

Abstract
In recent decades “saliva” has emerged as an important non-invasive biofluid for diagnostic purposes in both human and animal health sectors. However, with the rapid evolution of molecular detection technologies, the limitation has been the lack of an efficient method for the facile amplification of target RNA from such a complex matrix. Herein, we demonstrate the novel application of hydrogel microparticles of primer-immobilized networks (PIN) for direct quantitative reverse transcription PCR (dirRT-qPCR) of viral RNA from saliva samples without prior RNA purification. Each of these highly porous PIN particles operates as an independent reactor. They filter in micro-volumes of the analyte solution. Viral RNA is captured and converted to complementary DNA (cDNA) through the RT step using covalently incorporated RT primers. The PIN with cDNA of the viral target will be ready for subsequent highly specific qPCR. Preceded by heat-treatment for viral lysis, we were able to conduct PIN dirRT-qPCR with 95% efficiency of the matrix (M) gene for influenza A virus (IAV) and 5’ untranslated region (5’ UTR) for chicken coronavirus spiked into saliva samples. The addition of reverse transcriptase enzyme (RTase) and 10% dilution of the matrix improved the assay sensitivity considerably. PIN particles’ compatibility with microfluidic PCR chip technology has significantly reduced total sample processing time to 50 min, instead of an average of 120 min that are normally used by other assays. We anticipate this technology will be useful for other viral RNA targets by changing the incorporated RT primer sequences and can be adapted for onsite diagnostics.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13206-022-00065-0.
Keywords: Primer-immobilized particles, Direct RT-qPCR, Saliva, Influenza A virus, Chicken coronavirus
Introduction
The presence of viral nucleic acids, deoxyribonucleic acid (DNA), and ribonucleic acid (RNA) as diagnostic biomarkers in saliva, has upgraded their utilization as an excellent non-invasive diagnostic matrix for molecular assays [1, 2]. The use of oral fluid for the detection of infectious diseases in humans and domestic animals has come to attention in recent decades. In domestic animals, it followed a report on pen side collection of porcine oral fluid using a cotton rope “rope saliva” for the detection of porcine reproductive and respiratory syndrome virus [2]. Since then, the use of rope saliva for the detection of both DNA and RNA viruses using quantitative polymerase chain reaction (qPCR) and reverse transcription (RT)-qPCR assays, respectively, have grown very fast globally [3]. Their sampling procedure minimizes the risk to practitioners of exposure to zoonotic diseases and is easily accessible, cheap, and simple to collect [2, 4–6]. For the same reason, some sample preparation kits for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) utilize saliva samples. They also promote easier disease surveillance and are particularly useful for herd screening, and whenever repeated sampling is required [3, 7, 8].
However, using this matrix for viral RNA separation and detection by molecular assays is challenging. Several obstacles have to be defeated to utilize saliva samples for diagnostic purposes: Limited amount of selected biomarkers [4, 7], rapid degradation of the matrix and instability of RNA [9–11], and PCR inhibitors [12–15]. All together must be outclassed to obtain reliable testing results. The use of RNase inhibitors and stabilizers [6, 7, 11, 16], bovine serum albumin [17], heat treatment [18–21], matrix dilution [12, 14, 17, 22], and many others have been in use to revamp PCR amplification outcomes in case of poor PCR performances.
One of the critical steps in most molecular assays is to obtain high-quality RNA from complex matrices, required for high efficiencies of downstream applications [23]. The common RNA extraction methods used today
are either liquid–liquid extraction (e.g. phenol–chloroform extraction) or solid-phase extraction (e.g. silica matrices, magnetic beads, and many others). Despite their success, they remain to be either complicated or labor-intensive, with several separate sample-processing steps [24, 25]. Despite all these impediments, we still desired to develop a microparticle-based dirRT-qPCR to detect RNA viruses suitable for the onsite application. Direct RT-qPCR provides a simplified process for sample analysis, with very low sample volume and turnaround time of results, reduced chances for cross-contamination, exposure to infectious materials, and toxic reagents [26, 27]. Recently, several dirRT-qPCR methods have been reported for the detection of SARS-CoV-2 in the current pandemic, using the conventional RT-qPCR platforms and heat treatment for viral lysis without RNA purification [19, 20]. We believe this is the right direction toward early detection of infectious diseases, with all the above features for dirRT-qPCR supportive for point of care diagnostics.
In this study, we are reporting a novel and efficient dirRT-qPCR assay for the detection of two RNA viruses from saliva, a non-invasive and complex animal matrix. Previously, we showed that PIN particles have high selectivity and efficiency for RT-qPCR [28, 29]. In addition, we have managed to utilize the PIN particles as a simple way for RNA preparation from complex matrices in this study. Using PIN fabricated from polyethylene glycol resulted in hydrophilic, non-fouling, and porous PIN particles. These features permit the particles to filter-in micro-volumes of the analyte solution, into a 3D reaction chamber for dirRT-qPCR using TaqMan probe chemistry. Taking the advantage of immobilized target-specific primer to a cross-linking polymer, they specifically capture the target RNA from the matrix, for highly specific dirRT-qPCR. Coupling the PIN particles with dirRT-qPCR in a microfluidic PCR chip, we were able to detect the M gene of IAV and 5’UTR of chicken CoV using an ultra-fast qPCR platform in less than an hour.
Materials and Methods
Ethical Statement
All animal experiments conducted in this study observed the laid down protocols and regulations. The study protocol was approved by the Institutional Animal Care and Use Committee of Konkuk University (permit number: KU18107). Anesthesia, euthanasia, and animal killing were not applied during the execution of this study.
Study Design
We applied a spike-in test scheme, whereby both synthetic DNA and RNA templates for the IAV M gene were spiked into negative pure and rope saliva samples (Fig. SI1), collected from healthy pig farms at the Quarantine Office, Daejeon, Republic of Korea. The pseudo-samples created were used to set up the experiment to obtain optimized values with consistent results. This provided a protocol guideline for inactivated IAV strain H1N1 and chicken CoV spiked in rope saliva samples for method evaluation. All the samples were analyzed using qPCR and RT-qPCR for DNA and RNA, respectively. Conventional solution-based RT-qPCR was used as our standard method against the PIN particles dirRT-qPCR, using ultra-fast real-time PCR (G2-4) System (Micobiomed Co. Ltd, Seoul, Korea). The effects of the following variables on RT-qPCR performance were evaluated; heat-treatment duration for viral lysis, RT conditions including the addition of RTase enzyme, and the use of RNase inhibitors. The threshold (Ct) values which were automatically generated by the instrument software and detection signal intensity data were collected and analyzed to evaluate the tested variables and method performance.
Sample Collection and Preparation
Sample Preparation
A total of four negative rope saliva samples were used in the first stage of the experiment to optimize all the steps of the experiment using pseudo samples. The rope saliva samples were collected as previously described [2]. One batch of rope saliva samples was centrifuged at 3000 rpm for 15 min to remove debris, with another batch of samples left without centrifugation to examine the contribution of centrifuge when using PIN particles dirRT-qPCR. All the samples were aliquoted and stored at − 20 °C until further analyses.
Viral RNA Extraction
Two different protocols to avoid bias when comparing PIN dirRT-qPCR with QIAamp Viral RNA Mini Kit-based assay were used for IAV. At first, we considered equal ratio and sample input volume for both methods. Samples were prepared by mixing 1 µL of saliva + 1 µL of virus + 138 µL PBS to make a total volume of 140 µL, which was used for extraction. Second, we considered an equal sample ratio between saliva and virus (1:1), with increased sample input volume (70 µL of saliva + 70 µL of virus) to make 140 µL recommended by the kit protocol. The QIAamp Viral RNA Mini Kit (Cat. No 52904) (Qiagen, Hilden, Germany) was used as a standard kit for RNA extraction, according to manufacturer instructions with the above-stated modifications.
Aging Test
To assess the aging for matrix stability, a set of five aliquoted samples were stored at 4 °C for up to 30 days. Samples were analyzed using conventional solution-based one-step RT-qPCR on days 0, 1, 3, 5, 7, 10, and then, after every 2 days. Each time we recorded the Ct values and examined their trend for evidence of matrix degradation.
Primers and TaqMan Probe Design
All oligonucleotides were designed to detect the conserved regions for the M gene of IAV and 5’ UTR of chicken CoV (Table 1). The oligos were purified using PAGE purification and purchased from (Integrated DNA Technologies, Inc., USA).
Table 1.
Oligonucleotide primers, probes, and templates used in this study
| Species | Type | Sequence information (5’–3’) | Size |
|---|---|---|---|
| IAV H1N1, M gene | F: | Acrydite-GACCRATCCTGTCACCTCTGAC | 106 bp |
| R: | AGGGCATTYTGGACAAAKCGTCTA | ||
| P: | 6-FAM/CACCGTGCCCAGTGAGCGAGGACT/BHQ-1 | ||
| T: | AGGGCATTTTGGACAAAGCGTCTACGCTGCAGTCCTCGCTCACTGGGCACGGTGAGCGTGAACACAAACCCCAAAATCCCCTTAGTCAGAGGTGACAGGATTGGTC | ||
| Chicken CoV, 5’UTR | F: | Acrydite-GCTTTTGAGCCTAGCGTT | 143 bp |
| R: | GCCATGTTGTCACTGTCTATTG | ||
| P: | FAM-CACCACCAGAACCTGTCACCTC-BHQ_1 | ||
| T: | GCTTTTGAGCCTAGCGTTGGGCTACGTTCTCGCACA AGGTCGGCTATACGACGTTTGTAGGGGGTAGTGCCA AACAACCCCTGAGGTGACAGGTTCTGGTGGTGTTTA GTGAGCAGACATACAATAGACAGTGACAACATGGC |
F is a forward primer, R is a reverse primer and P is a single quenched TaqMan probe consisting of 6-FAM molecule as reporter dye at 5’end and BHQ-1 as a quencher at the 3’ end of the oligonucleotide. T is amplicon information of the targets
Preparation of PIN Particles
The pre-polymer solution and spotting procedures for PIN preparation were carried out as previously described [28, 29]. The manufactured PIN particles for the M gene were used for both synthetic and virus experiments. Each viral-specific PIN particle was functionalized with 5’ acrydite modified gene-specific forward primers for the M gene and 5’UTR for IAV and chicken CoV, respectively (Table 1). Experiments involving freeze-dried (FD-PIN) particles aimed at increasing PIN absorption affinity of the analyte solution, with an interest in examining its contribution to dirRT-qPCR performance and its suitability for point of care applications. The particles were frozen at − 80 °C for 1 to 4 h to allow the assessment of freezing time that gives stable Ct value and better detection signal strength over time at room temperature storage. The process was followed by drying PIN particles at 5 millitorrs using a freeze drier FD-1000 Eyela (Rikakikai Co. Ltd, Tokyo, Japan) for 2 h.
Synthetic RNA Template and Virus Preparation
Synthetic RNA (1.39 × 109 copies/µL) was synthesized and purchased from (Integrated DNA Technologies, Inc., USA) (Table 1), while (0.2%) formalin-inactivated IAV (108.8 EID50/mL) and chicken CoV (106.5 EID50/mL) stock solutions were kindly provided by the Avian disease and infectious disease laboratory, Konkuk University, Seoul, Korea. Samples spiked with RNA template were mixed with 20 U/µL of RNA inhibitor, RiboLock RNase Inhibitor (Thermal Fisher Scientific Inc. Vilnius, Lithuania) in a ratio of 1:1:1 (1 µL RNA + 1 µL saliva + 1 µLRNA inhibitor). The mixture was then heat-treated at 100 °C for 1, 3, 5, and 10 min for pseudo samples to assess possible debris and PCR inhibitors reduction, and 100 ºC for 10, 30, 60, 120, and 180 s, for viral lysis time. In the case of the addition of RTase enzyme experiments, 200 U/µL of RevertAid™ RTase (Thermal Fisher Scientific Inc. Vilnius, Lithuania) was used, in the same ratios (1 µL RNA + 1 µL saliva + 1 µL RNA inhibitor) + 1 µL RTase. RNase-free water was used as a no template control (NTC). The T100™ Thermal cycler (Bio-Rad, Inc. Korea) was used for heat treatment and the resulted solutions were used as templates for RT-qPCR. The dirRT-qPCR was performed and performance evaluated to identify suitable holding time for debris reduction and virus lysis. In addition, the roles played by RNA inhibitor and RTase enzyme to improve the assay performance were observed.
Direct RT-qPCR
All the amplification reactions for both solution and PIN particles were TaqMan probe-based assays. The reaction mix for solution RT-qPCR contained 1.6 µL of 10 µM for each forward and reverse primers, 1.6 µL of 10 µM TaqMan probe, 2 µL of template, 1.2 µL distilled water, and 8 µL of one-step 2 × RT-PCR Master mix (TaqMan, RNA) (Micobiomed Co. Ltd, Seoul, Korea). For PIN particles, 4 particles were inserted into the microfluidic Lab Chip channels, Veri-Q PCR 204 (Micobiomed Co. Ltd, Seoul, Korea). Then, was incubated at 4 °C for 30 min with 1.6 µL of 10 µM free reverse primer, 1.6 µL of 10 µM TaqMan probe, 3 µL of template, 1.8 µL distilled water, and 8 µL of similar PreMix. Except for FD-PIN particles, the RT-qPCR was set directly without 30-min incubation. We covered particles with 16 µL of mineral oil to contain fluorescence of amplification signals within individual particles. Thermal cycling conditions were as follows: 42 °C for 10 min, followed by 40 cycles at 95 °C for 8 s, 95 °C for 4 s, and 58 °C for 30 s, with combined annealing and extension steps.
Mass Sample Testing for Assay Evaluation
Rope saliva samples (n = 25) were prepared for analysis similar to pseudo-samples and spiked with inactivated IAV. The dirRT-qPCR performance using FD-PIN particles was evaluated by running in parallel with conventional solution-based RT-qPCR for samples extracted using the column-based nucleic acid extraction method (QIAamp viral RNA Mini Kit) as a standard assay. We run (n = 12) samples with the modified protocol for PIN dirRT-qPCR to enhance its performance following the increased sample input volume for QIAamp Kit. During heat treatment samples contained 1:1:0.1 ratios for the virus, saliva, and 2 × RNA inhibitor, respectively. The dirRT-qPCR reaction mixture contained 1 μL of 10 μM for each reverse primer and probe, 1 μL of RTase, 5 μL of template, and 8 μL of one-step 2 × RT-PCR Master mix (TaqMan, RNA) (Micobiomed Co. Ltd, Seoul, Korea).
Chicken CoV Testing for Assay Validation
To confirm this newly developed testing protocol, we decided to use another RNA virus instead of IAV. We used inactivated chicken CoV (strain M41) to spike into a similar matrix, and specific FD-PIN particles were produced to detect the 5’ UTR of chicken CoV. We tested all the variables as we did for IAV, followed by amplicon size verification using conventional PCR and nucleotide sequencing to confirm the specificity of the assay.
PIN Particle’s Amplicon Verification
To remove the mineral oil, the PIN particles used for dirRT-qPCR were washed five times with PBS containing 0.05% Tween 20. The particles were used to provide template DNA for conventional PCR to confirm the size of amplicons generated in the PIN particles. PCR was done using a T100™ Thermal cycler (Bio-Rad, Inc. Kaki Bukit, Singapore), with thermal cycling conditions as follows: 95 °C for 5 min followed by 34 cycles at 95 °C for 1 min, 56 °C for 30 s, and 72 °C for 1 min. The reaction mix contained one PIN particle, 2 μL of 10 µM for each forward and reverse primer, and 16 μL of distilled water into AccuPower® PCR PreMix (Bioneer Corp, Daejeon, Korea). Then, 5 μL of the PCR product were loaded into 3% agarose gel stained with 3 μL of SYBR® Safe DNA gel stain (Invitrogen, Thermal Fisher Scientific, Carlsbad, CA, USA), and pictured under the UV light.
Nucleotide Sequencing
To confirm the identity of the dirRT-qPCR products, we sent the 143 bp DNA bands for the 5’UTR gene of chicken CoV obtained to Bioneer corporation (Daejeon, Korea) for nucleotide sequencing. The chromatograms for both forward and reverse primers were read and assessed for their quality using Sequence Scanner software v2.0 (Applied Biosystems, Foster, CA, USA), and the consensus sequences were assembled using BioEdit software v7.2. The nucleotide identity of the viral sequences was verified using BLASTn search against the NCBI GenBank database.
Data Analysis
All the raw data were automatically extracted in Microsoft Office-Excel 2016 (Microsoft, USA), from the Ultra-fast real-time PCR (G2-4) System (Micobiomed Co. Ltd, Seoul, Korea) computer software. We applied simple descriptive statistics such as mean and range to analyze our data. We also applied linear regression to find a correlation between Ct values and template concentration. The originLab®2020 software, Northampton, USA, was used for data analysis and drawing the curves.
Results and Discussion
Comparison Between PIN and Solution Phase dirRT-qPCR
In (Fig. 1), we use the schematic diagram to demonstrate the steps for PIN dirRT-qPCR. These are sample pre-processing through heat treatment, followed by amplification and detection in real-time within PIN particles loaded into a microfluidic qPCR chip. The PIN particles are highly porous 3-D hemispheres, with a diameter of approximately 500 µm and a height of 120 µm. They possess the nanopores well permeable for nanoparticles of radius around 10 nm, based on the hydrodynamic radius of fluorescein isothiocyanate (FITC)-labeled dextran nanoparticles of 150 kDa used for diffusivity experiments (Fig. SI2) [30]. The nanopores filter in to fill approximately 80% of PIN volume with dilute analyte solution that includes reagents and target RNA for amplification [29]. The PIN particles were made as previously described by our group [28, 29]. In the same way, we developed specific PIN particles for two viral targets used in this study.
Fig. 1.

Schematic presentation of PIN particle dirRT-qPCR of viral RNA from saliva samples. The sample is heat-treated to release viral RNA for RT, amplification, and detection inside PIN particles. Following viral lysis, the mixture is mixed with one-step RT-qPCR reagents and loaded into a microfluidic qPCR chip containing PIN particles. The target viral RNAs are captured and selectively reverse-transcribed using immobilized acrydite forward primer, followed by 40 cycles of qPCR using PCR primer (reverse primer) and detection in real-time
We observed different amplification curve characteristics when saliva samples spiked with synthetic RNA for IAV were directly analyzed by both methods (Fig. 2). The saliva matrix delayed the RT-qPCR amplification signals substantially (ΔCt > > 7) (Fig. SI3b), corresponding to reports suggesting saliva components interfere with the PCR process [13, 14]. However, DNA qPCR of the saliva matrix underwent no meaningful negative effects (Fig. SI3a). Contrary to those reports, the findings suggest that the RT process is mainly vulnerable to the components of the matrix, and their effects on the RT step have not been extensively explored 14. This finding suggests the need for a special pre-processing method for this particular matrix or technology to improve assay sensitivity, especially with RNA viruses in real samples. In addition, jagged amplification curves were observed following conventional solution-based one-step RT-qPCR with saliva samples spiked with synthetic RNA (Fig. 2b). Contrary to conventional assay, the pattern of amplification signals showed consistency in terms of clear Ct values, increased signal intensity, and typical sigmoidal curves with PIN dirRT-qPCR (Fig. 2b). We believe the physical and chemical characteristics of PIN particles together with other parameters optimized in this work have enabled the method to outclass the challenges observed in (Fig. 2b) for solution-based one-step RT-qPCR. The hydrophilic and porous nature enhances similar efficiency and amplification rates with typical qPCR sigmoid curves when using PIN particles as to aqueous media qPCR platforms. The filtering process of the analyte solution by the PIN particle plays a great role in keeping enzyme reaction stable, as observed during diffusivity experiments done to assess the sieving properties of the particles (Fig. SI2). Bulky materials could not approach the particles’ inner volume, such as dextran nanoparticles. At the same time, it enables the covalently immobilized RT primers to capture the desired concentration of high-quality RNA from dilute micro-volumes of the analyte solution, ready the for RT process of the viral RNA. Due to the higher concentration of RT primers involved during this process, an enormous amount of double-stranded cDNA is made available as templates for subsequent qPCR. This process is an added advantage for the PIN method, as the actual initial template concentration increases in PIN volume for efficient qPCR performance [31].
Fig. 2.

Differences between solution-based and PIN dirRT-qPCR using synthetic RNA without any pre-treatment procedures. a Synthetic RNA only. b Rope saliva spiked with synthetic RNA in a 1:1 ratio
Heat-Treatment Pre-processing
Generally, RT-qPCR was succeeded after the introduction of heat treatment as a pre-process for viral lysis, which exposes out target RNA for both RT and the amplification process. Since 1981 [21], it was demonstrated that brief exposure of bacterial cells to 100 °C for a few seconds releases bacterial plasmid, reduces debris, and inactivates nuclease enzymes. More recently, several dirRT-qPCR protocols preceded by heat treatment for viral lysis have been suggested for COVID-19 diagnosis [19, 20, 32]. For the same reasons, we had to find an optimum temperature and holding time for effective viral lysis by assimilation using synthetic RNA. Heat treatment played a role as the solemnly viral lysis method as well as to reduce the effects of debris and heat susceptible inhibitors [18–20]. Contrarily, the assay sensitivity never improved following heat treatment at 100 °C, despite a time increase from 1 to 10 min. There was no significant difference in Ct values between 1 and 3 min of heat treatment, while the amplification curves for 5 and 10 min heat treatment of spiked samples were far behind that of control by an average Ct difference of 3.36 units from 22.18 to 25.54 (Fig. 3). This kind of either gradual or heat-induced inhibition was consistently observed when either rope or pure saliva collected from pigs and human saliva samples were used. A similar observation was made by Ochert et al. [13], when working with human saliva and linked the inhibition with polysaccharides. Apart from Ochert’s report, there are no recent reports for saliva containing a specific PCR inhibitory ingredient [14]. As in (Fig. SI3b), it is obvious that RT is a delicate step and vulnerable to the saliva matrix; at the same time longer heat treatment incubation aggravated more delayed Ct values, while shorter heat treatment time was advantageous in terms of reducing the effect of saliva matrix. To avoid RNA degradation if it occurs during heat treatment, an RNase inhibitor was added (data shown in ‘use of RNase inhibitor’). The amplification was consistent regardless of RNase inhibitors. It was inferred that the slowed amplification is not mainly due to RNA degradation during the period of heat treatment.
Fig. 3.
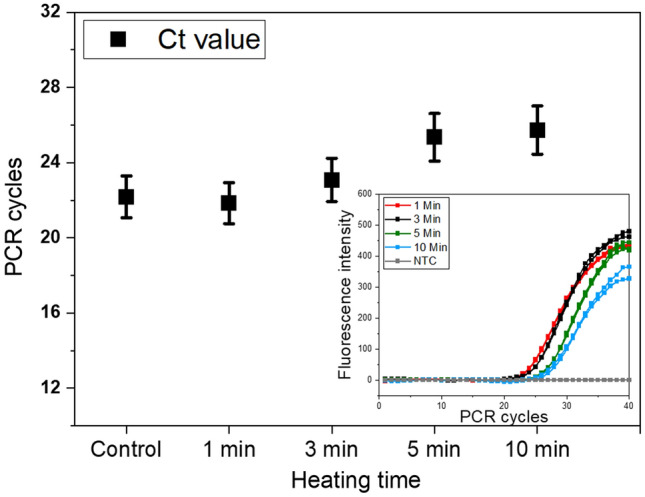
Heat treatment of rope saliva samples at 100 °C for 3 min had consistent Ct values. The control sample contained (RNA + saliva + RNase inhibitor), with no heat treatment
Virus Lysis
The use of RNA virus came after having collected enough reference values from synthetic RNA template experiments. This experiment was important to evaluate the performance of direct amplification of viral RNA using PIN particles. Natural viruses required a brief holding time to completely lyse to release viral RNA for amplification. Using solution-based one-step RT-qPCR, we found that at 100 °C heat treatment and a holding time ranging from 10 to 180 s were sufficient for viral lysis. We adopted 100 °C for 30 s (Table 2), for the sake of utilizing the minimum effective holding time, although there was no sensitivity difference between 10 and 30 s when analyzed with PIN dirRT-qPCR. A rope saliva sample spiked with virus stock without heat treatment was used as a control to evaluate the effectiveness of viral heat treatment.
Table 2.
Evaluation of viral lysis holding time following heat treatment at 100 °C
| Heat treatment for viral lysis | |||||||
|---|---|---|---|---|---|---|---|
| Time in (sec) | 10 | 30 | 60 | 120 | 180 | RNA | Control |
| Average Ct values | 16.48 | 17.26 | 18.57 | 19.02 | 17.70 | 16.12 | 27.11 |
Use of RNase Inhibitors
The harsh environment that RNA was subjected to during heat treatment amid a complex matrix prompted the use of RNase inhibitor to protect the unstable RNA and to improve the sensitivity. We adjusted enzyme concentration to 20 U/µL, which seemed to provide protective outcomes to RNA in the absence of rope saliva. On the other hand, when kept under the pressure of the matrix, there was no clear evidence for the change in assay sensitivity. Out of four samples tested in duplicate (n = 8), only one sample had a significantly improved Ct difference of an average of 3.16 Ct units from 19.40 to 16.24. The mean Ct value was 15.61 ranging from 13.9 to 16.24 as compared to samples tested without RNase inhibitor had a mean Ct value of 16.7 ranging from 13.9 to 19.4. In such a scenario, where both heat treatment and RNase inhibitor do not significantly show a change in the quality of the assay sensitivity, we opted to use the enzyme in all the experiments for protective purposes. There are evidences that suggests the use of either RNase inhibitors or stabilizers protects the unstable RNA during analysis [1, 6, 33].
Optimization of RT Process
As the RT step is sensitive to saliva, stabilization of the RT step is very important, especially for those with very low concentrations of target RNA. The resolution of RT-qPCR depends highly on priming strategy, RT temperature and time, RNA concentration, and its conversion rate to cDNA by the RTase enzyme [34]. The RT temperature and time were carefully examined,where 42, 50, and 55 °C for either 5 or 10 min were evaluated. Finally, we opted for 42 °C for 10 min that had consistent better amplification results. We did not observe any change in qPCR performance when DNA spiked with saliva was analyzed using conventional solution-based qPCR (Fig. SI3a), as compared to RNA spiked with saliva in RT-qPCR. Where, low sensitivity with significantly pushed Ct values and reduced signal strength to RT-qPCR was observed (Fig. SI3b). We decided to introduce a chemical intervention to see whether sensitivity will improve significantly, with reports suggesting higher enzyme concentrations reduce inhibition effects [15]. The addition of 200 U/µL of RTase from an external source made a considerable improvement in RT-qPCR sensitivity. Out of four samples tested in duplicate (n = 8), 3/4th of the samples had pulled Ct values to different extents (Fig. 4). The mean Ct difference between samples with and without RTase was 3.91 ranging from 0.88 to 6.63. The mean Ct value was 13.43 ranging from 12.12 to 14.79 for samples with added RTase as compared to samples tested without the addition of RTase, mean Ct value of 17.34 ranging from 13.39 to 20.91 (Fig. 4). Perhaps, there were limited resources for optimum reaction in the selected PCR PreMix, while considering the small reaction volume available in PIN particles. We think the following must be considered for better assay output, PCR PreMix or RTase selection, optimum enzyme concentrations, RNA input quantity, and integrity. This is also suggestive of the presence of active inhibitory ingredients interfering with the enzymatic reaction of the RT step. We have been working with both rope saliva [2] and partly human saliva. The two types of saliva behave differently; with early findings, we think there might be more of the inhibitors/RNases in human saliva than we have observed in swine saliva (Unpublished data). As we are looking for future use of this matrix for diagnostic purposes, we desire to see a thorough exploration of these PCR inhibitors accomplished to enhance its utilization for molecular diagnostics.
Fig. 4.

Addition of an external RTase enzyme bolstered the PIN particle’s dirRT-qPCR performance. About 3/4 th of the samples (a–c) had pulled Ct values away from the control (saliva + RNA)
Stability of the Matrix
Before optimization of all the parameters, we needed assurance of matrix stability for consistent evaluation. We used an RNase inhibitor throughout this experiment, which supported the stability of the matrices. Understanding the advantages of adhering to proper matrix storage procedures when considering RNA purification, ensures both the matrix and RNA remain intact during analysis. We found that matrix could stay active and reliable in terms of inhibitory effects within an average shaking zone of ± 1 Ct value for up to 10 days when stored at 4 °C without using an RNase inhibitor (Fig. 5). Beyond that, we experienced unstable Ct values as the matrix effect changed and method evaluation becomes less reliable [11]. Our findings are in line with several other studies on matrix or RNA stability studies. It was previously reported that porcine reproductive and respiratory syndrome virus RNA was stable under similar conditions for more than 7 days [6]. Therefore, in all experiments, we used freshly collected samples within 10 days from the date of collection.
Fig. 5.
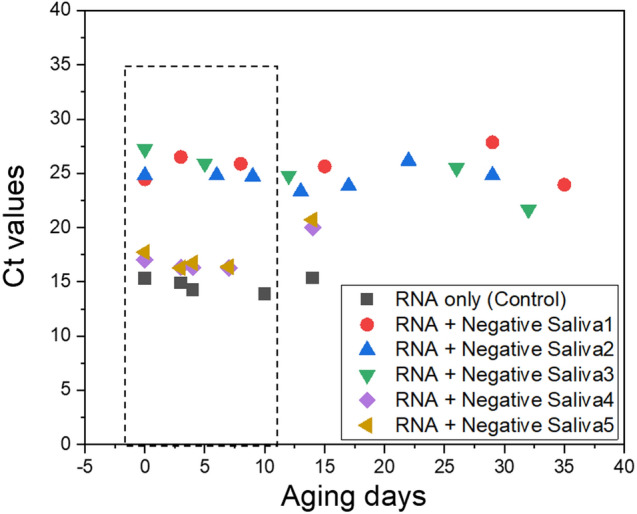
Aging test results for rope saliva samples (n = 5) using solution-based one-step RT-qPCR. The storage period should not exceed 10 days at 4 °C
Thus, PIN particles have efficiently managed to perform dirRT-qPCR of synthetic RNA spiked into this complex matrix. The PIN dirRT-qPCR succeeded after the introduction of heat treatment incubation at 100 °C for 30 s for viral lysis. Addition of both RNase inhibitors for RNA protection and external RTase enzyme to increase the rate of cDNA synthesis provided obvious signs of improved assay performance.
Method Evaluation Using RNA Viruses
Mass Sample Testing
Following its potential for onsite application, we analyzed non-centrifuged mass samples aiming for more method evaluation. We did a parallel testing experiment to assess the performance of FD-PIN dirRT-qPCR against the QIAamp Viral RNA Mini Kit viral RNA extraction, followed by conventional solution-based one-step RT-qPCR. We experienced PCR inhibition of up to 40% of samples (n = 25) when saliva was used undiluted, as compared to synthetic RNA experiments where the inhibition was not evident. The matrix dilution ratio of about 1:9 (10% v/v) broadened viral detection coverage to 100%, with improved Ct value by an average of 2.79 units. The Ct drooped from an average of 30.98 Ct units (ranging from 22.08 to 40.0) to 28.08 (ranging from 25.59 to 31.1) as a result of 10% dilution of the matrix (Fig. 6). These results differ from those of other studies where the saliva matrix was analyzed undiluted, or diluted during RNA purification with lysis reagents when column-based RNA purification kits are used (e.g. QIAamp Mini Viral Kit). Our findings are exactly similar to observations reported by Ambers and his group [14] on human saliva samples for forensic investigations. This means that the type of technology used for sample analysis is critical in deciding whether matrix dilution is necessary.
Fig. 6.
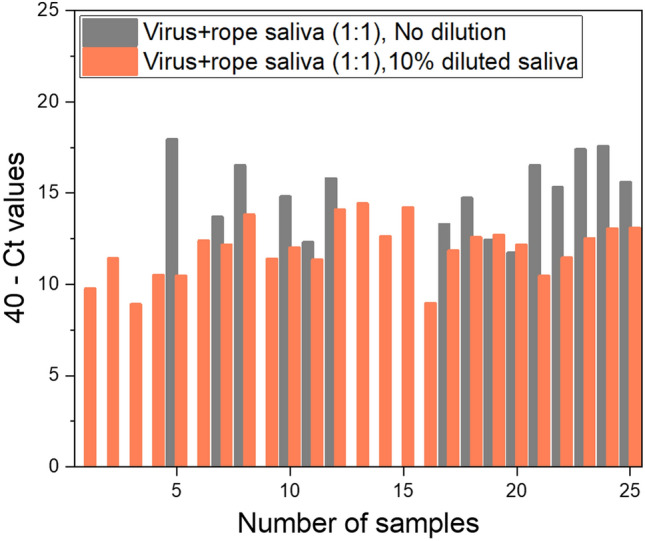
Forty percent of the undiluted rope saliva samples had RT-qPCR inhibited with PIN dirRT-qPCR (n = 25). The trend resolved following 10% v/v dilution of the matrix
Method Validation with IAV
We ran a parallel experiment using FD-PIN particles for dirRT-qPCR, against QIAamp Kit as the standard method to detect inactivated IAV spiked into rope saliva. The FD-PIN dirRT-qPCR displayed a better performance, with an average Ct difference of 3.05 units lower than the QIAamp Kit (Fig. 7a). The mean Ct values were 25.76 ranging from 23.71 to 27.79, and 28.81 ranging from 28.15 to 29.7 for dirRT-qPCR with FD-PIN particles and QIAamp Kit, respectively. The addition of RTase resulted in an improvement of 1.72 Ct units, from an average Ct difference of 1.33 units between the methods when similar samples were analyzed without the addition of RTase (Fig. SI4). The enhanced performance is a result of the increased rate of converting viral RNA to cDNA within the PIN volume, meaning that thorough optimization of the RT step is critical. Considering the smaller reaction volume for the PIN particles, it is important for RTase concentration, RT temperature, and holding time to be at the optimum for good performance of the RT step before qPCR [31, 34, 35]. However, with increased sample input volume, the sensitivity of the QIAamp Kit was improved with an average of 2 Ct units ahead of PIN dirRT-qPCR (Fig. SI5). This result depicts the reason for a tenfold difference in LOD between the methods (Fig. SI6). Despite its relatively low sensitivity, which depends on sample input volume, PIN dirRT-qPCR stands to be a simpler method with reduced sample processing steps for direct amplification without prior RNA purification process. The method has outclassed several challenges related to the existing nucleic acid extraction methods. Taking advantage of immobilized affinity bait, the 5’ acrylate modified primer acts as both RT and a gene-specific primer. Apart from its amplification roles, the primer is also responsible to capture and protect the target RNA within PIN particles [28, 36] fabricated from biologically friendly pre-polymers (PEG and PEGDA), which do not permit non-specific interactions, making them safer for use [36]. In addition, the method has a shorter protocol by 30 min, as compared to normal PIN particles and QIAamp Kit methods. It looks more promising and suitable onsite diagnostic technology for direct viral RNA amplification from complex matrices.
Fig. 7.

Comparison between FD-PIN method and QIAamp Viral Mini Kit during mass samples testing. a Average Ct values for the two methods, with FD-PIN particle presenting with an average of 3.05 units ahead of QIAamp kit. Gel electrophoresis results show 106 bp bands from rope saliva samples spiked with IAV using centrifuge off samples. b FD-PIN dirRT-qPCR. c QIAamp Viral Mini Kit followed by solution-based one-step RT-qPCR
The average signal strength was remarkably higher for FD-PIN dirRT-qPCR as compared to both the QIAamp Kit method and the normal PIN particles as control. The mean signal strength was 316.7 and 76.3 a.u, for FD-PIN particles and QIAamp Mini Viral Kit, respectively (Fig. 8a). Normally, there is a direct correlation between signal intensity and PCR product concentration, the phenomenon that enhances the assay sensitivity [37, 38]. We assume the huge difference in signal strength is a result of the physical characteristics of the hydrogels and higher concentration of amplicons within 3D FD-PIN particles (Fig. 8b, c). Sorokin and his group reported collecting more fluorescence signals from 3D surfaces, which enhanced assay sensitivity as compared to 2D reaction planes [36, 39].
Fig. 8.

Detection signal strength between the two methods. a The difference in amplification signal intensities between FD-PIN particles and QIAamp Kit, with the normal PIN particles standing as the control (n = 25). b, c Middle section of the microfluidic chip showing detection signal strength differences between PCR cycles 1 and 40. b FD-PIN dirRT-qPCR; c Solution-based one-step RT-qPCR following QIAamp Kit viral RNA extraction
Method Validation with Chicken CoV
The developed method was also validated using chicken CoV, another RNA virus of the genus Gamma coronavirus. Using the same sample ratio and volume, specific target detection was achieved with an average of 3.02 Ct units lower than the QIAamp Kit for chicken CoV using FD-PIN dirRT-qPCR (Fig. 9a). The mean Ct value was 24.23 ranging from 23.46 to 24.79 for PIN dirRT-qPCR as compared to QIAamp Kit with a mean Ct value of 27.25 ranging from 25.7 to 28.8. These results look similar to those observed for IAV when the same sample input protocol was used. The PIN method has displayed reproducibility of test results between the two viral targets, showing its capability to detect other lethal RNA viral pathogens of both human and animal importance. In the era, where we are experiencing an increased number of emerging pathogens, like the current pandemic by SARS-CoV-2 of the COVID-2019, and the former MERS-CoV both from the genus Beta coronavirus and many others, they can be easily diagnosed using this method. Referring to the two model viruses, having relatively the same size and structural similarities, we think the method will be suitable for other viruses with such similar features. The specific target gene for chicken CoV was confirmed following conventional second PCR, where DNA bands of 143 bp specific to the 5’ UTR gene were recovered, purified, and sequenced (Fig. 9b, c). Following BLASTn for identity search, all the sequences were 100% identical to chicken CoV strain M41 from China with an accession number MK937830. This has proved that dirRT-qPCR of viral RNA from a complex matrix was very specific and suitable for other downstream applications regardless of the use of a centrifuge.
Fig. 9.

Method validation using chicken CoV with the same protocols as for IAV. a Ct values obtained from rope saliva samples spiked with inactivated chicken CoV for both FD-PIN and QIAamp Viral Mini Kit RNA. b Gel electrophoresis results showing 143 bp amplicon for 5’ UTR of chicken CoV detected using FD-PIN. c Gel results for viral RNA extraction from similar samples with QIAamp Viral Mini Kit, using both centrifuged (Lane 1 and 2) and non-centrifuge samples (Lane 3 to 5)
Limitations of the Study
There are several notable limitations in the current study which are worth to be shared. Among others is the use of a low sample size to optimize various variables in this study as a proof of concept, as well as the failure to obtain and test field clinical samples to further the validation process. The final validation step using virus stock solution was only performed in moderate to high viral loads. The low viral loads may have demonstrated different results considering the low LOD for the QIAamp Kit. Finally, the use of chicken CoV was only for the sake of observing reproducibility and suitability for use with other RNA viruses. We understand the virus was not the right model to use in swine rope saliva, as the virus is normally diagnosed using cloacal or stool samples.
Conclusion
To our understanding, this is the first report on the application of PIN particles for dirRT-qPCR of viral RNA from non-invasive and a complex matrix. The assay has enabled efficient amplification and detection of a specific target from dilute sample solution without the use of toxic reagents, RNA carriers, and other PCR enhancers and is capable without sample clarification by using a centrifuge. Integration with a highly specific detection system and compatibility with microfluidic chip technologies has facilitated rapid pathogen detection in less than an hour. The technology will be ideal for on-site diagnostic applications with the availability of a simple and compact device suitable for decentralized amplification process, as compared to the currently used platform.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank the technical support of Bok-Kyung Ku, the Veterinary Researcher at the Quarantine Office, Daejeon, Korea.
Author Contributions
Conceptualization: SKK; methodology: EGK, MJK, SJ, J-YN, C-SS, and SKK; formal analysis: EGK and MJK; validation: EGK, MJK, SJ; data curation: EGK, and MJK; writing original draft preparation: EGK; writing—review and editing: GM and SKK; Supervision: GM, and SKK; project administration: SKK; all authors have read and approved the final manuscript.
Funding
The work was partly funded by the National Research Foundation of Korea (NRF) (Grant no. 2018r1a2a1a05077112, 2016M3A9B6918639) and the Korean government (MSIP) through the R&D Convergence Program of the National Research Council of Science & Technology of the Republic of Korea (CAP-16-02-KIST). EGK is a recipient of a scholarship from the Government of Tanzania through the World Bank (WB-ACE II Grant PAD1436, IDA Credit 5799-TZ) and the Regional Scholarship and Innovation Fund (RSIF) of the Partnership for Skills in Applied Sciences, Engineering, and Technology (PASET).
Declarations
Conflict of interest
The authors declare to have no Conflict of interest/Competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Park NJ, Li Y, Yu T, Brinkman BM, Wong DT. Characterization of RNA in saliva. Clin. Chem. 2006;52(6):988–994. doi: 10.1373/clinchem.2005.063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prickett JR, Kim W, Simer R, Yoon K-J, Zimmerman J. Production. Oral-fluid samples for surveillance of commercial growing pigs for porcine reproductive and respiratory syndrome virus and porcine circovirus type 2 infections. JoSHP. 2008;16(2):86–91. [Google Scholar]
- 3.Henao-Diaz A, Gimenez-Lirola L, Baum DH, Zimmerman J. Guidelines for oral fluid-based surveillance of viral pathogens in swine. Porcine Health Manage. 2020;6:28. doi: 10.1186/s40813-020-00168-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong DT. Salivary Diagnostics: amazing as it might seem, doctors can detect and monitor diseases using molecules found in a sample of spit. Am. Sci. 2008;96(1):37–43. doi: 10.1511/2008.69.3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Detmer SE, Patnayak DP, Jiang Y, Gramer MR, Goyal SM. Detection of Influenza A virus in porcine oral fluid samples. J. Vet. Diagn. Invest. 2011;23(2):241–247. doi: 10.1177/104063871102300207. [DOI] [PubMed] [Google Scholar]
- 6.Decorte I, Van der Stede Y, Nauwynck H, De Regge N, Cay AB. Effect of saliva stabilizers on detection of porcine reproductive and respiratory syndrome virus in oral fluid by quantitative reverse transcriptase real-time PCR. Vet. J. 2013;197(2):224–228. doi: 10.1016/j.tvjl.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Fabryova H, Celec P. On the origin and diagnostic use of salivary RNA. Oral Dis. 2014;20(2):146–152. doi: 10.1111/odi.12098. [DOI] [PubMed] [Google Scholar]
- 8.Tvarijonaviciute A. Saliva in health and disease: the present and future of a unique sample for diagnosis. Cham: Springer Nature; 2020. [Google Scholar]
- 9.Martinez H, Beaudry G, Veer J, Robitaille M, Wong D, Iverson B, et al. Ambient temperature storage of RNA in GenTegra™ for use in RT-qPCR. Biotechniques. 2010;48(4):328–329. doi: 10.2144/000113409. [DOI] [Google Scholar]
- 10.Relova D, Rios L, Acevedo AM, Coronado L, Perera CL, Perez LJ. Impact of RNA degradation on viral diagnosis: an understated but essential step for the successful establishment of a diagnosis network. Vet. Sci. 2018;5(1):19. doi: 10.3390/vetsci5010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sullivan R, Heavey S, Graham DG, Wellman R, Khan S, Thrumurthy S, et al. An optimised saliva collection method to produce high-yield, high-quality RNA for translational research. PLoS ONE. 2020;15(3):e0229791. doi: 10.1371/journal.pone.0229791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schrader C, Schielke A, Ellerbroek L, Johne R. PCR inhibitors - occurrence, properties and removal. J Appl Microbiol. 2012;113(5):1014–1026. doi: 10.1111/j.1365-2672.2012.05384.x. [DOI] [PubMed] [Google Scholar]
- 13.Ochert A, Boulter A, Birnbaum W, Johnson N, Teo C. Inhibitory effect of salivary fluids on PCR: potency and removal. PCR Methods Appl. 1994;3(6):365–368. doi: 10.1101/gr.3.6.365. [DOI] [PubMed] [Google Scholar]
- 14.Ambers A, Wiley R, Novroski N, Budowle B. Direct PCR amplification of DNA from human bloodstains, saliva, and touch samples collected with microFLOQ((R)) swabs. Forensic. Sci. Int. Genet. 2018;32:80–87. doi: 10.1016/j.fsigen.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Chittick WA, Stensland WR, Prickett JR, Strait EL, Harmon K, Yoon K-J, et al. Comparison of RNA extraction and real-time reverse transcription polymerase chain reaction methods for the detection of porcine reproductive and respiratory syndrome virus in porcine oral fluid specimens. J. Vet. Diagn. Invest. 2011;23(2):248–253. doi: 10.1177/104063871102300208. [DOI] [PubMed] [Google Scholar]
- 16.Park NJ, Yu T, Nabili V, Brinkman BM, Henry S, Wang J, et al. RNAprotect saliva: an optimal room-temperature stabilization reagent for the salivary transcriptome. Clin. Chem. 2006;52(12):2303–2304. doi: 10.1373/clinchem.2006.075598. [DOI] [PubMed] [Google Scholar]
- 17.Scipioni A, Mauroy A, Ziant D, Saegerman C, Thiry E. A SYBR Green RT-PCR assay in single tube to detect human and bovine noroviruses and control for inhibition. Virol. J. 2008;5:94. doi: 10.1186/1743-422X-5-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rames E, Roiko A, Stratton H, Macdonald J. DNA heat treatment for improving qPCR analysis of human adenovirus in wastewater. Food Environ. Virol. 2017;9(3):354–357. doi: 10.1007/s12560-017-9294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mancini F, Barbanti F, Scaturro M, Errico G, Iacobino A, Bella A, et al. Laboratory management for SARS-CoV-2 detection: a user-friendly combination of the heat treatment approach and rt-Real-time PCR testing. Emerg. Microbes Infect. 2020;9(1):1393–1396. doi: 10.1080/22221751.2020.1775500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fomsgaard AS, Rosenstierne MW. An alternative workflow for molecular detection of SARS-CoV-2—escape from the NA extraction kit-shortage, Copenhagen, Denmark, March 2020. Euro Surveill. 2020 doi: 10.2807/1560-7917.ES.2020.25.14.2000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holmes DS, Quigley M. A rapid boiling method for the preparation of bacterial plasmids. Anal. Biochem. 1981;114(1):193–197. doi: 10.1016/0003-2697(81)90473-5. [DOI] [PubMed] [Google Scholar]
- 22.King C, Debruyne R, Kuch M, Schwarz C, Poinar H. A quantitative approach to detect and overcome PCR inhibition in ancient DNA extracts. Biotechniques. 2009;47(5):941–949. doi: 10.2144/000113244. [DOI] [PubMed] [Google Scholar]
- 23.Barbosa, C., Nogueira, S., Gadanho, M., Chaves, S.: DNA extraction: finding the most suitable method. In: Molecular Microbial Diagnostic Methods, pp. 135–54 (2016)
- 24.Cheng HR, Jiang N. Extremely rapid extraction of DNA from bacteria and yeasts. Biotechnol. Lett. 2006;28(1):55–59. doi: 10.1007/s10529-005-4688-z. [DOI] [PubMed] [Google Scholar]
- 25.Tan SC, Yiap BC. DNA, RNA, and protein extraction: the past and the present. J. Biomed. Biotechnol. 2009;2009:574398. doi: 10.1155/2009/574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, He JA, Wang W, Xia Y, Song L, Chen ZH, et al. Development of a direct reverse-transcription quantitative PCR (dirRT-qPCR) assay for clinical Zika diagnosis. Int. J. Infect. Dis. 2019;85:167–174. doi: 10.1016/j.ijid.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Kang K, Yang K, Zhong J, Tian Y, Zhang L, Zhai J, et al. A direct real-time polymerase chain reaction assay for rapid high-throughput detection of highly pathogenic North American porcine reproductive and respiratory syndrome virus in China without RNA purification. J. Anim. Sci. Biotechnol. 2014;5(1):45. doi: 10.1186/2049-1891-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh EH, Jung S, Kim WJ, Kim KP, Kim SK. Microparticle-based RT-qPCR for highly selective rare mutation detection. Biosens. Bioelectron. 2017;87:229–235. doi: 10.1016/j.bios.2016.08.057. [DOI] [PubMed] [Google Scholar]
- 29.Jung S, Kim J, Lee DJ, Oh EH, Lim H, Kim KP, et al. extensible multiplex real-time PCR of microRNA using microparticles. Sci. Rep. 2016;6:22975. doi: 10.1038/srep22975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen H, Hao J, Li SK. Characterization of human sclera barrier properties for transscleral delivery of bevacizumab and ranibizumab. J. Pharm. Sci. 2013;102(3):892–903. doi: 10.1002/jps.23387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim MY, Jung S, Kim J, Lee HJ, Jeong S, Sim SJ, et al. Highly sensitive and multiplexed one-step RT-qPCR for profiling genes involved in the circadian rhythm using microparticles. Sci. Rep. 2021;11(1):6463. doi: 10.1038/s41598-021-85728-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kriegova E, Fillerova R, Kvapil P. Direct-RT-qPCR detection of SARS-CoV-2 Without RNA extraction as part of a COVID-19 testing strategy: from sample to result in one hour. Diagnostics (Basel) 2020;10(8):605. doi: 10.3390/diagnostics10080605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reck M, Tomasch J, Deng Z, Jarek M, Husemann P, Wagner-Dobler I, et al. Stool metatranscriptomics: A technical guideline for mRNA stabilisation and isolation. BMC Genomics. 2015;16:494. doi: 10.1186/s12864-015-1694-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bustin SA, Nolan T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech.: JBT. 2004;15(3):155. [PMC free article] [PubMed] [Google Scholar]
- 35.Bustin S, Nolan T. Talking the talk, but not walking the walk: RT-qPCR as a paradigm for the lack of reproducibility in molecular research. Eur. J. Clin. Invest. 2017;47(10):756–774. doi: 10.1111/eci.12801. [DOI] [PubMed] [Google Scholar]
- 36.Pregibon DC, Doyle PS. Optimization of encoded hydrogel particles for nucleic acid quantification. Anal. Chem. 2009;81(12):4873–4881. doi: 10.1021/ac9005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, Wang J, Deng F, Yan Z, Xia Y, Wang Z, et al. TqPCR: a touchdown qPCR assay with significantly improved detection sensitivity and amplification efficiency of SYBR green qPCR. PLoS ONE. 2015;10(7):e0132666. doi: 10.1371/journal.pone.0132666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi B, Li Y, Wu D, Wu W. A handheld continuous-flow real-time fluorescence qPCR system with a PVC microreactor. Analyst. 2020;145(7):2767–2773. doi: 10.1039/C9AN01894H. [DOI] [PubMed] [Google Scholar]
- 39.Sorokin NV, Chechetkin VR, Livshits MA, Pan'kov SV, Donnikov MY, Gryadunov DA, et al. Discrimination between perfect and mismatched duplexes with oligonucleotide gel microchips: role of thermodynamic and kinetic effects during hybridization. J. Biomol. Struct. Dyn. 2005;22(6):725–734. doi: 10.1080/07391102.2005.10507039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.