

SUMMARY
Proliferating cells known as neoblasts include pluripotent stem cells (PSCs) that sustain tissue homeostasis and regeneration of lost body parts in planarians. However, the lack of markers to prospectively identify and isolate these adult PSCs has significantly hampered their characterization. We used single-cell RNA sequencing (scRNA-seq) and single cell transplantation to address this long-standing issue. Large-scale scRNA-seq of sorted neoblasts unveiled a novel subtype of neoblast (Nb2) characterized by high levels of PIWI-1 mRNA and protein, and marked by a conserved cell-surface protein coding gene, tetraspanin 1 (tspan-1). tspan-1-positive cells survived sub-lethal irradiation, underwent clonal expansion to repopulate whole animals, and when purified with an anti-TSPAN-1 antibody, rescued the viability of lethally irradiated animals after single-cell transplantation. The first prospective isolation of an adult PSC bridges a conceptual dichotomy between functionally and molecularly defined neoblasts, shedding light on mechanisms governing in vivo pluripotency and a source of regeneration in animals.
INTRODUCTION
Pluripotent stem cells (PSCs) are normally embryonic cells essential for the generation of all three embryonic germ layers and resulting mature cell types. However, planarians are among the few known organisms capable of indefinitely maintaining PSCs beyond embryogenesis. First described in the late 1800’s, these adult undifferentiated cells are known today as neoblasts (Newmark and Sánchez Alvarado, 2002 and references therein), originate from early embryonic blastomeres (Davies et al 2017), persist throughout adult life, and collectively produce all cell types found in the planarian body plan, including the germ line in sexually reproducing animals (Roberts-Galbraith and Newmark, 2015). Planarians are renowned for their ability to restore damaged body parts to full function, and to regenerate complete animals even from tiny tissue fragments. For over a century, neoblasts have been known to play an essential role in the regenerative capabilities of planarians, and detailed morphological studies have helped identify these cells in the planarian body plan (Newmark and Sánchez Alvarado, 2002 and references therein). Molecular studies have uncovered specific markers for neoblasts, particularly the expression of piwi-1 (Reddien et al., 2005; Sánchez Alvarado et al., 2002), while Fluorescence Activated Cell Sorting (FACS) of cells derived from wild-type and lethally-irradiated animals have helped identify and purify cell fractions (X1 and X2) enriched in neoblasts and progenitors (Hayashi et al., 2006; Reddien et al., 2005).
More recent work involving partial irradiation and transplantation methods (Guedelhoefer and Sánchez Alvarado, 2012 and references therein) showed neoblast repopulation and migration are additional properties associated with pluripotency. A series of experiments involving single-cell transplantations demonstrated that a single neoblast is capable of rescuing lethally irradiated hosts by reconstituting all cell types in the animal’s body, albeit at very low frequencies (Wagner et al., 2011). Only 7 out of 120 injected neoblasts resulted in successful restoration of host viability, suggesting extensive functional heterogeneity in the neoblast population. To differentiate neoblasts capable of rescuing lethally irradiated animals from those that failed to do so, the cells were named clonogenic neoblasts (cNeoblasts). Additionally, single-cell gene expression analyses in the related species Dugesia japonica (Hayashi et al., 2010) and the detection of expression of 96 genes in 176 individual neoblasts isolated by FACS (van Wolfswinkel et al., 2014) demonstrated that neoblasts also display significant molecular heterogeneity. Three classes of neoblasts (σ, γ, ζ) were defined on the basis of relatively limited expression profiles and functional assays. For instance, the σ-class neoblasts which proliferate in response to injury, possess broad lineage capacity, and give rise to the ζ-neoblasts, suggesting that pluripotency and cNeoblasts are likely associated with this neoblast class. However, because σ-class neoblasts are defined empirically and thus cannot be isolated prospectively, it has not been possible to determine via single cell transplantation if all neoblasts in this class are pluripotent. While previous studies provided important insights into key aspects of neoblast biology, the present dichotomy between the functional (cNeoblast) and molecular definitions (σ, γ, ζ) of neoblasts continues to significantly hinder our understanding of pluripotency regulation in adult animals. Hence, it remains difficult to precisely define: 1) how many functional types or states of neoblasts exist in the large proliferative cell population of adult planarians; 2) the properties and expressed genetic programs of cNeoblasts; and 3) how pluripotent neoblasts respond specifically to regeneration signals to produce all the cell types needed to restore form and function.
Prospective cell sorting followed by transplantation has been the gold standard to assess functional properties of stem cells in multiple systems (Reya et al., 2001), but this technique has lacked adequate molecular markers and reagents for specifically labeling and isolating unique planarian cell types. Although gene expression profiles of multiple stem cell populations have been described (Molinaro and Pearson, 2016; van Wolfswinkel et al., 2014; Wurtzel et al., 2015), no single family of cell-surface proteins has yet been found in which members are differentially expressed in a way that correlate with pluripotency. We show for the first time the identification and prospective isolation of adult PSCs. By enriching for cells high in both transcript and protein levels of the neoblast-specific marker PIWI-1, followed by single-cell RNA-seq (scRNA-seq) of 7,088 of these cells, we uncovered a novel PIWI-1high neoblast subtype (Nb2) characterized by expression of a tetraspanin family member homolog we termed tspan-1. RNAi of tspan-1 affected both neoblast repopulation and neoblast mobilization. Importantly, tspan-1-positive cells survive sub-lethal irradiation and undergo clonal expansion to repopulate whole animals, fulfilling the operational criteria defining cNeoblasts. Moreover, an antibody developed against TSPAN-1 allowed us to prospectively isolate adult PSCs: a single transplanted TSPAN-1+ neoblast into lethally irradiated hosts restored viability and fully reconstituted regenerative capacities with high efficiency. Thus, scRNA-seq coupled with new prospective cell sorting assays has opened the door for a detailed dissection of the underlying molecular and cellular mechanisms regulating in vivo pluripotency and whole-body regeneration.
RESULTS
Simultaneous detection of piwi-1 expression and PIWI-1 protein levels in single cells define a functional spectrum of neoblast states in vivo
Different levels of PIWI-1 protein and transcript occur in neoblasts and their progeny (Molinaro and Pearson, 2016). Using super-resolution microscopy, we noted piwi-1+ cells displayed a heterogeneous number of piwi-1 transcripts (Fig. 1A) and sought to test this observation by quantifying piwi-1 expression in cell populations via image-based flow cytometry (Porichis et al., 2014). A piwi-1high subpopulation encompassing 41% of the total number of cells, and a piwi-1low subpopulation representing 6% of the total cells (Figs. 1B; S1A). Signal specificity and observed cell subpopulations were confirmed via Imagestream (Fig. S1B). An anti-PIWI-1 antibody also revealed two PIWI-1-positive cell subpopulations (Figs. 1C; S1C, D), and Hoechst co-staining indicated that 68.4% of PIWI-1high cells were in S/G2/M phase (X1 gate), while 81.6% of all PIWI-1low cells were in G0/G1 phase (X2, Xins gates) (Fig. S1E). Therefore, high levels of PIWI-1 protein appear primarily associated with actively dividing cells, whereas low levels of PIWI-1 protein are associated with interphase cells (G0/G1) (Guo et al., 2006).
Figure 1. Piwi-1 mRNA and protein measurements in neoblast populations.
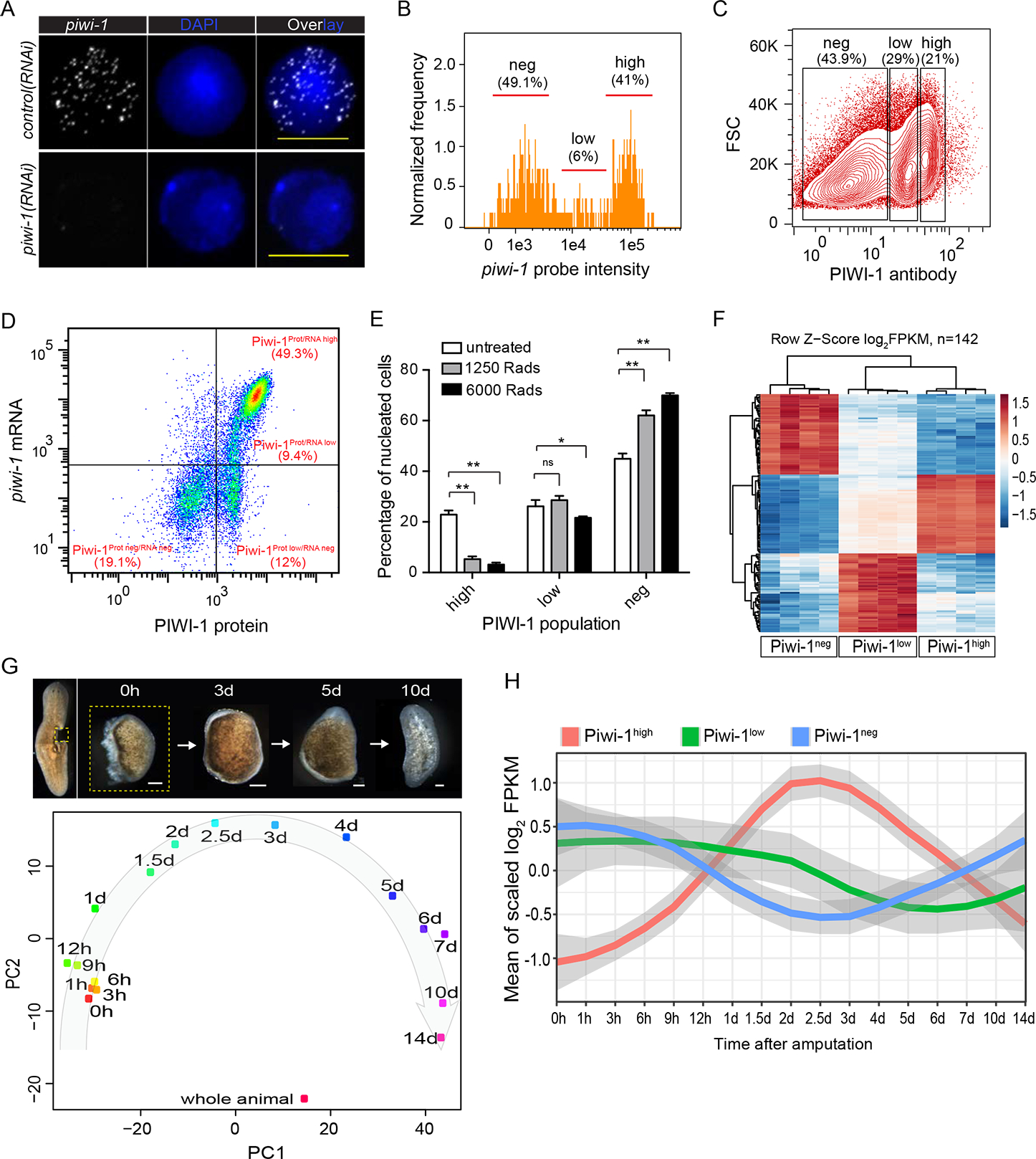
(A) Super resolution images of FISH staining of piwi-1 transcripts on single X1 cells from control or piwi-1(RNAi) animals. Representative cells shown. n>10 for each condition. Scale bar, 10 μm.
(B) piwi-1 transcript distribution by ImageStream flow cytometric analysis. piwi-1 high cells, high; piwi-1 low cells, low; piwi-1 negative cells, neg. Positive cell population determined by distribution of negative control probe stained cells shown in S1A. Representative of 3 independent experiments shown.
(C) PIWI-1 antibody intracellular staining followed by flow cytometric analyses. PIWI-1 high cells, high; PIWI-1 low cells, low; PIWI-1 negative cells, neg. Representative of more than 3 independent experiments shown.
(D) Representative FACS plot and frequency of pre-sorted cells after co-staining with piwi-1 probe and PIWI-1 antibody. Positive cell population determined by distribution of negative control probe or isotype antibody stained cells shown in S1F. Representative of 3 independent experiments shown.
(E) Comparison of PIWI-1 signal levels in cells from planarian 1-day after treatment with different irradiation dosages. Each column represents percent of indicated cells on total nucleated cells. Error bars: SD. **p-value <0.001, *p-value <0.05.
(F) Differential expression heatmap of signature genes for Piwi-1high, Piwi-1low and Piwi-1neg populations. Shown are log2FPKM with row scaled (z-score) based on RNA-seq data of cell populations shown in (C). Shown are 142 genes enriched for each cell population (Table S1).
(G) Principal component analysis (PCA) of all regeneration time points profiled. Data were from time-course of small tissue fragments undergoing a full cycle of whole-body regeneration. Each dot represents the average of 4 replicates. Scale bar, 100 μm. (H) Dynamic changes of signature genes specifically expressed in each of the three PIWI-1 groups (shown in F) over the RNA-seq of whole-body regeneration time courses. See also Figs. S1, S2; Table S1.
piwi-1 RNA and PIWI-1 protein co-staining followed by flow cytometry defined a spectrum of cells that broadly binned into 4 subpopulations (Figs. 1D; S1F). Cells possessing higher PIWI-1-protein and piwi-1 RNA levels (Piwi-1Prot/RNA high), designated as Piwi-1high. Two subpopulations displaying either low (Piwi-1Prot/RNA low) or undetectable piwi-1 RNA levels (Piwi-1Prot low/RNA neg), referred to as Piwi-1low. And a final Piwi-1neg subset with undetectable levels of either protein or RNA (Piwi-1Prot neg/RNA neg). Piwi-1high cells were mainly cycling cells. Piwi-1low cells are homogeneous in size and found mostly in G0/G1 phase of the cell cycle, and which we propose to represent both the non-cycling stem cells readying for cell cycle re-entry and fate-determined progenitor cells. Lastly, Piwi-1neg cells were heterogeneous in size, likely representing cells that have exited the cell cycle (Fig. S1E).
Piwi-1high cells are enriched in stem cell activity, yet are transcriptionally distinct from X1 cells
Sublethal (1,250 rads) or lethal (6,000 rads) irradiation caused little alteration or a mild reduction (~10% loss for lethal) of Piwi-1low cells, respectively (Fig. 1E). Yet, Piwi-1high cells were almost entirely eliminated 1 day post irradiation (dpi) at lethal dosages, with ~20% of cells surviving sublethal dosages. Because PIWI-1+ residual cells retained a similar pre-irradiation distribution (Fig. S1G), changes in PIWI-1 levels caused by irradiation are likely not caused by a reduction of protein levels in individual cells, but rather by elimination of Piwi-1high cells (Fig. S1H). Sublethally irradiated animals eventually recover, showing a requirement for a small number of Piwi-1high cells for maintaining pluripotency. While consistent with observations of residual cNeoblasts (Wagner et al., 2011), the data indicated that not all Piwi-1high cells have an equal response to irradiation and that a cohort of Piwi-1high cells that are better at surviving irradiation may exist. Transcriptional differences between Piwi-1high, Piwi-1low, Piwi-1neg and Hoechst-sorted X1, X2 and Xins cell fractions support this conclusion (Fig. S2A–D; Method Details). We found Piwi-1high cells to be similar to X1 cells, while Piwi-1low cells were more akin to X2 and Xins cells (Fig. S2E). Although expression profiles of Piwi-1high and X1 cells were highly similar (Fig. S2E), Piwi-1high cells remained transcriptionally distinct from X1 cells (Fig. S2F) and cannot be readily resolved using σ, γ, and ζ neoblast class markers (Fig. S2G), suggesting that Piwi-1high cells are distinct from previously reported X1 cells, and may be a specific subset of stem cells.
Piwi-1high cells encompass spectrum of neoblast types that likely include cNeoblasts
We identified specific markers for Piwi-1high, Piwi-1low and Piwi-1neg cells and defined their expression under irradiation and regeneration (Fig. 1F; Table S1). Consistent with flow cytometry data (Fig. 1E), sub-lethal irradiation drastically reduced the expression of Piwi-1high signature genes, leaving Piwi-1low and Piwi-1neg gene expression signatures unaltered by 1 dpi (Fig. S2H). Irradiation sensitivity and recovery of Piwi-1high, but not of Piwi-1low cells after 4 dpi (Fig. S2H), indicated that Piwi-1high cells were likely major contributors to neoblast repopulation after sublethal irradiation. As expected, lethal irradiation diminished high and low signature gene expressions (Fig. S2I), and led to animal demise. We then followed the expression of Piwi-1high, Piwi-1low and Piwi-1neg signature markers throughout the full RNAseq regeneration cycle of complete animals from minute body fragments (Figs. 1G, H). Piwi-1high markers increased dramatically after amputation, reached a peak 2 days after amputation, and decreased afterward, consistent with PSC responses to amputation (van Wolfswinkel et al., 2014; Wenemoser and Reddien, 2010). However, Piwi-1low and Piwi-1neg markers were all down-regulated by 4 to 7 days, and gradually recovered accompanied by downregulation of Piwi-1high gene signatures (Fig. 1G, H). Our data indicate that Piwi-1high cells are key players in the initial regeneration response and that cNeoblast activity likely resides with Piwi-1high cells.
We determined how Piwi-1high and Piwi-1low cells may relate to cells obtained via X1(FS) flow cytometric gates used to detect cNeoblasts (Wagner et al., 2011). Since Piwi-1high cells are slightly bigger in size (FSC parameter, Fig. 1C), we devised a sorting strategy to isolate Piwi-1high and Piwi-1low enriched subpopulations (Fig. S2J). Immunofluorescence staining for PIWI-1 showed Piwi-1high cells populating the X1(FS)-L gate, while the X2(FS)-R gate was populated with Piwi-1low cells (Fig. S2J, K). We measured their respective proliferative capacities via F-ara-EdU labeling followed by sorting X1(FS)-L and X2(FS)-R subpopulations and co-staining with PIWI-1. We found that F-ara-EdU signal was mainly found in X1(FS)-L gate Piwi-1high cells (Fig. S2K, L). We conclude that high levels of both piwi-1 gene expression and PIWI-1 protein positively correlate with a neoblast subpopulation encompassing a defined spectrum of functional states, including cNeoblasts.
Single cell RNA-seq resolves cellular diversity of Piwi-1high cells.
Intrigued by the properties of Piwi1high cells, we isolated X1 cells enriched in high piwi-1 expression (Fig. S3A, B), and profiled ~7,614 individual cells via scRNA-seq (Fig. 2). Unsupervised analyses that did not rely on known markers of neoblast subtypes uncovered 12 distinct classes from 7,088 high-quality cells after QC filter (Method Details). We designated these classes Nb1 to Nb12 and ordered them based on high (Nb1) to low (Nb12) piwi-1 expression levels (Figs. 2A; S3C). We further defined groups of genes that best classified the cells parsed into 12 distinct cell clusters to generate a scaled expression heatmap of discriminative gene sets for each cluster (Method Details; Fig. 2B; see Table S2 for a list of markers). Expression of each cluster’s gene signatures was validated using multiplex FISH co-stained with piwi-1 (Fig. 2C, D), and largely confirmed the cell clusters revealed by scRNA-seq (Fig. 2E, F). We also systematically compared the differentially expressed markers of each major cell type to gene signatures extracted from existing scRNA-seq datasets (Table S2) (Molinaro and Pearson, 2016; Wurtzel et al., 2015). We found 7 of the 12 clusters identified were readily characterized by post-hoc expression overlap of previously characterized cell-type specific markers, suggesting that they are progenitors. For instance, clusters with high piwi-1 expression (Fig. S3C) such as Nb1 and Nb5 map to ζ- and γ-class neoblasts, respectively, while clusters Nb4 and Nb6 were enriched in muscle gene expression (e.g., collagen, foxd, myosin light chain) (Witchley et al., 2013). Although the expression of known ζ and γ class markers (p53 and hnf-4) readily segregated into distinct clusters (e.g., Nb1 and Nb5, respectively), expression of the σ-class marker soxP-1 did not (Fig. S3D, E). Instead, soxP-1 was found highly expressed in at least 6 neoblast cell clusters, albeit at different levels (Fig. S3D, E), suggesting soxP-1 expression might be akin to piwi-1 in showing quantitative differences across cells. The observed soxP-1 expression dispersion across the population of cells sampled is consistent with past observations (Molinaro and Pearson, 2016), suggesting soxP-1 is expressed in multiple lineage progenitors and PSCs.
Figure 2. Single cell RNA-seq resolves the cellular complexity of Piwi-1high cells.
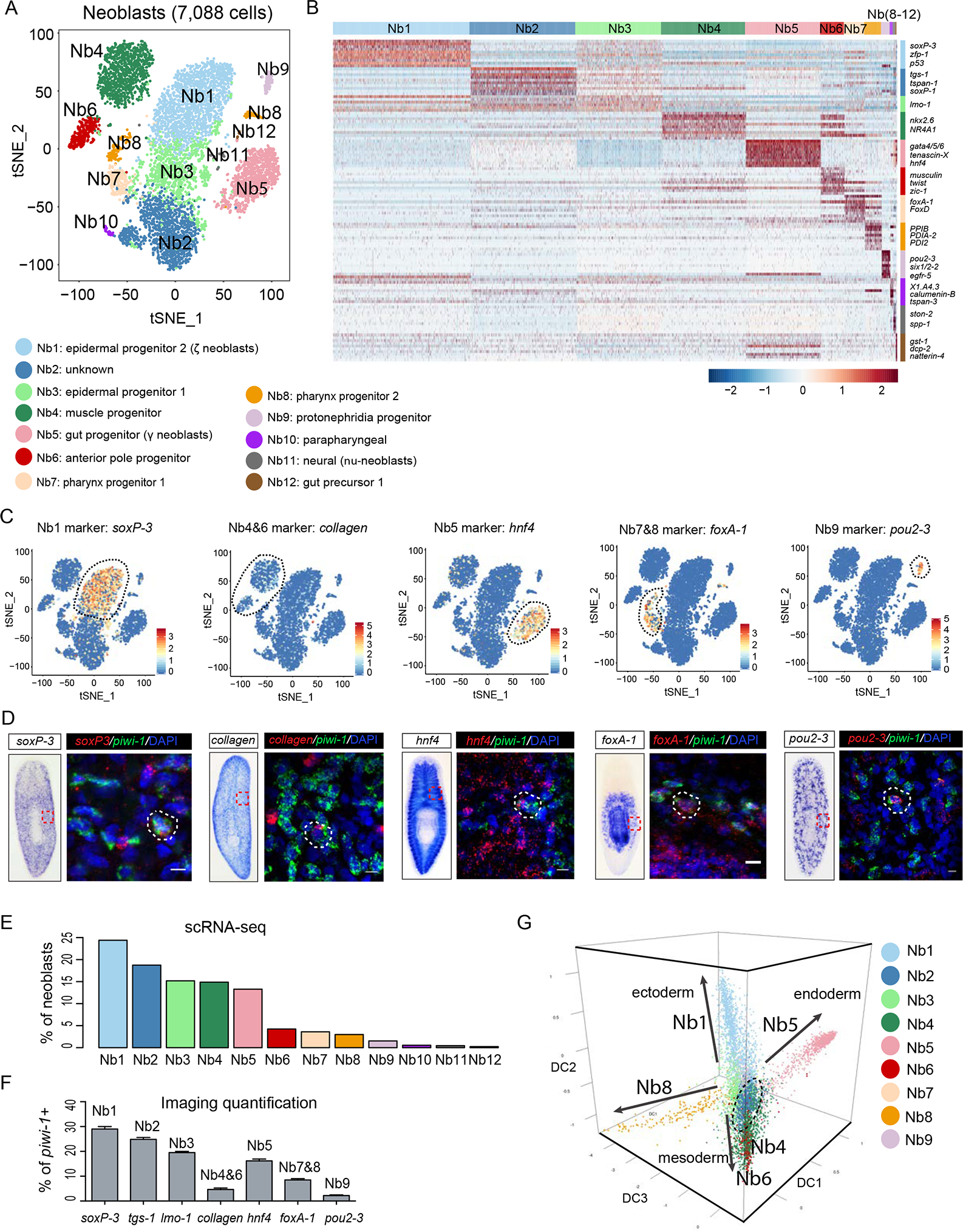
(A) t-SNE plot shows two-dimensional representation of global gene expression relationships among all neoblasts (n=7,088 after filter). Cluster identity was assigned based on the top 10 marker genes of each cluster (Table S2), followed by inspection of RNA in situ hybridization patterns. Neoblast groups, Nb.
(B) Scaled expression heatmap of discriminative gene sets for each cluster defined in (A). Color scheme shows z-score distribution from −2.5 (Blue) to 2.5 (Red). Right margin color bars highlight gene sets specific to respective Nb cluster.
(C-D) Neoblast cluster marker expression in the t-SNE clustered cells (C). Representative images of Whole-mount In Situ Hybridization (WISH) (left) and multiplex FISH (right) for common neoblast marker (piwi-1) and five major Nb cluster markers (D). White-dashed shapes outline piwi-1+ cells that co-express different cell cluster markers. Scale bar, 10 μm.
(E) Percentage of each neoblast cluster (C) by single-cell RNA-seq.
(F) Percentage Quantification of co-FISH of indicated marker gene with piwi-1 shown in panel (D) for each Nb cluster. Error bar, SD; n>3 animals for each cell cluster marker.
(G) Neoblast and progenitor cell cluster visualization using first three components of diffusion map. Cells colored by t-SNE clusters. 4 main branches are indicated with solid arrows, and two sets of clusters at root are circled with dashed lines. Nb10 through Nb12 were not included due to low cell number.
Clusters with lower piwi-1 expression (Fig. S3C) could be likewise characterized. Nb10 and 12 were found to be enriched in parapharyngeal (ascl-4) and mature gut (gst-1) markers, respectively (Fig. S3F, H). We also recovered a proposed new class of neoblasts (ν-neoblasts) previously defined by the expression of ston-2 and the neuronal marker ChAT (Molinaro and Pearson, 2016; Wenemoser and Reddien, 2010) in cluster Nb11 (Fig. S3G). The remaining 3 clusters (Nb2, Nb3, and Nb8) could not be readily assigned to previously described neoblast classes. Examples are, the Piwi-1high Nb2 cluster defined by the expression of a poorly characterized member of the tetraspanin gene family (tspan-1), and cluster Nb8, which is closely associated to the pharyngeal progenitor cluster Nb7 (Figs. 2C, D; S3I).
Nb2 is a novel Piwi-1high neoblast group possessing pluripotent cell properties
To assess the significance of the identified neoblast clusters (Fig. 2A), we defined the potential developmental trajectories of the thousands of cells sampled using statistical methods, followed by comparative analyses of the expression behaviors of the identified signature genes for each of the clusters (Nb1 to Nb12) during regeneration, sublethal and lethal irradiation. Statistically, we applied a computational approach for dimensional reduction based on the concept of diffusion pseudotime (DPT) (Haghverdi et al., 2016). DPT analyses uncovered 4 differentiation paths (Fig. 2G; Movie S1). Projecting known markers onto each path revealed 4 branches representing ectodermal (neuronal and epidermal), mesodermal (muscle), and endodermal (gut) lineages (Fig. S3I). Interestingly, the novel cell clusters Nb2, and Nb3 located to the root of the trajectory. We then parsed all the scRNA-seq-defined clusters (Nb1 to Nb12) using 4 criteria to identify which one(s) may contain PSC activity.
First, we reasoned cNeoblasts should be in the Piwi-1high cell population and express markers of σ-class cells under homeostatic conditions. cNeoblasts should also express high levels of bruli, ezh and sz12–1 self-renewal regulators (Wagner et al., 2012). These conditions specified clusters Nb1 through 9 (Fig. S4A). Second, we removed defined progenitor cell classes from clusters Nb1 through Nb9 to avoid potential overlap of self-renewal genes with cNeoblasts. Two cell clusters expressing high levels of epidermal and gut progenitor markers, zfp-1 (Nb1) and hnf4 (Nb5) (Fig. 2C) (van Wolfswinkel et al., 2014), were readily recovered. Four other clusters were likewise singled out: muscle progenitor and anterior pole markers were expressed in clusters Nb4 (myoD+) (Cowles et al., 2013) and Nb6 (zic-1+) (Vasquez-Doorman and Petersen, 2014; Vogg et al., 2014), respectively; pharynx progenitors (foxA1+) were detected in clusters Nb7 and Nb8 (Adler et al., 2014; Scimone et al., 2014); and protonephridia markers (pou2–3+ and egfr5+) in cluster Nb9 (Figs. 2C, D; S3I) (Rink et al., 2011; Scimone et al., 2011). Subtraction of these six cell clusters identified Nb2 and Nb3 clusters as potentially encompassing pluripotent neoblasts (Fig. 3A).
Figure 3. Nb2 is a novel Piwi-1high neoblast group possessing pluripotent cell properties.
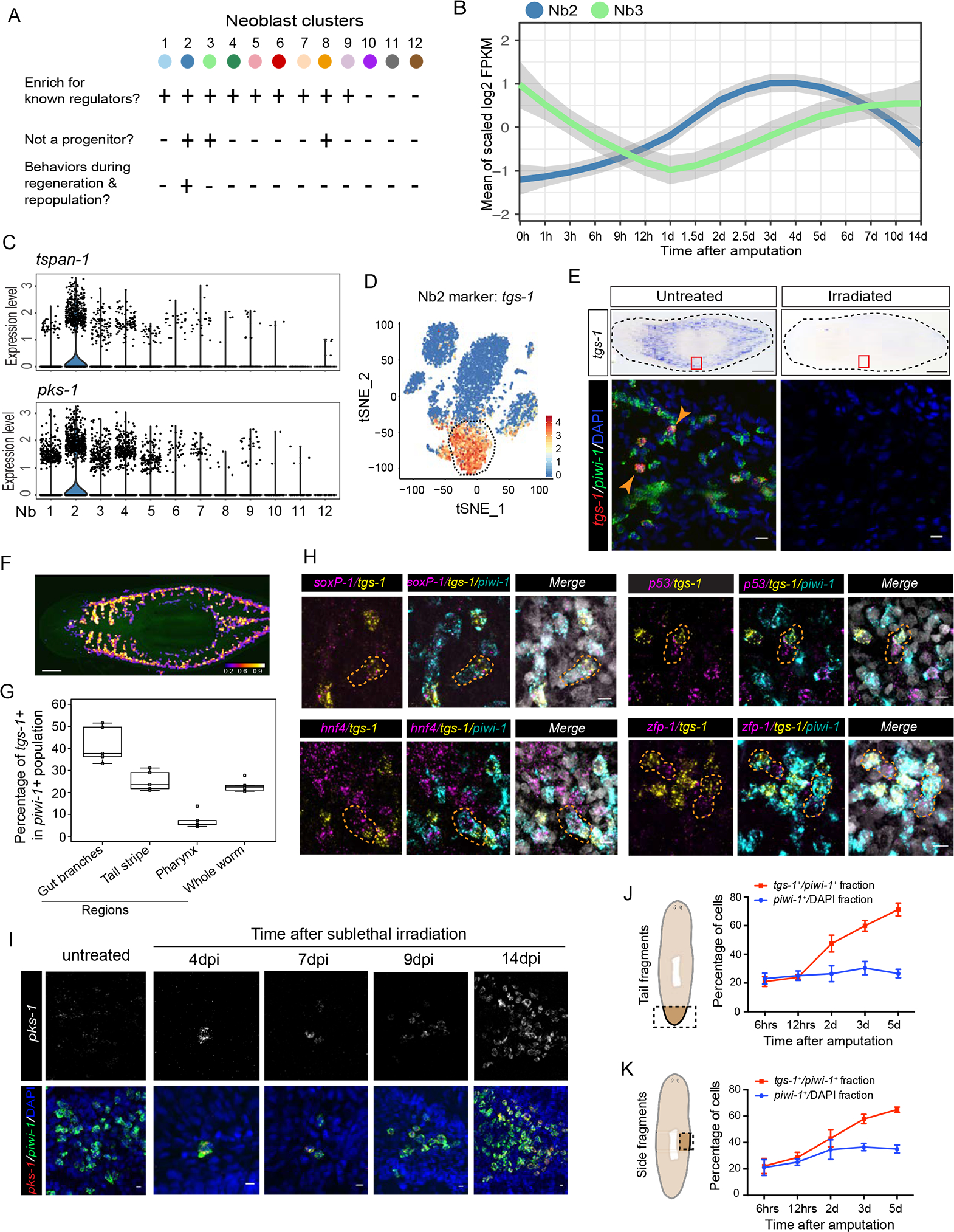
(A) Criteria for identifying cell cluster(s) associated with pluripotency properties.
(B) Nb2 and Nb3 signature gene expression is dynamic during RNA-seq regeneration time-course shown in Fig. 1G.
(C) Violin plots show distribution of expression levels for two Nb2 marker genes in cells (dots) of each of the 12 neoblast clusters.
(D) Expression of tgs-1 in t-SNE clustered cells.
(E) Representative WISH of tgs-1 in response to irradiation (top) and double-labeled FISH for tgs-1 and piwi-1 mRNA expression (bottom). White arrowheads highlight subset of tgs-1+ and piwi-1+ cells. Representative animals shown, n>6 for each condition. Scale bars, 250 μm for WISH data (top row), and 10 μm for FISH data (bottom row).
(F) tgs-1 and piwi-1 mRNA co-expression heatmap. Color scale indicates percentage of piwi-1+ cells that are also tgs-1+ across whole body. Scale bar, 250 μm.
(G) tgs-1+ cell quantification in (F) indicates enrichment proximal to gut branches but removed from pharyngeal region.
(H) Triple FISH of piwi-1, tgs-1 and known neoblast-class markers, soxP-1 (σ neoblast), p53 and zfp-1 (ζ neoblasts), and hnf4 (γ neoblasts). Orange-dashed shapes outline two neighboring piwi-1+ cells each expressing different level of tgs-1 and known neoblast markers. Scale bar, 10 μm.
(I) pks-1 with piwi-1 coexpression time-course after sublethal (1,250 rads) irradiation. Scale bar, 10 μm.
(J and K) Quantification of tgs-1 and piwi-1 coexpression during regeneration time-courses of either whole tail fragments (J) or whole side fragments (K) as shown in illustrations (dashed rectangle insets). Roughly 48.5K to 1 million cells from 4–8 animals were quantified for (J); and 14K to 68.3K cells from 2–4 animals were quantified for (K). See also Fig. S4.
Third, because PSC numbers increase after injury, changes in PSCs should be reflected by discrete alterations in gene expression. We analyzed RNA-seq data from regeneration datasets (Fig. 1G) and asked which cell clusters responded during regeneration. After amputation, only Nb2 marker genes showed increased expression within hours after amputation, while changes in Nb3 marker expression could only be detected ~2 days later (Figs 3B; S4B). Fourth, cNeoblasts previously characterized by surviving sub-lethal irradiation have been proposed to be PSCs (Wagner et al., 2011). To distinguish the cell group most likely to represent cNeoblasts, we followed all Nb cluster signatures across a published whole-animal transcriptional profile defined after sublethal irradiation (Lei et al., 2016), and found the Nb2 cluster signature gene expression profile to behave according to the following expectations: decline in expression up to 6 dpi with a marked increased and sustained expression from 6dpi onwards (Fig. S4C), and a sharp decline in expression of the Nb2 cluster signature genes in transcriptomic analyses of whole animals subjected to lethal irradiation (Fig. S4D). However, the Nb3 group signature genes, including an epidermal cell marker lmo-1 (Cheng et al., 2017), did not show differences between two irradiation doses (Fig. S4C, D). Out of all cells transcriptionally sampled, only the Nb2 cluster satisfied all four selection criteria (Fig. S4E). Therefore, the Nb2 cluster likely included pluripotent neoblasts prompting us to investigate this cluster of cells in vivo.
Piwi-1high Nb2 neoblasts are broadly distributed in the planarian body plan, are sensitive to irradiation and respond to sublethal irradiation and wounding.
We cloned Nb2 signature genes (Table S2) to visualize the distribution of these cells in animals by FISH. Even though the Piwi-1high Nb2 cluster is characterized by transcripts of the gene coding for the cell-surface protein tetraspanin-1 (tspan-1), its expression was difficult to detect by whole mount in situ hybridization under homeostatic conditions. However, we readily detected the expression of two other genes in this cluster: a homolog of the polyketide synthase gene (pks-1) (Fig. 3C), and a novel gene we refer to as tetraspanin group specific gene 1 (tgs-1) (Fig. 3D). Consistent with scRNA-seq data, tgs-1 is sensitive to irradiation treatment (Fig. 3E) and is expressed in ~25% of total piwi-1+ cells (Fig. 3F). The distribution of tgs-1 expression resembles the known location of neoblasts (Figs. 3E; S4F), but is restricted to neoblasts found in close proximity to gut branches, but away from the pharyngeal region (Fig. 3F, G). Interestingly, co-staining with either ζ (zfp-1, p53), γ (hnf-4), or σ (soxP-1) class and tgs-1 probes revealed tgs-1 co-expression in a subset of soxP-1+ (σ) neoblasts, but not in neighboring cells that were either ζ (zfp-1+ or p53+) or γ (hnf-4+) neoblasts (Fig. 3H). While supporting a restricted expression of tgs-1 in σ neoblasts, these data also suggest that tgs-1+ cells may have the potential to produce ζ or γ neoblast via asymmetric divisions.
cNeoblasts undergo clonal expansion to repopulate sublethally irradiated animals (Wagner et al., 2012; Wagner et al., 2011). Most neoblast colonies analyzed at an early stage of expansion (4dpi) expressed high levels of the Nb2 marker pks-1 (Fig. 3I). Colony size increased dramatically afterward, yielding dozens of piwi-1+ cells at 14 dpi with some of cells at the colony edge becoming pks-1− (Fig. 3I). These data suggested that Nb2 neoblasts survived and recovered after sublethal irradiation, consistent with the behaviors of Nb2 markers in whole-animal transcriptional profile following sublethal irradiation (Fig. S4C).
tspan-1 promotes neoblast repopulation and migration after sublethal irradiation
We sought to functionally characterize Nb2 cell markers, particularly putative membrane associated proteins. We identified at least 5 potential membrane associated protein candidates (Fig. 4A), but focused on tspan-1 (Fig. S5A) as it appeared highly specific to Nb2 cells (Fig. 4A). As stated previously, tspan-1 was difficult to detect under homeostatic conditions; however, its expression became readily apparent soon after amputation (Fig. 4B) and was only found in cells expressing high piwi-1 (Figs. 4C; S5B). Multiplex FISH analyses further demonstrated an increase of tspan-1+ cells during regeneration, with the expanded tspan-1+ cells predominantly co-expressing the σ neoblast marker soxP-1 (Fig. S5C). Because tspan-1 knockdown showed no discernable defect in animals, we interrogated its function via RNAi in both sublethal and partial irradiation conditions. Importantly, tspan-1 knockdown significantly reduced the efficiency of repopulation of piwi-1+ cells (Figs. 4D, E; S5D), and particularly soxP-1+ neoblasts (Fig. 4H, I) in sublethally irradiated animals compared to those treated with control RNAi. Because tetraspanin family members have been implicated in cell migration (Hemler, 2005), we also tested the ability of neoblasts to mobilize to wounds after partial irradiation (Guedelhoefer and Sánchez Alvarado, 2012) and found that cells in tspan-1(RNAi) animals failed to migrate to the wound site at comparable rates to control animals (Figs. 4F, G; S5E). We conclude from these experiments that tspan-1 expression is modulated in response to injury and that it plays a role in wound-induced stem cell repopulation and mobilization.
Figure 4. RNAi depletion of Nb2 marker genes affects Nb2 cell repopulation and mobilization after sublethal and partial irradiation.
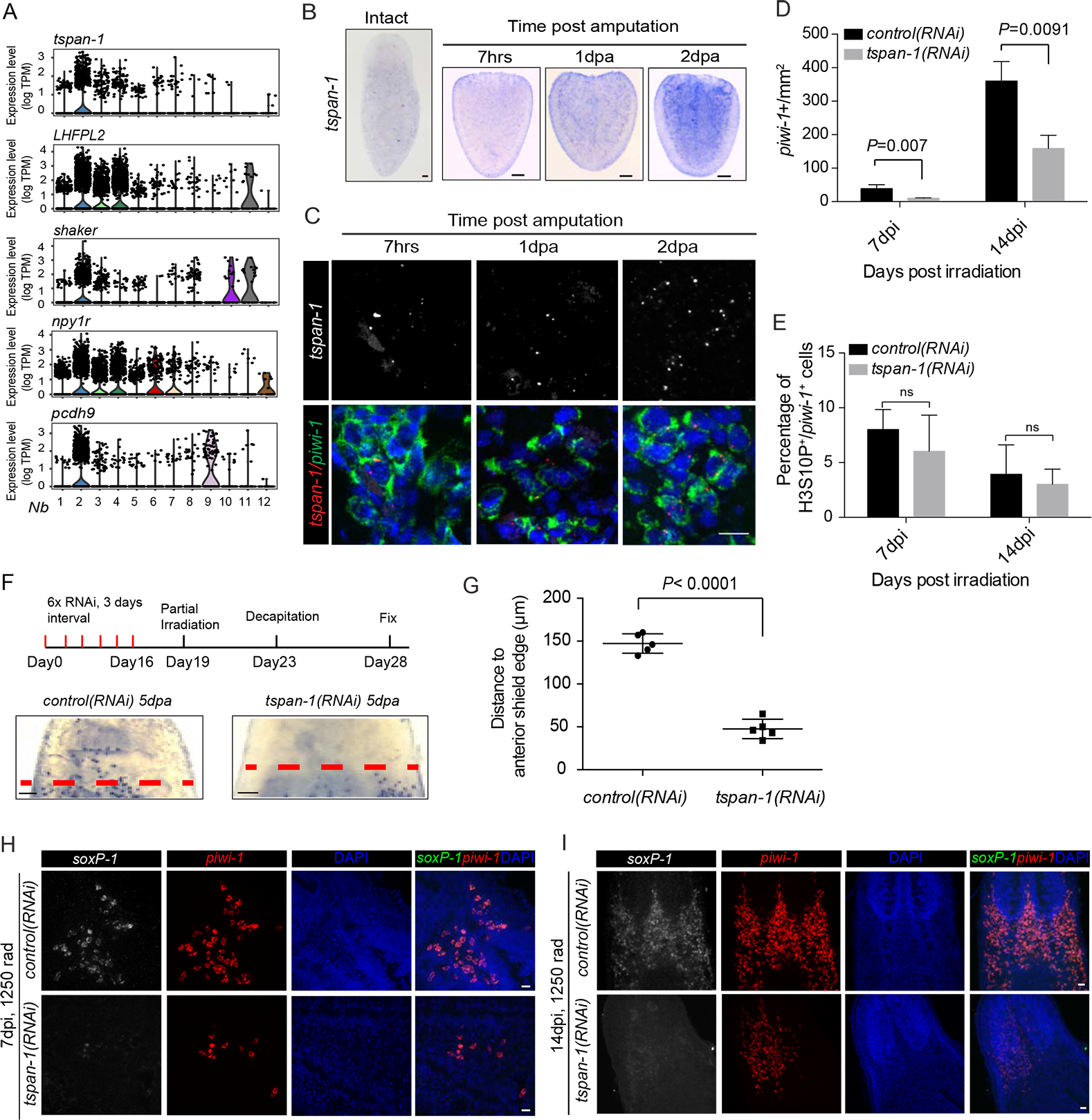
(A) Violin plots show distribution of expression levels for each of top-five predicted cell-surface protein coding genes enriched in Nb2 group.
(B) tspan-1 temporal expression assessed by in situ hybridization in tail fragments post amputation. Representative animals shown, n>5 for each condition. Scale bar, 100 μm.
(C) Double-labeled FISH using RNAscope for piwi-1 and tspan-1 mRNA in tail fragments fixed 7 hours (7hrs), 1-day, and 2-day post-amputation (dpa). Representative areas shown. n≥ 5 animals per condition. Scale bar, 10 μm.
(D-E) Quantification of piwi-1+ cells (D) and mitotic index (E) in control(RNAi) and tspan-1(RNAi) animals at indicated time points after sub-lethal irradiation. Error bar, SD.
(F-G) Cell dispersion assessed by piwi-1 staining (E) and quantified at anterior boundaries in decapitated animals at 9dpi, corresponding to 5 dpa. Representative animals shown. n≥5 animals per condition. Error bar, SD. Scale bar, 100 μm.
(H and I) tspan-1(RNAi) knockdown impairs σ-class cell repopulation after sub-lethal irradiation. Representative areas shown. n≥5 animals per condition. Scale bar, 10 μm. See also Fig. S5.
Single cell transplantation of TSPAN-1+ neoblasts rescues lethally-irradiated animals
To determine if single TSPAN-1+ Nb2 cells are pluripotent, we developed a specific polyclonal antibody against the EC2 region of TSPAN-1 (Fig. S6A–C) to prospectively isolate these cells. TSPAN-1-labeled cells clustered as a clear population during flow cytometry, representing ~7% of total cells (Fig. 5A). By co-staining with TSPAN-1 and Hoechst, we observed ~23% of the TSPAN-1 signal was from the X1 gating window (proliferative state), while ~41.2% was from X2 (Fig. S6D). After sorting cells into wells, we confirmed cell surface localization of the fluorescent signal and noted a morphological resemblance to putative cNeoblasts (Fig. 5B) (Wagner et al., 2012). Strikingly, live TSPAN-1+ cells displayed very active protrusions (Movie S2).
Figure 5. Single cell transplantation of single TSPAN-1 positive cell rescues lethally irradiated animals.
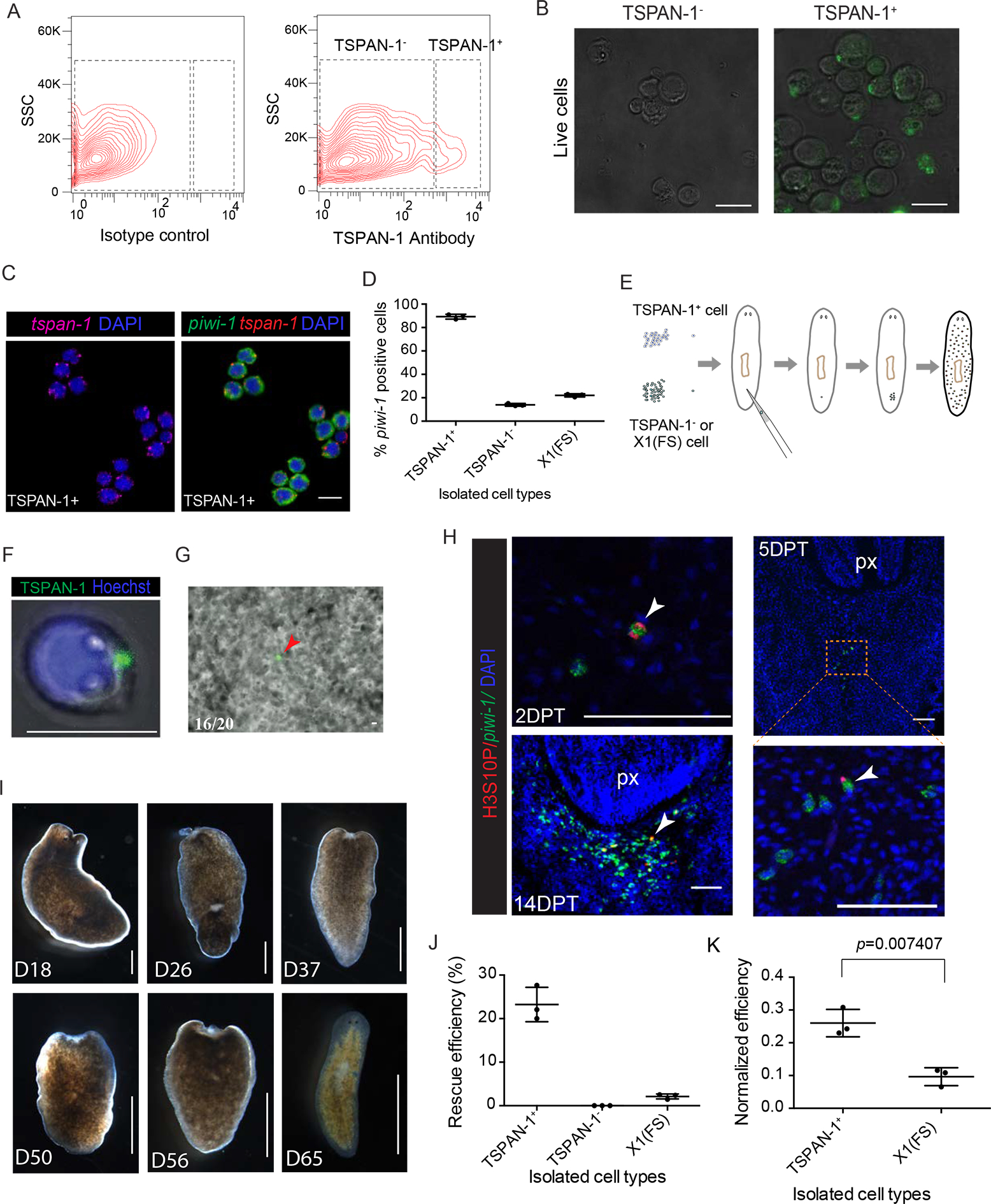
(A) Flow cytometric analyses showing staining of TSPAN-1 antibody. Shown is a representative of more than 3 independent experiments.
(B) Representative freshly sorted TSPAN-1− (left) and TSPAN-1+ (right) cells bright-field and fluorescence images. Scale bar, 10 μm.
(C-D) Representative double FISH of piwi-1 and tspan-1 probes on sorted TSPAN-1+ cells (C). Scale bar, 10 μm. Comparison of percentage of piwi-1+ cells in the indicated isolated cell populations (D).
(E) Single-cell transplantation assay. Sexual hosts were irradiated to eliminate all stem cells 2 days before transplantation. Individual cell was transplanted to assess repopulation/rescue efficiency.
(F) Freshly sorted single TSPAN-1+ cell live imaging verified membrane localization of antibody signal. Cell shows obvious cytoplasmic processes. Shown are maximal projections of Movie S2. Scale bar, 10 μm.
(G) Single transplanted cell live imaging immediately after single cell transplantation. Scale bar, 10 μm.
(H) Colony formation assayed by mitotic marker H3S10P (red) and neoblast marker piwi-1 (green) at indicated time point after transplantation. Anterior, up. Ventral shown. Colonies of dividing piwi-1+ cells (arrowhead). px, pharynx. Scale bar, 100 μm.
(I) Representative images of transplanted hosts at time points after single cell transplantation. Different animals shown in each panel. Scale bar, 1 mm.
(J and K) Rescue efficiency quantification (J), and piwi-1+ percentage normalized efficiency (K). piwi-1+ percentage obtained from (D).
Next, we tested whether TSPAN-1+ cells were also positive for piwi-1 and tspan-1 expression. We similarly tested purified X1(FS) cells, a fraction previously shown to contain rare cNeoblasts (Wagner et al., 2011) (Figs. 5C; S6E). 89.2 ± 2% of TSPAN-1+ cells were also piwi-1+, whereas only 22 ± 1.3% of X1(FS) cells were piwi-1+. Further, only 13.9 ± 1.2% of TSPAN-1− cells displayed piwi-1 expression, suggesting that the TSPAN-1 antibody does not enrich for piwi-1+ cells, but a subset of piwi-1+ cells (Figs. 5C, D; S6E). We then performed cell sorting followed by single cell transplantation of TSPAN-1+, TSPAN-1−, and X1(FS) cells into lethally-irradiated planarians (Fig. 5E). Because the TSPAN-1+ cells were still decorated with the fluorescent antibody (Fig. 5F), we could readily follow and confirm their injection into hosts (Fig. 5G; Movie S3). We also used piwi-1 expression soon after transplantation to confirm single cell injections of TSPAN-1+, TSPAN-1− and X1(FS) cells (Fig. S6F) and throughout the duration of the rescue experiments as an indicator of expansion. TSPAN-1+ cells were mitotically active 2 days post-transplantation (DPT) and formed robust colonies by 14 DPT (Fig. 5H). Of the combined 224 animals from three different replicates receiving TSPAN-1+ single-cell transplants, a total of 52 animals survived lethal irradiation for half a year, i.e., a rescue rate of ~23.2% (Fig. 5I, J). In marked contrast, only 5 out of a total of 232 animals injected with X1(FS) cells survived, corresponding to a rescue rate of ~2.0% (Fig. 5J). None of the non-injected control animals (n=82) (Wagner et al., 2011), nor the TSPAN-1− injected (n=194) irradiated hosts survived beyond 50 days.
To compare the different cell preparations (Fig. 5D, E), and given the low numbers of piwi-1+ cells in the X1(FS) cells (Fig. 5D), we normalized the single-cell rescue data to the number of piwi-1+ cells per injected cell by dividing rescue efficiency (Fig. 5J) by piwi-1+ percentage as assessed by FISH (Fig. 5D). If there is no difference between TSPAN1+ sorted piwi-1+ cells and randomly picked piwi-1+ cells from the X1(FS) sort gate, an improved rescue efficiency following normalization should not occur. The Welch Two Sample t-test, yielded a p-value of 0.007407 after normalization, therefore rejecting the null hypothesis and suggesting TSPAN-1+ cells are statistically enriched for pluripotent piwi-1+ cells, rather than piwi-1+ cells in general (Fig. 5K). As such, injections of TSPAN-1+ isolated cells result in a remarkable 14-fold rescue improvement over X1(FS) cell injections. Therefore, the raised TSPAN-1 antibody can be used to prospectively isolate functional, pluripotent neoblasts.
Transcriptional response of pluripotent neoblasts differs between homeostasis, repopulation and amputation conditions
The response of pluripotent neoblasts to irradiation and amputation plays key roles in promoting regeneration. Yet, the precise transcriptional response of the pluripotent subset of neoblasts to these perturbations remains poorly defined. Having transcriptionally and functionally defined a specific PSC population encompassed by the Nb2 cells (Figs. 2–5), we reasoned that the Nb2 markers may help inform the transcriptional dynamics of PSCs under different experimental conditions (Fig. 6A). First, we tested sub-lethal irradiation exposure. To profile rare PSCs and avoid interference from immediate progenitor cells, we determined a time point after sub-lethal irradiation (7dpi) with minimal piwi-1+ cells (Fig. S7A), followed by isolation and single-cell RNA-seq of 1,200 individual cells derived from X1 (Piwi-1high) and X2 (Piwi-1low) cell populations (Fig. S7B).
Figure 6. Response of pluripotent stem cells to regeneration and repopulation signals.
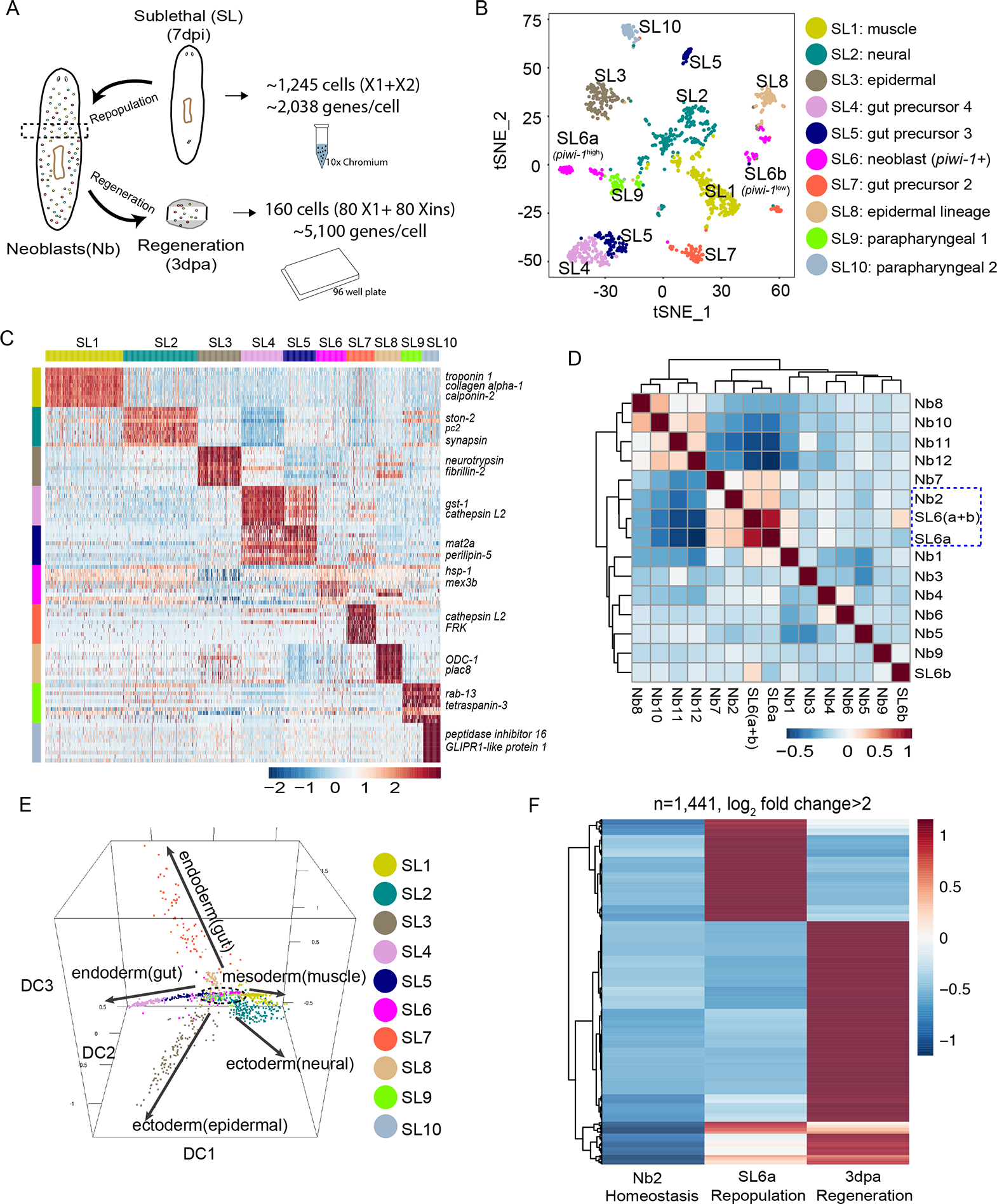
(A) Comparison strategy of single-cell transcriptomic data from homeostatic, repopulation and regeneration conditions. Droplet-based 3’ or plate-based full-length scRNA-seq were used.
(B) t-SNE plot of surviving X1 and X2 cells (n=1,039 after QC filter) after sub-lethal irradiation. Colors indicate unbiased cell classification via graph-based clustering. Sub-Lethal irradiated cell groups, SL.
(C) Scaled expression heatmap of discriminative gene sets for each cluster defined in (B) Color scheme based on z-score distribution from −2.5 (Blue) to 2.5 (Red). Left margin color bars highlight gene sets (right side) specific to respective cell subsets.
(D) Spearman rank correlation heatmap for all pairwise comparisons of indicated cell types. Spearman correlations were calculated using normalized read counts across the entire transcriptome (n=31,253 genes) for all RNA-seq experiments.
(E) Neoblast and progenitor cell cluster visualization using diffusion map. Five main branches are indicated with solid arrows, with one cluster (SL6) sitting at the root (dashed lines).
(F) Scaled expression heatmap of discriminative gene sets upregulated (Log2 fold change > 2) in regeneration (3dpa, tspan-1+ cells), repopulation (7dpi, SL6a cells) or both when compared to homeostatic Nb2 group.
Unsupervised clustering and tSNE analyses identified 10 sub-lethal (SL) cell clusters (Fig. 6B). Cross-referencing their respective signature genes to known marker genes resolved defined clusters with distinct cell types, e.g., SL1 to muscle cell types (troponin 1+ and collagen+) (Witchley et al., 2013), and SL2 to neural cell types (pc2+, spp-4+ and npp-2+) (Collins et al., 2010) (Fig. 6B, C; Table S3). A single cell cluster (SL6) was found with both cell cycle and piwi-1 expression signatures (Fig. S7C). Inspection of SL6 revealed extensive transcriptional similarity to Nb2 cells (Fig. 6D), including the Nb2 marker genes tgs-1, pks-1 (Fig. S7D) and tspan-1 (Fig. S7E), further confirming that Nb2 cells encompass cNeoblasts. Moreover, pseudo-temporal differentiation trajectories generated from the SL cell clusters by diffusion maps (Fig. 6E) revealed a general topology similar to the trajectories defined for homeostasis (Fig. 2G), with SL6 (like Nb2) at the root of the trajectory models, and the remaining groups projecting into branches (Figs. 6E; S7F; Movie S4). Transcriptional profiling defined the SL6 cluster as a discrete cell group, yet the t-SNE plot distributed this cluster into two subsets we termed SL6a and SL6b (Fig. 6B). SL6a is characterized by higher piwi-1 expression than SL6b (Figs. S7C; 6D). When SL6a and SL6b are compared to Piwi-1high and Piwi-1low RNA-seq profiles, we noted that SL6a is transcriptionally similar to Piwi-1high and SL6b most similar to Piwi-1low cells (Fig. S7G), indicating the spectrum of piwi-1 expression is maintained during repopulation. Moreover, the expression of most of the Nb2 marker genes is also enriched in the SL6a subset, but not in the SL6b subset (Figs. 6D; S7D), suggesting SL6a is likely the major cell cluster responsible for repopulation.
Next, we profiled the pluripotent state during regeneration. Because Nb2 cells undergo clonal expansion after amputation at 3dpa (Figs. 3J, K; 4C), we isolated cells at this time point from small, regenerating tissue fragments for scRNA-seq (Fig. 6A). Given the expansion of PSCs and the complexity of whole-body regeneration, we profiled ~160 cells at higher sequencing coverage per cell in order to capture broader single-cell transcriptional variations (Fig. 6A). We identified a cohort of Piwi-1high, tspan-1+ cells and defined discriminative Nb2 markers for repopulation (7dpi, SL6a cells) and regeneration (3dpa, tspan-1+ cells). We compared them by taking the average expression of each gene for a given cell population, and performed quantile normalization to minimize possible technical variation across different platforms (Bullard et al., 2010). The analysis uncovered differential expression of Nb2 markers for all three conditions (Fig. 6F), a variation reflected by the discrete quantitative responses of PSC-associated genes induced by each of the conditions investigated (Table S3).
DISCUSSION
Neoblasts are the cellular source of planarian regeneration, but their composition and behaviors in response to injury and physiological homeostasis have been under constant debate. Neoblasts share common attributes, but remain a complex mixture of both PSCs and lineage progenitors. Even after the unequivocal demonstration that neoblasts contain pluripotent stem cells (Wagner et al., 2011), the complexity of this cell type has impeded the development of prospective stem cell isolation methods to enrich for neoblast sub-classes, a pre-requisite to carefully study their properties. By simultaneously and quantitatively detecting both PIWI-1 protein and piwi-1 mRNA levels, we were able to both distinguish PSC (Piwi-1high) and lineage progenitor cells (Piwi-1low) from each other, and to systematically resolve some of the heterogeneity of the neoblast compartment. scRNA-seq demonstrated Piwi-1high cells encompassed no fewer than 12 discrete subpopulations of neoblasts. One of these, the novel tspan-1 expressing Nb2 group recapitulated several pluripotent cell properties. A new TSPAN-1 antibody facilitated the prospective isolation of these cells, and transplanted TSPAN-1+ single cells rescued lethally-irradiated animals with higher efficacy than previously tested dissociated cell populations. Our work refines the existing classification of neoblasts and demonstrates that pluripotent stem cells can be identified prospectively and efficiently purified from complex tissues.
PIWI-1 levels define a continuum of pluripotent stem cells and lineage progenitors in the neoblast compartment
To date, the use of piwi-1 expression to identify neoblasts has been qualitative in nature, with neoblasts remaining heterogeneous, such that certain subpopulations were largely characterized through retrospective analysis of reconstitution patterns in irradiated planarians (Eisenhoffer et al., 2008; van Wolfswinkel et al., 2014; Wagner et al., 2012). By developing quantitative methods to measure both piwi-1 mRNA and protein, we found that piwi-1 is differentially expressed across neoblasts in a way that readily distinguished two functionally distinct piwi-1+ subpopulations (Fig. 1). Both Piwi-1high and Piwi-1low cells responded differently to lethal and sublethal irradiation, displayed specific gene expression profiles, and behaved differently during key periods of regeneration (Fig. 1E, F, H). The correlation between piwi-1 expression levels (transcript and protein) and cellular properties indicate neoblasts exist in dynamic undifferentiated and lineage transition states, and that this characteristic coexistence of pluripotent stem cells and lineage progenitors may be a property required for maintaining robust homeostasis and regenerative capabilities (Fig. 7). The quantitative methods reported here open the door for mechanistic studies to disect a dynamic state of pluripotency in which a population of cells perpetuates itself through under diverse biological and environmental demands.
Figure 7. Proposed lineage composition model of planarian piwi-1+ cells.

12 major classes representing 6 cell lineages of all 3 germ layers were found in the neoblast compartment of adult planaria. Nb2 and SL6a can self-renew, and collectively give rise to a wide range of tissue types in single cell transplantation and repopulation, respectively. As differentiation ensues, piwi-1 expression is downregulated, and tissue-associated transcription factors upregulated.
Self-renewal and modulation of lineage progenitor composition in the neoblast compartment.
The expression of tissue-associated transcription factors involved in specifying progenitors in neoblasts has been attributed to neoblast specification during regeneration (Scimone et al., 2014). Interestingly, scRNA-seq analysis revealed that even under homeostatic conditions, Piwi-1high cells encompass not only pluripotent stem cells, but also subsets of cells expressing lineage-specific progenitor genes representing all 3 germ layers (Fig. 2). Because tissue-specific injuries can lead to the upregulation of tissue-specific transcription factors in neoblasts (Adler et al., 2014), the cell class composition of the neoblast compartment must be tightly regulated. It opens the question as to how regeneration signals instruct PSCs to produce the appropriate number and type of progenitor neoblasts in response to different regeneration requirements. Such signals may involve multi-lineage communication, such as feedback from lineage-primed progenitors, or post-mitotic cells (Tu et al., 2015) to initiate exit from pluripotency and to restrict cells to specific differentiation paths. This question can now be addressed by measuring gene expression profiles specifying the Piwi-1high neoblast classes Nb1 through NB12 under dynamic experimental conditions.
Nb2 cells encompass pluripotent, self-renewing stem cells that can be prospectively isolated using the membrane associate protein TSPAN-1
By systematically interrogating the properties of scRNA-seq-defined neoblast clusters NB1 through Nb12, we discovered that cluster Nb2 featured a rare population of pluripotent stem cells marked by tspan-1 expression (Figs. 2, 3; S4A). The tetraspanins were first cloned in leukocytes to characterize cell-membrane associated antigens to purify cells from complex mixtures (Boucheix and Rubinstein, 2001). These proteins are expressed by all metazoans, with 33 members in mammals, 37 in Drosophila melanogaster, 20 in Caenorhabditis elegans (Huang et al., 2005), and 48 in S. mediterranea (Fig. S5A). Members of this family of proteins have received special attention due to their involvement in regulating the migration and invasion of cancer cells (Hemler, 2014; Zöller, 2009). Interestingly, knockdown of tspan-1 in S. mediterranea resulted in an inhibition of mobilization of neoblasts to sites of amputation in partially irradiated animals (Fig. 4F, G). Also, the cells isolated using anti-TSPAN-1 antibodies displayed numerous and active protrusions (Movie S3) supporting a role for this family of proteins in membrane folding (Hemler, 2005). Importantly, prospective sorting of TSPAN-1+ cells followed by single-cell transplantation demonstrated that these cells actively divide to form colonies and rescue stem-cell depleted animals (Fig. 5). Therefore, these data not only provide a framework for characterizing rare stem cell populations, but may also shed light on the function of the deeply evolutionary conserved tetraspanin family of proteins in stem cell-mediated regeneration.
scRNA-seq and lineage reconstruction of an adult pluripotent stem cell compartment in different biological and experimental contexts
By combining single cell sequencing experiments post hoc, we can make predictions regarding the possible relationships and dynamics that may exist between different cell classes identified under different conditions. For example, the signature gene expressions of Nb cells can be used to discriminate cell groups in the Piwi-1low SL cells that survived sublethal irradiation treatment (Fig. S7H); however, the converse is not possible as the signatures from Piwi-1low SL cells fail to project back to the Piwi-1high Nb cells (Fig. S7I). Because the projection is unidirectional, i.e., from Piwi-1high to Piwi-1low, it is likely that Piwi-1low cells are descendants of Piwi-1high cells. The failure of Piwi-1low cell expression signatures to project back to Piwi-1high cells likely reflect the acquisition of expression profiles associated with new cell types arising from either lineage bifurcation or differentiation. Therefore, we propose a model in which TSPAN-1+ Nb2 cells generate all the lineage branches associated with maintaining homeostasis, restoring viability after lethal and sublethal irradiation, and regenerating missing body parts lost to amputation (Fig. 7). Lineage tracing as well as simultaneous localization of these cell types within tissues will be needed to test this model.
Implications for understanding the source of regenerative ability in animals
Although the neoblast concept has been around since the late 19th century, the nature and identity of the pluripotent cells capable of maintaining the remarkable regenerative capacities of planarians have remained unclear. By being able to prospectively identify and purify pluripotent neoblasts, we have shown these cells to be remarkably dynamic, constantly occupying diverse states of continuous fate determination and capable of specifically adapting their genomic output to different injuries, such as irradiation or amputation (Fig. 6F). Because pluripotent stem cells are generally assumed to be only present transiently in early embryogenesis, and can only be perpetuated artificially in vitro, our findings that pluripotent stem cells can be maintained in adult animals despite showing distinct transcriptional changes dictated by either physiological homeostasis and/or injury, are all the more provocative.
Identifying specific cell-surface markers for neoblast subtypes (e.g., tspan-1 for Nb2 and fgfr-1 for epidermal lineage) enables the purification of desired neoblast subtypes to investigate their biological properties, or for developing cell culture techniques. Moreover, tetraspanin surface markers are present in multiple mammalian stem cells (Karlsson et al., 2013; Kwon et al., 2015), indicating potential developmental conservation. Indeed, the tetraspanins are an important family of proteins that recruit other proteins at the cell membrane including integrins and cell adhesion molecules, thus initiating important cell decisions (Hemler, 2005). Hence, we postulate that TSPAN-1 and the other proteins associated with the neoblast classes reported here, likely play key roles in interpreting environmental changes to guide the genomic output and functions of neoblasts and their progenitors. As such, our findings demarcate a dynamic biological context where the identities and behaviors of self-renewing adult PSCs responsible for homeostasis and tissue regeneration can be both quantitatively measured in the context of changing windows of developmental competence, and studied in greater mechanistic detail.
STAR+METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Alejandro Sánchez Alvarado (asa@stowers.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Planarian culture and irradiation treatment
Asexual S. mediterranea (strain CIW4) animals were maintained at 20°C without antibiotics as previously described (Newmark and Sánchez Alvarado, 2000). For sexual S2F8b strain used for transplantation (Guo et al., 2016), animals were maintained in a recirculating water system without antibiotics. Animals were transferred to static culture one day before irradiation, and maintained in static culture thereafter supplemented with gentamicin (100 μg/mL gentamicin sulfate). For all experiments, animals were starved for at least 7 days. A GammaCell 40 Exactor irradiator was used to expose animals to either 1,250 or 6,000 rads for sublethal and lethal irradiations, respectively.
METHOD DETAILS
Gene identification and phylogenetic tree
To comprehensively identify planarian Tetraspanin proteins, we followed four steps to profile planarian Tetraspanin family members (Hemler, 2005): 1) we translated S. mediterranea transcriptome smed_20140614 (accession number GEO:GSE72389), and use hmmscan (HMMER) to find Tetraspanin domains (Pfam hit: PF00335) with an e-value of 0.001 as cutoff; 2) we selected candidates based on the presence of exactly 4 transmembrane domains (TMHMM); 3) we searched for the presence of CCG and at least 4 total cysteines; and 4) we searched genome using blastn, verifying the presence in genome assembly (Robb et al., 2015). To generate a phylogenetic tree, all identified sequences were multi-aligned with MUSCLE. The alignment was trimmed with trimAI. The tree was calculated using RAxML, and was further visualized by ITOL. The SMART sequence analysis tool was used to identify conserved domains in SMED homologs and for comparison with the Mus musculus (mm), Drosophila melanogaster (dm), Schistosoma mansoni (Smp), and Schmidtea mediterranea (SMED) proteins (http://smart.embl-heidelberg.de/).
Gene cloning and gene knockdown by RNAi feeding
Genes were cloned from a CIW4 whole-animal cDNA library into the pPR-T4P vector as previously described (Adler et al., 2014). RNAi was performed by feeding as previously described (Reddien et al., 2005) with modifications. Briefly, cloned gene vectors were transformed into bacterial strain HT115, cultured in 2XYT to O.D.= 0.6–0.8, induced to express dsRNA for 2 hours with 1mM IPTG at 37°C shaking at 250 rpm. Bacterial pellets were mixed with a weight ratio of 4:1 to beef liver paste. dsRNA food was given to the animals every 3 days for 3–6 feedings as needed.
Generation of TSPAN-1 antibody
The second extracellular loop (AA104-196) of TSPAN-1 was cloned into the pET21a vector for recombinant protein induction in E. coli. Two New Zealand Rabbits were immunized with peptide conjugated to KLH (GeneScript). Rabbit polyclonal antibodies titers against peptide were determined by indirect ELISA. After three immunizations, the anti-peptide antibody was purified from antiserum by protein A column (GeneScript). Antibody was preserved in Phosphate Buffered Saline (PBS, pH 7.4) with 0.02% Sodium Azide, and the concentration was measured by A280nm. The specificity of the antibody was confirmed using western blots (used at 1:1000) or immunofluorescence (used at 1:250) using RNAi sample as control. Diluted antibody (1:250–300) was used for FACS analyses. For western blots, total protein was extracted using modified RIPA lysis buffer (Santa Cruz Biotechnology), and protein concentration was determined by BCA Protein Assay Kit (Pierce). After denaturing by boiling for 5 minutes in sample buffer, 30 μg of proteins were analyzed by immunoblotting, and immunoblot signals were detected by enhanced chemiluminescence as previously described (Zeng et al., 2013).
FACS by Hoechst 33342 and cell-surface marker expression
Flow cytometry of Hoechst stained cells was conducted largely as previously described (Hayashi et al., 2006; Reddien et al., 2005). Planarians were diced and dissociated using CMFB (CMF+0.5% BSA) on a cold plate (4°C). Cells were washed off the plate with CMFB buffer, and pelleted by centrifugation (290g for 5 minutes at 4°C). To reduce clumping at downstream steps, dissociated cells were strained through a 40 μm filter prior to pelleting, and stained with Hoechst 33342 (ThermoFisher) for 45 minutes at room temperature. Before loading onto Influx sorter, cells were strained again and then Propidium iodide (PI) was added to discriminate dead cells. For TSPAN-1 antibody-mediated cell sorting, cell pellets were prepared as described above, and were directly re-suspended in CMFB buffer containing pre-diluted either primary antibody (1:300) or isotype control antibody (Resource Table), thoroughly triturated to ensure a single cell suspension, and primary antibody staining was conducted for 30 minutes to 1 hour at room temperature. Afterwards, cells were washed with an excess of CMFB buffer and pelleted again, and this was conducted one more time. Cells were then stained with fluorescent-labeled secondary antibody (1:300) for 30 minutes. Finally, washed cell pellets were re-suspended in CMFB buffer containing Propidium iodide (PI), and were strained through a 30 μm filter. Flow cytometry and sorting was conducted on a BD Influx cell sorter (Stowers FACS Core).
Intracellular flow cytometry and cell sorting for RNA extraction
To quantitatively analyze the presence of PIWI-1 in neoblast cells, intracellular flow cytometry was conducted using a custom PIWI-1 antibody (Clone#6116, kind gift from Qing Jing) generated using the same peptide sequence described before (Guo et al., 2006). After dissociation, cells were collected by centrifugation (300g for 5 minutes at 4°C) the supernatant aspirated, followed by fixation in 4% PFA on ice. Subsequently, cells were washed, and permeabilized in ice-cold 90% methanol. After wash by centrifugation, cells were re-suspended in 300 μl of primary antibody (prepared in incubation buffer at 1:300 with the addition of Ribonuclease inhibitors) for 1 hour at room temperature. Cells were washed by centrifugation in 2–3 ml incubation buffer and re-suspended in fluorochrome-conjugated secondary antibody in incubation buffer at the recommended dilution. Diluted DAPI solution (ThermoFisher, 1:500) was used to stain nuclear DNA and incubated for 30 min at room temperature. Cells were washed twice by centrifugation in 2–3 ml incubation buffer, and finally re-suspended in CMFB buffer before straining through a 40 μm cell strainer (BD). Flow cytometry was conducted on a BD Influx cell sorter (Stowers FACS Core).
PrimeFlow analysis of RNA/protein expression
Cells prepared under the same conditions as the FACS samples were processed with the PrimeFlow RNA Assay kit (ThermoFisher) following the manufacturers protocol, with minor modifications. Briefly, 1 million presorted nucleated (Hoechst 33342+) planarian cells were fixed, permeabilized, and intracellularly stained for detection of the PIWI-1 protein first when required (1 hour at 4°C). After an additional fixation step, cells were ready for 3–6 h of hybridization at 40 ± 1°C with a high-sensitivity target-specific set of probes (below). The cells were then subjected to two amplification steps (sequential 2-h incubations at 40°C with the PreAmp mix and Amp Mix solutions). After proper wash, diluted label probes were hybridized with the specific amplifiers for 1 h at 40°C. Negative controls were included in all experiments with the Bacterial DapB gene. Custom designed piwi-1 (Type 1 Alexa Fluor 647 or Type 6 Alexa Fluor 750), agat-1(Type 1 Alexa Fluor 647), tspan-1(Type 1 Alexa Fluor 647), and soxP-1 (Type 4 Alexa Fluor 488) probes were synthesized by Affymetrix (eBioscience). High throughput image acquisition at 60x magnification with an ImagestreamX Mark II allows for analysis of high-resolution images of single-cells. Genes of interest were targeted by different fluorophore-conjugated probes, and nuclei were stained with DAPI. Single cells were selected based on cell properties like area, aspect ratio (bright-field images) and nuclear staining. As a negative control, we used the Bacterial DapB gene (Type 1 probe), in addition to piwi-1, tspan-1 and soxP-1. Isotype antibody rabbit IgG was used to set the negative gate for PIWI-1 antibody staining. Spot counting was performed with the Amnis IDEAS software to obtain the expression distributions.
In situ hybridizations and antibody staining
Whole mount in situ hybridizations were performed as previously described (King and Newmark, 2013; Pearson et al., 2009) with some modifications. Animals were killed in 5% NAC in PBS, and fixed for 45 min in 4% formaldehyde in PBSTx (PBS+0.5% Triton X-100). Formamide bleach was performed for multiplex FISH, followed by proteinase K (Ambion) treatment for 10 min, post-fixation and pre-hybridization for 2 hours. Hybridization was done at 56°C in the probe (1:1000) containing Hyb buffer for >16 hours, followed by extensive wash and antibody development using tyramide signal amplification system. For double/triple-color fluorescence in situ hybridization (FISH), HRP inactivation was performed between labelings in 100mM NaN3 for 90min. To improve optical clarity after signal development, we used ScaleA2 with 80% glycerol and 4 M urea for colorimetric WISH, and glycerol (20%), DABCO (2.5%) (Sigma-Aldrich), and 4 M urea ScaleA2 for FISH (Adler et al., 2014). Anti-phospho-histone H3 (Ser10) (H3P) antibody (1:1,000; Abcam, ab32107) and Alexa Fluor 555-conjugated goat anti-rabbit secondary antibodies (1:1,000; Abcam, ab150086) were used to stain proliferating cells at the G2/M phase of cell cycle. For FISH on single cell, cells were dissociated and macerated in CMFB media and labeled with Hoechst 33342 or antibodies, sorted as described above. Cells were then sorted into Microplate 96-Well F-Bottom Plates with Coverglass Base (Greiner Bio-One) in CMF buffer, followed by fixation in 4% paraformaldehyde in CMF. Cell FISH was performed as described above, with slight modifications: all hybridization washes were limited to alternating 20 minute intervals, and all other washes were limited to 10 minutes. Western blocking reagent (Roche; 5% in MABT) or 5% horse serum was used as a blocking agent before antibody development. Only robust expression of transcription factors or markers was counted as positive cells using Fiji on maximal intensity projections. Fluorescent images were taken with a Zeiss LSM700 Confocal Microscope or a Perkin Elmer Ultraview spinning disk. Images were adjusted for brightness and contrast using Fiji.
F-ara-EdU labeling followed by X1(FS)-L and X2(FS)-R sorting
F-ara-EdU (Sigma) was fed to animals in calf liver paste at a concentration of 0.5mg/ml for a 10-hour chase. Animals were then diced and dissociated using CMFB (CMF+0.5% BSA) on a cold plate (4°C). Cells were sorted as described above, and FACS gates were set as indicated in Figure S2J. Cells were fixed in 4%PFA for 2 hours at 4 °C and were permeabilized in PBSTx(0.5%) solution for 20 minutes at room temperature. After wash, cells were incubated in freshly prepared 10uM Azide development buffer (ALEXA FLUOR 488 AZIDE 10 μM, CuSO4 1 mM, (+)-Sodium-L-Ascorbic 100 mM, in 1xPBS) 20 minutes at room temperature in the dark while gently shaking, followed by washing and further FISH staining as described above.
Single-molecule FISH (smFISH) by RNAscope
Flatworms were fixed with 4% PFA (in PBSTx) at room temperature (25 ± 2 °C) for 4 hours, followed by tissue processing in a Pathos Delta processor (Milestone Medical, in MI, USA) and paraffin embedding. tissue sections in 10-μm thickness were cut for RNA situ hybridization using the RNAscope v2 multiplex fluorescent detection kit (Advanced Cell Diagnostics) with a couple of modifications: Antigen retrieval was done at 100°C for eight minutes and digestion was performed with proteinase plus for 15 minutes at 40°C in a HybEZ hybridization oven (Advanced Cell Diagnostics), followed by probe hybridization for 2–3 hours at 40°C, signal amplification steps, and DAPI counterstaining. Probes used for single-molecule RNAscope (Advanced Cell Diagnostics) were: tspan-1 (C1 and C2, 11ZZ probe targeting 56–656 of provided sequence), tgs-1 (C2, 20ZZ probe targeting 302–1391 of provided sequence), and piwi-1 (C1, 19ZZ probe named targeting 4–2458 of DQ186985.1 deposited in NCBI). The scrambled probe provided with the RNAscope kit was used as a negative control, and 1day lethally irradiated animals were used as experimental negative control of neoblast expression.
Bulk RNA-seq of live cells and fixed cells
For untreated and 1-day irradiated animals, total RNA was purified using TRIzol as described (Zeng et al., 2013). For sorted X1, X2 and Xins populations based on Hoechst staining, 1×105 cells were sorted into 800 μl Trizol LS, and RNA was extracted according to the manufacturer’s manual. RNA from whole worms was treated with RNase-free DNase on QIAGEN RNAeasy columns and was eluted in nuclease-free water (Ambion). For each replicate, 1 μg RNA from five worms or 100 ng RNA from 1×105 sorted cells were used to generate RNA-Seq libraries using the Illumina TruSeq Stranded mRNA LT Kit. Libraries were sequenced in 50bp single reads using the Illumina HiSeq 2500 sequencer. RNA-Seq analysis was carried out by mapping reads to the transcriptome as previously described (version: smed_20140614) (Cheng et al., 2017), and was further processed using the in-house RNA analysis pipeline. For anti-PIWI-1 antibody sorted fixed cells, RNA was extracted using the RecoverAll Total Nucleic Acid Isolation kit (Ambion) with some modifications. Briefly, after sorting, cells were pelleted by centrifugation at 3000 g for 5′ at 4°C. The supernatant was discarded. Total RNA was isolated from the pellet using the RecoverAll Total Nucleic Acid Isolation kit, starting at the protease digestion stage of manufacturer-recommended protocol. The following modification to the isolation procedure was made: instead of incubating cells in digestion buffer for 15 minutes at 50 °C and 15 minutes at 80°C, we carried out the incubation for 1 hour at 50°C. Cell lysates were frozen at −80°C overnight before continuing the RNA isolation. The RNA quality is assessed using Agilent 2100 bioanalyzer on a RNA specific chip.
Bioinformatic analyses of bulk RNA-seq data
RNAseq reads were aligned to the Schmidtea mediterranea transcriptome smed_20140614 (GEO accession number: GSE72389) using Bowtie2 v2.2.9 default parameters. Counts were the sum of reads of each transcript. FPKM values were generated using the rpkm function from R package ‘edgeR’. R package ‘edgeR’ was used for differentially expressed gene test. For the regeneration time-courses study, genes were selected as significantly changed genes during time course study by R package ‘maSigPro’ with first step fitting selection only, q<1e-30. For the RNA-seq of time-course of wildtype sublethal irradiated animals, raw data containing prefix “Unc22_SR” from GSE84025 (sublethal irradiation treatment) were downloaded, and were processed in the same way as described above. For the public data deposited in NCBI, processed data table from GSE80540 (lethal irradiation treatment) was downloaded for the downstream analysis.
Single-cell RNA-seq library construction
For single cell RNA-seq from 10x Chromium platform, Hoechst stained X1 neoblasts (200,000 cells) from wildtype animals, and from X1 plus X2 cells from 7-day sublethally irradiated animals were collected on ice using Influx sorter. Approximately 5200 counted cells were loaded per channel. The libraries were made using the Chromium platform and Chromium Single Cell 3’ v2 chemistry. Briefly, cellular suspensions were loaded on a Chromium Single Cell Instrument (10x Genomics, Pleasanton, CA) to generate single cell GEMs. Single cell RNA-Seq libraries were prepared using the Chromium Single Cell 3’ Gel Bead and Library Kit (P/N 120236, 120237, 120262, 10x Genomics). Sequencing libraries were loaded on an Illumina HiSeq 2500 Rapid flowcell with two 50bp paired-end kits using the following read length: 26 bp Read1, 8 bp I7 Index and 98 bp Read2. On average, 73,824 reads and 284,823 reads were obtained for neoblasts and sublethal irradiated cells, respectively. For plate-based single cell RNA-seq assay, cells were harvested into 96-well plate directly, and cDNA libraries were prepared using SMART-Seq v4 Ultra Low Input RNA Kit (Takara) as per manufacturer’s recommendations using 18 cycles of amplification. Specifically, reactions were run at the recommended volume. Libraries were made using the Nextera XT kit (Illumina). After the PCR step, the libraries were individually cleaned with 0.8x AMPure XP SPRI beads (Beckman Coulter), and eluted in Resuspension Buffer. The libraries were quantified using Quant-IT DNA High-Sensitivity Assay Kit (Invitrogen) and examined using a high sensitivity DNA chip (Agilent). All libraries were then combined to one pool at equimolar concentrations for sequencing. Finally, deep sequencing of samples (2–4 million reads per cell) was performed using a HiSeq 2500 sequencer.
Single cell RNA-seq data analysis
Sequence reads are aligned to Schmidtea mediterranea transcriptome smed_20140614 (GEO accession number: GSE72389) using bowtie2 with default parameters. Read counts for each transcript are generated using Samtools idxstats (version 1.3). For plate based deep single cell RNA-seq data, only samples that have the total mapped reads more than 1.5millions and the number of detected genes larger than 1,000 but less than 10,000 were kept for further analysis. All mitochondria and ribosomal RNA related genes were excluded. Read counts were then normalized using TMM method for read depth and CPM (counts per million) were calculated (R package edge). For single-cell RNA-seq data generated using 10x Genomics, data were processed using Cell Ranger pipeline (version 1.3) and aligned to the same S. mediterranea transcriptome smed_20140614. Filtered UMI counts were further normalized using Seurat (v1.4) to generate normalized expression values. The diffusion maps were generated by R package ‘destiny’. Cluster enriched marker genes with p-value<1e-20 in the single cell analysis were used for diffusion map. Only the first 9 clusters were analyzed for the neoblast dataset. We also identified 2,141 genes with high expression variability across single cells, reduced the dimensionality of these data with PCA, and identified 14 significant PCs using a permutation test (Satija et al., 2015). The PCs were used as input for t-distributed stochastic neighbor embedding (t-SNE) analyses (Laurens van der Maaten and Hinton, 2008) and for visualization and clustering of cells using a graph-based approach (Satija et al., 2015; Wurtzel et al., 2015). We further identified 116 genes that best classified the cells parsed into 12 distinct cell clusters (see Table S2 for a list of markers).
Microscopy
Colorimetric WISH images were captured using a Leica M205A stereomicroscope (Leica, Wetzlar, Germany). Confocal images (e.g., cells and sections) were generally captured using either a Zeiss LSM-700 Falcon or a Perkin Elmer Ultraview spinning disk. For whole-animal quantitative imaging, full slide with 4–6 worm tiled image sets were acquired on a Nikon Eclipse Ti equipped with a Yokogawa W1 spinning disk head and a Prior PLW20 Well Plate loader. Images were captured using a Plan Apo 20X 0.8NA air objective, and were stitched in FiJi. Tiled images of individual worms were stitched using stitching plugins in FiJi with customized batch processing macros or wrapper plugins where necessary.
Structured illumination microscopy
SIM images were acquired at OMX Blaze V4 (GE Healthcare) with a 60× 1.42 NA Olympus Plan Apo oil objective. Alexa647 was excited using a 640-nm laser, and emission was collected by a PCO Edge sCMOS. SIM reconstruction was done with softWoRx software (GE Healthcare), with a Wiener filter of 0.001. SIM images shown in the figure were maximum projections of selected z sections.
Single cell transplantation
Single cell transplantation experiment was essentially adapted from previous reports with modifications (Davies et al., 2017; Wagner et al., 2011). Host animals (8–12 mm in length, ≥7 day starved) were selected from the clonally derived Smed sexual strains based on a careful comparisons of resistance to irradiation in the lab (unpublished), S2F8b (Guo et al., 2016) animals were selected. After optimizing the culture condition, we found that a recirculation system could maintain better health of the irradiated animals, so we normally culture S2F8b in the recirculation culture system in 1x Montjuic water without antibiotics before transplantation. Animals were starved for at least one week before pulling out from recirculating water for irradiation treatment. The host neoblast population was ablated by exposure to 6,000 rads on a GammaCell 40 Exactor irradiator; cohorts of un-irradiated animals were reserved to verify complete elimination of the neoblast population by WISH with riboprobes against piwi-1. The irradiated host were then transferred into static culture supplemented with gentamicin (100 μg/mL gentamicin sulfate) until transplantation. Cell sorting was performed as described above, except for only tail fragments were dissociated for the transplantation experiment, and all the sorting was done using Influx sorter. About 10,000 cells were sorted into CMFB at 4°C, and cells were then further washed briefly to remove debris and potential non-nucleated cells (vesicles), and were further concentrated and titrated into ~500 cells per microliter. Coverslip (24×50) was pre-treated with Sigmacote (Sigma, SL2), and air dried in the hood. One droplet of cell containing medium was loaded on coverslip under 20x, and single cell was loaded by mouth pipetting into borosilicate glass needles (Sutter Instrument Co., #B100–75-15) pulled using a flaming/brown micropipette puller (Sutter Instrument Co., Model P-97). Individual cell was injected using an Eppendorf FemtoJet at 0.9–1.5 psi, as described (Davies et al., 2017; Wagner et al., 2011). Hosts were immobilized on a cold peltier plate, ventral side up, and cell were injected into the tail stripe (i.e., the medial, post-pharyngeal parenchymal space between the two posterior branches of the intestine). Hosts were injected at 2-day post-irradiation (dpi) for rescue experiments. Transplanted animals and uninjected, 6,000-Rad-irradiated hosts for rescue experiments were maintained individually in 6-well plates at 20°C in the dark supplemented with gentamicin containing water, with water exchanges and visual inspection of animals performed every 2–3 days. Animals slated for fixation were reared in 10 cm petri dishes after transplantation, with 10 or fewer animals per dish, with water exchanges every 2–3 days.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data in figure panels reflect several independent experiments performed on different days. An estimate of variation within each group of data is indicated using standard deviation (SD). For statistical analysis GraphPad Prism (V6) was used.
Quantification of PIWI-1 and F-ara-EdU signal intensity
All fluorescence intensity quantification was done with FIJI. For quantitative analysis of PIWI-1 and F-ara-EdU intensity (Figures S2K and S2L), only PIWI-1+ cells were examined. To find PIWI-1+ cells, background was substracted with a rolling filter of 50, and images were threshold with “Huang Dark” to find the edge of individual cells and a list of ROIs (Region of interest) was generated by “analyzing particles”. For measuring fluorescence intensities of the PIWI-1 and F-ara-EdU, the summed z-sections were used. Integrated intensity was calculated from both channel within ROIs. A similar procedure was applied to quantify PIWI-1 intensity alone on sorted cells (Figure S1G).
Quantification of co-expression on whole animals
For quantification of FISH co-expression, spots counted as described previously (Adler et al., 2014). Custom plugins were used to segment the DAPI labeled worms and the ‘Find Maxima’ function was used to count spots, both wrapped in batch processing macros. For whole-animal quantitative imaging, prepared slides were loaded on a Nikon Eclipse Ti equipped with a Yokogawa W1 spinning disk head and a Prior PLW20 Well Plate loader. Custom routines written in Nikon Elements Jobs and FiJi were used to automatically find worms with a lower magnification objective and take tiled images using a Plan Apo 20X 0.8NA air objective. Images were stitched in FiJi. Background subtraction and depth attenuation were performed, and individual nuclei were found using a combination of Laplacian of Gaussian and three dimensional find maximum. Three dimensional spheres were centered on each detected nucleus and integrated over in multiple channels to provide co-localization data for piwi-1+/tgs-1+. For markers where signal was weaker or for where there was poor DAPI signal, individual piwi-1+ cells were manually annotated by an independent researcher and quantified in other channels. All quantification macros and plugins can be found at https://github.com/jouyun.
Dimensionality reduction and clustering of single cell RNA-seq
Dimensionality reduction and clustering Single-cell RNA-seq yields high dimensional gene expression data. To visualize and interpret these data, we obtained two-dimensional projections of the cell population by first reducing the dimensionality of the gene expression matrix using principal component analysis (PCA), then further reducing the dimensionality of these components using t-distributed Stochastic Neighbor Embedding (tSNE). We performed PCA on a reduced gene expression matrix composed of the 2141 over-dispersed genes (as described below). To identify significant principal components (PCs), we examined the distribution of eigenvalues obtained by performing PCA after shuffling the gene expression matrix (with 100 replicates). A PC was considered significant if the magnitude of its associated eigenvalue exceeded the maximum magnitude of eigenvalues observed in the shuffled data. Significant components (14 PCs) were used for further analysis. We further reduced these components using tSNE to project them into a two-dimensional space. To measure the global transcriptional response of pluripotent neoblasts in response to different treatments, we took the average expression of each gene for a given cell population, and did quantile normalization to minimize the possible technical variation across different platforms (Bullard et al., 2010).
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the bulk RNA-seq data of the sorted cells or whole animals and the single cell RNA-seq analyses reported in this paper have been submitted to NCBI GEO (GSE107875, GSE107874 and GSE107873). Full-length sequences of genes cloned for this study are deposited in GenBank, accession numbers: MG551539 (tspan-1), MG551540 (tgs-1) and MG551541 (pks-1). Original data underlying this manuscript can be accessed from the Stowers Original Data Repository (ODR) at: http://www.stowers.org/research/publications/libpb-1288.
Supplementary Material
Figure S6. Developing antibody against TSPAN-1, Related to Figure 5
(A) Schematic of the SMED-TSPAN-1 protein. TSPAN-1 contains 4 transmembrane (TM) domains and two extracellular loops (EC1 and EC2). The second extracellular loop contains a highly conserved CCG motif and two conserved cysteine residues. The proline and two glycine residues located in second extracellular loop are also conserved. The EC2 loop (AA104–196) was used for antigen design.
(B) IF analysis showing TSPAN-1 localization in whole-mount control animals (left) or in tspan-1(RNAi) animals (right). Nuclear regions were counterstained with DAPI. Representative areas shown. At least 6 animals for each condition were assayed. Scale bar, 10 μm.
(C) Western blot results show changes of TSPAN1 protein in response to RNAi.
(D) Hoechst staining of TSPAN-1+ cell population shows cell cycle status.
(E) Representative images of piwi-1 and tspan-1 FISH staining on sorted cell populations. Representative areas shown. At least 5 animals for each condition were assayed. Scale bar, 10 μm.
(F) Representative ISH of piwi-1 after bulk cell transplantation. Representative animals shown, n>6 per condition. Scale bar, 500 μm.
Figure S7. Resolving cNeoblast complexity by single cell RNAseq, Related to Figure 6
(A) Quantification of number of piwi-1+ cells in whole animals at different time points after sub-lethal (1,250 rads) irradiation. Each square in each time-point represents an individual worm.
(B) To be inclusive for cNeoblast cells, the combination of X1 (enriched for piwi-1high) and X2 (enriched for piwi-1low) cells were collected by sorting Hoechst-stained cells in 7-day post-sub-lethally irradiated animals.
(C-D) Expression of proliferative marker (pcna), neoblast marker piwi-1 and Nb2 markers (e.g., tgs-1 and pks-1) in the t-SNE clustered cells sub-lethally irradiated X1 and X2 cells from (B).
(E) Representative images of FISH co-staining of Nb2 marker tspan-1, and neoblast marker piwi-1 in the sorted X1 cell population at 7-day (7dpi) and 11-day (11dpi) post irradiation (1,250 rads). Representative cells shown, n>100 cells per condition. Scale bar, 10 μm.
(F) Diffusion map visualization of lineage markers in the 10 SL clusters identified by single-cell RNAseq.
(G) Correlation matrix between indicated cell groups and PIWI-1 populations. Heatmap displaying spearman rank correlations between indicated pairwise comparisons. Spearman correlations were calculated using normalized read counts across the entire transcriptome identified for all RNA-seq experiments (n=31,253 genes).
(H) Heatmap of scaled expression of Nb discriminative gene sets against each cluster that survived sublethal irradiation. Color scheme is based on z-score distribution from −2.5 (Blue) to 2.5 (Red). Left margin color bars highlight gene sets specific to the respective subset of Nb populations.
(I) Heatmap of scaled expression of SL discriminative gene sets against each cluster of neoblast cells. Color scheme is based on z-score distribution from −2.5 (Blue) to 2.5 (Red). Left margin color bars highlight gene sets specific to the respective subset of sublethal (SL) survived populations.
Figure S5. characterization of tspan-1 gene, Related to Figure 4
(A) Circular phylogenetic tree of TSPAN-1 proteins from Mus musculus (mm), Drosophila melanogaster (dm), Schistosoma mansoni (Smp), and Schmidtea mediterranea (SMED). The tree was generated with RAxML and viewed in ITOL.
(B) Co-expression of piwi-1 and tspan-1 assessed by Flow-FISH after amputation. Representative images from ImageStream flow cytometric analysis shown. Scale bar, 10 μm.
(C) Representative images of double-labeled FISH for tspan-1 and soxP-1 mRNA expression in regenerating planarians. Representative animals shown, n>6 per condition. Scale bar, 100 μm unless otherwise indicated.
(D) control(RNAi) and tspan-1(RNAi) planarians at 7 and 14 dpi stained for piwi-1 probe. Representative animals shown, n=5 per condition. Scale bar, 100 μm.
(E) Dispersion was assessed at anterior boundaries in anteriorly amputated animals at 9 dpi, corresponding to 5 dpa. Representative animals shown, n=5 per condition. Scale bar, 1mm.</SI_Caption>
Figure S2. Gene expression profiling of PIWI-1+ populations, Related to Figure 1
(A) Quality of RNA extracted from sorted cell populations after fixation assessed by virtual gel images using Agilent RNA Pico chip. RNA extracted from live cells was used for side-by-side comparison.
(B-D) Quality control of RNA-seq. Read level alignment statistics (B), and whole transcriptome-wide gene expression correlation before (C) and after (D) removing batch effects for each sample.
(E) Spearman correlation heatmap of mRNA profiles generated from anti-PIWI-1 antibody sorted cell populations, and Hoechst stained X1, X2 and Xins cell populations.
(F) Volcano plot of differentially expressed mRNAs in PIWI-1high cells versus X1 cells. Vertical lines indicate threshold for a relative expression fold change of 4- or −4-fold compared to X1 cells. 2,368 genes showing differential expression in these two samples were highlighted in red.
(G) Heatmap of scaled expression of markers of known neoblast classes. Left margin color bars highlight gene sets specific to the respective known neoblast classes.
(H) Whole-mount in situ hybridization (WISH) of piwi-1 on planarians exposed to sublethal irradiation (1,250 rads) at indicated days post irradiation (dpi) (top). Expression of signature genes for each PIWI-1 subgroup were plotted at indicated time points after sub-lethal (1,250 rads) irradiation. Scale bar, 250 μm.
(I) piwi-1 WISH of planarians exposed to lethal irradiation (6,000 rads) at indicated days post irradiation (dpi) (top). Expression of signature genes for each PIWI-1 subgroup were plotted at indicated time points after lethal (6,000 rads) irradiation. Scale bar, 250 μm.
(J) Flow cytometry gating strategy for X1(FS)-L and X2(FS)-R cells. X1 and X2 cells were identified by Hoechst 33342 staining (upper left) and back-gated to set the X1(FS) and X2(FS) gate based on size (FS) and complexity(SS) parameters (upper right). Since Piwi-1high cells are bigger in size (Figure 1C), we set X1(FS)-L and X2(FS)-R gates to avoid the overlay of X1(FS) and X2(FS). Shown is a representative of 3 independent experiments.
(K-L) Representative images of F-ara-EdU and PIWI-1 co-staining on sorted cell populations. Small starved animals were fed with F-ara-EdU, and cells were isolated 10 hours later for fixation followed by F-ara-EdU/PIWI-1 antibody detection. Quantification of signal intensity of each channel was done on PIWI-1+ cells (L) (n=239).
Figure S4. Characterization of Nb2 group signature genes, Related to Figure 3
(A) Violin plots show distribution of expression levels for each previously reported self-renewal genes in the cells (points) of each of the 12 neoblast clusters.
(B-D) Expression of signature genes for each neoblast subgroup at specific time points of regeneration (B), sub-lethal irradiation treatment (C) and lethal irradiation treatment
(D). Top-ten signature genes for each Nb clusters were used to generate the plots.
(E) Diagram describing the five-step screen that identified Nb2 as a cNeoblast containing candidate cell group in the single cell transcriptome.
(F) Representative images of co-expression of tgs-1 with piwi-1 across the whole animal from dorsal to ventral side. The percentage of tgs-1+ cells in piwi-1+ population was shown in color scale. Scale bar, 250 μm.
Figure S1. Simultaneous detection of piwi-1 expression and PIWI-1 protein levels in single cells, Related to Figure 1
(A) Expression distributions by RNA Flow-FISH of planarian cells stained with negative control bacterial DapB gene probe.
(B) Bright-field and fluorescence images of RNA Flow-FISH on piwi-1 negative (left), piwi-1 low (middle) and piwi-1 high (right) cells by ImageStream flow cytometric analysis.
(C) Representative density plot diagrams of PIWI-1 negative cells for isotype antibody stained planarian cells.
(D) Images of representative PIWI-1 low cells (left) and PIWI-1 high cells (right). Shown is a representative of >3 independent experiments. Scale bar, 10 μm.
(E) Representative cell cycle profiles determined by FACS analysis of Hoechst staining. Graphs illustrate cell counts in different phases of cell cycle, which correlate to Hoechst intensity. Cell cycle profile of PIWI-1high cells: 15.7% found in G0/G1, 24.7% in S and 42.2% in G2/M. Cell cycle profile of PIWI-1low cells: 86.6% found in G0/G1, 3.75% in S and 5.41% in G2/M.
(F) Expression distributions by RNA Flow-FISH using either single probes or negative probes. Negative control against bacterial DapB gene showed similar level of background staining as no probe control. Probes against piwi-1 and agat-1 showed distinct staining patterns, further indicating probe specificity.
(G) Signal intensity quantification reveals PIWI-1 distribution in untreated and 1-day irradiated animals. Each dot represents an individual cell.
(H) Representative contour plots of FACS analyses show irradiation mainly eliminated PIWI-1high/G2/M phase cells. Shown is a representative of >3 independent experiments.
Figure S3. Verification of single cell RNA-seq of neoblasts, Related to Figure 2
(A) Gating strategy of neoblast purification for single cell RNA-seq. By sorting Hoechst stained X1 and X2 cells, piwi-1high and piwi-1low cells, respectively, were enriched. Only X1 cells were subjected to scRNA-seq.
(B) Representative images of FISH on sorted cells showing the signal intensity of piwi-1 transcript in sorted X1 (left) and X2 cells (right). Scale bar, 10 μm.
(C) The distribution of expression levels for known neoblast marker piwi-1 in cells (points) in each of 12 neoblast clusters displayed in FeaturePlot (left) and Violin plots (right).
(D) Heatmap reports scaled expression of markers associated with three known neoblast-classes. Dashed rectangles represent the only two Nb clusters that could be unambiguously distinguished by known neoblast class markers (Nb1 and Nb5 by ζ and γ markers, respectively).
(E) Violin plots show distribution of expression levels for three known neoblast-class markers in the cells (points) of each of the 12 neoblast clusters.
(F-G) Violin plots show distribution of expression levels for the Nb10 (ascl4) and Nb11 (ston-2 and chat) markers in the cells (points) of each of the 12 neoblast clusters.
(H) Violin plots (top) and in situ hybridization (bottom) show expression pattern for gst-1 (Nb12 marker). Scale bar, 100 μm.
(I) Visualization of expression of five lineage progenitor markers in diffusion maps.
Movie S3. Image of a host after single cell transplantation, Related to Figure 5.
After loading a single cell into a needle, the cell was injected/transplanted into the medial, postpharyngeal, tail stripe of host. To ascertain the loading and injection, individual Hoechst labeled cell was checked with fluorescent channel after transplantation. Zero (n = 4/20 animals) or one (n = 16/20 animals) Hoechst+ cells were observed in all cases, with expected size and morphology.
Movie S2. Image of lively sorted single TSPAN-1+ cell, Related to Figure 5.
TSPAN-1 antibody signal (green) shows asymmetric distribution on one side of membrane. Blue, Hoechst 33342. The cell shows obvious blebs and active cytoplasmic processes (scale bar, 10μm).
Movie S4. Diffusion map of sublethal irradiated cell clusters, Related to Figure 6.
Diffusion pseudotime analysis uncovered five differentiation paths from which cell populations branched out. One cluster likely sitting at the root is SL6 (magenta).
Movie S1. Diffusion map of neoblast clusters, Related to Figure 2.
Diffusion pseudotime analyses uncovered four differentiation paths from which cell populations branched out. Two sets of clusters likely sitting at the root are Nb2 and Nb3. Nb10 through Nb12 were not included in the analyses due to low cell number.
Table S3. Signature genes of Sublethal groups and Genes upregulated during regeneration and repopulation, Related to Figure 6
Tab “Signature genes for SL groups” Neoblast cluster: cluster ID. ave_diff: the Log fold change of average expression in the current cluster against the rest of clusters. pct.1: the percentage of cells which express the marker gene in the current cluster. pct.2: the percentage of cells which express the marker gene in the rest of the clusters.
Tab “Repopulation_upregulated genes” contains genes that are upregulated (at least 4 folds) in sublethally-irradiated SL6a group versus Nb2 group during homeostasis. Tab “Regeneration_upregulated genes” contains genes that are upregulated (at least 4 folds) in regenerating 3dpa group versus Nb2 group during homeostasis.
Tab “Repopulation_specific_Up” contains genes that are upregulated (at least 4 folds) only during Repopulation but not during regeneration when compared to Nb2 group. Tab “Regeneration_specific_Up” contains genes that are upregulated (at least 4 folds) only during regeneration but not during repopulation when compared to Nb2 group. Tab “Upregulated in both” contains genes that are upregulated (at least 4 folds) in both regeneration and repopulation contexts. For all tabs, SMEDID: Transcript ID in smed20140614 transcriptome.
Table S2. Signature genes of Neoblast groups, Related to Figure 2
SMEDID: Transcript ID in smed20140614 transcriptome. Neoblast cluster: cluster ID. ave_diff: the Log fold change of average expression in the current cluster against the rest of clusters. pct.1: the percentage of cells which express the marker gene in the current cluster. pct.2: the percentage of cells which express the marker gene in the rest of the clusters.
Table S1. Signature genes of PIWI-1 groups, Related to Figure 1
Table S1 contains the list of signature genes for each PIWI-1 groups. SMEDID: Transcript ID in smed20140614 transcriptome.
ACKNOWLEDGEMENTS
We thank Linheng Li, Brian Slaughter, Blair Benham-Pyle, Biff Mann, Tatjana Piotrowski for comments; all members of the A.S.A. laboratory for discussion and advice; Lu Wang, Shasha Zhang, Wenyu Zhang, Yan Li, Kirsten Gotting, Chris Seidel for assistance in data analysis and sample preparation; Jeff Haug, Jay Unruh, Donald Little for valuable input on the manuscript; Jim Jenkin for antibody generation and transplantation; Qing Jing for providing PIWI-1 antibody (#6116); Stephanie Nowotarski for comments and graphical abstract. We are grateful to the Stowers Planarian, Cytometry, Molecular Biology, Histology and Tissue Culture core facilities. A.S.A. is a Howard Hughes Medical Institute and Stowers Institute for Medical Research investigator. This work was supported in part by NIH grant R37GM057260.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes 7 figures, 4 movies, and 3 tables found with this article online.
REFERENCES
- Adler CE, Seidel CW, McKinney SA, and Sánchez Alvarado A (2014). Selective amputation of the pharynx identifies a FoxA-dependent regeneration program in planaria. Elife 3, e02238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucheix C, and Rubinstein E (2001). Tetraspanins. Cell Mol Life Sci 58, 1189–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, and Dudoit S (2010). Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LC, Tu KC, Seidel CW, Robb SMC, Guo F, and Sánchez Alvarado A (2017). Cellular, ultrastructural and molecular analyses of epidermal cell development in the planarian Schmidtea mediterranea. Dev Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JJ 3rd, Hou X, Romanova EV, Lambrus BG, Miller CM, Saberi A, Sweedler JV, and Newmark PA (2010). Genome-wide analyses reveal a role for peptide hormones in planarian germline development. PLoS Biol 8, e1000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles MW, Brown DD, Nisperos SV, Stanley BN, Pearson BJ, and Zayas RM (2013). Genome-wide analysis of the bHLH gene family in planarians identifies factors required for adult neurogenesis and neuronal regeneration. Development 140, 4691–4702. [DOI] [PubMed] [Google Scholar]
- Davies EL, Lei K, Seidel CW, Kroesen AE, McKinney SA, Guo L, Robb SM, Ross EJ, Gotting K, and Sánchez Alvarado A (2017). Embryonic origin of adult stem cells required for tissue homeostasis and regeneration. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhoffer GT, Kang H, and Sánchez Alvarado A (2008). Molecular analysis of stem cells and their descendants during cell turnover and regeneration in the planarian Schmidtea mediterranea. Cell Stem Cell 3, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedelhoefer OC, and Sánchez Alvarado A (2012). Amputation induces stem cell mobilization to sites of injury during planarian regeneration. Development 139, 3510–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Peters AH, and Newmark PA (2006). A Bruno-like gene is required for stem cell maintenance in planarians. Dev Cell 11, 159–169. [DOI] [PubMed] [Google Scholar]
- Haghverdi L, Buttner M, Wolf FA, Buettner F, and Theis FJ (2016). Diffusion pseudotime robustly reconstructs lineage branching. Nat Methods 13, 845–848. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Asami M, Higuchi S, Shibata N, and Agata K (2006). Isolation of planarian X-ray-sensitive stem cells by fluorescence-activated cell sorting. Dev Growth Differ 48, 371–380. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Shibata N, Okumura R, Kudome T, Nishimura O, Tarui H, and Agata K (2010). Single-cell gene profiling of planarian stem cells using fluorescent activated cell sorting and its “index sorting” function for stem cell research. Dev Growth Differ 52, 131–144. [DOI] [PubMed] [Google Scholar]
- Hemler ME (2005). Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol 6, 801–811. [DOI] [PubMed] [Google Scholar]
- Hemler ME (2014). Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer 14, 49–60. [DOI] [PubMed] [Google Scholar]
- Huang S, Yuan S, Dong M, Su J, Yu C, Shen Y, Xie X, Yu Y, Yu X, Chen S, et al. (2005). The phylogenetic analysis of tetraspanins projects the evolution of cell-cell interactions from unicellular to multicellular organisms. Genomics 86, 674–684. [DOI] [PubMed] [Google Scholar]
- Karlsson G, Rorby E, Pina C, Soneji S, Reckzeh K, Miharada K, Karlsson C, Guo Y, Fugazza C, Gupta R, et al. (2013). The tetraspanin CD9 affords high-purity capture of all murine hematopoietic stem cells. Cell Rep 4, 642–648. [DOI] [PubMed] [Google Scholar]
- King RS, and Newmark PA (2013). In situ hybridization protocol for enhanced detection of gene expression in the planarian Schmidtea mediterranea. BMC Dev Biol 13, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HY, Bajaj J, Ito T, Blevins A, Konuma T, Weeks J, Lytle NK, Koechlein CS, Rizzieri D, Chuah C, et al. (2015). Tetraspanin 3 Is Required for the Development and Propagation of Acute Myelogenous Leukemia. Cell Stem Cell 17, 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten Laurens, and Hinton G (2008). Visualizing Data using t-SNE. Journal of Machine Learning Research 9, 2579–2605. [Google Scholar]
- Lei K, Thi-Kim Vu H, Mohan RD, McKinney SA, Seidel CW, Alexander R, Gotting K, Workman JL, and Sánchez Alvarado A (2016). Egf Signaling Directs Neoblast Repopulation by Regulating Asymmetric Cell Division in Planarians. Dev Cell 38, 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinaro AM, and Pearson BJ (2016). In silico lineage tracing through single cell transcriptomics identifies a neural stem cell population in planarians. Genome Biol 17, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmark PA, and Sánchez Alvarado A (2000). Bromodeoxyuridine specifically labels the regenerative stem cells of planarians. Dev Biol 220, 142–153. [DOI] [PubMed] [Google Scholar]
- Newmark PA, and Sánchez Alvarado A (2002). Not your father’s planarian: a classic model enters the era of functional genomics. Nat Rev Genet 3, 210–219. [DOI] [PubMed] [Google Scholar]
- Pearson BJ, Eisenhoffer GT, Gurley KA, Rink JC, Miller DE, and Sánchez Alvarado A (2009). Formaldehyde-based whole-mount in situ hybridization method for planarians. Dev Dyn 238, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porichis F, Hart MG, Griesbeck M, Everett HL, Hassan M, Baxter AE, Lindqvist M, Miller SM, Soghoian DZ, Kavanagh DG, et al. (2014). High-throughput detection of miRNAs and gene-specific mRNA at the single-cell level by flow cytometry. Nat Commun 5, 5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Oviedo NJ, Jennings JR, Jenkin JC, and Sánchez Alvarado A (2005). SMEDWI-2 is a PIWI-like protein that regulates planarian stem cells. Science 310, 1327–1330. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, and Weissman IL (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105. [DOI] [PubMed] [Google Scholar]
- Rink JC, Vu HT, and Sánchez Alvarado A (2011). The maintenance and regeneration of the planarian excretory system are regulated by EGFR signaling. Development 138, 3769–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb SM, Gotting K, Ross E, and Sánchez Alvarado A (2015). SmedGD 2.0: The Schmidtea mediterranea genome database. Genesis 53, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Galbraith RH, and Newmark PA (2015). On the organ trail: insights into organ regeneration in the planarian. Curr Opin Genet Dev 32, 37–46. [DOI] [PubMed] [Google Scholar]
- Sánchez Alvarado A, Newmark PA, Robb SM, and Juste R (2002). The Schmidtea mediterranea database as a molecular resource for studying platyhelminthes, stem cells and regeneration. Development 129, 5659–5665. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimone ML, Kravarik KM, Lapan SW, and Reddien PW (2014). Neoblast specialization in regeneration of the planarian Schmidtea mediterranea. Stem Cell Reports 3, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimone ML, Srivastava M, Bell GW, and Reddien PW (2011). A regulatory program for excretory system regeneration in planarians. Development 138, 4387–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu KC, Cheng LC, H, T.K.V., Lange JJ, McKinney SA, Seidel CW, and Sánchez Alvarado A (2015). Egr-5 is a post-mitotic regulator of planarian epidermal differentiation. Elife 4, e10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wolfswinkel JC, Wagner DE, and Reddien PW (2014). Single-cell analysis reveals functionally distinct classes within the planarian stem cell compartment. Cell Stem Cell 15, 326–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez-Doorman C, and Petersen CP (2014). zic-1 Expression in Planarian neoblasts after injury controls anterior pole regeneration. PLoS Genet 10, e1004452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogg MC, Owlarn S, Perez Rico YA, Xie J, Suzuki Y, Gentile L, Wu W, and Bartscherer K (2014). Stem cell-dependent formation of a functional anterior regeneration pole in planarians requires Zic and Forkhead transcription factors. Dev Biol 390, 136–148. [DOI] [PubMed] [Google Scholar]
- Wagner DE, Ho JJ, and Reddien PW (2012). Genetic regulators of a pluripotent adult stem cell system in planarians identified by RNAi and clonal analysis. Cell Stem Cell 10, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DE, Wang IE, and Reddien PW (2011). Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration. Science 332, 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenemoser D, and Reddien PW (2010). Planarian regeneration involves distinct stem cell responses to wounds and tissue absence. Dev Biol 344, 979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witchley JN, Mayer M, Wagner DE, Owen JH, and Reddien PW (2013). Muscle cells provide instructions for planarian regeneration. Cell Rep 4, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel O, Cote LE, Poirier A, Satija R, Regev A, and Reddien PW (2015). A Generic and Cell-Type-Specific Wound Response Precedes Regeneration in Planarians. Dev Cell 35, 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng A, Li YQ, Wang C, Han XS, Li G, Wang JY, Li DS, Qin YW, Shi Y, Brewer G, et al. (2013). Heterochromatin protein 1 promotes self-renewal and triggers regenerative proliferation in adult stem cells. J Cell Biol 201, 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zöller M (2009). Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer 9, 40–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S6. Developing antibody against TSPAN-1, Related to Figure 5
(A) Schematic of the SMED-TSPAN-1 protein. TSPAN-1 contains 4 transmembrane (TM) domains and two extracellular loops (EC1 and EC2). The second extracellular loop contains a highly conserved CCG motif and two conserved cysteine residues. The proline and two glycine residues located in second extracellular loop are also conserved. The EC2 loop (AA104–196) was used for antigen design.
(B) IF analysis showing TSPAN-1 localization in whole-mount control animals (left) or in tspan-1(RNAi) animals (right). Nuclear regions were counterstained with DAPI. Representative areas shown. At least 6 animals for each condition were assayed. Scale bar, 10 μm.
(C) Western blot results show changes of TSPAN1 protein in response to RNAi.
(D) Hoechst staining of TSPAN-1+ cell population shows cell cycle status.
(E) Representative images of piwi-1 and tspan-1 FISH staining on sorted cell populations. Representative areas shown. At least 5 animals for each condition were assayed. Scale bar, 10 μm.
(F) Representative ISH of piwi-1 after bulk cell transplantation. Representative animals shown, n>6 per condition. Scale bar, 500 μm.
Figure S7. Resolving cNeoblast complexity by single cell RNAseq, Related to Figure 6
(A) Quantification of number of piwi-1+ cells in whole animals at different time points after sub-lethal (1,250 rads) irradiation. Each square in each time-point represents an individual worm.
(B) To be inclusive for cNeoblast cells, the combination of X1 (enriched for piwi-1high) and X2 (enriched for piwi-1low) cells were collected by sorting Hoechst-stained cells in 7-day post-sub-lethally irradiated animals.
(C-D) Expression of proliferative marker (pcna), neoblast marker piwi-1 and Nb2 markers (e.g., tgs-1 and pks-1) in the t-SNE clustered cells sub-lethally irradiated X1 and X2 cells from (B).
(E) Representative images of FISH co-staining of Nb2 marker tspan-1, and neoblast marker piwi-1 in the sorted X1 cell population at 7-day (7dpi) and 11-day (11dpi) post irradiation (1,250 rads). Representative cells shown, n>100 cells per condition. Scale bar, 10 μm.
(F) Diffusion map visualization of lineage markers in the 10 SL clusters identified by single-cell RNAseq.
(G) Correlation matrix between indicated cell groups and PIWI-1 populations. Heatmap displaying spearman rank correlations between indicated pairwise comparisons. Spearman correlations were calculated using normalized read counts across the entire transcriptome identified for all RNA-seq experiments (n=31,253 genes).
(H) Heatmap of scaled expression of Nb discriminative gene sets against each cluster that survived sublethal irradiation. Color scheme is based on z-score distribution from −2.5 (Blue) to 2.5 (Red). Left margin color bars highlight gene sets specific to the respective subset of Nb populations.
(I) Heatmap of scaled expression of SL discriminative gene sets against each cluster of neoblast cells. Color scheme is based on z-score distribution from −2.5 (Blue) to 2.5 (Red). Left margin color bars highlight gene sets specific to the respective subset of sublethal (SL) survived populations.
Figure S5. characterization of tspan-1 gene, Related to Figure 4
(A) Circular phylogenetic tree of TSPAN-1 proteins from Mus musculus (mm), Drosophila melanogaster (dm), Schistosoma mansoni (Smp), and Schmidtea mediterranea (SMED). The tree was generated with RAxML and viewed in ITOL.
(B) Co-expression of piwi-1 and tspan-1 assessed by Flow-FISH after amputation. Representative images from ImageStream flow cytometric analysis shown. Scale bar, 10 μm.
(C) Representative images of double-labeled FISH for tspan-1 and soxP-1 mRNA expression in regenerating planarians. Representative animals shown, n>6 per condition. Scale bar, 100 μm unless otherwise indicated.
(D) control(RNAi) and tspan-1(RNAi) planarians at 7 and 14 dpi stained for piwi-1 probe. Representative animals shown, n=5 per condition. Scale bar, 100 μm.
(E) Dispersion was assessed at anterior boundaries in anteriorly amputated animals at 9 dpi, corresponding to 5 dpa. Representative animals shown, n=5 per condition. Scale bar, 1mm.</SI_Caption>
Figure S2. Gene expression profiling of PIWI-1+ populations, Related to Figure 1
(A) Quality of RNA extracted from sorted cell populations after fixation assessed by virtual gel images using Agilent RNA Pico chip. RNA extracted from live cells was used for side-by-side comparison.
(B-D) Quality control of RNA-seq. Read level alignment statistics (B), and whole transcriptome-wide gene expression correlation before (C) and after (D) removing batch effects for each sample.
(E) Spearman correlation heatmap of mRNA profiles generated from anti-PIWI-1 antibody sorted cell populations, and Hoechst stained X1, X2 and Xins cell populations.
(F) Volcano plot of differentially expressed mRNAs in PIWI-1high cells versus X1 cells. Vertical lines indicate threshold for a relative expression fold change of 4- or −4-fold compared to X1 cells. 2,368 genes showing differential expression in these two samples were highlighted in red.
(G) Heatmap of scaled expression of markers of known neoblast classes. Left margin color bars highlight gene sets specific to the respective known neoblast classes.
(H) Whole-mount in situ hybridization (WISH) of piwi-1 on planarians exposed to sublethal irradiation (1,250 rads) at indicated days post irradiation (dpi) (top). Expression of signature genes for each PIWI-1 subgroup were plotted at indicated time points after sub-lethal (1,250 rads) irradiation. Scale bar, 250 μm.
(I) piwi-1 WISH of planarians exposed to lethal irradiation (6,000 rads) at indicated days post irradiation (dpi) (top). Expression of signature genes for each PIWI-1 subgroup were plotted at indicated time points after lethal (6,000 rads) irradiation. Scale bar, 250 μm.
(J) Flow cytometry gating strategy for X1(FS)-L and X2(FS)-R cells. X1 and X2 cells were identified by Hoechst 33342 staining (upper left) and back-gated to set the X1(FS) and X2(FS) gate based on size (FS) and complexity(SS) parameters (upper right). Since Piwi-1high cells are bigger in size (Figure 1C), we set X1(FS)-L and X2(FS)-R gates to avoid the overlay of X1(FS) and X2(FS). Shown is a representative of 3 independent experiments.
(K-L) Representative images of F-ara-EdU and PIWI-1 co-staining on sorted cell populations. Small starved animals were fed with F-ara-EdU, and cells were isolated 10 hours later for fixation followed by F-ara-EdU/PIWI-1 antibody detection. Quantification of signal intensity of each channel was done on PIWI-1+ cells (L) (n=239).
Figure S4. Characterization of Nb2 group signature genes, Related to Figure 3
(A) Violin plots show distribution of expression levels for each previously reported self-renewal genes in the cells (points) of each of the 12 neoblast clusters.
(B-D) Expression of signature genes for each neoblast subgroup at specific time points of regeneration (B), sub-lethal irradiation treatment (C) and lethal irradiation treatment
(D). Top-ten signature genes for each Nb clusters were used to generate the plots.
(E) Diagram describing the five-step screen that identified Nb2 as a cNeoblast containing candidate cell group in the single cell transcriptome.
(F) Representative images of co-expression of tgs-1 with piwi-1 across the whole animal from dorsal to ventral side. The percentage of tgs-1+ cells in piwi-1+ population was shown in color scale. Scale bar, 250 μm.
Figure S1. Simultaneous detection of piwi-1 expression and PIWI-1 protein levels in single cells, Related to Figure 1
(A) Expression distributions by RNA Flow-FISH of planarian cells stained with negative control bacterial DapB gene probe.
(B) Bright-field and fluorescence images of RNA Flow-FISH on piwi-1 negative (left), piwi-1 low (middle) and piwi-1 high (right) cells by ImageStream flow cytometric analysis.
(C) Representative density plot diagrams of PIWI-1 negative cells for isotype antibody stained planarian cells.
(D) Images of representative PIWI-1 low cells (left) and PIWI-1 high cells (right). Shown is a representative of >3 independent experiments. Scale bar, 10 μm.
(E) Representative cell cycle profiles determined by FACS analysis of Hoechst staining. Graphs illustrate cell counts in different phases of cell cycle, which correlate to Hoechst intensity. Cell cycle profile of PIWI-1high cells: 15.7% found in G0/G1, 24.7% in S and 42.2% in G2/M. Cell cycle profile of PIWI-1low cells: 86.6% found in G0/G1, 3.75% in S and 5.41% in G2/M.
(F) Expression distributions by RNA Flow-FISH using either single probes or negative probes. Negative control against bacterial DapB gene showed similar level of background staining as no probe control. Probes against piwi-1 and agat-1 showed distinct staining patterns, further indicating probe specificity.
(G) Signal intensity quantification reveals PIWI-1 distribution in untreated and 1-day irradiated animals. Each dot represents an individual cell.
(H) Representative contour plots of FACS analyses show irradiation mainly eliminated PIWI-1high/G2/M phase cells. Shown is a representative of >3 independent experiments.
Figure S3. Verification of single cell RNA-seq of neoblasts, Related to Figure 2
(A) Gating strategy of neoblast purification for single cell RNA-seq. By sorting Hoechst stained X1 and X2 cells, piwi-1high and piwi-1low cells, respectively, were enriched. Only X1 cells were subjected to scRNA-seq.
(B) Representative images of FISH on sorted cells showing the signal intensity of piwi-1 transcript in sorted X1 (left) and X2 cells (right). Scale bar, 10 μm.
(C) The distribution of expression levels for known neoblast marker piwi-1 in cells (points) in each of 12 neoblast clusters displayed in FeaturePlot (left) and Violin plots (right).
(D) Heatmap reports scaled expression of markers associated with three known neoblast-classes. Dashed rectangles represent the only two Nb clusters that could be unambiguously distinguished by known neoblast class markers (Nb1 and Nb5 by ζ and γ markers, respectively).
(E) Violin plots show distribution of expression levels for three known neoblast-class markers in the cells (points) of each of the 12 neoblast clusters.
(F-G) Violin plots show distribution of expression levels for the Nb10 (ascl4) and Nb11 (ston-2 and chat) markers in the cells (points) of each of the 12 neoblast clusters.
(H) Violin plots (top) and in situ hybridization (bottom) show expression pattern for gst-1 (Nb12 marker). Scale bar, 100 μm.
(I) Visualization of expression of five lineage progenitor markers in diffusion maps.
Movie S3. Image of a host after single cell transplantation, Related to Figure 5.
After loading a single cell into a needle, the cell was injected/transplanted into the medial, postpharyngeal, tail stripe of host. To ascertain the loading and injection, individual Hoechst labeled cell was checked with fluorescent channel after transplantation. Zero (n = 4/20 animals) or one (n = 16/20 animals) Hoechst+ cells were observed in all cases, with expected size and morphology.
Movie S2. Image of lively sorted single TSPAN-1+ cell, Related to Figure 5.
TSPAN-1 antibody signal (green) shows asymmetric distribution on one side of membrane. Blue, Hoechst 33342. The cell shows obvious blebs and active cytoplasmic processes (scale bar, 10μm).
Movie S4. Diffusion map of sublethal irradiated cell clusters, Related to Figure 6.
Diffusion pseudotime analysis uncovered five differentiation paths from which cell populations branched out. One cluster likely sitting at the root is SL6 (magenta).
Movie S1. Diffusion map of neoblast clusters, Related to Figure 2.
Diffusion pseudotime analyses uncovered four differentiation paths from which cell populations branched out. Two sets of clusters likely sitting at the root are Nb2 and Nb3. Nb10 through Nb12 were not included in the analyses due to low cell number.
Table S3. Signature genes of Sublethal groups and Genes upregulated during regeneration and repopulation, Related to Figure 6
Tab “Signature genes for SL groups” Neoblast cluster: cluster ID. ave_diff: the Log fold change of average expression in the current cluster against the rest of clusters. pct.1: the percentage of cells which express the marker gene in the current cluster. pct.2: the percentage of cells which express the marker gene in the rest of the clusters.
Tab “Repopulation_upregulated genes” contains genes that are upregulated (at least 4 folds) in sublethally-irradiated SL6a group versus Nb2 group during homeostasis. Tab “Regeneration_upregulated genes” contains genes that are upregulated (at least 4 folds) in regenerating 3dpa group versus Nb2 group during homeostasis.
Tab “Repopulation_specific_Up” contains genes that are upregulated (at least 4 folds) only during Repopulation but not during regeneration when compared to Nb2 group. Tab “Regeneration_specific_Up” contains genes that are upregulated (at least 4 folds) only during regeneration but not during repopulation when compared to Nb2 group. Tab “Upregulated in both” contains genes that are upregulated (at least 4 folds) in both regeneration and repopulation contexts. For all tabs, SMEDID: Transcript ID in smed20140614 transcriptome.
Table S2. Signature genes of Neoblast groups, Related to Figure 2
SMEDID: Transcript ID in smed20140614 transcriptome. Neoblast cluster: cluster ID. ave_diff: the Log fold change of average expression in the current cluster against the rest of clusters. pct.1: the percentage of cells which express the marker gene in the current cluster. pct.2: the percentage of cells which express the marker gene in the rest of the clusters.
Table S1. Signature genes of PIWI-1 groups, Related to Figure 1
Table S1 contains the list of signature genes for each PIWI-1 groups. SMEDID: Transcript ID in smed20140614 transcriptome.
Data Availability Statement
The accession numbers for the bulk RNA-seq data of the sorted cells or whole animals and the single cell RNA-seq analyses reported in this paper have been submitted to NCBI GEO (GSE107875, GSE107874 and GSE107873). Full-length sequences of genes cloned for this study are deposited in GenBank, accession numbers: MG551539 (tspan-1), MG551540 (tgs-1) and MG551541 (pks-1). Original data underlying this manuscript can be accessed from the Stowers Original Data Repository (ODR) at: http://www.stowers.org/research/publications/libpb-1288.