

Abstract
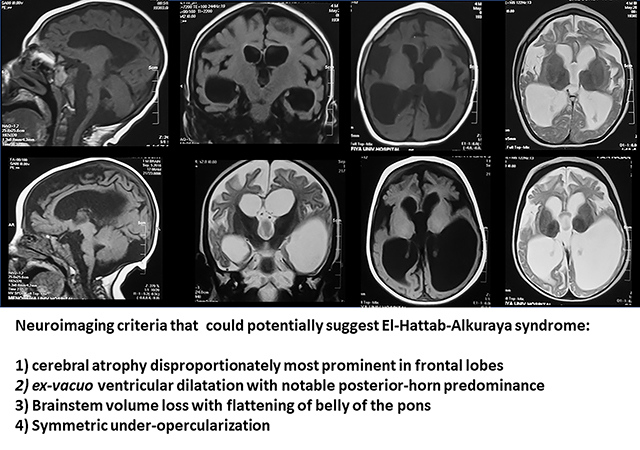
Homozygous pathogenic variants in WDR45B were first identified in six subjects from three unrelated families with global development delay, refractory seizures, spastic quadriplegia, and brain malformations. Since the initial report in 2018, no further cases have been described. In this report, we present 12 additional individuals from seven unrelated families and their clinical, radiological, and molecular findings. Six different variants in WDR45B were identified, five of which are novel. Microcephaly and global developmental delay were observed in all subjects, and seizures and spastic quadriplegia in most. Common findings on brain imaging include cerebral atrophy, ex-vacuo ventricular dilatation, brainstem volume loss, and symmetric under-opercularization. El-Hattab-Alkuraya syndrome is associated with a consistent phenotype characterized by early onset cerebral atrophy resulting in microcephaly, developmental delay, spastic quadriplegia, and seizures. The phenotype appears to be more severe among individuals with loss-of-function variants whereas those with missense variants were less severely affected suggesting a potential genotype-phenotype correlation in this disorder. A brain imaging pattern emerges which is consistent among individuals with loss-of-function variants and could potentially alert the neuroradiologists or clinician to consider WDR45B-related El-Hattab-Alkuraya syndrome.
Keywords: WDR45B, autophagy, neurodevelopmental disorders (NDD), brain atrophy, autosomal recessive (AR) trait
Graphical Abstract
INTRODUCTION
Neurodevelopmental disorders (NDD) are extremely heterogeneous in term of underlying etiology, involved pathways, neurobiology of the disease process, and associated manifestations1. These disorders are characterized by abnormal cognition, language, behavior and/or motor skills often because of abnormal brain development2.There are different mutational and molecular mechanisms associated with NDD including copy number variants (CNV) alleles, abnormal trinucleotide repeat expansions, secondary structure mutagenesis, single nucleotide variants (SNV), and others3. It is estimated that the diagnostic yield for clinical genetic testing, including chromosomal microarray (CMA) and exome sequencing (ES), can approach 50% in such disorders4.With exome reanalysis, further genomic studies and computational tools, NDD can be molecularly diagnosed in close to 80% of select populations1. Establishing a genetic etiology and molecular diagnosis in such cases may help in prognostication, in risk assessment for other family members, and increasingly allow for genotype-direct therapeutics. Given that a large number of NDD-associated genes are yet to be discovered, broad testing with exome or genome sequencing, complemented by functional studies, is essential to uncover the complete list of NDD-associated genes5.
Recent advances in genetic testing and the widespread utilization of next generation sequencing (NGS) personal genome clinical testing are enabling physicians and scientists to understand the molecular basis of NDD at an unprecedented pace and molecular precision. One example is the WDR45B gene that was first linked to NDD in humans by large scale studies that identified four subjects with two different variants in this gene6,7. In 2011, Najmabadi et al. performed homozygosity mapping, exon enrichment and NGS in 136 consanguineous families with autosomal-recessive (AR) intellectual disability (ID). Three siblings with ID and microcephaly were homozygous for a missense variant allele in WDR45Bc.326G>A:p.R109Q6. In 2017, Anazi et al. performed genomic evaluation with molecular karyotyping, multi-gene panels and ES on a cohort of 337 individuals with ID. A child with global developmental delay (GDD) and microcephaly, who was homozygous for nonsense variant in WDR45B (c.673C>T; p.R225*), was identified7.
The first detailed clinical report of WDR45B-associated NDD was published in 2018. Six individuals from three unrelated families who shared overlapping phenotypes were described. Common features include profound development delay (DD), early-onset refractory epilepsy, progressive spastic quadriplegia, and contractures. Findings on brain magnetic resonance imaging (MRI) included reduced cerebral volume, thinning of gray matter, and ventriculomegaly. Two different homozygous nonsense variants, c.799C>T (p.Q267*) and c.673C>T (p.R225*), were identified in WDR45B in these individuals8.This rare disease trait is now known as El-Hattab-Alkuraya syndrome (MIM#617977).
Since the first report, no additional patients have been described in the scientific or medical literature, suggesting the potential rarity of this syndrome. Herein, we present the detailed clinical, molecular, and neuroradiological features of 12 previously unreported subjects from seven unrelated, world-wide ascertained families with El-Hattab-Alkuraya syndrome.
MATERIALS AND METHODS
Retrospective chart review of 12 individuals belonging to seven unrelated families from diverse ethnic backgrounds and countries of origin including Saudi Arabia, Egypt, the United States, and Iran was conducted. Written informed consent was obtained from the legal guardians of the subjects included in this study for enrollment in the research study and publication of photographs. Additional ethical approval was obtained by the institutional review board for human subjects of the Baylor College of Medicine (BCM: IRB H-29697). This research was done in accordance with the Declaration of Helsinki.
Brain MRIs of 10 individuals were available for review by a single board-certified neuroradiologist (JVH). The molecular diagnosis of El-Hattab-Alkuraya syndrome was made by clinical or research ES. Family 1 had research ES analysis at BCM as previously described1. Family 2 had clinical ES at Prevention Genetics, USA using IlluminaNovaSeq6000 as per manufacturer’s instructions. Family 3 had clinical ES at Centogene GmbH, Rostock, Germany as previously described9. Family 4 had research ES at Universtiy of California, San Diego. Families 5 and 6 had clinical ES at King Fahad Medical City (KFMC), Riyadh, Saudi Arabia using Illumina HiSeq 4000 as per manufacturer’s instructions. Family 7 had research ES analysis at University College London (IoN UCL 07/Q0512/26) as previously described10. We used four splicing predictors (SpliceAI, HumanSpliceFinder (HSF), dbscSNV, and MaxEntScan) to characterize the impact of the four splice-site variants11–14.
RESULTS
CLINICAL AND NEURORADIOLOGICALDESCRIPTION
We describe 12 subjects from seven unrelated families with biallelic WDR45B deleterious variants (Figure 1 and Table 1). Detailed clinical information is available as supplementary file 1.
Figure 1.
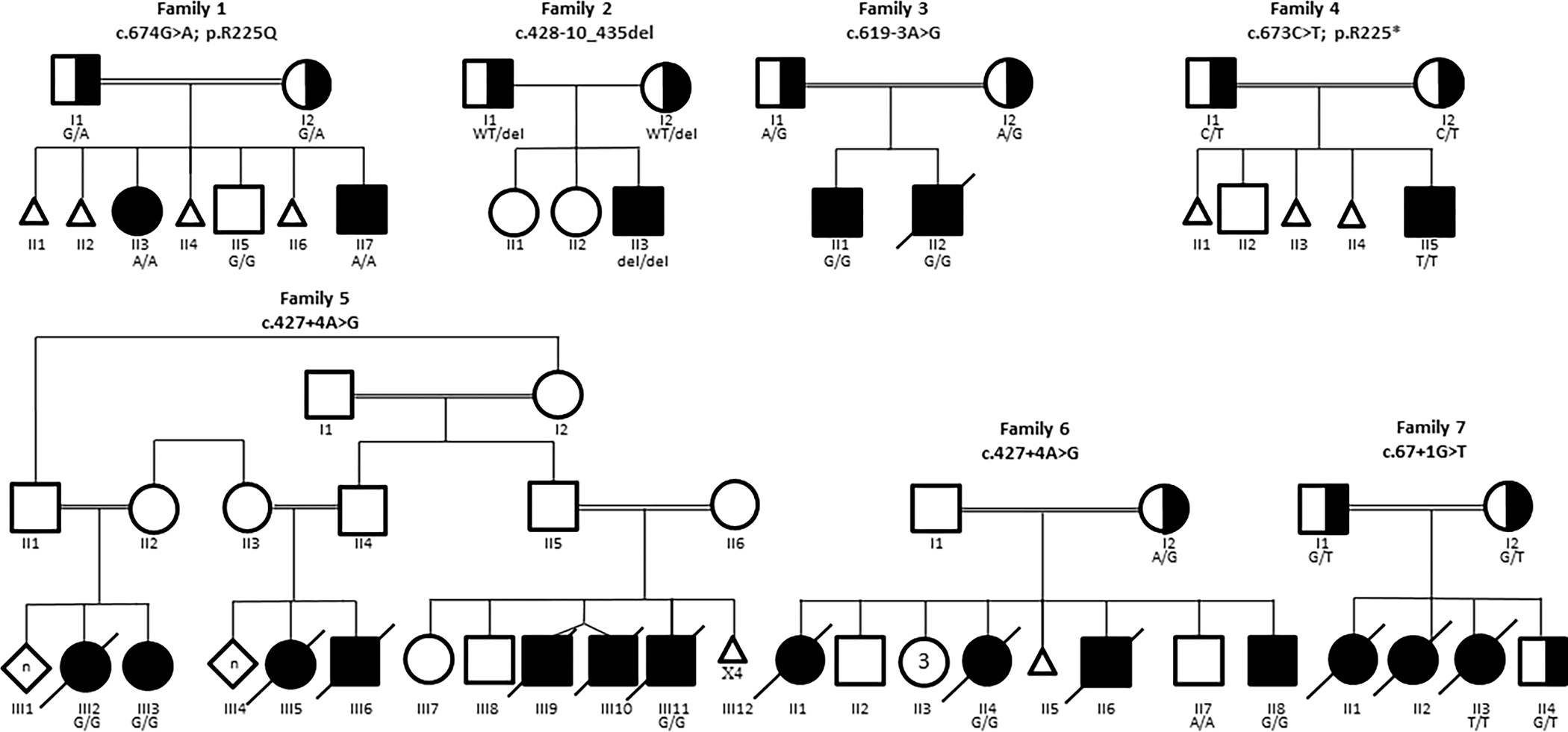
Pedigrees of families included in this report. When available, genotype for family members is indicated.
Table 1.
Characteristics of subjects included in this report
| Subject | 1 (F1-II3) |
2 (F1-II7) |
3 (F2-II3) |
4 (F3-II1) |
5 (F3-II2) |
6 (F4-II5) |
7 (F5-III2) |
8 (F5-III3) |
9 (F5-III11) |
10 (F6-II4) |
11 (F6-II8) |
12 (F7-II3) |
Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gender | Female | Male | Male | Male | Male | Male | Female | Female | Male | Female | Male | Female | 7 males: 5 females |
| Ethnicity | Arab | Arab | Cree | Arab | Arab | Arab | Arab | Arab | Arab | Arab | Arab | Iranian | |
| Country of origin | Egypt | Egypt | USA | Egypt | Egypt | Egypt | Saudi Arabia | Saudi Arabia | Saudi Arabia | Saudi Arabia | Saudi Arabia | Iran | |
| Consanguinity | + | + | − | + | + | + | + | + | + | + | + | + | 11/12 |
| WDR45B variant | c.674G>A; p.R225Q | c.674G>A; p. R225Q | c.428–10_435del18 | c.619–3A>G | c.619–3A>G | c.673C>T; p.R225* | c.427+4A>G | c.427+4A>G | c.427+4A>G | c.427+4A>G | c.427+4A>G | c.67+1G>T | |
| Age at last evaluation | 14 years | 4 years | 21 months | 6 years | 22 months | 7 months | 8 years | 10 years | 11 years | 15 years | 3 years | 3 years | 7 months-15 years |
| Outcome | Alive | Alive | Alive | Alive | Deceased | Alive | Deceased | Alive | Deceased | Deceased | Alive | Deceased | 5 deceased: 7 alive |
| Growth at birth Weight in kg (SD) Height in Cm (SD) OFC in Cm (SD) |
3 (−2) NA NA |
2.15(−2.2) NA NA |
2.45(−2) NA NA (Microcephaly) |
3(−1) 49(−0.5) 30(−3.2) |
NA NA NA |
2.25(−2.5) 48(−1.1) 31(−2.6) |
NA NA NA |
NA NA NA |
2.6(−1.5) NA 33(−1.3) |
NA NA NA (Brain atrophy) |
NA NA NA |
NA NA NA |
|
| Growth at last evaluation Weight in kg (SD) Height in Cm (SD) OFC in Cm (SD) |
46 (−0.7) 147.5(−2.2) 49.5(−3.7) |
16 (−1) 99(−0.8) 48(−2.3) |
12.1(0.4) 77(−2.8) 40.5(−5.5) |
12.7(−3.5) 88 (−5.4) 40 (−8.7) |
7(−4.7) 73(−4.1) 36(−8.9) |
6.8 (−1.8) 67(−0.6) 37.5(−5) |
15.1(−5.6) 115(−3.6) 46(−4) |
16.6(−4.7) 115(−3.6) 44.5(−4.2) † |
13.5 (−8.3) 102(−6) 44(−7) |
NA 103(−7.7) 42 (−9.1) |
7(−5.4) 62(−9.1) 43.5(−3.3) |
6(−11) 81(−3.3) 39(−6) |
|
| Microcephaly | + | + | + | + | + | + | + | + | + | + | + | + | 12/12 |
| Developmental delay | + | + | + | + | + | + | + | + | + | + | + | + | 12/12 |
| Epilepsy (Semiology) |
+(GTC) | − | + | + (Myoclonic) | + (Myoclonic, tonic) | + (Myoclonic, GTC) | + (Focal, GTC, myoclonic) | + (Focal, GTC, myoclonic) | + (Focal, GTC) | +(GTC) | +(GTC) | + | 11/12 |
| Epilepsy onset | 6 months | − | 3 months | 2 months | 1 month | Few days after birth | 12 days | 1.5 months | 2 months | 2 months | Neonatal period | NA | Few days-6 months |
| Drug-resistant epilepsy | + | − | + | + | + | + | + | + | + | + | + | + | 11/12 |
| EEG | Theta background with no sharp waves or spikes | Normal | Sharp waves in the left central and right frontal regions | Generalized epileptogenic activity | Generalized epileptogenic activity | Generalized epileptogenic activity | Diffuse slowing and attenuation | Frequent sharp wave discharges over the right posterior quadrant | Diffuse nonspecific slowing with poorly organized background with no epileptiform discharges | NA | NA | NA | |
| Spastic Quadriplegia | − (lower limbs only) | − | + | + | + | + | + | + | + | + | + | NA | 9/11 |
| Facial Features | |||||||||||||
| Bitemporal narrowing | + | − | NA | + | NA | − | NA | NA | + | − | + | NA | 4/7 |
| Thick and arched eyebrows | + | − | NA | + | NA | + (Arched only) | NA | NA | + (with synophrys) | − | + | NA | 5/7 |
| Large ears | − | − | NA | + | NA | + | NA | NA | + | + | + | NA | 5/7 |
| Long philtrum | − | − | NA | + | NA | + | NA | NA | + | − | + (smooth) | NA | 4/7 |
| Others | Facial asymmetry, upslanted and narrow PF, hypertelorism, fullness of upper eyelids, full cheeks | Broad forehead, epicanthal folds | NA | Long face, downslanted PF, thin upper lip, broad chin |
NA (Reported similar to Subject four) |
Narrow PF, hypertelorism, thick lips, retromicrognathia |
NA | NA | Elongated face, epicanthal folds, hypertelorism, depressed nasal bridge, maxillary hypoplasia, elongated jaw | Triangular face, hypertelorism, gum hyperplasia, | Elongated face, depressed nasal bridge, maxillary hypoplasia, elongated jaw | NA | |
Cm: Centimeters; EEG: electroencephalogram; GTC: generalized tonic clonic; Kg: Kilograms; NA: not available; PF: palpebral fissure; SD: standard deviation.
OFC obtained at 5 years of age
Subject 1 (F1-II3) and Subject 2 (F1-II7) were siblings (Family 1; Figure 1). Subject 1 was a 14-year-old girl with GDD, spastic paraplegia, microcephaly, seizures, aggressive and self-harming behaviors, strabismus, and dysmorphic features (Figure 2a). Subject 2 was a four-year-old boy who had GDD, axial hypotonia, microcephaly, strabismus, and dysmorphic facial features (Figure 2b). Subject 3 (F2-II3) (Family 2; Figure 1) was a 21-month-old boy who had GDD, hypotonia, microcephaly, and seizures. Subject 4 (F3-II1) and Subject 5 (F3-II2) were siblings (Family 3; Figure 1). Subject 4 was a six-year-old boy who had GDD, spastic quadriplegia, microcephaly, seizures, optic atrophy, failure to thrive (FTT), and dysmorphic facial features (Figure 2c–2d). Subject 5 had GDD, spastic quadriplegia, microcephaly, visual impairment, and FTT, and died at two years of age due to pneumonia with respiratory failure. Subject 6 (F4-II5) (Family 4; Figure 1) was a seven-month-old male child who had GDD, spastic quadriplegia, microcephaly, seizures, optic atrophy, and dysmorphic features (Figure 2e–2f).
Figure 2.
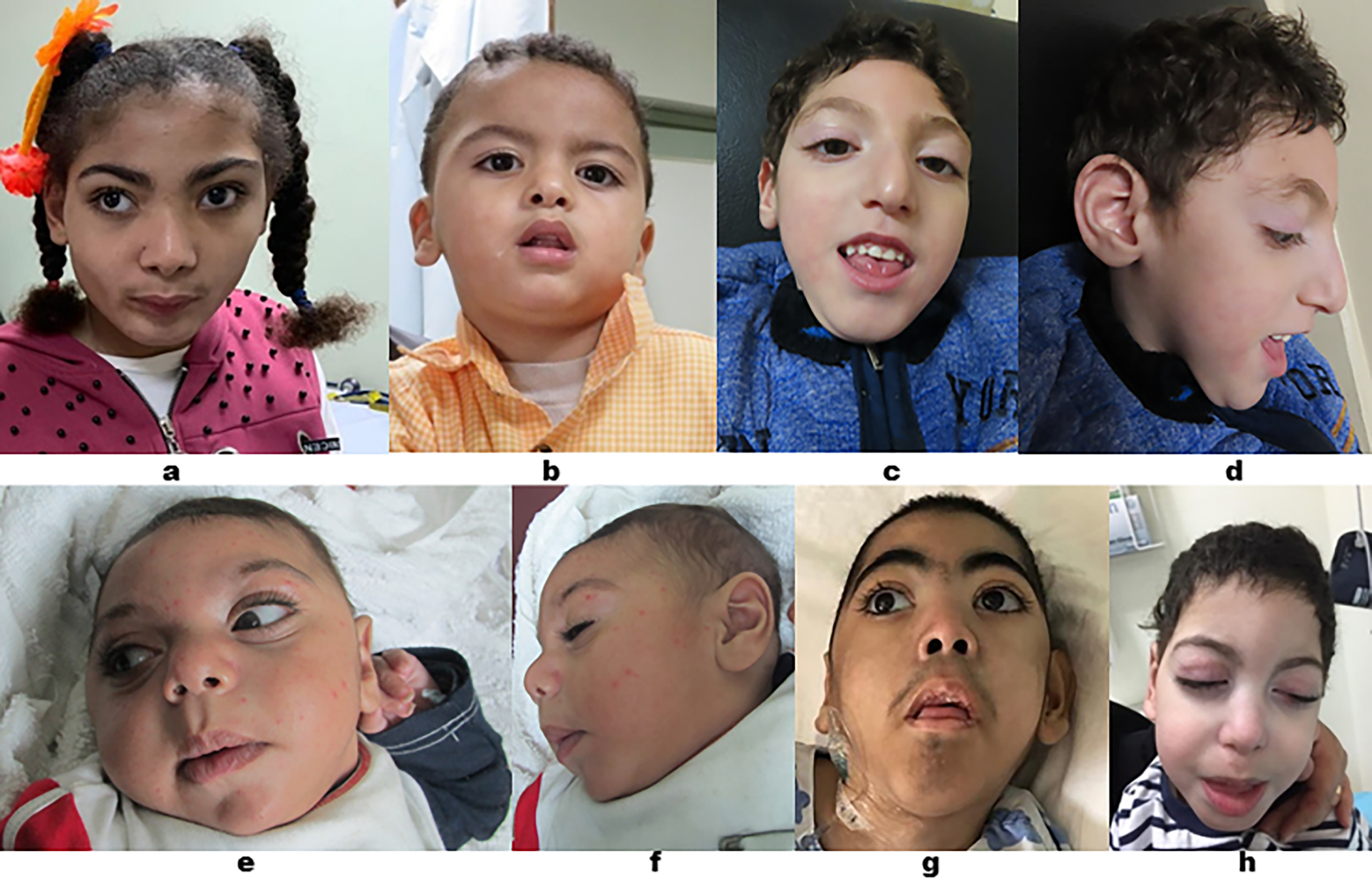
a: frontal view of Subject 1 (F1-II3) showing: facial asymmetry, bitemporal narrowing, thick and arched eyebrows, upslanted and narrow palpebral fissures, hypertelorism, fullness of upper eyelids, and full cheeks. b: frontal view of Subject 2 (F1-II7) showing: Broad forehead and epicanthal folds. c and d: frontal and lateral views of Subject 4 (F3-II1): long face, bitemporal narrowing, thick and arched eyebrows, downslanted palpebral fissures, large ears, long philtrum, thin upper lip, and broad chin. e and f: frontal and lateral views of Subject 6 (F4-II5) showing narrow palpebral fissures, hypertelorism, large ears, thick lips, long philtrum, and retromicrognathia. g: frontal view of Subject 9 (F5-III11) showingelongated face, bitemporal narrowing, thick and arched eyebrows with synophrys, epicanthal folds, hypertelorism, large ears, depressed nasal bridge, maxillary hypoplasia, long philtrum, and elongated jaw. h: frontal view of Subject 11 (F6-II8) showing elongated face, bitemporal narrowing, thick and arched eyebrows, hypertelorism, large ears, depressed nasal bridge, maxillary hypoplasia, long and smooth philtrum, and elongated jaw.
Subject 7 (F5-III2) and Subject 8 (F5-III3) were siblings (Family 5; Figure 1). Both had GDD, spastic quadriplegia, microcephaly, seizures, and FTT. Subject 7 died at eight years of age while Subject 8 was 10 years old at the time of this report. Subject 9 (F5-III11) was their cousin (Family 5; Figure 1). He had GDD, spastic quadriplegia, microcephaly, seizures, FTT requiring gastrostomy tube feeding, and dysmorphic facial features (Figure 2g). He died at 10 years of age. Subject 10 (F6-II4) and Subject 11 (F6-II8) were siblings (Family 6; Figure 1). Subject10 had GDD, spastic quadriplegia, microcephaly, seizures, cortical blindness, FTT requiring gastrostomy tube feeding, and dysmorphic facial features. She died at 15 years of age. Subject 11 was a three-year-old boy who had GDD, spastic quadriplegia, microcephaly, seizures, FTT, and dysmorphic facial features (Figure 2h). Subject 12 (F7-II3) (Family 7; Figure 1) had GDD, hypotonia, seizure, and FTT. She died at age of three and a half years.
Brain MRI images are available in 10 subjects. Common neuroradiological findings include cerebral atrophy, giant cisterna magna, corpus callosum thinning, brainstem volume loss with flattening of belly of pons, and ex-vacuo ventricular dilatation, disproportionate atrophy of the frontal lobe, symmetric under-opercularization, a posterior-horn predominance pattern of the ventriculomegaly, and dysplastic hippocampi (Table 2; Figure 3).
Table 2.
Brain MRI findings of subjects included in this report
| Subject ID | Subject 1 | Subject 2 | Subject 3 | Subject 4 | Subject 5 | Subject 6 | Subject 7 | Subject 8 | Subject 9 | Subject 10 | Subject 11 | Subject 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | F1 | F1 | F2 | F3 | F3 | F4 | F5 | F5 | F5 | F6 | F6 | F7 | Total |
| Subject | II3 | II7 | II3 | II1 | II2 | II5 | III2 | III3 | III11 | II4 | II8 | 113 | |
| Variant | Missense | LoF/Null | |||||||||||
| Cerebral atrophy | + | + | N/A | + | + | + | + | + | + | N/A | + | + | 10/10 |
| Disproportionate frontal lobe atrophy | − | − | N/A | + | + | + | + | + | + | N/A | + | + | 8/10 |
| Under-opercularization | − | − | N/A | + | + | + | + | + | + | N/A | + | + | 8/10 |
| Corpus callosum thinning | − (only foreshortening) | + | N/A | + | + | + | + | + | + | N/A | + | + | 9/10 |
| Ex-Vacuo ventricular dilatation | − | + | N/A | + | + | + | + | + | + | N/A | + | + | 9/10 |
| with posterior-horn predominance | − | − | N/A | + | + | + | + | + | + | N/A | + | + | 8/10 |
| Brainstem volume loss with flattening of belly of pons | − | + (subtle) | N/A | + | + | + | + | + | + | N/A | + | + | 9/10 |
| Dysplastic hippocampus | − | − | N/A | + | + | N/A | + | + | + | N/A | + | + | 7/9 |
| Cerebellar atrophy | − | + (Mild) | N/A | + (mild) | + | − | − | − | − | N/A | + (Mild) | + (Mild) | 5/10 |
| Cervical spine atrophy | − | − | N/A | + | + | − | + (mild) | − | − | N/A | − | − | 3/10 |
| Giant cisterna magna | + | + | N/A | + | + | + | + | + | + | N/A | + | + | 10/10 |
| Others | Abnormal signal in posterior limb of internal capsule | ||||||||||||
LoF: loss of function; N/A: not available
Figure 3. Brain MRI images for subjects with biallelic variants in WDR45B.
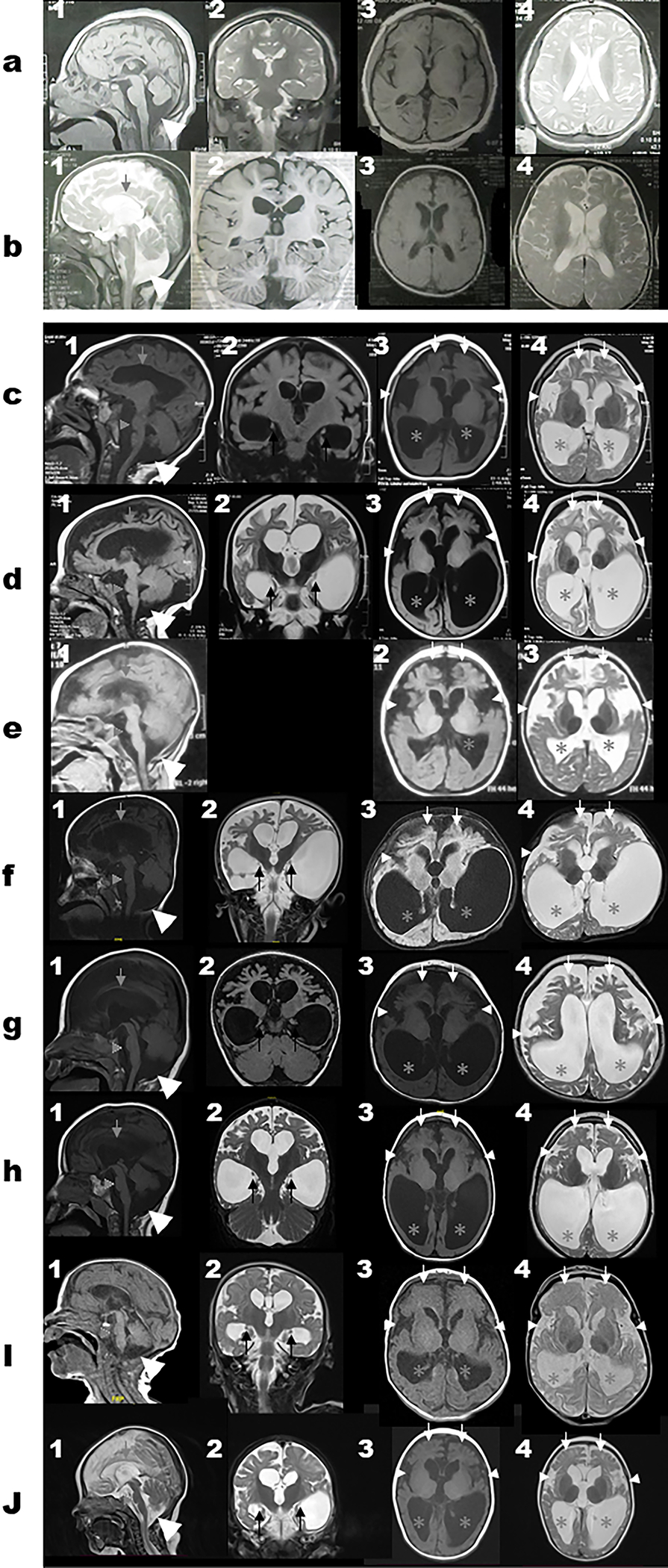
Select MRI brain images of Subject 1 (a1–4), Subject 2 (b1–4), Subject 4 (c1–4), Subject 5 (d1–4), Subject 6 (e1–3), Subject 7 (f1–4), Subject 8 (g1–4), Subject 9 (h1–4), Subject 11 (i1–4), and Subject 12 (j1–4). First column represents T1-weighted mid-sagittal images; Second column represents T1-weight or T2-weight coronal images; Third column represents T1-weight axial images while the fourth column represents T2-weight axial images. The white horizontal line separates panels a and b (for Subjects 1–2 with missense variant (c.674G>A; p.R225Q) and panels c-h (for the subjects with the loss of function variants) to show the differences in the MRI features seen in both groups. The cerebral atrophy and giant cisterna magna (large white arrow heads) are seen on panels 1a-1j. Corpus callosum thinning (grey arrows) and brainstem volume loss with flattening of belly of pons (grey arrow heads) seen in 1b-1j. Dysplastic hippocampi (black arrows) can be seen on the coronal images on panels 2c-2j. The disproportionate frontal lobe atrophy (white arrows), symmetric under-opercularization (small white arrow heads), and posterior-horn predominance pattern of the ventriculomegaly (grey asterisks) are best seen on panels 3c-3j and 4c-4j.
MOLECULAR FINDINGS
Six different variants in WDR45B [NM_019613.4] were identified in the seven unrelated families. Subjects 1 and 2 (Family 1) were found to be homozygous for a novel missense variant (c.674G>A; p.R225Q). This variant is predicted to be deleterious by multiple in silico prediction models (Supplementary Table 1). Subject 3 (Family 2) was found to be homozygous for a novel 18 bp deletion at an intron-exon junction (c.428–10_435del) which is predicted to affect splicing (Supplementary Table 1). The nature of this potential in-frame deletion which includes part of the neighboring exon in addition to an acceptor site strongly support the pathogenicity of this variant. Subjects 4 and 5 (Family 3) were found have a novel homozygous splice-site c.619–3A>G variant. This variant is predicted to alter the splice acceptor site based on multiple splicing algorithms (Supplementary Table 1). Subject 6 (Family 4) was homozygous for the previously reported nonsense variant (c.673C>T; p.R225*). Subjects 7,8, and 9 (Family 5) and subjects 10 and 11 (Family 6) were all homozygous for a novel splice-site variant (c.427+4A>G). Multiple splicing algorithms suggest alteration of the splice-donor site (Supplementary Table 1). Subject 12 (Family 7) was homozygous for splice-site variant (c.67+1G>T) that affects canonical splice-donor site and is predicted to impact splicing by all algorithms (Supplementary Table 1).
All variant alleles are ultra-rare (minor allele frequency [MAF]≤ 1/10,000) and absent in the homozygous state from the databases of BCM GREGoR (Genomics Research to Elucidate the Genetics of Rare diseases) and from the Genome Aggregation Database (GnomAD v.2.1.1)15,16. Only the missense variant (c.674G>A: p.R225Q) was detected in heterozygous state in one control subject in GnomAD with a MAF of 0.000003 and in the unaffected parents of Family 1 in BCM GREGoR database with a MAF of 0.000172.
Sanger dideoxy sequencing confirmed the variant alleles and segregation with disease in all families except for one (Family 5) as samples of additional family members were not available for segregation (Figure 1).
DISCUSSION
In this report, we present detailed clinical, molecular, and radiological features of 12 individuals from seven unrelated families with El-Hattab-Alkuraya syndrome. Ten subjects were of Arab ancestry (from Saudi Arabia and Egypt), one was Iranian, and the other was of Cree. Consanguinity was historically reported in 11 subjects. Six different variants in WDR45B were identified in the seven unrelated families including one missense variants (c.674G>A: p.R225Q) and five loss-of-function (LoF) variants: one frameshift variant (c.428–10_435del), one nonsense variant (c.673C>T: p.R225*) and three different splice-site variants (c.619–3A>G; c.427+4A>G, and c.67+1G>T). Variants were novel except for the nonsense variant (c.673C>T: p.R225*) which was recurrent and identified previously in three unrelated families. Such recurrent SNV alleles can occur by CpG dinucleotide mutagenesis; 4 of 6 Arg codons have CpG dinucleotides.
All subjects exhibited microcephaly (average z-score = −5.6; range −2.3 to −9.1 for the subject with data available) and severe to profound GDD with dependency on their caregivers for activities of daily living. The two siblings with missense variants showed slow gain of some skills and regression was not evident. Microcephaly was noted at birth in most individuals for whom measurements are available (4/5). In fact, evidence suggest microcephaly develops in utero and could be identified prenatally during the third trimester in two subjects (Subject 5 and Subject 10). When available, hearing assessment was normal while most subjects have poor visual tracking and one of them was diagnosed with cortical blindness. Optic atrophy was reported in two subjects while two other subjects have strabismus. Neurobehavioral problems, with aggressive and self-harming behavior were reported in one subject. All subjects but two displayed spastic quadriplegia that was associated with axial hypotonia in some of them.
Seizures were evident in 11 out of the 12 subjects. All subjects presented clinically with infantile-onset epilepsy (average age of onset of seizures 1.9 months; range: few days after birth- 6 months). The most common seizure form was generalized tonic clonic (GTC). Other seizure forms reported are myoclonic (5/12 subjects) and focal (3/12 subjects). Epilepsy was drug-resistant (requiring ≥ 2 antiepileptic drugs) in all subjects.
Subtle dysmorphic features were reported in most individuals (Figure 2). The most common reported features are thick and arched eyebrows and large ears (noted in five subjects); bitemporal narrowing, and long philtrum; each noted in four individuals.
Recurrent, unexplained miscarriages were noted in three families. Most subjects were born at full term. Two subjects were born with a low birth weight. FTT was noted in eight individuals and two subjects required gastrotomy tube feeding. Five subjects are deceased at the time of this report (age of death range 2–15 years; mean 7.9 years). When available, the most common cause for death was aspiration pneumonia which is an expected complication in patients with neurodegenerative diseases.
The consistent phenotype in this cohort further supports that El-Hattab-Alkuraya syndrome is a recognizable pattern of human malformation, i.e., a specific syndrome; the clinical synopsis of this rare disease trait is characterized by GDD, spastic quadriplegia, seizures, microcephaly, FTT, and distinctive facial features.
Common neuroradiological findings include cerebral atrophy and giant cisterna magna, both noted in all 10 subjects. Corpus callosum thinning, brainstem volume loss with flattening of belly of pons, and ex-vacuo ventricular dilatation were noted in most individuals (9/10). Notably, disproportionate atrophy of the frontal lobe (8/10), symmetric under-opercularization (8/10), posterior-horn predominance pattern of the ventriculomegaly (8/10), and dysplastic hippocampi (7/9) were additional neuroradiographic features that were only observed in the subjects with the LoF alleles. Finally, cerebellar atrophy and cervical spine atrophy were noted in five and three individuals, respectively. Notably, the neuroradiological findings appeared homogenously consistent among all current and previously reported subjects with the LoF variant alleles8, while those with the missense variants lacked the characteristic imaging findings (Figure 3 and Table 2). Cerebral atrophy, thin corpus callosum and ex-vacuo dilatation with cervical cord atrophy are reflective of white matter loss while the disproportionate frontal lobe atrophy suggests a frontal and temporal potential neurodegenerative process. This anterior-predominant cerebral atrophy could explain the bitemporal narrowing noted in several individuals. Given the striking similarities in brain MRI findings, we propose the following four neuroimaging criteria that when observed could potentially suggest the radiologist or clinician consider El-Hattab-Alkuraya syndrome: 1) cerebral atrophy disproportionately most prominent in frontal lobes, 2) ex-vacuo ventricular dilatation with notable posterior-horn predominance, 3) brainstem volume loss with flattening of belly of the pons, and 4) symmetric under-opercularization.
WDR45B encodes for WD (tryptophan-aspartic acid) repeat-containing protein 45B and is a member of the WIPI protein family. WIPI (WD-repeat protein Interacting with Phospho Inositides) proteins play an important role in autophagy through functioning as scaffold building units that interconnect autophagy signal control and autophagosome formation17,18. These proteins can be classified into two subgroups;WIPI1 and WIPI2 in the first group, and WDR45B (WIPI3) and WDR45 (WIPI4) in the second group19. WIPI proteins consist of a seven-bladed β-propeller structure encoded by seven WD40 repeats20. Blades 5 and 6 encompass two distinct functionally-conserved selective phosphoinositide(PIP)-binding sites that recognize two different PIPs20,21; PI(3)P is recognized by Site I on blade 5 while PI(3,5)P2 is recognized by Site II on blade 620.Embedded between Sites 1 and 2 is a highly-conserved signature L/FRRG motif that plays a major role in PIP recognition20,21 and autophagy function. Autophagy is essential for the maintenance of cellular homeostasis. Deregulation of autophagy has been implicated in a number of diseases, including neurodegenerative diseases22. Moreover, autophagy and apoptosis appear to have a major role in ‘shaping the brain’ during neurodevelopment and neuron migration.
Wdr45b knockout mice developed motor deficits and learning and memory defects. Their brains showed swollen axons and cerebellar atrophy with autophagy substrates accumulation in various brain regions. Interestingly, wdr45b and wdr45 double knockout mice had more severe autophagy defects and died early suggesting epistasis and that these two genes act together in autophagy19. Subsequently, the same group showed that WDR45/45B are specifically required for neural autophagy where they are involved in the progression of autophagosomes into autolysosomes19. This is achieved through WDR45/45B interaction with tether protein EPG5 and then targeting it to late endosomes/lysosomes to promote autophagosome maturation23. Interestingly, inhibiting O-GlcNAcylation facilitates the fusion of autophagosomes with late endosomes/lysosomes in Wdr45/45b double knockout (DKO) cells, indicating a potential therapeutic target23. Furthermore, pathogenic variants in the related gene, WDR45, are associated with beta-propeller protein-associated neurodegeneration (BPAN)24.This disorder has an overlapping phenotype with El-Hattab-Alkuraya syndrome including early-onset seizures and intellectual disability. With age seizures become less prominent, perhaps as epileptic neuronal circuits degenerate, whereas cognitive decline and movement disorders emerge as characteristic findings25.
The early-onset microcephaly noted in most patients, including in utero findings, indicates a very early onset disease process. For individuals in whom more than one measurement is available, and as shown in one of the individuals in the initial report, microcephaly was progressive8. One of the possible mechanisms for the disease process is accelerated programmed cell death given the role of WDR45B protein in autophagy. In fact, apoptosis was documented in wdr45 knockout (KO) mouse model26. Loss of WDR45 resulted in elevated endoplasmic reticulum (ER) stress which triggered the unfolded protein response (UPR) leading to neuronal apoptosis26. Such a mechanism has been well established in the peripheral nervous system neurons with therapeutic targeting, like curcumin, showing symptomatic improvement and abrogation and amelioration of the disease process in animal models for Charcot-Marie-Tooth disease (CMT)27–30.
Subject 1 and Subject 2 harbored the missense variant (c.674G>A: p.R225Q). An early genotype-phenotype correlation shows that these two siblings only lacked the characteristic neuroradiological findings (as discussed above) but also had milder clinical features compared to the LoF alleles. These patients had mainly dysmorphism and GDD, but with absence of seizures and spastic quadriplegia.The R225 amino acid localizes to the L/FRRG motif on WIPI3 (WDR45B) and introducing a p.R225A mutation at this locus that has been shown to abolish lipid binding capacity of WIPI320. Thus, we suggest that the missense variant herein (c.647G>A: p.R225Q) is a hypomorphic variant allele resulting in a configurational change that partially impairs the L/FRRG binding motif function and leads to an attenuated phenotype. Remarkably, the recurrent variant observed here (c.673C>T: p.R225*) also affects same amino acid but likely results in complete LoF. The splicing and frameshift variants also likely result in complete LoF (amorphic alleles); their resultant transcripts are predicted or anticipate to introduce premature termination codons (PTC) that are either degraded by non-sense-mediated decay (NMD) or result in a truncated protein31. Further functional assessment of the variant might further substantiate this hypothesis.
Two different families (Families 5 and 6) have the same splicing variant (c.427+4A>G). These families belong to the same tribe in Saudi Arabia, indicating a possible tribal founder allele for this private variant, which is well known in geographic areas of the world with a high rate of consanguinity32. Additionally, the recurrent nonsense variant c.673C>T: p.A225* observed in an Egyptian family and was previously reported in three unrelated families in two papers from Saudi Arabia7,8. It is likely this represents a mutation hotspot33, occurring by CpG dinucleotide mutagenesis. An Arab founder allele for c.673C>T: p.A225* is a less likely possibility given this variant is absent from several control databases including gnomAD 2.1.1, Greater Midde East Variome database34 with >1,100 control of Middle East origin (igm.ucsd.edu/gme/), and also from BCM GREGoR database enriched for Arab/Middle Eastern families.
In conclusion, here we present the clinical, molecular, and radiological features of El-Hattab-Alkuraya syndrome in 12 subjects with five novel WDR45B variants and one recurrent variant. This disorder is characterized by a predominant neurological phenotype with GDD, spastic quadriplegia, microcephaly, and early onset and refractory epilepsy. OurWDR45B allelic series further defines the trait, provides insights into the neurodevelopment and neurobiology of disease, and potentially implicates approaches to molecular therapies to including drugs like curcumin used already in many clinical trials. We suggested a possible genotype-phenotype correlation with evidence that LoF variants are associated with severe disease and characteristic potentially recognizable neuroimaging findings that should trigger clinicians and radiologist to consider the diagnosis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the families for their participation in the study.
FUNDING INFORMATION
This study was supported in part by the U.S. National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHBLI) to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542), NHGRI Baylor College of Medicine Genomics Research Elucidates Genetics of Rare (BCM-GREGoR; U01 HG011758). Also partially funded by U.S. National Institute of Neurological Disorders and Stroke (NINDS) (R35NS105078), National Institute of General Medical Sciences (NIGMS, R01GM106373), the Muscular Dystrophy Association (MDA;512848), and Spastic Paraplegia Foundation (SPF)to J.R.L. D.M. was supported by a Medical Genetics Research Fellowship Program through the United States National Institute of Health (T32 GM007526–42). J.E.P. was supported by NHGRI K08 HG008986. D.G.C. was supported by NIH - Brain Disorders and Development Training Grant (T32 NS043124–19) and MDA Development Grant (873841).
Footnotes
COMPETING INTERESTS
J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG); J.R.L. serves on the Scientific Advisory Board (SAB) of BG. WKC is a paid consultant for Regeneron Genetics Center.
Other authors declare no conflict of interest
DATA AVAILABILITY
The data that supports the findings of this study are available in the supplementary material of this article
REFERENCES
- 1.Mitani T, Isikay S, Gezdirici A, et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am J Hum Genet. 2021;108(10):1981–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullin AP, Gokhale A, Moreno-De-Luca A, Sanyal S, Waddington JL, Faundez V. Neurodevelopmental disorders: mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl Psychiatry. 2013;3:e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lupski JR. Clan genomics: From OMIM phenotypic traits to genes and biology. Am J Med Genet A. 2021;185(11):3294–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savatt JM, Myers SM. Genetic Testing in Neurodevelopmental Disorders. Front Pediatr. 2021;9:526779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardoso AR, Lopes-Marques M, Silva RM, et al. Essential genetic findings in neurodevelopmental disorders. Hum Genomics. 2019;13(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478(7367):57–63. [DOI] [PubMed] [Google Scholar]
- 7.Anazi S, Maddirevula S, Faqeih E, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2017;22(4):615–624. [DOI] [PubMed] [Google Scholar]
- 8.Suleiman J, Allingham-Hawkins D, Hashem M, Shamseldin HE, Alkuraya FS, El-Hattab AW. WDR45B-related intellectual disability, spastic quadriplegia, epilepsy, and cerebral hypoplasia: A consistent neurodevelopmental syndrome. Clin Genet. 2018;93(2):360–364. [DOI] [PubMed] [Google Scholar]
- 9.Bauer P, Kandaswamy KK, Weiss MER, et al. Development of an evidence-based algorithm that optimizes sensitivity and specificity in ES-based diagnostics of a clinically heterogeneous patient population. Genet Med Off J Am Coll Med Genet. 2019;21(1):53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu N, Welch CL, Wang J, et al. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med. 2018;10(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014;42(22):13534–13544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176(3):535–548.e24. [DOI] [PubMed] [Google Scholar]
- 13.Valenzuela-Palomo A, Bueno-Martínez E, Sanoguera-Miralles L, et al. Splicing predictions, minigene analyses, and ACMG-AMP clinical classification of 42 germline PALB2 splice-site variants. J Pathol. Published online November 30, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen AW, Murugan M, Li H, et al. A Genocentric Approach to Discovery of Mendelian Disorders. Am J Hum Genet. 2019;105(5):974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Proikas-Cezanne T, Takacs Z, Dönnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128(2):207–217. [DOI] [PubMed] [Google Scholar]
- 18.Bakula D, Müller AJ, Zuleger T, et al. WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat Commun. 2017;8:15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji C, Zhao H, Li D, et al. Role of Wdr45b in maintaining neural autophagy and cognitive function. Autophagy. 2020;16(4):615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang R, Ren J, Zhang Y, Feng W. Structural Conservation of the Two Phosphoinositide-Binding Sites in WIPI Proteins. J Mol Biol. 2019;431(7):1494–1505. [DOI] [PubMed] [Google Scholar]
- 21.Ren J, Liang R, Wang W, Zhang D, Yu L, Feng W. Multi-site-mediated entwining of the linear WIR-motif around WIPI β-propellers for autophagy. Nat Commun. 2020;11(1):2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. [DOI] [PubMed] [Google Scholar]
- 23.Ji C, Zhao H, Chen D, Zhang H, Zhao YG. β-propeller proteins WDR45 and WDR45B regulate autophagosome maturation into autolysosomes in neural cells. Curr Biol CB. 2021;31(8):1666–1677.e6. [DOI] [PubMed] [Google Scholar]
- 24.Haack TB, Hogarth P, Kruer MC, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91(6):1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gregory A, Kurian MA, Haack T, Hayflick SJ, Hogarth P. Beta-Propeller Protein-Associated Neurodegeneration. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews®. University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 26.Wan H, Wang Q, Chen X, et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy. 2020;16(3):531–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pennuto M, Tinelli E, Malaguti M, et al. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57(3):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khajavi M, Lupski JR. Balancing between adaptive and maladaptive cellular stress responses in peripheral neuropathy. Neuron. 2008;57(3):329–330. [DOI] [PubMed] [Google Scholar]
- 29.Khajavi M, Inoue K, Wiszniewski W, Ohyama T, Snipes GJ, Lupski JR. Curcumin treatment abrogates endoplasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am J Hum Genet. 2005;77(5):841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khajavi M, Shiga K, Wiszniewski W, et al. Oral curcumin mitigates the clinical and neuropathologic phenotype of the Trembler-J mouse: a potential therapy for inherited neuropathy. Am J Hum Genet. 2007;81(3):438–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coban-Akdemir Z, White JJ, Song X, et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am J Hum Genet. 2018;103(2):171–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health. 2009;6(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nesta AV, Tafur D, Beck CR. Hotspots of Human Mutation. Trends Genet TIG. 2021;37(8):717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott EM, Halees A, Itan Y, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48(9):1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article