

Abstract
The Sigma-1 receptor (S1R) is an endoplasmic reticulum (ER) chaperone protein that has been implicated in attenuating inflammatory stress-mediated brain injuries. Selective S1R agonists represent a new class of therapeutic agent for treating neuropsychiatric and neurodegenerative disorders, however, to date, no S1R ligand has been approved for therapeutic purposes. We used three potential methods on known and potential S1R ligands to develop an unambiguous high-throughput cell screen for S1R activity. We screened known and potential S1R ligands using radioligand binding and previously reported markers of S1R activity including BDNF release, modulation of IP3 mediated calcium release, and modulation of NGF-induced neurite sprouting. Here, we present results several prototypical S1R compounds and some compounds with the potential for drug repurposing. Using an in-situ ELISA approach we demonstrated that these compounds could stimulate S1R-mediated BDNF release, which is a valuable therapeutic property since BDNF plays a critical role in neuronal support. These compounds were classified as S1R agonists because the BDNF response was comparable to the prototypical agonist 4-PPBP and because it could be reversed by a S1R selective concentration of the antagonist BD1063. When modulation of IP3 mediated calcium response and NGF-induced neurite sprouting were used as a measure of S1R activation, we were unable to reproduce the published results and determined that they are not reliable measures for evaluating functional properties of S1R ligands.
Keywords: Sigma 1 Receptor, Sigma 2 Receptor, Drug Repurposing, BDNF, screening
1. Introduction
The Sigma-1 receptor (S1R) is a stress and ligand-regulated endoplasmic reticulum chaperone protein that shuttles lipids and proteins to the plasma membrane (Su, Hayashi, Maurice, Buch, & Ruoho, 2010). High densities of the S1R are found in brain tissue, including the cerebral cortex, various limbic structures, the hypothalamus, and the hippocampus (Alonso, et al., 2000; Hashimoto, Scheffel, & London, 1995). Within the nervous system, the S1R is located predominantly in the gray matter both in neurons (Alonso, et al., 2000; Klette, DeCoster, Moreton, & Tortella, 1995; Peviani, et al., 2014) and a variety of glial cell types (Gekker, et al., 2006; Hayashi & Su, 2004; Jiang, et al., 2006; Palacios, et al., 2003; Palacios, Muro, Verdu, Pumarola, & Vela, 2004; Peviani, et al., 2014; Robson, et al., 2014). In addition to modulating the actions of neurotransmitter receptors, ion channels, and synaptic function, S1Rs are involved in the regulation of diverse processes such as neuroprotection, neurorestoration, neuroplasticity, and neurotransmitter release (Kourrich, Su, Fujimoto, & Bonci, 2012; Ruscher, et al., 2012; Su, et al., 2010; Zheng, 2009).
Based on in vitro and in vivo animal studies, the S1R has gained considerable attention as a therapeutic target for treating neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Parkinson’s disease, Huntington’s disease, stroke, and traumatic brain injury (Ryskamp, Korban, Zhemkov, Kraskovskaya, & Bezprozvanny, 2019). For example, S1R-agonists such as (+)-pentazocine and PRE-084 are neuroprotective against amyloid β (Aβ) toxicity in vitro and in vivo (Jin, Fang, Zhao, & Liu, 2015). They attenuate memory deficits and neurotoxicity associated with acute Aβ25–35 intracerebroventricular injection in mice, and this effect is prevented by the S1R antagonist NE-100 (Marrazzo, et al., 2005; Maurice, Su, & Privat, 1998). S1R agonists also have anti-amnesic properties in amnesia models, including amyloid induced AD models (Maurice, et al., 1998; Villard, et al., 2009), as well as in a cholinergic deficit model (Zou, Yamada, Sasa, Nakata, & Nabeshima, 2000). Similarly, S1R agonists such as PRE-084 (Allahtavakoli & Jarrott, 2011; Shen, et al., 2008), cutamesine (Ruscher, et al., 2011), and other drugs with S1R agonist activity, such as fluvoxamine (Sato, Kawamata, Kobayashi, & Okada, 2014), are neuroprotective in rat models of stroke. Interestingly, compounds classified as S1R antagonists are also neuroprotective in experimental stroke including LS-127 and LS-137 (Luedtke, et al., 2012), BD1047 and BD1063 (Rodriguez-Munoz, Onetti, Cortes-Montero, Garzon, & Sanchez-Blazquez, 2018), and haloperidol (Schetz, et al., 2007).
The broad protective effects of S1R ligands likely result from the pleiotropic nature of the S1R including not only modulation of ion channels and neurotransmitter receptors, but also intracellular calcium and endoplasmic reticulum (ER) stress, reductions in nitrosative stress, increases in brain-derived neurotropic factor (BDNF) and its receptor TrkB, and increases in antiapoptotic proteins such as Bcl2 (Ryskamp, et al., 2019). Regulation of BDNF is particularly important as it is involved in the maintenance and repair of neurons, synaptic plasticity, and learning and memory (Cunha, Brambilla, & Thomas, 2010; Korte, et al., 1995; Lewin & Barde, 1996). In animal models, chronic administration of the S1R agonist SA4503 increases the level of BDNF protein in the rat hippocampus (Kikuchi-Utsumi & Nakaki, 2008). Similarly, in vitro SA4503 is associated with enhanced secretion of BDNF into the extracellular environment (Fujimoto, Hayashi, Urfer, Mita, & Su, 2012). S1Rs can also regulate neurite outgrowth as shown in in vitro studies using PC12 cells that demonstrated that numerous S1R ligands including (+)-pentazocine, imipramine, fluvoxamine, donepezil, SA4503, and 4-PPBP facilitate NGF-induced neurite sprouting, an effect that is reversed by S1R antagonists NE100 and BD1063 (Ishima, Fujita, & Hashimoto, 2014; Ishima & Hashimoto, 2012; Ishima, Nishimura, Iyo, & Hashimoto, 2008; Nishimura, Ishima, Iyo, & Hashimoto, 2008; Rossi, et al., 2011). Given the multitude of processes involving the S1R and the ability of certain S1R ligands to promote BDNF synthesis/secretion, facilitate NGF-induced neurite outgrowth and reduce nitrosative stress, the S1R is an attractive druggable target for potentially treating or slowing the progression of neurodegenerative disorders. However, despite promising preclinical studies, clinical trials of several S1R ligands for treating stroke (Urfer, et al., 2014), Alzheimer’s disease (Schneider, et al., 2019), schizophrenia (Niitsu, et al., 2012), and peripheral neuropathy (Bruna, et al., 2018) have not been successful. One possibility limiting the translatability of studies is how such compounds are screened and categorized as ligands based on different assays.
There are two known subtypes of sigma receptors, S1R and the sigma-2 receptor (S2R). Both S1R and S2R have high affinity for haloperidol, DTG, and (+)-3-PPP, however, the S1R has high selectivity for the positive enantiomer of benzomorphans, whereas the S2R has low affinity for (+)-benzomorphans and high affinity for (−)-benzomorphans (Hellewell and Bowen 1990). The S2R has been reported to be involved in regulation of cell survival, morphology and differentiation (Guitart, Codony, & Monroy, 2004; Huang, Lu, Zhang, & Wu, 2014; Vilner, de Costa, & Bowen, 1995), and is highly expressed in cancer cells (Al-Nabulsi, et al., 1999; Vilner, John, & Bowen, 1995). Further S2R agonists have been reported to induce cell death via induction of apoptosis (Bowen, 2000; Hornick, et al., 2012; Zeng, et al., 2012; Zeng, et al., 2014; Zeng, Vangveravong, McDunn, Hawkins, & Mach, 2013), increase reactive oxygen species and induce lysosomal permeabilization (Hornick, et al., 2012; Ostenfeld, et al., 2005). S2R agonists also have immunosuppressive and anti-inflammatory effects (Iniguez, et al., 2013) and can modify cardiac repolarization by blocking inward rectifying K+ channel in the heart, which has the potential to cause cardiac sudden death (Monassier, et al., 2007). Although activation of some of these pathways may be attractive targets from a cancer therapeutic perspective, from a neurodegenerative disease perspective, activation of these pathways could exacerbate the disease pathology. Hence, in this study the goal was to discover S1R selective compounds, without any S2R affinity and evaluate their functional profile using multiple functional assays. The primary current means of qualifying S1R ligands as agonists involves behavioral readouts: 1) memory and cognitive function improvement, 2) antidepressant-like effects and 3) psychostimulant-induced behavior modulation (Hayashi & Su, 2004). This approach is not high throughput nor cost effective for evaluating large numbers of compounds. However, there are some reports which suggest that S1R agonists potentiate the IP3 mediated calcium response (Hayashi, Maurice, & Su, 2000; Hayashi & Su, 2007; Hong, Nuwayhid, & Werling, 2004; Wu & Bowen, 2008), facilitate BDNF release (Fujimoto, et al., 2012; Malik, et al., 2015), and potentiate NGF-induced neurite sprouting (Ishima, et al., 2014; Ishima & Hashimoto, 2012; Ishima, et al., 2008; Nishimura, et al., 2008; Rossi, et al., 2011; Takebayashi, Hayashi, & Su, 2002). In this study, all three measures were utilized to develop robust high-throughput assays for evaluating S1R compounds. Based on the findings of this study, only BDNF release appears to be a reproducible measure for evaluating S1R ligands, specifically, the BDNF assay utilized here can be used to identify functionally selective agonists that activate the BDNF secretion pathway.
2. Materials and Methods
2.1. Chemicals and reagents
Compounds were purchased from the following sources: 4-PPBP, BD1063, (+)-igmesine, PRE-084 from Tocris Biosciences (Minneapolis, MN); butamirate, carbetapentane, and NE-100 were from Santa Cruz Biotechnology (Dallas, TX); and donepezil oxeladin, promethazine was from Sigma-Aldrich (St. Louis, MO). Radioligands were purchased from PerkinElmer: ([3H] - (+)-pentazocine ((+)-Pentazocine, [RING-1,3-3H], 33.9 Ci/mmol, NET1056), [3H]-DTG (1,3-Di-o-tolylguanidine, [p-RING-3H]-, 50 Ci/mmol, NET986) (Saint Louis, MO). All compounds were prepared in DMSO at concentrations ranging from 10–100 mM. Stocks were then diluted 1:1000 (v/v) in the final assay solution.
2.2. Cell culture
Human MCF-7 cells (American Type Cell Culture, Manassas, VA) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Fisher Scientific, Pittsburgh, PA) supplemented with 10% Fetal Bovine Serum (FBS, Fisher Scientific, Pittsburgh, PA), 100 μg/ml nonessential amino acids (Hyclone, Logan, UT), 2 mM L-glutamine (Sigma-Aldrich, St. Louis, MO), and 10 μg/L Bovine Insulin (Sigma-Aldrich, Sigma-Aldrich, St. Louis, MO). MCF-7 cells served as the source of human Sigma-2 receptors (S2R) as they express the S2R but lack detectable levels of the S1R (Schetz, et al., 2007; Vilner, John, et al., 1995). MCF-7 cells stably expressing the human S1R (MCF7-hS1R) were prepared as described previously Schetz, et al. (Schetz, et al., 2007) and kept under constant selective pressure with 100 μg/mL G-418 (Invitrogen, San Diego, CA). Rat PC12 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and PC-6-15 cells, a variant of the PC12 cells overexpressing the trkA receptor (Hempstead, et al., 1992), cells were obtained Dr. Moses Chao. Both PC12 and PC-6-15 cells were maintained in RPMI1640 media supplemented with 10% horse serum and 5% FBS. The PC-6-15 cells were grown under constant selective pressure with 100 μg/mL G-418. MN9D cells were obtained from ATCC and were maintained in DMEM complete with 10% FBS. All culture media were additionally supplemented by 100 IU/mL of penicillin and streptomycin (Corning, Manassas, VA) and 1 mM sodium pyruvate (Sigma-Aldrich, St. Louis, MO), and grown at 37 °C under 95% air, 95% humidity, and 5% CO2.
2.3. Measurement of BDNF secretion via in situ ELISA
The in-situ ELISA format utilized here has been shown to have improved sensitivity related to its rapid capture of secreted BDNF (Balkowiec & Katz, 2000; Dalwadi, Kim, & Schetz, 2017). The in-situ ELISA improves on traditional ELISA by seeding BDNF-secreting cells directly into the wells pre-coated with an anti-BDNF primary antibody. BDNF secreted by these cells is immediately captured by the primary antibody, drastically increasing the sensitivity and reproducibility of the ELISA assay. Intact cells are removed subsequent to the addition of the secondary and tertiary antibodies. The amount of BDNF secreted from the neuronal MN9D cells was quantified using an in-situ ELISA assay developed using the BDNF Emax ImmunoAssay kit (Cat. No. G7611, Promega, Madison, WI). Briefly, a Nunc MaxiSorp flat-bottom, polystyrene, 96-well immunoplate was coated for 48 hrs at 4°C with an anti-BDNF monoclonal antibody diluted 1:1000 v/v in carbonate buffer containing 25 mM sodium bicarbonate and 25 mM sodium carbonate, pH 9.7. Unbound antibody was removed by washing 5 times with 150 μL of TBST buffer (20 mM Tris-HCl, pH7.6, 150 mM NaCl, and 0.05% (v/v) Tween 20), before blocking non-specific sites first with blocking buffer for 1 hr and then with culture medium for 2 hrs. Cells were seeded at 35,000 cells per well and incubated overnight at 37°C in a humidified CO2 incubator. The following day, the wells were replaced with fresh culture media containing either experimental compounds or vehicle controls, then incubated for an additional 24 hours. In addition, on the same plate, but in separate wells, BDNF standards were added ranging in concentration from 15.6–250 pg/mL. After 24 hrs incubation with experimental compounds, the media was aspirated and 100 μL of Dulbecco’s phosphate saline (D-PBS without Ca2+ and Mg2+ supplemented with 5 mM EDTA) was added to each well and incubated for 15 min at 37°C to promote cell lifting. Cells were then detached from the bottom of wells by triturating in the center and around the edges of the well. After removing all cell debris, the wells were rinsed five times with 150 μL of TBST. The plate was then incubated with 1:500 v/v diluted polyclonal anti-human BDNF antibody for 2 hrs at room temperature. This antibody was removed, and wells were washed five times with 150 μL of TBST, before incubating with 1:200 v/v diluted polyclonal Anti-IgY HRP conjugate for 2 hrs at room temperature. Wells were then washed five times with 150 μL of TBST and the remaining specifically bound polyclonal antibody was detected with the 50 μL colorimetric HRP substrate 3,3’,5,5’-Tetramethylbenzidine (TMB). The reaction was terminated with 50 μL of 1 M HCl and the color intensity was quantified by measuring the absorbance at 450 nm using a Flex Station 3 plate reader (Molecular Devices, Sunnyvale, CA). Measurements from multiple experiments were normalized to maximal BDNF responses achieved by stimulating the prototypical sigma ligand 4-PPBP. A one-way ANOVA with a Bonferroni multiple comparisons post-hoc analysis (P < 0.05) was applied to determine significant differences between groups. When converted to pg/mL averaged BDNF values ± SEM (n = 3–10 experiments) were: 75.4 ± 6.9 for baseline (vehicle control) and 128.6 ± 12.8 for maximal stimulation by 10 μM 4-PPBP.
2.4. Measuring receptor density with radioligand binding
The density or maximum number of binding sites (Bmax) for the S1R in PC-6-15 cells was estimated by employing 10 nM of [3H]- (+)-pentazocine as the radioligand followed by calculation of the Bmax at saturation using a square hyperbola model: Bmax = (Y • (KD+X))/X, where Y is [specifically bound radioligand] and X = [radioligand concentration]. The affinity (KD) for [3H]-(+)-pentazocine at the cloned human S1R had been previously determined to be 3.7 nM (Schetz, et al., 2007).
2.5. Measuring the ligand affinities at S1R and S2R receptors by radioligand binding
The affinity (Ki) values of compounds interacting with the S1R and S2R were determined by displacement of 0.5 nM [3H] -(+)-pentazocine from MCF-7-S1R and 2.5 nM [3H]-DTG from untransfected MCF-7 cells, respectively. Binding conditions were the same as described previously for the S1R (Schetz, et al., 2007): binding buffer (Tris 50 mM, pH = 8.1 at 37°C), ice-cold wash buffer (Tris 10 mM, pH = 8.1 at 0–2°C), and incubation time (3 hrs at 37°C) with shaking. Non-specific binding for the S1R and S2R was determined in the presence of 5 μM BD1063 and 15 μM haloperidol, respectively. Following incubation, receptors were collected via rapid filtration through GF/C filters (Brandel, Gaithersburg, MD) followed by washing three times with 3 mL of ice-cold wash buffer. Dried filters were transferred to vials filled with 3.5 ml of scintillation fluid, and the radioactivity was quantified on a liquid scintillation analyzer (Tri Carb 2800TR) from Perkin Elmer (Saint Louis, MO). Mean values from duplicate or triplicate determinations are reported along with their associated standard error of the mean (SEM). Ki values were calculated from IC50 values using the Cheng-Prusoff equation. The concentration of membrane protein was determined using a BCA Protein Assay kit (Life Technologies, Grand Island, NY) following the manufacturer’s protocol.
2.6. Measurement of the effect of sigma ligands on IP3 induced change in intracellular calcium
MCF7 and MCF7-hS1R cells were plated on Poly-L-Lysine coated black walled 96 well plates at a density of 120,000 cells per well in complete media and incubated overnight in a humidified CO2 cell culture incubator. The following day, the media was removed, and cells were loaded with the FLIPR Calcium-6 QF dye (Molecular Devices, Sunnyvale CA) dissolved in Hank’s buffered saline supplemented with 20 mM HEPES pH = 7.4, for three hours in the dark. To test the effect of sigma ligands on IP3 induced calcium response, the loading dye was supplemented with the sigma ligands or vehicle (DMSO, 1:1000). Following dye loading and any pretreatments, rapid fluorescent signals in response to changes in intracellular calcium was measured at 485 nm excitation, 525 nm emission with the cutoff filter set at 515 nm using the Flexstation 3 (Molecular Devices, Sunnyvale, CA). The baseline calcium signal was measured for 120 seconds followed by stimulation of the endogenous bradykinin receptors with bradykinin to activate the Gq-PLC-IP3-Ca2+ pathway, and measured changes in the calcium signal for an additional 780 seconds. At the termination of the experiment, all signals were baseline subtracted and any change in intracellular calcium were quantified as the area under the curve (AUC), calculated by integration (Graphpad Prism version 4.0). For the bradykinin concentration response curves, the AUC was normalized to the maximal functional response defined by the asymptote and plotted as a sigmoidal dose response curve. AUCs for sigma ligand treated groups were normalized to the bradykinin response in the presence of vehicle. If the presence of the sigma ligand potentiated the bradykinin calcium response, it would be categorized as an agonist, and if it suppressed the response then it would be categorized as an inverse agonist. Ligands that suppressed the effects of an agonist sigma ligand without having effects on its own would be categorized as an antagonist.
2.7. Effect of sigma ligands on NGF induced neurite sprouting
The PC-6-15 variant of PC-12 cells (gift from Dr. Moses Chao), that overexpress the trkA receptor was used to shorten the duration of the assay (Hempstead, et al., 1992). Untransfected PC12 normally take 5 to 7 days to respond to NGF treatment and have a tendency to clump. The PC-6-15 cells respond to NGF within 24 hours and do not clump. Cells were maintained in RPMI1640 media supplemented with 50 units/mL streptomycin, 50 μg/mL penicillin, 1 mM sodium pyruvate, 0.1 mg/mL G418 to maintain selection, and 10% horse serum and 5% FBS. On day 1 of the experiment, cells were plated in type I collagen (EMD Millipore, Massachusetts) coated 96 well plates in low serum media (0.5% serum) and treated with vehicle (DMSO), S1R ligands, NGF (Cat# 01–125, βNGF, EMD Millipore, Massachusetts) (EC50 concentration) or S1R ligand + NGF; each treatment was performed in duplicate. Cells were the incubated at 37oC in a humidified incubator with 5% CO2 for 24 hrs. After the 24 hrs treatment, 10x bright field images were obtained from three different fields of view per well. The neurite length was quantified by tracing the neurites using ImageJ (Schmitt & Dirsch, 2009) and presented as total neurite length divided by the total number of cells.
2.8. Statistical analyses
Each experiment was performed in triplicate and then each repeated two or more times. The data for all experiments were averaged and plotted as means ± SEM. To evaluate statistical significance, a one-way ANOVA, followed by Bonferroni’s post hoc analysis was performed. Statistical significance of the Hill slope was assessed by extra sum-of-squares F test. A P-value less than 0.05 was considered significant.
In order to evaluate the pseudo-Hill slope parameter in competition binding of Sigma receptor ligands with [3H] -(+)-pentazocine at the human S1R and with [3H]-DTG at the human S2R, at the S1R, all the slopes were treated as a variable (instead of a fixed value) for the purpose of curve fitting. Using this approach, variable slope was the preferred model for 4-PPBP and NE-100, with a pseudo-Hill slope of −3.1 (P < 0.05) whereas a pseudo-Hill slope equal to unity was the preferred model for BD1063 (P > 0.05) for the S1R. At the S2R, both 4-PPBP, PRE-084, NE-100, and BD1063 had pseudo-Hill slopes near unity, thus a pseudo-Hill slope equal to unity was the preferred model (P > 0.05).
A pseudo-Hill slope equal to unity was the preferred model for all three antitussive agents at the S1R and S2R (P>0.05).
3. Results
In this study we report on prototypical and potential S1R ligands that have selectivity over the S2R and facilitate BDNF release in a neuronal cell line. We first empirically evaluated known S1R ligands and determined their selectivity over the S2R. Known S1R agonists include 4-PPBP, PRE-084, donepezil and (+)-igmesine, and known antagonists include BD1063 and NE-100, which served as reference compounds in this study. Human MCF-7 cells were used as the source of the S2R because these cells express the endogenous S2R (Vilner, John, et al., 1995; Wu & Bowen, 2008) but lack detectable levels of the S1R (Schetz, et al., 2007). [3H]- DTG was used as the radioligand to probe for the S2R. MCF-7 cells lack S1R, allowing unambiguous detection of S2R activity in untransfected cells. MCF-7 cells stably expressing the cloned human S1R were used as the source of S1R and the high affinity S1R selective radioligand [3H]-(+)-pentazocine was used to probe for the S1R (Schetz, et al., 2007). The affinities for the prototypical agonists at the S1R ranged from 1 to 92 nM. 4-PPBP had the highest affinity and PRE-084 had the lowest affinity (Figure 1, Table 1). At the S2R, the affinities for the same agonists ranged from 0.8 to 13,070 nM. 4-PPBP had the highest affinity and PRE-084 had the lowest affinity for S2R. Although PRE-084 had the greatest selectivity for S1R over S2R and 4-PPBP had no selectivity, in our pilot functional studies using BDNF secretion as the functional output, 4-PPBP treatment resulted in a robust BDNF secretion whereas PRE-084 had no effect (Figure 3). Hence, 4-PPBP was used as the reference agonist for all BDNF functional assays reported here. Affinities of two prototypical antagonists were also evaluated at the S1R and S2R (Figure 1, Table 1). Both NE-100 and BD1063 had low nanomolar affinities for the S1R (1.9 and 5.6 nM, respectively), and high nanomolar affinities for the S2R (41.4 and 210.2 nM, respectively). Both NE-100 and BD1063 had greater selectivity for S1R over the S2R, however, BD1063 had slightly greater selectivity than NE100 (38- vs 21-fold). The selective concentration of BD1063 was calculated to be 15 nM because it corresponds to a calculated S1R and S2R occupancy of 74% and 7%, respectively (Fractional Occupancy = [ligand]/([ligand]+Ki)), hence was used as the preferred antagonist in our subsequent functional assays.
Figure 1. Prototypical sigma receptor ligands PRE-084, NE-100 and BD1063 have greater selectivity for S1R over S2R, whereas 4-PPBP does not.

A, Competition binding of prototypical sigma receptor ligands with [3H] -(+)-pentazocine at the human S1R. The rank order affinities were as follows: 4-PPBP ≈ NE-100 ≈ BD1063 > PRE-084. 4-PPBP, NE100 and BD1063 had low nanomolar affinities for the S1R (range: 1.0 to 5.6 nM) whereas PRE-084 had high nanomolar affinity the S1R (92 nM) A variable slope was the preferred model for 4-PPBP and NE-100. B, Competition binding of prototypical sigma receptor ligands with [3H]-DTG at the human S2R. The affinities ranged from 0.8 nM to 13,070 nM, and the rank order affinities were as follows: 4-PPBP > NE-100 > BD1063 > PRE-084. A pseudo–Hill slope equal to unity was the preferred model for all ligands. Affinity values are listed in Table 1. Values are averages of three biological replicates ± SEM data and each value was calculated from three technical replicates.
Table 1.
PRE-084 is the most selective prototypical agonist and BD1063 is the most selective prototypical antagonist.
| Compound | Structure | S1R affinity (Ki, nM) | S2R affinity (Ki, nM) | S2R/S1R ratio (Ki S2R / Ki S1R) |
|---|---|---|---|---|
| 4-PPBP |

|
1.0 ± 0.3 | 0.8 ± 0.2 | 0.8 |
| PRE-084 |

|
92 ± 15 | 13070 ± 1725 | 142 |
| (+)-Igmesine |

|
9.2 ± 2.2 | 164 ± 29 | 18 |
| BD1063 |
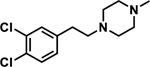
|
5.6 ± 0.5 | 210 ± 23 | 38 |
| NE-100 |

|
1.9 ± 0.4 | 41.4 ± 4.6 | 21 |
Affinity (Ki) values presented as mean ± SEM
Figure 3. S1R-selective compounds facilitate S1R mediated BDNF secretion from the neuronal MN9D cell line.

Levels of secreted BDNF were measured using in situ ELISA. All compounds were tested at 10 μM and were reversed with 15 nM BD1063. Data was normalized to the response produced by 10 μM 4-PPBP. Values are averages of three biological replicates ± SEM data and each value was calculated from three technical replicates. Statistical significance at P < 0.05 was determined by ANOVA followed by a Bonferroni’s post-hoc test and are marked with an asterisk. Efficacy of S1R ligands tested here can be found in Table 3.
Activation of the S1R has also been linked to antitussive effects and carbetapentane is a known antitussive agent that is commonly used in over-the-counter cough medication (Drugs.com 2014). Carbetapentane is also classified as an S1R agonist (Brown, Fezoui, Selig, Schwartz, & Ellis, 2004), and other antitussive agents like oxeladin and butamirate belong to the same structural class. Since all three antitussive agents only differ by a single modification, it allowed us to evaluate structure-activity relationship (SAR) of these compounds at the S1R in the context of BDNF release. The antitussive agents all had nanomolar affinities for both S1R and S2R demonstrating only weak selectivity over the S2R (Figure 2 and Table 2). Carbetapentane was only 3-fold selective over the S2R, and oxeladin and butamirate were 9- and 10-fold selective over S2R, respectively. The antihistamine promethazine was previously found to bind S1R in our lab (unpublished results) and was also included as a potential candidate drug. Promethazine was 5-fold selective for S1R over S2R with an affinity of 157 nM.
Figure 2. Antitussives are moderately selective for the S1R.

Competition binding of sigma receptor ligands with [3H] -(+)-pentazocine at the human S1R and with [3H]-DTG at the human S2R. Carbetapentane, butamirate and oxeladin had only 3 to 10-fold greater selectivity for the S1R over the S2R. Affinity values are listed in Table 2. Values are averages of three biological replicates ± SEM data and each value was calculated from three technical replicates.
Table 2.
Effect of specific substructural modifications on selectivity for S1R over S2R.
| Compound | Substructural feature | S1R affinity (Ki, nM) | S2R affinity (Ki, nM) | S2R/S1R ration (Ki S2R / Ki S1R) |
|---|---|---|---|---|
| Carbetapentane |

|
19.8 ± 2.3 | 55.7 ± 3.8 | 3 |
| Oxeladin |

|
25 ± 4.3 | 148 ± 64 | 6 |
| Butamirate |

|
17.2 ± 1.7 | 214 ± 41 | 10 |
Functional selectivity of these compounds was first tested with an assay of BDNF release as the prototypical S1R agonist SA4503, and a novel S1R agonist LS-1–137 stimulate BDNF secretion from neuronal and glial cells, respectively (Fujimoto, et al., 2012; Malik, et al., 2015). The neuronal MN9D cell line was selected because preliminary data generated in our lab suggested that MN9D cells produce a more robust BDNF release in response to SA4503 compared to the B104 neuronal cell line, which was previously used for this assay (Fujimoto, et al., 2012). An in-situ ELISA approach was utilized because of its improved sensitivity related to its rapid capture of secreted BDNF (Balkowiec & Katz, 2000; Dalwadi, et al., 2017). In addition to producing robust response, the MN9D neuronal cell line also expresses BDNF (data not shown), S1R (Bmax = 1.5 ± 0.26 pmol/mg membrane protein), and releases BDNF in response to the S1R agonist 4-PPBP. Further, with the MN9D cells, secreted BDNF could be detected following 24 hr treatment with S1R ligand, whereas the B104 cell line takes 7 days (Fujimoto, et al., 2012). All compounds were tested at an equimolar concentration of 10 μM, which represents maximum receptor occupancy (Figure 1 and 2). 4-PPBP was used as the reference agonist and represented the maximum BDNF secretion response; all compounds were normalized to the 4-PPBP response (Figure 3). Another reference agonist, PRE-084, failed to stimulate BDNF secretion. Reference compounds with BDNF secreting activity included (+)-igmesine, donepezil, and 2-Butoxyethyl 2-(diethylamino)-2-phenylacetate (Cas#1796909–31-3). The amount of BDNF secreted ranged from 24 to 103% of the maximum response (Table 3), where promethazine had the highest response. All other compounds had significantly lower efficacy than 4-PPBP (P < 0.05, one-way ANOVA, Bonferroni’s post-hoc). The responses of donepezil and (+)-igmesine could be reversed at a selective concentration of BD1063 (15 nM). With the exception of butamirate, all other compounds were able to stimulate BDNF release, and their efficacy ranged from 54% to 144%.
Table 3.
Efficacy of S1R ligands that function as agonists of BDNF secretion.
| Compound | Efficacy of BDNF secretion |
|---|---|
| 4-PPBP | 1 |
| Promethazine | 1.09 ± 0.34 |
| Cas# 1796909-31-3 | 0.24 ± 0.04* |
| (+)-igmesine | 0.42 ± 0.03* |
| Donepezil | 0.53 ± 0.08* |
| Carbetapentane | 0.73 ± 0.05* |
| Oxeladin | 0.55 ± 0.04* |
Values are normalized to the maximal response of 10 μM 4-PPBP. Efficacies significantly different from 4-PPBP are marked with an asterisk (P < 0.05, one-way ANOVA, Bonferroni’s post-hoc). All other efficacies were not significantly different. Values represent mean ± SEM.
At the intracellular level, S1R agonists have been reported to potentiate IP3-induced calcium mobilization (Hayashi, et al., 2000; Hayashi & Su, 2007; Hong, et al., 2004; Wu & Bowen, 2008) which offers another approach for evaluating the function of S1R ligands. MCF7 cells lacking S1R and MCF7 cells stably transfected with the human S1R (MCF7-hS1R) cells were used to evaluate S1R mediated potentiation of IP3-induced calcium mobilization as previously described by Wu et al. (Wu & Bowen, 2008). Since these cells express the endogenous Gq-coupled bradykinin B2 receptor (Searovic, et al., 2009), bradykinin was used to stimulate the IP3 pathway. A bradykinin concentration response curve was generated by measuring the change in intracellular calcium as a measure of Gq-PLC-IP3-[Ca2+]i signaling pathway activation and bradykinin potencies were obtained for each cell line (Figure 4A). The potencies (EC50) for bradykinin were 2.8 ± 0.3 nM and 0.9 ± 0.1 nM in the MCF7-hS1R and MCF7 untransfected cell lines, respectively, and were not significantly different from each other (P > 0.01, t-test). These bradykinin parameters were important to know because a submaximal IP3 response would allow for the detection of small changes in calcium mobilization induced by S1R ligands, whereas these small changes are likely to be masked if a saturating bradykinin concentration is used. We expected that when the cells were pre-treated with the S1R agonists they would be a potentiate bradykinin-induced calcium mobilization, and that this response could be reversed by the S1R antagonists; however, no potentiation was observed (Figure 4A). Similarly, there was no potentiation of the bradykinin response in untransfected MCF7 cells lacking S1R.
Figure 4. S1R does not appear to modulate Gq-PLC-IP3-[Ca2+]i mediated calcium release.

A, MCF7 cells express the endogenous bradykinin receptor B2 (Searovic et al. 2009). Bradykinin (BDK) concentration response curves were generated for the untransfected MCF7 cells and MCF7-hS1R by measuring changes in intracellular calcium as a measure of Gq activation. The potencies were 2.8 ± 0.3 nM for the MCF7-hS1R cell line and 0.9 ± 0.1 nM for the untransfected MCF7 cells. The potencies were not significantly from one another (P > 0.01, t-test). B, Activation of the S1R by prototypical sigma ligands did not have any effect on the Gq-PLC-IP3 mediated calcium response. Values are averages of three biological replicates ± SEM data and each value was calculated from three technical replicates.
In the PC12 cell line, S1R agonists like (+)-pentazocine, SA4503 and 4-PPBP facilitate NGF-induced neurite sprouting (Ishima, et al., 2014; Ishima & Hashimoto, 2012; Ishima, et al., 2008; Nishimura, et al., 2008; Rossi, et al., 2011; Takebayashi, et al., 2002). In an effort to reduce the assay duration, we used PC12-derived PC-6-15 cells which overexpress the trkA receptor (Hempstead, et al., 1992). Unlike the PC12 cells, PC-6-15 do not clump when plated which makes neurite quantification more reproducible and reliable, and they are also potently activated by NGF within 12 hours. In contrast, it takes approximately 4–6 days for PC12 cells to produce similar levels of sprouting (Ishima, et al., 2014; Rossi, et al., 2011; Takebayashi, et al., 2002). We first measured the density of the S1R protein in PC-6-15 cell membranes utilizing the [3H] -(+)-pentazocine radioligand, and the Bmax (maximum number of binding site) was determined to be 0.24 ± 0.01 pmoles/mg membrane protein. To identify a submaximal concentration of NGF that induced neurite sprouting, cells were treated with 0.3 to 100 ng/mL NGF, and the neurite length/cell was quantified by tracing individual neurites to obtain the total neurite length and dividing this number by the total number of cells (Figure 5A). NGF increased the total neurite length in a concentration dependent manner, and since 0.3 ng/mL was the lowest concentration tested that induced neurite sprouting, it was used to test the potentiation effects of S1R ligands. Ligands with the highest BDNF-releasing activity were selected. When these cells were treated with the S1R ligands alone, there was no significant effect on neurite sprouting. When co-treated with 0.3 ng/mL NGF, neither 4-PPBP or promethazine enhanced NGF induced sprouting (Figure 5B). Examples of neurite outgrowth in response to NGF and NGF cotreatment with 4-PPBP and promethazine are shown in Figure 6. Overall, out of the three functional assays tested, in our hands, only BDNF secretion seems to be a reliable measure for assessing functionally selective S1R agonist activity.
Figure 5. S1R activation has no effect on neurite sprouting, nor does it potentiate NGF induced neurite sprouting in PC-6-15.

A, NGF induces neurite sprouting in concentration dependent manner in the PC-6-15 cells. B, PC-6-15 cells were maintained in media with 0.5% serum and treated with either vehicle (1:1000 v/v DMSO), 0.3 ng/mL NGF, sigma ligands, or sigma ligands + 0.3 ng/mL NGF for 24 hours. Treatment with 0.3 ng/mL NGF resulted in a significant increase neurite sprouting, but sigma ligands had no effect on neurite sprouting nor did they potentiate NGF induced neurite sprouting. Data presented as mean ± SEM. * P < 0.05 by One-way ANOVA, Bonferroni’s post-hoc test. Values are averages of three biological replicates ± SEM data and each value was calculated from three technical replicates.
Figure 6. Examples of neurite outgrowth.

A, Dose response of neurite outgrowth in PC-6-15 cells in response to 24 hours treatment with NGF. B, Examples of neurite tracing performed on images in A. C, Raw images and neurite tracing for cells treated with 0.3 ng/mL NGF and either 10 μM 4-PPBP (top), or 10 μM Promethazine. Scale bars = 100 μm.
4. Discussion
In this study we explored three different in vitro functional assays to evaluate the functions of prototypical and potential compounds at the S1R. Most, but not all, of the S1R ligands tested stimulated BDNF secretion indicating that these ligands are activating a functionally selective pathways that is responsible for BDNF secreting activity.
Though the Ki for 4-PPBP, PRE-084, NE-100 and BD1063 at the S1R and S2R have been previously reported (Garces-Ramirez, et al., 2011; Lee, Chen, & Schetz, 2008; Okuyama & Nakazato, 1996; Whittemore, Ilyin, & Woodward, 1997), they were repeated in this report to validate our assay model and to do a parallel comparison of the affinities using the same cell background (See Results section, 1st paragraph for model detail on assay model). To evaluate the selectivity of these compounds’ inhibition curves were generated at the S1R and S2R. The S1R and S2R affinity values for 4-PPBP, PRE-084, NE-100 and BD1063 reported here are comparable to those reported previously (Garces-Ramirez, et al., 2011; Lee, et al., 2008; Okuyama & Nakazato, 1996; Whittemore, et al., 1997), thus validating the approach. It is interesting to note that though 4-PPBP had no selectivity over the S2R, at the S1R it had a very steep pseudo-Hill slope (−3.1) which indicates positive cooperativity, hence the S1R has more than one binding site for 4-PPBP. In contrast, PRE-084 had a pseudo-Hill of slope of −0.98 which was not different from unity suggesting binding to a single site on the S1R. Both 4-PPBP and PRE-084 are considered S1R agonists, however, when BDNF secretion was used as a measure of S1R activation, 4-PPBP induced a robust BDNF secretion response, whereas PRE-084 had no activity. One might speculate then that 4-PPBP is likely interacting with a S1R binding site differently from PRE-084 which may account for the functionally selective profile of these compounds. Further, though 4-PPBP is not a S1R-selective compound, its effect on BDNF secretion could be reversed by a S1R-selective concentration of BD1063 suggesting that the response is likely being mediated by the S1R and not the S2R.
To begin to identify different structural features that impact selectivity and have the desired functional output, the structure-activity space three antitussive agents were evaluated. The structures of carbetapentane, butamirate and oxeladin only differ by the type of alkyl modification at one location (Table 2). Carbetapentane has a cyclopentyl group, is only threefold selective over the S2R, and its efficacy for BDNF secretion is 72% of the maximum response. Changing the cyclopentyl (carbetapentane) group to a diethyl (oxeladin) or an ethyl (butamirate) group increased the selectivity by 2 and 3-fold, respectively, over carbetapentane. However, these changes also reduced the BDNF secretion activity. Oxeladin had slightly reduced BDNF secretion activity (55%), thus, the presence of the diethyl group caused an 18% reduction in efficacy relative to carbetapentane. Butamirate has an ethyl group, and this completely abolished the BDNF secretion activity. These modifications had very little to no effect on the affinity at the S1R, but moderately decrease the affinity at the S2R suggesting that this region exerts only modest control on selectivity for S1R over S2R. Further, bulky alkyl groups (e.g. cyclopentyl or diethyl) at that position may be necessary for activating the BDNF secretion pathway.
In addition to modulating BDNF secretion, classical sigma agonists haven been reported to modulate IP3 induced calcium response (Hayashi, et al., 2000; Hayashi & Su, 2007; Hong, et al., 2004; Wu & Bowen, 2008). In NG108 (neuroblastoma-glioma hybrid cell line) and SH-SY5Y (neuronal) cells, (+)-pentazocine and PRE-084 were reported to potentiate bradykinin-induced increases in intracellular calcium and this effect was reversed by treatment with the antagonists NE-100 and haloperidol (Hayashi, et al., 2000; Hong, et al., 2004). Similarly, in the MCF7 cells transfected with the S1R, (+)-pentazocine increased bradykinin induced calcium release and this response was reduced in the presence of BD1063 (Wu & Bowen, 2008). Further, BD1063 on its own was able to reduce the bradykinin induced calcium response suggesting that it functions as an inverse agonist at the S1R (Wu & Bowen, 2008). When some of the same sigma ligands were tested in the untransfected MCF7 cells, no effect on bradykinin induced calcium mobilization was observed (Figure 4B). The lack of an effect makes sense if the effects are due to the S1R, since the untransfected MCF7 cells lack detectable levels of the S1R (Schetz, et al., 2007; Vilner, John, et al., 1995; Wu & Bowen, 2008). However, when the effects of the sigma reference ligands 4-PPBP, PRE-084, (+)-SKF10047, BD1063 and NE100 were tested, in the MCF7-hS1R cell line, no significant potentiation or suppression of the bradykinin response by the reference sigma agonists or antagonists, respectively, was detected. In other words, the results from the MCF7-hS1R cell line looked similar to the untransfected MCF7 cell line. The lack of any S1R response in this system is not due to low expression of the S1R because the Bmax for S1R in the MCF7-hS1R cell line was 109 ±23.7 pmol/mg (Lee, et al., 2008) whereas the S1R Bmax in Wu and Bowen’s model was 31.4 ± 4.5 pmol/mg (Wu & Bowen, 2008). The major difference between our study and Wu and Bowen study is that they used (+)-pentazocine as that their reference agonist. It is possible that the reference agonists we tested do not have the same functionally selective profile as (+)-pentazocine, which might explain why the S1R mediated potentiation of the bradykinin response was not detected in this study. However, Wu and Bowen also showed that BD1063 can suppress the bradykinin response in the S1R-expressing cell line but not in the untransfected cell line. When the same experiment was performed in our study, the BD1063 inverse agonist response in the S1R expressing cell line was not reproducible. Further, since the MCF7-hS1R had a higher Bmax than Wu and Bowen’s S1R expressing MCF7 cell line, it would be expected that the inverse agonist response would stronger than that observed by Wu ad Bowen. Also, in the NG108 cell line, PRE-084 was reported to potentiate the bradykinin-induced calcium response (Hayashi, et al., 2000), but the result was not reproducible in our laboratory. It is possible that the MCF7 cells do not have the same machinery as the NG108 cell line which is why it might not be able to produce this response. If the S1R mediated modulation of the IP3-induced calcium response only requires the presence of BiP, IP3R and the S1R, then the MCF7-hS1R model should have all the necessary components to elicit the same response as the NG108 cell line. The MCF7 cells express BiP (Fu, Li, & Lee, 2007), IP3R as evidenced by the bradykinin induced Gq response, and S1R was overexpressed in these cells (Schetz, et al., 2007),
The idea of S1R agonists potentiating IP3 mediated calcium response by stabilizing the IP3 receptor (IP3R) originates from the work of Hayashi and Su, 2007. In that report they demonstrated that S1R agonists (+)-pentazocine, PRE-084, and SKF10047 (all previously characterized as agonists based on their in vivo behavioral profile) caused the S1R to dissociate from the chaperone protein BiP and interact with the type 3 IP3R (IP3R3). Treatment with the S1R antagonists NE-100 and haloperidol had no effect on the BiP-S1R interaction (Hayashi & Su, 2007). Further they showed that there was a correlation between IP3R3-S1R interaction and an increase in calcium mobilization from the ER to the mitochondria in the presence of IP3. Since the IP3R3s are predominantly localized at the mitochondria-associated ER membranes (MAM) and provide direct connection for ER-mitochondria calcium signaling (Mendes, et al., 2005), it seems reasonable that stabilization of the IP3R3 by the S1R would potentiate calcium mobilization into the mitochondria. Cytosolic calcium mobilization is mediated primarily via the type 1 IP3R (IP3R1) (Mendes, et al., 2005) and there is no evidence that the activated S1R interacts with and stabilizes IP3R1. Since cytosolic calcium was measured in this study, it is possible that the S1R mediated potentiation on the IP3 response cannot be detected by measuring change in cytoplasmic calcium because the S1R is not interacting with IP3R1 but rather with the IP3R3 and mobilizing calcium from the ER to the mitochondria. Additionally, since the IP3R1-S1R interaction has not been studied, it is possible that the S1R does not interact with or stabilize IP3R1. The other possibility is that S1R could be interacting with IP3R1, but this interaction alone may not be enough to cause an increase in calcium mobilization, and an additional stimulus like ER stress may be required. If a strong correlation between S1R agonists and an increase in cytosolic calcium mobilization was established here, then it would have been a useful tool for identifying S1R agonists. However, it appears that the S1R-IP3R-Ca2+ relationship is not as simple as predicted and cannot be used as a reliable measure for evaluating S1R functional pharmacology (i.e., agonist/antagonist/inverse agonist properties) using changes cytosolic calcium as the outcome measure.
S1R agonists have been reported to modulate neurite sprouting (Ishima, et al., 2014; Ishima & Hashimoto, 2012; Ishima, et al., 2008; Nishimura, et al., 2008; Rossi, et al., 2011; Takebayashi, et al., 2002). For example, (+)-pentazocine, 4-PPBP, and PRE-084 were shown to potentiate NGF-induced neurite sprouting in the PC12 cell line (Nishimura, et al., 2008; Rossi, et al., 2011; Takebayashi, et al., 2002). PC12 cells take approximately 5 days to sprout measurable neurites following chronic exposure to NGF and all studies to date using PC12 cells involved 4–5 days treatments. In an effort to reduce the duration of the assay and make it higher throughput, we utilized a variant of the PC12 cell line (PC-6-15) that overexpresses the trkA receptor (Hempstead, et al., 1992). The PC-6-15 cell line produces long neurites (neurite length greater than two cell bodies) within 2 hours of NGF treatment, and 90% of cells formed dense neuronal processes within 24 hours. The expectation was that this cell line could be used to shorten the assay time for evaluating the effects of S1R ligands on neurite sprouting. However, the reference S1R agonist 4-PPBP was unable to stimulate neurite sprouting, nor did it potentiate NGF induced sprouting. Other S1R agonists like PRE-084, carbetapentane and fluvoxamine were also tested, and none of them had any effect on sprouting on their own or on NGF-induced sprouting (data not shown) in the PC-6-15 cell line. This response is not due the lack of S1R in these cells, because the radioligand binding data using the S1R selective [3H] -(+)-pentazocine as the radioligand clearly shows that the S1R protein is present in these cells. It is possible that the S1R is not a major contributor in the neurite sprouting process (i.e., it only has a minor effect on neurite sprouting), and that the neurite tracing method is not sensitive enough to detect those small changes. The other possibility is that overexpressing the trkA receptor is decreasing the expression of the S1R by decreasing gene expression, translation, and/or targeting the protein for degradation. As a result, the S1R levels may not high enough to produce a measurable change in neurite sprouting. Regardless of what the explanation may be, the PC-6-15 is not a suitable model for evaluating the role of the S1R on neurite sprouting, however it is an excellent model for evaluating small NGF-like molecules targeting the trkA receptor.
5. Conclusion
Using an in-situ ELISA approach we demonstrated that S1R ligands can facilitate BDNF secretion, and that this response can be reversed by a S1R-selective concentration of the antagonist BD1063. Thus, the S1R ligands examined in this study exhibit S1R agonist properties when BDNF secretion is used a measure of S1R activation. We wanted to evaluate the agonist activity of these compounds using other measures of activation, and since there were reports of S1R agonists potentiating IP3-induced ER to cytoplasm calcium mobilization and potentiating NGF induced neurite sprouting, we examined the suitability of these assays. However, when we attempted to reproduce the S1R-IP3-Ca2+ response using prototypical S1R agonists, we were unable to reproduce the results and eventually reached the conclusion that it is not reliable measure for evaluating S1R ligands using changes in cytosolic calcium as the outcome measure. For measuring the effects of S1R ligands on neurite sprouting we utilized a modified PC12 cell model that overexpresses the trkA receptor because a 5-day assay is not a time productive option for evaluating large numbers of compounds and the overexpression system we used allowed us to reduce the duration of the assay to one day. We could not detect potentiation of NGF-induced neurite sprouting for any of the reference S1R agonists tested. This may be because the PC-6-15 cell model used is not appropriate for testing this parameter.
Highlights:
Current Sigma 1 receptor (S1R) ligands have limited selectivity
High throughput screening is useful for identifying new selective ligands
BDNF secretion is superior to Ca2+ mobilization and neurite outgrowth for screening of S1R ligands
Antitussives show enhanced selectivity for S1R
Funding Source:
This work was supported by the National Institutes of Health [NIH – R21NS095271-01] award to J. A. Schetz and D. A. Schreihofer
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Nabulsi I, Mach RH, Wang LM, Wallen CA, Keng PC, Sten K, Childers SR, & Wheeler KT (1999). Effect of ploidy, recruitment, environmental factors, and tamoxifen treatment on the expression of sigma-2 receptors in proliferating and quiescent tumour cells. British journal of cancer, 81, 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahtavakoli M, & Jarrott B. (2011). Sigma-1 receptor ligand PRE-084 reduced infarct volume, neurological deficits, pro-inflammatory cytokines and enhanced anti-inflammatory cytokines after embolic stroke in rats. Brain Res Bull, 85, 219–224. [DOI] [PubMed] [Google Scholar]
- Alonso G, Phan V, Guillemain I, Saunier M, Legrand A, Anoal M, & Maurice T. (2000). Immunocytochemical localization of the sigma(1) receptor in the adult rat central nervous system. Neuroscience, 97, 155–170. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, & Katz DM (2000). Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci, 20, 7417–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen WD (2000). Sigma receptors: recent advances and new clinical potentials. Pharm Acta Helv, 74, 211–218. [DOI] [PubMed] [Google Scholar]
- Brown C, Fezoui M, Selig WM, Schwartz CE, & Ellis JL (2004). Antitussive activity of sigma-1 receptor agonists in the guinea-pig. Br J Pharmacol, 141, 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna J, Videla S, Argyriou AA, Velasco R, Villoria J, Santos C, Nadal C, Cavaletti G, Alberti P, Briani C, Kalofonos HP, Cortinovis D, Sust M, Vaque A, Klein T, & Plata-Salaman C. (2018). Efficacy of a Novel Sigma-1 Receptor Antagonist for Oxaliplatin-Induced Neuropathy: A Randomized, Double-Blind, Placebo-Controlled Phase IIa Clinical Trial. Neurotherapeutics, 15, 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha C, Brambilla R, & Thomas KL (2010). A simple role for BDNF in learning and memory? Front Mol Neurosci, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalwadi DA, Kim S, & Schetz JA (2017). Activation of the sigma-1 receptor by haloperidol metabolites facilitates brain-derived neurotrophic factor secretion from human astroglia. Neurochem Int, 105, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Li J, & Lee AS (2007). GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res, 67, 3734–3740. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Hayashi T, Urfer R, Mita S, & Su TP (2012). Sigma-1 receptor chaperones regulate the secretion of brain-derived neurotrophic factor. Synapse, 66, 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garces-Ramirez L, Green JL, Hiranita T, Kopajtic TA, Mereu M, Thomas AM, Mesangeau C, Narayanan S, McCurdy CR, Katz JL, & Tanda G. (2011). Sigma receptor agonists: receptor binding and effects on mesolimbic dopamine neurotransmission assessed by microdialysis. Biol Psychiatry, 69, 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekker G, Hu S, Sheng WS, Rock RB, Lokensgard JR, & Peterson PK (2006). Cocaine-induced HIV-1 expression in microglia involves sigma-1 receptors and transforming growth factor-beta1. Int Immunopharmacol, 6, 1029–1033. [DOI] [PubMed] [Google Scholar]
- Guitart X, Codony X, & Monroy X. (2004). Sigma receptors: biology and therapeutic potential. Psychopharmacology (Berl), 174, 301–319. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Scheffel U, & London ED (1995). In vivo labeling of sigma receptors in mouse brain with [3H]4-phenyl-1-(4-phenylbutyl)piperidine. Synapse, 20, 85–90. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Maurice T, & Su TP (2000). Ca(2+) signaling via sigma(1)-receptors: novel regulatory mechanism affecting intracellular Ca(2+) concentration. J Pharmacol Exp Ther, 293, 788–798. [PubMed] [Google Scholar]
- Hayashi T, & Su TP (2004). Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc Natl Acad Sci U S A, 101, 14949–14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, & Su TP (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell, 131, 596–610. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, & Kaplan DR (1992). Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron, 9, 883–896. [DOI] [PubMed] [Google Scholar]
- Hong W, Nuwayhid SJ, & Werling LL (2004). Modulation of bradykinin-induced calcium changes in SH-SY5Y cells by neurosteroids and sigma receptor ligands via a shared mechanism. Synapse, 54, 102–110. [DOI] [PubMed] [Google Scholar]
- Hornick JR, Vangveravong S, Spitzer D, Abate C, Berardi F, Goedegebuure P, Mach RH, & Hawkins WG (2012). Lysosomal membrane permeabilization is an early event in Sigma-2 receptor ligand mediated cell death in pancreatic cancer. J Exp Clin Cancer Res, 31, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YS, Lu HL, Zhang LJ, & Wu Z. (2014). Sigma-2 receptor ligands and their perspectives in cancer diagnosis and therapy. Med Res Rev, 34, 532–566. [DOI] [PubMed] [Google Scholar]
- Iniguez MA, Punzon C, Nieto R, Burgueno J, Vela JM, & Fresno M. (2013). Inhibitory effects of sigma-2 receptor agonists on T lymphocyte activation. Front Pharmacol, 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishima T, Fujita Y, & Hashimoto K. (2014). Interaction of new antidepressants with sigma-1 receptor chaperones and their potentiation of neurite outgrowth in PC12 cells. Eur J Pharmacol, 727, 167–173. [DOI] [PubMed] [Google Scholar]
- Ishima T, & Hashimoto K. (2012). Potentiation of nerve growth factor-induced neurite outgrowth in PC12 cells by ifenprodil: the role of sigma-1 and IP3 receptors. PLoS One, 7, e37989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishima T, Nishimura T, Iyo M, & Hashimoto K. (2008). Potentiation of nerve growth factor-induced neurite outgrowth in PC12 cells by donepezil: role of sigma-1 receptors and IP3 receptors. Prog Neuropsychopharmacol Biol Psychiatry, 32, 1656–1659. [DOI] [PubMed] [Google Scholar]
- Jiang G, Mysona B, Dun Y, Gnana-Prakasam JP, Pabla N, Li W, Dong Z, Ganapathy V, & Smith SB (2006). Expression, subcellular localization, and regulation of sigma receptor in retinal muller cells. Investigative ophthalmology & visual science, 47, 5576–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JL, Fang M, Zhao YX, & Liu XY (2015). Roles of sigma-1 receptors in Alzheimer’s disease. Int J Clin Exp Med, 8, 4808–4820. [PMC free article] [PubMed] [Google Scholar]
- Kikuchi-Utsumi K, & Nakaki T. (2008). Chronic treatment with a selective ligand for the sigma-1 receptor chaperone, SA4503, up-regulates BDNF protein levels in the rat hippocampus. Neurosci Lett, 440, 19–22. [DOI] [PubMed] [Google Scholar]
- Klette KL, DeCoster MA, Moreton JE, & Tortella FC (1995). Role of calcium in sigma-mediated neuroprotection in rat primary cortical neurons. Brain Res, 704, 31–41. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, & Bonhoeffer T. (1995). Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A, 92, 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Su TP, Fujimoto M, & Bonci A. (2012). The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci, 35, 762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IT, Chen S, & Schetz JA (2008). An unambiguous assay for the cloned human sigma1 receptor reveals high affinity interactions with dopamine D4 receptor selective compounds and a distinct structure-affinity relationship for butyrophenones. Eur J Pharmacol, 578, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, & Barde YA (1996). Physiology of the neurotrophins. Annu Rev Neurosci, 19, 289–317. [DOI] [PubMed] [Google Scholar]
- Luedtke RR, Perez E, Yang SH, Liu R, Vangveravong S, Tu Z, Mach RH, & Simpkins JW (2012). Neuroprotective effects of high affinity Sigma1 receptor selective compounds. Brain Res, 1441, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Rangel-Barajas C, Sumien N, Su C, Singh M, Chen Z, Huang RQ, Meunier J, Maurice T, Mach RH, & Luedtke RR (2015). The effects of sigma (sigma1) receptor-selective ligands on muscarinic receptor antagonist-induced cognitive deficits in mice. Br J Pharmacol, 172, 2519–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrazzo A, Caraci F, Salinaro ET, Su TP, Copani A, & Ronsisvalle G. (2005). Neuroprotective effects of sigma-1 receptor agonists against beta-amyloid-induced toxicity. Neuroreport, 16, 1223–1226. [DOI] [PubMed] [Google Scholar]
- Maurice T, Su TP, & Privat A. (1998). Sigma1 (sigma 1) receptor agonists and neurosteroids attenuate B25–35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience, 83, 413–428. [DOI] [PubMed] [Google Scholar]
- Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, & Leite MF (2005). The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem, 280, 40892–40900. [DOI] [PubMed] [Google Scholar]
- Monassier L, Manoury B, Bellocq C, Weissenburger J, Greney H, Zimmermann D, Ehrhardt JD, Jaillon P, Baro I, & Bousquet P. (2007). sigma(2)-receptor ligand-mediated inhibition of inwardly rectifying K(+) channels in the heart. J Pharmacol Exp Ther, 322, 341–350. [DOI] [PubMed] [Google Scholar]
- Niitsu T, Fujisaki M, Shiina A, Yoshida T, Hasegawa T, Kanahara N, Hashimoto T, Shiraishi T, Fukami G, Nakazato M, Shirayama Y, Hashimoto K, & Iyo M. (2012). A randomized, double-blind, placebo-controlled trial of fluvoxamine in patients with schizophrenia: a preliminary study. J Clin Psychopharmacol, 32, 593–601. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Ishima T, Iyo M, & Hashimoto K. (2008). Potentiation of nerve growth factor-induced neurite outgrowth by fluvoxamine: role of sigma-1 receptors, IP3 receptors and cellular signaling pathways. PLoS One, 3, e2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuyama S, & Nakazato A. (1996). NE-100: A novel sigma receptor antagonist. CNS Drug Reviews, 2, 226–237. [Google Scholar]
- Ostenfeld MS, Fehrenbacher N, Hoyer-Hansen M, Thomsen C, Farkas T, & Jaattela M. (2005). Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res, 65, 8975–8983. [DOI] [PubMed] [Google Scholar]
- Palacios G, Muro A, Vela JM, Molina-Holgado E, Guitart X, Ovalle S, & Zamanillo D. (2003). Immunohistochemical localization of the sigma1-receptor in oligodendrocytes in the rat central nervous system. Brain Res, 961, 92–99. [DOI] [PubMed] [Google Scholar]
- Palacios G, Muro A, Verdu E, Pumarola M, & Vela JM (2004). Immunohistochemical localization of the sigma1 receptor in Schwann cells of rat sciatic nerve. Brain Res, 1007, 65–70. [DOI] [PubMed] [Google Scholar]
- Peviani M, Salvaneschi E, Bontempi L, Petese A, Manzo A, Rossi D, Salmona M, Collina S, Bigini P, & Curti D. (2014). Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol Dis, 62, 218–232. [DOI] [PubMed] [Google Scholar]
- Robson MJ, Turner RC, Naser ZJ, McCurdy CR, O’Callaghan JP, Huber JD, & Matsumoto RR (2014). SN79, a sigma receptor antagonist, attenuates methamphetamine-induced astrogliosis through a blockade of OSMR/gp130 signaling and STAT3 phosphorylation. Exp Neurol, 254, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Munoz M, Onetti Y, Cortes-Montero E, Garzon J, & Sanchez-Blazquez P. (2018). Cannabidiol enhances morphine antinociception, diminishes NMDA-mediated seizures and reduces stroke damage via the sigma 1 receptor. Mol Brain, 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Pedrali A, Urbano M, Gaggeri R, Serra M, Fernandez L, Fernandez M, Caballero J, Ronsisvalle S, Prezzavento O, Schepmann D, Wuensch B, Peviani M, Curti D, Azzolina O, & Collina S. (2011). Identification of a potent and selective sigma(1) receptor agonist potentiating NGF-induced neurite outgrowth in PC12 cells. Bioorg Med Chem, 19, 6210–6224. [DOI] [PubMed] [Google Scholar]
- Ruscher K, Inacio AR, Valind K, Rowshan Ravan A, Kuric E, & Wieloch T. (2012). Effects of the sigma-1 receptor agonist 1-(3,4-dimethoxyphenethyl)-4-(3-phenylpropyl)piperazine dihydro-chloride on inflammation after stroke. PLoS One, 7, e45118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscher K, Shamloo M, Rickhag M, Ladunga I, Soriano L, Gisselsson L, Toresson H, Ruslim-Litrus L, Oksenberg D, Urfer R, Johansson BB, Nikolich K, & Wieloch T. (2011). The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke. Brain : a journal of neurology, 134, 732–746. [DOI] [PubMed] [Google Scholar]
- Ryskamp DA, Korban S, Zhemkov V, Kraskovskaya N, & Bezprozvanny I. (2019). Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front Neurosci, 13, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Kawamata T, Kobayashi T, & Okada Y. (2014). Antidepressant fluvoxamine reduces cerebral infarct volume and ameliorates sensorimotor dysfunction in experimental stroke. Neuroreport, 25, 731–736. [DOI] [PubMed] [Google Scholar]
- Schetz JA, Perez E, Liu R, Chen S, Lee I, & Simpkins JW (2007). A prototypical Sigma-1 receptor antagonist protects against brain ischemia. Brain Res, 1181, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, & Dirsch VM (2009). Modulation of endothelial nitric oxide by plant-derived products. Nitric Oxide, 21, 77–91. [DOI] [PubMed] [Google Scholar]
- Schneider LS, Thomas RG, Hendrix S, Rissman RA, Brewer JB, Salmon DP, Oltersdorf T, Okuda T, Feldman HH, & Alzheimer’s Disease Cooperative Study, T. S. G. (2019). Safety and Efficacy of Edonerpic Maleate for Patients With Mild to Moderate Alzheimer Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. [DOI] [PMC free article] [PubMed]
- Searovic P, Alonso M, Oses C, Pereira-Flores K, Velarde V, & Saez CG (2009). Effect of tamoxifen and retinoic acid on bradykinin induced proliferation in MCF-7 cells. J Cell Biochem, 106, 473–481. [DOI] [PubMed] [Google Scholar]
- Shen YC, Wang YH, Chou YC, Liou KT, Yen JC, Wang WY, & Liao JF (2008). Dimemorfan protects rats against ischemic stroke through activation of sigma-1 receptor-mediated mechanisms by decreasing glutamate accumulation. J Neurochem, 104, 558–572. [DOI] [PubMed] [Google Scholar]
- Su TP, Hayashi T, Maurice T, Buch S, & Ruoho AE (2010). The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci, 31, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi M, Hayashi T, & Su TP (2002). Nerve growth factor-induced neurite sprouting in PC12 cells involves sigma-1 receptors: implications for antidepressants. J Pharmacol Exp Ther, 303, 1227–1237. [DOI] [PubMed] [Google Scholar]
- Urfer R, Moebius HJ, Skoloudik D, Santamarina E, Sato W, Mita S, Muir KW, & Cutamesine Stroke Recovery Study, G. (2014). Phase II trial of the Sigma-1 receptor agonist cutamesine (SA4503) for recovery enhancement after acute ischemic stroke. Stroke, 45, 3304–3310. [DOI] [PubMed] [Google Scholar]
- Villard V, Espallergues J, Keller E, Alkam T, Nitta A, Yamada K, Nabeshima T, Vamvakides A, & Maurice T. (2009). Antiamnesic and neuroprotective effects of the aminotetrahydrofuran derivative ANAVEX1–41 against amyloid beta(25–35)-induced toxicity in mice. Neuropsychopharmacology, 34, 1552–1566. [DOI] [PubMed] [Google Scholar]
- Vilner BJ, de Costa BR, & Bowen WD (1995). Cytotoxic effects of sigma ligands: sigma receptor-mediated alterations in cellular morphology and viability. J Neurosci, 15, 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilner BJ, John CS, & Bowen WD (1995). Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res, 55, 408–413. [PubMed] [Google Scholar]
- Whittemore ER, Ilyin VI, & Woodward RM (1997). Antagonism of N-methyl-Daspartate receptors by sigma site ligands: potency, subtype-selectivity and mechanisms of inhibition. J Pharmacol Exp Ther, 282, 326–338. [PubMed] [Google Scholar]
- Wu Z, & Bowen WD (2008). Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: constitutive enhancement of calcium signaling in MCF-7 tumor cells. J Biol Chem, 283, 28198–28215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Rothfuss J, Zhang J, Chu W, Vangveravong S, Tu Z, Pan F, Chang KC, Hotchkiss R, & Mach RH (2012). Sigma-2 ligands induce tumour cell death by multiple signalling pathways. British journal of cancer, 106, 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Rothfuss JM, Zhang J, Vangveravong S, Chu W, Li S, Tu Z, Xu J, & Mach RH (2014). Functional assays to define agonists and antagonists of the sigma-2 receptor. Anal Biochem, 448, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Vangveravong S, McDunn JE, Hawkins WG, & Mach RH (2013). Sigma-2 receptor ligand as a novel method for delivering a SMAC mimetic drug for treating ovarian cancer. British journal of cancer, 109, 2368–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P. (2009). Neuroactive steroid regulation of neurotransmitter release in the CNS: action, mechanism and possible significance. Prog Neurobiol, 89, 134–152. [DOI] [PubMed] [Google Scholar]
- Zou LB, Yamada K, Sasa M, Nakata Y, & Nabeshima T. (2000). Effects of sigma(1) receptor agonist SA4503 and neuroactive steroids on performance in a radial arm maze task in rats. Neuropharmacology, 39, 1617–1627. [DOI] [PubMed] [Google Scholar]