

Abstract
Epinephrine (EPI), an endogenous catecholamine involved in the body’s fight-or-flight responses to stress, activates α1-adrenergic receptors (α1ARs) expressed on various organs to evoke a wide range of physiological functions, including vasoconstriction. In the smooth muscle of human bronchi, however, the functional role of EPI on α1ARs remains controversial. Classically, evidence suggests that EPI promotes bronchodilation by stimulating β2-adrenergic receptors (β2ARs). Conventionally, the selective β2AR agonism of EPI was thought to be, in part, due to a predominance of β2ARs and/or a sparse, or lack of α1AR activity in human airway smooth muscle (HASM) cells. Surprisingly, we find that HASM cells express a high abundance of ADRA1B (the α1AR subtype B) and identify a spontaneous “switch-like” activation of α1ARs that evokes intracellular calcium, myosin light chain phosphorylation, and HASM cell shortening. The switch-like responses, and related EPI-induced biochemical and mechanical signals, emerged upon pharmacological inhibition of β2ARs and/or under experimental conditions that induce β2AR tachyphylaxis. EPI-induced procontractile effects were abrogated by an α1AR antagonist, doxazosin mesylate (DM). These data collectively uncover a previously unrecognized feed-forward mechanism driving bronchospasm via two distinct classes of G protein-coupled receptors (GPCRs) and provide a basis for reexamining α1AR inhibition for the management of stress/exercise-induced asthma and/or β2-agonist insensitivity in patients with difficult-to-control, disease subtypes.
Keywords: adrenergic receptors, airway smooth muscle, asthma, bronchospasm, catecholamines
INTRODUCTION
Asthma defines a syndrome characterized by recurrent airway inflammation and nonspecific airway hyperresponsiveness (AHR) to a wide range of endogenous or exogenous stimuli (1, 2). According to the Centers for Disease Control and Prevention, over 25 million people in the United States had asthma in 2019, with more than half of the individuals exhibiting uncontrolled disease. As a cornerstone therapy, inhaled bronchodilators (short- and long-acting combined) that target β2-adrenoceptors (β2ARs), which are G protein-coupled receptors (GPCRs) expressed on human airway smooth muscle (HASM) cells, reverse or prevent airflow obstruction—a root cause of asthma morbidity and mortality (3, 4). Of note, for patients with difficult-to-control, severe asthma and/or β2-agonist insensitivity, parenteral epinephrine (EPI) is considered as a supplementary add-on therapy, especially to treat anaphylaxis in children (5).
Epinephrine (or adrenaline), a stress hormone released through the hypothalamic-pituitary-adrenal axis, binds both β2- and α1-adrenoceptors (α1ARs) with a high affinity (6, 7). Classically, in the body’s fight-or-flight responses, EPI activates α1ARs expressed on various organs to evoke wide-ranging physiological functions (8, 9). The functional role of EPI on α1ARs in HASM remains unclear, however, and decreased plasma EPI levels contribute to AHR by decreasing basal β2AR stimulation (10, 11). Indeed, reversal of HASM shortening, either with β2-agonists or EPI, occurs by binding to the β2AR that couples to a stimulatory G protein (Gαs), activating adenylate cyclase to generate 3′,5′-cyclic adenosine monophosphate (cAMP) (12). Increased cAMP stimulates protein kinase A that then mediates multiple downstream signals to decrease intracellular calcium ([Ca2+]i) and myosin light chain phosphorylation (pMLC) levels—the latter by the actions of decreased myosin light chain kinase (MLCK) activity and/or increased myosin light chain phosphatase (MLCP) activity-promoting airway smooth muscle relaxation (13–15).
Historically, EPI has served as an add-on bronchodilator for asthma exacerbations (16), but EPI can also activate the α1AR to induce airway constriction (17–19). Furthermore, activation of α1AR with methoxamine evokes bronchoconstriction in subjects with asthma that is abrogated with prazosin, an α1AR antagonist (20, 21). Whether α1AR activation plays a role in asthma or whether blocking the α1AR has therapeutic value remains unclear (22–24).
Given the limitations of β2-agonist therapy in asthma, including drug tolerance or β2AR tachyphylaxis with repeated use of β2-agonists (25–28), and because EPI can signal through both the β2AR and α1AR (7, 8), here we considered a plausible therapeutic value of α1AR antagonism in preclinical models of β2-agonist insensitivity (29–31). Specifically, we posited that EPI induces bronchoconstriction by preferentially activating α1ARs expressed on HASM cells, following β2AR tachyphylaxis. We demonstrated that, under experimental conditions that induce β2AR inhibition or desensitization, EPI evoked [Ca2+]i, pMLC, and HASM cell shortening. Pretreatment with an α1AR antagonist doxazosin mesylate (DM) prevented EPI-induced [Ca2+]i, pMLC, and HASM cell shortening. These data suggest that EPI-induced HASM cell shortening is α1AR-dependent within the context of β2AR desensitization. Collectively, our results support that α1AR inhibition could serve as a therapeutic target for asthma subtypes, including β2-agonist insensitive and stress/exercise-induced obstructive lung disease.
METHODS
HASM RNA-Seq Data
We used RNA-seq results from a previously published study that is available in the Gene Expression Omnibus under accession number GSE94335 (32). Briefly, this data set consisted of primary HASM cells derived from age- and sex-matched fatal asthma (n = 9) and nonasthma (n = 8) lung donors. Normalized read counts for the vehicle control condition based on DESeq2 output of the matrix of raw mapped read counts were obtained for the α1AR and β2AR genes.
Materials
Unless otherwise stated, all chemicals and drugs were obtained from Sigma Aldrich (St. Louis, MO). Primary antibodies against pMLC (phosphorylation at Thr18/Ser19) and GAPDH were purchased from Cell Signaling Technologies (Danvers, MA), MLC from EMD Millipore (Burlington, MA), and α1AR from Abcam (Cambridge, UK). For Western blot analyses, IRDye 800CW donkey anti-rabbit and IRDye 680RD donkey anti-mouse were purchased from LI-COR Biosciences (Lincoln, NE). For immunofluorescence analyses, Alexa Fluor 488-conjugated AffiniPure Donkey anti-rabbit IgG [711-545-152] and Rhodamine Red-X conjugated AffiniPure Donkey anti-mouse IgG [715-295-151] were purchased from Jackson ImmunoResearch (West Grove, PA).
Isolation and Culture of HASM
HASM cells were derived from tracheas obtained from de-identified lung donors procured from the International Institute for the Advancement of Medicine (Edison, NJ) or from the National Disease Research Interchange (Philadelphia, PA); these are not subjected to Rutgers IRB approval. Cell isolation was performed as previously described (33). HASM cells were cultured in Ham’s F-12 medium supplemented with 10% FBS, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 2.5 mg/mL amphotericin B. For all experiments, HASM cells were serum-deprived for 24 h and used within the first four passages to ensure the proper smooth muscle phenotype (33, 34). Donor demographics for all studies are summarized in Supplemental Table S1.
Immunofluorescent Staining of Human Bronchus
Immunofluorescent studies were performed on 7-µm thick cryosections of human bronchial tissue. The cold acetone/methanol fixed sections were washed with PBS and treated with a blocking solution (3% BSA and 1:20 diluted FcR Blocking Reagent, MACS Miltenyi Biotec Cat. No: 120-000-442) for 30 min. The sections were then incubated with rabbit anti-α1AR (Abcam [ab3462]) and mouse anti-α-smooth muscle actin (Sigma [A2547])) at 4°C overnight (diluted in 1% BSA/PBS) and detected with Alexa Fluor 488-conjugated AffiniPure Donkey anti-rabbit IgG and Rhodamine Red-X conjugated AffiniPure Donkey anti-mouse IgG, respectively. Nonimmune serum served as a negative control, and DAPI was used to stain the nucleus. The stained sections were visualized with a Nikon epifluorescence microscope. All experiments were performed in tissue from a minimum of five distinct donor lungs.
Immunoblot Analysis
For pharmacological inhibitions of β2ARs, HASM cells were treated for 10 min with 10 μM propranolol, before stimulation with increasing concentration of EPI (0.01–10 μM). To induce β2AR desensitization, HASM cells were treated for 18 h with 10 μM albuterol (29–31); the cell monolayers were washed to remove albuterol and then stimulated for 10 min with 10 μM EPI or carbachol (CCh). To assess the mechanistic action of EPI on α1AR, cells were treated for 10 min with 1 μM doxazosin mesylate (DM), before EPI and CCh stimulation. The cell monolayers were then scraped and collected after adding 0.1% final concentration of perchloric acid. Cells were pelleted, lysed, and incubated overnight at 4°C with NuPage reducing agent and sample buffer. Proteins were separated using SDS-PAGE and transferred to nitrocellulose membranes. Phosphorylation of MLC (pMLC at Thr18/Ser19) was assessed and normalized to total MLC band densities. Protein bands were detected using near-infrared conjugated secondary antibodies with Li-Cor Odyssey CLX, and the intensity of the protein bands was calculated using Image Studio v. 5.2. A total of five individual nonasthma donor cell lines were used in all experiments.
Calcium Mobilization Analysis
Cells were grown in 48-well plates and cytosolic calcium levels were measured, as previously described (35). All measurements were performed in serum-free conditions after treating the cell monolayers with or without either propranolol (10 μM, 10 min) or albuterol (10 μM, 18 h). Briefly, cells were incubated with calcium-binding Fluo 8 dye for 1 h (kept in the dark). In some experiments, DM (1 μM, 10 min) or diluent was added 10 min before EPI (25 μM). Real-time fluorescence intensity was then measured in a fluorescent plate reader over 120 s (Clariostar BMG Labtech). Five donor cell lines were run with four technical replicates per experimental condition.
Magnetic Twisting Cytometry
Magnetic twisting cytometry (MTC) was used to assess the single-cell contractility (i.e., HASM cellular contraction and relaxation), as previously described (34, 36, 37). Briefly, an RGD-coated ferrimagnetic microbead (4.5 μm in diameter) functionalized to the cytoskeleton through cell surface integrin receptors was magnetized and twisted by an external magnetic field that varied sinusoidally in time. Forced bead motions (lateral bead displacements in response to the resulting oscillatory torque) were detected optically with a spatial resolution of ∼5 nm, and their changes were monitored, in real-time, in response to 10 μM EPI. Cell stiffness is computed as the ratio of specific torque to lateral bead displacements and expressed in units of Pascal per nanometer (Pa/nm). For each individual HASM cell, changes in stiffness in response to EPI were normalized to their respective stiffness before EPI addition.
Bronchodilation Assay
Human precision-cut lung slices (hPCLS) were prepared as previously described (29), and agonist-induced changes in the airway luminal area were measured using Image-Pro Plus software (v. 6.0; Media Cybernetics). For these studies, hPCLS were first contracted with carbachol (10 μM, 10 min), washed, and allowed to return to baseline followed by stimulation with EPI (10 μM). hPCLS were desensitized with Salmeterol (1 μM, 48 h), then stimulated with EPI (10 μM), washed, and then stimulated with carbachol (10 μM). hPCLS were washed and treated with the β2AR inhibitor propranolol (10 μM, 30 min), and then stimulated with EPI (10 μM). Bronchodilation and bronchoconstriction were calculated as the percent increase or decrease from the most recent baseline measurement. Refer to Supplemental Fig. S1 for a time course depiction.
Statistical Analysis
GraphPad Prism software (GraphPad) was used to determine if samples were normally distributed using the Kolmogorov–Smirnov test. Unless otherwise stated, statistical comparisons were done with two-tailed, paired Student’s t tests for comparison between two conditions, or an analysis of variance (ANOVA) followed by post hoc t tests with Tukey’s correction for multiple comparisons. P values of <0.05 were considered statistically significant. Data are represented as means ± SE with a minimum of three biological replicates per condition.
RESULTS
HASM Cells Express α1-Adrenoceptors
According to RNA-Seq results for HASM cells derived from age- and sex-matched fatal asthma or nonasthma lung donors, both the ADRA1A and ADRA1B genes (encoding α1AR subtypes A and B) were expressed. As shown in Fig. 1A, ADRA1B had mean normalized counts of 212 in asthma-donor-derived HASM cells and 247 in nonasthma-donor-derived HASM cells, values that were approximately 10-fold higher than those of ADRB2, which encodes the β2AR (mean normalized counts of 28 and 17 in asthma- and nonasthma-donor-derived HASM cells, respectively). Of note, levels for ADRA1B and ADRB2 varied according to donor, but were not significantly different by asthma status. In all samples, expression levels for ADRA1A were low (mean normalized counts were ∼1).
Figure 1.

Bronchial tissue expression of α1AR on HASM. A: α1ar (adra1b) (n = 9 asthma and 8 nonasthma donors) mRNA transcripts from RNAseq (represented as normalized read count). B: immunofluorescence staining of human bronchus tissue. C: comparison of nonasthma and fatal asthma derived donor bronchial tissue. HAE, human airway epithelial cells; HASM, human airway smooth muscle; SMA, antismooth muscle actin = red, anti-α1AR = green, DAPI = blue. α1AR, α1-adrenergic receptor.
To assess the distribution of α1ARs in an intact airway, and their localization to HASM cells, bronchial tissue sections were co-stained for α1AR and α-smooth muscle actin. As shown in Fig. 1B, we detected protein levels of α1ARs on epithelial and smooth muscle cells of human airways. Staining of the α1AR was comparable in both cell types, with little difference between asthma- and nonasthma-donor-derived airways (Fig. 1C). Collectively, these data demonstrate that α1ARs are present on HASM cells.
Epinephrine Evokes Calcium Signal and pMLC upon β2AR Blockade in HASM Cells
Stimulation of α1ARs can induce airway constriction, but the cellular mechanisms remain unclear. The role for α1ARs in asthma pathogenesis is equally unclear. Here we posited that, under a specific condition of β2AR tachyphylaxis in asthma pathobiology, endogenous or exogenous catecholamine(s) can induce bronchoconstriction by activating α1ARs expressed on HASM. To test this hypothesis, we used EPI, an agonist of both α1- and β2-adrenoceptors, in HASM cells treated with and without a pharmacological inhibitor of β2AR, propranolol (prop).
As phosphorylation of myosin light chain (pMLC) is a pivotal signaling event mediating agonist-induced HASM shortening, we first measured pMLC in response to increasing doses of EPI. As expected, in HASM cells treated with diluent (controls), EPI had little effect on pMLC levels (Fig. 2A); EPI evokes bronchodilation via preferentially activating the β2AR expressed on HASM cells (12, 16). In contrast, prop (10 μM, 10 min) appreciably increased pMLC levels in response to EPI; the increases were dose-dependent, and maximum pMLC levels were achieved with 1–10 μM EPI (Fig. 2A).
Figure 2.
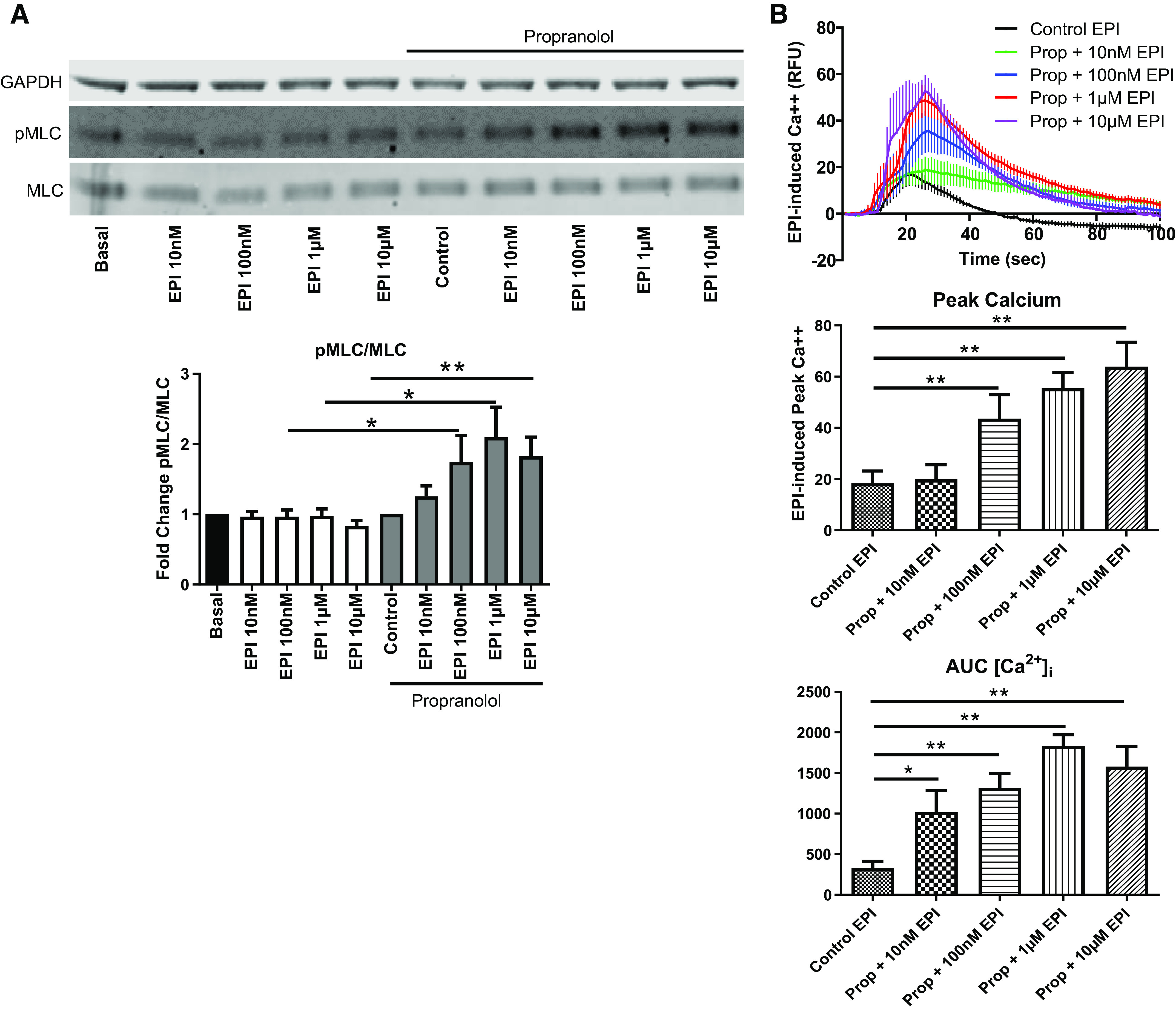
EPI induces MLC phosphorylation and cytosolic calcium flux in HASM after β2AR blockade. A: EPI (10 nM–10 µM, 10 min) does not increase pMLC in diluent treated HASM; however as EPI concentration increases, pMLC increases in propranolol-pretreated (10 nm–10 μM, 10 min) HASM (values are means ± SE, n = 8) (*P ≤ 0.05, **P ≤ 0.01). B: EPI-induced cytosolic calcium levels are increased after β2AR blockade with propranolol (Prop) in HASM as measured by peak calcium and AUC (values are means ± SE, n = 4–9) (*P ≤ 0.05, **P ≤ 0.01). AUC, area under the curve; β2AR, β2-adrenergic receptor; EPI, epinephrine; HASM, human airway smooth muscle; pMLC, myosin light chain phosphorylation.
Previous studies showed that α1AR couples to Gαq/11 and evokes intracellular calcium mobilization via phospholipase C (38–40). As such, we measured EPI-induced intracellular calcium levels ([Ca2+]i) using Fluo 8. As shown in Fig. 2B, in HASM cells pretreated with prop, EPI (0.01–10 μM) markedly increased [Ca2+]i in a dose-dependent manner. A small, transient increase in [Ca2+]i was also detected with 10 μM EPI in cells treated with diluent controls. Compared with control cells, EPI-induced [Ca2+]i, as shown by peak relative fluorescence units (RFUs) and the integrated area under the curve (AUC), was significantly greater in prop-treated cells, at all tested doses of EPI (Fig. 2B). Collectively, these results suggest that EPI can act as a procontractile agonist within the context of β2AR inhibition.
Epinephrine-Induced pMLC Is Mediated via α1ARs
To mimic the effect of β2AR tachyphylaxis in vitro, HASM cells were treated with 10 μM albuterol for 18 h (29–31). As shown in Fig. 3, HASM cells treated with diluent control showed no appreciable increase in pMLC in response to increasing doses of EPI. In contrast, in HASM cells treated with albuterol (i.e., β2-agonist insensitive cellular model), EPI markedly increased pMLC, in a concentration-dependent manner (Fig. 3). Of note, EPI-induced increases in pMLC levels were inhibited by pretreating the β2-agonist insensitive cells with DM, an antagonist to α1AR. Incubation with DM had little effect on carbachol (CCh)-induced pMLC levels (Fig. 4). These results suggest that EPI-induced HASM contraction is mediated by selective activation of α1ARs expressed on HASM cells.
Figure 3.

EPI induces MLC phosphorylation in HASM under β2AR desensitizing conditions. As EPI (0.1–100 µM, 10 min) concentration increases, pMLC increases in β2AR desensitized [Alb-PT (albuterol pretreated) 10 μM, 24 h] HASM but not in nondesensitized (basal). Carbachol (CCh; 20 µM, 10 min) was used as a positive control to elicit pMLC (values are mean ± SEM, n = 4 cell lines) (*P ≤ 0.05). β2AR, β2-adrenergic receptor; EPI, epinephrine; HASM, human airway smooth muscle; pMLC, myosin light chain phosphorylation.
Figure 4.
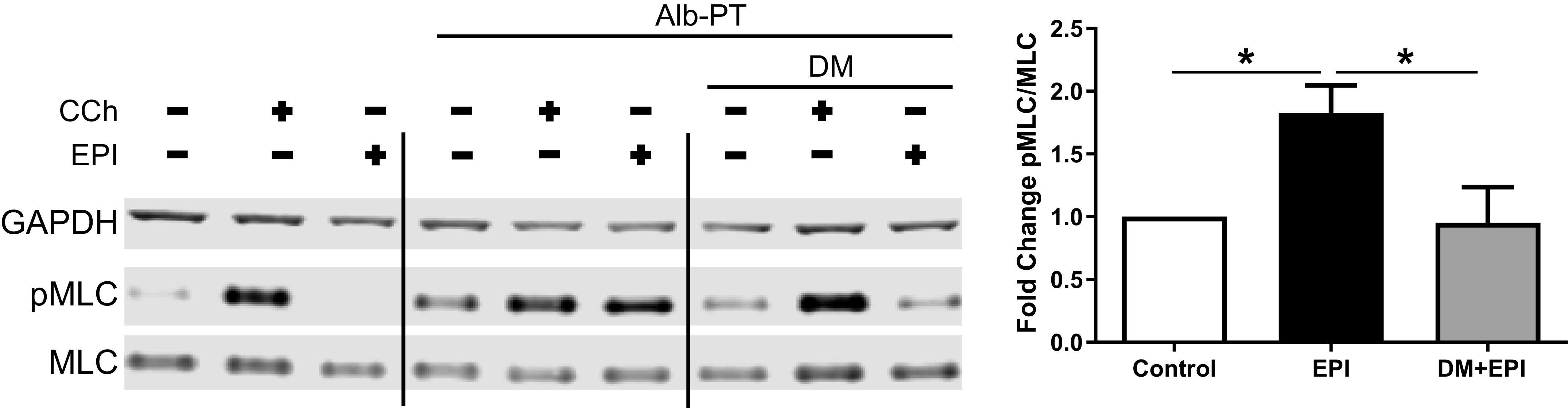
Inhibition of α1AR abrogates EPI-induced pMLC in HASM following β2AR desensitization. Representative immunoblot of HASM pretreated with DM (1 µM, 10 min pretreatment) before EPI (10 µM, 10 min) stimulation. Band densities measured by fold change pMLC relative fluorescence units normalized to total MLC relative to Alb-PT control (values are means ± SE, n = 5–13 experiments from 5 unique cell lines) (*P ≤ 0.05). β2AR, β2-adrenergic receptor; CCh, carbachol; EPI, epinephrine; HASM, human airway smooth muscle; pMLC, myosin light chain phosphorylation.
Inhibition of α1ARs Abrogates EPI-Induced HASM Shortening
To ascertain whether EPI can act as a procontractile agonist via activating α1ARs expressed on HASM cells, we assessed single-cell calcium and single-cell shortening methods as surrogates for HASM contraction (37). Under experimental conditions that induce β2AR desensitization (as above), we found that pharmacological inhibition of α1ARs with DM significantly attenuated EPI-induced intracellular calcium mobilization (Fig. 5); DM had little effect on EPI-induced [Ca2+]i in nondesensitized HASM cells. To address the physiological consequences of α1AR activation with EPI, magnetic twisting cytometry (MTC) was applied to quantify changes in the cytoskeletal stiffness of HASM cells (34, 36). In nondesensitized HASM cells, EPI decreased the cell stiffness (i.e., HASM cell relaxation); and, EPI-induced relaxation was not affected by DM pre-treatment (Fig. 6). In contrast, upon β2AR desensitization, EPI increased the cell stiffness (i.e., HASM cell contraction), which was abrogated by pretreating the cells with DM (Fig. 6). These results suggest that, under specific conditions of β2AR tachyphylaxis in asthma, endogenous or exogenous EPI can preferentially activate α1ARs and evoke intracellular calcium, myosin light chain phosphorylation, and HASM cell shortening. These data also suggest a feed-forward mechanism via two distinct classes of GPCRs to reinforce bronchoconstriction (e.g., exercise-induced or emotional stress) in patients with β2-agonist insensitivity.
Figure 5.
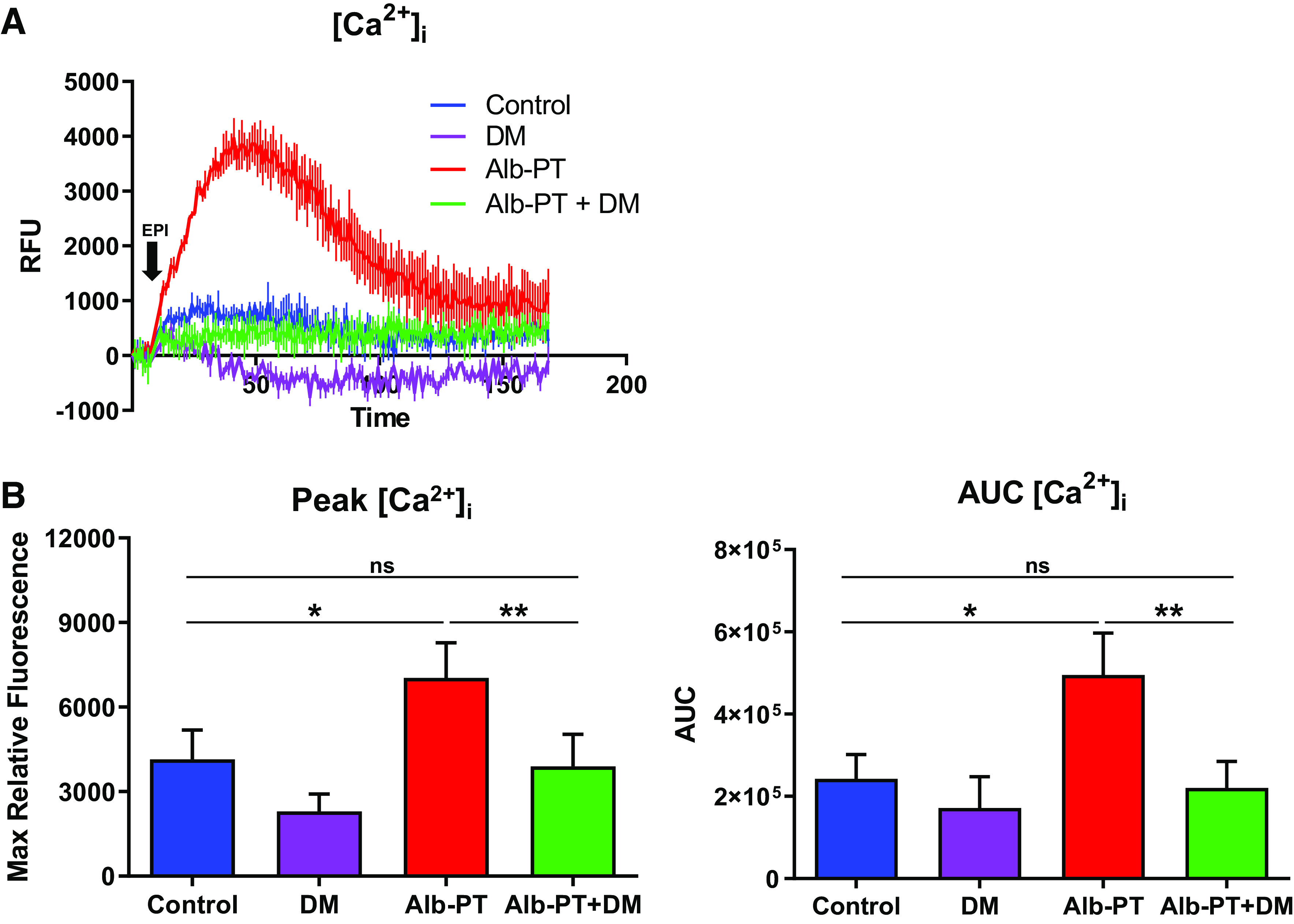
Inhibition of α1AR decreases EPI-induced cytosolic calcium levels following β2AR desensitization of HASM. A: representative graph of EPI-induced cytosolic calcium flux. B: inhibition of α1AR (DM, 1 µM, 10 min pretreatment) before EPI (25 µM) stimulation decreases cytosolic calcium levels ([Ca2+]i) as measured by peak relative fluorescence units (peak RFUs) and integrated area under the curve (AUC) after albuterol-induced β2AR desensitization (values are means ± SE, n = 5 unique cell lines run in triplicate) (*P ≤ 0.05, **P ≤ 0.01). α1AR, α1-adrenergic receptor; β2AR, β2-adrenergic receptor; DM, doxazosin mesylate; EPI, epinephrine; HASM, human airway smooth muscle.
Figure 6.

EPI induces HASM cell shortening under experimental conditions that induce β2AR desensitization. HASM cells were treated for 18 h with or without 10 µM albuterol. After 18 h, cells were washed 3× with fresh media, incubated for 20 min with magnetic beads, and washed again 3× with fresh media to remove unbound beads, before stimulation with 10 µM EPI. For each individual cell, baseline stiffness was measured for first 60 s, and changes in the stiffness in response to EPI were measured continuously for the next 240 s (EPI was added at 60 s). For α1AR blockade, Alb-PT and diluent treated cells were treated for 10 min with 1 µM DM, followed by stimulation with 10 µM EPI. Data are presented as means ± SE (n = 295–389 individual cells per condition). Analyses were done by using one-way ANOVA, followed by Tukey’s multiple comparison tests. ****P = 0.0001. α1AR, α1-adrenergic receptor; β2AR, β2-adrenergic receptor; DM, doxazosin mesylate; EPI, epinephrine; HASM, human airway smooth muscle.
Epinephrine Induces Bronchoconstriction after Desensitization of the β2AR
To examine whether EPI induced bronchoconstriction in an intact airway, human small airways in hPCLS from one nonasthma and one fatal asthma donor were treated with EPI before and after β2AR desensitization. EPI induced bronchodilation at baseline; however, after salmeterol-induced β2AR desensitization or β2AR blockade with propranolol, EPI induced bronchoconstriction (Fig. 7A). EPI induced greater bronchoconstriction in the fatal asthma donor-derived hPCLS as seen in Fig. 7B. These data suggest that EPI works as a bronchodilator at baseline, but switches to a bronchoconstrictor after β2AR tachyphylaxis or blockade.
Figure 7.

Epinephrine induces bronchoconstriction after β2AR tachyphylaxis. EPI (10 μM, 10 min) induced bronchodilation at baseline and EPI-induced bronchoconstriction after salmeterol-induced β2AR tachyphylaxis (1 μM, 48 h pretreatment) or β2AR blockade with propranolol (10 μM, 30 min pretreatment) in both nonasthma hPCLS (A) and fatal asthma hPCLS (B). Carbachol (CCh)-induced bronchoconstriction (10 μM, 10 min) used as a positive control to confirm contractility of hPCLS (values are means ± SEM, n = 8 slices from one representative nonasthma donor, n = 5 slices from one representative fatal asthma donor) (*P ≤ 0.05, **P ≤ 0.01). β2AR, β2-adrenergic receptor; DM, doxazosin mesylate; EPI, epinephrine; HASM, human airway smooth muscle; hPCLS, human precision cut lung slices; Prop, propranolol; Sal, salmeterol.
DISCUSSION
Here we demonstrated that, under experimental conditions that induce β2AR tachyphylaxis, EPI evoked calcium mobilization, myosin light chain phosphorylation, and bronchoconstriction by activating α1ARs expressed on HASM cells (Fig. 8). Immunohistochemistry studies showed that α1ARs are expressed in the smooth muscle layers of intact human airways. Of note, in isolated primary HASM cells, we detected a high abundance of ADRA1B transcripts (the α1AR subtype B)—but not ADRA1A (the α1AR subtype A)—which were approximately 10× higher than that of ADRB2 (encoding β2AR). These results are consistent with selective β2AR agonism of EPI (i.e., generated endogenously or administered parenterally) and support the concept of spare α1-adrenoceptors on HASM cells (41, 42). Our findings also address a long-standing question of α1AR expression and function in HASM cells (22–24) and provide a basis for reexamining α1AR inhibition for the management of stress/exercise-induced asthma and/or β2-agonist insensitivity in patients with difficult-to-control, disease subtypes.
Figure 8.

Alterations in β2AR and α1AR signaling pathways. Under normal, nondesensitized conditions, EPI preferentially binds the β2AR, leading to increases in cAMP and dephosphorylation of MLC to induce relaxation. Following desensitization of the β2AR, EPI preferentially binds the α1AR, inducing increased cytosolic calcium flux and pMLC, inducing bronchoconstriction. α1AR, α1-adrenergic receptor; β2AR, β2-adrenergic receptor; cAMP, 3′,5′-cyclic adenosine monophosphate; EPI, epinephrine; pMLC, myosin light chain phosphorylation.
Epinephrine (or adrenaline), an endogenous catecholamine involved in the body’s fight-or-flight responses to stress, activates α1ARs expressed on various organs to evoke a wide range of physiological functions, including vasoconstriction (8, 9). In the smooth muscle of human bronchi, however, the physiological effect of EPI on α1ARs remains unclear. Evidence suggests that EPI promotes bronchodilation by stimulating β2ARs expressed on HASM (12, 39). As shown in Figs. 3, 4, 5, and 6, under normal, non-β2AR-desensitized conditions, EPI induced HASM cell relaxation, which was not affected by an α1AR antagonist, doxazosin mesylate. In contrast, upon β2AR desensitization, EPI evoked HASM cell contraction in an α1AR-dependent manner (Figs. 3, 4, 5, and 6). In intact human small airways, EPI also evoked airway constriction in the face of β2 desensitization or blockade (Fig. 7). This “switch-like” activation of α1ARs, and related EPI-induced biochemical and mechanical signals, also emerged upon pharmacological inhibition of β2ARs (Fig. 2). These results support previous studies examining the effects of α1AR activation on smooth muscle contraction (20, 21). Furthermore, our findings are consistent with the findings of the study by Naline et al. (43), showing decreased adrenaline-induced relaxation of human bronchi after 1 h salmeterol pretreatment. Taken together, these studies now identify specific conditions, in which an endogenous catecholamine (EPI) can induce a “switch-like” activation of α1ARs, suggesting a previously unrecognized feed-forward mechanism driving bronchospasm via two distinct classes of G protein-coupled receptors (GPCRs).
Interestingly, HASM cells were enriched with transcript levels of ADRA1B (the α1AR subtype B), but not ADRA1A (the α1AR subtype A) (Fig. 1A). Since α1AR subtype-specific antibodies are unavailable (44), whether α1-adrenoceptor subtype protein expression exists on HASM cells remains unknown. We, however, chose a commercially available antibody against α1ARs and performed immunohistochemistry studies. Staining of the α1AR was readily visualized in both intact airways and isolated HASM cells (Fig. 1B). Using intact airways, we detected appreciable α1AR expression in both epithelial and smooth muscle layers that was unaltered in asthma. Our future studies will investigate the distribution of α1AR subtypes and, using siRNA-mediated knockdown and/or subtype-specific inhibition approaches, determine the specificity and selectivity of receptor subtype that is activated by EPI in HASM cells. Our data collectively showed the presence of α1ARs on HASM cells and EPI-induced mechanical reinforcement of HASM shortening is α1AR-dependent.
Previous studies have implicated the α1AR in exercise-induced asthma (22, 23, 45), as exercise increases the release of adrenalines (epinephrine and norepinephrine) that, like the fight-or-flight responses to stress, evoke a wide range of physiological responses, including vasoconstriction. Of note, an asthma exacerbation is a high-stress event. Accordingly, overflow of EPI and NE in circulation could stimulate airway smooth muscle given the spatial proximity of the airways to the vasculature, including the nerves that innervate them (46). EPI levels can also be elevated in individuals who receive EPI injections as part of the treatment for anaphylaxis. Normally, circulating EPI would induce bronchodilation via the β2AR; however, if an individual has been repeatedly using β2-agonists (short- and long-acting combined) for relief of bronchospasm, tachyphylaxis of the β2AR can occur, increasing the probability of EPI activating the α1AR system and evoking bronchoconstriction.
Our study suggests that stress/exercise-induced release of EPI may evoke bronchoconstriction in patients with difficult-to-control or β2-agonist insensitivity. Of note, Inman and O’Byrne (47) in their study found that four times daily use of albuterol for 1 wk worsened exercise-induced bronchoconstriction compared with placebo, which may, in part, be due to endogenous EPI release and activation of the α1AR due to repeated albuterol-induced desensitization of the β2AR. We posit that mortalities resulting from this may be underreported due to EPI being reported as ineffective rather than potentially worsening asthma. As such, further studies are warranted to investigate the therapeutic value of α1AR blockade, specifically in individuals exhibiting airflow obstruction due to stress/exercise and/or therapy-resistance to β2-agonists.
Interestingly, glucocorticoids increase α1bAR mRNA (48), suggesting that inhaled steroids may contribute to altering the balance between β2AR- and α1AR-dependent signaling in airway smooth muscle in response to EPI. In addition, activation of α1AR and/or α2AR has been shown to induce the proliferation of airway smooth muscle in rabbits that was inhibited by adenylate cyclase activation (49). Taken together, these studies suggest that multiple mechanisms by which α1AR may play a role in asthma.
In conclusion, we demonstrated that blockade of the α1AR abrogated EPI-induced calcium flux, pMLC, and HASM shortening, under experimental conditions that induce β2AR desensitization, providing a basis for reexamining α1AR inhibition for the management of stress/exercise-induced asthma and/or β2-agonist insensitivity in patients with difficult-to-control, disease subtypes.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Table S1 and Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.19617252.
GRANTS
This work was supported by the New Jersey Alliance for Clinical and Translational Science (UL1TR0030117) and the National Institutes of Health grants (P01HL114471 and R56HL155937).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.T.D., G.C., S.S.A., and R.A.P. conceived and designed research; B.T.D., G.C., S.O., J.L., M.K., B.E.H., and V.P. performed experiments; B.T.D., G.C., S.O., J.L., M.K., B.E.H., C.J.K.-W., S.S.A., and R.A.P. analyzed data; B.T.D., G.C., S.O., J.L., V.P., C.J.K.-W., S.S.A., and R.A.P. interpreted results of experiments; B.T.D., G.C., J.L., S.S.A., and R.A.P. prepared figures; B.T.D., G.C., C.J.K.-W., S.S.A., and R.A.P. drafted manuscript; B.T.D., G.C., S.O., J.L., M.K., B.E.H., V.P., C.J.K.-W., S.S.A., and R.A.P. edited and revised manuscript; B.T.D., G.C., S.O., J.L., M.K., B.E.H., V.P., C.J.K.-W., S.S.A., and R.A.P. approved final version of manuscript.
REFERENCES
- 1.Woolcock AJ, Peat JK. Epidemiology of bronchial hyperresponsiveness. Clin Rev Allergy 7: 245–256, 1989. doi: 10.1007/BF02914477. [DOI] [PubMed] [Google Scholar]
- 2.Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 18: 716–725, 2012. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 3.Barnes PJ. New drugs for asthma. Nat Rev Drug Discov 3: 831–844, 2004. doi: 10.1038/nrd1524. [DOI] [PubMed] [Google Scholar]
- 4.Wendell SG, Fan H, Zhang C. G protein-coupled receptors in asthma therapy: pharmacology and drug action. Pharmacol Rev 72: 1–49, 2020. doi: 10.1124/pr.118.016899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fergeson JE, Patel SS, Lockey RF. Acute asthma, prognosis, and treatment. J Allergy Clin Immunol 139: 438–447, 2017. doi: 10.1016/j.jaci.2016.06.054. [DOI] [PubMed] [Google Scholar]
- 6.Meier KE, Snavely MD, Brown SL, Brown JH, Insel PA. alpha 1- and beta 2-adrenergic receptor expression in the Madin-Darby canine kidney epithelial cell line. J Cell Biol 97: 405–415, 1983. doi: 10.1083/jcb.97.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldie RG, Paterson JW, Lulich KM. Adrenoceptors in airway smooth muscle. Pharmacol Ther 48: 295–322, 1990. doi: 10.1016/0163-7258(90)90051-3. [DOI] [PubMed] [Google Scholar]
- 8.Bulbring E, Tomita T. Catecholamine action on smooth muscle. Pharmacol Rev 39: 49–96, 1987. [PubMed] [Google Scholar]
- 9.Minneman KP. Alpha 1-adrenergic receptor subtypes, inositol phosphates, and sources of cell Ca2+. Pharmacol Rev 40: 87–119, 1988. [PubMed] [Google Scholar]
- 10.Ind PW, Causon RC, Brown MJ, Barnes PJ. Circulating catecholamines in acute asthma. Br Med J (Clin Res Ed) 290: 267–269, 1985. doi: 10.1136/bmj.290.6464.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welsh L, Roberts RG, Kemp JG. Fitness and physical activity in children with asthma. Sports Med 34: 861–870, 2004. doi: 10.2165/00007256-200434130-00001. [DOI] [PubMed] [Google Scholar]
- 12.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3: 639–650, 2002. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 13.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature 372: 231–236, 1994. [Erratum in Nature 372:812, 1994]. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 14.Komalavilas P, Penn RB, Flynn CR, Thresher J, Lopes LB, Furnish EJ, Guo M, Pallero MA, Murphy-Ullrich JE, Brophy CM. The small heat shock-related protein, HSP20, is a cAMP-dependent protein kinase substrate that is involved in airway smooth muscle relaxation. Am J Physiol Lung Cell Mol Physiol 294: L69–L78, 2008. doi: 10.1152/ajplung.00235.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan SJ, Deshpande DA, Tiegs BC, Misior AM, Yan H, Hershfeld AV, Rich TC, Panettieri RA, An SS, Penn RB. beta-Agonist-mediated relaxation of airway smooth muscle is protein kinase A-dependent. J Biol Chem 289: 23065–23074, 2014. doi: 10.1074/jbc.M114.557652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sears MR, Lotvall J. Past, present and future–beta2-adrenoceptor agonists in asthma management. Respir Med 99: 152–170, 2005. doi: 10.1016/j.rmed.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 17.De La Mata RC, Penna M, Aviado DM. Reversal of sympathomimetic bronchodilation by dichloroisoproterenol. J Pharmacol Exp Ther 135: 197–203, 1962. [PubMed] [Google Scholar]
- 18.Mathe AA, Astrom A, Persson NA. Some bronchoconstricting and bronchodilating responses of human isolated bronchi: evidence for the existence of -adrenoceptors. J Pharm Pharmacol 23: 905–910, 1971. doi: 10.1111/j.2042-7158.1971.tb09891.x. [DOI] [PubMed] [Google Scholar]
- 19.Patel KR, Kerr JW. The airways response to phenylephrine after blockade of alpha and beta receptors in extrinsic bronchial asthma. Clin Allergy 3: 439–448, 1973. doi: 10.1111/j.1365-2222.1973.tb01351.x. [DOI] [PubMed] [Google Scholar]
- 20.Black JL, Salome C, Yan K, Shaw J. The action of prazosin and propylene glycol on methoxamine-induced bronchoconstriction in asthmatic subjects. Br J Clin Pharmacol 18: 349–353, 1984. doi: 10.1111/j.1365-2125.1984.tb02475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Black JL, Salome CM, Yan K, Shaw J. Comparison between airways response to an alpha-adrenoceptor agonist and histamine in asthmatic and non-asthmatic subjects. Br J Clin Pharmacol 14: 464–466, 1982. doi: 10.1111/j.1365-2125.1982.tb02012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnes PJ, Ind PW, Dollery CT. Inhaled prazosin in asthma. Thorax 36: 378–381, 1981. doi: 10.1136/thx.36.5.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bleecker ER, Chahal KS, Mason P, Permutt S. The effect of alpha adrenergic blockade on non-specific airways reactivity and exercise induced asthma. Eur J Respir Dis Suppl 128: 258–265, 1983. [PubMed] [Google Scholar]
- 24.Shiner RJ, Molho MI. Comparison between an alpha-adrenergic antagonist and a beta 2-adrenergic agonist in bronchial asthma. Chest 83: 602–606, 1983. doi: 10.1378/chest.83.4.602. [DOI] [PubMed] [Google Scholar]
- 25.Sears MR, Taylor DR, Print CG, Lake DC, Li QQ, Flannery EM, Yates DM, Lucas MK, Herbison GP. Regular inhaled beta-agonist treatment in bronchial asthma. Lancet 336: 1391–1396, 1990. doi: 10.1016/0140-6736(90)93098-A. [DOI] [PubMed] [Google Scholar]
- 26.Lipworth BJ. Airway subsensitivity with long-acting beta 2-agonists. Is there cause for concern? Drug Saf 16: 295–308, 1997. doi: 10.2165/00002018-199716050-00002. [DOI] [PubMed] [Google Scholar]
- 27.Dohlman HG. Diminishing returns. Nature 418: 591, 2002. doi: 10.1038/418591a. [DOI] [PubMed] [Google Scholar]
- 28.Ortega VE, Peters SP. Beta-2 adrenergic agonists: focus on safety and benefits versus risks. Curr Opin Pharmacol 10: 246–253, 2010. doi: 10.1016/j.coph.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Cooper PR, Kurten RC, Zhang J, Nicholls DJ, Dainty IA, Panettieri RA. Formoterol and salmeterol induce a similar degree of beta2-adrenoceptor tolerance in human small airways but via different mechanisms. Br J Pharmacol 163: 521–532, 2011. doi: 10.1111/j.1476-5381.2011.01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.An SS, Wang WCH, Koziol-White CJ, Ahn K, Lee DY, Kurten RC, Panettieri RA, Liggett SB. TAS2R activation promotes airway smooth muscle relaxation despite beta(2)-adrenergic receptor tachyphylaxis. Am J Physiol Lung Cell Mol Physiol 303: L304–L311, 2012. doi: 10.1152/ajplung.00126.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D, Tokmakova A, Lujan LK, Strzelinski HR, Kim N, Beidokhti MN, Giulianotti MA, Mafi A, Woo J-AA, An SS, Goddard WQ, Liggett SB. Identification and characterization of an atypical Galphas-biased beta2AR agonist that fails to evoke airway smooth muscle cell tachyphylaxis. Proc Natl Acad Sci USA 118: e2026668118, 2021. doi: 10.1073/pnas.2026668118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kan M, Koziol-White C, Shumyatcher M, Johnson M, Jester W, Panettieri RA, Himes BE. Airway smooth muscle-specific transcriptomic signatures of glucocorticoid exposure. Am J Respir Cell Mol Biol 61: 110–120, 2019. doi: 10.1165/rcmb.2018-0385OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panettieri RA, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI. A human airway smooth muscle cell line that retains physiological responsiveness. Am J Physiol Cell Physiol 256: C329–C335, 1989. doi: 10.1152/ajpcell.1989.256.2.C329. [DOI] [PubMed] [Google Scholar]
- 34.An SS, Mitzner W, Tang W-Y, Ahn K, Yoon A-R, Huang J, Kilic O, Yong HM, Fahey JW, Kumar S, Biswal S, Holgate ST, Panettieri RA, Solway J, Liggett SB. An inflammation-independent contraction mechanophenotype of airway smooth muscle in asthma. J Allergy Clin Immunol 138: 294–297, 2016. doi: 10.1016/j.jaci.2015.12.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orfanos S, Jude J, Deeney BT, Cao G, Rastogi D, van Zee M, Pushkarsky I, Munoz HE, Damoiseaux R, Di Carlo D, Panettieri RA. Obesity increases airway smooth muscle responses to contractile agonists. Am J Physiol Lung Cell Mol Physiol 315: L673–L681, 2018. doi: 10.1152/ajplung.00459.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the microrheology of living cells. Phys Rev Lett 87: 148102, 2001. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]
- 37.Huang J, Lam H, Koziol-White C, Limjunyawong N, Kim D, Kim N, Karmacharya N, Rajkumar P, Firer D, Dalesio NM, Jude J, Kurten RC, Pluznick JL, Deshpande DA, Penn RB, Liggett SB, Panettieri RA, Dong X, An SS. The odorant receptor OR2W3 on airway smooth muscle evokes bronchodilation via a cooperative chemosensory tradeoff between TMEM16A and CFTR. Proc Natl Acad Sci USA 117: 28485–28495, 2020. doi: 10.1073/pnas.2003111117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu D, Katz A, Lee CH, Simon MI. Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem 267: 25798–25802, 1992. doi: 10.1016/S0021-9258(18)35680-1. [DOI] [PubMed] [Google Scholar]
- 39.Billington CK, Penn RB. Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir Res 4: 2, 2003. doi: 10.1186/1465-9921-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carr R, Koziol-White C, Zhang J, Lam H, An SS, Tall GG, Panettieri RA, Benovic JL. 3rd,. Interdicting Gq Activation in Airway Disease by Receptor-Dependent and Receptor-Independent Mechanisms. Mol Pharmacol 89: 94–104, 2016. doi: 10.1124/mol.115.100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fox AW, Friedman PA. Evidence for spare alpha 1-adrenoceptors for the accumulation of inositol phosphates in smooth muscle. J Pharm Pharmacol 39: 68–71, 1987. doi: 10.1111/j.2042-7158.1987.tb07169.x. [DOI] [PubMed] [Google Scholar]
- 42.Marunaka Y, Niisato N, Miyazaki H. New concept of spare receptors and effectors. J Membr Biol 203: 31–39, 2005. doi: 10.1007/s00232-004-0729-0. [DOI] [PubMed] [Google Scholar]
- 43.Naline E, Zhang Y, Qian Y, Mairon N, Anderson GP, Grandordy B, Advenier C. Relaxant effects and durations of action of formoterol and salmeterol on the isolated human bronchus. Eur Respir J 7: 914–920, 1994. [PubMed] [Google Scholar]
- 44.Jensen BC, Swigart PM, Simpson PC. Ten commercial antibodies for alpha-1-adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch Pharmacol 379: 409–412, 2009. doi: 10.1007/s00210-008-0368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berkin KE, Walker G, Inglis GC, Ball SG, Thomson NC. Circulating adrenaline and noradrenaline concentrations during exercise in patients with exercise induced asthma and normal subjects. Thorax 43: 295–299, 1988. doi: 10.1136/thx.43.4.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doidge JM, Satchell DG. Adrenergic and non-adrenergic inhibitory nerves in mammalian airways. J Auton Nerv Syst 5: 83–99, 1982. doi: 10.1016/0165-1838(82)90030-3. [DOI] [PubMed] [Google Scholar]
- 47.Inman MD, O'Byrne PM. The effect of regular inhaled albuterol on exercise-induced bronchoconstriction. Am J Respir Crit Care Med 153: 65–69, 1996. doi: 10.1164/ajrccm.153.1.8542164. [DOI] [PubMed] [Google Scholar]
- 48.Sakaue M, Hoffman BB. Glucocorticoids induce transcription and expression of the alpha 1B adrenergic receptor gene in DTT1 MF-2 smooth muscle cells. J Clin Invest 88: 385–389, 1991. doi: 10.1172/JCI115315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noveral JP, Grunstein MM. Adrenergic receptor-mediated regulation of cultured rabbit airway smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 267: L291–L299, 1994. doi: 10.1152/ajplung.1994.267.3.L291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 and Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.19617252.
Data Availability Statement
Data will be made available upon reasonable request.