

1. Introduction
Opioids have long been the standard of care for moderate-to-severe acute pain [44] and have also been used to temper chronic pain symptoms [36]. However, given their high abuse liability and other deleterious side effects, there is an urgent need for alternatives. The voltage gated sodium channel isoform Nav1.7 is highly expressed on nociceptive sensory afferents [4] and critically involved in transmission of pain signals from the site of injury to the spinal cord [16,20]. The role of Nav1.7 in pain is validated by human genetics as loss-of-function mutations result in a congenital insensitivity to pain [12], an effect that has been recapitulated in rodents [21,22], while gain-of-function mutations cause extreme pain syndromes [13,17,52].
Voltage-gated sodium channel isoforms (Nav1.x) share a common topology, with 4 homologous domains, each with 6 transmembrane alpha helices [6]. The nine mammalian isoforms (Nav1.1–1.9) have high homology at known small molecule binding sites, making selective inhibition of a single isoform challenging. Several small molecule Nav1.7 inhibitors have successfully achieved selectivity by targeting the voltage sensor in domain IV (VSD IV). These compounds bind to and stabilize the inactivated state of the channel [1]. Potency is significantly enhanced when assayed in physiological protocols that bias the channel towards the inactivated state [29]. While these compounds have shown promising efficacy in preclinical studies, none have yet shown consistent efficacy in clinical trials [35].
The natural guanidinium toxins saxitoxin (STX) and tetrodotoxin (TTX) bind to voltage-gated sodium channels at a site comprised of the domain I–IV S5–S6 extracellular pore loops. Selective inhibition of NaV1.7 can be achieved by taking advantage of a two amino acid sequence motif in the domain III pore loop, T1398/I1399, which is specific to primate Nav1.7 [38,47]. Due to the presence of this motif, STX is >100X less potent against human Nav1.7 (hNav1.7) compared to other TTX-sensitive Nav isoforms (Nav1.1-1.4, Nav1.6) [48]. Through a series of chemical modifications, we identified ST-2262, a STX analog that is highly selective for Nav1.7 over the other human Nav1.x isoforms, and is equipotent in protocols that favor the resting and inactivated states [38]. While a promising candidate for therapeutic development, ST-2262 has insufficient potency against mouse or rat Nav1.7 to conduct a comprehensive pain behavior evaluation in rodents.
Further exploration of guanidinium toxins as selective inhibitors of Nav1.7 has led to the discovery of ST-2530, a fully synthetic analogue of STX that does not lose the same degree of potency against rodent Nav1.7 as earlier compounds. This finding has allowed us to evaluate the pharmacodynamic consequences of selective inhibition of Nav1.7 in a range of mouse pain behavior models and across different modalities, including aversive thermal, mechanical and chemical stimuli. ST-2530 was broadly efficacious in acute, postoperative incisional, and neuropathic pain models at doses that had no effect on general behavioral assays or olfaction, a potential liability of Nav1.7 inhibition [49]. These studies indicate that selective, pharmacological inhibition of Nav1.7 at the channel pore may be an effective strategy to reduce pain sensation.
2. Methods
2.1. Animals
C57Bl/6 mice were housed in a vivarium at Montana State University (Bozeman, MT) , University of New England (Biddeford, ME), or Murigenics (Vallejo, CA) in standard polycarbonate cages at up to 4 per cage with same-sex littermates and maintained on a 12h light/dark cycle with ad libitum access to food and water. Mice housed at Montana State University and University of New England were fed PicoLab Rodent Diet 20 (Purina LabDiet, St. Louis, MO) and 2018 Teklad Global 18% Protein Rodent Diet (Envigo, Indianapolis, IN), respectively, both of which contain soy. All experimental procedures were approved by an Institutional Animal Care and Use Committee in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals and conducted at an AAALAC-accredited facility.
2.2. Cell Culture
Whole cell recordings were carried out on human embryonic kidney 293 (HEK) or Chinese Hamster Ovary (CHO) cells that stably expressed one human NaV1.x isoform. The following cell lines were utilized: NaV1.1 – CHO, division arrested, purchased from Charles River Laboratories (CRL, ChanTest, Cleveland, OH), Catalog #CT4178; NaV1.2 – CHO, division arrested, CRL, Catalog #CT4010; NaV1.3 – CHO, division arrested, CRL, Catalog #CT4157; NaV1.4 – HEK, shared by academic resource [34]; NaV1.5 – HEK, SB Drug Discovery (Glasgow, UK), Catalog #SB-HEK-hNaV1.5; NaV1.6 - HEK, SB Drug Discovery. Catalog #SB-HEK-hNaV1.6; NaV1.7 - HEK, SB Drug Discovery. Catalog #SB-HEK-hNaV1.7; NaV1.8/β1 – HEK, Eurofins Discovery Services (St. Charles, MO), Catalog #CYL3025. Stably transfected competent cells were maintained at 37°C in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher, Waltham MA) supplemented with 10% fetal bovine serum (Thermo Fisher), 100 U/mL penicillin G sodium, 100 μg/mL streptomycin sulfate (VW, Radnor, PA), 500 μg/mL G418 (Thermo Fisher), and blasticidin S hydrochloride (for cell lines from SB Ion Channels; VWR). Division arrested cells were maintained at 37°C for 2–3 days in Ham’s Nutrient Mixture F-12 (VWR) media supplemented with 10% fetal bovine serum, 100 U/mL penicillin G sodium, 100 μg/mL streptomycin sulfate.
For recordings with mouse NaV1.7, CHO cells were transiently transfected with plasmids encoding the corresponding genes. The following transgenes were utilized: Scn9a Mus musculus – TransOMIC (Huntsville, AL), Clone #BC172147; pcDNA3.1(+)IRES GFP – Addgene (Watertown, MA), Catalog #51406. CHO cells were plated into a 96-well plate at 80–90% confluency. The cells were rinsed in phosphate-buffered saline (PBS) followed by the addition of opti-MEM media (Thermo Fisher) and the transfection mix with reagents from the Lipofectamine 3000 kit (Thermo Fisher). Transfection mix contained the plasmid encoding the gene of interest along with the plasmid encoding green fluorescent protein (GFP). Cells were incubated for 3 days to allow the heterologous expression. GFP expression was used to identify transfected cells on the day of the experiment.
2.3. Manual Patch Clamp Electrophysiology
On the day of testing, cells were rinsed with PBS, lifted from the plate with Detachin (VWR), resuspended in fresh media, and 30 μl were plated onto 5 mm glass cover slips (Bellco Glass, Vineland, NJ). After cells adherence, the cover slip was transferred to the testing chamber and continuously superfused with extracellular solution, which contained (in mM): NaCl (135), KCl (4.5) CaCl2 (2), MgCl2 (1), HEPES (10); pH 7.4. Cells were patch clamped with borosilicate glass pipettes pulled to a tip diameter yielding a resistance of 1.0–2.0 MΩ, and filled with an internal solution that contained (in mM): CsF (125), NaCl (10), EGTA (10), HEPES (10), pH 7.2. In some experiments ST-2530 was dissolved in DMSO in a 10 mM stock and then subsequently was serially diluted in the extracellular solution. In other experiments, ST-2530 was dissolved directly in extracellular solution and serially diluted. Alone, the maximum DMSO concentration of 1% had no effect on sodium currents (data not shown).
Channel currents were measured using whole-cell patch-clamp electrophysiology with either a HEKA EPC 9 amplifier with built-in ITC-16 interface (HEKA Elektronik Dr. Schulze GmbH, Germany) or Axon CNS Multiclamp 700B amplifier with Axon Digidata 1550A data acquisition system (Molecular Devices, San Jose, CA). The output was filtered with a low-pass, four pole Bessel filter with a cutoff frequency of 10 kHz and was sampled at 20–50 kHz. Pulse stimulation and data acquisition were controlled with Pulse software (v8.40; HEKA Elektronik) or with the pClamp Clampex data acquisition module (v11.0; Molecular Devices). All measurements were performed at room temperature (20–22°C). Recordings began at least 5 min after establishing the whole-cell and voltage-clamp configuration. Cells with access resistance < 5 MΩ were used for the study. Series resistance compensation circuit was turned on and set at 80% and 100 μsec.
Cells were held at −110 mV, and the voltage dependence of activation (Vact) was determined with a series of voltage steps. The voltage step that elicited the maximal current (−20 to +10 mV depending on the isoform) was subsequently used to evoke a channel current from its resting state. Cells were hyperpolarized to -140 mV for 105 ms to deactivate all channels, and then currents were evoked from the resting state every 3 seconds with a depolarizing step to the voltage of maximal activation for 10 ms.
Once the baseline evoked current was stable in amplitude, ST-2530 was washed onto the cell. Following stable inhibition, a half-log higher concentration was perfused onto the cell. Between 1 and 5 concentrations were washed onto each cell, with 100 μM being the maximum concentration tested. Drug was then washed out until the cell returned to a stable current. Saxitoxin (Millipore Sigma, Burlington, MA) or tetrodotoxin (Abcam, Cambridge, MA) was used as a positive control for all NaV1.x isoforms except Nav1.8; a single concentration near the IC50 for each isoform was tested either prior to or following ST-2530 perfusion or washout. For Nav1.8 recordings, tetrodotoxin (1 μM) was added to the extracellular solutions to inhibit any tetrodotoxin-sensitive Nav isoforms potentially expressed as background in the cell line.
Recorded peak current data were baseline-normalized. In some cells, the steady state current following test article washout differed from the initial baseline current. For these recordings, drift correction was applied to account for the change in baseline by fitting the initial baseline current and washout steady state current to a linear equation. The steady state current before and after test article application was used to calculate the percentage of current inhibited at each concentration. Percent inhibition as a function of compound concentration was pooled from all recorded cells and the resulting data set was fit to a four-parameter logistic curve using least squares regression in GraphPad Prism, version 7 (San Diego, CA) to produce a single IC50 curve and asymmetrical (likelihood) 95% confidence interval. If less than 50% inhibitory response was observed at the highest concentration tested, the IC50 was reported to be greater than this concentration.
Association and dissociation kinetics were estimated using the Solver add-in for Excel (Microsoft, Redmond, WA). The association constant (Kon in min−1M−1) and dissociation constant (Koff in min−1) were estimated by minimizing the sum of squared errors between the recorded peak current values and the following kinetic model [28]:
where I(t) is the estimated peak current at time t (min), I(t−1) is the estimated peak current at the previous time point, I(t0) is the baseline peak current before addition of the drug, C is the drug concentration (M). The kinetic model relies on the following assumptions: i) drug association is a pseudo-first order process, dependent on the concentrations of the free drug and of the unbound ion channel, which is represented by the amplitude of the peak current, and ii) drug dissociation is a first order process, dependent on the concentration of the drug–ion channel complex, which is represented by the inhibited current. Kd was then determined by dividing Koff by Kon. Pore blockers of ion channels are generally non-competitive inhibitors of ion flux, and therefore %inhibition of current accurately reflects ligand binding. A similar approach has been used to estimate the association and dissociation rates of STX and TTX against variants of rat NaV1.4 [39]. In this study, Kd estimates from kinetics closely matched equilibrium IC50. For msNav1.7 recordings, IC50 and Kd were determined in most cells. In 2 recordings, the washout period was not sufficient to calculate the Koff; in these cases only IC50 was determined.
2.4. Behavioral Assays
Male C57Bl/6 mice aged 6-15 weeks old were used for all experiments. All behavioral assessments were performed in lights-on conditions and the investigator was blinded as to treatment group with the exception of the tail flick experiment and the Hargreaves experiment with PF-05089771. ST-2530 was administered by subcutaneous (SC) injection to the nape of the neck. PF-05089771 was administered by oral gavage (PO). Group sizes were 7-12 animals per condition. Studies were conducted at Montana State University except where noted.
2.4.1. Thermal Plantar Test (Hargreaves)
This model was performed as previously described [23]. Mice were placed in a small transparent acrylic enclosure on a glass floor warmed to 30°C (IITC Life Science, Woodland Hills, CA). Prior to thermal stimulation, mice were allowed at least 45 minutes in the enclosure for acclimation. The hind paw was stimulated with a focused, radiant heat light source until animals withdrew the paw away from the thermal stimulus. For each testing time point, there were two stimuli, one per hind paw, that were averaged together to determine the withdrawal latency. A 20 second cutoff time was used to prevent tissue damage. For the study assessing multiple doses of ST-2530, the experiment was conducted using a within-subjects design with at least 3 days between test sessions, with dosing order varied per animal. For the study examining the interaction between ST-2530 and naloxone, a between-subjects design was used.
2.4.2. Tail Immersion
This model was performed similar to previously described and conducted at the University of New England COBRE Behavioral Core [26]. Following a 30 minute acclimation period in the test room, mice were grasped by the nape of the neck, the distal 1/3 of the tail was immersed into a water bath warmed to 52°C, and the latency for the mouse to flick the tail out of water was measured. A 10 seconds cut-off was used to prevent tissue damage.
2.4.3. Tail Pinch
This model was performed as previously described [20]. Mice were placed in a novel cage with bedding. The test article, ST-2530, or vehicle was administered SC. After 30 minutes, a 1.5-inch bulldog clamp that supplies 500–900 grams pressure (Roboz, RS-7440-35, Gaithersburg, MD) was placed on the proximal end of an unrestrained mouse’s tail. The latency to bring the nose within 1 cm of the clamp was measured. To avoid tissue damage, the maximum trial time was 15 seconds.
2.4.4. Formalin Test
This study was performed similar to previously described and conducted at the University of New England COBRE Behavioral Core [25]. 10% Neutral Buffered Formalin, 200 grade, was purchased from Sigma (St. Louis, MO) and diluted prior to use. Mice were allowed to acclimate to the test room for 30 minutes. Subjects were administered test compound SC, and then 15 minutes later were placed into the novel test chamber. Following 15 minutes of acclimation in the chamber, mice received 10 μl of 2.5% formalin into the dorsal surface of the left hind paw. Subjects were videotaped for 45 minutes in the chamber and time spent licking or biting the affected paw was measured. Data was recorded in 5 minute bins.
2.4.5. Incisional Pain Assay
This study was performed as previously described [41]. Briefly, under isoflurane anesthesia and following sterile preparation, a 5 mm incision was made with a No. 11 scalpel blade through the skin and fascia on the plantar surface of the left hind paw, starting around 2 mm from the heel and extending towards the toes. The underlying muscle was slightly pulled with curved forceps, and then the skin was sutured using a single 6-0 nylon suture and topical antibiotic ointment was applied. Mice were assessed on the Hargreaves assay on both hind paws prior to the surgical procedure and beginning 48 h following the procedure for 5 consecutive days. At each test time point, there were 2 stimuli per paw that were averaged together, with an intra stimulus interval of at least 2 minutes.
2.4.6. Spared Nerve Injury
This model was performed as previously described [40]. Briefly, under isoflurane anesthesia the left thigh was shaved and after sterile preparation, the skin and the underlying biceps femoris muscle was incised and the sciatic nerve at its trifurcation point was exposed. The tibial and common peroneal nerves were ligated using 8-0 silk and then severed, leaving the sural nerve intact. The muscle and skin were sutured using 6-0 polyglycolic acid and 5-0 nylon sutures, respectively, and then topical antibiotic was applied.
To assess mechanical sensitivity, paw withdrawal threshold to Von Frey monofilaments (0.04, 0.07, 0.16, 0.4, 0.6, 1, 1.4, 2, and 4 gram filaments) was determined using the modified up-down method [7]. Prior to testing, subjects were habituated on the mesh wire floor for at least 45 minutes. Mechanical withdrawal testing was conducted prior to and 10 days following the SNI procedure to determine baseline withdrawal thresholds before and after injury. The mechanical withdrawal threshold of the contralateral paw was assessed as a control for nerve injury. On days 14, 18, 22 and 26 post-SNI procedure, withdrawal threshold was assessed before and after dosing. On all testing days and at each time point, the mechanical withdrawal threshold was determined for each paw.
2.4.7. Open arena assessment
This model was performed similar to previously described [27]. Locomotor activity and rearing were measured in a 45 X 45 cm open field with a grid of 16 X 16 infrared beams, spaced 2.5 cm apart (Panlab, Harvard Apparatus, Barcelona, Spain). A lower and upper frame of infrared beams tracked horizontal movement (spontaneous locomotion) and vertical movement (rearing), respectively. Subjects were administered ST-2530 or vehicle SC, placed into the open arena after 30 minutes and movement was tracked for 5 minutes. For each dose tested, the experiment was conducted using a crossover-design in which half of the subjects received active compound and half received vehicle on test session 1. Treatment was reversed for test session 2.
2.4.8. Rotarod
The experiment was conducted as previously described [45]. Motor coordination was assessed using a rotarod (IITC Life Science) that accelerated from 4 to 40 RPM over 5 minutes. Subjects had one training session on the rotarod for 5 minutes at a constant speed of 5 RPM. On the following day, subjects received an SC dose of ST-2530 or vehicle and then 30 minutes later were challenged to the accelerating rotarod. A single trial was used in an effort to capture the effect of the drug on motor coordination at time point near maximum plasma whole blood exposure.
2.4.9. Olfaction
The experiment was conducted as previously described [51]. Subjects were fasted overnight and the following day received vehicle or ST-2530 SC 30 minutes prior to the buried food test. The latency to find a buried palatable food pellet (piña colada mini supreme treat, Bio-Serv) was determined with a maximum test time of 15 minutes.
2.5. Pharmacokinetic Study
The in-life portion of this study was conducted at Murigenics (Vallejo, CA). Mice received 3 mg/kg ST-2530 SC. Blood draws (100 μl via retro-orbital puncture) were taken up to 2 times per mouse and there were 4 mice per time point (pre-dose, 5, 15, 30, 60 120 min), for an n=12. Whole blood was collected into K2-EDTA coated vials that contained 10 mM ammonium acetate buffer (1:1 v/v).
Whole blood samples were analyzed by Quintara Biosciences (Hayward, CA). A 20 μl aliquot of the blood/buffer sample was treated with 100 μl acetonitrile containing internal standard (verapamil). The mixture was vortexed on a shaker for 15 min and then centrifuged at 4000 rpm for 15 min. An aliquot of 70 μl of the supernatant was mixed with 70 μl water and injected for LC-MS/MS analysis. Samples were analyzed on a Qtrap 6500+ triple quadrupole mass spectrometer (Sciex, Framingham, MA) using a Turbo Spray IonDrive electrospray ion source equipped with an ExionLC system. Chromatographic separation was performed on an Acquity UPLC BEH C18, 50 X 2.1 mm, 1.7 μm column (Waters, Milford, MA). A gradient method consisting of mobile phase A (water with 0.1% formic acid) and mobile phase B (acetonitrile with 0.1% formic acid) was initiated at 15% B, increased to 95% B from 0.2 to 1.2 minutes, and remained at 95% through 1.7 minutes. The flow rate was 0.5 ml/min. Calibration standards and quality control samples were prepared by adding 2 μl of the test compound standard solution to 18 μl of blank 1:1 whole blood:buffer matrix and were processed with the unknown samples in the same batch.
2.6. Protein Binding Assay
This study was conducted at Quintara Biosciences (Hayward, CA). Rapid equilibrium dialysis (RED) device inserts along with a Teflon base plate (Pierce, Rockford, IL) were used for binding studies. Mouse whole blood was obtained from BioIVT (Westbury, NY) and diluted with an equal volume of PBS, pH 7.4, for ease of handling. DMSO stock (1 mM) of ST-2530 was spiked into whole blood to a concentration of 1 μM. An aliquot (100 μL) was transferred to into the sample chamber and PBS buffer, pH 7.4, was placed into the adjacent chamber. The plate was sealed with a self-adhesive lid and incubated at 37°C on an orbital shaker (250 rpm) for 4 hours. After 4 hours, aliquots were taken from both the sample and buffer chamber. An equal volume of buffer was added to the diluted blood samples and diluted blood was added to the buffer samples. The resulting samples were analyzed by LC-MS/MS.
2.7. Drugs
ST-2530 was prepared by SiteOne Therapeutics by methods similar to those previously described [38] (Figure 1A). For in vivo studies, ST-2530 was dissolved in 4.5% mannitol/10 mM sodium acetate, pH 5.6 and sterile filtered, and was administered SC at a volume of 10 ml/kg. Naloxone HCl (Patterson Veterinary Supply, Devens, MA) was purchased as a 0.4 mg/ml solution in saline, diluted to 0.2 mg/ml in sterile saline, and administered IP at a volume of 10 ml/kg. PF-05089771 (Alomone Labs, Jerusalem, Israel) was dissolved in 30% 2-hydroxypropyl-beta-cyclodextrin (HPBCD), pH 10 with KOH, and administered PO at a volume of 10 ml/kg.
Figure 1.
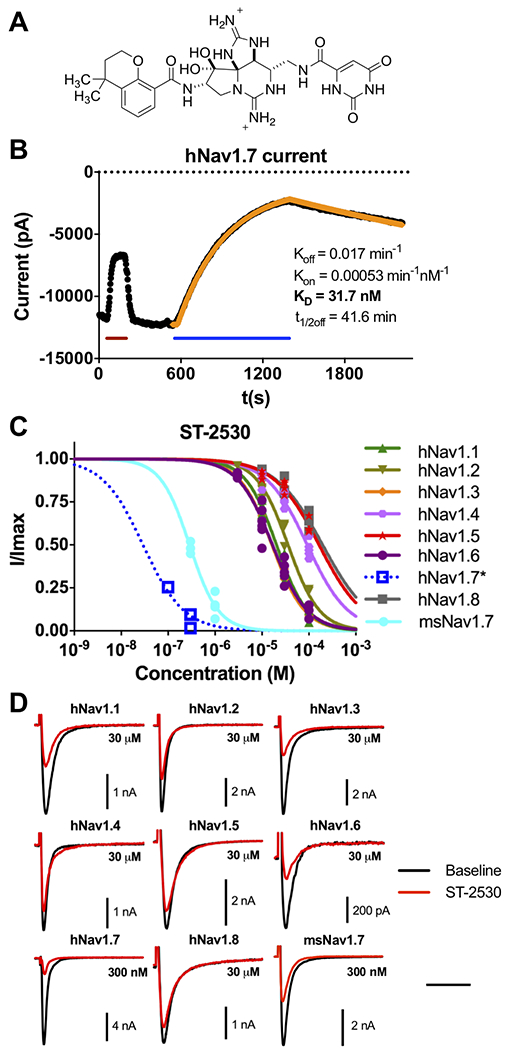
Electrophysiological characterization of ST-2530 against Nav isoforms. (A) Chemical structure of ST-2530. (B) Representative current trace from a recording with ST-2530 against hNav1.7, showing recorded peak current values (black) and estimated association and dissociation kinetics (yellow). During recording STX (200 nM) was perfused onto the cell (red line) as a positive control, and following washout ST-2530 (300 nM) was perfused (blue line). Koff and Kon were estimated by fitting first-order and second-order rate constants to the observed washout and wash-in kinetics, respectively. Kd was determined from their ratio. (C) Dose-response relationship of ST-2530 against hNav1.1–1.8 and msNav1.7. *hNav1.7 association kinetics were slow and consequently steady state inhibition was not achieved. I/Imax was estimated (blue open squares) by modeling association and dissociation kinetics, and determining the asymptote of the numerical model. A dose-response curve (dotted blue line) was fit with Hill coefficient equal to 1. (D) Representative waveforms of hNav1.1–1.8 and msNav1.7 at baseline (black) and following exposure to ST-2530 (red; 300 nM for hNav1.7 and msNav1.7, 30 μM for all other isoforms). Time scale – 5 ms for all isoforms except hNav1.8 (10 ms).
2.8. Statistical Analysis
Statistical comparisons were run using GraphPad Prism, version 7. In the open arena assessment, a within-subjects design was used to compare vehicle to ST-2530, and significance was determined with a paired-t-test. In the rotarod, buried food test, and tail pinch, data were analyzed with a one-way ANOVA, with Dunnett’s post-hoc multiple comparison test to compare dose groups to vehicle. For the Hargreaves assay with ST-2530, a within-subjects design was used and significance was determined with a two-way repeated measure ANOVA, with Dunnett’s multiple comparison test to compare latency at time points post dose to latency at baseline within the same dose group. In the Hargreaves assay with naloxone/saline dosing, a two-way ANOVA with Bonferroni multiple comparison was used. For the Hargreaves with PF-05089771, tail flick, and formalin assays, significance was assessed with a mixed ANOVA with Dunnett’s test, comparing post-dose time points to baseline in the tail flick and Hargreaves, and dose groups to vehicle in the formalin test. In the incision and SNI studies, to compare effect on the injured paw, a two-way repeated measures was used with Bonferroni multiple comparison test. To assess effect of ST-2530 on each test session, a mixed ANOVA with Dunnett’s test was used, comparing post-dose time points to baseline values within the same dose group. Data are expressed ± SEM with the exception of IC50 values, where variability is represented by the 95% CI. In statistical assessments, the α value was set to 0.05.
3. Results
3.1. ST-2530 is a potent and selective Nav1.7 inhibitor
The potency of ST-2530 against Nav1.7 and selectivity over off-target sodium channel isoforms was assessed with whole cell patch clamp electrophysiology on recombinant cell lines stably or transiently expressing the channel of interest. ST-2530 was evaluated with a resting state protocol in which there was a single depolarizing step from a hyperpolarized resting membrane potential to the voltage of max activation for each channel, ranging from −20 to +10 mV. This protocol contrasts with voltage clamp protocols frequently used to evaluate other selective Nav1.7 inhibitors, such as the arylsulfonamide class, in which a conditioning pre-pulse is incorporated to bias the channel toward the inactivated state [29]. Against human Nav1.7 (hNav1.7), ST-2530 had slow association and dissociation kinetics (Figure 1B), making accurate determination of IC50 challenging due to the long wash-in time required to achieve steady state inhibition. Therefore, for hNav1.7 recordings, association and dissociation rate constants were estimated by fitting a curve to recorded peak value currents following a single ST-2530 concentration and washout, respectively, an analysis that has been used for acylsulfonamide Nav1.7 inhibitors [3]. With this approach, the Kd was determined to be 25 ± 7 nM (n=4, Table 1). The dissociation rate of ST-2530 extrapolates to a ligand/protein half-life (t1/2) of 63 ± 16 min. Against mouse Nav1.7 (msNav1.7), the dissociation and association rates were faster than against the human channel, with the t1/2 off of 7.6 ± 0.9 min. The Kd at msNav1.7 was greater than at the human channel (250 ± 40 nM, n=5, Table 1). Since binding kinetics against msNav1.7 were faster, steady state inhibition and consequently accurate determination of IC50 was feasible. The IC50 was 290 nM (95% CI 220–340, n=7), Table 2, Figure 1C), which is in close alignment with the Kd value.
Table 1:
ST-2530 Kd values for hNav1.7 and msNav1.7
| Isoform (# cells) | Kd (nM) | Koff (min−1) | Kon (min−1nM−1) | t1/2off (min) |
|---|---|---|---|---|
| hNav1.7 (4) | 25 ± 7 | 0.013 ± 0.002 | 0.00068 ± 0.00022 | 63 ± 16 |
| msNav1.7 (5) | 250 ± 40 | 0.097 ± 0.013 | 0.00041 ± 0.00003 | 7.6 ± 0.9 |
Values are mean ± SEM
Table 2:
ST-2530 IC50s against Nav1.1-1.6, 1.8 and msNav1.7
| Isoform | IC50 (μM) | 95% CI | # of cells |
|---|---|---|---|
| hNav1.1 | 20 | 18–23 | 6 |
| hNav1.2 | 36 | 31–42 | 5 |
| hNav1.3 | 16 | 15–17 | 5 |
| hNav1.4 | 92 | 80–110 | 6 |
| hNav1.5 | >100 | 5 | |
| hNav1.6 | 17 | 14–20 | 6 |
| hNav1.8 | >100 | 4 | |
| msNav1.7 | 0.29 | 0.22–0.34 | 7 |
Note: 100 μM was highest concentration tested
ST-2530 was highly selective for hNav1.7 over all other human Nav isoforms tested, with >500X selectivity over Nav1.1 (20 μM, 95% CI 18–23, n=6), Nav1.3 (16 μM, 15–17, n=5) and Nav1.6 (17 μM, 14–20, n=6) and >1000X over the over tested Nav isoforms (Table 2, Figure 1C). All off-target isoforms had fast association and dissociation kinetics allowing for standard IC50 determination using multiple test concentrations. ST-2530 was also evaluated on a commercially available radioligand binding assay panel against 68 different rat and human protein targets, including μ-, δ-, and κ-opioid receptors, and L- and N-type voltage-gated calcium channels (LeadProfilingScreen, Eurofins, Taipei, Taiwan). ST-2530 (10 μM) did not produce >50% inhibition against any of the targets in the panel (Table S1). These results indicate that ST-2530 is a highly potent and selective human Nav1.7 inhibitor and while the test article is less potent against msNav1.7, the reduction in potency is less dramatic than other compounds in the series [38].
3.2. Effect of ST-2530 on locomotion and olfaction
ST-2530 was evaluated on a range of in vivo assays. Initially, the dose at which there were no confounding effects on general behavior was determined. Mouse spontaneous locomotion and rearing 30 minutes after SC dosing of ST-2530 was assessed in an open field arena. Behavior was evaluated for 5 minutes. Compared to vehicle control, 3 mg/kg ST-2530 did not significantly affect locomotor activity (paired t-test: t(11)=0.74, p=0.47, Figure 2A) or rearing (paired t-test: t(11)=0.49, p=0.63, Figure 2B), whereas 10 mg/kg significantly attenuated these behaviors (paired t-test: locomotion – t(7)=9.94, p<0.001; rearing – t(7)=9.09, p<0.001; Figure 2A,B). Furthermore, while 3 mg/kg ST-2530 did not impact motor coordination on the accelerating rotarod, 10 mg/kg reduced the latency to fall (one-way ANOVA: F(2,21)=6.32, p=0.007; Figure 2C). These assays suggest that 3 mg/kg, but not 10 mg/kg, does not perturb spontaneous activity or motor coordination.
Figure 2.
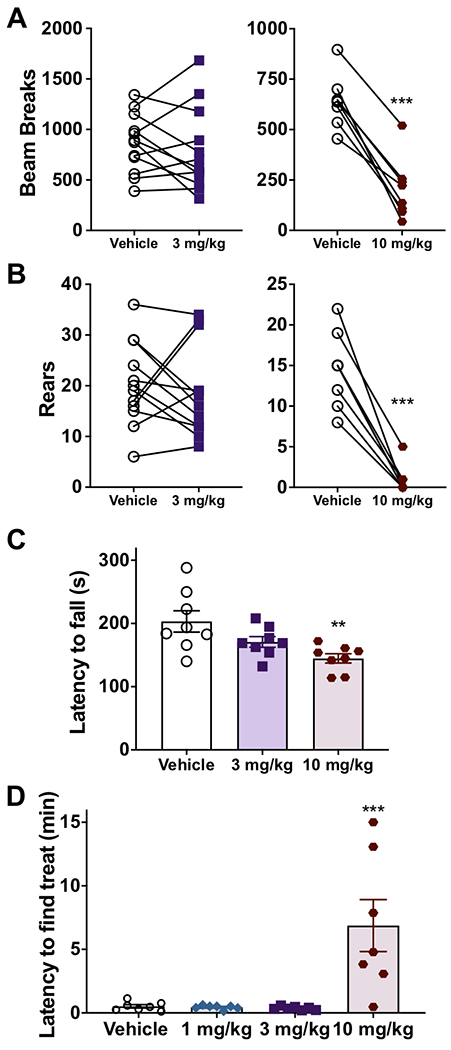
Effect of ST-2530 on locomotor behavior and olfaction. (A,B) Spontaneous locomotion, as quantified by number of beam breaks (A), and number of rears (B) 30 minutes after SC dosing of ST-2530 (3 mg/kg – purple square, n=12; 10 mg/kg – red circle, n=8) or vehicle (open circle). *** - p<0.001. (C) Latency (in seconds) to fall off a rotarod accelerating from 4 to 40 RPM over 5 minutes (n=8 per group). Dunnett’s multiple comparison test, comparing dose groups to vehicle: ** - p<0.01. (D) Latency (in minutes) to find a buried food treat in a novel cage (n=7 per group). Dunnett’s multiple comparison test, comparing dose group to vehicle: *** - p<0.001.
Loss of function mutations and genetic knockout of Nav1.7 in humans and mice, respectively, cause anosmia [21,49]. We therefore assessed whether ST-2530 produced anosmia in mice. ST-2530 or vehicle was dosed SC 30 minutes prior to the buried food test. While neither 1 nor 3 mg/kg ST-2530 altered subjects’ ability to find a buried treat, 10 mg/kg significantly increased the time required to find the hidden treat (one-way ANOVA: F(3,24)=9.90, p=0.002, Figure 2D). While motor deficits may confound the results of 10 mg/kg ST-2530 on the buried food test, these assays indicate that 3 mg/kg ST-2530 does not affect general mouse behavior.
3.3. ST-2530 reduces behavioral responses to aversive thermal, mechanical and chemical stimuli
ST-2530 was assessed on a number of mouse acute pain models with different stimulus modalities. Based on the results from the locomotor and olfaction assays (Figure 2), the highest dose tested was 3 mg/kg. ST-2530 was first evaluated on the Hargreaves assay in which the latency to remove a hind paw from an aversive thermal stimulus is measured. ST-2530 dose-dependently increased the withdrawal latency to the thermal stimulus, with a peak effect lasting from 15 to 60 minutes post-dose (two-way RM ANOVA: time – F(5,55)=7.19, p<0.001; dose – F(3,33)=19.5, p<0.001; time X dose interaction – F(15,165)=3.36, p<0.001; Figure 3A). As a comparator, we assessed PF-05089771, an arylsulfonamide Nav1.7 inhibitor that has a state-dependent IC50 against hNav1.7 of 11 nM and near equal potency against mNav1.7 [2], on the Hargreaves assay. Following PO administration, PF-05089771 at a dose of 100 mg/kg, but not 30 mg/kg, increased thermal withdrawal latency, an effect that persisted through the final testing time point of 120 minutes (mixed ANOVA: time – F(4,128)=4.27, p=0.003; dose – F(2,32)=7.45, p=0.002; time X dose interaction – F(8,128) = 2.30, p=0.025; Figure 3B). While efficacy can be achieved in the Hargreaves assay with PF-05089771, a much higher dose is required when compared to ST-2530.
Figure 3.
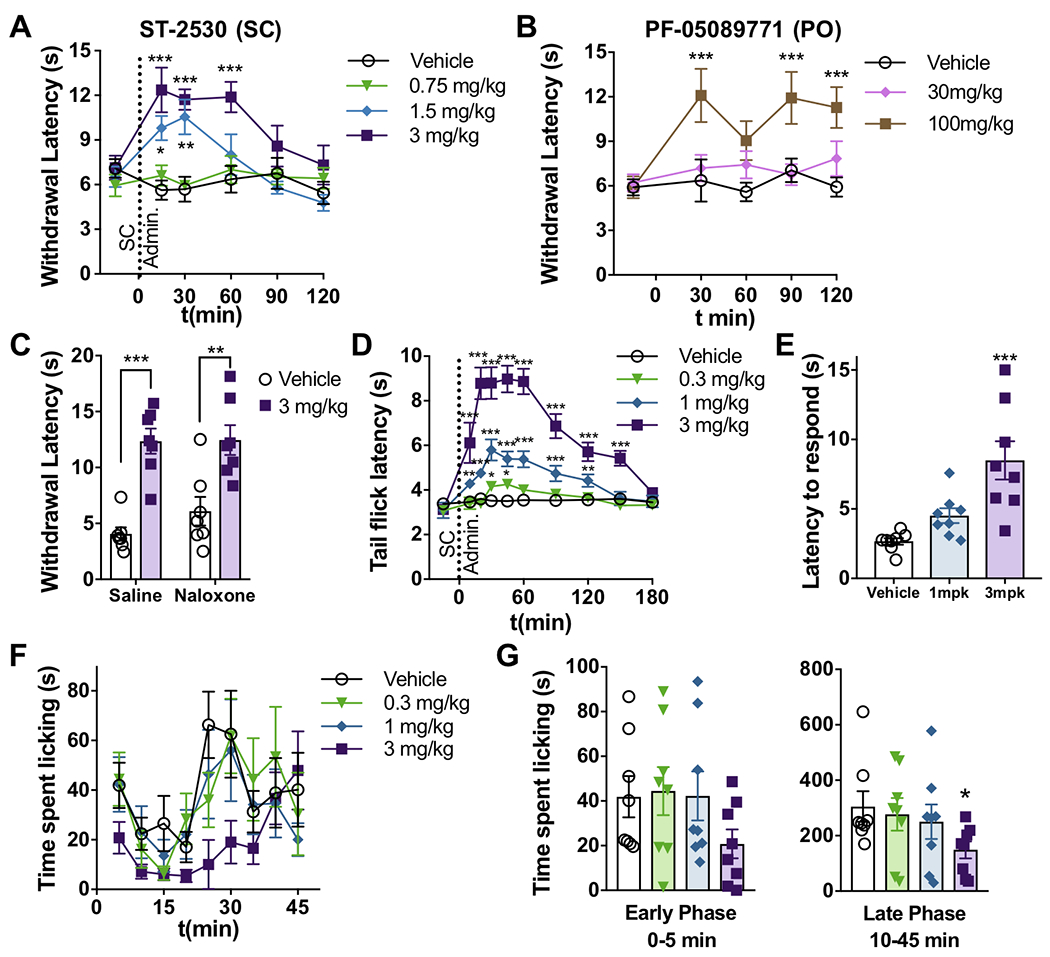
Effect of ST-2530 on acute pain assays. (A) Withdrawal latency (in seconds) to an aversive thermal stimulus on the hind paw (Hargreaves assay, n=12) before and after SC administration of ST-2530. Dunnett’s multiple comparison test, comparing post-dose timepoint to baseline value for each dose group: * - p<0.05, ** - p<0.01, *** - p<0.001. (B) Withdrawal latency in the Hargreaves assay (n=11-12) before and after PO dosing of PF-05089771. Dunnett’s: *** - p<0.001. (C) The Hargreaves assay 30 minutes after SC administration of ST-2530 (3 mg/kg) or vehicle (SC) and IP naloxone (2 mg/kg) or saline (IP) (n=7 per group). Bonferroni’s multiple comparison test: ** - p<0.01; *** - p<0.001. (D) Latency (in seconds) to flick tail out of 52°C water (n=8 per group). Dunnett’s, comparing post-dose timepoint to baseline value for each dose group: * - p<0.05, ** - p<0.01, ***- p<0.001. (E) Latency to bring nose within 0.5 cm of a bulldog clamp placed on the proximal end of the tail. Dunnett’s: *** - p<0.001. (F,G) Time (in seconds) spent licking hind paw following formalin injection, shown in 5 minute bins (F) or in the early (0–5 min) and late (10–45min) phases (G) (n=8 per group). Dunnett’s comparing dose groups to vehicle: * - p<0.05.
Previously, it has been shown that transgenic mice with loss of Nav1.7 expression in sensory neurons have higher expression of Met-enkephalin and that treatment with the mu opioid receptor antagonist naloxone reverses the phenotype of insensitivity to aversive thermal stimuli [33]. To test whether μ-opioid receptor activation is required for the analgesic effect of ST-2530, naloxone (2 mg/kg) or saline was administered IP along with 3 mg/kg ST-2530 or vehicle SC, 30 minutes prior to the Hargreaves assay. Naloxone had no effect on the increase in withdrawal latency to the thermal stimulus mediated by ST-2530 (Two-way ANOVA: ST-2530 – F(1,24)= 43.0, p<0.001; no naloxone effect or interaction; Figure 3C), indicating that analgesia in the Hargreaves assay is independent of μ-opioid activation.
The effect of ST-2530 was evaluated on the tail immersion assay. ST-2530 dose-dependently increased the latency to flick the tail out of 52°C water (mixed ANOVA: time – F(9,252)=44.1, p<0.001; dose – F(3,28)=33.8, p<0.001; time X dose interaction – F(27,252)=14.0, p<0.001, Figure 3D). The maximum effect was observed from 20 to 60 minutes post dose and, at a dose of 3 mg/kg SC, the increase in latency was apparent for up to 150 minutes post dose. These findings indicate that ST-2530 reduces sensitivity to different types of aversive thermal stimuli.
To evaluate whether ST-2530 affects behavioral responses to an aversive mechanical stimulus, mice were assessed on the tail pinch assay. Thirty minutes following SC administration of ST-2530 or vehicle a bulldog clamp was placed on the proximal end of the tail. The latency to bring the nose within 0.5 cm of the clamp was measured. ST-2530 dose-dependently increased the latency to respond to the clamp (one-way ANOVA: F(2,21)=11.8, p<0.001, Figure 3E), indicating that ST-2530 has an antinociceptive effect on an aversive mechanical stimulus. Next, we evaluated whether ST-2530 affects behavioral responses to the chemical irritant formalin (37% w/w formaldehyde). Formalin was injected into the plantar surface of the hind paw of mice administered ST-2530 or vehicle, and the time spent licking the paw over the next 45 minutes was counted. Formalin causes a biphasic response with an early, acute phase and a late, inflammatory phase [42]. At the dose of 3 mg/kg, ST-2530 significantly reduced the time spent attending to the formalin-injected paw in the late phase (10–45 minutes after formalin injection; mixed ANOVA: time – F(1,28)=75.7, p<0.001, Figure 3G). There was also a reduction for the early phase (0–5 minutes), but this did not meet statistical significance. These results are consistent with findings on other Nav1.7 inhibitors that have been assessed on the formalin assay [3,19,50]. In summary, ST-2530 was broadly analgesic to acute aversive thermal, mechanical and chemical stimuli.
3.4. ST-2530 is analgesic in an incisional pain model
The effect of ST-2530 on thermal hypersensitivity was assessed in a postoperative pain model. The hind paw incision in mice is an established model of postoperative pain that causes reversible edema around the wound site and increases sensitivity to both mechanical and thermal stimuli [10,41]. Under anesthesia, a 5 mm incision was made on the plantar surface of the left hind paw, and following the procedure subjects were allowed 48 hours to recover. Hargreaves testing commenced after the recovery period and was conducted once a day for five consecutive days. The thermal sensitivity of both the left and right hind paws was evaluated.
Overall, the incisional procedure produced a robust thermal hypersensitivity in the left, injured hind paw when compared to the right, uninjured hind paw (two-way RM ANOVA: test day – F(5,115)=11.8, p<0.001; paw – F(1,23)=131, p<0.001; test day X paw interaction – F(5,115)=11.4, p<0.001. Figure 4A) At 48 hours post-procedure, prior to administration of ST-2530 or vehicle, a significant hypersensitivity to thermal stimuli was evident in the left hind paw when compared to the right paw. The hypersensitivity was apparent until 6 days post-procedure, at which time there was no significant difference in withdrawal latency at the left versus right paw (Figure 4A).
Figure 4.
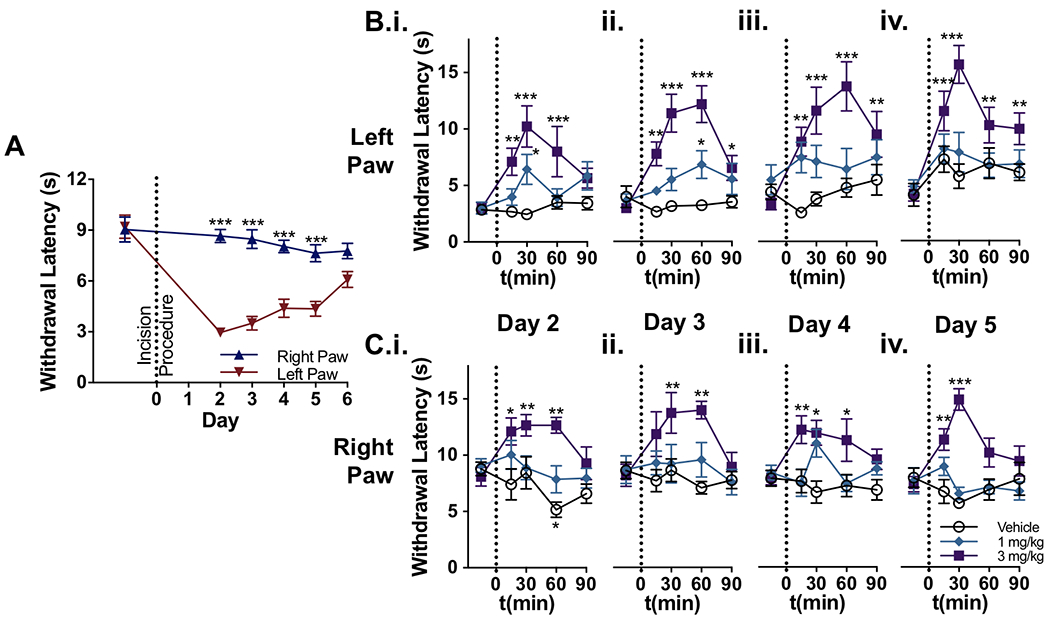
Effect of ST-2530 on the thermal hypersensitivity in an incisional pain model. (A) Withdrawal latency to a thermal stimulus on the left and right hind paw before and after the incision procedure on the left hind paw (n=24 subjects; all 3 groups combined during baseline, pre-dosing timepoint). Bonferroni’s multiple comparison test comparing left and right paw: *** - p<0.001. (B,C) Withdrawal latency to a thermal stimulus on the left (B) and right hind paw (C) on test sessions post-incisional procedure (n=8 per group). Dunnett’s multiple comparison test, comparing post-dose timepoint to baseline value for each dose group: * - p<0.05, ** - p<0.01, *** - p<0.001.
ST-2530 or vehicle was dosed once on each day after the recovery period in which thermal hypersensitivity was present. On each test session, ST-2530 dose-dependently reversed the thermal hypersensitivity induced by the incisional injury to the left hind paw (Figure 4B). An antinociceptive effect was also observed at the uninjured hind paw, evidenced by a significant increase in the thermal withdrawal latency over the baseline response (Figure 4C). At 48 hours post-incision, 3 mg/kg ST-2530 significantly increased thermal withdrawal latency in both the left injured paw (mixed ANOVA: time – F(4,84)=6.87, p<0.001; dose group – F(2,21)=6.59, p=0.006; dose group X time interaction – F(8,84)=3.78, p<0.001; Figure 4B.i.) and the right uninjured paw (mixed ANOVA: dose group – F(2,21)=10.3, p<0.001; dose group X time interaction – F(8,84)=2.66, p=0.012; Figure 4C.i.). At 6 days post-incision on test day 5, the effect of ST-2530 on withdrawal latency to thermal stimulus persisted on both the injured left paw (mixed ANOVA: time – F(4,84)=9.79, p<0.001; dose group – F(2,21)=8.68, p=0.002; dose group X time interaction – F(8,84)=2.83, p=0.008; Figure 4B.iv.) and the uninjured right paw (mixed ANOVA: dose group – F(2,21)=12.9, p<0.001; dose group X time interaction – F(8,84)=5.27, p<0.001, Figure 4C.iv.). These results demonstrate that ST-2530 reverses thermal hypersensitivity induced by an incisional injury, and consistently produces an analgesic response to an aversive thermal stimulus over multiple consecutive test days.
3.5. ST-2530 reverses mechanical allodynia induced by the spared nerve injury
To determine whether ST-2530 affects allodynia associated with a peripheral nerve injury, we utilized the spared nerve injury (SNI) in mice, a neuropathic pain model that causes robust and long-lasting mechanical allodynia in the affected limb [40]. Under anesthesia, two of the three branches of the left sciatic nerve, the tibial and common peroneal nerves, were severed, leaving the sural nerve intact. This nerve injury produces mechanical allodynia that is localized to the lateral portion of the affected hind paw [5]. To verify that the SNI procedure produced mechanical allodynia, both subjects’ hind paws were tested using Von Frey monofilaments before and 10 days following the SNI procedure, and the 50% withdrawal threshold was determined using the modified up-down method [7]. The SNI procedure on the left hind limb produced a significant reduction in the withdrawal threshold in the left hind paw when compared to the right hind paw, and this allodynia was present for the entirety of the experiment, up to 26 days post-procedure (mixed ANOVA: test day – F(5,75)=10.97, p<0.001; paw – F(1,15)=111, p<0.001; test day X paw interaction – F(5,75)=9.062), Figure 5A).
Figure 5.
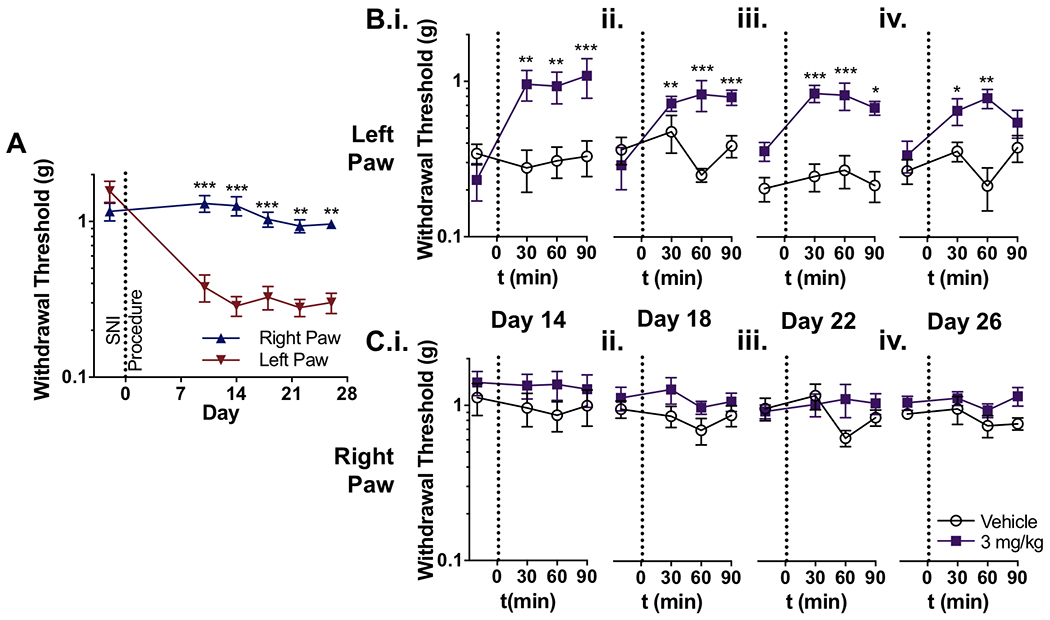
Effect of ST-2530 on mechanical allodynia produced by SNI. (A) Withdrawal threshold to a mechanical stimulus on the left and right hind paw before and after the SNI procedure on the left hind limb (n=16 subjects; 2 groups combined during baseline, pre-dosing timepoint). Bonferroni’s multiple comparison test comparing left and right paw: ** - p<0.01 *** - p<0.001. (B,C) Withdrawal threshold to a mechanical stimulus on the left (B) and right hind paw (C) on test sessions on days 14 (i), 18 (ii), 22 (iii), and 26 days (iv) post-SNI procedure (n=8 per group). Dunnett’s multiple comparison test, comparing post-dose timepoint to baseline value for each dose group: * - p<0.05, ** - p<0.01, *** - p<0.001. Y-axes in all graphs are in logarithmic scale.
Beginning on day 14 and continuing through day 26 post-SNI procedure, the withdrawal threshold on both paws was assessed every four days before and after mice received either 3 mg/kg ST-2530 or vehicle. Mice that received ST-2530 exhibited an increased withdrawal threshold to mechanical stimulus in the left hind paw (Figure 5B). This effect was apparent on all test sessions including day 14 (mixed ANOVA: time – F(3,42)=2.94, p=0.043; dose group – F(1,14)=13.0, p=0.003; dose group X time interaction – F(3,42) = 3.48, p=0.024; Figure 5B.i.) and day 26 post-SNI procedure (mixed ANOVA: dose group – F(1,14)=14.8, p=0.002; dose group X time interaction – F(3,42)=3.44, p=0.025, Figure 5B.iv.). In contrast, ST-2530 had no effect on the mechanical withdrawal threshold on the uninjured right hind paw (Figure 5C). These data indicate that ST-2530 reverses mechanical allodynia caused by SNI without affecting sensitivity to non-aversive mechanical stimuli, as demonstrated by the absence of an effect on the mechanical withdrawal threshold on the uninjured side.
3.6. ST-2530 Pharmacokinetics
In a group of mice that did not undergo behavioral testing a pharmacokinetic (PK) experiment was performed to measure exposure to ST-2530 following administration of 3 mg/kg SC (n=4 per time point, Table 3). The maximum concentration in whole blood (Cmax) was 2,850 ± 210 ng/ml and occurred at the 30 minute time point. In a separate experiment the mouse whole blood protein binding was determined to be 79.1 ± 1.4% (mean ± SD, n=2 samples). Taking into account the bound fraction of drug in whole blood, which according to the free drug hypothesis is not available to engage the target [46], the unbound blood concentration at Cmax was estimated to be 1.00 ± 0.07 μM, approximately 4x the msNav1.7 Kd. At this estimated level of target occupancy, we found that ST-2530 was efficacious on all pain assays that were performed with no effects on locomotion, motor coordination, or olfaction.
Table 3:
Whole blood concentration levels of ST-2530 following 3 mg/kg SC administration
| t(min) | Blood Conc. (ng/ml) | Cu (μM)* |
|---|---|---|
| 5 | 1110 ± 30 | 0.39 ± 0.01 |
| 15 | 2100 ± 150 | 0.73 ± 0.05 |
| 30 | 2850 ± 210 | 1.00 ± 0.07 |
| 60 | 1020 ± 220 | 0.36 ± 0.08 |
| 120 | 220 ± 60 | 0.08 ± 0.02 |
Values are mean ± SEM (n=4 per timepoint)
Cu – unbound blood concentration
79.1% mouse whole blood protein binding
4. Discussion
In this report, we describe pharmacological characteristics of ST-2530, a selective inhibitor of Nav1.7 discovered by rational modification of the natural product saxitoxin. Compared to a previously disclosed analog [38], ST-2530 exhibits superior potency against msNav1.7, allowing for characterization of pharmacodynamic effects on a range of mouse pain models. At a dose of 3 mg/kg SC, ST-2530 was broadly efficacious in mouse models of acute pain evoked by aversive thermal, mechanical and chemical stimuli, postoperative pain, and neuropathic pain. Consistent with reports of genetic NaV1.7 loss-of-function, an increase in the withdrawal latency to noxious heat was observed in uninjured animals [21,22]. 3 mg/kg ST-2530 had no effects on locomotion, motor coordination, or olfaction. Based on the results of an independent PK study, 3 mg/kg ST-2530 produces a mean (SEM) peak blood concentration of 2,850 ± 210 ng/ml at 30 minutes post-dose, corresponding to an unbound blood concentration of 1.0 μM, or approximately 4x the in vitro IC50. The requirement to achieve exposure of 4x Nav1.7 IC50 for analgesia is consistent with previous results with acylsulfonamide inhibitors that bind to VSD IV [3]. Further work is required to explore the pharmacology of ST-2530 on DRG neurons, both from naïve mice and mice after injury, in order to correlate results in pain behavior models with effects on native DRG excitability.
Nav1.7 inhibitors described here and in a previous report [38] are differentiated from other isoform -selective small molecules in that they bind to the extracellular pore of the channel, i.e the same binding site as STX and TTX. These compounds take advantage of a 2 amino acid variation in hNav1.7 that is not present in other hNav isoforms or in rodent Nav1.7 to enhance isoform selectivity [48]. Whereas the earlier compound displayed >30x reduced potency against msNav1.7 compared to hNav1.7 [38], the ~10x species variation in potency of ST-2530 combined with greater potency against hNav1.7 makes it possible to assess of pharmacodynamic effects in rodent pain models. Further work is required to understand why ST-2530 retains superior potency against msNav1.7 compared to earlier compounds and to reveal the molecular interactions responsible for selectivity over off-target hNav isoforms in the absence of the domain III TI sequence motif in the mouse channel [48]. It is also unclear why the association and dissociation kinetics of ST-2530 are slower in comparison to other small molecules that bind to the extracellular pore including ST-2262 [38], STX and TTX. However, given that the Kd on msNav1.7 was nearly equal to the IC50, and that the Hill slope of other small molecule antagonists that bind to the extracellular pore of hNav1.x is consistently ~1 [9,37], Kd is likely a good approximation of the IC50 of ST-2530 at equilibrium against hNav1.7. While the molecular interactions that account for Nav isoform selectivity are not fully understood, ST-2530 is a potent and selective Nav1.7 inhibitor that, in contrast with other small molecule inhibitors that bind to VSD IV, retains potency and selectivity when evaluated using a resting-state protocol [2,3,19,27,31,43]. Furthermore, while the state-dependent Nav1.7 inhibitor PF-05089771 had an IC50 against msNav1.7 of 8 nM when using a protocol that biases the channel towards the inactivated state [2], an efficacy signal in the Hargreaves assay was only achieved with a 100 mg/kg dose delivered PO (Figure 3B). In contrast, ST-2530 had an IC50 against msNav1.7 of 290 nM (Table 2) and showed consistent efficacy on a range of pain models at 3 mg/kg administered SC (Figures 3–5). The stark difference in efficacious dose between these two compounds may be partly explained by the fact that PF-05089771 is highly protein bound, with a free fraction of around 0.01 [3]; however, studies have shown that the PF-05089771 unbound concentration required to achieve efficacy is >30X over the IC50 [3] which greatly contrasts with ST-2530’s estimated free concentration requirement of ~4X over the msNav1.7 IC50. It is possible that the ability to inhibit the channel in its resting state reduces the occupancy requirements for efficacy on pain models.
ST-2530 was efficacious in a range of pain behavior models believed to represent acute, subacute and chronic neuropathic pain states, and against different pain modalities including thermal, mechanical and chemical. The observation of an analgesic pharmacodynamic effect within 10–15 minutes after subcutaneous dosing suggests that the change in behavioral responses is a direct effect of inhibition of the channel. It has previously been shown that selective deletion of Nav1.7 from sensory neurons increases the expression of proenkephalin (PENK), and that pretreatment with naloxone prior to the thermal plantar test reverses the pro-analgesic phenotype of the Nav1.7 mutant [33]. However, this finding was not replicated in a rat Nav1.7 loss-of-function mutant on the hot plate test [8], in a mouse tamoxifen inducible Nav1.7 knockout on the Hargreaves test [14], or in wild-type mice treated with a sulfonamide Nav1.7 inhibitor on the tail flick assay [24]. Furthermore, in a healthy subject induced pluripotent stem cell (iPSC) line with a frameshift mutation in Nav1.7 that renders the channel functionless, there was no change in PENK mRNA expression compared to wild-type iPSCs, and naloxone did not reverse the reduced excitability found in the hNav1.7 mutant iPSCs [30]. Consistent with these findings, the present study demonstrates that naloxone, at a dose that reversed the pro-analgesic phenotype of mutant Nav1.7 mice [33], has no effect on the analgesic effect of an isoform-selective Nav1.7 inhibitor in the thermal plantar test in mice. While it is possible that repeat dosing with ST-2530 could affect the endogenous opioid system, these results indicate that ST-2530 is acutely analgesic, and that antinociceptive effects are mediated directly by inhibition of Nav1.7.
There is an especially urgent need for more effective pharmacotherapies for the treatment of neuropathic pain [18]. A key finding in the present study is that ST-2530 reversed mechanical allodynia following SNI without affecting the withdrawal threshold to non-noxious mechanical stimuli in the uninjured hind limb. As there are CNS circuit maladaptations associated with neuropathic pain [11], it has been unclear whether Nav1.7 inhibition would be an effective strategy for treatment. In a transgenic mouse model with adult-onset tamoxifen-inducible Nav1.7 deletion, wild-type and inducible knockout mice that underwent SNI showed similar levels of mechanical allodynia following treatment with tamoxifen, whereas cold allodynia was reversed in the inducible knockout [45]. Other evidence indicates that Nav1.7 is required for mechanical allodynia in neuropathic pain phenotypes. In a rat Nav1.7 loss-of-function model, mechanical allodynia did not develop following spinal nerve ligation [22]. While It is unclear why these transgenic rodent models have divergent effects on the development of mechanical allodynia, there may be subtle differences in the location and extent of Nav1.7 deletion or loss-of-function [22,45]. Interestingly, in a neuropathic pain model in which the spinal nerve was transected at the L5 segment, mechanical allodynia developed normally in sensory nerve specific Nav1.7 knockout mice, but did not develop in mice with Nav1.7 ablated from both sensory and sympathetic nerves [32]. Collectively these results indicate that Nav1.7 is required for mechanical allodynia in certain neuropathic pain phenotypes and that inhibition by ST-2530 is sufficient to reverse allodynia associated with neuropathic insult.
While doses of ST-2530 evaluated in pain behavior models did not affect locomotion, motor coordination or olfaction, a higher dose reduced locomotor activity, rearing and performance on the rotarod and olfactory test. The increased latency on the buried food test could be attributed to reduced olfactory sensitivity, but the reduction in motor activity with 10 mg/kg ST-2530 confounds interpretation. The mechanism responsible for the decrease in locomotion is unclear. Assuming dose-proportional pharmacokinetics, Cmax following the 10 mg/kg dose (~3.3 μM) would remain well below the IC50 against off-target hNav isoforms, however the IC50s against off-target msNav isoforms were not determined. Given the results from the radioligand binding assay panel of rat and human protein targets, there is no evidence that ST-2530 has significant affinity for non-Nav1.x off-target proteins (Table S1) but, again, the possibility of species variation in potency confounds interpretation. Further studies are needed to determine the mechanism underlying the effect on locomotion at a supra-pharmacological dose.
The present study demonstrates that ST-2530, a selective inhibitor of Nav1.7 derived from saxitoxin, is analgesic in a range of mouse behavioral models that represent acute and chronic pain. While the success of Nav1.7 inhibitors that have reached the clinic to date has been limited, these drug candidates were compromised by poor pharmacological characteristics, such as insufficient selectivity over off-target Nav1.x isoforms [15], or poor pharmaceutical properties, such as very high protein binding that led to high exposure requirements [3]. Our work indicates that selective pharmacological inhibition of Nav1.7 is possible with agents that likely bind to the extracellular pore, the so-called ‘site 1’ binding site shared by saxitoxin and tetrodotoxin [38]. With adequate Nav isoform selectivity and unbound drug concentrations around 4x Nav1.7 IC50, analgesic efficacy is possible in a broad range of acute and pain models by inhibition of Nav1.7.
Supplementary Material
Acknowledgments
We would like to thank the veterinarians and the animal research technicians at Montana State University’s Animal Resources Center for their assistance in the behavioral studies. The UNE COBRE Behavioral Core is supported by NIH NIGMS (P20GM103643). Other behavioral studies were partially funded by federal grant USAMRMC W81XWH-17-1-0672.
Footnotes
Conflict of Interest: JTB, HP, GL, SK, AD, DM, XZ, JCH and JVM are employees of SiteOne Therapeutics. DCY is a shareholder and consultant of SiteOne Therapeutics.
References
- [1].Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H, Shriver S, Young C, Lin S, Johnson JP, Wu P, Li J, Coons M, Tam C, Brillantes B, Sampang H, Mortara K, Bowman KK, Clark KR, Estevez A, Xie Z, Verschoof H, Grimwood M, Dehnhardt C, Andrez J-C, Focken T, Sutherlin DP, Safina BS, Starovasnik MA, Ortwine DF, Franke Y, Cohen CJ, Hackos DH, Koth CM, Payandeh J. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 2015;350:aac5464–aac5464. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- [2].Alexandrou AJ, Brown AR, Chapman ML, Estacion M, Turner J, Mis MA, Wilbrey A, Payne EC, Gutteridge A, Cox PJ, Doyle R, Printzenhoff D, Lin Z, Marron BE, West C, Swain NA, Storer RI, Stupple PA, Castle NA, Hounshell JA, Rivara M, Randall A, Dib-Hajj SD, Krafte D, Waxman SG, Patel MK, Butt RP, Stevens EB. Subtype-Selective Small Molecule Inhibitors Reveal a Fundamental Role for Nav1.7 in Nociceptor Electrogenesis, Axonal Conduction and Presynaptic Release. PLOS ONE 2016;11:e0152405. doi: 10.1371/journal.pone.0152405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bankar G, Goodchild SJ, Howard S, Nelkenbrecher K, Waldbrook M, Dourado M, Shuart NG, Lin S, Young C, Xie Z, Khakh K, Chang E, Sojo LE, Lindgren A, Chowdhury S, Decker S, Grimwood M, Andrez J-C, Dehnhardt CM, Pang J, Chang JH, Safina BS, Sutherlin DP, Johnson JP, Hackos DH, Robinette CL, Cohen CJ. Selective NaV1.7 Antagonists with Long Residence Time Show Improved Efficacy against Inflammatory and Neuropathic Pain. Cell Rep 2018;24:3133–3145. doi: 10.1016/j.celrep.2018.08.063 [DOI] [PubMed] [Google Scholar]
- [4].Black JA, Frézel N, Dib-Hajj SD, Waxman SG. Expression of Nav1.7 in DRG Neurons Extends from Peripheral Terminals in the Skin to Central Preterminal Branches and Terminals in the Dorsal Horn. Mol Pain 2012;8:1744-8069-8–82. doi:10.1186/1744-8069-8-82. doi: 10.1186/1744-8069-8-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bourquin AF, Süveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse: Pain 2006;122:14e1–14e14. doi: 10.1016/j.pain.2005.10.036. [DOI] [PubMed] [Google Scholar]
- [6].Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol Rev 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- [7].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- [8].Chen L, Effraim PR, Carrara J, Zhao P, Dib-Hajj FB, Dib-Hajj SD, Waxman SG. Pharmacological characterization of a rat Nav1.7 loss-of-function model with insensitivity to pain: PAIN 2020;161:1350–1360. doi: 10.1097/j.pain.0000000000001807. [DOI] [PubMed] [Google Scholar]
- [9].Choudhary G, Shang L, Li X, Dudley SC. Energetic localization of saxitoxin in its channel binding site. Biophys J 2002;83:912–919. doi: 10.1016/S0006-3495(02)75217-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Clark JD, Qiao Y, Li X, Shi X, Angst MS, Yeomans DC. Blockade of the Complement C5a Receptor Reduces Incisional Allodynia, Edema, and Cytokine Expression: Anesthesiology 2006;104:1274–1282. doi: 10.1097/00000542-200606000-00024. [DOI] [PubMed] [Google Scholar]
- [11].Costigan M, Scholz J, Woolf CJ. Neuropathic Pain: A Maladaptive Response of the Nervous System to Damage. Annu Rev Neurosci 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci Off J Soc Neurosci 2004;24:8232–8236. doi: 10.1523/JNEUROSCI.2695-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Deng L, Shields S, Reese R, Kaminker J, Hackos D. Poster PTH446: Examination of the Role of Endogenous Opioids in the Insensitivity-to-pain Phenotype of An Adult-onset Nav1.7 Knockout Mouse. 16th World Congress on Pain 2016, Yokohama, Japan. [Google Scholar]
- [15].Deuis J, Wingerd J, Winter Z, Durek T, Dekan Z, Sousa S, Zimmermann K, Hoffmann T, Weidner C, Nassar M, Alewood P, Lewis R, Vetter I. Analgesic Effects of GpTx-1, PF-04856264 and CNV1014802 in a Mouse Model of NaV1.7-Mediated Pain. Toxins 2016;8:78. doi: 10.3390/toxins8030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The NaV1.7 sodium channel: from molecule to man. Nat Rev Neurosci 2013;14:49–62. doi: 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]
- [17].Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006 [DOI] [PubMed] [Google Scholar]
- [18].Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpää M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice ASC, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015;14:162–173. doi: 10.1016/S1474-4422(14)70251-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Focken T, Liu S, Chahal N, Dauphinais M, Grimwood ME, Chowdhury S, Hemeon I, Bichler P, Bogucki D, Waldbrook M, Bankar G, Sojo LE, Young C, Lin S, Shuart N, Kwan R, Pang J, Chang JH, Safina BS, Sutherlin DP, Johnson JP, Dehnhardt CM, Mansour TS, Oballa RM, Cohen CJ, Robinette CL. Discovery of Aryl Sulfonamides as Isoform-Selective Inhibitors of NaV1.7 with Efficacy in Rodent Pain Models. ACS Med Chem Lett 2016;7:277–282. doi: 10.1021/acsmedchemlett.5b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fouillet A, Watson JF, Piekarz AD, Huang X, Li B, Priest B, Nisenbaum E, Sher E, Ursu D. Characterisation of Nav1.7 functional expression in rat dorsal root ganglia neurons by using an electrical field stimulation assay. Mol Pain 2017;13:174480691774517. doi: 10.1177/1744806917745179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gingras J, Smith S, Matson DJ, Johnson D, Nye K, Couture L, Feric E, Yin R, Moyer BD, Peterson ML, Rottman JB, Beiler RJ, Malmberg AB, McDonough SI. Global Nav1.7 Knockout Mice Recapitulate the Phenotype of Human Congenital Indifference to Pain. PLoS ONE 2014;9:e105895. doi: 10.1371/journal.pone.0105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Grubinska B, Chen L, Alsaloum M, Rampal N, Matson D, Yang C, Taborn K, Zhang M, Youngblood B, Liu D, Galbreath E, Allred S, Lepherd M, Ferrando R, Kornecook T, Lehto S, Waxman S, Moyer B, Dib-Hajj S, Gingras J. Rat NaV1.7 loss-of-function genetic model: Deficient nociceptive and neuropathic pain behavior with retained olfactory function and intra-epidermal nerve fibers. Mol Pain 2019;15:174480691988184. doi: 10.1177/1744806919881846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- [24].Houghton A, Ballard J, Burgey C, John C, Kim R, Jochnowitz N, Kraus R, Klein B, Layton M. Poster PT0290: A Selective Nav1.7 Inhibitor is Antinociceptive, and Potentiates Opioid Antinociception, in a Mouse Tail Flick Assay. 16th World Congress on Pain 2016, Yokohama, Japan. [Google Scholar]
- [25].Hunskaar S, Fasmer OB, Hole K. Formalin test in mice, a useful technique for evaluating mild analgesics. J Neurosci Methods 1985;14:69–76. doi: 10.1016/0165-0270(85)90116-5. [DOI] [PubMed] [Google Scholar]
- [26].Janssen PA, Niemegeers CJ, Dony JG. The inhibitory effect of fentanyl and other morphine-like analgesics on the warm water induced tail withdrawl reflex in rats. Arzneimittelforschung 1963;13:502–507. [PubMed] [Google Scholar]
- [27].Kornecook TJ, Yin R, Altmann S, Be X, Berry V, Ilch CP, Jarosh M, Johnson D, Lee JH, Lehto SG, Ligutti J, Liu D, Luther J, Matson D, Ortuno D, Roberts J, Taborn K, Wang J, Weiss MM, Yu V, Zhu DXD, Fremeau RT, Moyer BD. Pharmacologic Characterization of AMG8379, a Potent and Selective Small Molecule Sulfonamide Antagonist of the Voltage-Gated Sodium Channel NaV1.7. J Pharmacol Exp Ther 2017;362:146–160. doi: 10.1124/jpet.116.239590. [DOI] [PubMed] [Google Scholar]
- [28].Krause A, Lowe PJ. Visualization and communication of pharmacometric models with berkeley madonna. CPT Pharmacomet Syst Pharmacol 2014;3:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, Krambis MJ, Antonio BM, Zellmer SG, Printzenhoff D, Padilla KM, Lin Z, Wagoner PK, Swain NA, Stupple PA, de Groot M, Butt RP, Castle NA. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci 2013;110:E2724–E2732. doi: 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, Clark AJ, Millar V, Peck LJ, Ebner D, Tracey I, Serra J, Bennett DL. Defining the Functional Role of NaV1.7 in Human Nociception. Neuron 2019;101:905–919.e8. doi: 10.1016/j.neuron.2019.01.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McKerrall SJ, Nguyen T, Lai KW, Bergeron P, Deng L, DiPasquale A, Chang JH, Chen J, Chernov-Rogan T, Hackos DH, Maher J, Ortwine DF, Pang J, Payandeh J, Proctor WR, Shields SD, Vogt J, Ji P, Liu W, Ballini E, Schumann L, Tarozzo G, Bankar G, Chowdhury S, Hasan A, Johnson JP, Khakh K, Lin S, Cohen CJ, Dehnhardt CM, Safina BS, Sutherlin DP. Structure- and Ligand-Based Discovery of Chromane Arylsulfonamide Nav1.7 Inhibitors for the Treatment of Chronic Pain. J Med Chem 2019;62:4091–4109. doi: 10.1021/acs.jmedchem.9b00141. [DOI] [PubMed] [Google Scholar]
- [32].Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, Malcangio M, Wood JN. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun 2012;3:791. doi: 10.1038/ncomms1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Minett MS, Pereira V, Sikandar S, Matsuyama A, Lolignier S, Kanellopoulos AH, Mancini F, Iannetti GD, Bogdanov YD, Santana-Varela S, Millet Q, Baskozos G, MacAllister R, Cox JJ, Zhao J, Wood JN. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat Commun 2015;6:8967. doi: 10.1038/ncomms9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mitrovic N, Lerche H, Heine R, Fleischhauer R, Pika-Hartlaub U, Hartlaub U, George AL, Lehmann-Horn F. Role in fast inactivation of conserved amino acids in the IV/S4-S5 loop of the human muscle Na+ channel. Neurosci Lett 1996;214:9–12. doi: 10.1016/0304-3940(96)12866-4. [DOI] [PubMed] [Google Scholar]
- [35].Mulcahy JV, Pajouhesh H, Beckley JT, Delwig A, Du Bois J, Hunter JC. Challenges and Opportunities for Therapeutics Targeting the Voltage-Gated Sodium Channel Isoform NaV1.7. J Med Chem 2019;62:8695–8710. doi: 10.1021/acs.jmedchem.8b01906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nicholson B Responsible Prescribing of Opioids for the Management of Chronic Pain: Drugs 2003;63:17–32. doi: 10.2165/00003495-200363010-00002. [DOI] [PubMed] [Google Scholar]
- [37].Noda M, Suzuki H, Numa S, Stühmer W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett 1989;259:213–216. doi: 10.1016/0014-5793(89)81531-5. [DOI] [PubMed] [Google Scholar]
- [38].Pajouhesh H, Beckley J, Delwig A, Hajare HS, Luu G, Monteleone D, Zhou X, Ligutti J, Amagasu S, Moyer B, Yeomans DC, Du Bois J, Mulcahy JV. Discovery of a selective, state-independent inhibitor of NaV1.7 by modification of guanidinium toxins. Sci Rep 2020;10(1):14791. doi: 10.1038/s41598-020-71135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Penzotti JL, Fozzard HA, Lipkind GM, Dudley SC. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys J 1998;75:2647–2657. doi: 10.1016/S0006-3495(98)77710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pertin M, Gosselin R-D, Decosterd I. The Spared Nerve Injury Model of Neuropathic Pain. In: Luo ZD, editor. Pain Research. Methods in Molecular Biology. Totowa, NJ: Humana Press, 2012, Vol. 851. pp. 205–212. doi: 10.1007/978-1-61779-561-9_15. [DOI] [PubMed] [Google Scholar]
- [41].Pogatzki EM, Raja SN. A mouse model of incisional pain. Anesthesiology 2003;99:1023–1027. doi: 10.1097/00000542-200310000-00041. [DOI] [PubMed] [Google Scholar]
- [42].Porro CA, Cavazzuti M. Spatial and temporal aspects of spinal cord and brainstem activation in the formalin pain model. Prog Neurobiol 1993;41:565–607. doi: 10.1016/0301-0082(93)90044-S. [DOI] [PubMed] [Google Scholar]
- [43].Roecker AJ, Egbertson M, Jones KLG, Gomez R, Kraus RL, Li Y, Koser AJ, Urban MO, Klein R, Clements M, Panigel J, Daley C, Wang J, Finger EN, Majercak J, Santarelli V, Gregan I, Cato M, Filzen T, Jovanovska A, Wang Y-H, Wang D, Joyce LA, Sherer EC, Peng X, Wang X, Sun H, Coleman PJ, Houghton AK, Layton ME. Discovery of selective, orally bioavailable, N-linked arylsulfonamide Nav1.7 inhibitors with pain efficacy in mice. Bioorg Med Chem Lett 2017;27:2087–2093. doi: 10.1016/j.bmcl.2017.03.085. [DOI] [PubMed] [Google Scholar]
- [44].Rosenblum A, Marsch LA, Joseph H, Portenoy RK. Opioids and the treatment of chronic pain: Controversies, current status, and future directions. Exp Clin Psychopharmacol 2008;16:405–416. doi: 10.1037/a0013628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shields SD, Deng L, Reese RM, Dourado M, Tao J, Foreman O, Chang JH, Hackos DH. Insensitivity to Pain upon Adult-Onset Deletion of Nav1.7 or Its Blockade with Selective Inhibitors. J Neurosci 2018;38:10180–10201. doi: 10.1523/JNEUROSCI.1049-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Smith DA, Di L, Kerns EH. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 2010;9:929–939. doi: 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- [47].Thomas-Tran R, Du Bois J. Mutant cycle analysis with modified saxitoxins reveals specific interactions critical to attaining high-affinity inhibition of hNaV1.7. Proc Natl Acad Sci 2016;113:5856–5861. doi: 10.1073/pnas.1603486113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Walker JR, Novick PA, Parsons WH, McGregor M, Zablocki J, Pande VS, Du Bois J. Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (Nav1.7). Proc Natl Acad Sci U S A 2012;109:18102–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Weiss J, Pyrski M, Jacobi E, Bufe B, Willnecker V, Schick B, Zizzari P, Gossage SJ, Greer CA, Leinders-Zufall T, Woods CG, Wood JN, Zufall F. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature 2011;472:186–190. doi: 10.1038/nature09975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wu Y-J, Guernon J, McClure A, Luo G, Rajamani R, Ng A, Easton A, Newton A, Bourin C, Parker D, Mosure K, Barnaby O, Soars MG, Knox RJ, Matchett M, Pieschl R, Herrington J, Chen P, Sivarao DV, Bristow LJ, Meanwell NA, Bronson J, Olson R, Thompson LA, Dzierba C. Discovery of non-zwitterionic aryl sulfonamides as Nav1.7 inhibitors with efficacy in preclinical behavioral models and translational measures of nociceptive neuron activation. Bioorg Med Chem 2017;25:5490–5505. doi: 10.1016/j.bmc.2017.08.012. [DOI] [PubMed] [Google Scholar]
- [51].Yang M, Crawley JN. Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci 2009;Chapter 8:Unit 8.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 2004;41:171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.