, metastatic renal cell carcinoma; NSCLC, non-small cell lung cancer; ORR, objective response rate; OS, overall survival; PFS, progression free survival; RCC, renal cell carcinoma
In the phase 3 KEYNOTE-426 trial, mRCC patients treated with combined axitinib and pembrolizumab exhibited significantly increased PFS, 47% reduced risk of death, and an ORR of 59.3% compared with 35.7% in the sunitinib monotherapy group [178, 179]. The overall efficacy and toxicity profiles prompted FDA approval of axitinib/pembrolizumab as a first-line strategy for treatment-naïve mRCC patients in 2019 [180]. The phase 3 JAVELIN Renal 101 trial also reported significantly improved median PFS (13.8 months vs. 7 months) in advanced RCC patients taking avelumab plus axitinib compared to sunitinib alone [181, 182], which led the FDA to approve the regimen as a first-line treatment in 2019 [183]. Additionally, a separate phase 3 trial (IMmotion151) incorporating atezolizumab and bevacizumab provided significant increases to PFS (11.2 months vs. 7.7 months) for mRCC patients relative to sunitinib treatment [184, 185].
A variety of combination therapies have also been designed to make use of the observed preclinical relationship between VEGFR2 disruption and PD-L1 upregulation in HCC as a result of TME-produced IFN-γ [176, 186]. One such combination therapy involving lenvatinib (a multi-kinase angiogenesis inhibitor [e.g., targeting VEGFRs, FGFRs, and PDGFRα]) and pembrolizumab in a phase 1b evaluation demonstrated significantly improved ORRs while maintaining an acceptable safety profile in individuals with unresectable HCC [187, 188]. These results prompted the LEAP-002 phase 3 trial (NCT03713593) to evaluate lenvatinib/pembrolizumab as a viable first-line option for unresectable HCC [189] although the regimen achieved FDA approval in 2021 for mRCC [190]. Cabozantinib (e.g., blocking VEGFRs, FLT-3, and TIE2) represents another distinct kinase inhibitor with immunomodulatory potential and has been previously approved as a monotherapy against sorafenib-refractory HCC [191]. Accordingly, cabozantinib is currently being evaluated in combination with ICIs like atezolizumab in the phase 3 COSMIC-312 trial (NCT03755791) following early trial successes [192]. Lastly, a major advance for unresectable HCC came with the recent phase 3 IMbrave150 trial that demonstrated improved median PFS (6.8 months vs. 4.3 months) and OS at 1-year (67.2% vs. 54.6%) with acceptable safety profiles compared to the standard sorafenib treatment [193]. Atezolizumab/bevacizumab was approved by the FDA in 2020 as a first-line therapy for advanced HCC [194].
The treatment of NSCLC has similarly benefited from the application of combined anti-angiogenic/ICI therapeutic strategies. The Impower150 phase 3 trial comparing combined therapeutic strategies. The Impower150 phase 3 trial comparing combined atezolizumab/bevacizumab/carboplatin/paclitaxel (ABCP) to bevacizumab/carboplatin/paclitaxel demonstrated significantly increased median PFS (8.3 months vs. 6.8 months) and OS (19.2 months vs. 14.7 months) with comparable AE rates [195]. These clinical endpoints were found to be significant regardless of PD-L1 expression and EGFR or ALK genetic mutation status [196]. As a result, the ABCP combination was approved by the FDA in 2018 as first-line treatment for patients with metastatic non-squamous NSCLC.
Yet, combination therapies are not without their shortcomings nor created equal. The endogenous/acquired resistance mechanisms in the TME (as explained earlier) can predispose individuals to inefficacious therapies while other patients may suffer from significant AEs as a result of drug combinations. Although such problems could be lessened/mitigated by the use of predictive circulating biomarkers and/or TME-based pharmacogenomics, they cannot entirely safeguard the patient experience [197, 198]. This has proven beneficial in the administration of atezolizumab and bevacizumab in comparison to sunitinib monotherapy (i.e., IMmotion151 trial), wherein, a significant increase in PFS was observed in the sarcomatoid variant of mRCC and PD-L1 expressing tumors [168, 184]. On the contrary, treatment with the TKI pazopanib (that is approved as a single-agent for RCC) alongside nivolumab or pembrolizumab was found to be associated with significant hepatoxicity in mRCC patients and not deemed safe for further study [199]. The addition of axitinib/pembrolizumab has also fostered high grade 3 and 4 elevations of hepatic enzymes without any attributable mechanistic causes (KEYNOTE-426 trial) [184, 199]. Additionally, combinations with ICI antibodies can even present problems such as in the case of dual PD-1/CTLA-4 blockade for melanoma patients where over 50% of individuals experienced a significant immune-related AE [200]. Certainly the bulk of such toxicities may be manageable in the short-term (through, for example, clinically inducing immune suppression), but longer lasting immune-mediated issues could arise in the end [201].
Sustained treatment efficacy of any multipronged regimen represents another towering challenge to the patient. Although this problem is inherent to oncology in general, the improvements experienced with many of the “successful” combination treatments described above (evidenced by enhanced PFS and OS metrics) does not typically translate into durable patient responses for a majority of individuals. Optimistically, many of the common challenges that face immunotherapeutic/anti-angiogenic therapies can be eventually answered by, for example, a deeper mechanistic understanding of the interplay between components of the TME alongside therapeutic interventions [164, 202]. Yet, a shorter path to action also involves identifying new and novel therapeutic agents that can effectively incite tumor regressions while theoretically circumventing known impediments to lasting protection. The appropriation of bispecific antibodies (bsAbs) may represent one such approach to safely optimizing complex anti-angiogenic and ICI combinations in patients. BsAbs retain distinct Fab binding sites directed at different epitopes and are generally classified to be either IgG-or non-IgG-like molecules [203]. Their modular design permits an array of immune effector mechanisms due to the potential interplay between targeted sites. In 2014, the FDA approved blinatumomab (Blincyto®) as the first bispecific T-cell engager, a non-IgG like bsAb for the treatment of Philadelphia chromosome-negative relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Blinatumomab is composed of linked scFv domains that work to incite CD3+ T cells killing against tethered CD19+ tumor cells [204]. However, the common bridged engagement phenomenon of bsAbs is not limited to T cell interactions. An additional FDA-approved bsAb emicizumab (Hemlibra®) treats factor VIII deficiency in hemophilia A by connecting factors IXa and X [205, 206]. BsAb-based therapeutics may also mediate agonist (i.e., mimicking the activity of natural ligands and cofactors) and/or antagonist (i.e., inactivating or blocking receptors/ligands to blunt downstream cellular events) effects [207]. In May 2021, the FDA approved the new bsAb amivantamab (Rybrevant™) for treatment of NSCLC. Amivantamab incites tumor destruction by binding and blocking the function of surface-retained MET and mutant EGFR [208, 209]. These aforementioned approved molecules ultimately help rationalize a burgeoning therapeutic pipeline of over 100 bsAbs in clinical development for cancer [203, 210, 211]. BsAbs may also be uniquely suited to neutralize multiple candidate ICI and/or angiogenic targets more effectively within the TME (and with drastically reduced toxicities) than previous clinical attempts using additive monotherapies (as hypothesized in Figure 4) – although this area is still in its infancy and requires clinical validation.
Figure 4. Potential for bispecific antibodies enhancing anti-angiogenic and ICI strategies for cancer.
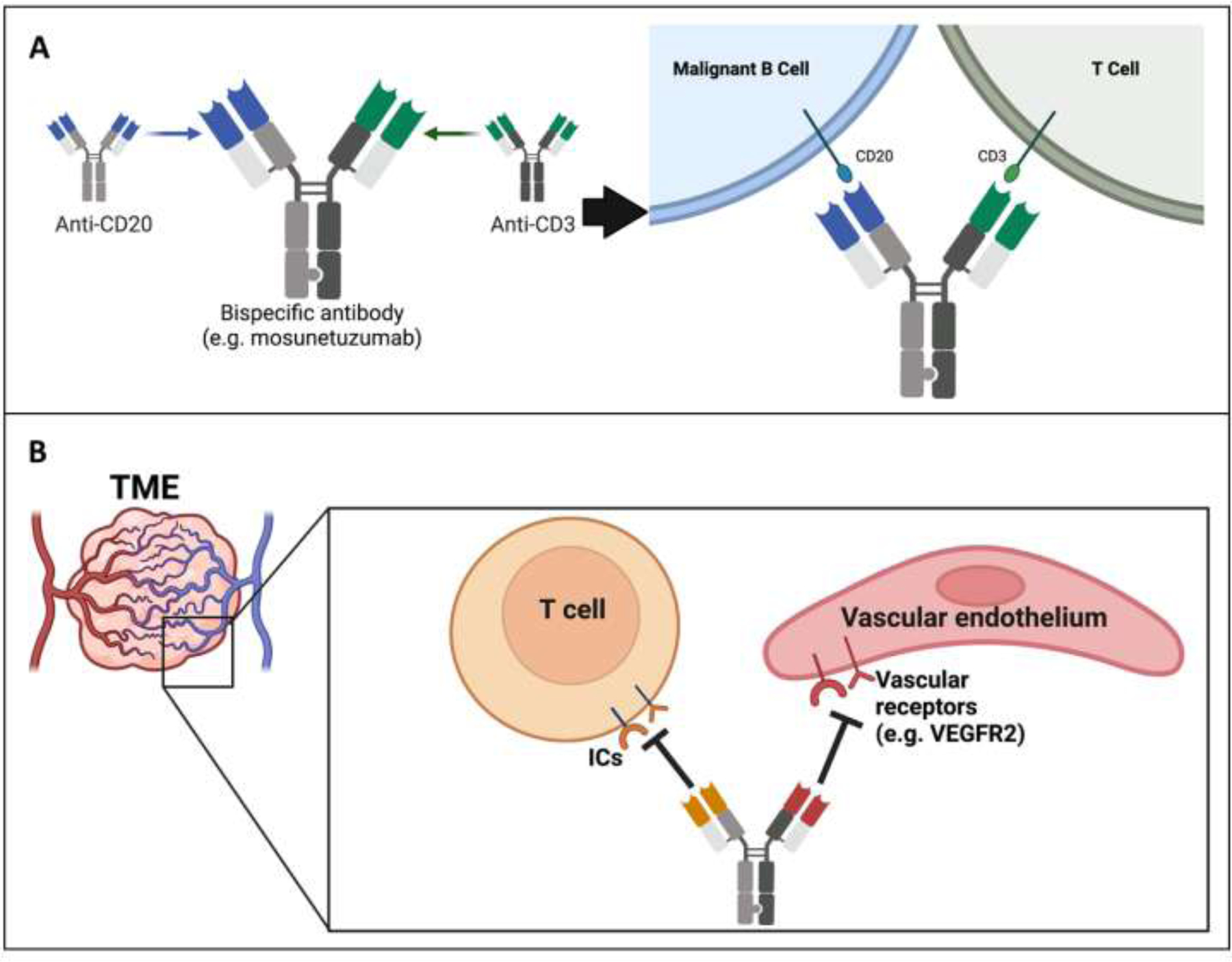
(Panel A) Generation of a standard bsAb format that specifically targets tumor cells and subsequently engages/activates locoregional CD3+ T cells. The provided example is the emerging bsAb mosunetuzumab for lymphoma, where variable domains from disparate antibodies have been attached to an Fc silenced heavy chain. (Panel B) Hypothetical utility of bsAbs to perform complex neutralization schemes within the TME that drives potent immune-driven protection and circumvents cancer evasion strategies. Ultimately, bsAbs could be instituted (along with other targeting agents) to synergistically block ICs and/or vascular targets to better achieve tumor localization that minimizes toxicities in patients. Abbreviations used: BsAb, bispecific antibody; ICs, immune checkpoints; ICI, immune checkpoint inhibitor; TME, tumor microenvironment. Images created with Biorender.com.
Regarding tumor-derived angiogenesis, Boehringer Ingelheim is evaluating a humanized VEGF x Ang-2 bsAb (designated BI 836880) in patients with solid tumors. Previously, a separate murinized VEGF x Ang-2 bsAb demonstrated improved therapeutic efficacy versus either anti-angiogenic antibody alone in transplant tumor models, and combined PD-1 neutralization provided enhanced bsAb activity (since the bsAb upregulated PD-L1 on tumor vessels) [212]. Similarly, in a preclinical brain metastasis model, BI 836880 prolonged animal survival and reduced brain metastasis formation that was linked to vascular disruption [213]. The molecule has also demonstrated manageable safety profiles in early phase clinical trials when used alone (NCT02674152, NCT026895053) and anti-tumor activity in combination with PD-1 blockade in NSCLC patients, with studies ongoing (NCT0346842) [214]. A DLL4 x VEGF bsAb (dilpacimab) is also being developed by AbbVie. In preclinical testing, dilpacimab achieved superior in vivo tumor protection over individual monotherapies through disruption of tumor angiogenesis, and the inclusion of chemotherapy alongside dilpacimab provided enhanced therapeutic benefits [215]. Preliminary phase 1 testing (NCT01946074) supported a good safety profile, with clinical activity occurring in a subset of advanced solid tumor patients [216]. Dilpacimab is currently under phase 2 evaluation in individuals with mCRC (NCT03368859). There is also intense commercial interest in designing bsAbs that engage multiple ICs. Some prominent examples include co-targeting PD-1 x CTLA-4 (Macrogenics, AstraZeneca, Xencor), PD-1 x LAG-3 (Macrogenics, Roche), and PD-1 x PD-L1 (Eli Lilly). As tumor infiltrating lymphocytes tend to express both PD-1 and CTLA-4, combined IC abrogation from a bsAb within the TME is hypothesized to better sustain T cell effector functions, which has been confirmed preclinically [217, 218] and led to several ongoing clinical trials (NCT03761017, NCT04522323, NCT05005728). Similarly, a PD-1 x LAG-3 bsAb would aim to resuscitate exhausted T cells in cancer lesions of patients as currently being clinically investigated (e.g., NCT04082364, NCT04785820). Eli Lilly’s PD-1 x PD-L1 bsAb (designated LY3434172) works to bridge PD-1+ T cells and PD-L1+ tumor cells, thereby, preferentially saturating PD-1 on effector T cells in the TME (opposed to peripheral lymphocytes). Although LY3434172 is under phase 1 evaluation (NCT03936959), in preclinical studies, the bsAb promoted superior in vitro T cell functionality and in vivo anti-tumor responses that were not possible with individual or combined parent antibody treatments [219]. Lastly, a noteworthy bsAb (from the company Akesobio) under clinical trial evaluation outside the United States actually combines angiogenic and IC targeting. This unique tetrameric bsAb engages both VEGF and PD-1 and presupposes that since these immunosuppressive targets are upregulated in the TME, bsAb-directed blockade will enhance anti-tumor activities. Initial phase 1 testing has determined a safe maximum tolerated dose with encouraging ORRs in patients with solid cancers (NCT04047290) [220]. The company is currently recruiting patients for various expansion trials (e.g., NCT04900363). Ultimately, one could envision bsAbs and other standard targeted agents being utilized together to deliver safe and multi-potent regimens that instill lasting immunity by ablating tumor angiogenesis and IC consequences. The inclusion of bsAbs to antagonize and/or stimulate multiple targets while tethering disparate cells in close proximity has numerous clinical applications with a favorable outlook of improved treatment efficacy for the patient.
Conclusions
ICIs have helped position immunotherapy at the forefront of cancer treatment. It is clear that for select patients and malignancies, IC blockade serves to best elicit protective immune effector responses against cancer growth and progression. However, a number of daunting challenges still face the field in order to more effectively and safely provide durable responses in substantial patient segments. Namely, improved strategies most be adopted that overcome the immunosuppressive and vascularized properties of the TME that ingratiates initially intractable or relapsed lesions. Additive therapies (such as FDA approved co-applied anti-vascular drugs and ICIs) have already provided the proof-of-concept of improved efficacy for certain malignancies but can be severely held back by inefficiencies in tumor penetrance and increased adverse events. The emerging precedence for engineered antibodies such as bsAbs provides a potential solution for developing and integrating complex therapeutic regimens in individuals with cancer, and, indeed, relevant molecules are currently in early phases of clinical evaluation. In the coming years, it will be exciting to observe how these novel agents may help further springboard the already favorable outcomes of ICI/anti-angiogenic strategies in patients.
ACKNOWLEDGMENTS
DBL is supported in part by funds from the Cancer Prevention and Research Institute of Texas (CPRIT) (RP210154), NIH (R15 CA215874), DOD (W81XWH-18–1-0293), and Dodge Jones Foundation-Abilene.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors have no competing interests to declare that would negatively influence the work.
REFRENCES
- 1.Kubli SP, Berger T, Araujo DV, Siu LL, Mak TW. Beyond immune checkpoint blockade: emerging immunological strategies. Nat Rev Drug Discov 2021. Epub 2021/03/10. doi: 10.1038/s41573-021-00155-y. [DOI] [PubMed]
- 2.Couzin-Frankel J Breakthrough of the year 2013. Cancer immunotherapy. Science 2013;342(6165):1432–3. Epub 2013/12/21. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015;27(4):450–61. Epub 2015/04/11. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers (Basel) 2020;12(3). Epub 2020/04/05. doi: 10.3390/cancers12030738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol 2018;29(1):71–83. Epub 2017/10/27. doi: 10.1093/annonc/mdx686. [DOI] [PubMed] [Google Scholar]
- 6.Jia Y, Liu L, Shan B. Future of immune checkpoint inhibitors: focus on tumor immune microenvironment. Annals of Translational Medicine 2020;8(17):1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 2018;8(9):1069–86. Epub 2018/08/18. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 8.Fraser JH, Rincon M, McCoy KD, Le Gros G. CTLA4 ligation attenuates AP-1, NFAT and NF-kappaB activity in activated T cells. Eur J Immunol 1999;29(3):838–44. Epub 1999/03/26. doi: . [DOI] [PubMed] [Google Scholar]
- 9.Jutz S, Leitner J, Schmetterer K, Doel-Perez I, Majdic O, Grabmeier-Pfistershammer K, et al. Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: Simultaneous measurement of NF-kappaB, NFAT and AP-1. J Immunol Methods 2016;430:10–20. Epub 2016/01/19. doi: 10.1016/j.jim.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci Adv 2020;6(5):eaay4458. Epub 2020/02/18. doi: 10.1126/sciadv.aay4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Q, Qu J, Zhao M, Xu Q, Sun Y. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol Res 2020;152:104595. Epub 2019/12/16. doi: 10.1016/j.phrs.2019.104595. [DOI] [PubMed] [Google Scholar]
- 12.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017;355(6332):1428–33. Epub 2017/03/11. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagchi S, Yuan R, Engleman EG. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol 2021;16:223–49. Epub 2020/11/17. doi: 10.1146/annurev-pathol-042020-042741. [DOI] [PubMed] [Google Scholar]
- 14.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999;11(2):141–51. Epub 1999/09/15. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 15.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 2007;8(3):239–45. Epub 2007/02/17. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 2017;19(6):1189–201. Epub 2017/05/13. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philips EA, Garcia-Espana A, Tocheva AS, Ahearn IM, Adam KR, Pan R, et al. The structural features that distinguish PD-L2 from PD-L1 emerged in placental mammals. J Biol Chem 2020;295(14):4372–80. Epub 2019/12/29. doi: 10.1074/jbc.AC119.011747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butte MJ, Pena-Cruz V, Kim MJ, Freeman GJ, Sharpe AH. Interaction of human PD-L1 and B7–1. Mol Immunol 2008;45(13):3567–72. Epub 2008/07/01. doi: 10.1016/j.molimm.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12(4):252–64. Epub 2012/03/23. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol 2018;8:86. Epub 2018/04/13. doi: 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol 2016;39:44–51. Epub 2016/01/23. doi: 10.1016/j.coi.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin S, Bhattacharya R, Cabral F. Human mutations that confer paclitaxel resistance. Mol Cancer Ther 2010;9(2):327–35. Epub 2010/01/28. doi: 10.1158/1535-7163.MCT-09-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nami B, Wang Z. Genetics and Expression Profile of the Tubulin Gene Superfamily in Breast Cancer Subtypes and Its Relation to Taxane Resistance. Cancers (Basel) 2018;10(8). Epub 2018/08/22. doi: 10.3390/cancers10080274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haist M, Stege H, Grabbe S, Bros M. The Functional Crosstalk between Myeloid-Derived Suppressor Cells and Regulatory T Cells within the Immunosuppressive Tumor Microenvironment. Cancers (Basel) 2021;13(2). Epub 2021/01/13. doi: 10.3390/cancers13020210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang PY, Guo SS, Zhang Y, Lu JB, Chen QY, Tang LQ, et al. Tumor CTLA-4 overexpression predicts poor survival in patients with nasopharyngeal carcinoma. Oncotarget 2016;7(11):13060–8. Epub 2016/02/27. doi: 10.18632/oncotarget.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salvi S, Fontana V, Boccardo S, Merlo DF, Margallo E, Laurent S, et al. Evaluation of CTLA-4 expression and relevance as a novel prognostic factor in patients with non-small cell lung cancer. Cancer Immunol Immunother 2012;61(9):1463–72. Epub 2012/02/10. doi: 10.1007/s00262-012-1211-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu S, Qin T, Liu Z, Wang J, Jia Y, Feng Y, et al. anlotinib alters tumor immune microenvironment by downregulating PD-L1 expression on vascular endothelial cells. Cell Death Dis 2020;11(5):309. Epub 2020/05/06. doi: 10.1038/s41419-020-2511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marhelava K, Pilch Z, Bajor M, Graczyk-Jarzynka A, Zagozdzon R. Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers (Basel) 2019;11(11). Epub 2019/11/14. doi: 10.3390/cancers11111756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wherry EJ. T cell exhaustion. Nat Immunol 2011;12(6):492–9. Epub 2011/07/09. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 30.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 2011;332(6029):600–3. Epub 2011/04/09. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 2013;210(9):1695–710. Epub 2013/07/31. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du X, Tang F, Liu M, Su J, Zhang Y, Wu W, et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res 2018;28(4):416–32. Epub 2018/02/24. doi: 10.1038/s41422-018-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Song X, Li K, Zhang T. FcgammaR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front Immunol 2019;10:292. Epub 2019/03/14. doi: 10.3389/fimmu.2019.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcgammaRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 2015;28(3):285–95. Epub 2015/09/17. doi: 10.1016/j.ccell.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Brito ABC, Camandaroba MPG, de Lima VCC. Anti-PD1 versus anti-PD-L1 immunotherapy in first-line therapy for advanced non-small cell lung cancer: A systematic review and meta-analysis. Thorac Cancer 2021;12(7):1058–66. Epub 2021/02/16. doi: 10.1111/1759-7714.13867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366(26):2455–65. Epub 2012/06/05. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019;51(6):1059–73 e9. Epub 2019/11/24. doi: 10.1016/j.immuni.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711–23. Epub 2010/06/08. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res 2012;18(7):2039–47. Epub 2012/01/25. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16(4):375–84. Epub 2015/03/22. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 41.Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, et al. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat Struct Mol Biol 2015;22(12):953–8. Epub 2015/11/26. doi: 10.1038/nsmb.3129. [DOI] [PubMed] [Google Scholar]
- 42.Robert C, Ribas A, Hamid O, Daud A, Wolchok JD, Joshua AM, et al. Three-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Journal of Clinical Oncology 2016;34(15_suppl):9503-. doi: 10.1200/JCO.2016.34.15_suppl.9503. [DOI] [Google Scholar]
- 43.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014;384(9948):1109–17. Epub 2014/07/19. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 44.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol 2019;30(4):582–8. Epub 2019/02/05. doi: 10.1093/annonc/mdz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015;372(26):2521–32. Epub 2015/04/22. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 46.Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol 2015;16(8):908–18. Epub 2015/06/28. doi: 10.1016/S1470-2045(15)00083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flynn JP, Gerriets V. Pembrolizumab StatPearls. Treasure Island (FL)2021. [Google Scholar]
- 48.Guo L, Zhang H, Chen B. Nivolumab as Programmed Death-1 (PD-1) Inhibitor for Targeted Immunotherapy in Tumor. J Cancer 2017;8(3):410–6. Epub 2017/03/07. doi: 10.7150/jca.17144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N Engl J Med 2018;379(4):341–51. Epub 2018/06/05. doi: 10.1056/NEJMoa1805131. [DOI] [PubMed] [Google Scholar]
- 50.Migden MR, Khushalani NI, Chang ALS, Lewis KD, Schmults CD, Hernandez-Aya L, et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: results from an open-label, phase 2, single-arm trial. Lancet Oncol 2020;21(2):294–305. Epub 2020/01/19. doi: 10.1016/S1470-2045(19)30728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Romero D Cemiplimab is a new option in BCC. Nat Rev Clin Oncol 2021;18(7):400. Epub 2021/05/27. doi: 10.1038/s41571-021-00528-7. [DOI] [PubMed] [Google Scholar]
- 52.Sezer A, Kilickap S, Gumus M, Bondarenko I, Ozguroglu M, Gogishvili M, et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet 2021;397(10274):592–604. Epub 2021/02/15. doi: 10.1016/S0140-6736(21)00228-2. [DOI] [PubMed] [Google Scholar]
- 53.Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol 2016;17(10):1374–85. Epub 2016/09/07. doi: 10.1016/S1470-2045(16)30364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol 2016;34(26):3119–25. Epub 2016/06/09. doi: 10.1200/JCO.2016.67.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Powles T, van der Heijden MS, Castellano D, Galsky MD, Loriot Y, Petrylak DP, et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol 2020;21(12):1574–88. Epub 2020/09/25. doi: 10.1016/S1470-2045(20)30541-6. [DOI] [PubMed] [Google Scholar]
- 56.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N Engl J Med 2018;379(24):2342–50. Epub 2018/10/04. doi: 10.1056/NEJMoa1809697. [DOI] [PubMed] [Google Scholar]
- 57.Necchi A, Joseph RW, Loriot Y, Hoffman-Censits J, Perez-Gracia JL, Petrylak DP, et al. Atezolizumab in platinum-treated locally advanced or metastatic urothelial carcinoma: post-progression outcomes from the phase II IMvigor210 study. Ann Oncol 2017;28(12):3044–50. Epub 2017/09/28. doi: 10.1093/annonc/mdx518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 2018;379(22):2108–21. Epub 2018/10/23. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 59.Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer 2019;18(1):155. Epub 2019/11/07. doi: 10.1186/s12943-019-1091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang X, Liu G, Li Y, Pan Y. Immune checkpoint: The novel target for antitumor therapy. Genes Dis 2021;8(1):25–37. Epub 2019/12/20. doi: 10.1016/j.gendis.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. LAG-3: from molecular functions to clinical applications. J Immunother Cancer 2020;8(2). Epub 2020/09/16. doi: 10.1136/jitc-2020-001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009;10(1):29–37. Epub 2008/12/02. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol 2009;182(11):6659–69. Epub 2009/05/21. doi: 10.4049/jimmunol.0804211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richter K, Agnellini P, Oxenius A. On the role of the inhibitory receptor LAG-3 in acute and chronic LCMV infection. Int Immunol 2010;22(1):13–23. Epub 2009/11/03. doi: 10.1093/intimm/dxp107. [DOI] [PubMed] [Google Scholar]
- 65.Qi Y, Chen L, Liu Q, Kong X, Fang Y, Wang J. Research Progress Concerning Dual Blockade of Lymphocyte-Activation Gene 3 and Programmed Death-1/Programmed Death-1 Ligand-1 Blockade in Cancer Immunotherapy: Preclinical and Clinical Evidence of This Potentially More Effective Immunotherapy Strategy. Front Immunol 2020;11:563258. Epub 2021/01/26. doi: 10.3389/fimmu.2020.563258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016;44(5):989–1004. Epub 2016/05/19. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8(+) T cells induced by melanoma vaccines. Cancer Res 2014;74(4):1045–55. Epub 2013/12/18. doi: 10.1158/0008-5472.CAN-13-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010;207(10):2187–94. Epub 2010/09/08. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol 2014;44(9):2703–11. Epub 2014/05/20. doi: 10.1002/eji.201344392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu J, Zhang S, Hu Y, Yang Z, Li J, Liu X, et al. Targeting PD-1 and Tim-3 Pathways to Reverse CD8 T-Cell Exhaustion and Enhance Ex Vivo T-Cell Responses to Autologous Dendritic/Tumor Vaccines. J Immunother 2016;39(4):171–80. Epub 2016/04/14. doi: 10.1097/CJI.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 71.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res 2011;71(10):3540–51. Epub 2011/03/25. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 72.Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011;117(17):4501–10. Epub 2011/03/10. doi: 10.1182/blood-2010-10-310425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol 2011;186(3):1338–42. Epub 2011/01/05. doi: 10.4049/jimmunol.1003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer 2020;8(2). Epub 2020/09/10. doi: 10.1136/jitc-2020-000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). Journal of Clinical Oncology 2020;38(15_suppl):9503-. doi: 10.1200/JCO.2020.38.15_suppl.9503. [DOI] [Google Scholar]
- 76.Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest 2015;125(11):4053–62. Epub 2015/09/29. doi: 10.1172/JCI81187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77.Slater BT, Han X, Chen L, Xiong Y. Structural insight into T cell coinhibition by PD-1H (VISTA). Proc Natl Acad Sci U S A 2020;117(3):1648–57. Epub 2020/01/11. doi: 10.1073/pnas.1908711117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang X, Zhang X, Li E, Zhang G, Wang X, Tang T, et al. VISTA: an immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J Hematol Oncol 2020;13(1):83. Epub 2020/07/01. doi: 10.1186/s13045-020-00917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mulati K, Hamanishi J, Matsumura N, Chamoto K, Mise N, Abiko K, et al. VISTA expressed in tumour cells regulates T cell function. Br J Cancer 2019;120(1):115–27. Epub 2018/11/02. doi: 10.1038/s41416-018-0313-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pilones KA, Hensler M, Daviaud C, Kraynak J, Fucikova J, Galluzzi L, et al. Converging focal radiation and immunotherapy in a preclinical model of triple negative breast cancer: contribution of VISTA blockade. Oncoimmunology 2020;9(1):1830524. Epub 2020/11/06. doi: 10.1080/2162402X.2020.1830524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu W, Dong J, Zheng Y, Zhou J, Yuan Y, Ta HM, et al. Immune-Checkpoint Protein VISTA Regulates Antitumor Immunity by Controlling Myeloid Cell-Mediated Inflammation and Immunosuppression. Cancer Immunol Res 2019;7(9):1497–510. Epub 2019/07/26. doi: 10.1158/2326-6066.CIR-18-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J Clin Invest 2014;124(5):1966–75. Epub 2014/04/20. doi: 10.1172/JCI74589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory functions of VISTA. Immunol Rev 2017;276(1):66–79. Epub 2017/03/05. doi: 10.1111/imr.12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu W, Hieu T, Malarkannan S, Wang L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell Mol Immunol 2018;15(5):438–46. Epub 2018/01/30. doi: 10.1038/cmi.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RPM, et al. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol 2017;30(12):1666–76. Epub 2017/08/05. doi: 10.1038/modpathol.2017.89. [DOI] [PubMed] [Google Scholar]
- 86.Zhang M, Pang HJ, Zhao W, Li YF, Yan LX, Dong ZY, et al. VISTA expression associated with CD8 confers a favorable immune microenvironment and better overall survival in hepatocellular carcinoma. BMC Cancer 2018;18(1):511. Epub 2018/05/04. doi: 10.1186/s12885-018-4435-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Loeser H, Kraemer M, Gebauer F, Bruns C, Schroder W, Zander T, et al. The expression of the immune checkpoint regulator VISTA correlates with improved overall survival in pT1/2 tumor stages in esophageal adenocarcinoma. Oncoimmunology 2019;8(5):e1581546. Epub 2019/05/10. doi: 10.1080/2162402X.2019.1581546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang Q, Lei Y, Li X, Guo F, Liu M. A Highlight of the Mechanisms of Immune Checkpoint Blocker Resistance. Front Cell Dev Biol 2020;8:580140. Epub 2020/12/22. doi: 10.3389/fcell.2020.580140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Valero C, Lee M, Hoen D, Zehir A, Berger MF, Seshan VE, et al. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors With 10 or More Mutations per Megabase. JAMA Oncol 2021. Epub 2021/02/19. doi: 10.1001/jamaoncol.2020.7684. [DOI] [PMC free article] [PubMed]
- 90.Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med 2020;383(23):2207–18. Epub 2020/12/03. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 91.Almquist DR, Ahn DH, Bekaii-Saab TS. The Role of Immune Checkpoint Inhibitors in Colorectal Adenocarcinoma. BioDrugs 2020;34(3):349–62. Epub 2020/04/05. doi: 10.1007/s40259-020-00420-3. [DOI] [PubMed] [Google Scholar]
- 92.Kim JY, Kronbichler A, Eisenhut M, Hong SH, van der Vliet HJ, Kang J, et al. Tumor Mutational Burden and Efficacy of Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Cancers (Basel) 2019;11(11). Epub 2019/11/17. doi: 10.3390/cancers11111798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21. Epub 2013/08/16. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 2020;20(3):174–86. Epub 2020/01/26. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klein D The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front Oncol 2018;8:367. Epub 2018/09/27. doi: 10.3389/fonc.2018.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hu-Lieskovan S, Malouf GG, Jacobs I, Chou J, Liu L, Johnson ML. Addressing resistance to immune checkpoint inhibitor therapy: an urgent unmet need. Future Oncol 2021;17(11):1401–39. Epub 2021/01/22. doi: 10.2217/fon-2020-0967. [DOI] [PubMed] [Google Scholar]
- 97.Popovic A, Jaffee EM, Zaidi N. Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J Clin Invest 2018;128(8):3209–18. Epub 2018/08/02. doi: 10.1172/JCI120775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer 2017;5:16. Epub 2017/02/28. doi: 10.1186/s40425-017-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu G, Ren D, Lei X, Shi R, Zhu S, Zhou N, et al. Mutations Associated with No Durable Clinical Benefit to Immune Checkpoint Blockade in Non-S-Cell Lung Cancer. Cancers (Basel) 2021;13(6). Epub 2021/04/04. doi: 10.3390/cancers13061397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2017;6(1):e1249561. Epub 2017/02/16. doi: 10.1080/2162402X.2016.1249561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun 2017;8:15440. Epub 2017/06/01. doi: 10.1038/ncomms15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 2017;8(1):1136. Epub 2017/10/27. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 2016;375(9):819–29. Epub 2016/07/20. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dunn GP, Sheehan KC, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res 2005;65(8):3447–53. Epub 2005/04/19. doi: 10.1158/0008-5472.CAN-04-4316. [DOI] [PubMed] [Google Scholar]
- 105.Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med 2017;9(389). Epub 2017/05/12. doi: 10.1126/scitranslmed.aal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hammers HJ, Plimack ER, Infante JR, Rini BI, McDermott DF, Lewis LD, et al. Safety and Efficacy of Nivolumab in Combination With Ipilimumab in Metastatic Renal Cell Carcinoma: The CheckMate 016 Study. J Clin Oncol 2017;35(34):3851–8. Epub 2017/07/06. doi: 10.1200/JCO.2016.72.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373(1):23–34. Epub 2015/06/02. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18(9):1182–91. Epub 2017/07/25. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372(21):2006–17. Epub 2015/04/22. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2017;377(14):1345–56. Epub 2017/09/12. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang Y, Jin G, Pang Y, Huang Y, Wang W, Zhang H, et al. Comparative Efficacy and Safety of Nivolumab and Nivolumab Plus Ipilimumab in Advanced Cancer: A Systematic Review and Meta-Analysis. Front Pharmacol 2020;11:40. Epub 2020/03/03. doi: 10.3389/fphar.2020.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sharma P, Siefker-Radtke A, de Braud F, Basso U, Calvo E, Bono P, et al. Nivolumab Alone and With Ipilimumab in Previously Treated Metastatic Urothelial Carcinoma: CheckMate 032 Nivolumab 1 mg/kg Plus Ipilimumab 3 mg/kg Expansion Cohort Results. J Clin Oncol 2019;37(19):1608–16. Epub 2019/05/18. doi: 10.1200/JCO.19.00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.D’Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol 2018;19(3):416–26. Epub 2018/01/27. doi: 10.1016/S1470-2045(18)30006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen L, Qiu X, Wang X, He J. FAP positive fibroblasts induce immune checkpoint blockade resistance in colorectal cancer via promoting immunosuppression. Biochem Biophys Res Commun 2017;487(1):8–14. Epub 2017/03/18. doi: 10.1016/j.bbrc.2017.03.039. [DOI] [PubMed] [Google Scholar]
- 115.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554(7693):538–43. Epub 2018/02/15. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 116.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554(7693):544–8. Epub 2018/02/15. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huinen ZR, Huijbers EJM, van Beijnum JR, Nowak-Sliwinska P, Griffioen AW. Anti-angiogenic agents-overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol 2021;18(8):527–40. Epub 2021/04/10. doi: 10.1038/s41571-021-00496-y. [DOI] [PubMed] [Google Scholar]
- 118.Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol 2018;15(5):310–24. Epub 2018/02/13. doi: 10.1038/nrclinonc.2018.9. [DOI] [PubMed] [Google Scholar]
- 119.Gimbrone MA Jr., Leapman SB, Cotran RS, Folkman J. Tumor dormancy in vivo by prevention of neovascularization. J Exp Med 1972;136(2):261–76. Epub 1972/08/01. doi: 10.1084/jem.136.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer 2017;17(8):457–74. Epub 2017/07/15. doi: 10.1038/nrc.2017.51. [DOI] [PubMed] [Google Scholar]
- 121.Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle 2006;5(16):1779–87. Epub 2006/08/26. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- 122.Abhinand CS, Raju R, Soumya SJ, Arya PS, Sudhakaran PR. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal 2016;10(4):347–54. Epub 2016/09/14. doi: 10.1007/s12079-016-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev 2011;37(1):63–74. Epub 2010/06/24. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Paduch R The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell Oncol (Dordr) 2016;39(5):397–410. Epub 2016/04/30. doi: 10.1007/s13402-016-0281-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wong SY, Hynes RO. Lymphatic or hematogenous dissemination: how does a metastatic tumor cell decide? Cell Cycle 2006;5(8):812–7. Epub 2006/04/22. doi: 10.4161/cc.5.8.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Teleanu RI, Chircov C, Grumezescu AM, Teleanu DM. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J Clin Med 2019;9(1). Epub 2020/01/08. doi: 10.3390/jcm9010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Duran CL, Howell DW, Dave JM, Smith RL, Torrie ME, Essner JJ, et al. Molecular Regulation of Sprouting Angiogenesis. Compr Physiol 2017;8(1):153–235. Epub 2018/01/23. doi: 10.1002/cphy.c160048. [DOI] [PubMed] [Google Scholar]
- 128.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009;15(3):220–31. Epub 2009/03/03. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017;544(7649):250–4. Epub 2017/04/04. doi: 10.1038/nature21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zirlik K, Duyster J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol Res Treat 2018;41(4):166–71. Epub 2018/03/22. doi: 10.1159/000488087. [DOI] [PubMed] [Google Scholar]
- 131.Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther 2020;5(1):22. Epub 2020/04/17. doi: 10.1038/s41392-020-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ricciuti B, Foglietta J, Bianconi V, Sahebkar A, Pirro M. Enzymes involved in tumor-driven angiogenesis: A valuable target for anticancer therapy. Semin Cancer Biol 2019;56:87–99. Epub 2017/11/13. doi: 10.1016/j.semcancer.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 133.Marme D Tumor Angiogenesis: A Key Target for Cancer Therapy. Oncol Res Treat 2018;41(4):164. Epub 2018/03/28. doi: 10.1159/000488340. [DOI] [PubMed] [Google Scholar]
- 134.Comunanza V, Bussolino F. Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front Cell Dev Biol 2017;5:101. Epub 2017/12/23. doi: 10.3389/fcell.2017.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 2014;26(5):605–22. Epub 2014/12/18. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350(23):2335–42. Epub 2004/06/04. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 137.Loupakis F, Stein A, Ychou M, Hermann F, Salud A, Osterlund P. A Review of Clinical Studies and Practical Guide for the Administration of Triplet Chemotherapy Regimens with Bevacizumab in First-line Metastatic Colorectal Cancer. Target Oncol 2016;11(3):293–308. Epub 2015/12/22. doi: 10.1007/s11523-015-0400-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bennouna J, Sastre J, Arnold D, Osterlund P, Greil R, Van Cutsem E, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol 2013;14(1):29–37. Epub 2012/11/22. doi: 10.1016/S1470-2045(12)70477-1. [DOI] [PubMed] [Google Scholar]
- 139.Aghajanian C, Goff B, Nycum LR, Wang YV, Husain A, Blank SV. Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol Oncol 2015;139(1):10–6. Epub 2015/08/15. doi: 10.1016/j.ygyno.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Miles DW, Chan A, Dirix LY, Cortes J, Pivot X, Tomczak P, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol 2010;28(20):3239–47. Epub 2010/05/26. doi: 10.1200/JCO.2008.21.6457. [DOI] [PubMed] [Google Scholar]
- 141.Gray R, Bhattacharya S, Bowden C, Miller K, Comis RL. Independent review of E2100: a phase III trial of bevacizumab plus paclitaxel versus paclitaxel in women with metastatic breast cancer. J Clin Oncol 2009;27(30):4966–72. Epub 2009/09/02. doi: 10.1200/JCO.2008.21.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Robert NJ, Dieras V, Glaspy J, Brufsky AM, Bondarenko I, Lipatov ON, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol 2011;29(10):1252–60. Epub 2011/03/09. doi: 10.1200/JCO.2010.28.0982. [DOI] [PubMed] [Google Scholar]
- 143.Eskens FALM, Sleijfer S. The use of bevacizumab in colorectal, lung, breast, renal and ovarian cancer: Where does it fit? European Journal of Cancer 2008;44(16):2350–6. doi: 10.1016/j.ejca.2008.07.042. [DOI] [PubMed] [Google Scholar]
- 144.Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med 2011;365(26):2484–96. Epub 2011/12/30. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- 145.Garon EB, Ciuleanu TE, Arrieta O, Prabhash K, Syrigos KN, Goksel T, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet 2014;384(9944):665–73. Epub 2014/06/17. doi: 10.1016/S0140-6736(14)60845-X. [DOI] [PubMed] [Google Scholar]
- 146.Qin S, Li A, Yi M, Yu S, Zhang M, Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol 2019;12(1):27. Epub 2019/03/15. doi: 10.1186/s13045-019-0718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356(2):125–34. Epub 2007/01/12. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 148.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359(4):378–90. Epub 2008/07/25. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 149.Bennasroune A, Gardin A, Aunis D, Cremel G, Hubert P. Tyrosine kinase receptors as attractive targets of cancer therapy. Crit Rev Oncol Hematol 2004;50(1):23–38. Epub 2004/04/20. doi: 10.1016/j.critrevonc.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 150.Rini BI, Hutson TE, Figlin RA, Lechuga MJ, Valota O, Serfass L, et al. Sunitinib in Patients With Metastatic Renal Cell Carcinoma: Clinical Outcome According to International Metastatic Renal Cell Carcinoma Database Consortium Risk Group. Clin Genitourin Cancer 2018;16(4):298–304. Epub 2018/06/02. doi: 10.1016/j.clgc.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schmid TA, Gore ME. Sunitinib in the treatment of metastatic renal cell carcinoma. Ther Adv Urol 2016;8(6):348–71. Epub 2016/12/03. doi: 10.1177/1756287216663979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Motzer RJ, Escudier B, Gannon A, Figlin RA. Sunitinib: Ten Years of Successful Clinical Use and Study in Advanced Renal Cell Carcinoma. Oncologist 2017;22(1):41–52. Epub 2016/11/04. doi: 10.1634/theoncologist.2016-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fan LC, Teng HW, Shiau CW, Tai WT, Hung MH, Yang SH, et al. Regorafenib (Stivarga) pharmacologically targets epithelial-mesenchymal transition in colorectal cancer. Oncotarget 2016;7(39):64136–47. Epub 2016/09/01. doi: 10.18632/oncotarget.11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ettrich TJ, Seufferlein T. Regorafenib. Recent Results Cancer Res 2018;211:45–56. Epub 2018/08/03. doi: 10.1007/978-3-319-91442-8_3. [DOI] [PubMed] [Google Scholar]
- 155.Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):295–302. Epub 2012/11/28. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol 2006;3(1):24–40. Epub 2006/01/13. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 157.Funakoshi T, Latif A, Galsky MD. Safety and efficacy of addition of VEGFR and EGFR-family oral small-molecule tyrosine kinase inhibitors to cytotoxic chemotherapy in solid cancers: a systematic review and meta-analysis of randomized controlled trials. Cancer Treat Rev 2014;40(5):636–47. Epub 2014/03/19. doi: 10.1016/j.ctrv.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 158.Patel A, Sun W. Ziv-aflibercept in metastatic colorectal cancer. Biologics 2014;8:13–25. Epub 2013/12/26. doi: 10.2147/BTT.S39360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Singh SR, Dogra A, Stewart M, Das T, Chhablani J. Intravitreal Ziv-Aflibercept: Clinical Effects and Economic Impact. Asia Pac J Ophthalmol (Phila) 2017;6(6):561–8. Epub 2017/10/04. doi: 10.22608/APO.2017263. [DOI] [PubMed] [Google Scholar]
- 160.Tang PA, Moore MJ. Aflibercept in the treatment of patients with metastatic colorectal cancer: latest findings and interpretations. Therap Adv Gastroenterol 2013;6(6):459–73. Epub 2013/11/02. doi: 10.1177/1756283X13502637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ciombor KK, Berlin J, Chan E. Aflibercept. Clin Cancer Res 2013;19(8):1920–5. Epub 2013/02/28. doi: 10.1158/1078-0432.CCR-12-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet 2016;388(10043):518–29. Epub 2016/02/09. doi: 10.1016/S0140-6736(15)01088-0. [DOI] [PubMed] [Google Scholar]
- 163.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 2011;8(4):210–21. Epub 2011/03/03. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 2018;15(5):325–40. Epub 2018/03/07. doi: 10.1038/nrclinonc.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ciciola P, Cascetta P, Bianco C, Formisano L, Bianco R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. J Clin Med 2020;9(3). Epub 2020/03/07. doi: 10.3390/jcm9030675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res 2010;70(15):6171–80. Epub 2010/07/16. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res 2007;13(13):3951–9. Epub 2007/07/04. doi: 10.1158/1078-0432.CCR-07-0374. [DOI] [PubMed] [Google Scholar]
- 168.Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun 2016;7:12624. Epub 2016/08/31. doi: 10.1038/ncomms12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tao L, Huang G, Shi S, Chen L. Bevacizumab improves the antitumor efficacy of adoptive cytokine-induced killer cells therapy in non-small cell lung cancer models. Med Oncol 2014;31(1):777. Epub 2013/11/26. doi: 10.1007/s12032-013-0777-3. [DOI] [PubMed] [Google Scholar]
- 170.Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, et al. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer 2011;129(9):2158–70. Epub 2010/12/21. doi: 10.1002/ijc.25863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bose A, Lowe DB, Rao A, Storkus WJ. Combined vaccine+axitinib therapy yields superior antitumor efficacy in a murine melanoma model. Melanoma Res 2012;22(3):236–43. Epub 2012/04/17. doi: 10.1097/CMR.0b013e3283538293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhao S, Ren S, Jiang T, Zhu B, Li X, Zhao C, et al. Low-Dose Apatinib Optimizes Tumor Microenvironment and Potentiates Antitumor Effect of PD-1/PD-L1 Blockade in Lung Cancer. Cancer Immunol Res 2019;7(4):630–43. Epub 2019/02/14. doi: 10.1158/2326-6066.CIR-17-0640. [DOI] [PubMed] [Google Scholar]
- 173.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A 2012;109(43):17561–6. Epub 2012/10/10. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ren S, Xiong X, You H, Shen J, Zhou P. The Combination of Immune Checkpoint Blockade and Angiogenesis Inhibitors in the Treatment of Advanced Non-Small Cell Lung Cancer. Front Immunol 2021;12:689132. Epub 2021/06/22. doi: 10.3389/fimmu.2021.689132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med 2017;9(385). Epub 2017/04/14. doi: 10.1126/scitranslmed.aak9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology 2020;71(4):1247–61. Epub 2019/08/06. doi: 10.1002/hep.30889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yasuda S, Sho M, Yamato I, Yoshiji H, Wakatsuki K, Nishiwada S, et al. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin Exp Immunol 2013;172(3):500–6. Epub 2013/04/23. doi: 10.1111/cei.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res 2008;14(20):6674–82. Epub 2008/10/18. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 179.Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019;380(12):1116–27. Epub 2019/02/20. doi: 10.1056/NEJMoa1816714. [DOI] [PubMed] [Google Scholar]
- 180.Chau V, Bilusic M. Pembrolizumab in Combination with Axitinib as First-Line Treatment for Patients with Renal Cell Carcinoma (RCC): Evidence to Date. Cancer Manag Res 2020;12:7321–30. Epub 2020/09/05. doi: 10.2147/CMAR.S216605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Choueiri TK, Motzer RJ, Rini BI, Haanen J, Campbell MT, Venugopal B, et al. Updated efficacy results from the JAVELIN Renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann Oncol 2020;31(8):1030–9. Epub 2020/04/28. doi: 10.1016/j.annonc.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019;380(12):1103–15. Epub 2019/02/20. doi: 10.1056/NEJMoa1816047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.FDA. Approved Drug Products: Labels for BAVENCIO (avelumab): FDA; 2019. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761049s006lbl.pdf.
- 184.Garje R, An J, Greco A, Vaddepally RK, Zakharia Y. The Future of Immunotherapy-Based Combination Therapy in Metastatic Renal Cell Carcinoma. Cancers (Basel) 2020;12(1). Epub 2020/01/16. doi: 10.3390/cancers12010143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019;393(10189):2404–15. Epub 2019/05/14. doi: 10.1016/S0140-6736(19)30723-8. [DOI] [PubMed] [Google Scholar]
- 186.Kimura T, Kato Y, Ozawa Y, Kodama K, Ito J, Ichikawa K, et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1–6 hepatocellular carcinoma model. Cancer Sci 2018;109(12):3993–4002. Epub 2018/11/18. doi: 10.1111/cas.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wu C-J, Hung Y-W, Lee PC, Lee C, Chen MH, Chao Y, et al. Real-world experience of pembrolizumab plus lenvatinib in unresectable hepatocellular carcinoma in Taiwan. Journal of Clinical Oncology 2020;38(15_suppl):e16627–e. doi: 10.1200/JCO.2020.38.15_suppl.e16627. [DOI] [Google Scholar]
- 188.Finn RS, Ikeda M, Zhu AX, Sung MW, Baron AD, Kudo M, et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J Clin Oncol 2020;38(26):2960–70. Epub 2020/07/28. doi: 10.1200/JCO.20.00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Llovet JM, Kudo M, Cheng A-L, Finn RS, Galle PR, Kaneko S, et al. Lenvatinib (len) plus pembrolizumab (pembro) for the first-line treatment of patients (pts) with advanced hepatocellular carcinoma (HCC): Phase 3 LEAP-002 study. Journal of Clinical Oncology 2019;37(15_suppl):TPS4152–TPS. doi: 10.1200/JCO.2019.37.15_suppl.TPS4152. [DOI] [Google Scholar]
- 190.Motzer R, Alekseev B, Rha SY, Porta C, Eto M, Powles T, et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N Engl J Med 2021;384(14):1289–300. Epub 2021/02/23. doi: 10.1056/NEJMoa2035716. [DOI] [PubMed] [Google Scholar]
- 191.Deeks ED. Cabozantinib: A Review in Advanced Hepatocellular Carcinoma. Target Oncol 2019;14(1):107–13. Epub 2019/02/16. doi: 10.1007/s11523-019-00622-y. [DOI] [PubMed] [Google Scholar]
- 192.Kelley RK, J WO, Hazra S, Benzaghou F, Yau T, Cheng AL, et al. Cabozantinib in combination with atezolizumab versus sorafenib in treatment-naive advanced hepatocellular carcinoma: COSMIC-312 Phase III study design. Future Oncol 2020;16(21):1525–36. Epub 2020/06/04. doi: 10.2217/fon-2020-0283. [DOI] [PubMed] [Google Scholar]
- 193.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 2020;382(20):1894–905. Epub 2020/05/14. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 194.Vogel A, Rimassa L, Sun H-C, Abou-Alfa GK, El-Khoueiry AB, Pinato DJ, et al. Clinical value of atezolizumab + bevacizumab for first-line unresectable hepatocellular carcinoma (HCC): A network meta-analysis. Journal of Clinical Oncology 2020;38(15_suppl):4585-. doi: 10.1200/JCO.2020.38.15_suppl.4585. [DOI] [Google Scholar]
- 195.Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med 2019;7(5):387–401. Epub 2019/03/30. doi: 10.1016/S2213-2600(19)30084-0. [DOI] [PubMed] [Google Scholar]
- 196.Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 2018;378(24):2288–301. Epub 2018/06/05. doi: 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 197.Radpour R, Forouharkhou F. Single-cell analysis of tumors: Creating new value for molecular biomarker discovery of cancer stem cells and tumor-infiltrating immune cells. World J Stem Cells 2018;10(11):160–71. doi: 10.4252/wjsc.v10.i11.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Louie AD, Huntington K, Carlsen L, Zhou L, El-Deiry WS. Integrating Molecular Biomarker Inputs Into Development and Use of Clinical Cancer Therapeutics. Front Pharmacol 2021;12:747194. Epub 2021/11/06. doi: 10.3389/fphar.2021.747194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lee CH, Motzer R. Combination VEGFR/immune checkpoint inhibitor therapy: a promising new treatment for renal cell carcinoma. Br J Cancer 2018;119(8):911–2. Epub 2018/10/18. doi: 10.1038/s41416-018-0175-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2019;381(16):1535–46. doi: 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 201.Burton EM, Tawbi HA. Bispecific Antibodies to PD-1 and CTLA4: Doubling Down on T Cells to Decouple Efficacy from Toxicity. Cancer Discov 2021;11(5):1008–10. Epub 2021/05/06. doi: 10.1158/2159-8290.CD-21-0257. [DOI] [PubMed] [Google Scholar]
- 202.Lee WS, Yang H, Chon HJ, Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med 2020;52(9):1475–85. Epub 2020/09/12. doi: 10.1038/s12276-020-00500-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zhang X, Yang Y, Fan D, Xiong D. The development of bispecific antibodies and their applications in tumor immune escape. Exp Hematol Oncol 2017;6:12. Epub 2017/05/05. doi: 10.1186/s40164-017-0072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhao J, Song Y, Liu D. Recent advances on blinatumomab for acute lymphoblastic leukemia. Exp Hematol Oncol 2019;8:28. Epub 2019/11/12. doi: 10.1186/s40164-019-0152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kitazawa T, Igawa T, Sampei Z, Muto A, Kojima T, Soeda T, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med 2012;18(10):1570–4. Epub 2012/10/02. doi: 10.1038/nm.2942. [DOI] [PubMed] [Google Scholar]
- 206.Brinkmann U, Kontermann RE. Bispecific antibodies. Science 2021;372(6545):916–7. Epub 2021/05/29. doi: 10.1126/science.abg1209. [DOI] [PubMed] [Google Scholar]
- 207.Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today 2015;20(7):838–47. Epub 2015/03/03. doi: 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 208.Vijayaraghavan S, Lipfert L, Chevalier K, Bushey BS, Henley B, Lenhart R, et al. Amivantamab (JNJ-61186372), an Fc Enhanced EGFR/cMet Bispecific Antibody, Induces Receptor Downmodulation and Antitumor Activity by Monocyte/Macrophage Trogocytosis. Mol Cancer Ther 2020;19(10):2044–56. Epub 2020/08/05. doi: 10.1158/1535-7163.MCT-20-0071. [DOI] [PubMed] [Google Scholar]
- 209.Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs 2021;13(1):1860476. Epub 2021/01/19. doi: 10.1080/19420862.2020.1860476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yu S, Liu Q, Han X, Qin S, Zhao W, Li A, et al. Development and clinical application of anti-HER2 monoclonal and bispecific antibodies for cancer treatment. Exp Hematol Oncol 2017;6:31. Epub 2017/12/07. doi: 10.1186/s40164-017-0091-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol 2020;17(7):418–34. Epub 2020/04/04. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 212.Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med 2017;9(385). Epub 2017/04/14. doi: 10.1126/scitranslmed.aak9670. [DOI] [PubMed] [Google Scholar]
- 213.Kovalchuk B, Berghoff AS, Karreman MA, Frey K, Piechutta M, Fischer M, et al. Nintedanib and a bi-specific anti-VEGF/Ang2 nanobody selectively prevent brain metastases of lung adenocarcinoma cells. Clin Exp Metastasis 2020;37(6):637–48. Epub 2020/09/13. doi: 10.1007/s10585-020-10055-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Girard N, Wermke M, Barlesi F, Landsteiner HT, Jayadeva G, Alt J, et al. Phase Ib study of BI 836880, a VEGF/Ang2-blocking nanobody, in combination with BI 754091, an anti-PD-1 antibody: Initial results in patients (pts) with advanced non-small cell lung cancer (NSCLC). Journal of Clinical Oncology 2020;38(15_suppl):9566-. doi: 10.1200/JCO.2020.38.15_suppl.9566. [DOI] [Google Scholar]
- 215.Li Y, Hickson JA, Ambrosi DJ, Haasch DL, Foster-Duke KD, Eaton LJ, et al. ABT-165, a Dual Variable Domain Immunoglobulin (DVD-Ig) Targeting DLL4 and VEGF, Demonstrates Superior Efficacy and Favorable Safety Profiles in Preclinical Models. Mol Cancer Ther 2018;17(5):1039–50. Epub 2018/03/30. doi: 10.1158/1535-7163.MCT-17-0800. [DOI] [PubMed] [Google Scholar]
- 216.Gordon MS, Nemunaitis J, Barve M, Wainberg ZA, Hamilton EP, Ramanathan RK, et al. Phase I Open-Label Study Evaluating the Safety, Pharmacokinetics, and Preliminary Efficacy of Dilpacimab in Patients with Advanced Solid Tumors. Mol Cancer Ther 2021;20(10):1988–95. Epub 2021/07/29. doi: 10.1158/1535-7163.MCT-20-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Berezhnoy A, Sumrow BJ, Stahl K, Shah K, Liu D, Li J, et al. Development and Preliminary Clinical Activity of PD-1-Guided CTLA-4 Blocking Bispecific DART Molecule. Cell Rep Med 2020;1(9):100163. Epub 2020/12/31. doi: 10.1016/j.xcrm.2020.100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dovedi SJ, Elder MJ, Yang C, Sitnikova SI, Irving L, Hansen A, et al. Design and Efficacy of a Monovalent Bispecific PD-1/CTLA4 Antibody That Enhances CTLA4 Blockade on PD-1(+) Activated T Cells. Cancer Discov 2021;11(5):1100–17. Epub 2021/01/10. doi: 10.1158/2159-8290.CD-20-1445. [DOI] [PubMed] [Google Scholar]
- 219.Kotanides H, Li Y, Malabunga M, Carpenito C, Eastman SW, Shen Y, et al. Bispecific Targeting of PD-1 and PD-L1 Enhances T-cell Activation and Antitumor Immunity. Cancer Immunol Res 2020;8(10):1300–10. Epub 2020/09/03. doi: 10.1158/2326-6066.CIR-20-0304. [DOI] [PubMed] [Google Scholar]
- 220.Coward J, Mislang ARA, Frentzas S, Lemech CR, Nagrial A, Jin X, et al. Safety and efficacy of AK112, an anti-PD-1/VEGF-A bispecific antibody, in patients with advanced solid tumors in a phase I dose escalation study. Journal of Clinical Oncology 2021;39(15_suppl):2515-. doi: 10.1200/JCO.2021.39.15_suppl.2515. [DOI] [Google Scholar]