

Abstract
Bacteroides fragilis toxin (BFT) is a protein secreted by enterotoxigenic (ETBF) strains of B. fragilis. BFT is synthesized as a proprotein (proBFT) that is predicted to be a lipoprotein and that is cleaved into two discrete fragments by a clostripain-like protease called fragipain (Fpn). In this study, we obtained evidence that Fpn cleaves proBFT following its transport across the outer membrane. Remarkably, we also found that the disruption of the fpn gene led to a strong reduction in the level of >100 other proteins, many of which are predicted to be lipoproteins, in the culture medium of an ETBF strain. Experiments performed with purified Fpn provided direct evidence that the protease releases at least some of these proteins from the cell surface. The observation that wild-type cells outcompeted an fpn− strain in co-cultivation assays also supported the notion that Fpn plays an important role in cell physiology and is not simply dedicated to toxin biogenesis. Finally, we found that purified Fpn altered the adhesive properties of HT29 intestinal epithelial cells. Our results suggest that Fpn is a broad-spectrum protease that not only catalyzes the protein secretion on a wide scale but that also potentially cleaves host cell proteins during colonization.
Keywords: Bacteroides, lipoprotein, microbiome, outer membrane, protease, protein secretion
1 |. INTRODUCTION
The Bacteroides are a group of commensal Gram-negative bacteria that are widely distributed and highly abundant in the human gut microbiome. One member of this genus, B. fragilis, is of particular interest because it is involved in a variety of activities that influence human health including polysaccharide digestion, gut development, and modulation of the immune system (Erturk-Hasdemir and Kasper, 2018; Wexler and Goodman, 2017). B. fragilis is also an opportunistic pathogen that is a major cause of gastrointestinal abscesses and anaerobic sepsis if there is a disruption of the intestinal epithelium (Redondo et al., 1995; Wexler, 2007). In addition, specific enterotoxigenic (ETBF) strains of B. fragilis contain a pathogenicity island that encodes B. fragilis toxin (BFT), also known as fragilysin (Pierce and Bernstein, 2016; Sears et al., 1995). Production of this protein causes diarrheal disease and is associated with an increased risk of colon cancer (Dejea et al., 2018; Myers et al., 1987; Sears et al., 2008; Wu et al., 2009). Available evidence suggests that ~30% of the human population carries ETBF strains asymptomatically (Sears et al., 2014). The absence of symptoms may be due to the suppression of BFT expression by a recently described mechanism (Hecht et al., 2017).
Although proteins secreted by B. fragilis or localized to the cell surface play important roles in host colonization, microbial interactions, and pathogenicity, the mechanisms of protein secretion are poorly understood. B. fragilis has only a few of the well characterized secretion systems found in E. coli and other Proteobacteria (Abby et al., 2016; Wilson et al., 2015). One of these systems, the type VI secretion system (T6SS), is involved in intraspecies competition and prevents closely related B. fragilis strains from colonizing the same niche in the gut (Hecht et al., 2016; Wexler et al., 2016). B. fragilis also appears to produce multiple type 1 secretion systems (T1SSs) simultaneously (Wilson et al., 2015), but the proteins that are secreted through these systems are unknown. Interestingly, unlike E. coli, B. fragilis has been shown to produce a large number of lipoproteins that are exposed on the cell surface and/or secreted into the extracellular milieu (Wilson et al., 2015). This observation suggests that extracellular lipoproteins play an important role in B. fragilis physiology. While the Lol pathway is known to transport lipoproteins from the outer leaflet of the inner membrane (IM) to the inner leaflet of the outer membrane (OM) in E. coli, the mechanisms by which lipoproteins are transported across the OM have not been widely investigated. A novel integral membrane protein called SLAM promotes the translocation of lipoproteins across the OM in Neisseria (Hooda et al., 2016), but a counterpart has yet to be identified in the Bacteroides.
BFT is the only toxin secreted by B. fragilis that is known to target host cells (Pierce and Bernstein, 2016; Rhee et al., 2009). BFT is presumably a lipoprotein because it contains a lipobox motif [(LVI) (ASTVI)(GAS)|C] (Babu et al., 2006) that spans the signal peptidase II (SP II) cleavage site. The cysteine in this motif is typically acylated and becomes the N-terminal residue of a lipoprotein following signal peptide cleavage. BFT is synthesized as a 397 residue prepro-protein with a large N-terminal pro domain. This domain interfaces with and presumably promotes the folding of a C-terminal catalytic domain, which functions as a metalloprotease (Franco et al., 1997, 2007; Goulas et al., 2011). Activation of the catalytic domain requires its release from the pro domain by proteolytic cleavage. The active toxin has been shown to cleave several host proteins including E-cadherin, actin, and fibrinogen (Moncrief et al., 1995; Wu et al., 1998). Cleavage of E-cadherin alters the integrity of the epithelial cell layer and leads to cell rounding. BFT has also been implicated in the formation of colon tumors in a mouse model (Wu et al., 2009) and is associated with colorectal cancer in humans (Boleij et al., 2015; Haghi et al., 2019).
A member of the C11 family of cysteine proteases designated fragipain (Fpn) was recently shown to mediate the proteolytic maturation of proBFT at R211 (Choi et al., 2016). The prototypical C11 proteases are the arginine-specific clostripains produced by the Clostridia. C. histolyticum clostripain has been well studied for its bio-technology applications and its possible role in disease progression (Chakravorty et al., 2011). The C11 family also includes the gingipains produced by Porphyromonas gingivalis, a dental pathogen that belongs to the same order as B. fragilis (the Bacteroidales). The gingipains are major virulence factors that promote nutrient acquisition, adhesion, and manipulate the host immune response (Fitzpatrick et al., 2009; Guo et al., 2010; Li and Collyer, 2011). These proteases are also arginine-specific and have been shown to cleave a variety of host proteins including hemoglobin, complement, and defensins, but only a few bacterial proteins. Their bacterial targets include fimbrial proteins that are involved in adherence to epithelial cells (Kadowaki et al., 1998; Shoji et al., 2004). Like other members of the C11 family of proteases, Fpn is itself a proenzyme that is activated by an auto-cleavage reaction (Choi et al., 2016; Herrou et al., 2016). While Fpn is also predicted to be a lipoprotein, the cellular location where it cleaves proBFT is unknown.
In this study, we show that both proBFT and Fpn are located on the cell surface of B. fragilis and provide evidence that Fpn cleaves BFT following its translocation across the OM. These results, together with the observation that Fpn homologs are encoded in the genomes of many members of the Bacteroidetes that do not produce BFT, led us to conjecture that Fpn cleaves additional cell surface proteins. Consistent with our hypothesis, we found that the level of the majority of the proteins found in the secretome of a wild-type ETBF strain, which includes many lipoproteins, was strongly reduced in an fpn− strain. Interestingly, we also found that purified Fpn can release at least some of these proteins from the cell surface. Finally, we found that Fpn promotes morphological changes in epithelial cells through the cleavage of E-cadherin. Our results indicate that Fpn is a broad-spectrum protease that promotes the secretion of a wide variety of proteins and raise the possibility that it also acts on host cell proteins during colonization.
2 |. RESULTS
2.1 |. Fpn mediates the proteolytic processing of BFT on the cell surface
As in a previous study (Choi et al., 2016), we used a transposon mutagenesis strategy to identify genes that are required for the production of active BFT. After transforming B. fragilis ETBF strain 20656-2-1 (ATCC 43860) with a Mariner-based transposon delivery plasmid (Ichimura et al., 2014), we incubated the culture medium from ~4,000 individual insertion mutants with HT29 intestinal epithelial cells and screened for the absence of cell rounding (intoxication) that is normally caused by BFT. Transposon insertion sites were identified by sequencing DNA obtained after consecutive rounds of semi-random PCR. Genes disrupted by the transposon were identified by BLAST using both the NCBI database and the genome sequence of strain 20656-2-1 (Pierce and Bernstein, 2016). One particularly interesting mutation mapped to gene BF60_25670, which encodes the same clostripain-like protease that was identified in the earlier screen and designated Fpn (Choi et al., 2016) (Figure S1a,b). Western blot analysis using a polyclonal antiserum raised against a C-terminal BFT peptide (Pierce and Bernstein, 2016) showed that the cleaved, mature ~20 kD toxin was present in the culture media of the “high producing” ETBF strain 86-5443-2-2 and wild-type 20656-2-1 cells, but not in the culture media of an isogenic bft− strain or the fpn mutant (Figure S1c). Consistent with previous results (Choi, et al., 2016), we observed only unprocessed BFT (“proBFT”) in the culture medium of the mutant strain.
To further characterize Fpn, we replaced its signal peptide with an N-terminal His-tag and overproduced the derivative in E. coli. After purifying the protein by Ni-NTA chromatography, we observed two major ~18 and ~24 kD bands (Figure S2a). In light of previous evidence that the active form of Fpn (like the active form of the related clostripain protease) is generated by an autocatalytic cleavage (Dargatz et al., 1993; Herrou et al., 2016; Manabe et al., 2010), these two polypeptides are presumably N- and C-terminal fragments of Fpn that form a complex and co-purify. Consistent with this interpretation, only the smaller polypeptide was recognized by an anti-His antiserum (Figure S2b). As expected, a ~43 kD polypeptide eluted from the column when the predicted catalytic cysteine residue (C180) was mutated to alanine and was used to generate a polyclonal antiserum that recognized both of the smaller polypeptides (Figure S2c).
Two observations provided strong evidence that both proBFT and Fpn are surface-localized lipoproteins. In addition to containing a typical lipobox, both proteins contain a conserved acidic (and asparagine-rich) sequence motif between residues +2 and +6 that has been shown to promote the surface localization of lipoproteins in several different members of the Bacteroidetes (Lauber et al., 2016; Valguarnera et al., 2018). To examine the localization of proBFT and Fpn experimentally, we treated intact fpn− and fpn+ cells with proteinase K (PK) and conducted Western blots. The finding that the protease degraded both proteins, but not the periplasmic chaperone SurA, showed that they were exposed on the cell surface (Figure 1a,b). In the case of Fpn, the C-terminal ~24 kD fragment was digested more rapidly than the lipid-anchored ~18 kD fragment presumably because it extends farther from the cell surface.
FIGURE 1.
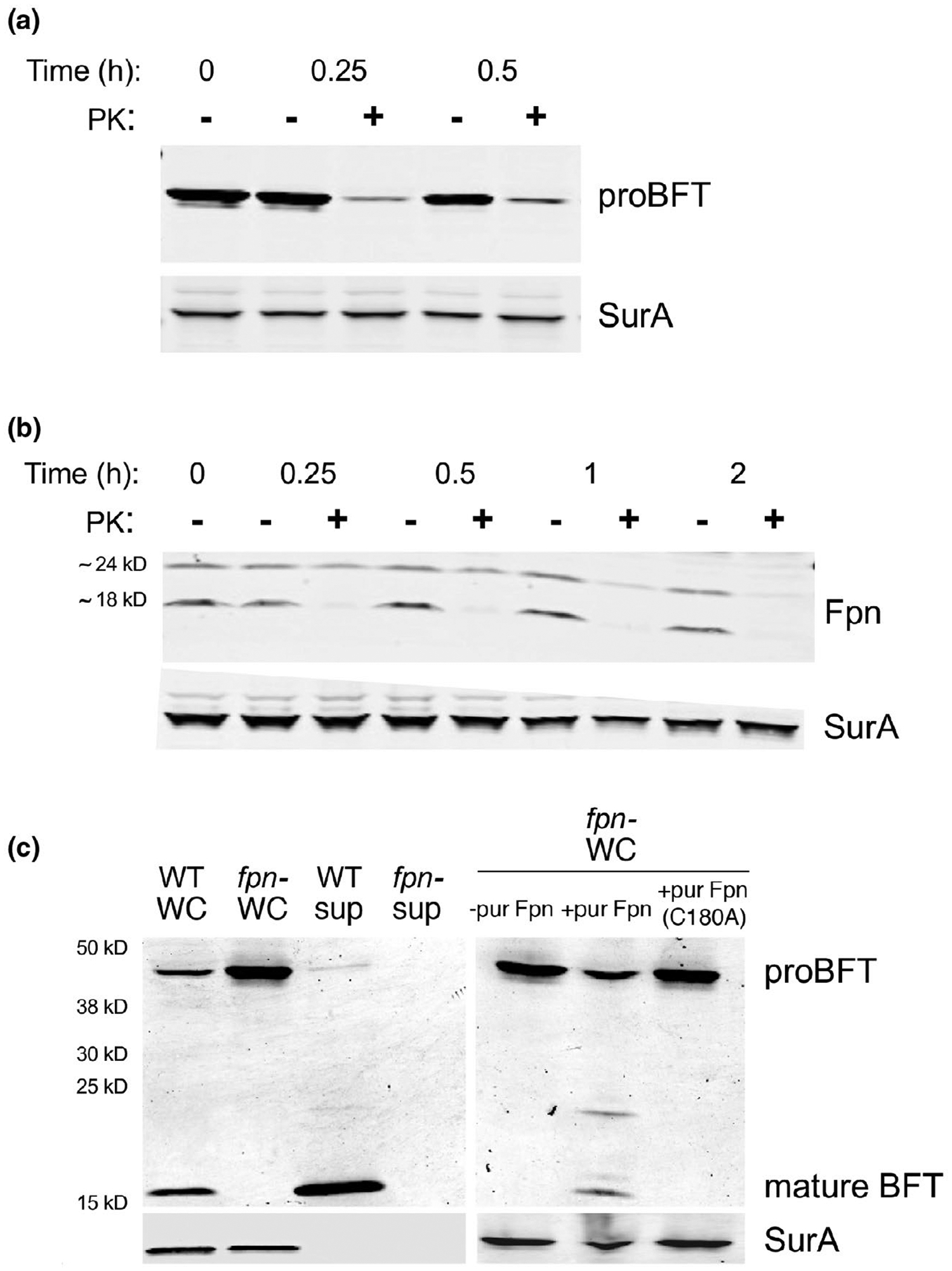
Fpn cleaves BFT on the cell surface. An fpn− ETBF strain (a) and an isogenic wild-type strain (b) were grown to mid-log phase. Intact cells were then treated with proteinase K (PK) for the indicated time, and Western blots were performed using antisera against SurA and either BFT (a) or Fpn (b). (c) Cells were grown to mid-log phase and separated from the culture medium by centrifugation. The intact fpn− cells were mock treated or treated with purified Fpn or the Fpn (C180A) mutant for 30 min at 37°C. Whole-cell (WC) and culture medium (sup) fractions were then analyzed by Western blot using antisera against BFT and SurA
The results strongly suggested that Fpn cleaves proBFT after the two proteins are localized on the cell surface. This idea was corroborated by the observation that the incubation of intact fpn− cells with purified Fpn, but not with the Fpn C180A mutant, promoted the proteolytic maturation of the toxin (Figure 1c). The finding that the cleavage was blocked by the cysteine protease inhibitors TLCK and leupeptin provided additional evidence that the reaction was mediated by Fpn rather than a contaminating protease (Figure S3). Interestingly, mature BFT was also released from proBFT in a concentration-dependent fashion by clostripain produced by C. histolyticum (Figure S3). This result raises the possibility that proBFT is susceptible to cleavage at or near residue R211 by a broad range of related C11 family members. Finally, we found that the mutation of the cysteine residue in the BFT lipobox or the replacement of the native signal peptide with a generic signal peptide blocked both the membrane localization and the proteolytic processing of the protein in an fpn+ strain (Figure S4). This finding shows that a soluble (and presumably periplasmically localized) form of proBFT is not subject to proteolytic maturation. Taken together, the results support a model in which proBFT is lipidated, transferred to the cell surface, and then cleaved after it interacts with Fpn.
2.2 |. The loss of Fpn dramatically affects the composition of the secretome
Intriguingly, a homology search revealed that Fpn-like proteases are widely distributed throughout the Bacteroidetes and are found in an abundance of organisms (such as NTBF strains of B. fragilis) that do not appear to produce BTF or any related toxins. We identified 238 homologs of the Fpn protein produced by ETBF strain 20656-2-1 using BLASTp (default settings). All of the homologs are found in the Bacteroidetes and 222 of them are in the Bacteroidaceae family. In addition, a query of the Eggnog Database showed that Fpn is a member of an orthologous group of 39 closely related peptidases found in 35 species, all but one of which belongs to the Bacteroidetes (Figure 2). Almost all of the members of this family contain a conserved cysteine residue near the N-terminus and 85% are predicted to be lipoproteins. The Fpn homologs are also members of the C11 superfamily of proteases, which includes the clostripains and gingipains, all share a similar structure despite considerable sequence diversity (Barrett and Rawlings, 2001; Choi et al., 2016). The gingipains, however, form a unique group of divergent proteins that do not contain a lipobox motif and are secreted by the type IX pathway (Sato et al., 2013) despite the fact that Porphyromonas is a member of the Bacteroidetes. In any case, the broad distribution of the Fpn homologs strongly suggests that these proteins are not simply dedicated to the proteolytic maturation of BTF.
FIGURE 2.
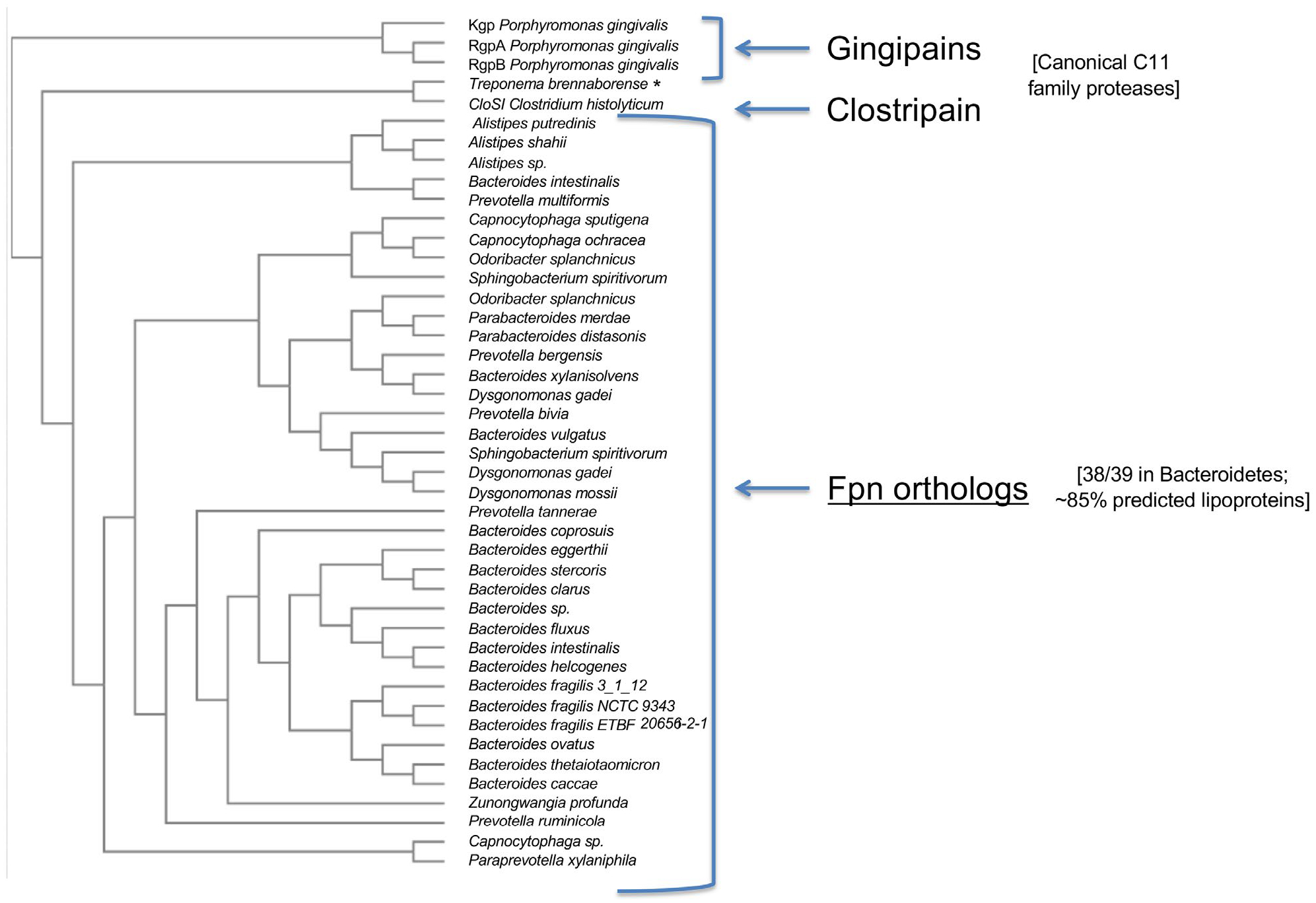
Distribution of Fpn proteases among the Bacteroidetes. An evolutionary tree of closely related orthologs of the Fpn protease produced by ETBF strain 20656-2-1 identified in the EggNOG database was constructed using Clustal Omega (http://www.clustal.org/omega/) and Phylogeny. Several P. gingivalis gingipains and the C. histolyticum CloSI protein were included for comparison. A taxonomic profile of the 39 closely related orthologs that were identified (group ENOG4108ZPA, 7.42e-142, score = 476.2) shows that all but one are produced by members of the Bacteroidetes. The 39th ortholog is produced by a Treponema species (*). Most of the closely related orthologs (~85%) are predicted by LipoP (http://www.cbs.dtu.dk/servi ces/LipoP/) to contain a lipobox motif
To test the possibility that Fpn might promote the release of multiple proteins from the cell surface, we analyzed the proteins that are present in the culture medium of wild-type and isogenic fpn− ETBF strains by SDS-PAGE. Interestingly, the results showed that many proteins found in the culture medium of wild-type cells are absent from the culture medium of fpn− cells or present at greatly reduced levels (Figure 3a). In contrast, the disruption of bft produced a much more modest effect on protein secretion under similar conditions (Figure S5). Although we cannot rule out the possibility that some of the proteins were released at least in part from outer membrane vesicles (OMVs), the results suggest that the Fpn protease plays an especially important role in the generation of the secretome. Initially we identified some of the most abundant proteins secreted by the wild-type strain by excising bands from a gel and subjecting them to mass spectrometry. The most likely candidate proteins include factors that are predicted to be involved in carbohydrate utilization, a MACPF domain protein, and several proteins of unknown function (Figure 3b). Seven of the nine proteins contain a lipobox motif and are, therefore, predicted to be lipoproteins. Interestingly, the N-terminus of most of these proteins resembles the acidic sequence motif that is a hallmark of exported lipoproteins in B. fragilis (Table S1). Most of the proteins ran slightly faster on SDS-PAGE than their predicted molecular weight (calculated without the signal peptide). The identification of peptides derived from the N-terminal regions of these proteins by mass spectrometry provided additional evidence that they were cleaved near the membrane anchor.
FIGURE 3.
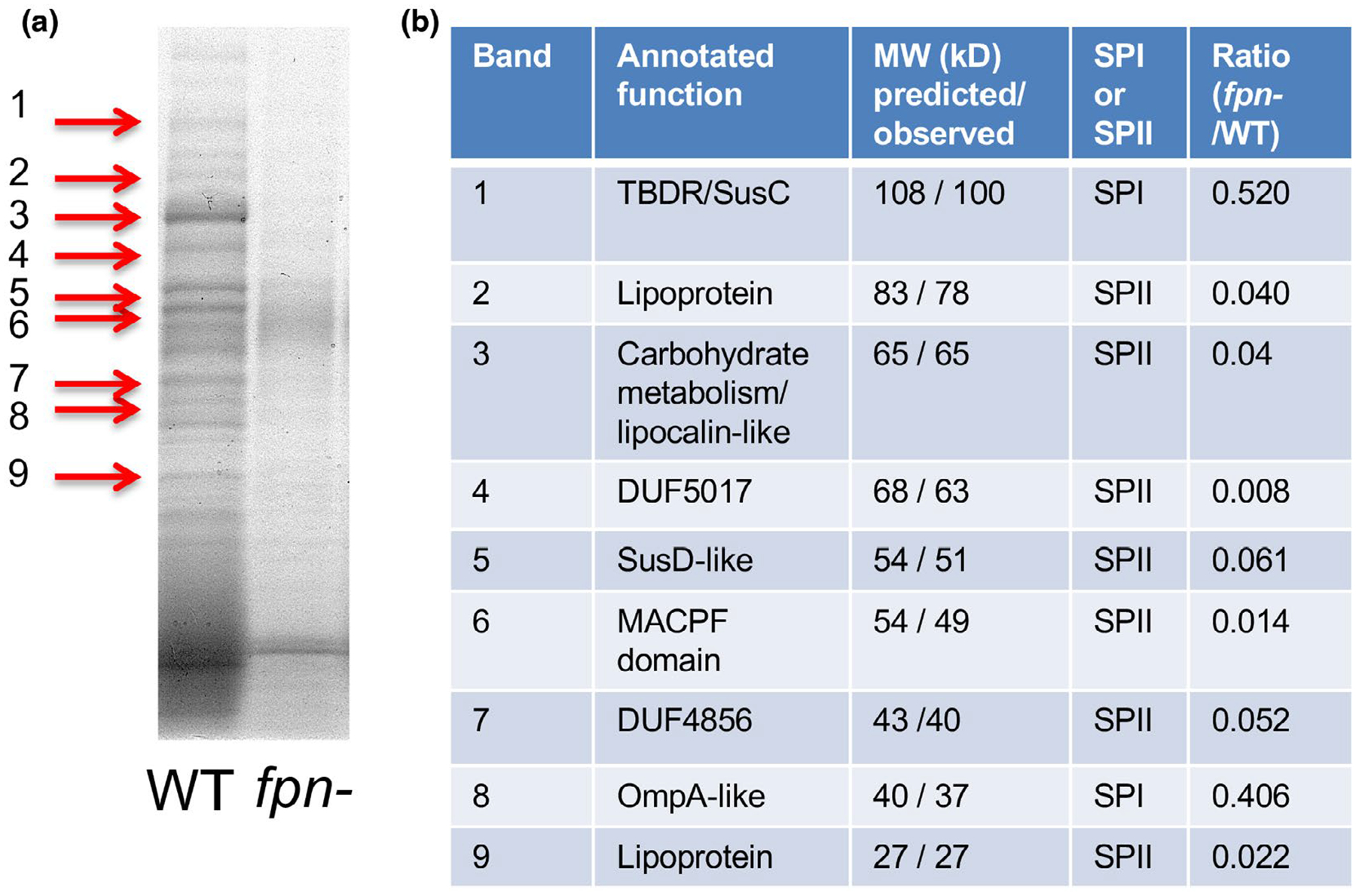
The level of many secretome components is strongly reduced in an fpn− strain. (a) Proteins present in the culture medium of wild-type (WT) ETBF and isogenic fpn− strains were resolved by SDS-PAGE and visualized using Colloidal Blue. (b) Nine proteins that were enriched in the culture medium of wild-type cells (red arrows) were excised from the gel and identified by mass spectrometry. The predicted size of each protein (lacking the signal peptide) and the observed size (based on mobility on SDS-PAGE) is indicated. The presence of a signal peptidase I (SP I) or signal peptidase II (SP II) cleavage site as predicted by LipoP is shown. The ratio of each protein in the secretome of the two strains is based on an analysis of the culture media by quantitative mass spectrometry
We next performed a quantitative comparison of the complete secretome of the wild-type and fpn− strains using dual-labeled isotopes for direct comparison of peptide abundance. A large number of proteins (221) were designated to be potential components of the wild-type secretome after low confidence peptides and cytoplasmic and periplasmic proteins that were identified using CELLO (Yu et al., 2004) were filtered out (Table S2). Orthologs of many of these proteins that were identified in a recent proteomic analysis of strain NTBF 9343 were located in the secretome or OM fraction (Wilson et al., 2015). The intracellular proteins were likely detected as a result of a low level of cell lysis. About half (105) of the potential secretome components were between 1.5- and 10-fold less abundant in the supernatant of the fpn− strain (mutant: wild-type ratio 0.67–0.1) (Figure 4a). An additional 99 proteins were 10- to 100-fold less abundant (ratio 0.1–0.01), and five proteins were more than 100-fold less abundant (ratio < 0.01). Only five proteins were present at a significantly higher level (ratio > 1.5-fold) in the culture medium of the fpn− strain (Figure 4a,c; Table S2). Essentially the same ratios were observed in multiple independent experiments (Figure 4b). All of the proteins identified in gel slices (Figure 3) were also detected in the quantitative comparison and found to be less abundant in the culture medium of the mutant strain. Like these abundant proteins, a majority of the proteins detected in the quantitative comparison are predicted to contain a SPII cleavage site and are, therefore, likely to be secreted lipoproteins (Figure 4a). In addition, RT-PCR experiments confirmed that the strong reduction in the level of several proteins in the secretome of the fpn− strain was not due to indirect effects on transcription (Figure S6).
FIGURE 4.
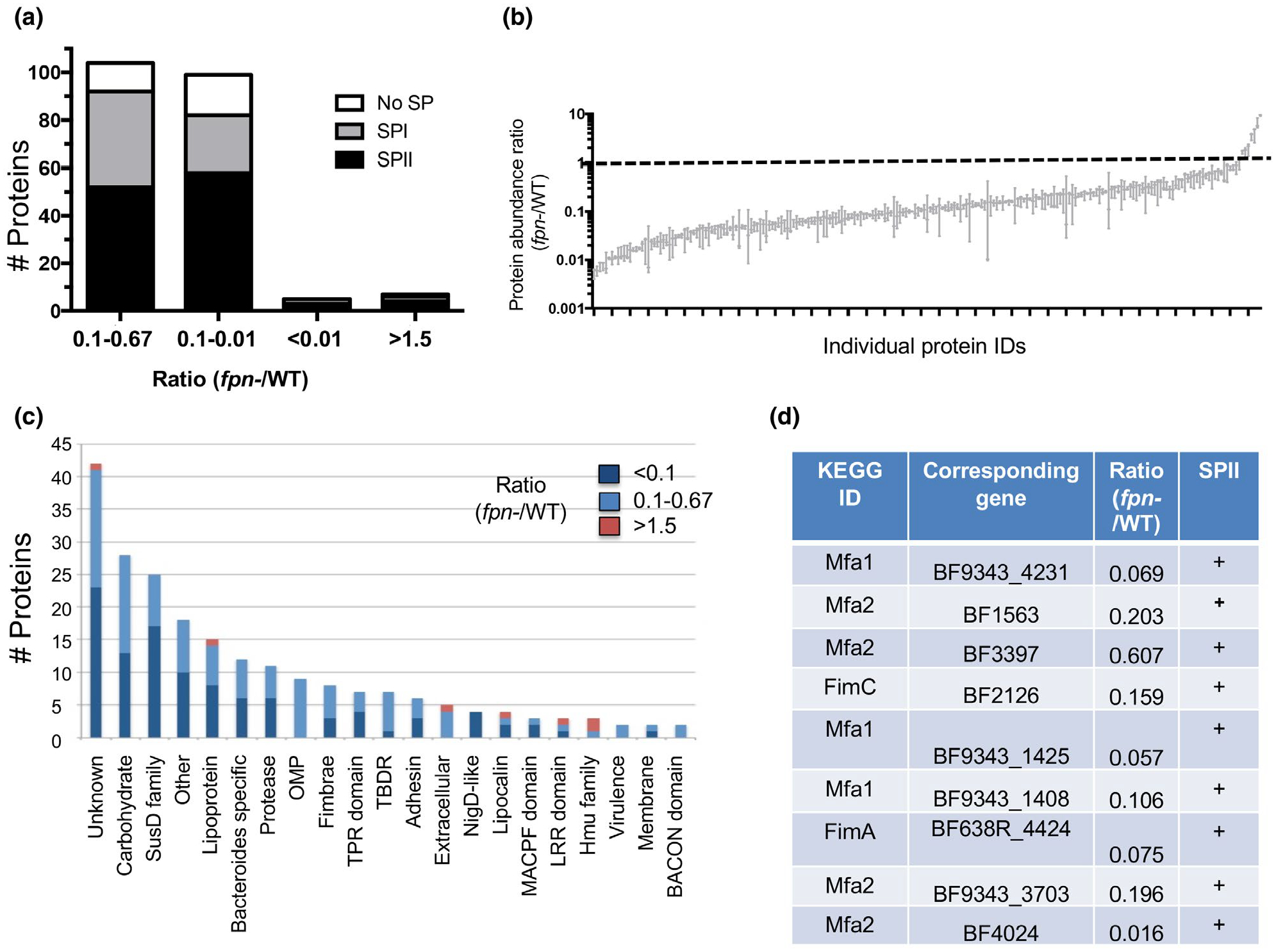
Quantitative comparison of the secretome of wild-type and fpn− strains. The relative abundance of proteins secreted into the culture medium of an fpn− strain and its parental wild-type (WT) ETBF strain was determined by quantitative mass spectrometry. (a) Proteins were binned based on relative abundance and analyzed for the presence of a signal peptidase I (SP I) or signal peptidase II (SP II) cleavage site by LipoP. Some proteins had no detectable signal peptide (no SP). The number of proteins in each category is shown. (b) Min to max box plot showing the relative abundance of individual secretome components in fpn− and WT strains identified by quantitative mass spectrometry in three replica experiments. (c) Categorization of secretome components based on their annotation in the KEGG database. (d) Putative fimbrial proteins that are significantly less abundant in the secretome of the fpn− strain were identified based on homology to proteins in the KEGG database. The presence of a signal peptidase II (SP II) cleavage site was determined by LipoP
The proteins that were present at significantly reduced levels in the culture medium of the fpn− strain vary widely in function (Figure 4c). A large subset of these proteins are involved in carbohydrate metabolism, including components of the Sus pathway (Foley et al., 2016; Shipman et al., 2000) and BACON domain proteins (Mello et al., 2010). Another group of proteins are found predominantly in the Bacteroides but have no known function. These proteins, as well as a distinct set of putative antibacterial proteins that contain MACPF domains (Chatzidaki-Livanis et al., 2014; Roelofs et al., 2016), may play important roles in survival in the gut. Furthermore, homologs of the P. gingivalis major and minor pilus proteins including subunits of both long fimbriae (FimA and FimC) and short fimbriae (Mfa1 and Mfa2) were significantly reduced in the culture medium of the mutant strain (Figure 4c,d). FimA is a surface exposed lipoprotein that is added to growing fimbriae following cleavage by the gingipains (Kadowaki et al., 1998; Shoji et al., 2004). The results suggest that Fpn and the gingipains may play a similar role in the biogenesis of fimbriae in Bacteroides and Porphyromonas. The Hmu family of proteins, which are also known targets of the gingipains, were among the five proteins that were more abundant in the secretome of the fpn− strain. These proteins promote heme uptake after their release from the cell surface (Guo et al., 2010; Olczak et al., 2008) and provide iron that is essential for the growth of B. fragilis. If Fpn cleaves the Hmu homologs in B. fragilis, then cells may compensate for the loss of the protease by increasing their production.
We next wished to obtain direct evidence that proteins that were present at greatly reduced levels in the culture medium of the fpn− strain are Fpn substrates. To this end we first attached an epitope tag (FLAG-tag) to the C-terminus of three proteins, Mfa1 (BF9343_4231), Mfa2 (BF9343_3703) and a NigD-like lipoprotein (BF9343_2956). All three proteins are lipoproteins that contain the acidic N-terminal motif associated with surface localization. The proteins were produced in wild-type and fpn− ETBF strains and detected by Western blot using an anti-FLAG-tag antiserum. In an initial experiment, we examined the localization of each protein. Interestingly, we detected two forms of each protein in wild-type cultures that likely correspond to full-length and proteolytically processed species (Figure 5a). The unprocessed form was found exclusively in a whole-cell extract while ~50%–100% of the processed form was found in the culture medium. In contrast, we observed only the full-length form of each protein in fpn− cultures. All of the Mfa2 homolog and the NigD-like protein was located in the whole-cell extract while about half of the Mfa1 homolog was located in the culture medium, possible due to an association with OMVs. All of the full-length protein was sensitive to PK digestion and was, therefore, exposed to the environment (Figure 5b). Taken together, the results strongly suggest that Fpn facilitates the release of each of the proteins from the cell surface. To test this idea further, we added purified Fpn or the Fpn (C180A) mutant to fpn− cells that expressed each protein. Consistent with our prediction, the native protease (but not the C180A mutant) effectively cleaved the Mfa1 and Mfa2 homologs and released a fragment that co-migrated with the proteolytically processed species observed in the wild-type ETBF strain (Figure 5c), Curiously, the purified Fpn protease did not cleave the NigD-like protein. This observation suggests that the NigD-like protein (and presumably other proteins as well) are released into the culture medium via a cascade in which Fpn activates a second protease.
FIGURE 5.
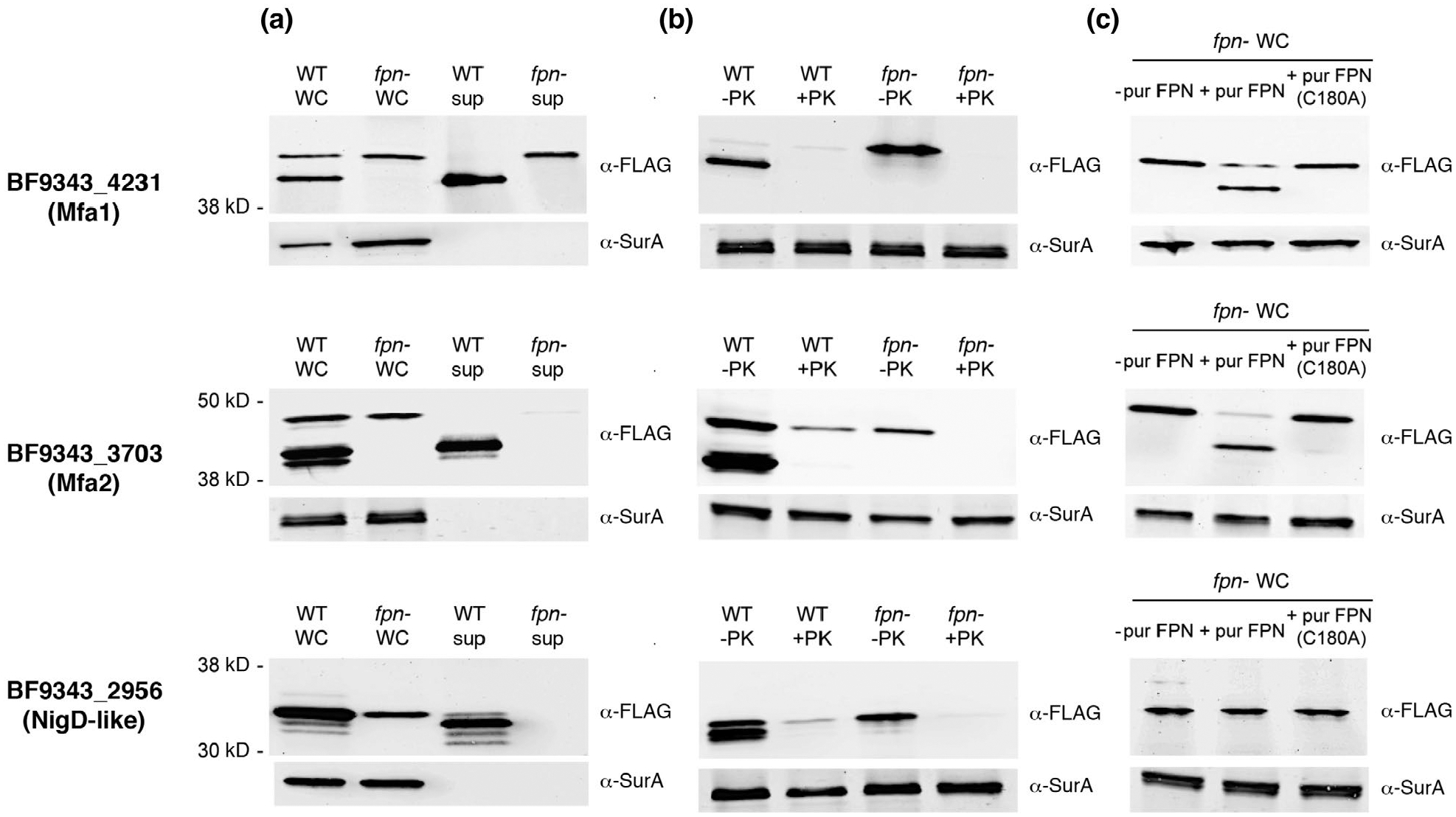
Purified Fpn releases two fimbrial proteins from the cell surface. Wild-type (WT) and isogenic fpn− ETBF strains transformed with a derivative of pFD340 that encodes a FLAG-tagged version of Mfa1 (BF9343_4231), Mfa2 (BF9343_3703) or a NigD-like protein (BF9343_2956) were grown to mid-log phase. In (a) cells were separated from the culture medium by centrifugation. In (b) intact cells were pelleted and mock treated or treated with PK for 30 min at 37°C. In (c) intact fpn− cells were mock treated or treated with purified Fpn or the Fpn (C180A) mutant for 30 min at 37°C. Whole cell (WC) and culture medium (sup) fractions were then analyzed by Western blot using anti-FLAG-tag and anti-SurA antisera
Our observations on the proteolytic processing of the Mfa1 and Mfa2 homologs together with a bioinformatic analysis led to the intriguing possibility that Fpn cleaves a significant subset of its substrates at a common consensus sequence. The small molecular weight shifts that we observed in the presence of Fpn strongly suggest that the cleavage sites—like those in abundant secretome components (Figure 3)—are located near the N-terminus. Interestingly, although the nine fimbrial proteins we identified in the secretome differ greatly in size and sequence (~10%–20% pairwise identity), each protein contains an arginine embedded in a conserved sequence motif [(AXTR(A/G) (A/G)] 27–51 residues from the putative lipidated cysteine residue (Figure S7a). This arginine is the most N-terminal arginine in seven of the nine proteins. Cleavage of the Mfa1 and Mfa2 homologs at this site would remove 26 or 37 amino acids, respectively, and create a mobility shift that is consistent with our experimental results (Figure 5). The first arginine in the mature region of abundant secretome components 3 and 6 (Figure 3) as well as some of the secreted lipoproteins we identified in our broader mass spectrometry analysis (Figure 4 and Table S2) are likewise embedded in the same sequence motif (Figure S7b). Remarkably, one of these lipoproteins (BF1567) contains only a single, centrally located arginine (residue 198 out of 430 amino acids) that is embedded in a closely related sequence. In this regard it is notable that Fpn has been shown to cleave fragilysin at a centrally located arginine (R211) that is located within a similar motif (Herrou et al., 2016). It should be emphasized that although the NigD-like protein we analyzed that does not appear to be released directly by Fpn lacks this sequence motif, many of the other secretome components we identified also lack the motif. If at least some of these proteins are released by Fpn, then the recognition of the conserved sequence motif is not the only factor that governs Fpn cleavage.
2.3 |. Fpn enhances cell fitness
The evidence that Fpn plays a major role in shaping the secretome of an ETBF strain suggested that the loss of the protein might produce deleterious effects on cell physiology. Consistent with previous results (Choi et al., 2016), we found that the fpn− strain grew as well as the parental ETBF strain in rich medium (Figure 6a). Additionally, we did not observe a difference in growth when the two strains were grown in minimal medium containing either glucose or starch as a carbon source or a difference in cell viability during stationary phase (Figure S8; data not shown). When we inoculated the wild-type and fpn− strains into a single culture at a 1:1 ratio, however, wild-type cells outnumbered mutant cells by a factor of ~100 after 24 hr (Figure 6b). This result suggests that while Fpn is not required for growth, it provides a fitness advantage in a mixed population. Because proteins released by wild-type cells would have the potential to diffuse throughout the culture and complement defects in trans, it seems likely that the loss of fitness in the fpn− strain results from the accumulation of uncleaved proteins on the cell surface or the loss of a surface localized activity.
FIGURE 6.
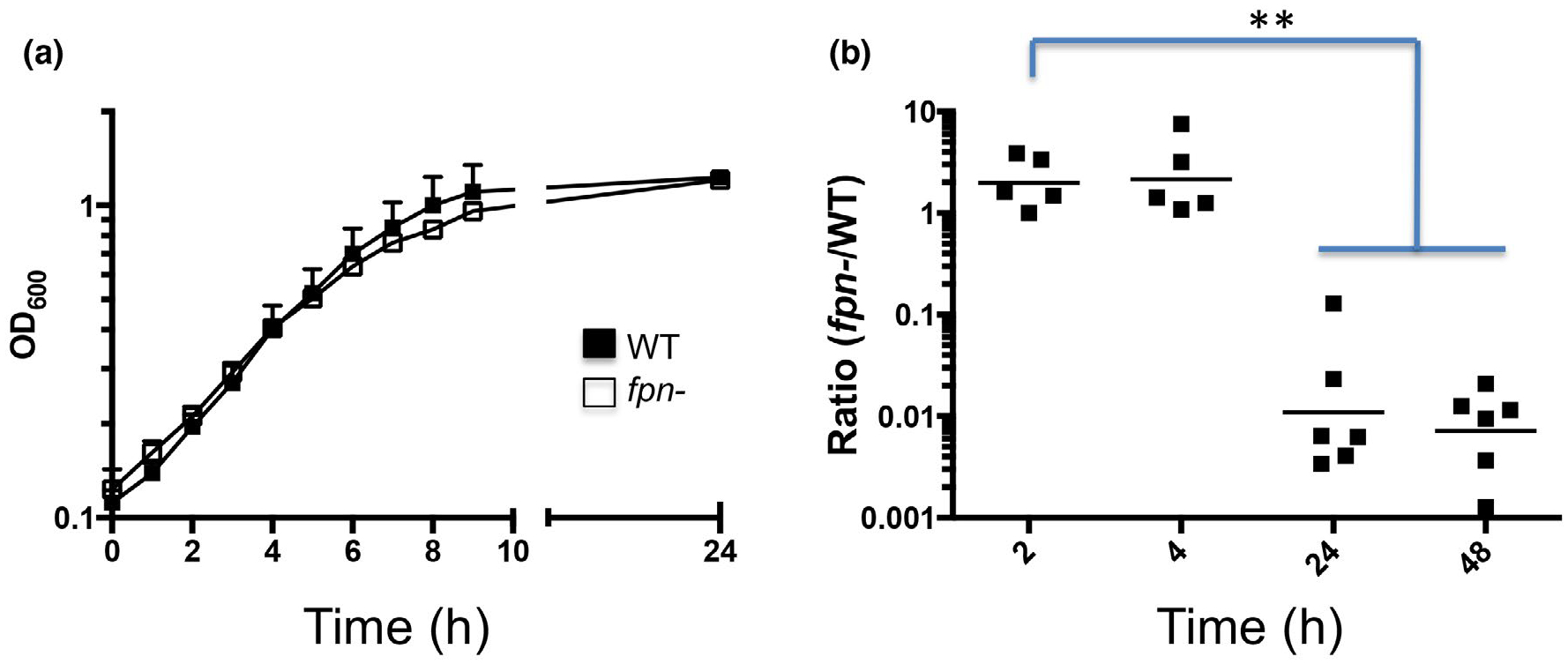
Fpn enhances the relative fitness of B. fragilis. (a) The growth of wild-type (WT) and isogenic fpn− ETBF strains in rich medium was monitored at OD600. The standard deviation from four cultures is shown. (b) The WT and fpn− strains were inoculated at a 1:1 ratio into rich medium and co-cultured. The ratio of the two strains at various times post-inoculation was determined by replica plating CFUs. Each symbol is an individual culture and the geometric mean is shown (**p < .01 unpaired student’s t test comparing T = 2 to either T = 24 or 48 hr)
2.4 |. Fpn cleaves proteins on the surface of HT29 intestinal epithelial cells
Because the gingipains produced by P. gingivalis are major virulence factors that target multiple mammalian proteins (Carlisle et al., 2009; Guo et al., 2010; Sheets et al., 2005; Sroka et al., 2001), we hypothesized that Fpn may likewise cleave one or more host cell proteins. To test this possibility, HT29 intestinal epithelial cells were incubated with wild-type Fpn, the Fpn C180A mutant or clostripain, a protein that shares the same catalytic mechanism. After 3 hr, cells incubated with wild-type Fpn showed a significant and dose-dependent loss of adhesion both to adjacent cells and glass slides (Figure 7a). Clostripain produced a comparable, although somewhat more pronounced effect. The gingipains have also been shown to decrease cell adhesion at similar concentrations, which are thought to be considerably lower than in vivo concentrations (Guentsch et al., 2011; Katz et al., 2002; Ruggiero et al., 2013). In the same concentration range, BFT only causes more limited cell rounding (Remacle et al., 2014). In contrast, cells incubated with either the Fpn C180A mutant or tissue culture medium alone showed no loss of attachment. These results suggest that Fpn can cleave at least one surface protein that is required for cell adhesion.
FIGURE 7.
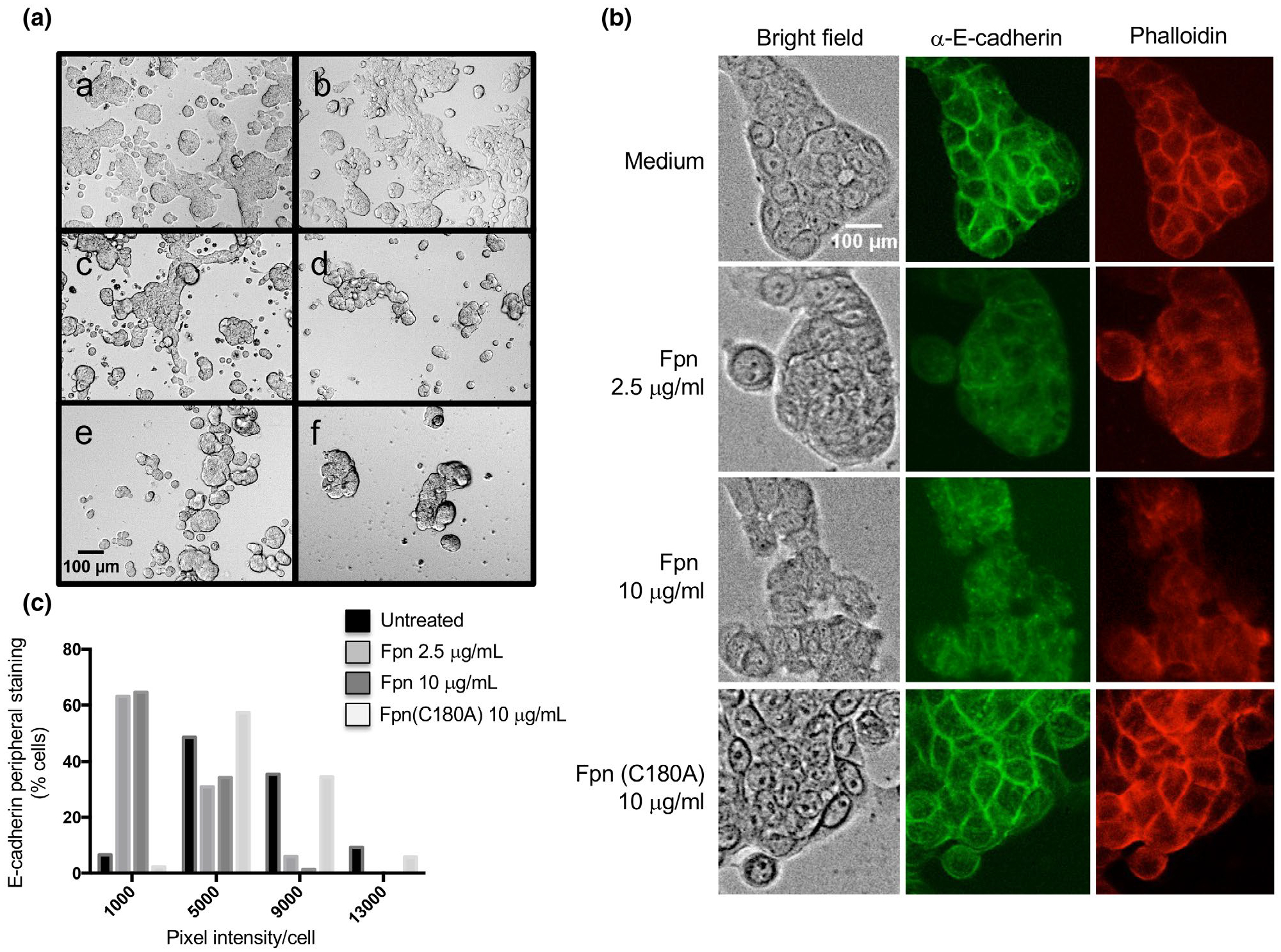
Incubation of HT29 cells with Fpn leads to a loss of adhesion and cell-cell junctions. (A) Live cell microscopy (bright field, 10X magnification) of HT29 cells incubated with culture medium alone (a), 10 μg/ml Fpn (C180A) (b), 2.5 μg/ml Fpn (c), 10 μg/ml Fpn (d), 2.5 μg/ml C. histolyticum Clo protease (e), or 10 μg/ml Clo (f). (B) HT29 cells were incubated with culture medium alone, Fpn (2.5 or 10 μg/ml) or Fpn (C180A) (10 μg/ml). Cells were fixed, stained with either FITC-conjugated anti-E-cadherin or rhodamine-conjugated phalloidin, and visualized at 20X magnification. (C) The intensity of peripheral E-cadherin staining in individual cells was determined, and the results were binned based on the percent of cells within the population that showed different levels of staining (arbitrary units)
We next sought to gain insight into the mechanism by which Fpn impairs cell adhesion. Because previous work has shown that toxins such as BFT impair the attachment of HT29 cells to solid surfaces through the cleavage of E-cadherin and the rearrangement of the actin cytoskeleton (Koshy et al., 1996; Rhee et al., 2009; Saidi et al., 1997; Wu et al., 1998), we fixed cells that remained attached to the slides after protease treatment and stained them with either an FITC labeled anti-E-cadherin antibody or rhodamine-labeled phalloidin, a reagent that detects F-actin. Both qualitative and quantitative analyses clearly showed that cells treated with Fpn had lower peripheral levels of E-cadherin and F-actin than cells treated with tissue culture medium or the Fpn C180A mutant (Figure 7b,c). A similar loss of E-cadherin and F-actin at cell-cell junctions was observed when cells were incubated with clostripain (data not shown). Although the results do not show whether Fpn acts directly or indirectly on E-cadherin, they do provide evidence that the protease can cleave host proteins as well as bacterial proteins. Because only very low levels of Fpn appear to be released from the cell surface (Figure S9), the protease would probably be most active against targets on the surface of cells that interact directly with B. fragilis or in the local environment where its concentration would be relatively high.
3 |. DISCUSSION
In this study, we show that the Fpn protease plays a key role in the generation of the secretome of an ETBF strain. We initially obtained evidence that both proBFT and Fpn are surface localized lipoproteins and that Fpn cleaves proBFT on the extracellular side of the OM rather than in the periplasmic space. In light of evidence that many lipoproteins are exposed on the surface of B. fragilis (Wilson et al., 2015), that Fpn is conserved in many Bacteroides species that do not produce BFT and that other C11 proteases cleave multiple substrates, we conjectured that Fpn might cleave other proteins in addition to BFT. Indeed we found that the disruption of fpn led to a strong reduction in the level of the majority of the components of the secretome. The large scale effect on secretion strongly suggests that Fpn is a broad-spectrum protease that releases a wide variety of functionally and structurally diverse cell surface proteins into the extracellular space. Consistent with this notion, we obtained direct evidence that Fpn cleaves two fimbriae from the cell surface. Interestingly, our results suggest that Fpn cleaves at least a subset of its substrates at an arginine residue that is embedded in a short sequence motif. Although the presence of this motif may help to identify Fpn substrates, Fpn might also cleave many proteins that lack the motif at alternative arginine residues. In any case, based on the finding that purified Fpn did not release a NigD-like lipoprotein from the cell surface, it seems likely that some of the proteins we identified in the secretome were released into the culture medium indirectly through the activation of uncharacterized surface localized proteases. Furthermore, it is also conceivable that some of the proteins were cleaved from OMVs rather than directly from the cell surface. Finally, we found that purified Fpn alters the adhesive properties and morphology of cultured epithelial cells by cleaving surface proteins. Although further studies are needed to determine the functions of Fpn in vivo, our results raise the possibility that it also promotes colonization or enhances survival by cleaving proteins produced by other microbes.
Because many of the secretome components that are potential Fpn substrates contain both a lipobox motif and an acidic motif between residues +2 and +6 that is associated with extracellular localization, our results suggest that Fpn plays an especially important role in the release of lipoproteins from the cell surface. The detection of peptides that reside near the extreme N-terminus of several secreted lipoproteins by mass spectrometry implies that the entire protein, including the acyl moiety, is transported across the OM prior to its release by Fpn. While the mechanism of translocation is unknown, it is possible that an unidentified factor transports lipoproteins from one side of the OM to the other. Indeed the Neisseria SLAM protein has been shown to facilitate the transport of at least one lipoprotein (TbpB) that is likely to be fully exposed on the cell surface (Noinaj et al., 2012) and might, therefore, function as a “flip-pase.” It is also conceivable that some surface exposed lipoproteins pass directly from the IM to the cell surface through the action of one of the multiple T1SSs produced by B. fragilis. By analogy, the Klebsiella PulA protein is a lipoprotein that is exposed on the cell surface by a type II secretion system (Francetic and Pugsley, 2005).
The conservation of Fpn in the Bacteroidetes strongly suggests that at least a subset of the proteins that it releases into the environment are highly beneficial. However, the presence of the protease on the cell surface may be a double-edged sword. The presence of fpn in the genome may facilitate the retention of genes acquired through horizontal transfer that encode substrates that enhance nutrient up-take, interactions with the host or survival in specific niches. Indeed its conservation in B. fragilis (Choi et al., 2016) may help to explain the introduction of bft into the genome of NTBF strains more than once during the course of evolution (Pierce and Bernstein, 2016). Although the substrate specificity of Fpn is unclear, like other C11 proteases it might cleave substrates at arginine residues relatively promiscuously. Indeed like the gingipains, it might even be able to cleave substrates into discrete peptides or degrade proteins non-specifically (Guo et al., 2010). The properties of Fpn may require that lipoproteins that remain on the cell surface fold into pro-tease-resistant conformations and may impose constraints on the conformation of other surface exposed polypeptides. Furthermore, the cleavage of proteins produced by host cells or other microbes may be either beneficial or deleterious. The seemingly paradoxical observation that the disruption of the B. thetaiotamicron fpn homolog increased fitness in a mouse model (Goodman et al., 2009) suggests that the protease may activate the immune system or produce other negative effects under some conditions. For these reasons there may be strong selective pressures that control the production and substrate specificity of Fpn and its release from the cell surface.
Our results raise the possibility that the functions of the gingipains and perhaps other extracellular bacterial proteases have been underestimated. Perhaps because the gingipains are known to be secreted virulence factors, their cleavage of a wide array of host proteins has been well documented. It is unclear, however, if they play a significant role in the cleavage of bacterial proteins other than a few fimbrial subunits (Kadowaki et al., 1998; Shoji et al., 2004). Likewise, members of the omptin family of proteases have been shown to cleave several host factors including antimicrobial peptides and plasminogen, but the range of known bacterial substrates is limited to a few autotransporters (Hritonenko and Stathopoulos, 2007). The finding that Fpn cleaves and thereby activates BFT, which itself is a protease, suggests that it can also promote protease cascades. The potential complexity of proteolysis in situ is illustrated by the observation that an fpn− mutant of B. fragilis was attenuated in anaerobic sepsis but not in a mouse model of colitis (Moncrief et al., 1995). This observation suggests that the toxin can be activated by a host protease or a bacterial protease that is produced in the gut in addition to Fpn. In any case, our proteomic data show that a single protease like Fpn has the potential to alter interactions with both host cells and other microorganisms in the gut microbiome by dramatically affecting the composition of the secretome.
4 |. EXPERIMENTAL PROCEDURES
4.1 |. Bacteria and tissue culture cells
B. fragilis ETBF strain 20656-2-1 was obtained from the ATCC (strain 43860). Strain 86-5443-2-2 and the isogenic bft deletion strain were obtained from Dr. Cindy Sears (Johns Hopkins University). NTBF strain NCTC 9343 was modified to express wild-type BFT toxin, BFT (C19A) or BFT (OmpASP) from plasmid pFD340 (Goodman et al., 2009) by triparental mating. All strains were grown in BHIS medium supplemented with 0.2% NaHCO3, 50 μg/ml gentamicin, and 10 μg/ml erythromycin at 37°C in anaerobic jars or Balch tubes (5% CO2, 95% N2). pFD340 derivatives that expressed FLAG-tagged lipoproteins were electroporated into competent cells (Ichimura et al., 2010) and plated on GAM agar containing cefoxitin (50 μg/ml). E. coli transformed with pET28 expression plasmids were grown in LB containing 30 μg/ml kanamycin. HT29 cells were obtained from the ATCC (HTB-38) and grown at 37°C with 5% CO2 in McCoy’s 5A medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FBS.
4.2 |. Antisera against B. fragilis proteins
A rabbit polyclonal antiserum raised against a C-terminal peptide of BFT has been previously described (Pierce and Bernstein, 2016). To generate a rabbit polyclonal antiserum against SurA (BF9343_3784), E. coli Rosetta (DE3) cells were transformed with a derivative of pET28b harboring surA lacking the signal peptide region. Each protein was overproduced, isolated from inclusion bodies and purified by Ni-NTA chromatography. The Fpn C180A mutant was produced as described below and also used to prepare polyclonal rabbit antibodies. B. fragilis GroEL was detected using a cross-reactive antiserum raised against the E. coli homolog (Sigma). An anti-His-tag antiserum (Qiagen) was used to detect His-tagged Fpn.
4.3 |. Transposon mutagenesis
A single colony of strain 20656-2-1 was inoculated into 4 ml reduced GAM broth and grown overnight at 37°C anaerobically. To make competent cells, a 0.1 ml aliquot of the overnight culture was diluted into 10 ml fresh GAM broth and grown to OD660 = 0.4–0.6 (~4 hr). A 0.5 ml aliquot of the log phase culture was then inoculated into 50 ml GAM broth and grown for an additional 48 hr. Cells were centrifuged for 10 min at 4,000g, washed twice with 50 ml ice-cold 10% glycerol and then, resuspended in 500 μl 10% glycerol (Ichimura et al., 2010). The transposon vector pMI07 (1 μg) was added to 100 μl competent cells in a 0.2 cm Biorad cuvette. Cells were pulsed at 12.5 KV/cm2, 200 Ω, 25 μF. After the addition of 0.9 ml pre-warmed GAM broth, cells were incubated for 9–12 hr at 37°C anaerobically and then, plated on GAM agar with erythromycin. Transposon insertion sites were identified by two rounds of semi-random PCR (Ichimura et al., 2014) using OneTaq 2X master mix (NEB). PCR products were purified with the Zymoclean Gel DNA Recovery Kit (Zymo Research) and eluted with water prior to sequencing with primer Mariner S (Ichimura et al., 2014).
4.4 |. Plasmid construction
Plasmid pFD340 was first modified to introduce a C-terminal 3× FLAG-tag followed by a stop codon. Equimolar concentrations of oligonucleotides 5ʹ- ATCGCTAGCGACTACAAA GACCATGACGGTGATTATAAAGATCATGACATCGATTAC AAGGATGACGAT-3ʹ and 5ʹ-CGTAGATCTTTCTTGTCATCGTCAT CC T TG TA ATCG ATG TC ATG ATC T T TATA ATC ACCG T CATGGTCT-3ʹ were heated to 95°C for 2 min and gradually cooled to 25°C over 45 min in a thermocycler to anneal. The annealed oligonucleotides were then cloned into the cognate Nhe I and Bgl II restriction sites. The pFD340-FLAG plasmid was further modified by removing the erythromycin-resistance cassette with Bam HI and Pme I and replacing it with a cefoxitin-resistance marker that was amplified by PCR using the primers 5ʹ-gtacctaggAAAATCAG TTCTTTAGCGA-3ʹ and 5ʹ-gctgtttaaacACACAGGCGGAACTTTG ATA-3ʹ and plasmid pLYL05 as a template (Ichimura et al., 2010). Genes encoding lipoproteins were amplified by PCR using genomic DNA from strain 20656-2-1 as a template and the oligonucleotides 5ʹ-CGAggatccCATTCTGTCTGAATCAAC-3ʹand 5ʹ-ctcGCTAGCaata ttgctatcccaaggtg-3ʹ (for BF9343_3703), 5ʹ-CGAggatccCATACTAT TATGAAGAAGA-3ʹ and 5ʹ-ctcGCTAGCcttaggagtctcagtgaa-3ʹ (for BF9343_4231) and 5ʹ-CGAggatccGTCTCTGCATTTACTTATT-3ʹ and 5ʹ-ctcGCTAGCttccgcagttttatattc-3ʹ (for BF9343_2956) and cloned into the pFD340-FLAG-cfx plasmid at the cognate Bam HI and Nhe I restriction sites.
4.5 |. BFT intoxication assay
B. fragilis reference strains were diluted 1:50 and grown anaerobically for 16 hr at 37°C. Cultures were centrifuged at 4,000g for 15 min at 4°C and the supernatant sterilized using a 0.22 μM filter. For the transposon screen, individual clones were grown overnight in 96 deep-well plates and then, centrifuged at 3,000g for 15 min at 4°C. The supernatant (culture medium fraction) was then analyzed for BFT activity. Each multi-well plate contained a medium alone control, NTBF strain NCTC 9343, NCTC 9343 expressing BFT from plasmid pFD340, and the WT ETBF strain 20656-2-1. HT29 cells were grown to ~70% confluence either on culture slides (Falcon) or in treated tissue culture plates (Corning Costar 3596) and washed with PBS. The cells were then incubated with an equal mixture of B. fragilis culture medium and tissue culture medium without FBS for 6 hr prior to visualization. Slides were fixed and permeabilized with 90% cold methanol and stained with 10% Giemsa.
4.6 |. Fpn production and purification
ETBF strain 20656-2-1 gene OCR32129 (fpn) was amplified by PCR without the signal peptide using the oligonucleotides 5ʹ-GGA GGAATTCTTGTCAGCAGGATGGGC-3ʹ and 5ʹ-TTATGCGGCCGCC TAATTCTTATACCAACCGGTATCC and cloned into the EcoR I and Not I sites of pET28b in-frame with a vector-encoded N-terminal 6 x His-tag. The C180A mutation was introduced using the Quik Change II kit (Agilent). Overnight cultures of BL21 (DE3) transformed with a plasmid encoding Fpn or Fpn (C180A) were diluted and then, grown to OD600 ~ 0.4 at 37°C prior to the addition of 1 mM IPTG. Following an incubation of 4 hr, cells were harvested and lysed using a Misonix sonicator. Lysates derived from cells that produced wild-type Fpn were then centrifuged twice at 17,000g for 20 min at 4°C. The protein was purified by passing the supernatant over a Ni-NTA column and eluting with 250 mM imidazole, concentrated using a 10K MWCO Amicon Ultra Centrifugal Filter Unit, diluted into activity buffer (50 mM Tris–HCl, pH 7.5) and re-concentrated. The final sample was incubated on ice for 1 hr following the addition of 2 mM CaCl2 and 2.5 mM DTT. The mutant protein was isolated from inclusion bodies obtained by centrifuging cell lysates as described above. Pellets were washed with PBS, resuspended in 8M urea and incubated at room temperature for 1 hr. The urea-solubilized material was centrifuged at 20,000g for 20 min at 4°C and the supernatant was passed over a Ni-NTA column. The Fpn (C180A) protein was then eluted using a pH gradient. Protein concentrations were determined by A280 measurements using a Nanodrop.
4.7 |. Cell fractionations and protein localization experiments
Overnight cultures were diluted 1:20, grown to mid-log phase grown in BHIS and centrifuged at 4,000g for 15 min at 4°C. The culture medium was then decanted. In experiments that involved analysis of the secretome, the medium was recentrifuged followed by concentration using an Amicon Ultra Centrifugal Filter Unit (10K MWCO). OMVs were removed from the culture medium by ultracentrifugation at 110,000g in a TLA100.4 rotor for 30 min at 4°C. In cell fractionation experiments, cell pellets were washed once with PBS, resuspended in PBS containing 1 mM PMSF, 0.1 mg/ml lysozyme, and 0.3% NaCl and disrupted by sonication. Cell lysates were centrifuged at 17,000g for 15 min at 4°C to remove unbroken cells. The resulting whole-cell lysate fraction was then centrifuged at 200,000g for 45 min at 4°C in a TLA100.4 rotor. The supernatant (soluble fraction) was removed and the pellet (membrane fraction) was washed twice before being resuspended in PBS. Equivalent amounts of protein from each fraction were then TCA precipitated. In surface localization experiments, overnight cultures were diluted and grown to mid-log phase. Cells were then centrifuged at 4,000g for 20 min at 4°C, washed once in PBS and resuspended in 1 ml PBS. Half of the cells were treated with PK (2 mg/ml) at 37°C for the indicated time. PK digestions were stopped by the addition of 10 mM freshly prepared PMSF and proteins were TCA precipitated. Proteins were resolved on 8%–16% minigels (Thermo Fisher Scientific) and detected by Western blot using 1:1,000 and 1:500 dilutions of anti-BFT and anti-FLAG-tag (mouse monoclonal F64R, Thermo Fisher Scientific) antibodies. Antibody-antigen complexes were detected using an Odyssey near infrared imaging instrument (Licor).
4.8 |. Fpn and clostripain activity assays
In experiments in which the proteolytic processing of proteins localized on the surface of intact cells was analyzed, 10 ml of fpn− cells grown in BHIS media to mid-log phase were washed twice in 10 mM Tris pH 7.6 and resuspended in 3 ml 10 mM Tris pH 7.6. Purified Fpn or Fpn (C180A) (1 μg) in activity buffer was then incubated with 1 ml cells for 30 min at 37°C. Cells were then spun at 4,000g for 10 min at 4°C, washed with 10 mM Tris 7.6/ 1 mM PMSF and TCA precipitated. In experiments in which the proteolytic processing of BFT in cell lysates was analyzed, fpn− cells grown to mid-log phase were resuspended in PBS containing 1 mM PMSF and sonicated. Purified Fpn or Fpn (C180A) or C. histolyticum clostripain (Sigma) (0.5–1.5 μg) in activity buffer was then incubated with 100 μg of protein from the cell lysate for 30 min at room temperature. In some experiments the protease inhibitors TLCK and leupeptin (50 μM–1 mM) were added to the reaction. BFT- and FLAG-tagged proteins were detected by Western blot as described above.
To analyze the effect of proteases on HT29 cells, cells were incubated with varying amounts of purified Fpn, Fpn (C180A) and C. histolyticum clostripain diluted in 200 μl of McCoy’s 5A medium with 5mM cysteine for 3 hr prior to imaging at 10X magnification. For immunofluorescence, cells were washed and fixed with 1:1 methanol:acetone for 10 min at −20°C, incubated with blocking buffer (1% BSA) at room temperature for 10 min and stained with FITC conjugated mouse anti E-cadherin (BD Biosciences) and ActinRed 555 (Life Technologies R37112) according to the manufacturer’s instructions. Cells were visualized at 20X magnification. Image analysis was performed using ImageJ to measure cellular fluorescence as pixel intensity. E-cadherin fluorescence at the cellular perimeter was determined using Corrected Total Cellular Fluorescence (CTCF) (McCloy et al., 2014) for 75–100 cells per sample from two independent replicates.
4.9 |. Mass spectrometry
Strains were grown to late log phase and centrifuged at 4,000g for 20 min at 4°C (with slow acceleration and deceleration settings) to isolate culture medium fractions. OMVs were removed by ultracentrifugation as described above. For total secretome analysis, 10% TCA/ 0.01% sarkosyl was used to precipitate proteins from three independent culture medium samples obtained from both the wild-type and fpn− mutant strains. Protein samples were dissolved in 300 μl 100 mM Tris–HCl pH 8/8M urea, reduced, alkylated, and subjected to proteolytic digestion essentially as described (Kulak et al., 2014). After thorough digestion of the protein sample (as determined by checking a small amount of sample at intermediate times) each sample was absorbed onto reverse phase stage tips and labeled using reductive dimethylation (Boersema et al., 2009; Lau et al., 2014). Subsamples (one wild type and one mutant) were then mixed and applied together to an SCX STAG tip and eluted in six fractions as described (Kulak et al., 2014). Each fraction was run on a 5 hr LC/MS/MS gradient and the resulting data analyzed using MaxQuant 1.5.3.28 (Cox and Mann, 2008). In one sample the wild-type components were labeled as light dimethyl isotopes and the mutant as heavy dimethyl isotopes, while in the other two this pattern was reversed (Kovanich et al., 2012). The inversion was managed using the group approach and the designation of labels in the Type pull-down menu of MaxQuant. For analysis of the data, nonbacterial contaminants and predicted cytoplasmic/periplasmic contaminants were removed. Additionally, any peptides that were not identified in all samples and proteins that were represented by only one or two peptides were eliminated from the analysis. The average of three independent samples was used to determine the difference in protein abundance between wild-type and fpn− strains. Interpro, PFAM, pSORT, and LipoP were used to determine the predicted function and localization of the identified proteins.
For in-gel identification, OMVs were removed from the culture medium of cells grown to late log phase as described above. Proteins were concentrated by 10% TCA/ 0.01% SDS precipitation and resuspended in 19 μl Laemmli sample buffer. Subsequently the samples were incubated with 1 μl 100 mM DTT at 60°C for 10 min and then, with 5 μl 100 mM iodoacetamide at room temperature for 30 min in the dark. Alkylated proteins were separated by SDS-PAGE and stained with Colloidal Blue (Invitrogen) according to the manufacturer’s instructions. The identification of bands by mass spectrometry has been described (Wilson et al., 2015). In brief, mixtures of soluble proteins and proteins contained in gel slices were digested into peptide fragments using standard procedures. Data were collected using a Waters NanoAcquity UHPLC system interfaced with a Thermo Elite mass spectrometer. Short gradients were used in gel-based experiments and analyzed using Mascot. The most abundant protein identified in each band that had the correct molecular weight and that was not present in the control lane (fpn− strain) was chosen as the most likely candidate.
4.10 |. Analysis of gene expression
RNA samples were isolated from log phase samples using an RNeasy purification kit (Qiagen) and treated with Turbo DNase I (Ambion). cDNA was prepared from 1 μg RNA and qRT-PCR was performed using POWER SYBR green PCR Mastermix (Applied Biosystems) and the oligonucleotides listed in Table S3 in a Bio-Rad C1000 Touch ThermoCycler. The expression of each gene relative to the expression of gapDH in the same sample was determined and normalized to a standard curve. Additionally, each sample was tested in parallel with a control that was not treated with reverse transcriptase.
4.11 |. Strain fitness assay
Co-culture experiments were performed by first diluting separate overnight cultures of two distinct strains 1:10 into BHIS. When the cultures reached OD600 = 0.2–0.4 each culture was adjusted to the same OD600 value prior to diluting both strains 1:20 into a single culture. Time points were taken at T = 0, 2, 4, and 24 hr. The 24 hr culture was passaged at a 1:50 dilution and allowed to grow further in order to obtain a 48 hr sample. Serial dilutions of each sample were plated on BHIS agar prior to replica plating onto BHIS erm. The ratio of fpn−/WT cells was determined as ermR CFU/(total CFU-ermR CFU) normalized to the input ratio as determined by CFU. Statistical significance was determined by analyzing the data using an unpaired student’s t test at various time points.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Cindy Sears for providing bacterial strains. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Funding information
Intramural Research Program, National Institute of Diabetes and Digestive and Kidney Diseases
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
All primary data including primary mass spectrometry data can be obtained from the corresponding author upon request.
REFERENCES
- Abby SS, Cury J, Guglielmini J, Neron B, Touchon M and Rocha EP (2016) Identification of protein secretion systems in bacterial genomes. Scientific Reports, 6, 23080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu MM, Priya ML, Selvan AT, Madera M, Gough J, Aravind L, et al. (2006) A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. Journal of Bacteriology, 188(8), 2761–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AJ and Rawlings ND (2001) Evolutionary lines of cysteine peptidases. Biological Chemistry, 382(5), 727–733. [DOI] [PubMed] [Google Scholar]
- Boersema PJ, Raijmakers R, Lemeer S, Mohammed S and Heck AJ (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nature Protocols, 4(4), 484–494. [DOI] [PubMed] [Google Scholar]
- Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, et al. (2015) The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clinical Infectious Diseases, 60(2), 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle MD, Srikantha RN and Brogden KA (2009) Degradation of human alpha- and beta-defensins by culture supernatants of Porphyromonas gingivalis strain 381. Journal of Innate Immunity, 1(2), 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty A, Awad MM, Hiscox TJ, Cheung JK, Carter GP, Choo JM, et al. (2011) The cysteine protease α-clostripain is not essential for the pathogenesis of Clostridium perfringens-mediated myonecrosis. PLoS One, 6(7), e22762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzidaki-Livanis M, Coyne MJ and Comstock LE (2014) An antimicrobial protein of the gut symbiont Bacteroides fragilis with a MACPF domain of host immune proteins. Molecular Microbiology, 94(6), 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi VM, Herrou J, Hecht AL, Teoh WP, Turner JR, Crosson S, et al. (2016) Activation of Bacteroides fragilis toxin by a novel bacterial protease contributes to anaerobic sepsis in mice. Nature Medicine, 22(5), 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J and Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology, 26(12), 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dargatz H, Diefenthal T, Witte V, Reipen G and von Wettstein D (1993) The heterodimeric protease clostripain from Clostridium histolyticum is encoded by a single gene. Molecular and General Genetics, 240(1), 140–145. [DOI] [PubMed] [Google Scholar]
- Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. (2018) Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science, 359(6375), 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erturk-Hasdemir D and Kasper DL (2018) Finding a needle in a haystack: Bacteroides fragilis polysaccharide A as the archetypical symbiosis factor. Annals of the New York Academy of Sciences, 1417(1), 116–129. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick RE, Wijeyewickrema LC and Pike RN (2009) The gingipains: scissors and glue of the periodontal pathogen, Porphyromonas gingivalis. Future Microbiology, 4(4), 471–487. [DOI] [PubMed] [Google Scholar]
- Foley MH, Cockburn DW and Koropatkin NM (2016) The Sus operon: a model system for starch uptake by the human gut Bacteroidetes. Cellular and Molecular Life Sciences, 73(14), 2603–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francetic O and Pugsley AP (2005) Towards the identification of type II secretion signals in a nonacylated variant of pullulanase from Klebsiella oxytoca. Journal of Bacteriology, 187(20), 7045–7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AA, Buckwold SL, Shin JW, Ascon M and Sears CL (2005) Mutation of the zinc-binding metalloprotease motif affects Bacteroides fragilis toxin activity but does not affect propeptide processing. Infection and Immunity, 73(8), 5273–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AA, Mundy LM, Trucksis M, Wu S, Kaper JB and Sears CL (1997) Cloning and characterization of the Bacteroides fragilis metalloprotease toxin gene. Infection and Immunity, 65(3), 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, et al. (2009) Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host & Microbe, 6(3), 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulas T, Arolas JL and Gomis-Rüth FX (2011) Structure, function and latency regulation of a bacterial enterotoxin potentially derived from a mammalian adamalysin/ADAM xenolog. Proceedings of the National Academy of Sciences of the United States of America, 108(5), 1856–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guentsch A, Kramesberger M, Sroka A, Pfister W, Potempa J and Eick S (2011) Comparison of gingival crevicular fluid sampling methods in patients with severe chronic periodontitis. Journal of Periodontology, 82(7), 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Nguyen KA and Potempa J (2010) Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontology 2000, 54(1), 15–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghi F, Goli E, Mirzaei B and Zeighami H (2019) The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer, 19(1), 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht AL, Casterline BW, Choi VM and Bubeck Wardenburg J (2017) A two-component system regulates Bacteroides fragilis toxin to maintain intestinal homeostasis and prevent lethal disease. Cell Host & Microbe, 22(4), 443–448 e445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht AL, Casterline BW, Earley ZM, Goo YA, Goodlett DR and Bubeck Wardenburg J (2016) Strain competition restricts colonization of an enteric pathogen and prevents colitis. EMBO Reports, 17(9), 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrou J, Choi VM, Bubeck Wardenburg J and Crosson S (2016) Activation mechanism of the Bacteroides fragilis cysteine peptidase, fragipain. Biochemistry, 55(29), 4077–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooda Y, Lai CC, Judd A, Buckwalter CM, Shin HE, Gray-Owen SD, et al. (2016) Slam is an outer membrane protein that is required for the surface display of lipidated virulence factors in Neisseria. Nature Microbiology, 1, 16009. [DOI] [PubMed] [Google Scholar]
- Hritonenko V and Stathopoulos C (2007) Omptin proteins: an expanding family of outer membrane proteases in Gram-negative Enterobacteriaceae. Molecular Membrane Biology, 24(5–6), 395–406. [DOI] [PubMed] [Google Scholar]
- Ichimura M, Nakayama-Imaohji H, Wakimoto S, Morita H, Hayashi T and Kuwahara T (2010) Efficient electrotransformation of Bacteroides fragilis. Applied and Environment Microbiology, 76(10), 3325–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura M, Uchida K, Nakayama-Imaohji H, Hirakawa H, Tada T, Morita H, et al. (2014) Mariner-based transposon mutagenesis for Bacteroides species. Journal of Basic Microbiology, 54(6), 558–567. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Nakayama K, Yoshimura F, Okamoto K, Abe N and Yamamoto K (1998) Arg-gingipain acts as a major processing enzyme for various cell surface proteins in Porphyromonas gingivalis. Journal of Biological Chemistry, 273(44), 29072–29076. [DOI] [PubMed] [Google Scholar]
- Katz J, Yang QB, Zhang P, Potempa J, Travis J, Michalek SM, et al. (2002) Hydrolysis of epithelial junctional proteins by Porphyromonas gingivalis gingipains. Infection and Immunity, 70(5), 2512–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy SS, Montrose MH and Sears CL (1996) Human intestinal epithelial cells swell and demonstrate actin rearrangement in response to the metalloprotease toxin of Bacteroides fragilis. Infection and Immunity, 64(12), 5022–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovanich D, Cappadona S, Raijmakers R, Mohammed S, Scholten A and Heck AJ (2012) Applications of stable isotope dimethyl labeling in quantitative proteomics. Analytical and Bioanalytical Chemistry, 404(4), 991–1009. [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N and Mann M (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods, 11(3), 319–324. [DOI] [PubMed] [Google Scholar]
- Lau HT, Suh HW, Golkowski M and Ong SE (2014) Comparing SILAC- and stable isotope dimethyl-labeling approaches for quantitative proteomics. Journal of Proteome Research, 13(9), 4164–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber F, Cornelis GR and Renzi F (2016) Identification of a new lipoprotein export signal in Gram-Negative bacteria. mBio, 7(5), e01232–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N and Collyer CA (2011) Gingipains from Porphyromonas gingivalis—complex domain structures confer diverse functions. European Journal of Microbiology and Immunology (Bp), 1(1), 41–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe S, Nariya H, Miyata S, Tanaka H, Minami J, Suzuki M, et al. (2010) Purification and characterization of a clostripain-like protease from a recombinant Clostridium perfringens culture. Microbiology, 156(Pt 2), 561–569. [DOI] [PubMed] [Google Scholar]
- McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A and Burgess A (2014) Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle, 13(9), 1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello LV, Chen X and Rigden DJ (2010) Mining metagenomic data for novel domains: BACON, a new carbohydrate-binding module. FEBS Letters, 584(11), 2421–2426. [DOI] [PubMed] [Google Scholar]
- Moncrief JS, Obiso R, Barroso LA, Kling JJ, Wright RL, Van Tassell RL, et al. (1995) The enterotoxin of Bacteroides fragilis is a metalloprotease. Infection and Immunity, 63(1), 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers LL, Shoop DS, Stackhouse LL, Newman FS, Flaherty RJ, Letson GW, et al. (1987) Isolation of enterotoxigenic Bacteroides fragilis from humans with diarrhea. Journal of Clinical Microbiology, 25(12), 2330–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N, Easley NC, Oke M, Mizuno N, Gumbart J, Boura E, et al. (2012) Structural basis for iron piracy by pathogenic Neisseria. Nature, 483(7387), 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olczak T, Sroka A, Potempa J and Olczak M (2008) Porphyromonas gingivalis HmuY and HmuR: further characterization of a novel mechanism of heme utilization. Archives of Microbiology, 189(3), 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JV and Bernstein HD (2016) Genomic diversity of enterotoxigenic strains of Bacteroides fragilis. PLoS One, 11(6), e0158171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo MC, Arbo MD, Grindlinger J and Snydman DR (1995) Attributable mortality of bacteremia associated with the Bacteroides fragilis group. Clinical Infectious Diseases, 20(6), 1492–1496. [DOI] [PubMed] [Google Scholar]
- Remacle AG, Shiryaev SA and Strongin AY (2014) Distinct interactions with cellular E-cadherin of the two virulent metalloproteinases encoded by a Bacteroides fragilis pathogenicity island. PLoS One, 9(11), e113896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, et al. (2009) Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infection and Immunity, 77(4), 1708–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs KG, Coyne MJ, Gentyala RR, Chatzidaki-Livanis M and Comstock LE (2016) Bacteroidales secreted antimicrobial proteins target surface molecules necessary for gut colonization and mediate competition in vivo. mBio, 7(4), e01055–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero S, Cosgarea R, Potempa J, Potempa B, Eick S and Chiquet M (2013) Cleavage of extracellular matrix in periodontitis: gingipains differentially affect cell adhesion activities of fibronectin and tenascin-C. Biochimica et Biophysica Acta, 1832(4), 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidi RF, Jaeger K, Montrose MH, Wu S and Sears CL (1997) Bacteroides fragilis toxin rearranges the actin cytoskeleton of HT29/C1 cells without direct proteolysis of actin or decrease in F-actin content. Cell Motility and the Cytoskeleton, 37(2), 159–165. [DOI] [PubMed] [Google Scholar]
- Sato K, Yukitake H, Narita Y, Shoji M, Naito M and Nakayama K (2013) Identification of Porphyromonas gingivalis proteins secreted by the Por secretion system. FEMS Microbiology Letters, 338(1), 68–76. [DOI] [PubMed] [Google Scholar]
- Sears CL, Geis AL and Housseau F (2014) Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. Journal of Clinical Investigation, 124(10), 4166–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears CL, Islam S, Saha A, Arjumand M, Alam NH, Faruque AS, et al. (2008) Association of enterotoxigenic Bacteroides fragilis infection with inflammatory diarrhea. Clinical Infectious Diseases, 47(6), 797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears CL, Myers LL, Lazenby A and Van Tassell RL (1995) Enterotoxigenic Bacteroides fragilis. Clinical Infectious Diseases, 20(Suppl 2), S142–148. [DOI] [PubMed] [Google Scholar]
- Sheets SM, Potempa J, Travis J, Casiano CA and Fletcher HM (2005) Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infection and Immunity, 73(3), 1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman JA, Berleman JE and Salyers AA (2000) Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. Journal of Bacteriology, 182(19), 5365–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M, Naito M, Yukitake H, Sato K, Sakai E, Ohara N, et al. (2004) The major structural components of two cell surface filaments of Porphyromonas gingivalis are matured through lipoprotein precursors. Molecular Microbiology, 52(5), 1513–1525. [DOI] [PubMed] [Google Scholar]
- Sroka A, Sztukowska M, Potempa J, Travis J and Genco CA (2001) Degradation of host heme proteins by lysine- and arginine-specific cysteine proteinases (gingipains) of Porphyromonas gingivalis. Journal of Bacteriology, 183(19), 5609–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valguarnera E, Scott NE, Azimzadeh P and Feldman MF (2018) Surface exposure and packing of lipoproteins into outer membrane vesicles are coupled processes in Bacteroides. mSphere, 3(6), e00559–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler AG, Bao Y, Whitney JC, Bobay LM, Xavier JB, Schofield WB, et al. (2016) Human symbionts inject and neutralize antibacterial toxins to persist in the gut. Proceedings of the National Academy of Sciences of the United States of America, 113(13), 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler AG and Goodman AL (2017) An insider’s perspective: Bacteroides as a window into the microbiome. Nature Microbiology, 2, 17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler HM (2007) Bacteroides: the good, the bad, and the nitty-gritty. Clinical Microbiology Reviews, 20(4), 593–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MM, Anderson DE and Bernstein HD (2015) Analysis of the outer membrane proteome and secretome of Bacteroides fragilis reveals a multiplicity of secretion mechanisms. PLoS One, 10(2), e0117732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Lim KC, Huang J, Saidi RF and Sears CL (1998) Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proceedings of the National Academy of Sciences of the United States of America, 95(25), 14979–14984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature Medicine, 15(9), 1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CS, Lin CJ and Hwang JK (2004) Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Science, 13(5), 1402–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All primary data including primary mass spectrometry data can be obtained from the corresponding author upon request.