

Summary
Hyaluronan (HA) is an acidic heteropolysaccharide of alternating N-acetylglucosamine and glucuronic acid sugars that is ubiquitously expressed in the vertebrate extracellular matrix1. The high molecular weight polymer modulates essential physiological processes in health and disease, including cell differentiation, tissue homeostasis, and angiogenesis2. HA is synthesized by a membrane-embedded processive glycosyltransferase, HAS, that catalyzes the synthesis and membrane translocation of HA from UDP-activated precursors3,4. Here, we describe five cryo-electron microscopy structures of a viral HAS homolog at different states during substrate binding and initiation of polymer synthesis. Combined with biochemical analyses and molecular dynamics simulations, our data reveal how HAS selects its substrates, hydrolyzes the first substrate to prime the synthesis reaction, opens a HA-conducting transmembrane (TM) channel, ensures alternating substrate polymerization, and coordinates HA inside its TM pore. Our work suggests a detailed model for the formation of an acidic extracellular heteropolysaccharide and provides the first insights into the biosynthesis of one of the most abundant and essential glycosaminoglycans in the human body.
Introduction
Hyaluronan (HA) is a ubiquitous acidic glycosaminoglycan of the vertebrate extracellular matrix (ECM), particularly enriched in connective tissues, the vasculature, and cartilage1. HA modulates a broad range of tissue remodeling processes, including wound healing, embryological development, angiogenesis, and tumorigenesis5. It is a linear polysaccharide of alternating β-1,3 and β-1,4-linked N-acetylglucosamine (GlcNAc) and glucuronic acid (GlcA) units, respectively, (Fig. 1a and b) that is typically megadaltons in size2,6. High molecular weight HA (>106 kDa) associates with healthy tissue homeostasis, while low molecular weight species exhibit pro-inflammatory and angiogenic properties2.
Fig. 1|. Structure of hyaluronan synthase.
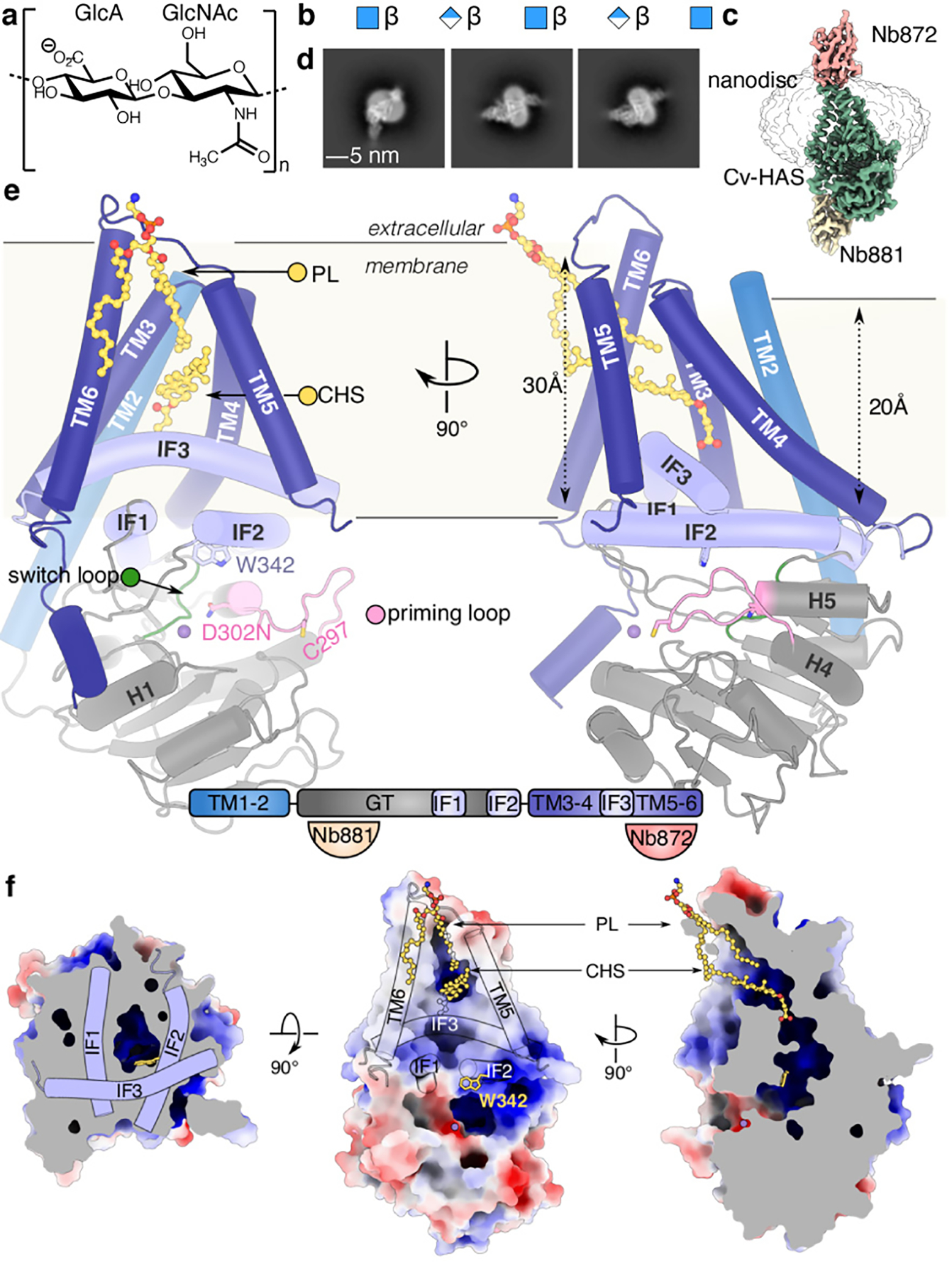
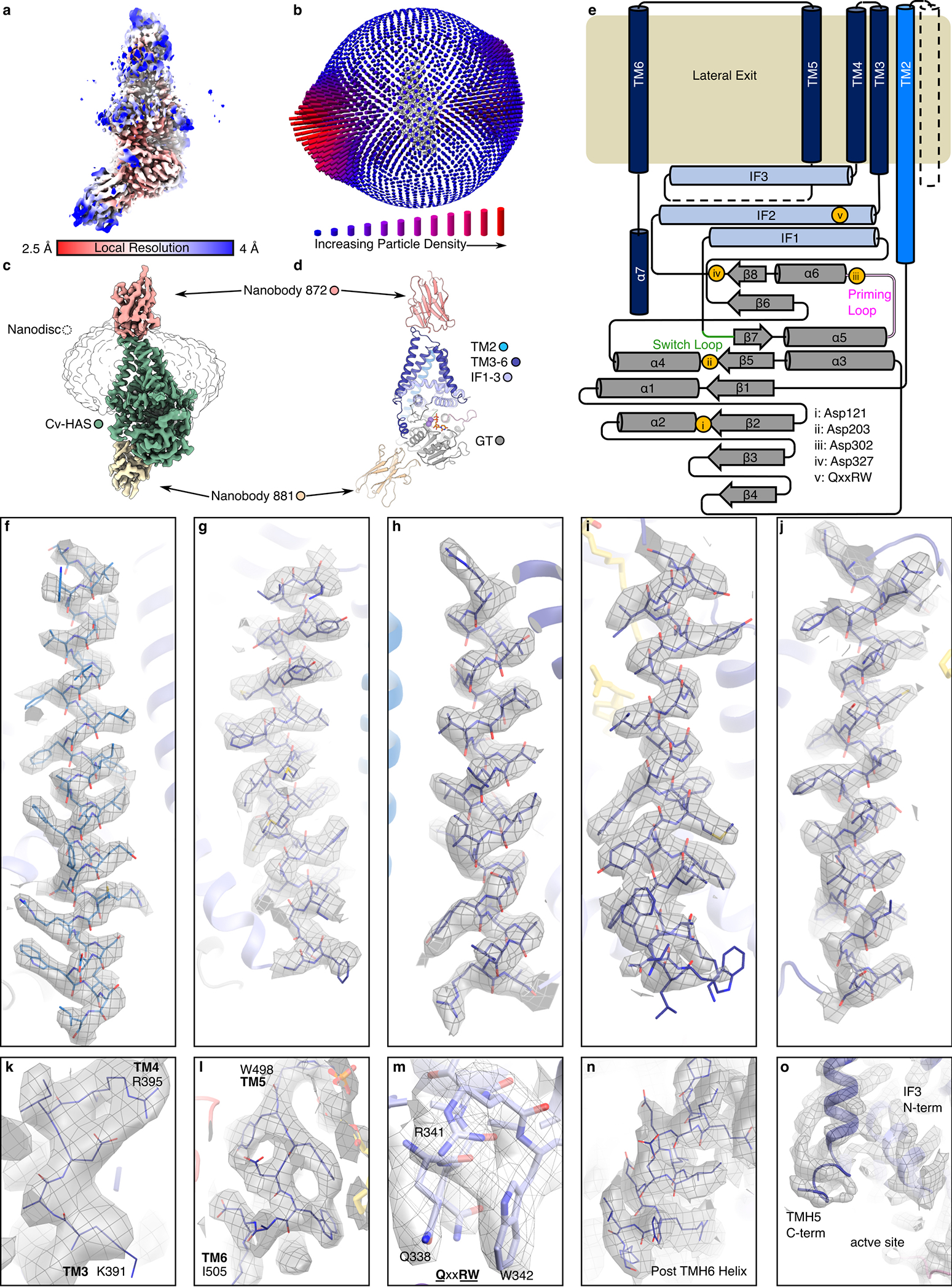
(a and b) HA GlcA-GlcNAc disaccharide repeat unit and representation of an HA polymer (blue square: GlcNAc, blue/white diamond: GlcA). (c) Volume of the Cv-HAS-Nb872/881 complex with Nb872 and 881 in salmon and yellow, respectively, Cv-HAS in green, with the nanodisc at lower contour represented as an outline. (d) Representative 2D class averages of nanobody-bound Cv-HAS. (e) Apo conformation of Cv-HAS. The glycosyltransferase (GT), interface (IF), and TM regions are colored green, light blue, and blue, respectively. Manganese ions are shown as purple spheres. Bottom panel: Schematic of Cv-HAS’ domain organization and Nb interactions. (f) Surface electrostatics of Cv-HAS. Partially ordered lipid and detergent molecules occupying the membrane exposed channel’s opening are shown as yellow ball-and-sticks. The electrostatic potential was calculated using the APBS plugin in PyMol (red – blue: −5 to +5 kT)30.
HA is synthesized from UDP-activated monosaccharides. Type-I HASs contain a single catalytic domain and secrete the nascent HA polymer during synthesis through a TM channel formed by their membrane-embedded segment7,3,4. Type-II HAS, however, is limited to select bacteria and is a bifunctional cytosolic glycosyltransferase (GT)8,9. Thus, Type-I HASs couple HA synthesis with translocation, utilizing a single catalytic domain to transfer two different donor sugars and to form substrate-specific glycosidic linkages.
Vertebrates express three HAS isoforms (Extended Data Fig. 1) that differ in tissue expression and catalytic activity; HAS-2 is essential10,11. HAS contains a cytosolic GT domain flanked by two N-terminal and four C-terminal TM helices (TMH). How HAS selects its substrates, catalyzes regio- and stereospecific glycosyl transfer, secretes HA, and controls polymer length are important unresolved questions.
Chlorella viruses (Cv) contain HAS enzymes homologous to vertebrate HAS that synthesize HA in vivo and in vitro12,13. Cv infect unicellular green algal endosymbionts of the ciliate Paramecium bursaria; HA production by the infected algae may benefit the endosymbiotic relationship. Cv-HAS shares with human HAS-2 ~45% sequence similarity, the same predicted number and distribution of TMHs, as well as a conserved GT domain (Extended Data Fig. 1).
We determined cryo-electron microscopy (EM) structures of Cv-HAS in apo, UDP-bound, UDP-GlcNAc-bound, and GlcNAc-primed states. Our analyses provide the structural and mechanistic basis for substrate selectivity and alternating polymerization, demonstrate that only GlcNAc can prime HA biosynthesis, and suggest a model for processive HA synthesis and translocation. These insights are corroborated by functional studies and molecular dynamics (MD) simulations.
Results
Cv-HAS was expressed in E. coli and purified as described in the Methods. For EM analysis, Cv-HAS was bound to two camelid nanobodies produced in vivo, Nb872 and Nb881 (Fig. 1c and d)14. Nb872, binding to the extracellular TMH5–6 loop, supports in vitro catalytic activity similar to un-complexed Cv-HAS and thermo-stabilizes the enzyme (Extended Data Fig. 2a and b). Nb881 binds the periphery of the GT domain and reduces HA biosynthesis for unknown reasons (Extended Data Fig. 2b–c).
Cryo-EM analyses of Cv-HAS were performed in E. coli lipid nanodiscs, see Methods. Samples were analyzed bound to UDP, a product and competitive inhibitor of Cv-HAS15, and the substrate UDP-GlcNAc. Three-dimensional sorting and variability analyses of the ‘UDP-GlcNAc-bound’ dataset revealed additional states: nucleotide-free ‘apo’, substrate-bound, and monosaccharide-bound ‘primed’ (Extended Data Fig. 2–4, Supplementary Video 1). The cryo-EM maps generated in cryoSPARC16 range in estimated resolutions from 3.1–2.7 Å (Extended Data Table 1) and resolve essentially all residues, with the exceptions of residues 1–37 (including TMH1), 452–464 connecting IF3 with TMH5, and 553–561 at the C-terminus.
Architecture of Cv-HAS
The Cv-HAS GT domain adopts a GT-A fold17 and packs against the five resolved TMHs (TMH2–6) as well as three amphipathic interface helices (IF1–3) (Fig. 1e). It contacts the TMHs via IF1 and IF2 (residues 238–263 and 331–357, respectively), of which IF2 contains a QxxRW motif characteristic of membrane-embedded processive family-2 GTs, according to the CAZy classification18 (residues 339–342). IF3, a C-terminal extension of TMH4, runs perpendicular and on top of the IF1/2 pair, framing the entrance to the TM channel.
The TM architecture resembles a teepee with helices straddling the cytosolic IF helices. TMH2 extends past the cytosolic water/lipid interface to interact with the GT domain. Past TMH6, 13 C-terminal residues (540–552) form a short α-helix that packs into a groove on the GT domain formed by its central β-sheet and helix 1, together with IF1 (Fig. 1e).
TMH1 is disordered in all cryo-EM maps. A MapPred co-evolution analysis19 suggests evolutionarily-coupled residues within TMH1 and TMH2 that position the helix in a groove between TMH2 and 4 (Extended Data Fig. 5a). Direct interactions with TMH2 are also supported by AlphaFold220 and RoseTTAfold21 Cv-HAS models, although they position TMH1 differently around TMH2 (Extended Data Fig. 5b–c).
TMH1 is also disordered when using two cytosolic Nbs for structure determination (Extended Data Fig. 5d), indicating that its flexibility is not induced by Nb872. The helix is not essential for function as TMH1-truncated Cv-HAS produces HA in vitro, albeit at reduced levels compared to the wild type enzyme (Extended Data Fig. 5e).
Helices 4 and 5 of the GT domain are connected by an 11-residue long ‘priming loop’ that forms one wall of the active site (residue 291–301). The loop precedes the putative base catalyst Asp302 within the GDD motif that facilitates glycosyl transfer. It contains a conserved cysteine at its tip (Cys297) and extends roughly along the GT’s central β-sheet towards the nucleotide-binding pocket (Fig. 1e). As demonstrated initially by the structure of the soluble enzyme SpsA and later cellulose synthase, the catalytic pocket of family-2 GTs includes characteristic acidic motifs that form a single substrate binding pocket17,22,23. For Cv-HAS, these include the nucleotide binding ‘DGD’ (121–123), the cation binding ‘DSD’ (201–203), and the putative catalytic ‘GDD’ motif (300–302) (Fig. 1e, Extended Data Fig. 1).
Cv-HAS forms a lipid-filled channel
Cv-HAS contains a short membrane-embedded region about 20 and 30 Å thick near TMH2 and above the active site (proximal to the TMH5–6 face), respectively (Fig. 1e and f). It forms a curved channel with a cytosolic entrance above the active site formed by Trp342 of the QxxRW motif within IF2 (Fig. 1f). Halfway across the membrane, IF3, TMH5, and TMH6 create a lateral channel opening towards the lipid bilayer. Two lipid molecules, assigned as a CHS molecule and a phospholipid tail based on their shapes, occupy and seal the portal (Fig. 1f and Extended Data Fig. 6). Atomistic MD simulations in a POPE bilayer corroborate that these lipid acyl chains indeed prevent water flux across the membrane (Extended Data Fig. 6b).
Substrate-bound conformation of Cv-HAS
Cv-HAS structures bound to UDP-GlcNAc were determined using an inactive enzyme in which Asp302 was replaced with Asn (Extended Data Fig. 1 and 7). Unexpectedly, the substrate was partially hydrolyzed during sample preparation, capturing views of the active site before and after priming (Fig 2a–c). Nucleotide binding displaces the priming loop from the active site towards the cytosolic water-lipid interface (Fig. 2d, Extended Data Fig. 7d and f, and Supplementary Video 2). This conformation is stabilized by the loop’s Phe292, interacting with Arg348 and Tyr352 of IF2, as well as Tyr299 occupying a hydrophobic pocket at the interface of IF2, IF3 and TMH5. Cys297 fits into a crevice at the water/lipid interface formed by Thr298 and Lys344 as well as Arg348 of IF2. Cys297 is critical for function; replacing it with Ala renders Cv-HAS inactive (Fig. 2e). Relative to the apo state, nucleotide binding induces a ~10 degree rigid body rotation of the GT domain towards the membrane, fostering the priming loop-IF2 interaction and narrowing the active site cleft (Fig. 2a, Supplementary Video 3).
Fig. 2|. Substrate-bound and primed Cv-HAS conformations.
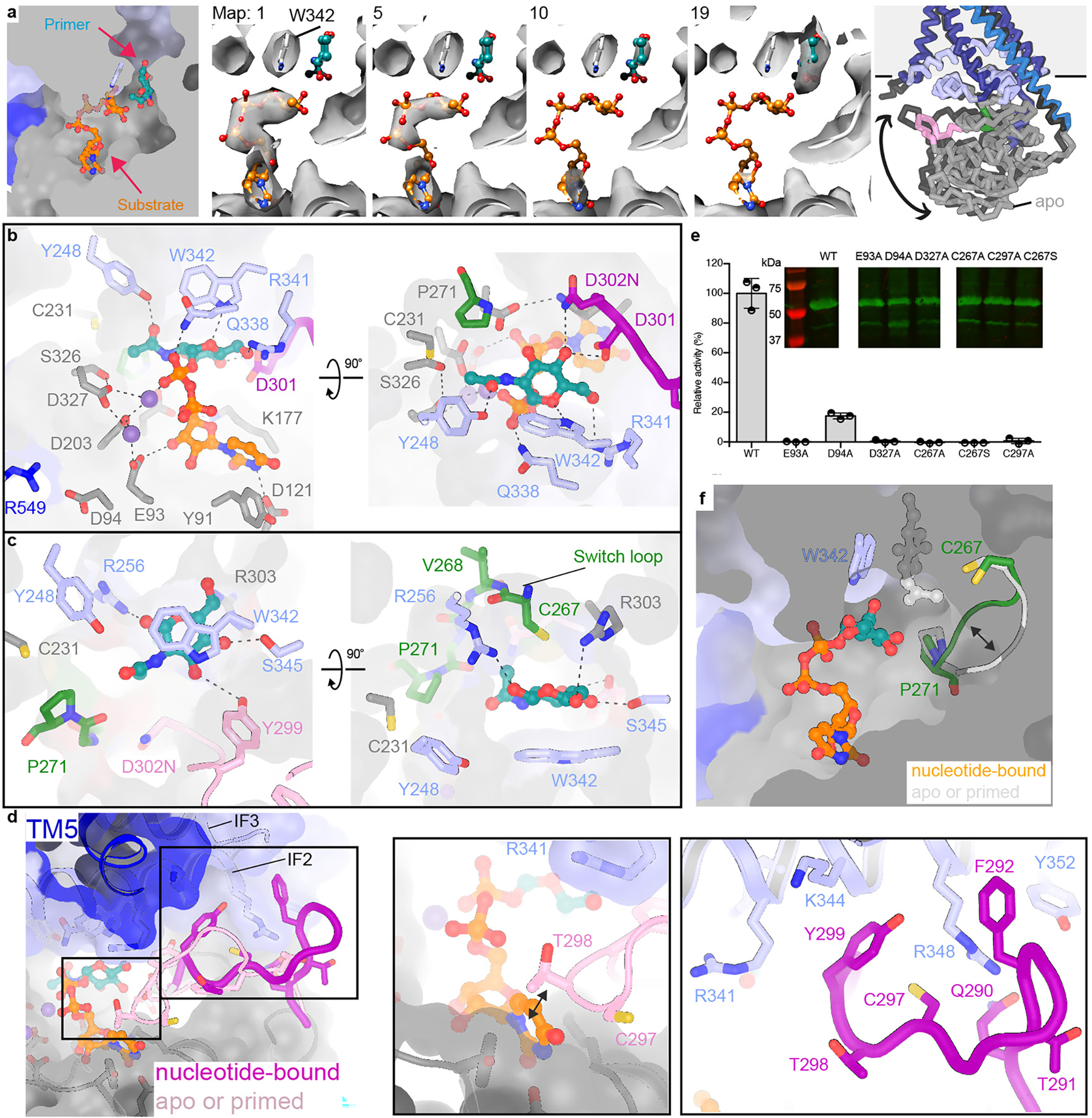
(a) 3D variability analysis of UDP-GlcNAc-incubated Cv-HAS particles. Shown are representative volumes of the generated ensemble (Supplementary Video 1). Left panel: Substrate and acceptor locations mapped onto one structure; Right panel: Rotation of the GT domain towards the TM region upon substrate binding. (b and c) Substrate coordination at the active site. Dashed lines indicate distances from ~2.5 to ~4.2Å. (d) Priming loop retraction in response to substrate binding. The loop is shown as a cartoon with the flanking residues shown as sticks and semi-transparent surfaces. (e) Contribution of conserved residues to catalytic activity normal to wild type (WT) levels and obtained by quantifying the production of 3H-labeled HA. Inset: Western blot of the inverted membrane vesicles used for activity measurements. Full scans are provided as Supplementary Fig. 1a. Error bars represent deviations from the means with n=3 independent experiments. (f) Conformational changes of the switch loop. The primer and substrate are shown as ball-and-sticks in the same structure.
Cv-HAS requires manganese for catalytic activity13, indicating that it binds Mn2+-complexed substrates. The UDP and UDP-GlcNAc-bound structures reveal similar UDP coordination (Extended Data Fig. 7a and b, Supplementary Discussion 1). Asp121 of the ‘DGD’ motif contacts the uracil’s N3 ring nitrogen, while the ribose’s C3 hydroxyl group interacts with Glu93 from the conserved ‘EDP’ motif (Fig. 2b, Extended Data Fig. 1). The UDP-GlcNAc diphosphate hydrogen-bonds with Gln338 of the QxxRW motif while the associated Mn2+ ion is coordinated by Asp203 and Asp327. A second Mn2+ ion is coordinated by Glu93 as well as Asp327 and Asp203 (Fig. 2b). Replacing Glu93 or Asp327 with alanine renders Cv-HAS inactive, thus both Mn2+ ions are functionally important (Fig. 2e). The additional Mn2+ ion may position the DxD motif (Asp201-Asp203) for substrate binding. Mutating Asp94 of the EDP motif to Ala reduces catalytic activity to about 20% relative to wild type. Asp94 does not directly coordinate Mn2+, but interacts with Arg549 near Cv-HAS’ C-terminus (Fig. 2b).
In the substrate-bound state, the donor sugar ring oxygen is proximal to Trp342’s N1 hydrogen (Fig. 2b), right below the acceptor-binding site. The GlcNAc acetamido group occupies a pocket formed by Asp203, Cys231, Tyr248, Pro271, Asp327, and Ser326, and its carbonyl oxygen hydrogen bonds with the side chain hydroxyl of Tyr248 (Fig. 2b). The sugar’s C4 hydroxyl interacts with Asp301 and Asp302, while its C6 hydroxyl points towards Arg341, positioned within a positively charged pocket containing Lys177. This pocket likely accommodates the C6 carboxylate when the substrate is UDP-GlcA (see below).
Compared to the nucleotide-free or primed conformations (see below), UDP or substrate binding repositions a partially conserved CVGGP loop (switch loop, residues 267–271) at the back of the nucleotide-binding pocket (Fig. 2f). The loop moves toward the membrane interface upon nucleotide binding, likely stabilized by unresolved water molecules. Cys267 at the beginning of the switch loop is necessary for function (Fig. 2e, Supplementary Discussion 2).
Priming of Cv-HAS
Substrate hydrolysis likely generated the GlcNAc monosaccharide primer that diffused to the acceptor-binding site at the TM channel entrance (Fig. 2a, Extended Data Fig. 7e, Supplementary Video 1). The primer’s acetamido group is surrounded by Pro271, Arg256, Cys231, Tyr248 as well as backbone atoms from the switch loop (Fig. 2c). The switch loop’s ‘down’ conformation positions the Cys267 sulfhydryl group ~ 4.2 Å away from the ring oxygen (Fig. 2f), while CH-π stacking interactions with Trp342 stabilize the sugar ring. Further, the conserved Ser345 (Extended Data Fig. 1) is in hydrogen bonding distance to the primer’s C5 hydroxyl, while the conserved Arg303 binds the C6 hydroxyl group. These interactions position the primer’s C3 hydroxyl towards the active site and the putative base catalyst (D302N) (Fig. 2c).
Upon priming, the priming loop re-inserts into the catalytic pocket (Fig. 2d, Extended Data Fig. 7f) and the GT domain relaxes away from the membrane (Fig. 2a, Supplementary Video 3). While an inserted loop position is also observed in the apo state, the loop is better resolved in the primed state, suggesting that the primer (or HA polymer) is stabilizing. Accordingly, the priming loop’s Tyr299, a conserved Phe in vertebrate HAS, rotates towards the primer-binding site in close proximity to GlcNAc’s C3 hydroxyl group (Fig. 2c).
Substrate hydrolysis initiates HA biosynthesis
The in situ generation of the GlcNAc primer suggests substrate hydrolysis as a general initiation mechanism of HAS-related enzymes. Accordingly, supplied monosaccharides should also prime HA biosynthesis. To test this model, wild type Cv-HAS was incubated with 14C-labeled GlcNAc or GlcA and unlabeled substrates. The synthesized polysaccharide revealed that the GlcNAc monosaccharide is readily incorporated into HA, while GlcA fails to prime the synthesis reaction (Extended Data Fig. 8).
Additionally, we used substrate hydrolysis experiments to verify the initiation mechanism. Cv-HAS hydrolyzes its substrates when exposed to only one of them without forming a homo-polysaccharide (Extended Data Fig. 8a, lane 1). This allows monitoring substrate hydrolysis rates in real time based on enzyme-coupled NADH oxidation (Extended Data Fig. 9)7. We observe that GlcNAc monosaccharide increases the UDP-GlcA hydrolysis rate ~30-fold and decreases UDP-GlcNAc hydrolysis ~2.5-fold. Conversely, hydrolysis rates of both substrates were insignificantly affected by comparable concentrations of GlcA (Extended Data Fig 9e–f). Thus, a GlcNAc primer facilitates the binding and/or turnover of UDP-GlcA to form an HA disaccharide, while a GlcA primer does not enhance UDP-GlcNAc hydrolysis.
HAS priming creates a continuous TM channel
Strikingly, we resolve two different TM architectures of the primed state – one similar to the apo and substrate-bound states, and another with the N-terminus of TMH2 tilted away from TMH3 (Fig. 3 and Extended Data Fig. 7h–i, Supplementary Video 4), creating a continuous TM channel. The newly formed channel overlaps with the curved, lipid-plugged channel above the active site to form a common vestibule, yet diverges halfway across the membrane (Fig. 3c). The newly created channel is lined with hydrophilic and apolar residues and its dimensions are sufficient to accommodate an HA polymer about 5 disaccharides long (Fig. 3d).
Fig. 3|. Structural rearrangements upon GlcNAc priming.
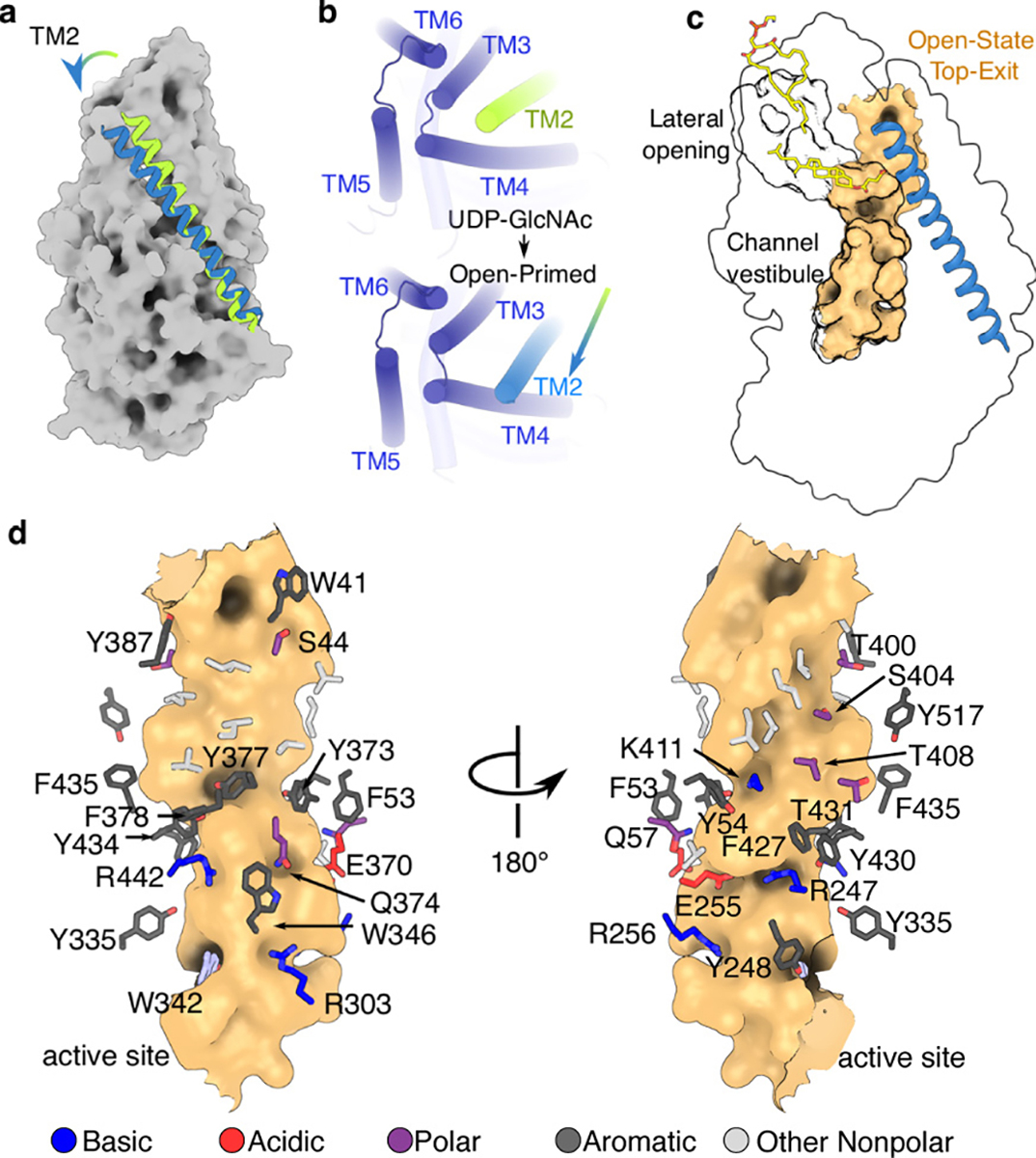
(a) Movement of TMH2 (green= substrate-bound, blue= primed-open) upon substrate hydrolysis. (b) Movement of TMH2. TMH1 is shown at its predicted location as a green cylinder. (c) Partial overlap of the lipid-plugged channel with the continuous TM channel formed upon priming (shown as an orange surface). The predicted location of TMH1 is indicated as a green cartoon helix. The channels were rendered in HOLLOW using a 1.2 Å probe31. (d) Residues lining the open TM-channel.
TMH2 kinks around its contact point with IF1, mediated by Gln57 and Phe60 of TMH2 and Val254, Ala258 and Leu261 of IF1 (Extended Data Fig. 7g). The conserved residues Arg256 and Gln259 on the opposite side of IF1 line the channel lumen and Arg256 contacts the primer’s C1 hydroxyl group (Fig. 2c). TMH2 could be further stabilized in this ‘open’ conformation by the predicted location of TMH1 (Extended Data Fig. 5a–c). Thus, HAS priming creates an exit path for the nascent HA at the interface of TMH1–2 and 3–4, similar to cellulose synthase (Extended Data Fig. 6c)23.
Dynamic coordination of HA inside the TM channel
We performed atomistic MD simulations to gain insights into HA coordination by Cv-HAS. Simulations were performed with a HA hexasaccharide manually placed within the channel vestibule, with the terminal non-reducing end sugar occupying the acceptor-binding site next to Trp342 (Fig. 2c).
We identify significant register-dependent differences in the positional stability of HA. While oligosaccharides starting with GlcNAc at the acceptor site were stable over the course of 5 simulations, polymers starting with GlcA were pulled into the TM channel by roughly one sugar unit (Fig. 4a). After this motion, the GlcA-ending polymer is coordinated similarly compared to the GlcNAc-ending chain, yet one sugar unit farther inside the channel (Fig. 4a–e, Supplementary Table 1).
Fig. 4|. Dynamics of HA-bound HAS.
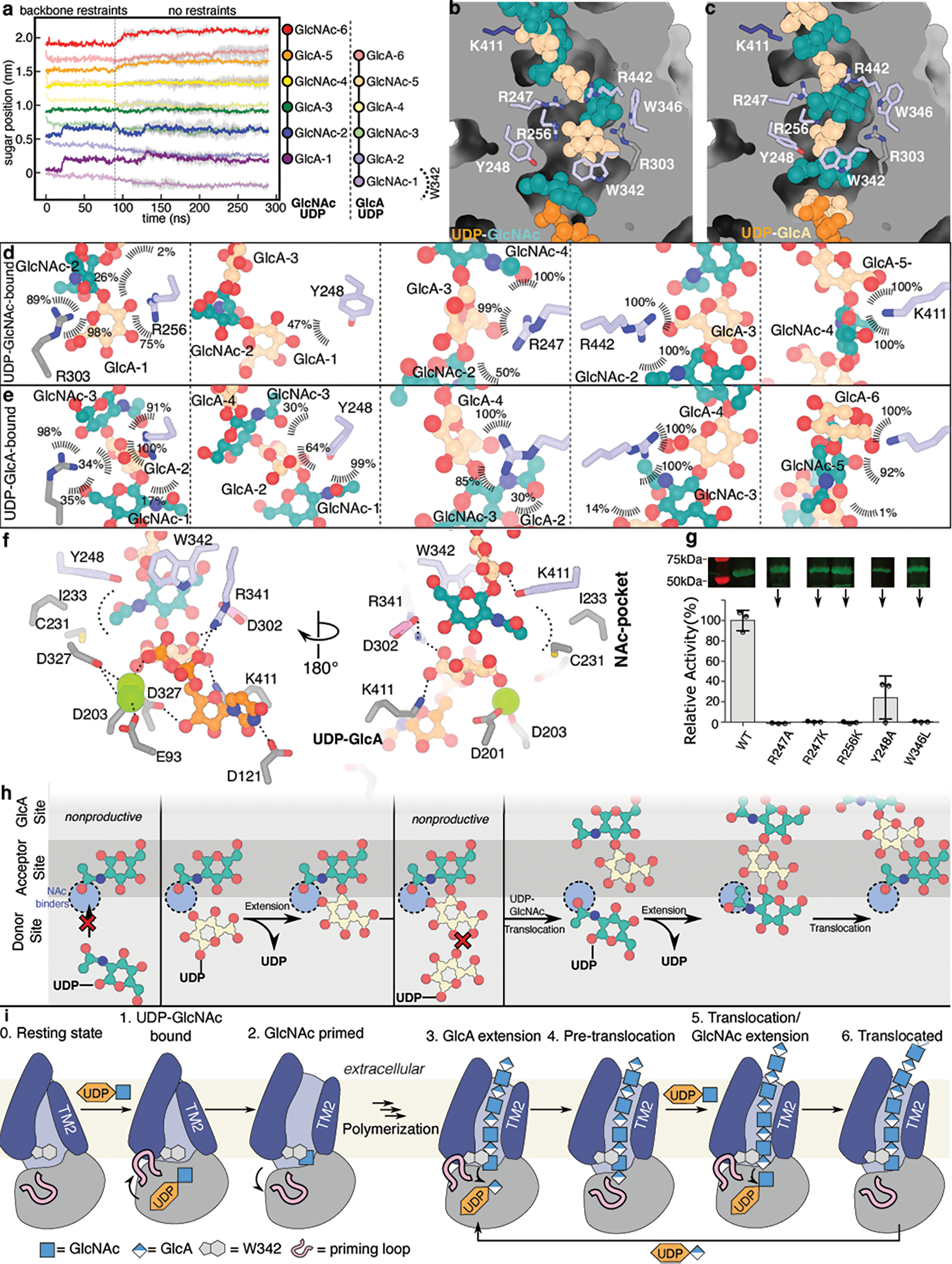
(a) MD simulations of Cv-HAS bound to either UDP-GlcNAc and a GlcA-ending HA hexasaccharide or UDP-GlcA and a GlcNAc-ending polymer. (b-c) Representative coordination of HA at different registers after 300 ns MD simulations. (d) Coordination of HA inside the TM channel in a UDP-GlcNAc bound state. Values represent contact probabilities in percent over 300 ns of simulation, averaged over 5 independent repeats. The N-acetyl binding pocket is highlighted in blue. (e) As in panel (d) but for an UDP-GlcA bound state and HA polymer with different register. (f) Coordination of UDP-GlcA at the active site. Shown is a representative pose from a 300 ns simulation. The contact analysis is shown in Supplementary Table 1. (g) Mutagenesis of HA coordinating residues and corresponding catalytic activity. Inset: Anti-His-tag Western blot of IMVs used for activity assays. Full scans are provided in Supplementary Fig. 1b. Error bars represent deviations from the means with n=3 independent experiments. (h and i) Model of HA elongation and membrane translocation. (h) HA’s terminal sugar unit determines the selectivity of the catalytic pocket. GlcNAc or GlcA acceptors only allow binding UDP-GlcA or UDP-GlcNAc substrate, respectively. (i) UDP-GlcNAc hydrolysis and outward movement of TMH2 to prime HAS. Right: Processive primer extension and translocation leads to HA secretion. HA translocation is likely coupled to substrate binding. The priming loop (shown in pink) retracts from the active site to enable substrate binding, while the GT contracts and relaxes upon substrate binding and HA-extension, respectively.
HA primarily interacts with conserved polar and charged side chains. Arg303 and Arg256 bind GlcA carboxylates in the terminal or the second position (Fig 4b–e), while Arg247 and Arg442 coordinate GlcA at the third or fourth positions. The GlcNAc groups are mainly coordinated by polar and aromatic residues, including Tyr248, which coordinates GlcNAc, but not GlcA, when in the terminal position. Trp346 interacts with GlcNAc when at the second or third position. All of these HA-coordinating interactions are also seen in simulations seeded from different initial HA poses albeit with some localized variability (Supplementary Table 1).
MD simulations of UDP-GlcA and HA-bound Cv-HAS also provide insights into the likely coordination of the UDP-GlcA substrate. While the UDP moiety is coordinated similarly to the experimental UDP-GlcNAc-bound state (Fig. 2b), the substrate’s carboxylate is coordinated by Lys177 as well as Arg341 of the QxxRW motif, which bridges the nucleotide’s α-phosphate and GlcA’s carboxylate (Fig 4f and Supplementary Table 1).
HA biosynthesis experiments with Cv-HAS carrying substitutions at HA coordinating residues reveal their functional importance. Replacing Arg247 with Ala or Lys and Arg256 with Lys inactivates the enzyme, supporting a critical role for GlcA coordination. Substituting Trp346 with Leu similarly abolishes activity, while an Ala substitution of Tyr248 supports activity at roughly 20% of wild type (Fig. 4g).
Discussion
Cellulose, chitin and HA synthases are membrane-embedded GTs that synthesize and secrete high molecular weight polysaccharides. Unlike cellulose and chitin, HA is a heteropolysaccharide of alternating GlcNAc and GlcA units linked via different glycosidic linkages. Our analyses provide molecular insights into how HAS combines these tasks.
Cv-HAS elongates HA’s non-reducing end, similar to cellulose and chitin synthases13,23,24. Considering the similarities of vertebrate HASs to Cv-HAS, we propose that Cv-HAS mechanistically and structurally represents the vertebrate enzymes. This is supported by the agreement of AlphaFold2 and RoseTTAfold models of human HAS-2 with the Cv-HAS structure (Extended Data Fig. 5a–c)20. The only discrepancies exist within the predicted location of TMH1, which appears to be flexible.
Catalytic activity of HAS may be modulated by post-translational modifications (Supplementary Discussion 3). Initiating HA biosynthesis requires the formation of a priming carbohydrate acceptor, likely by substrate hydrolysis (Extended Data Fig. 8 and 9). A similar mechanism has been proposed for cellulose synthase25,26 and experimentally tested for chitin synthase27. The observation that GlcA fails to prime the biosynthesis reaction is consistent with its instability at the acceptor-binding site in MD simulations.
HAS extends GlcNAc-ending HA polymers only with GlcA to ensure an alternating HA sequence. Our 3D variability analysis of Cv-HAS incubated with UDP-GlcNAc reveals that the enzyme either binds UDP-GlcNAc at the active site or GlcNAc at the acceptor position, but not simultaneously. This is likely due to clashes between the GlcNAc primer and substrate and corroborated by the reduced rate of UDP-GlcNAc hydrolysis in the presence of saturating GlcNAc monosaccharide (Extended Data Fig. 9). To investigate this phenomenon at the structural level, we performed atomistic MD simulations of HAS with GlcNAc both in the donor (UDP-GlcNAc) and acceptor (HA) positions. Quantifying the occupancy of the acetamido binding pocket reveals that either the donor or acceptor GlcNAc, but not both, are able to bind the pocket (Extended Data Fig. 10a and b).
We further propose that the GlcNAc and GlcA acceptors are differently coordinated and positioned at the acceptor site. This would ensure that only the C4 hydroxyl of GlcA and the C3 hydroxyl of GlcNAc serve as acceptors during glycosyl transfer due to their proximities to the base catalyst Asp302 of the GDD motif (Fig. 4h).
Compared to cellulose synthase, HAS’ TM channel is wider and rich in positively charged residues, reflecting differences of the conducted polysaccharides. Structural insights into cellulose synthase are limited to polysaccharide associated states, i.e. describing enzymes with an open TM channel, whereas our HAS analyses are predominantly of the closed state. The overall similarity of the HAS and cellulose synthase TM architectures (Extended Data Figure 6c and 10c) reinforce our assignment of the extracellular HAS channel exit at the TMH2–4 interface.
Our cryo EM structures do not resolve a conserved loop connecting IF3 with TMH5, right above the catalytic pocket (Fig. 1). The loop contains a WGTR/SG motif (Extended Data Fig. 1), which is important for function of bacterial HAS28. It resembles cellulose synthase’s gating loop, which transiently inserts into the catalytic pocket for substrate binding (Extended Data Fig. 10c)25. A similar process in HAS could reposition the substrate towards the acceptor sugar to facilitate HA elongation.
Following glycosyl transfer, cellulose synthase translocates the extended polysaccharide upon substrate binding through conformational changes of a conserved α-helix of the GT domain29. The same helix exists in HAS, with the GDD motif capping its N-terminus. A similar step-by-step HA translocation mechanism seems plausible (Fig. 4h and i). Because HA is negatively charged, the membrane’s electrochemical potential and the conserved positive charges lining the TM channel likely also contribute to translocation. Our MD simulations suggest that an unfavorable register with GlcA at the acceptor position may be a transient intermediate that facilitates polymer translocation upon UDP-GlcNAc binding (Fig. 4h and i).
Materials and Methods
Construct Design and Mutagenesis
The gene for Paramecium bursaria Chlorella virus CZ-2 hyaluronan synthase (Cv-HAS) was cloned as described13 and sub-cloned after restriction enzyme digest with NcoI/XhoI into a pET28a vector modified to encode a C-terminal 10x histidine tag (pHAScz2). Mutagenesis was performed by PCR with overlapping primers (Supplementary Table 2) on pHAScz2 using KOD Hot Start polymerase (Novagen). Constructs for deletions of the N-terminal TMH were generated by amplifying the vector and remaining Cv-HAS gene with primers containing complementary overhangs.
Protein Expression and Purification
Cv-HAS was expressed in E.coli C43. Cultures were grown in terrific broth (TB) supplemented with 4% v/v glycerol and 1XM buffer32. Each culture was inoculated from an overnight culture in lysogeny broth (LB), grown to an OD600 of ~0.8 at 30 °C, cooled 1 hour to 20 °C, then expression was induced with 100 mg/L Isopropyl β-D-1-thiogalactopyranoside (IPTG). Constructs were expressed ~18 hours at 20 °C, then cell pellets were harvested by centrifugation.
Cell pellets were resuspended in a buffer of 10% glycerol, 100mM NaCl, and 20mM Tris pH 7.5 (RB), then incubated for 1 hour at 4 °C with 1 mg/mL lysozyme (Alfa Aesar). Cell suspensions were processed by 3 passes through a microfluidizer, then intact cells and aggregated material was removed by low-speed centrifugation for 25 min at 12.5 kRPM in a JA-20 rotor (Beckman). Membranes were isolated from the resulting lysates by centrifugation for 2 hours at 200k × g in a Ti45 rotor (Beckman), then harvested and flash-frozen in liquid N2 and stored at −80 °C.
For in surfo samples, membranes were thawed and resuspended in 300 mM NaCl, 20 mM Tris 7.5, 10% glycerol, 5mM β-mercaptoethanol (BME) (SB), supplemented with 40 mM imidazole, 1% lauryl maltose neopentyl glycol (LMNG), and 0.2% cholesterol hemisuccinate (CHS), then incubated 1 hour with agitation. Aggregated material was removed by centrifugation at 200k × g for 30 min, then the supernatant was incubated with Ni-NTA resin for 1 hour. Resin was washed with SB supplemented with 80 mM imidazole and the protein was eluted with SB supplemented with 320 mM imidazole. The eluted fraction was concentrated and purified by size-exclusion chromatography (SEC) using an S200-increase column equilibrated in RB. Peak fractions were harvested and concentrated, followed by glycerol removal by a second SEC in 100 mM NaCl and 20 mM Tris pH 8.0 (FB) (all steps performed at 4°C).
Purification of nanodisc samples was similar, except membranes were solubilized in SB supplemented with 40 mM imidazole, 1 % n-Dodecyl β-D-maltoside (DDM), and 0.1 % CHS, the Ni-NTA wash and elution buffers were supplemented with 0.02 % DDM/0.002 % CHS, and the first SEC buffer was 100 mM NaCl, 20 mM Tris pH 7.5, 0.02 % DDM and 0.002 % CHS. Following the first SEC, protein was concentrated and reconstituted into nanodiscs in a 1:3:30 molar ratio of HAS:MSPE3D1:E.coli total lipid extract (solubilized in 60 mM DDM). To produce nanobody complexes, selected nanobodies were added in two-fold molar excess compared to HAS to the nanodisc reconstitution mixture. After a 30 minute incubation, BioBeads were added stepwise to a final volume of 0.5 mL and incubated overnight to sequester detergent. Properly formed HAS nanodiscs were purified by SEC in FB, then fractions were screened by SDS-PAGE and negative stain EM for presence of both MSP and HAS and particle quality (all steps performed at 4°C).
In vitro HA Synthesis Assays
HA synthesis was assayed by incorporation of 3H- or 14C-labeled sugars into HA produced by Cv-HAS in inverted membrane vesicles (IMVs), as described 13. Wild type or mutated HAS constructs were expressed as described above. Lysates resulting from the low-speed spin were floated on a 2 M sucrose cushion, then spun for 2 hours at 200k × g in a Ti45 rotor. IMVs were harvested, diluted in RB, and centrifuged again at 200k × g for 1.5 hours. The resulting IMV pellet was harvested, homogenized with a dounce in RB, and flash-frozen in liquid N2.
Cv-HAS levels in each IMV isolate were quantified by Western blotting. 200 μL IMVs were solubilized by addition of 800 μL SB containing 1 % DDM, 0.1 % CHS, and 20 mM Imidazole, then incubated 1 hour with Ni-NTA resin. The resin was washed twice in 250 μL SB containing 0.03 % DDM, 0.003 % CHS, and 20 mM Imidazole. The washed resin was then resuspended in Laemmli buffer and 1 M imidazole, then analyzed on a 12.5 % polyacrylamide gel, and transferred at 100 V to a nitrocellulose membrane. The membrane was washed 5 min in dH2O, blocked twice for 15 minutes with 5 % milk in Tris-buffered saline-tween buffer (TBST), and incubated with primary antibody for 1 hour (all at room temperature). The membrane was rinsed 15 mins in TBST, then incubated for 1 hour with IRDye800-conjugated secondary antibody, then rinsed again for 15 mins in TBST. Blots were imaged using an Odyssey Licor scanner at 700 and 800nm, and analyzed using imageJ to determine relative protein content across multiple Cv-HAS variants.
Prior to each assay, IMVs were diluted to normalize Cv-HAS concentration. 10 μL of each IMV were mixed with 10 μL 2X reaction buffer, containing 40 mM MnCl2, 80 mM Tris pH 7.5, 150 mM NaCl, 10 mM TCEP, 10 mM UDP-GlcNAc, 10 mM UDP-GlcA, and 0.2 μCi of the appropriate radiolabeled sugar (listed below with each experiment). Reaction mixtures were incubated 2 hours at 30 °C then analyzed.
For descending paper chromatography, the radiolabel was 3H-UDP-GlcNAc and each reaction was quenched with 2% SDS. 20 μL of each reaction mixture was spotted onto Whatman 2mm filter paper, developed with 65% 1 M (NH4)2SO4/35 % ethanol, and the origins were counted using a Beckman S Ls liquid scintillation counter. Activity of each variant was repeated in triplicate, from which background was subtracted as determined by a control reaction lacking UDP-GlcA.
For autoradiography comparison of HA sizes produced across Cv-HAS variants, the radiolabel was 14C-UDP-GlcA. Reactions were quenched by addition of loading buffer, then run at 100 V for 2 hours on a 12.5 % polyacrylamide gel. The gel was fixed using 30 % methanol/3 % glycerol for 15 mins and dried onto a Whatman 2mm filter paper using a Biorad model 583 gel dryer for 2 hours. The dried gel was exposed for ~1 week to a Kodak Biomax MS film. Autoradiography of the initiation experiments was identical, except the radiolabels were 14C-labeled GlcNAc or GlcA (not UDP activated). Hyaluronate lyase digestions were performed by adding 1 mg/mL final concentration of recombinantly expressed S. pneumoniae hyaluronate lyase33 to the relevant reaction mixture.
Nanobody generation
Camelid nanobodies against Cv-HAS were generated in vivo by immunizing a male llama weekly over a period of 6 weeks with purified wild type and catalytically inactive (D201A) Cv-HAS. Animal protocols were approved by the committee for ethical treatment of laboratory animals, Vrije Universiteit Brussel (VUB). Approximately 1 mg of total protein reconstituted into E. coli total lipid proteoliposomes was used for immunization. The isolation of high affinity Cv-HAS binders and nanobody cloning into the pMESy4 expression vector followed previously described procedures14. Briefly, Cv-HAS wild type, Cv-HAS wild type with synthesized hyaluronan, and Cv-HAS D201A reconstituted into E. coli total lipid proteoliposome were each solid phase coated and used as target for the biopanning in 25 mM MES pH 6.5, 0.2 M NaCl, 10 % glycerol, 5 mM beta-mercaptoethanol, 3.5 mM MnCl2. After rescue of the eluted phage, individual colonies were screened in ELISA using the same buffer conditions. Thirty-eight different Cv-HAS-specific nanobody families were discovered belonging to 28 families based on their CDR3 sequences.
Nanobody purification
Constructs for each nanobody gene cloned in pMESy4 were expressed in E.coli WK6 cultures grown in TB supplemented with 4% v/v glycerol and 1XM buffer. Periplasmic fractions of the harvested cell pellets were extracted by osmotic shock using a Tris-sucrose-EDTA buffer (TES). Periplasmic extracts were incubated with Ni-NTA resin, then nonspecifically bound material was removed by successive washes in 20 mM Tris pH 7.5 containing 1 M NaCl and 20 mM Imidazole, then 0.1 M NaCl and 40 mM Imidazole, and 0.1 M NaCl, 320 mM Imidazole to elute. Eluted protein was further purified by SEC using an S75 column equilibrated in FB, then concentrated and flash-frozen in liquid N2.
Thermo-stability assays
Stability measurements were based on monitoring Cv-HAS’ enzymatic activity following incubation at elevated temperatures. The activity was estimated by quantifying the release of UDP in real time using an enzyme-coupled reaction that oxidizes NADH, as previously described7. Cv-HAS was individually incubated with the 38 nanobodies in a thermo-cycler at temperatures ranging from 30 to 66°C for 2 hours in 1:3 molar ratio. Biosynthesis reactions were performed in 120 μL volumes containing 1 mM PEP, 0.75 mM NADH, and 1 U of pyruvate kinase and lactate dehydrogenase each.
The oxidation of NADH was monitored at room temperature based on its absorbance at 340 nm in a microplate spectrophotometer in 30 s intervals for 1 hour. Reaction rates were calculated based on Lambert-Beer’s law using an NADH extinction coefficient at 340 nm of 6220 (M cm)-1. All experiments were performed in triplicate and error bars represent the deviations from the means.
Substrate hydrolysis assays
Hydrolysis of UDP-GlcNAc or UDP-GlcA was measured using an enzyme-coupled assay as described above, Fig S11 A–B. 1 μM Cv-HAS in nanodiscs was incubated with 2 mM substrate and the indicated concentration of GlcNAc or GlcA monosaccharide. Background hydrolysis was measured by omitting Cv-HAS. For each combination of substrate and monosaccharide, Cv-HAS catalyzed and background hydrolysis rates were measured in triplicate (Extended Data Fig. 9c–f). Net hydrolysis rates were normalized separately for each substrate based on reactions with no monosaccharide (Extended Data Fig. 9f).
EM Sample Preparation and Data Collection
Protein samples were concentrated using a 50 kDa filter (Amicon) to 3–4 mg/mL. For relevant samples MnCl2 and UDP or UDP-GlcNAc were added to a final concentration of 2 mM. 3 μL samples in detergent were applied to glow-discharged C-flat holey carbon grids (Cu 1.2/1.3, 300 mesh), blotted for 10 seconds at 4°C and 100 % humidity, then plunge frozen in liquid ethane using a Vitrobot Mark IV (FEI). 3 μL samples in nanodiscs were applied to Quantifoil holey carbon grids (Cu 1.2/1.3, 300 mesh) glow discharged with amylamine, blotted for 4 s at 4 °C and 100 % humidity, then plunge frozen in liquid ethane.
Cryo-EM data were collected at the University of Virginia Molecular Electron Microscopy Core (MEMC) on a Titan Krios (FEI) 300-kV electron microscope using a Gatan Imaging Filter (GIF) and a K3 direct detection camera. Movies were collected in EPU (Thermofisher) at a magnification of 81,000× with an energy filter width of 10 eV, using counting mode with a total dose of 51 e−/Å over 40 frames, and with a target defocus of −1.0 to −2.0 μm, Table S1. Several earlier (unreported) datasets were collected under similar conditions at the National Cryo-Electron Microscopy Facility (NCEF) at the National Cancer Institute (NCI).
EM Data Processing and Model Building
All data processing steps were performed in cryoSPARC16, unless otherwise noted. Movies were imported and gain corrected, then subjected to patch-based motion-correction and contrast transfer function estimation (CTFFIND4)34. Initially, particles were manually picked from accepted micrographs to generate references for template-based particle picking. References for later datasets were based on 2D classes from earlier sets. Particles identified from template-based picking were extracted using 2-fold Fourier cropping for initial rounds of 2D classification and/or ab initio reconstruction followed by heterogeneous refinement to remove junk particles. Accepted particles were re-extracted at full scale then subjected to further rounds of classification. High-resolution reconstructions from clean particle sets were generated by non-uniform (NU) refinement, then masked local refinement (using NU algorithm), followed by local CTF estimation and refinement of the corrected particles. Finally, 3D variability analysis and subsequent clustering was used to separate populations within the high-resolution reconstructions belonging to distinct catalytic states. Clusters belonging to apo, UDP-bound, substrate bound, or primed states were individually pooled and used to generate final reconstructions from NU or local refinement. Two maps for the apo state were derived by 3D variability analyses from the dataset collected for wild-type Cv-HAS in the presence of UDP and from the dataset for D302N Cv-HAS collected in the presence of UDP-GlcNAc. The wild-type apo model (CC mask 0.68) and D302N apo model (CC mask 0.75) are virtually identical with an RMSD of 0.132 Å. Reported is the model derived from the UDP-GlcNAc dataset, which extends to 3.1 Å versus 3.3 Å.
The initial atomic model was built into a ~3.5 Å map, derived from collection on an apo Cv-HAS sample in surfo, starting from a poly-alanine model of the RsBcsA GT domain. The model was generated from iterative rounds of building in Coot35 and real-space refinement in PHENIX 36. Register was assigned based on well-resolved map regions and on the location of highly conserved catalytic motifs. TM helices were built de novo with register interpreted from density for bulky residues and based on connectivity to the GT domain. The initial model was refined and register errors corrected using subsequent higher resolution maps in surfo and in nanodiscs to generate the final models. All figures were prepared using PyMol and Chimera30,37.
Molecular Dynamics Simulations
The coordinates of Cv-HAS, without nanobodies, obtained in nanodiscs were used for building atomistic MD simulations. Where included, the substrate coordinates were taken from the resolved cryo-EM densities for UDP-GlcNAc, and the terminal residue of the HA chain placed according to the position of the primer monosaccharide. Three different HA poses were manually constructed in Coot35 for each register, to provide a broad degree of HA conformational sampling. The systems were described with the CHARMM36m force field 38 and built into model POPE membranes with TIP3P waters and K+ and Cl− to 150 mM, using CHARMM-GUI39,40. The bound Mn2+ were swapped to Mg2+. The final systems had approximately 300 lipids, 150 ions, and 27500 waters, for a total of ca. 125,000 atoms and a box size of 10 × 10 × 12.5 nm. All titratable side chains were set to their default protonation state, based on analysis run with propKa3.1 41,42.
Each system was minimized using the steepest descents method, then equilibrated in 6 rounds with restraints initially applied to protein backbone and sidechain atoms, lipid phosphate atoms and selected protein, lipid and sugar dihedrals, as per the standard CHARMM-GUI output. Where the HA chain was present, to optimise its positioning in the channel additional equilibration simulations of 90 ns were run with 50 kJ/mol/nm2 positional restraints applied to the protein backbone and to the bound substrate, but not to the HA chain. Production simulations were then seeded using the output frame of this final equilibration, with 3 or 5 repeats seeded for each system. Simulations were run using 2 fs time steps, with V-rescale thermostat at 303.15 K with a tau t of 1 ps, and semi-isotropic Parrinello-Rahman pressure coupling with tau p of 5 ps and a reference of 1 bar 40,43. All simulations were run in Gromacs 2019.443–45. In total, ca. 9 μs of data were generated, with full details in Supplementary Table 3.
Lipid and solvent densities in the apo MD simulations were computed over the 5 repeats for each system using the VolMap tool of VMD 46,47. HA-residue contact analysis was determined for each frame of the simulation based on a distance cut-off of 0.4 nm between each residue in the system and the specified sugar, and was run using MDAnalysis 47. Quantification of the occupancy of the binding pocket using in Extended Data Fig. 10 was performed by counting the number of frames that the acetamido group of the donor or acceptor GlcNAc was within 0.4 nm of any atom in Cys-231, and was run using the Gromacs tool gmx mindist. Analysis of the HA chain position in relation to the channel was calculated using the Gromacs tool gmx distance. The distance in the z-dimension between the centre-of-mass of each sugar and the centre-of-mass of the protein was computed, and plotted using NumPy48 and Matplotlib49.
Data availability
Raw EM movies and maps have been deposited at the PDB and EM data banks under accession codes 7SP7/EMD-25367, 7SP6/EMD-25366, 7SP8/EMD-25368, 7SP9/ EMD-25369, and 7SPA/EMD-25370 for the UDP-bound, D302N apo, UDP-GlcNAc-bound, primed (closed), and primed (open) states, respectively.
Extended Data
Extended Data Fig. 1|. Sequence alignment of HAS orthologs.

Comparison of HAS primary sequences from Chlorella virus (Cv), Homo sapiens (Hs) and Streptococcus equisimilis (Se). Naked mole rats (Nmr) contain an asparagine residue at Cv-HAS positions S165 and T289. Topology predictions were performed using TopCons50. Cylinders indicate secondary structure elements observed in Cv-HAS.
Extended Data Fig. 2|. Identification, data collection, and processing of Cv-HAS bound to two Nanobodies and UDP.
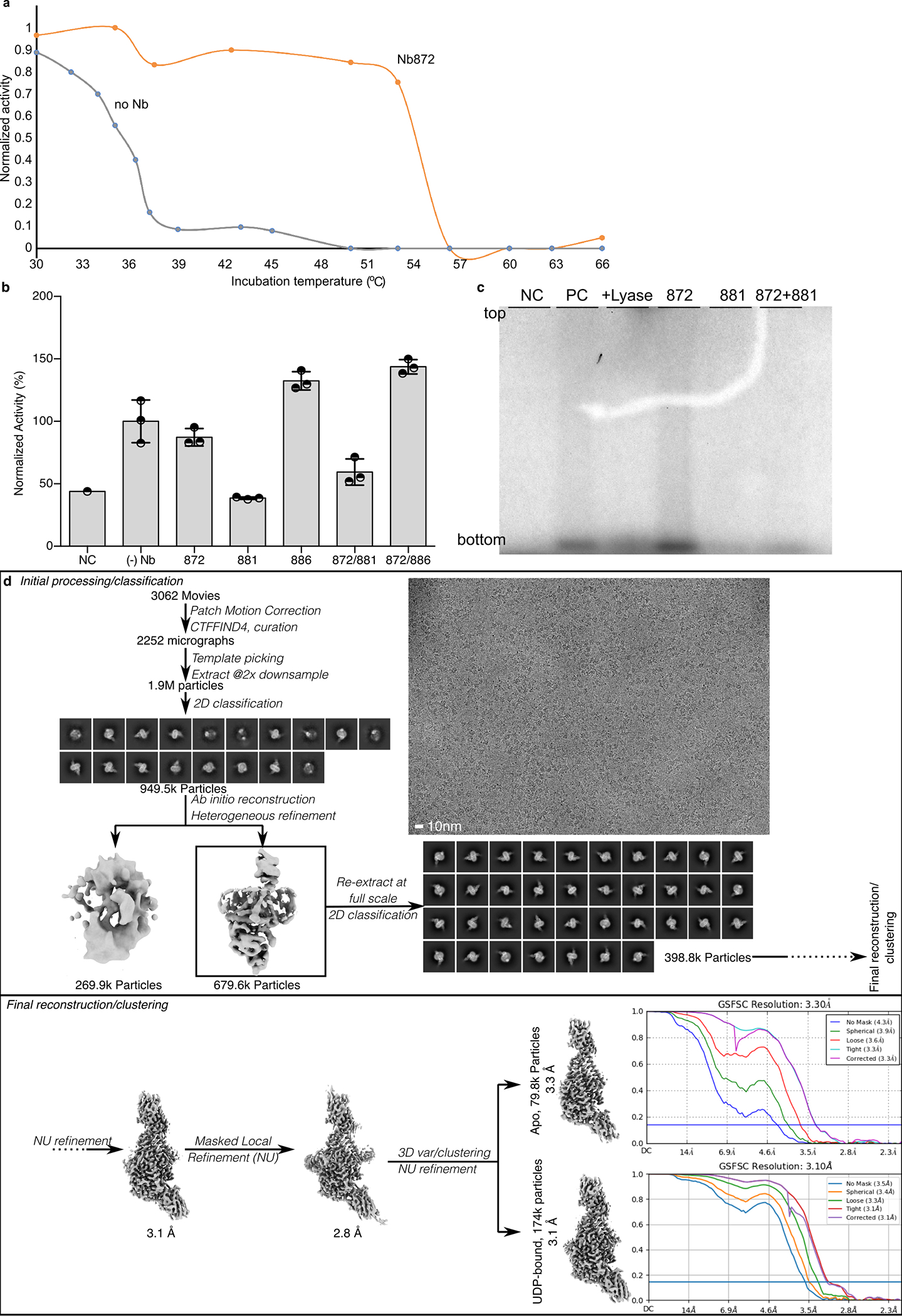
(a) Increased melting temperature of Cv-HAS in the presence of Nb872. Protein melting was measured based on enzymatic activity detected by quantifying the release of UDP in real time. (b) HA biosynthesis in the presence of the indicated nanobodies and based on quantification of 3H-labeled HA by scintillation counting. Data is normalized relative to product yields in the absence of nanobodies. Error bars represent deviations from the means with n=3 independent experiments. (c) Representative autoradiography of 14C-labeled HA produced in the presence of the indicated nanobodies. The experiment has been repeated at least 4 times with essentially identical results. NC: Negative control in the absence of UDP-GlcNAc substrate (for panel b) or UDP-GlcA (for panel c). PC: Positive control in the absence of nanobody. Lyase: Hyaluronan lyase treatment prior to SDS-PAGE. (d) This workflow produced the UDP-bound Cv-HAS structure.
Extended Data Fig. 3|. Cryo-EM data collection and processing of Cv-HAS D302N in the presence of substrate.
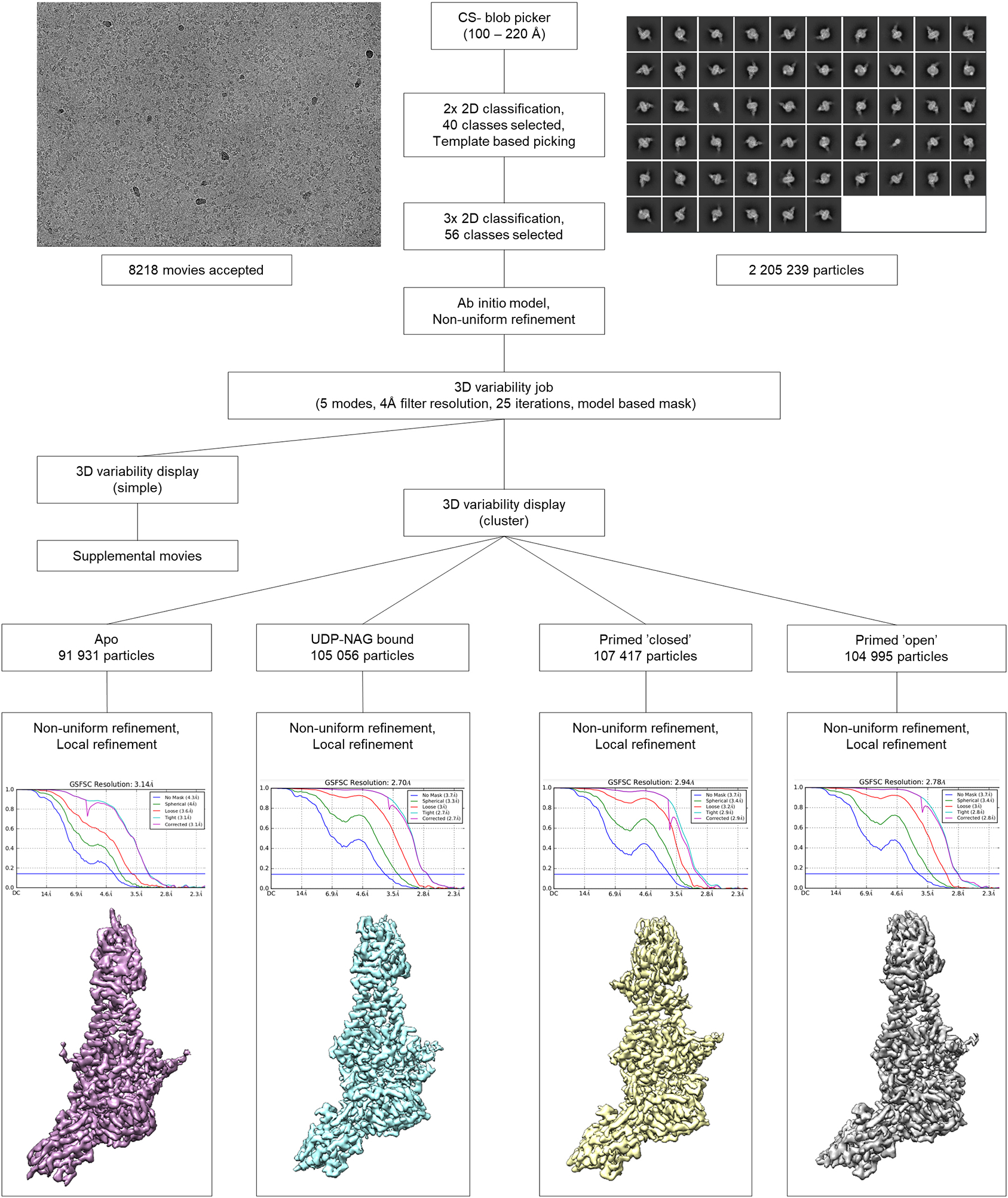
This workflow generated the apo, substrate-bound, primed, and primed with open channel Cv-HAS structures.
Extended Data Fig. 4|. Map quality and model building of UDP-bound Cv-HAS.
(a-d) Map overview, estimated resolution based on FSC, and particle orientation distribution. (e) Secondary structure elements and topology of Cv-HAS. (f-j) TM helices 2 to 6 of Cv-HAS. (k) TMH3–4 extracellular loop. (l) The extracellular TMH5–6 loop. (m) The QxxRW motif. (n) The C-terminal cytosolic helix. (o) The unresolved TMH5-IF3 loop. All maps are contoured at 7.0σ.
Extended Data Fig. 5|. Predicted location of TM helix 1.
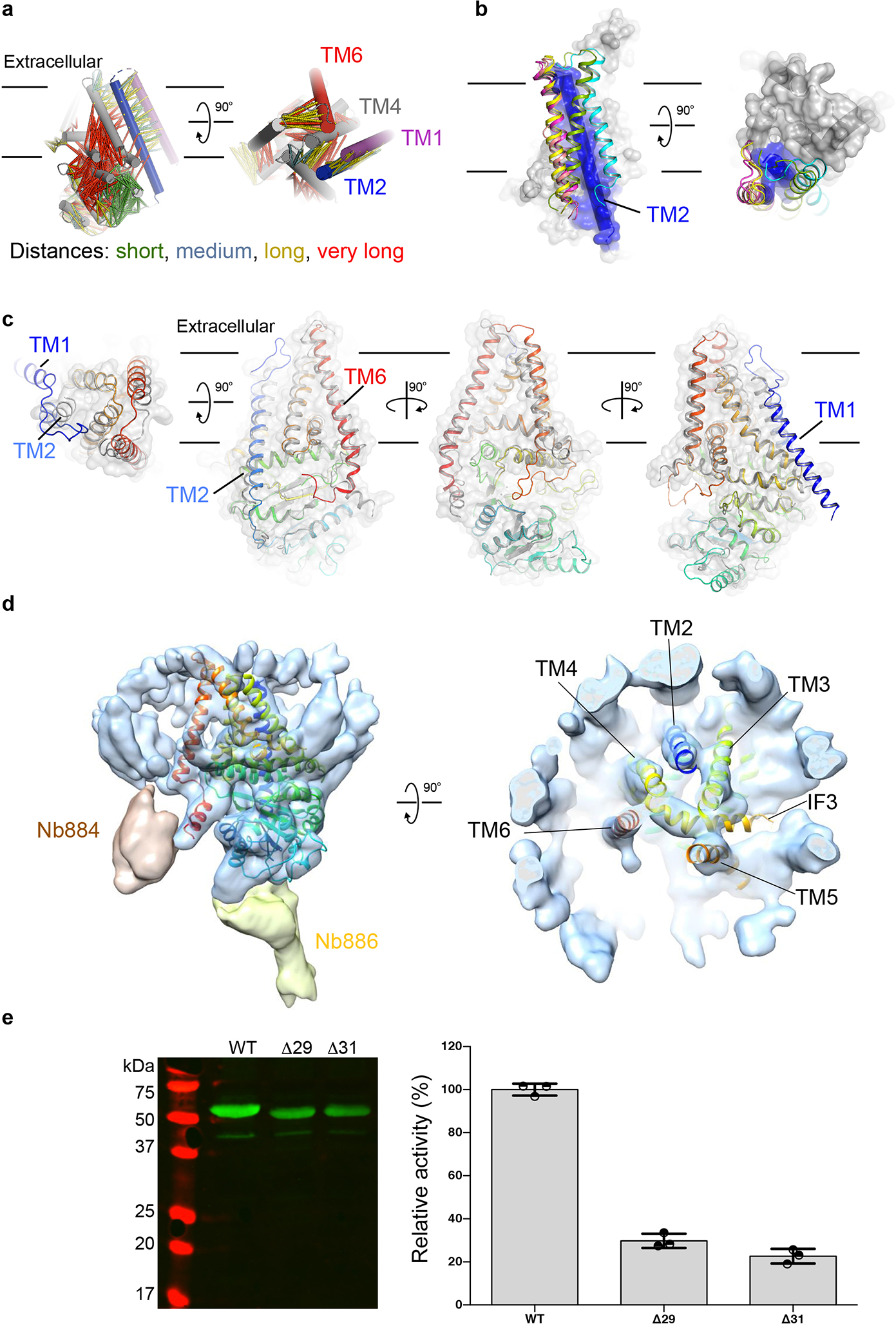
(a) Relationship of evolutionarily coupled residues within Cv-HAS’ TM and GT regions, generated in MapPred based on 65,535 sequences. TMH1 is shown at its predicted location as a violet cylinder. (b) RoseTTAfold models of full-length Cv-HAS. Cv-HAS is shown as a surface and its TMH 2 as a blue cylinder. TMH1 is shown as a cartoon at its predicted locations. (c) An AlphaFold2 predicted structure of human HAS-2 (colored blue to red from its N- to C-terminus) overlaid with the Cv-HAS structure shown as a gray cartoon and semi-transparent surface. (d) TMH1 remains disordered when two cytosolic Nbs are used for cryo-EM analyses. (e) Catalytic activity of TMH1 truncated Cv-HAS. Left: Western blot of IMVs used for in vitro activity measurements. Right: Catalytic activity of the indicated Cv-HAS mutants expressed relative to the wild type enzyme. The assay quantifies 3H-labeled HA by scintillation counting. Control reactions in the absence of UDP-GlcA served as background and are subtracted. Error bars represent deviations from the means with n=3 independent experiments..
Extended Data Fig. 6|. Lipids plug the lateral channel opening.
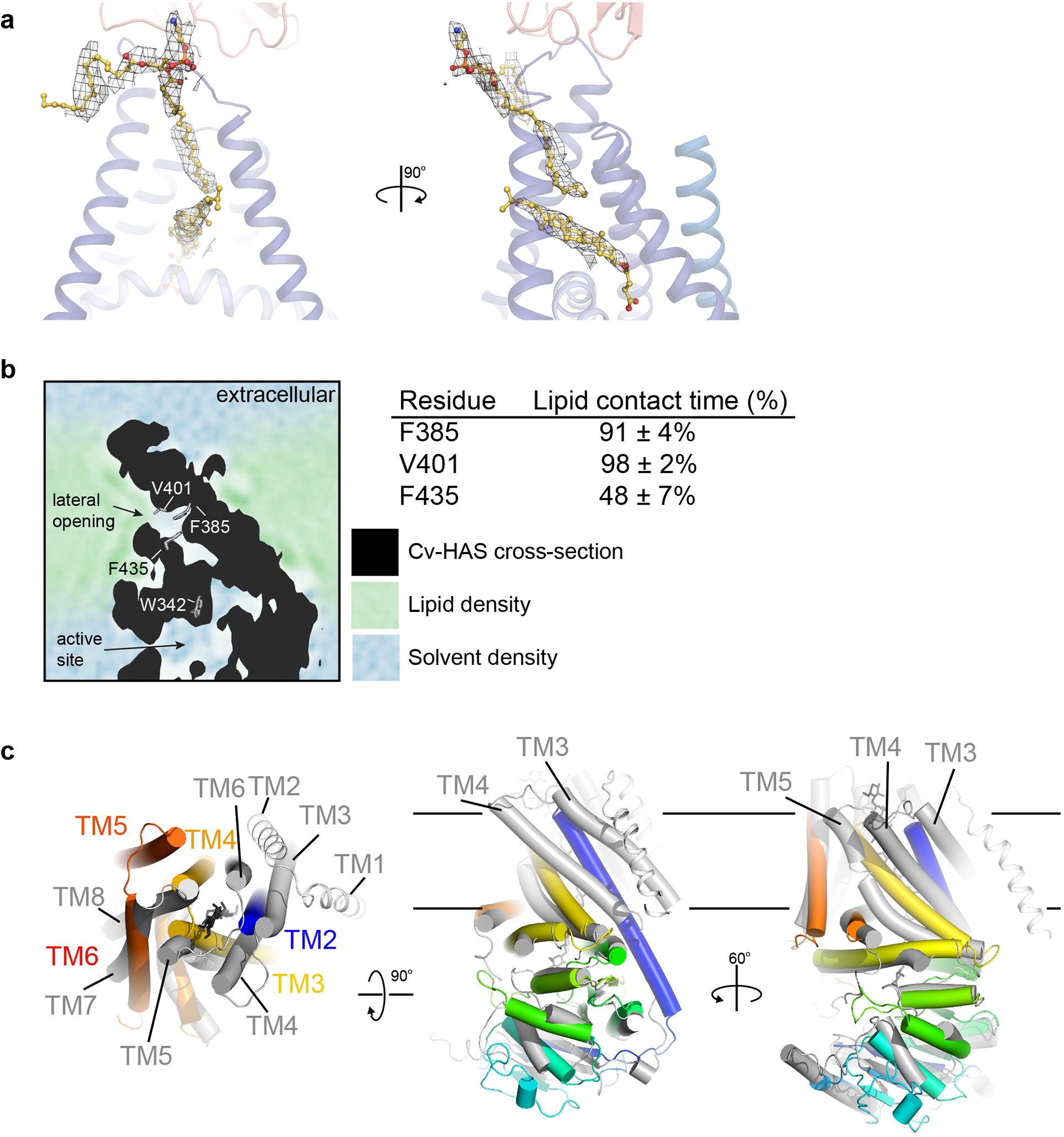
(a) Representative map regions for modeled lipids contoured at 7.0σ (from the UDP-GlcNAc bound set). (b) 2D slice from MD simulations of Cv-HAS (black area) within a POPE bilayer. Water and lipid densities are colored blue and green, respectively. Right panel: Lipid contact times with selected channel residues. (c) Comparison of the Cv-HAS (rainbow colored from the N- to C-terminus) and RsBcsA (gray, 4P00). Cellulose associated with BcsA is shown as black sticks. Helices are shown as cylinders except BcsA’s N-terminal two TMHs, which are shown as coils.
Extended Data Fig. 7|. Details of substrate-binding and of priming-induced conformational changes.
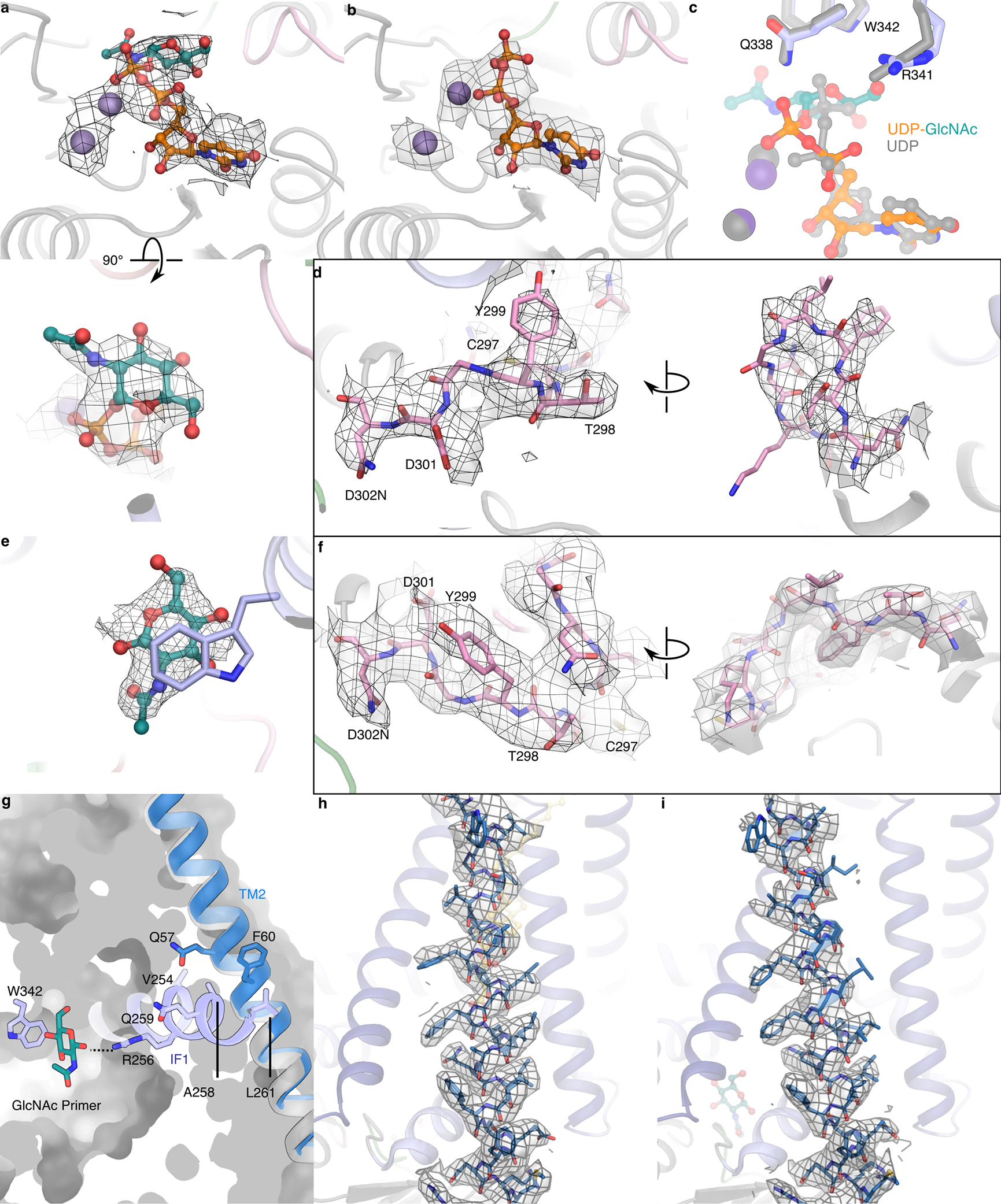
(a and b) Map quality for UDP-GlcNAc, UDP, and Mn2+ ligands. (c) Comparison of UDP and UDP-GlcNAc positions. (d) Map for the priming loop in nucleotide bound states. (e) Representative map for the GlcNAc primer. (f) Map for the priming loop in the primed states. (g) Contact point of TMH2 (open in blue, closed in gray) with IF1 in the primed state. (h and i) Map quality for TMH2 in a closed position (UDP-GlcNAc bound) and open position. All maps are contoured at 7.0σ.
Extended Data Fig. 8|.
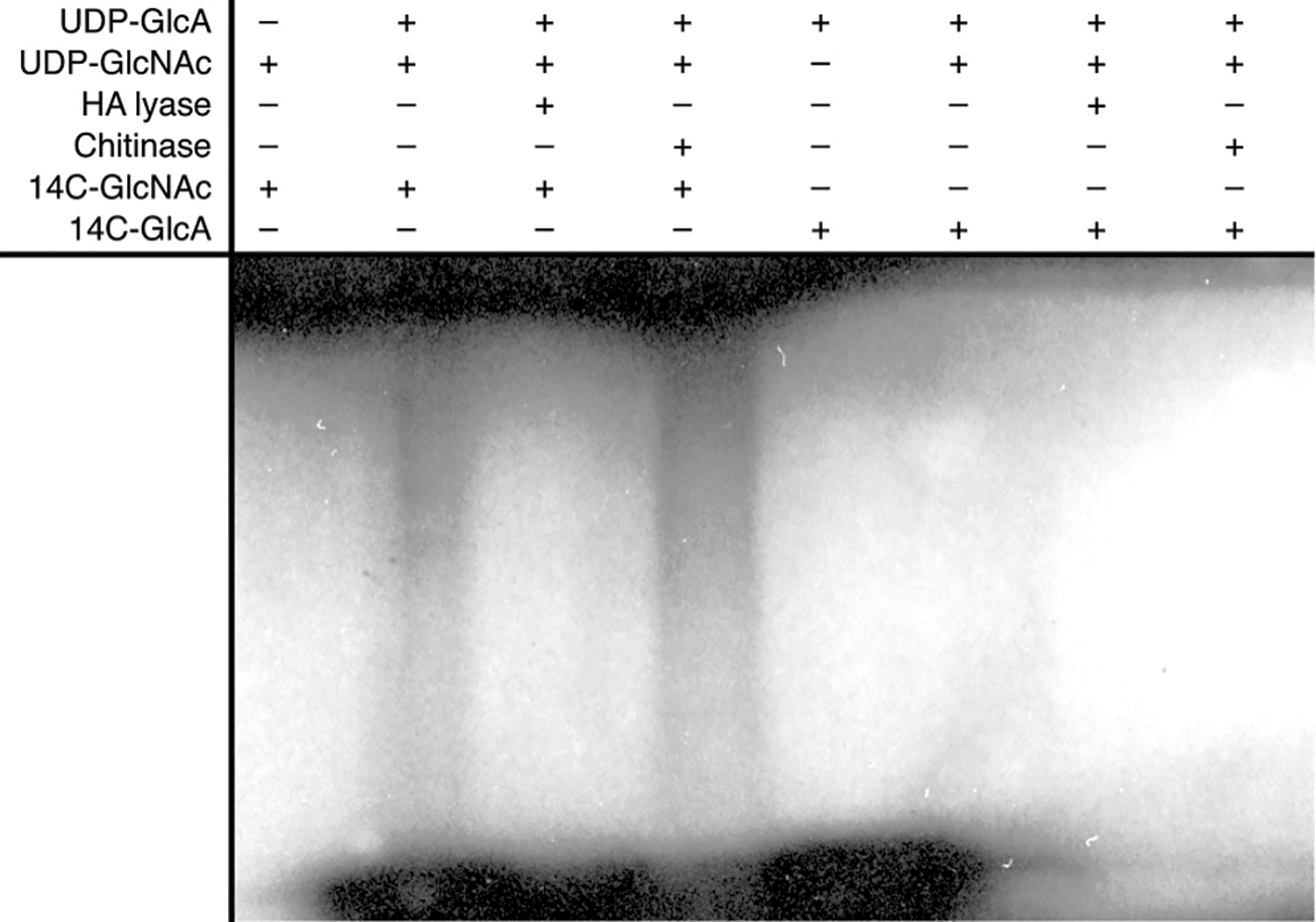
GlcNAc priming of HA biosynthesis. Shown is an autoradiogram of 14C-labeled HA after SDS-PAGE. The experiment has been repeated at least 3 times with essentially identical results.
Extended Data Fig. 9|. Effect of monosaccharides on substrate hydrolysis.
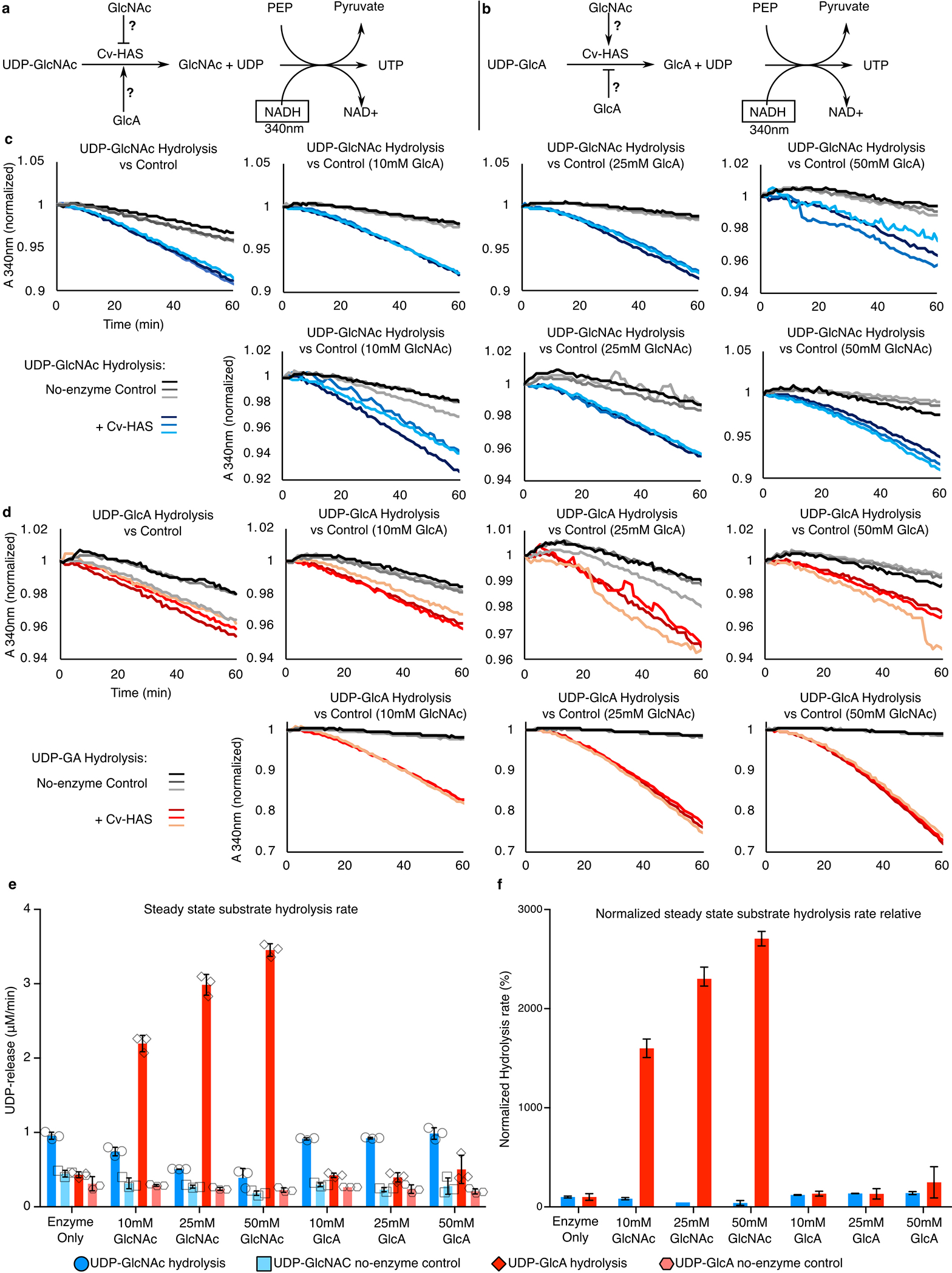
(a and b) Reaction schemes for UDP-GlcA and UDP-GlcNAc hydrolysis. (c and d) Raw absorbance measurements. (e) Quantification of hydrolysis rates in the presence of increasing monosaccharide concentrations. Blue and Red: Hydrolysis of UDP-GlcNAc and UDP-GlcA, respectively. Light and dark colors represent control reactions in the absence of enzyme. Right panel: Background subtracted hydrolysis rates. Error bars represent deviations from the means with n=3 independent experiments.
Extended Data Fig. 10|. Likely mechanism of alternating substrate polymerization and comparison with cellulose synthase.
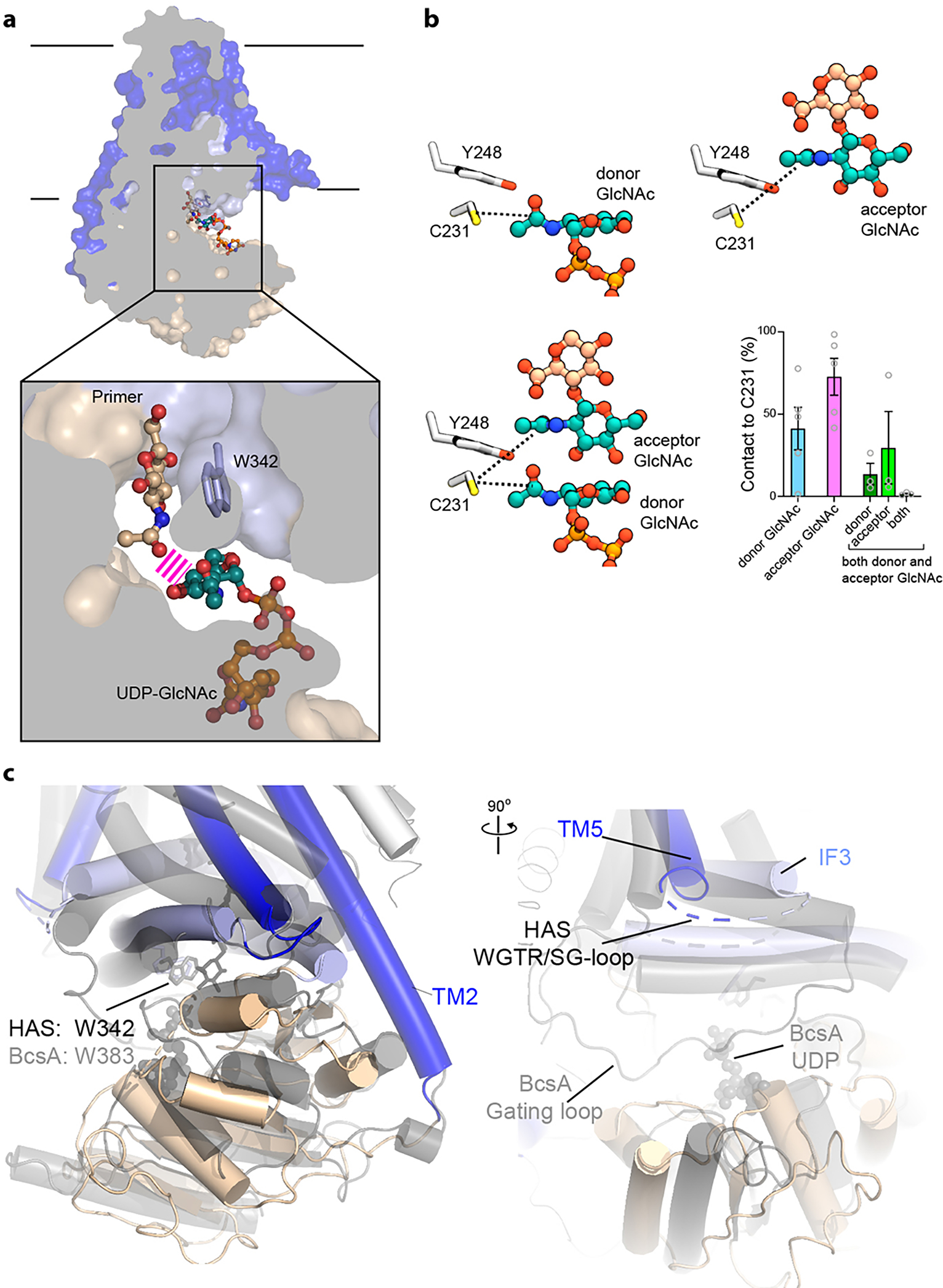
(a) Superimposition of substrate-bound and primed Cv-HAS structures. The close distance between the primer and donor sugar is indicated by gray bars. (b) Contact likelihood between C231 and GlcNAc for the systems in a over the last 125 ns of each simulation. In the case of GlcNAc being in both donor and acceptor positions, both GlcNAc units are less likely to bind C231, and exhibit very high variance regarding binding poses. Of particular note, the chance of both GlcNAc being in the C231 pocket at the same time is very low (ca. 1.5±0.8%). (c) Cv-HAS is superimposed with the Rhodobacter sphaeroides (Rs) BcsA-B complex (PDB: 4P00) based on secondary structure matching. Rs-BcsA-B is colored gray and Cv-HAS is colored blue and green for its TM and GT domains. The cellulose polymer associated with Rs-BcsA-B is shown as black sticks.
Extended Data Table 1 |. Data collection, processing, and refinement statistics.
EM statistics are listed for wild type (WT) and catalytically inactive (D302N) Cv-HAS.
| State | Apo (WT) (detergent) | UDP (WT) | D302N: Apo | UDP-GlcNAc | Primed (closed) | Primed (open) | Cytosolic Nbs |
|---|---|---|---|---|---|---|---|
| PDB EMDB | 7SP7 EMD-25367 | 7SP6 EMD-25366 | 7SP8 EMD-25368 | 7SP9 EMD-25369 | 7 SPA EMD-25370 | ||
|
| |||||||
| Data collection/ processing | |||||||
| Magnification | 81,000x | 81,000x | 81,000x | 81,000x | |||
| Voltage (kV) | 300 | 300 | 300 | 300 | |||
| Electron Exposure (e-/Å2) | 51 | 51 | 48 | 50 | |||
| Defocus range (pin) | −2.2 to−1.4 | −2.0 to−1.0 | −2.0 to−1.0 | −2.25 to−1.0 | |||
| Pixel size (Å2) | 1.08 | 1.08 | 1.08 | 1.08 | |||
| Symmetry imposed | PI | PI | PI | PI | |||
| Initial Particles | 1,548,653 | 1,897,858 | 4,391,341 | 2,065,933 | |||
| Final Particles | 114152 | 174,023 | 91931 | 105,056 | 107,417 | 104,995 | 387,019 |
| Map resolution (Å) | 3.4 | 3.1 | 3.1 | 2.7 | 2.9 | 2.8 | 5.7 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map Resolution Range (Å) | 10–3.3 | 50.6–2.74 | 51.1–2.83 | 45.5–2.41 | 42.2–2.58 | 26.4–2.48 | - |
| Refinement | |||||||
| Initial Model | 4P00 | Apo (det) | UDP | UDP | UDP | Closed | |
| Model resolution (Å) | 3.4 | 3.5 | 2.9 | 3.2 | 3.1 | ||
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | ||
| Map sharpening B factor (Å2) | 113.2 | 82.9 | 61.3 | 55.7 | 56.6 | 56.7 | 305.0 |
| Model composition | |||||||
| Non-hydrogen Atoms | 6052 | 5957 | 6090 | 5966 | 6017 | ||
| Protein Residues | 739 | 733 | 742 | 737 | 737 | ||
| Ligands | 5 | 3 | 5 | 2 | 3 | ||
| B factors (Å2) | |||||||
| Protein | 78.87 | 96.12 | 66.52 | 54.16 | 49.08 | ||
| Ligand | 83.23 | 104.60 | 74.96 | 65.46 | 59.66 | ||
| R.M.S. deviations | |||||||
| Bond lengths (Å) | 0.001 | 0.001 | 0.001 | 0.001 | 0.002 | ||
| Bond angles (°) | 0.352 | 0.419 | 0.406 | 0.399 | 0.409 | ||
| Validation | |||||||
| MolProbity Score | 1.12 | 1.29 | 1.30 | 1.29 | 1.34 | ||
| Clash Score | 3.34 | 5.33 | 5.47 | 5.43 | 6.2 | ||
| Poor rotomers (%) | 0 | 0 | 0.16 | 0 | 0 | ||
| Ramachandran plot | |||||||
| Favored (%) | 98.63 | 98.48 | 98.50 | 98.76 | 98.62 | ||
| Allowed (%) | 1.37 | 1.52 | 1.50 | 1.24 | 1.38 | ||
| Outliers (%) | 0 | 0 | 0 | 0 | 0 | ||
Supplementary Material
{kind=link}
Acknowledgments:
We thank K. Dryden and M. Purdy from the MEMC at the University of Virginia as well as Adam Wier and support staff at the NCI-NCEF. E.P. and J.S. acknowledge the support of Instruct-ERIC, part of the European Strategy Forum on Research Infrastructures (ESFRI), and the Research Foundation - Flanders (FWO) for supporting the nanobody discovery and thank Eva Beke for technical assistance. J.Z. and F.M. were supported by NIH grant R21AI148853. R.H. was supported by NIH grant R01GM101001 (awarded to J.Z.). R.A.C. and P.J.S. are supported by Wellcome (208361/Z/17/Z). P.J.S.’s lab is supported by awards from the BBSRC (BB/P01948X/1, BB/R002517/1 and BB/S003339/1) and MRC (MR/S009213/1). P.J.S. acknowledges the University of Warwick Scientific Computing Research Technology Platform for computational access.
Footnotes
Competing interests:
The authors declare no competing interests.
References:
- 1.Girish KS & Kemparaju K The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci 80, 1921–1943, (2007). [DOI] [PubMed] [Google Scholar]
- 2.Cyphert JM, Trempus CS & Garantziotis S Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int J Cell Biol 2015, 563818, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vigetti D et al. Hyaluronan: biosynthesis and signaling. Biochim Biophys Acta 1840, 2452–2459, (2014). [DOI] [PubMed] [Google Scholar]
- 4.DeAngelis P Hyaluronan synthases: fascinating glycosyltransferases from vertebrates, bacterial pathogens, and algal viruses. Cell Mol Life Sci 56, 670–682, (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sironen R et al. Hyaluronan in human malignancies. Exp Cell Res 317, 383–391, (2011). [DOI] [PubMed] [Google Scholar]
- 6.Cowman MK, Lee H-G, Schwertfeger KL, McCarthy JB & Turley EA The Content and Size of Hyaluronan in Biological Fluids and Tissues. Front Immunol 6, 261, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hubbard C, McNamara J, Azumaya C, Patel M & Zimmer J The hyaluronan synthase catalyzes the synthesis and membrane translocation of hyaluronan. J Mol Biol 418, 21–31, (2012). [DOI] [PubMed] [Google Scholar]
- 8.Weigel PH & Deangelis PL Hyaluronan synthases: a decade-plus of novel glycosyltransferases. J Biol Chem 282, 36777–36781, (2007). [DOI] [PubMed] [Google Scholar]
- 9.DeAngelis PL, Jing W, Drake RR & Achyuthan AM Identification and molecular cloning of a unique hyaluronan synthase from Pasteurella multocida. J Biol Chem 273, 8454–8458, (1998). [DOI] [PubMed] [Google Scholar]
- 10.Camenisch TD et al. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest 106, 349–360, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itano N et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J Biol Chem 274, 25085–25092, (1999). [DOI] [PubMed] [Google Scholar]
- 12.DeAngelis P, Jing W, Graves M, Burbank D & Van Etten J Hyaluronan synthase of chlorella virus PBCV-1. Science 278, 1800–1803, (1997). [DOI] [PubMed] [Google Scholar]
- 13.Blackburn MR et al. Distinct reaction mechanisms for hyaluronan biosynthesis in different kingdoms of life. Glycobiology 28, 108–121, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardon E et al. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc 9, 674–693, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tlapak-Simmons VL, Baron CA & Weigel PH Characterization of the purified hyaluronan synthase from Streptococcus equisimilis. Biochemistry 43, 9234–9242, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296, (2017). [DOI] [PubMed] [Google Scholar]
- 17.Lairson LL, Henrissat B, Davies GJ & Withers SG Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem 77, 521–555, (2008). [DOI] [PubMed] [Google Scholar]
- 18.Cantarel B, Coutinho P, Rancurel C & Bernard T The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37, D233–238, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Q et al. Protein contact prediction using metagenome sequence data and residual neural networks. Bioinformatics 36, 41–48, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tunyasuvunakool K, Adler J, Wu Z, Jumper J & Hassabis D Highly accurate protein structure prediction for the human proteome. Nature, 10.1038/s41586-41021-03828-41581, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baek M Accurate prediction of protein structures and interactions using a 3-track network. bioRxiv 10.1101/2021.06.14.448402 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charnock SJ & Davies GJ Structure of the nucleotide-diphospho-sugar transferase, SpsA from Bacillus subtilis, in native and nucleotide-complexed forms. Biochemistry 38, 6380–6385, (1999). [DOI] [PubMed] [Google Scholar]
- 23.Morgan J, Strumillo J & Zimmer J Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature 493, 181–186, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purushotham P, Ho R & Zimmer J Architecture of a catalytically active homotrimeric plant cellulose synthase complex. Science 369, 1089–1094, (2020). [DOI] [PubMed] [Google Scholar]
- 25.Morgan JLW, McNamara JT & Zimmer J Mechanism of activation of bacterial cellulose synthase by cyclic di-GMP. Nature Struct Mol Biol 21, 489–496, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McManus J, Yang H, Wilson L, Kubicki J & Tien M Initiation, Elongation, and Termination of Bacterial Cellulose Synthesis. ACS Omega 3, 2690–2698, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orlean P & Funai D Priming and elongation of chitin chains: Implications for chitin synthase mechanism. Cell Surf 5, 100017, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J et al. Key Role of the Carboxyl Terminus of Hyaluronan Synthase in Processive Synthesis and Size Control of Hyaluronic Acid Polymers. Biomacromolecules 18, 1064–1073, (2017). [DOI] [PubMed] [Google Scholar]
- 29.Morgan JL et al. Observing cellulose biosynthesis and membrane translocation in crystallo. Nature 531, 329–334, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.PyMol. PyMol, <https://pymol.org/2/> (https://pymol.org/2/). [Google Scholar]
- 31.Ho B & Gruswitz F HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct Biol 8, 49, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Studier F Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41, 207–234, (2005). [DOI] [PubMed] [Google Scholar]
- 33.Kelly SJ, Taylor KB, Li S & Jedrzejas MJ Kinetic properties of Streptococcus pneumoniae hyaluronate lyase. Glycobiology 11, 297–304, (2001). [DOI] [PubMed] [Google Scholar]
- 34.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132, (2004). [DOI] [PubMed] [Google Scholar]
- 36.Adams P et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612, (2004). [DOI] [PubMed] [Google Scholar]
- 38.Huang J et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature 14, 71–73, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jo S, Kim T, Iyer VG & Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865, (2008). [DOI] [PubMed] [Google Scholar]
- 40.Lee J et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theo Comput 12, 405–413, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sondergaard CR, Olsson MHM, Rostkowski M & Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pK(a) Values. J Chem Theor Comp 7, 2284–2295, (2011). [DOI] [PubMed] [Google Scholar]
- 42.Olsson MHM, Sondergaard CR, Rostkowski M & Jensen JH PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pK(a) Predictions. J Chem Theor Comp 7, 525–537, (2011). [DOI] [PubMed] [Google Scholar]
- 43.Bussi G, Donadio D & Parrinello M Canonical sampling through velocity rescaling. J Chem Phys 126, 014101, (2007). [DOI] [PubMed] [Google Scholar]
- 44.Parrinello M, Rahman A Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys 52, 7182–7190, (1981). [Google Scholar]
- 45.Berendsen HJC, van der Spoel D, van Drunen R GROMACS: A message-passing parallel molecular dynamics implementation. Comp Phys Comm 91, 43–56, (1994). [Google Scholar]
- 46.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38, (1996). [DOI] [PubMed] [Google Scholar]
- 47.Hunter JD Matplotlib: A 2D Graphics Environment. Comp Sci Engineering 9, 90–95, (2007). [Google Scholar]
- 48.Michaud-Agrawal N, Denning EJ, Woolf TB & Beckstein O MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J Comp Chem 32, 2319–2327, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harris CR et al. Array programming with NumPy. Nature 585, 357–362, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernsel A, Viklund H, Hennerdal A & Elofsson A TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res 37, W465–468, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bussi G, Donadio D & Parrinello M Canonical sampling through velocity rescaling. J Chem Phys 126, 014101, (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw EM movies and maps have been deposited at the PDB and EM data banks under accession codes 7SP7/EMD-25367, 7SP6/EMD-25366, 7SP8/EMD-25368, 7SP9/ EMD-25369, and 7SPA/EMD-25370 for the UDP-bound, D302N apo, UDP-GlcNAc-bound, primed (closed), and primed (open) states, respectively.