

Abstract
A bicuspid aortic valve (BAV) is the most prevalent congenital cardiac anomaly, in which the valve has only two leaflets, instead of the normal three. Patients with a BAV have an increased risk of aneurysm formation and the development of an aortic dissection. Vascular smooth muscle cells in both the non- and dilated aortic wall are characterized by a maturation defect in all BAV patients, as compared to patients with a tricuspid aortic valve, which can contribute to inherent developmental susceptibility. Besides structural abnormalities of the vascular wall, a turbulent blood flow, caused by bicuspid valve geometry, could expedite the pathological process in the aortic wall, leading to aortopathy. Although the risk for aortopathy is significant, not all BAV patients experience (acute) aortic complications in their lifespan, highlighting the complexity of the pathogenetic process. Recent studies have focused on the embryonic development of semilunar valves and the ascending aortic wall. Their findings highlight that a defect in the embryogenesis could not only explain the development of a malformed aortic valve but also the increased risk for ascending aorta and arch pathology. This review presents an overview of the normal and abnormal development of the aortic valve and the aortic wall: a common defect in early embryogenesis causes the development of a BAV and associated aortopathy.
Keywords: Bicuspid aortic valve (BAV), aortopathy, embryology, pathology, neural crest cells
Introduction
Bicuspid aortic valve (BAV) disease is the most common non-syndromic congenital cardiac anomaly. The majority of patients with a BAV experience complications in their clinical course due to valve related abnormalities such as aortic stenosis, aortic regurgitation and infective endocarditis. Moreover, BAV patients have a sharply increased risk for the development of a thoracic aortic aneurysm (1), defined as a permanent pathologic widening of the aorta greater than 1.5 times the normal diameter. Aneurysm formation is a potentially devastating process which is associated with a life-long risk of lethal adverse aortic events, being an acute thoracic aortic dissection (a tear of the inner layer of the aortic wall, causing the inner and middle layer of the aorta to split) or an aortic rupture.
Until now, the only way to detect the presence of a thoracic aortic aneurysm and prevent an acute aortic syndrome is with formal screening of the aortic diameter. Although considered as the only criterion in the aortic surgery guidelines, the aortic diameter as a selection criterion has a very low sensitivity, as the majority of dissections occur at a (sub)normal diameter (2,3), highlighting the importance of intrinsic pathology of the ascending aortic wall in predisposing adverse conditions. Treatment of thoracic aortic aneurysms and dissections (TAADs) therefore, remains a challenge in both the elective, as well as emergency setting. Traditionally, the decision of when and if to operate is based on the balance of expected surgical risk and hazard of aortic rupture. Difficulty arises in elective cases when only the aortic diameter is taken into consideration and, nothing is known about the intrinsic aortic pathology of the individual patient. To be able to predict the risk for aortic complications in the individual patient and design targeted therapy, more insight in the TAADs disease process is warranted.
The embryonic origin of the semilunar valves is closely related to the development of the ascending aorta. Hence, distortion of the early developmental pathways could not only lead to a BAV, but also the associated aortopathy.
Considering the clinical implications, comprehension of the development of the ascending aorta and both normal and abnormal aortic valves is mandatory.
In this review we summarize several aspects of normal and abnormal development of the aortic semilunar valves and the ascending aortic wall. Based on the current developmental knowledge, we propose a unified hypothesis that the increased vulnerability for aortic complications in BAV can be explained by a defect in the early development of the aortic valve and ascending aortic wall.
The normal development of the aortic valve and ascending aortic wall
The cardiovascular system, including the heart, is the primary functional organ to form during organogenesis. During valvulogenesis endocardial cushions are first formed in both the atrioventricular canal and outflow tract, which contribute to the atrioventricular (mitral and tricuspid) and semilunar (aortic and pulmonary) valve leaflets, respectively (4). Development of semilunar valves is a complex process in which three different cardiac progenitor cells play a role, being the neural crest, second heart field and endocardial cushion derived cells (Figure 1) (5). Animal models have shown that the two main facing cushions of the semilunar valves are filled by mesenchymal cells, derived both from the endocardium through epithelium-mesenchymal transition, and later arriving neural crest cells. The third leaflet has a more second heart field-related etiology, as demonstrated in a murine endothelial nitric oxide (eNOS) knock out model for bicuspidy (6).
Figure 1.
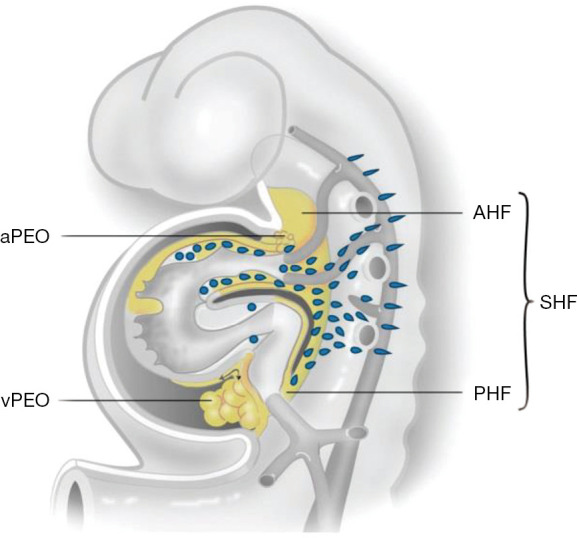
The developing heart tube. During embryogenesis the SHF contributes to the arterial pole which includes the great arteries and the right ventricle from the AHF. From the PHF, second heart field cells enter the venous pole. The myocardium and intrapericardial part of the aorta are covered by epicardial cells that are provided by a proepicardial organ of the vPEO and aPEO, respectively. Not shown in this figure: neural crest cells migrate from the neural tube primarily to the arterial pole of the heart [adapted from Grewal et al. 2014 (5)]. SHF, second heart field; AHF, anterior population; PHF, posterior heart field; vPEO, venous pole; aPEO, arterial pole.
Next to contributing to the aortic and pulmonary valve formation and outflow tract (7,8) the cardiac progenitor cells are important for the development of other elements of the heart and great arteries. Second heart field progenitor cells give origin to three specific cell lines, as has been demonstrated in lineage tracing studies employing Nkx2.5: (I) endothelial-derived endocardial cushion cells, which are in part derived from the endothelium, (II) outflow tract and right ventricular myocardium (9), and to (III) vascular smooth muscle cells (VSMCs) of the great arteries. Besides second heart field cells, neural crest cells also contribute to the VSMCs comprising the aorta. The aortic VSMCs thus have different embryonic origins, moreover migration of these VSMCs is site-specific: VSMCs in the aortic root are neural crest- and secondary heart field derived; in the ascending aorta and aortic arch VSMCs are neural crest derived and the VSMCs in the descending aorta are derived from the paraxial mesoderm (10) (Figure 2). Within the aortic root wall, the inner media is derived from neural crest cells (11), whereas the outer media/adventitia is second heart field-derived (12) (Figure 2). Other than neural crest and second heart field cells, arterial epicardium contribution to the VSMCs of the ascending aorta has also been described (13,14).
Figure 2.

Developmental origin of the BAV and aortic wall. Progenitor neural crest and second heart field cells contribute to the normal development of an aortic valve and the vascular smooth muscle cells in the ascending aortic wall. Mutations in genes related to the neural crest and second heart lead to the development of a BAV and a maturation defect of vascular smooth muscle cells in the aortic root, ascending aorta and arch. Type 1 RCC-NCC and type 1 RCC-LCC have been attributed to defects in the neural crest and second heart field cell signalling respectively. RCC-NCC: fusion between the RCC and NCC. LCC-NCC: fusion between the LCC and NCC. RCC-LCC: fusion between the RCC and LCC. TAD: reduced glutathione. BAV, bicuspid aortic valve; RCC, right coronary cusp; NCC, non-coronary cusp; LCC, left coronary cusp; VSMC, vascular smooth muscle cell; SHF, second heart field.
During embryogenesis, the regulation of cell migration, proliferation, and extracellular matrix deposition in the developing valves and aortic wall is orchestrated by several signalling pathways including Wnt/β-catenin, NOTCH, transforming growth factor β (TGFβ), bone morphogenetic protein, vascular endothelial growth factor, NFATc1 and MAPK and, transcription factors such as Twist1, Tbx20, Msx1/2, and Sox9 (15,16). Multiple cell population are thus involved in both aortic wall and semilunar valve formation. A defect in one of the signalling pathways can consequently lead to a malformed aortic valve.
Clinically, several BAV subtypes are distinguished based on the number of fused commissures and the raphe position. The Sievers classification identifies three major types: type 0 (no raphe), type 1 (one raphe) and type 2 (two raphes) (17). Type 1 can be further subdivided on basis of the raphe position: raphe between right coronary cusp (RCC) and left coronary cusp (LCC): RCC-LCC; between RCC and non-coronary cusp (NCC): RCC-NCC and raphe between LCC and NCC: LCC-NCC (Figure 2).
Several studies have investigated the role of cardiac progenitor cells in the development of different BAV forms. It has been proposed that type 1 RCC-LCC and type 1 RCC-NCC BAVs have a different developmental origin (18). This study demonstrated that the type 1 RCC-LCC BAVs are caused by an altered neural crest cell behaviour, which is supported by the findings of Phillips et al. in their Rock 1,2 deficient mouse model (7). Fernandez et al. further argue that an eNOS mutation is responsible for a type 1 RCC-NCC BAV (18), which is in line with our own findings (19). Our study further demonstrated that disrupted eNOS signalling in knockout mice causes aortic dilation and dissection, in addition to congenital BAV formation (19). The latter findings indicate a role of second heart field in the development of type 1 RCC-NCC BAVs as eNOS is expressed by endocardial cells (20), cardiomyocytes (20) and VSMCs (21), which are all second heart field-derived cell populations.
A myriad of murine studies has identified genetic mutations responsible for the development of a BAV, but unfortunately did not focus on the differentiation of the type of BAV. In these studies, markers of both the neural crest cells (Pax3) (22) and second heart field cells (Nkx2.5 and GATA5) (23) have been associated with the development of BAV.
Disturbed NOTCH signalling in both the neural crest (24) and second heart field has also been linked to the development of a BAV (22,25). Besides leading to a malformed semilunar valve, inhibition of NOTCH in the second heart field also causes impairment of the VSMCs of the great arteries (26).
The aforementioned studies, which have dealt with the embryonic development of an aortic valve and the aortic vascular smooth muscle cells, have taught us that there are many similarities in the normal as well as the abnormal development. Common developmental defects in the progenitor cells might therefore be responsible for both a BAV and the associated aortopathy (Figure 2). Furthermore, the progenitor cells are responsible for region specific VSMCs in the aortic wall. Therefore, defects in the neural crest and second heart field signalling in the BAV patients lead to BAV-related aortopathy in the proximal thoracic aorta and, is rarely found in the descending thoracic aorta (27). Whereas in Marfan patients, defects are seen in the neural crest, second heart field and paraxial mesoderm causing aortopathy in the ascending and descending aorta.
In the next paragraph, we discuss genetic defects related to the development of BAV and thoracic aortic dilation in human and their origin in embryogenesis.
Genetic basis of BAV and thoracic aortic dilation
BAV is a heterogeneous disorder with an autosomal dominant inheritance pattern with incomplete penetrance and variable expressivity (28). BAV can be found as a feature of some genetic syndromes such as Marfan (MFS), Loeys-Dietz and Turner. In non-syndromic forms, BAV inheritance is complex, as a myriad of genes encode transcription factors, extracellular matrix proteins and signalling pathways which regulate cell proliferation, differentiation, adhesion or apoptosis, all necessary for the development of a normal semilunar valve. Consequently, a single genetic mutation might increase the risk to develop a BAV however, this is insufficient to cause the disease by its own. This makes it challenging to determine the exact underlying genetic and epigenetic mechanism of BAV in the human population and murine models.
Table 1 gives an overview of several genetic mutations which have been linked to the development of a BAV. All identified mutations are associated with the cardiac progenitor cell lines. More specifically, neural crest signalling alterations lead to the most common type 1 RCC-LCC BAV (29), while second heart field-related genes are associated to the BAV type 1 RCC-NCC.
Table 1. Genetic mutations associated with a bicuspid aortic valve.
| Disorder | Genetic mutation |
|---|---|
| BAV | NOTCH1, 9q34 |
| AXIN1-PDIA2 | |
| Endoglin gene | |
| Marfan syndrome | FBN1, 15q21.1 |
| TGFBR2, 3p25-24.2 | |
| Loeys-Dietz | TGFBR1, 9q33-34 |
| TGFBR2, 3p24-25 | |
| Ehlers Danlos | COL3A1, 2q24.3-q31 |
| FTAAD | TAAD1, 5q13-14 |
| TAAD2, 3p24-25 | |
| TAAD3, 15q24-26 | |
| TAAD4, 10q23-24 | |
| TAAD5, 9q33-34 |
BAV, bicuspid aortic valve; FTAAD, familial thoracic aortic aneurysms and dissection.
NOTCH1, a marker of the neural crest and second heart field, is associated with both the development of a BAV, as well as associated aortopathy (30). Genetic haplotypes within the AXIN1-PDIA2 locus, integral in the Wnt pathway which regulates both heart valve formation (31) and cardiac neural crest development (32), are identified in the development of a BAV. Another haplotype within the Endoglin gene is associated with BAV and also, required for the differentiation of neural crest cells into VSMCs that populate the aorta (33).
Interestingly, mutations related to BAV have thus also been linked to thoracic aortic dilation. However, it remains to be elucidated whether, in the absence of BAV, thoracic aortopathy is more related to neural crest or second heart field signalling defects.
We describe the genetics of thoracic aortic dilation in syndromes including MFS, Loeys-Dietz, Ehlers Danlos and familial thoracic aortic aneurysms and dissection (FTAAD) and their association with embryonic development. The genetic defects and chromosomal location of these syndromes are summarized in Table 1.
MFS is a clinical diagnosis based of phenotypical cardiovascular, skeletal and ocular manifestations. This autosomal dominant connective tissue disorder has a very high risk for progressive aortopathy forming the major cause of morbidity and mortality in MFS patients. Genetic mutations underlying MFS are described in Table 1 (34). The described TGFβ receptor mutation results in an increased TGFβ activity.
Loeys-Dietz is caused by a defect in the TGFβ signalling pathway (Table 1). This disorder is characterized by aortic valvular and vessel wall defects. Bicuspidy is seen in approximately 10% of patients with Loeys-Dietz syndrome. Other phenotypic characteristics include craniosynostosis, cleft palate, bifid uvula, congenital heart disease and mental retardation.
Ehlers Danlos is caused by a defect in collagen encoding genes (Table 1) (35). Collagen fibres form an integral part of the medial and adventitial layer in the vascular wall. Besides providing mechanical strength to the vessel wall, the fibres also regulate local homeostasis. Patients with vascular Ehlers-Danlos have an increased risk for an aortic dissection and rupture, with a reported incidence of at least 10% (36).
FTAAD is inherited in an autosomal dominant matter. The condition is characterized by an increased risk for aortopathy, in the absence of syndromic features. Diverse genetic loci have been identified in FTAAD (Table 1) (34,37). Many TAAD mutations have been associated with a TGFβ receptor, with a resulting increased TGFβ pathway activity in the aorta (34).
In most of the syndromes associated with thoracic aortic dilation, an increased TGFβ activity has been identified. TGFβ is not specific for any progenitor cell line. It is involved in neural crest cells, second heart field cells and in the endothelium (38).
In the BAV population, type 1 RCC-LCC has the most pronounced aortic wall abnormalities. As discussed above, this BAV type is associated with neural crest defects and often has an increased TGFβ activity (39). But there are two main drawbacks to this theory. Firstly, in syndromes as MFS, Ehlers-Danlos and Loeys-Dietz, bicuspidy is not an obligatory clinical manifestation, indicating that a defective TGFβ signalling is at least not the main factor causing BAV formation. Moreover, the clinical course is not complicated with thoracic aortic dilation in all BAV patients. Thus, neural crest involvement and TGFβ activity together are not sufficient to explain the variability within the pathogenesis of BAV and associated aortopathy. Additional pathogenetic factors need to be taken into account, such as haemodynamics or a contribution of second heart field. The next sections highlight important histopathological features of the pathological ascending aortic wall in the adult BAV.
The ascending aortic wall in BAV
Aortic dilation begins at a young age in the BAV individual and is usually characterized by ‘mid-ascending’- type dilation. Over the past years histopathological analysis of non- and dilated ascending aortic wall samples in BAV and TAV patients have revealed many structural differences. One of the first studies which suggested differences in the ascending aorta of BAV and TAV was published in 1984 by Larson et al. (40). In subsequent years, several histopathological studies were published comparing the BAV and TAV population.
VSMCs are critical in the differences between BAV and TAV histopathology. Not only have these cells been found in apoptosis (41), VSMCs are also morphologically distinct in the BAV population. VSMCs in BAV patients are less well differentiated, due to a defect in phenotypic switch leading to a significantly lower expression of differentiated, contractile VSMC markers such as smoothelin, calponin and SM22alpha. Furthermore, Lamin A/C, which plays a key role in the differentiation of VSMCs, is significantly less expressed in the BAV, as compared to the TAV, population (42). These less differentiated, immature VSMCs are noted in both the non- and dilated BAV population, excluding aortic dilation as a cause of the observed phenotypic switch of the VSMCs. Also, as VSMC immaturity is seen in all BAV patients but not all develop aortic dilation, a secondary hit needs to be identified responsible for aortopathy in a subset of BAV. A panel of markers had recently been identified which could identify BAV patients susceptible for future dilation (21). This so-called susceptibility pathway included c-Kit, a marker for dedifferentiated VSMCs, and its phosphorylated state (pc-Kit) triggered by the presence of matrix metalloproteinase-9 (MMP9) influencing hypoxia-inducible-factor-1-alpha (HIF1α) and endothelial nitric oxide synthase (eNOS) (21). In this study, an increased MMP-9 activity was found exclusively in BAV patients susceptible for aortic complications (21). MMPs play an important role in vascular biology, and control degradation of extracellular matrix proteins, such as elastin and collagen. The integrity of the aortic extracellular matrix may therefore be compromised by an enhanced MMP activity. Inflammation, oxidative stress and matrix degradation products and an increased TGFβ-1 activity leads to an overproduction of MMP9 by VSMCs of the aorta and inflammatory cells (43). Such overexpression of MMPs is a common feature in aortic aneurysms (44). Particularly, MMP-2 and MMP-9 have been implicated in thoracic aortic disease (21,44). MMP-2 and MMP-9 are involved in the turnover of elastic matrix components; they are released from smooth muscle cells (44). Overactivity of MMPs, on the other hand is also possible by downregulation of their antagonists, the tissue inhibitors of MMPs (TIMP-1, TIMP-2). MMP/TIMP expression patterns reported for different types of thoracic aortic aneurysms vary significantly and can be influenced by other factors such as hypertension, cellular oxidative stress and reactive oxygen species (45).
Histopathological markers of cardiovascular ageing, which form the hallmark of TAV aortopathy, are found significantly less expressed in BAV as compared to TAV (42). For instance, BAV aortopathy is generally regarded as less/non-inflammatory (42,46). These findings are comparable with the clinical observation of BAV ascending aortic aneurysms not commonly being associated with systemic atherosclerosis (47-49). Differences in medial elastic fibers have also been described between BAV and TAV. In BAV elastin fiber fragmentation is significantly less profound as compared to the TAV (42), the fiber orientation is changed and the fiber mass is reduced (50). The reduction in fiber mass is in line with our earlier findings of elastic fiber thinning in BAV patients (42).
Besides the medial differences, in our previous studies we also found that the BAV population has a significantly thinner intimal layer and develops an increased thickness only under hemodynamic shear stress.
Pathogenesis of thoracic aortic dilation in BAV: role of haemodynamic factors and second heart field
The geometry of the bi-leaflet aortic valve creates a nonaxial transvalvular flow jet within the proximal aorta (51). An increased stroke volume due to a regurgitant aortic valve further increases the hemodynamic stress on the vascular wall. It has been suggested that the uneven wall stress distribution contributes to the development of aortic complications. Opinions are however divided on the predominance of shear stress in the development of aortopathy. Studies have confirmed that thoracic aortic aneurysms can develop in the absence of valve abnormalities (52). Also, after surgical replacement of the malformed BAV, progressive thoracic aortic dilation is reported (53). These studies suggest that hemodynamic factors are not sufficient to explain the onset and progression of aortic complications in BAV and instead, structural wall abnormalities are important in the pathogenesis of aortopathy. The non-hemodynamic theory is that genetically determined developmental abnormalities of the aortic wall lead to a defect in the cellular microenvironment, causing or at least, contributing to the aortic pathology.
To study the role of haemodynamics superimposed on structural wall abnormalities, we investigated non- and dilated ascending aorta of both valve types immunohistochemically and compared the aortic area with maximum shear stress to the opposite area. As described above, histopathologically BAV show a defect in VSMC maturation, decreased medial inflammation, elastin fragmentation and cystic medial necrosis, as compared to the TAV (54). Besides medial differences, BAV are characterized by a significantly thinner tunica intima (21,42,55). While comparing the jet and non-jet samples in both BAV and TAV, we did not find any difference in the pathologic features in the adventitia, middle and outer media even if corrected for aortic stenosis/regurgitation, aortic dilation and raphe position (56,57). A turbulent flow did lead to a significant increase in intimal thickness, which was however seen in all patients regardless of the valve type and aortic dilation. These results suggest that the fundamental difference in the aortic wall in BAV and TAV are independent of shear stress. Haemodynamic factors therefore, might play a role in the aortic complications, but superimposed on the already present structurally immature aortic wall seen in BAV.
Conclusions
This review gives an overview of the normal and abnormal development of the aortic semilunar valve and the ascending aortic wall. An altered neural crest cell and second heart field contribution, separately or in combination, can account for a structurally different aortic wall in combination with bicuspidy, as aortic vasculature and valve development are closely related and share common embryonic cell populations. Neural crest cell defects are mostly associated with type 1 RCC-LCC BAV and defects in second heart field with type 1 RCC-NCC BAV (Figure 2).
Aortic complications vary between the three different BAV types. For instance, in the type 1 RCC-LCC BAV, aortic root diameters are significantly larger as compared to the type 1 RCC-NCC BAV (17,18). Aortopathy further seems most outspoken in BAV type 1 RCC-LCC, which has been linked to a defect in neural crest cells. Conversely, patients with a type 1 RCC-NCC BAV experience valve dysfunction at a younger age (18). Current findings indicate that neural crest cell defects are related to most outspoken aortic wall abnormalities. It however, remains challenging to conclusively distinguish BAV patients susceptible for aortopathy (Figure 2). Future research therefore, needs to focus on identifying developmental defects which account for the additional pathology and increase susceptibility for aortopathy in a subset of BAV patients. These alternative, molecular biological markers are necessary to diagnose this subset of vulnerable patients.
It is interesting to note that a specific group of animals, being reptiles, normally present with bicuspid semilunar valves. Evidently, these have a different physiology, being ‘cold-blooded’ as well as anatomy, as they present two aortas. Nevertheless, here the bicuspid valves function efficiently, even until advanced ages (58).
Video.

Normal and abnormal development of the aortic valve and ascending aortic wall: an overview of the embryology and pathology of the bicuspid aortic valve
Acknowledgments
Funding: None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Tzemos N, Therrien J, Yip J, et al. Outcomes in adults with bicuspid aortic valves. JAMA 2008;300:1317-25. 10.1001/jama.300.11.1317 [DOI] [PubMed] [Google Scholar]
- 2.Neri E, Barabesi L, Buklas D, et al. Limited role of aortic size in the genesis of acute type A aortic dissection. Eur J Cardiothorac Surg 2005;28:857-63. 10.1016/j.ejcts.2005.10.013 [DOI] [PubMed] [Google Scholar]
- 3.Pape LA, Tsai TT, Isselbacher EM, et al. Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007;116:1120-7. 10.1161/CIRCULATIONAHA.107.702720 [DOI] [PubMed] [Google Scholar]
- 4.Gittenberger-de Groot AC, Bartelings MM, Deruiter MC, et al. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res 2005;57:169-76. 10.1203/01.PDR.0000148710.69159.61 [DOI] [PubMed] [Google Scholar]
- 5.Grewal N, DeRuiter MC, Jongbloed MR, et al. Normal and abnormal development of the aortic wall and valve: correlation with clinical entities. Neth Heart J 2014;22:363-9. 10.1007/s12471-014-0576-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soto-Navarrete MT, López-Unzu MÁ, Durán AC, et al. Embryonic development of bicuspid aortic valves. Prog Cardiovasc Dis 2020;63:407-18. 10.1016/j.pcad.2020.06.008 [DOI] [PubMed] [Google Scholar]
- 7.Phillips HM, Mahendran P, Singh E, et al. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc Res 2013;99:452-60. 10.1093/cvr/cvt132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poelmann RE, Gittenberger-de Groot AC. A subpopulation of apoptosis-prone cardiac neural crest cells targets to the venous pole: multiple functions in heart development? Dev Biol 1999;207:271-86. 10.1006/dbio.1998.9166 [DOI] [PubMed] [Google Scholar]
- 9.Harmon AW, Nakano A. Nkx2-5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genesis 2013;51:862-9. 10.1002/dvg.22721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawada H, Rateri DL, Moorleghen JJ, et al. Smooth Muscle Cells Derived From Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta-Brief Report. Arterioscler Thromb Vasc Biol 2017;37:1722-6. 10.1161/ATVBAHA.117.309599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson JC, Wisse LJ, Wirokromo V, et al. Disturbed nitric oxide signalling gives rise to congenital bicuspid aortic valve and aortopathy. Dis Model Mech 2020;13:dmm044990. 10.1242/dmm.044990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mifflin JJ, Dupuis LE, Alcala NE, et al. Intercalated cushion cells within the cardiac outflow tract are derived from the myocardial troponin T type 2 (Tnnt2) Cre lineage. Dev Dyn 2018;247:1005-17. 10.1002/dvdy.24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gittenberger-de Groot AC, Winter EM, Bartelings MM, et al. The arterial and cardiac epicardium in development, disease and repair. Differentiation 2012;84:41-53. 10.1016/j.diff.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 14.Grewal N, Goumans MJ, deRuiter M, et al. Aortopathy in bicuspid aortic valve and marfan syndrome is characterized by a lack of activation potential of the epicardium in the ascending aorta. Int J Pathol Clin Res 2017;3. doi: . 10.23937/2469-5807/1510051 [DOI] [Google Scholar]
- 15.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res 2009;105:408-21. 10.1161/CIRCRESAHA.109.201566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol 2011;73:29-46. 10.1146/annurev-physiol-012110-142145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sievers HH, Schmidtke C. A classification system for the bicuspid aortic valve from 304 surgical specimens. J Thorac Cardiovasc Surg 2007;133:1226-33. 10.1016/j.jtcvs.2007.01.039 [DOI] [PubMed] [Google Scholar]
- 18.Fernández B, Durán AC, Fernández-Gallego T, et al. Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. J Am Coll Cardiol 2009;54:2312-8. 10.1016/j.jacc.2009.07.044 [DOI] [PubMed] [Google Scholar]
- 19.Peterson JC, Chughtai M, Wisse LJ, et al. Nos3 mutation leads to abnormal neural crest cell and second heart field lineage patterning in bicuspid aortic valve formation. Dis Model Mech 2018;11:dmm034637. 10.1242/dmm.034637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bukowska A, Röcken C, Erxleben M, et al. Atrial expression of endothelial nitric oxide synthase in patients with and without atrial fibrillation. Cardiovasc Pathol 2010;19:e51-60. 10.1016/j.carpath.2008.12.014 [DOI] [PubMed] [Google Scholar]
- 21.Grewal N, Gittenberger-de Groot AC, DeRuiter MC, et al. Bicuspid aortic valve: phosphorylation of c-Kit and downstream targets are prognostic for future aortopathy. Eur J Cardiothorac Surg 2014;46:831-9. 10.1093/ejcts/ezu319 [DOI] [PubMed] [Google Scholar]
- 22.Thomas PS, Sridurongrit S, Ruiz-Lozano P, et al. Deficient signaling via Alk2 (Acvr1) leads to bicuspid aortic valve development. PLoS One 2012;7:e35539. 10.1371/journal.pone.0035539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain D, Dietz HC, Oswald GL, et al. Causes and histopathology of ascending aortic disease in children and young adults. Cardiovasc Pathol 2011;20:15-25. 10.1016/j.carpath.2009.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest 2011;121:2876-87. 10.1172/JCI44555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.High FA, Zhang M, Proweller A, et al. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest 2007;117:353-63. 10.1172/JCI30070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garg V. Molecular genetics of aortic valve disease. Curr Opin Cardiol 2006;21:180-4. 10.1097/01.hco.0000221578.18254.70 [DOI] [PubMed] [Google Scholar]
- 27.Granata A, Serrano F, Bernard WG, et al. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet 2017;49:97-109. 10.1038/ng.3723 [DOI] [PubMed] [Google Scholar]
- 28.Prakash SK, Bossé Y, Muehlschlegel JD, et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J Am Coll Cardiol 2014;64:832-9. 10.1016/j.jacc.2014.04.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mancini ML, Verdi JM, Conley BA, et al. Endoglin is required for myogenic differentiation potential of neural crest stem cells. Dev Biol 2007;308:520-33. 10.1016/j.ydbio.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aicher D, Urbich C, Zeiher A, et al. Endothelial nitric oxide synthase in bicuspid aortic valve disease. Ann Thorac Surg 2007;83:1290-4. 10.1016/j.athoracsur.2006.11.086 [DOI] [PubMed] [Google Scholar]
- 31.Padang R, Bannon PG, Jeremy R, et al. The genetic and molecular basis of bicuspid aortic valve associated thoracic aortopathy: a link to phenotype heterogeneity. Ann Cardiothorac Surg 2013;2:83-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res 2004;95:459-70. 10.1161/01.RES.0000141146.95728.da [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niessen K, Karsan A. Notch signaling in cardiac development. Circ Res 2008;102:1169-81. 10.1161/CIRCRESAHA.108.174318 [DOI] [PubMed] [Google Scholar]
- 34.Pannu H, Tran-Fadulu V, Milewicz DM. Genetic basis of thoracic aortic aneurysms and aortic dissections. Am J Med Genet C Semin Med Genet 2005;139C:10-6. 10.1002/ajmg.c.30069 [DOI] [PubMed] [Google Scholar]
- 35.Bergqvist D. Ehlers-Danlos type IV syndrome. A review from a vascular surgical point of view. Eur J Surg 1996;162:163-70. [PubMed] [Google Scholar]
- 36.van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 2011;43:121-6. 10.1038/ng.744 [DOI] [PubMed] [Google Scholar]
- 37.Guo D, Hasham S, Kuang SQ, et al. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13-14. Circulation 2001;103:2461-8. 10.1161/01.CIR.103.20.2461 [DOI] [PubMed] [Google Scholar]
- 38.Bartram U, Molin DG, Wisse LJ, et al. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation 2001;103:2745-52. 10.1161/01.CIR.103.22.2745 [DOI] [PubMed] [Google Scholar]
- 39.Gomez D, Al Haj Zen A, Borges LF, et al. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol 2009;218:131-42. 10.1002/path.2516 [DOI] [PubMed] [Google Scholar]
- 40.Larson EW, Edwards WD. Risk factors for aortic dissection: a necropsy study of 161 cases. Am J Cardiol 1984;53:849-55. 10.1016/0002-9149(84)90418-1 [DOI] [PubMed] [Google Scholar]
- 41.Mohamed SA, Misfeld M, Hanke T, et al. Inhibition of caspase-3 differentially affects vascular smooth muscle cell apoptosis in the concave versus convex aortic sites in ascending aneurysms with a bicuspid aortic valve. Ann Anat 2010;192:145-50. 10.1016/j.aanat.2010.02.006 [DOI] [PubMed] [Google Scholar]
- 42.Grewal N, Gittenberger-de Groot AC, Poelmann RE, et al. Ascending aorta dilation in association with bicuspid aortic valve: a maturation defect of the aortic wall. J Thorac Cardiovasc Surg 2014;148:1583-90. 10.1016/j.jtcvs.2014.01.027 [DOI] [PubMed] [Google Scholar]
- 43.Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother 2003;57:195-202. 10.1016/S0753-3322(03)00065-9 [DOI] [PubMed] [Google Scholar]
- 44.Fedak PW, de Sa MP, Verma S, et al. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. J Thorac Cardiovasc Surg 2003;126:797-806. 10.1016/S0022-5223(03)00398-2 [DOI] [PubMed] [Google Scholar]
- 45.Phillippi JA, Klyachko EA, Kenny JP, 4th, et al. Basal and oxidative stress-induced expression of metallothionein is decreased in ascending aortic aneurysms of bicuspid aortic valve patients. Circulation 2009;119:2498-506. 10.1161/CIRCULATIONAHA.108.770776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balistreri CR, Pisano C, Candore G, et al. Focus on the unique mechanisms involved in thoracic aortic aneurysm formation in bicuspid aortic valve versus tricuspid aortic valve patients: clinical implications of a pilot study. Eur J Cardiothorac Surg 2013;43:e180-6. 10.1093/ejcts/ezs630 [DOI] [PubMed] [Google Scholar]
- 47.Agmon Y, Khandheria BK, Meissner I, et al. Is aortic dilatation an atherosclerosis-related process? Clinical, laboratory, and transesophageal echocardiographic correlates of thoracic aortic dimensions in the population with implications for thoracic aortic aneurysm formation. J Am Coll Cardiol 2003;42:1076-83. 10.1016/S0735-1097(03)00922-7 [DOI] [PubMed] [Google Scholar]
- 48.Dolmaci OB, Driessen AHG, Klautz RJM, et al. Comparative evaluation of coronary disease burden: bicuspid valve disease is not atheroprotective. Open Heart 2021;8:e001772. 10.1136/openhrt-2021-001772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dolmaci OB, Legué J, Lindeman JHN, et al. Extent of Coronary Artery Disease in Patients With Stenotic Bicuspid Versus Tricuspid Aortic Valves. J Am Heart Assoc 2021;10:e020080. 10.1161/JAHA.120.020080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsamis A, Phillippi JA, Koch RG, et al. Extracellular matrix fiber microarchitecture is region-specific in bicuspid aortic valve-associated ascending aortopathy. J Thorac Cardiovasc Surg 2016;151:1718-1728.e5. 10.1016/j.jtcvs.2016.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Girdauskas E, Disha K, Borger MA, et al. Relation of bicuspid aortic valve morphology to the dilatation pattern of the proximal aorta: focus on the transvalvular flow. Cardiol Res Pract 2012;2012:478259. 10.1155/2012/478259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Braverman AC, Güven H, Beardslee MA, et al. The bicuspid aortic valve. Curr Probl Cardiol 2005;30:470-522. 10.1016/j.cpcardiol.2005.06.002 [DOI] [PubMed] [Google Scholar]
- 53.Yasuda H, Nakatani S, Stugaard M, et al. Failure to prevent progressive dilation of ascending aorta by aortic valve replacement in patients with bicuspid aortic valve: comparison with tricuspid aortic valve. Circulation 2003;108 Suppl 1:II291-4. 10.1161/01.cir.0000087449.03964.fb [DOI] [PubMed] [Google Scholar]
- 54.Matthias Bechtel JF, Noack F, Sayk F, et al. Histopathological grading of ascending aortic aneurysm: comparison of patients with bicuspid versus tricuspid aortic valve. J Heart Valve Dis 2003;12:54-9; discussion 59-61. [PubMed] [Google Scholar]
- 55.Fernandes SM, Khairy P, Sanders SP, et al. Bicuspid aortic valve morphology and interventions in the young. J Am Coll Cardiol 2007;49:2211-4. 10.1016/j.jacc.2007.01.090 [DOI] [PubMed] [Google Scholar]
- 56.Grewal N, Girdauskas E, deRuiter M, et al. The effects of hemodynamics on the inner layers of the aortic wall in patients with a bicuspid aortic valve. Integr Mol Med 2017;4. doi: 10.15761/IMM.1000308 [DOI] [Google Scholar]
- 57.Grewal N, Girdauskas E, DeRuiter M, et al. The role of hemodynamics in bicuspid aortopathy: a histopathologic study. Cardiovasc Pathol 2019;41:29-37. 10.1016/j.carpath.2019.03.002 [DOI] [PubMed] [Google Scholar]
- 58.Poelmann RE, Gittenberger-de Groot AC, Goerdajal C, et al. Ventricular Septation and Outflow Tract Development in Crocodilians Result in Two Aortas with Bicuspid Semilunar Valves. J Cardiovasc Dev Dis 2021;8:132. 10.3390/jcdd8100132 [DOI] [PMC free article] [PubMed] [Google Scholar]