

Keywords: diabetic complications, DNA methylation, epigenetics, metabolic memory
Abstract
Inherent and acquired abnormalities in gene regulation due to the influence of genetics and epigenetics (traits related to environment rather than genetic factors) underlie many diseases including diabetes. Diabetes could lead to multiple complications including retinopathy, nephropathy, and cardiovascular disease that greatly increase morbidity and mortality. Epigenetic changes have also been linked to diabetes-related complications. Genes associated with many pathophysiological features of these vascular complications (e.g., inflammation, fibrosis, and oxidative stress) can be regulated by epigenetic mechanisms involving histone posttranslational modifications, DNA methylation, changes in chromatin structure/remodeling, and noncoding RNAs. Intriguingly, these epigenetic changes triggered during early periods of hyperglycemic exposure and uncontrolled diabetes are not immediately corrected even after restoration of normoglycemia and metabolic balance. This latency in effect across time and conditions is associated with persistent development of complications in diabetes with prior history of poor glycemic control, termed as metabolic memory or legacy effect. Epigenetic modifications are generally reversible and provide a window of therapeutic opportunity to ameliorate cellular dysfunction and mitigate or “erase” metabolic memory. Notably, trained immunity and related epigenetic changes transmitted from hematopoietic stem cells to innate immune cells have also been implicated in metabolic memory. Hence, identification of epigenetic variations at candidate genes, or epigenetic signatures genome-wide by epigenome-wide association studies can aid in prompt diagnosis to prevent progression of complications and identification of much-needed new therapeutic targets. Herein, we provide a review of epigenetics and epigenomics in metabolic memory of diabetic complications covering the current basic research, clinical data, and translational implications.
INTRODUCTION
Diabetes encompasses a group of metabolic diseases characterized by hyperglycemia that are quite prevalent worldwide. As of 2021, there were ∼537 million adults with diabetes worldwide and this is projected to rise to 783 million by 2045 (1). The most common forms include type 1 diabetes (T1D), type 2 diabetes (T2D), and gestational diabetes.
T1D afflicts more than 1.2 million children and adolescents and is characterized by insulin deficiency due to destruction of pancreatic β cells by autoimmunity. In contrast, T2D accounts for over 90% of all individuals with diabetes. There is also a large population of obese and/or insulin resistant individuals with prediabetes. In T2D, carbohydrate, lipid, and protein metabolism are dysregulated along with impaired β-cell insulin secretion and insulin resistance in skeletal muscle, liver, and adipose tissue. Gestational diabetes, a common complication of pregnancy, can also predispose to the development of obesity and diabetes later in life. Even though etiologies vary across different types of diabetes, chronic hyperglycemia evident in all diabetes promotes organ dysfunction leading to microvascular complications including retinopathy, diabetic kidney disease (DKD), and neuropathy, and macrovascular complications that are primarily cardiovascular diseases (CVDs) such as coronary heart disease, atherosclerosis, myocardial infarction, hypertension, and stroke. Some of these complications can progress and result in loss of vision, heart and end-stage renal failure (ESRD), lower extremity amputations, and even death.
PATHOPHYSIOLOGY OF DIABETIC COMPLICATIONS
Various molecular and cellular mechanisms have been associated with pathophysiological changes seen in the development of most diabetic complications, including inflammation, fibrosis, and oxidative stress (2–5). Briefly, hyperglycemia and diabetes-associated risk factors such as dyslipidemia and hypertension enhance the production of reactive oxygen species (ROS), advanced glycation end products (AGEs), and oxidized low density lipoprotein (LDL) and activate signaling pathways such as the polyol, hexosamine, and protein kinase C (PKC) pathways, which further activate transcription factors such as nuclear factor κB (NF-κB) and subsequently induce proinflammatory cytokines [such as tumor necrosis factor-α (TNF-α), interleukin 1 (IL1), and interleukin 6 (IL6)], chemokines [e.g., C-C motif chemokine ligand 2 (CCL2)], and various disease/cell-specific fibrotic and growth factors such as transforming growth factor β1 (TGF-β1), angiotensin II (AngII), insulin-like growth factor (IGF), vascular endothelial growth factor (VEGF), angiopoietins, and collagens. These changes cause cellular dysfunction including mitochondrial dysfunction, oxidative stress, and endoplasmic reticulum (ER) stress, and pathological changes such as hypertrophy, inflammation with monocyte/macrophage infiltration, fibrosis [with extracellular matrix (ECM) accumulation], and apoptosis in diabetes-targeted cells/tissues/organs, leading to the development and progression of complications (6–9) (Fig. 1).
Figure 1.

Mediators of diabetic complications, involvement of epigenetic mechanisms and their persistence in metabolic memory. Hyperglycemia in diabetes or risk factors associated with diabetes such as obesity/insulin resistance, dyslipidemia, and hypertension alter the production or activity of multiple molecules including ROS, AGEs, oxidized LDL, and NO, activate signaling pathways such as the polyol, hexosamine, PKC, and NF-κB, and subsequently induce proinflammatory cytokines (e.g., TNF-α, IL1, and IL6), chemokines (e.g., CCL2), and various disease/cell-specific growth and fibrotic factors such as TGF-β1, AngII, IGF, VEGF, angiopoietins, collagens, and fibronectin. These diabetic stimuli, especially hyperglycemia, and the indicated subsequent downstream effects can also induce epigenetic changes including DNAme, histone PTMs, chromatin-remodeling, along with ncRNAs (lncRNAs and miRNAs) that can act via epigenetic mechanisms. Persistence of some of these changes even after glucose levels are normalized can cause long-term persistent cellular malfunction including mitochondrial dysfunction, oxidative stress, and ER stress. This results in prolonged pathological changes such as inflammation with monocyte/macrophage infiltration, fibrosis with extracellular matrix accumulation, hypertrophy, and cell death (apoptosis) in diabetes-targeted cells/tissues/organs, leading to development and/or uncontrolled progression of complications including retinopathy, DKD, neuropathy, and CVD, sometimes even after glycemic control. AGE, advanced glycation end product; AngII, angiotensin II; CCL2-C, C motif chemokine ligand 2; CVD, cardiovascular disease; DKD, diabetic kidney disease; DNAme, DNA methylation; ER, endoplasmic reticulum; histone PTM, histone posttranslational modification; IGF, insulin-like growth factor; IL1, interleukin 1; IL6-interleukin 6; LDL, low density lipoprotein; lncRNA, long noncoding RNA; miRNA, microRNAs; ncRNA, noncoding RNA; NF-κB, nuclear factor κB; NO, nitric oxide; PKC, protein kinase C; ROS, reactive oxygen species; TGF-β1, transforming growth factor β1; VEGF, vascular endothelial growth factor.
On the other hand, since diabetes and its complications are complex gene-environmental diseases, both genetic predisposition and epigenetics are involved in their etiology. However, only limited genetic variants have been associated with diabetic complications, implying that epigenetic regulation of gene expression in response to environmental factors plays a major role in diabetic complications (4, 5, 10, 11).
EPIGENETICS
Epigenetics refers to molecular processes and traits modified by lifestyles, behavior or environment factors that enable mitotically and/or meiotically heritable changes in gene expression and function without altering the underlying DNA sequence (12). Epigenetic marks include DNA methylation (DNAme) and histone posttranslational modifications (PTMs), which can affect chromatin remodeling, structure, accessibility, and nuclear organization and cooperate with noncoding RNAs (ncRNAs) and RNA modifications to alter gene expression in response to environmental triggers. The levels and functions of DNAme and histone PTMs at a specific gene locus are generally determined by three types of enzymes: writers (introducing specific modification to DNA or histone), erasers (removing the modification), and readers (containing specialized protein domains capable of recognizing specific modifications at a locus) (5, 13).
DNAme, the most stable and heritable epigenetic marks, refers to the methylation of DNA, especially on cytosines of CpG dinucleotides, to form 5-methylcytosine. DNA methyltransferases (DNMTs), as DNAme writers, mediate DNAme and include DNMT1, which mediates DNAme maintenance, and DNMT3a/3b, which mediates de novo methylation (14). The ten-eleven translocation proteins (TET1, TET2, and TET3) function as erasers of DNAme. They mediate demethylation by oxidizing 5-methylcytosine to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxylcytosine (15). Generally, DNAme changes at promoters regions can repress gene expression, whereas gene body DNAme can regulate transcription, elongation, and alternative splicing (14).
Chromatin is composed of subunits called nucleosomes, each comprising an octamer of histones wrapped by 147 base pairs of DNA and linked by histone H1. Histones undergo PTMs on their tails including methylation, acetylation, phosphorylation, and ubiquitination, which play significant and different roles in regulating DNA structure, gene transcription, chromatin accessibility, and chromosome condensation (16). The status of PTMs at specific loci is also determined by the dynamic interplay between writers and erasers. For example, histone acetylation is regulated by the balance between histone acetyltransferases (HATs, such as EP300) and histone deacetylases [HDACs, such as Sirtuin 1 (SIRT1)]. Histone lysine methylation may be mediated by lysine histone methyltransferases (KMTs) including SET domain-containing methyltransferases (such as SET enzymes for histone H3K4 methylation and SUV39H1/2 for H3K9 methylation) and erased by histone lysine demethylases including lysine-specific demethylase KDM (also called LSD) and Jumanji C-domain-containing proteins (JMJDs) (16). The bromodomain and extra-terminal-containing (BET) proteins, BRD2, BRD3, and BRD4, are PTM readers that bind acetylated histones and recruit transcription factors and transcription elongation complex at many genes including proinflammatory and immunoregulatory genes (17). Notably, specific histone modifications can distinguish regulatory regions in the genome and exert different functions on gene expression (16, 18). For examples, histone 3 lysine 9 acetylation (H3K9ac) and H3K4 tri-methylation (H3K4me3) are marks of active gene expression and enriched at active promoters, whereas repressive marks H3K9me3 and H3K27me3 are enriched at inactive or silent promoters. H3K36me3 marks gene bodies, whereas H3K4me1 and H3K27ac are enriched at poised and active enhancers respectively (16, 18). Combinations of several specific PTMs have been used to define “chromatin states,” with distinct biological roles in various cells (19).
In addition to these classic epigenetic marks, ncRNAs including microRNAs (miRNAs) and long ncRNAs (lncRNA, >200 nucleotides) can regulate gene expression via epigenetic mechanisms. Mature miRNAs are ∼22 nucleotide long small RNA duplexes, which, by specifically binding to target mRNAs (mainly 3′-UTRs), induce posttranscriptional gene silencing, translational repression, or RNA decay (20). LncRNAs exert cellular effects and gene regulation via various mechanisms including modulation of chromatin function by cis and trans interactions with chromatin factors, RNA binding proteins, enhancers, and/or modulating the actions of miRNAs (21, 22).
Histone PTMs together with DNAme, chromatin remodeling complexes, and ncRNAs remodel chromatin structure to euchromatin (open and accessible to transcription factors and RNA polymerase II for transcription) or heterochromatin (closed and less accessible for transcription) in cooperation with the actions of writers or erasers for each type of modification as indicated in Fig. 2, A–C (23). For euchromatin, the enrichment of active histone PTMs varies on promoters, enhancers, and transcribed regions as described earlier, whereas histone H3K9me3, H3K27me3, and DNAme are generally associated with heterochromatin (23). Chromatin is further organized into 3-dimensional structure, which is separated into discrete regions, called topologically associated domains (TADs) (24). Within these TADs, promoters and enhancers are linked to form chromosomal loops for gene activation with segregated genes sharing the same enhancers being coregulated (25).
Figure 2.
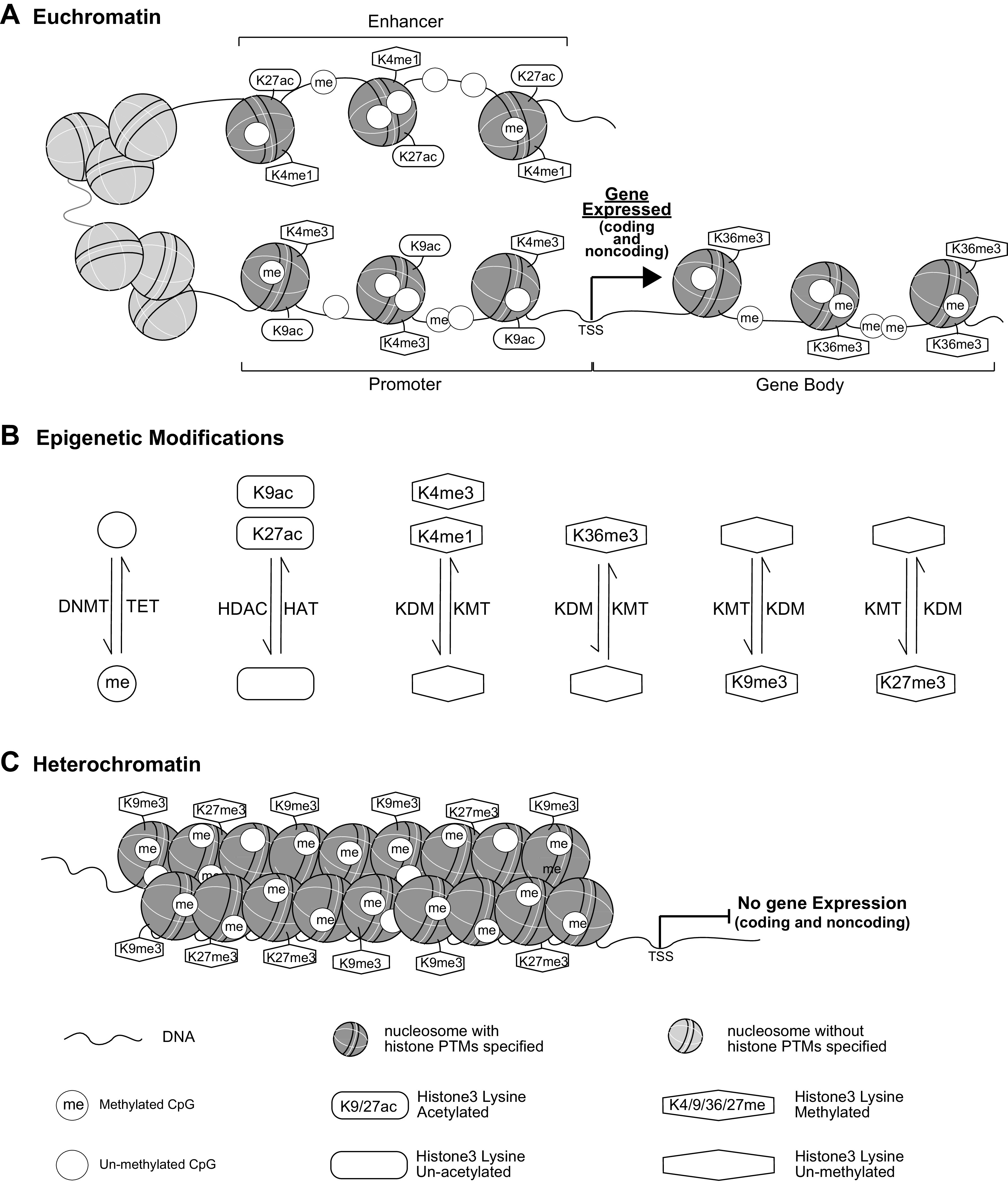
Epigenetic regulation of gene expression. Key epigenetic marks, including DNAme and histone PTMs, associated with euchromatin and heterochromatin are shown in A and C, respectively, and the interconversions between these two chromatin structures are shown in B. DNAme and histone PTMs interface with chromatin remodeling complexes, and noncoding (nc) RNAs to remodel chromatin into two major chromatin states, euchromatin and heterochromatin. At euchromatin (A) where loosely packed nucleosomes allow open chromatin states more easily accessible to transcription factors and RNA polymerases, gene promoters are usually enriched with marks such as histone H3K4me3 and H3K9ac, enhancers with H3K4me1 and H3K27ac, and gene bodies with H3K36me3. DNAme levels are low at enhancers and promoters, and high in gene bodies. With the binding of transcription factors to enhancers and promoters in euchromatin, RNA polymerases are recruited to either coding or noncoding (including pri-miRNAs and lncRNAs) genes for active transcription. Pri-miRNAs are processed to mature miRNAs via posttranscriptional processes in the cytoplasm, and lncRNA can modulate chromatin functions by interaction with chromatin factors, RNA binding proteins, and enhancers (not shown). On the other hand, nucleosomes at heterochromatin (C) are densely packed and the associated epigenetic marks include H3K9me3, H3K27me3 and DNAme. With reduced accessibility to transcription factors and RNAs polymerases at heterochromatin regions, gene transcription/expression is inhibited. The signals of each epigenetic mark enriched at euchromatin or heterochromatin regions can be modified by the dynamic actions of writers or erases, including histone and DNA methyltransferases, histone acetyltransferases, demethylases, and deactylases (shown in B) allowing for the remodeling of chromatin structure. TSS, transcription start site; DNMT, DNA methyltransferase; HAT, histone acetyltransferase; HDAC, histone deacetylase; KDM, histone lysine demethylase; KMT, histone lysine methyltransferase; PTMs, posttranslational modifications; TET, ten-eleven translocation enzyme.
Epigenetic Modifications in Diabetes and Diabetic Complications
Under normal conditions, epigenetic marks define cell identity and phenotypes and regulate important biological processes such as genomic imprinting, cell differentiation, and stem cell fate. They can be altered by environmental factors, nutritional states, lifestyles/behavior, metabolic, and genetic factors and hence modify the course of diseases including diabetes and its complications (26). In recent years, many epigenetic modifications (including DNAme and histone PTMs), miRNAs, and lncRNAs have been implicated in the regulation of genes, signaling pathways, and processes associated with diabetes and its various complications including retinopathy, DKD, neuropathy, and CVDs. These variations have been identified in in vitro and in vivo experimental studies as well as in humans, especially by epigenome-wide association studies (EWAS) on samples from clinical cohorts of diabetic subjects, many of which have been extensively reviewed (4, 5, 10, 11, 27). In this review, we focus more specifically on the role of epigenetics in the metabolic memory of diabetic complications.
METABOLIC MEMORY
Hyperglycemia, characterized by elevated hemoglobin A1c (HbA1c) levels, is a major risk factor for diabetic complications. Despite efforts to control glucose levels through diet, exercise, and medications including insulin, many subjects with diabetes continue to experience various harmful complications long after glucose normalization, implicating a “memory” of the prior glucose exposure in target cells (28). This phenomenon of glycemic or metabolic memory was observed in clinical trials and experimental models. The Diabetes Control and Complications Trial (DCCT) conducted on 1,441 subjects with T1D showed that, relative to conventional glycemic control (CONV), intensive glycemic control (INT) delayed progression of microvascular complications, including nephropathy and neuropathy (29). After DCCT ended in 1993, both groups of patients were advised to remain on INT therapy with insulin injections and followed for long term (1994 to present) in the observational Epidemiology of Diabetes Intervention and Complications (EDIC) study. During EDIC, both groups achieved similar mean HbA1c levels (∼8%) as illustrated in Fig. 3A (30). Remarkably, despite similar HbA1c levels, patients in the original DCCT INT treatment group to-date continue to have significantly lower risk of microvascular as well as macrovascular complications relative to those in the original CONV treatment arm of DCCT as shown in Fig. 3B. This persistent and enduring effects of INT therapy on reducing the risk of complications was attributed to prolonged hyperglycemia in the CONV therapy group during DCCT and has been termed metabolic memory in humans (30–32). The benefits of INT therapy were also shown in the UK Prospective Diabetes Study (called “legacy effect”) (33), Steno-2 study (34), and a meta-analysis study on CVD (35) in T2D patients. Data from the Pittsburgh Epidemiology of Diabetes Complications study (36) also support the importance of prior history of glycemic control in preventing complication development.
Figure 3.

Metabolic memory and underlying epigenetic mechanisms. A: schematic diagram depicting the association between glycemic control and the phenomenon of metabolic memory as derived from in vitro studies and in vivo studies with animals and diabetic patients. Depicted are glucose levels in the blood or cell growth medium between two groups of diabetic patients (e.g., patients with T1D in DCCT study), diabetic animals or cultured cells from target organs over two time periods (different or similar glycemic control periods as shown on the X-axis). During the “different-glycemic control” period, one group consisted of diabetic patients or animals experiencing high blood glucose (HG, e.g., high HbA1c levels of patients in DCCT CONV group) due to poor glycemic control, or cells grown in medium containing HG. The other group consisted of diabetic patients (e.g., DCCT INT group) or animals or cells maintained at normal glucose (NG) levels. At the end of this period, the glycemic control of HG group was changed to be similar to that in the group with NG, such that the glucose levels of both groups were normal or close-to-normal (“similar-glycemic control” Period). B: schematic diagram depicting different rates of incidences of diabetic complication development/progression or levels of complication-associated molecular changes between the two groups of diabetic patients/animals/cells over the same two time periods. During “different-glycemic control” period, HG group showed higher incidence of cell/organ dysfunction and complication development or progression compared with NG group. During the “similar-glycemic control” period, despite maintaining the glucose in both groups at the same normal or close-to-normal levels, the former HG treatment group continue to have higher rates of cellular dysfunction and complication development compared with the former NG group. This phenomenon has been called metabolic memory. C: evidence for the involvement of epigenetic mechanisms in metabolic memory. During the “different-glycemic control” period, HG decreased DNAme and/or histone PTMs associated with heterochromatin (e.g., H3K9me3 and H3K27me3) and increased euchromatin-associated PTMs (e.g., H3K4me3, H3K9ac, and H3K4me1) at key genes (gene symbols of some examples are listed below each modification) relevant to pathophysiological changes (such as inflammation, oxidative stress, and mitochondrial dysfunction) associated with diabetic complications in target cells/tissues. These epigenetic changes upregulate the expression of the corresponding genes. On the other hand, HG could also increase DNAme or H4K20me3 (another mark for heterochromatin) and decrease euchromatin-associated marks (H3K4me1 and H3K27ac) at some other “protective” genes (symbols listed), which downregulated the expression of the corresponding genes. Importantly, these epigenetic changes established during the “Different-glycemic control” period can persist in the second “similar-glycemic control” period, leading to persistent gene expression changes, organ dysfunction, and sustained complications, highlighting epigenetic mechanisms for metabolic memory. Besides DNAme and histone PTMs, the expression of some mature miRNAs was modified by HG during the first period with changes persisting during the second period. The potential involvement of some lncRNAs in metabolic memory has also been suggested in some studies. CONV, conventional glycemic control group; DCCT, Diabetes Control and Complications Trial; INT, intensive glycemic control; PTMs, posttranslational modifications; T1D, type 1 diabetes.
Similar findings of metabolic memory were noted in animal studies (Fig. 3, A and B). Diabetic dogs with poor glycemic control for 2.5 yr continued developing more retinopathy even after restoration of good glucose control (37). In zebrafish, wound healing deficits persisted even after restoration of normal glucose (NG) balance was achieved (38). In diabetic rats, high glucose (HG) induced upregulation of fibronectin persisted in endothelial cells, kidney, and heart after restoration of near-normoglycemia (39). Similarly, persistent retinal changes (including oxidative stress, inflammation, and cell death and features of retinopathy) and renal cortical changes (including oxidative stress) were observed in a model of metabolic memory in diabetic rats (40–42).
Mechanistically, persistent pathophysiological changes associated with diabetic complications (e.g., AGEs, oxidative stress, mitochondrial dysfunction, inflammation) were implicated in metabolic memory (5). More recently, the role of epigenetic changes (including DNAme, PTMs), and their interface with ncRNAs, in metabolic memory have been widely studied in in vitro and in vivo experimental models as well as in EWAS with clinical cohorts of diabetic subjects that we discuss in the ensuing sections with some key findings discussed later and also summarized in Fig. 3C.
DNAme in Experimental Models of Metabolic Memory
DNAme, the most well studied, stable, and heritable epigenetic mark, is a prime candidate to study connections between epigenetic and metabolic memory. Accordingly, it was found that the persistent global DNA hypomethylation induced by diabetic conditions can be transmitted across cell divisions in zebrafish (43). Indeed, reports also showed persistent HG-induced DNAme changes on genes linked to loss of mitochondrial homeostasis and/or mitochondrial DNA (mtDNA) mismatch related to diabetic retinopathy even after reinstitution of NG. This was functionally linked to mitochondrial dysfunction, oxidative stress, and cell death. The changes in retinal cells included hypermethylation of the regulatory region of DNA polymerase γ (POLG) that encodes an enzyme for mtDNA replication (44), hypermethylation of mtDNA displacement-loop regions containing mtDNA transcription and replication elements that induce mtDNA base mismatch (45, 46), hypomethylation of matrix metalloproteinase-9 (MMP-9) related to apoptosis of retinal capillary cells (47), and hypermethylation of genes encoding mitochondrial fusion and mismatch repair proteins (48). These DNAme changes were also associated with persistent binding of DNMT1 to mtDNA and TET2 to MMP-9 (49). In another study, global and genome-wide DNAme profiles identified sustained DNAme changes in foot fibroblasts cultured from patients (with and without foot ulcers) versus nondiabetic individuals despite prolonged cell culture in NG (50). Diabetes was also shown to alter DNAme at key genes associated with DKD in renal proximal tubular cells (51, 52) or endothelial cells (ECs) (53) in models of renal or vascular hyperglycemic memory. In these studies, along with DNAme, other epigenetic marks were also affected, and such interplay will be further discussed later.
Histone PTMs in Experimental Models of Metabolic Memory
Global or locus-specific PTMs are also implicated in metabolic memory. Report shows hyperglycemia increased HDAC and decreased HAT activities in the retina and retinal ECs, which resulted in decreased histone H3 acetylation with these changes persisting even after termination of hyperglycemia, suggesting a role for global histone acetylation in metabolic memory of diabetic retinopathy (54). On the other hand, persistent differential enrichment of PTMs induced by HG at key target inflammatory genes could induce sustained inflammation in metabolic memory. In one of the first such reports, levels of H3K9me3 and the corresponding repressive KMT Suv39h1 occupancy were significantly decreased at the promoters of key inflammatory genes including Il6, Ccl2, and colony stimulating factor 2 (Csf2), and in parallel, the repressive histone methyltransferase Suv39h1 was downregulated in vascular smooth muscle cells (VSMCs) cultured from diabetic db/db mice compared with nondiabetic db/+ mice. These changes remained evident even after culture of the diabetic VSMC for several passages in vitro under nondiabetic conditions along with increased inflammatory genes, proliferation, and sustained inflammatory phenotype (55). Similar PTMs mediated by Suv39h1 were also found at IL6 in cardiomyocytes treated with HG (56), suggesting common epigenetic mechanisms controlling inflammation across different cells. Moreover, in human ECs, transient HG treatment caused sustained alterations in H3K4me1 and increased binding of the corresponding KMT (Set7) at the promoter of the p65 subunit of NF-κB in vitro even after return to normoglycemia, with similar changes observed in aortas in vivo in a glucose-induced mouse model of memory. This resulted in increased expression of proatherogenic and inflammatory genes such as vascular cell adhesion protein 1 and CCL2 (57). In a follow-up study, the levels of H3K9me2 and H3K9me3 were found to be decreased whereas the demethylase LSD1 was increased at the same p65 promoter in aortic ECs cultured for short term in HG (58, 59). In addition, H3K4me1 KMT Set7 was associated with sustained inflammatory gene expression in response to prior hyperglycemia in ECs and therefore implicated as a key player in hyperglycemic memory (60). In another study, ECs from T1D mice cultured in NG exhibited persistent increase in the expression of Serpine1 (encoding the profibrotic protein plasminogen activator inhibitor-1 associated with EC dysfunction) and H3K4me3 at its promoter. Similar robust and persistent Serpine1 expression was observed in normal mouse ECs cultured in HG followed by glucose normalization (61). Interestingly, SERPIN1 was shown to be transregulated by a chromatin-associated lncRNA LINC00607 located at a super enhancer in human umbilical vein ECs treated with HG and TNFα suggesting epigenetic cross talk between chromatin and novel chromatin-associated lncRNAs leading to vascular dysfunction and metabolic memory (62).
Persistent PTMs can also impact oxidative stress in diabetic complications. HG or poor glycemic control were reported to increase SUV420h2-mediated H4K20me3 signals and decrease H3K4me1 and H3K4me2 signals at retinal Sod2 (encoding the antioxidant manganese superoxide dismutase). These changes persisted with the sustained downregulation of Sod2 after restoration of euglycemia (63, 64). In diabetic rodent retinas and retinal ECs, PTMs also regulated the activity of NF-E2-related factor 2 (Nrf2), a redox-sensitive transcription factor providing cellular defense against the cytotoxic ROS (65, 66). These epigenetic changes can adversely impact the antioxidant balance during diabetic retinopathy development.
Histone PTMs can also regulate multiple signaling pathways (4, 5). Thioredoxin interacting protein (TXNIP), a prooxidant that acts by binding to the antioxidant thioredoxin, upregulated the proinflammatory cyclooxygenase-2 gene in rat retinal ECs even after glucose normalization via chromatin remodeling involving increased promoter H3K9ac and decreased H3K9me3 (67). In renal mesangial cells and murine kidneys, TXNIP itself was upregulated by HG via epigenetic mechanisms involving increases in active histone marks (H3K9ac, H3K4me1, and H3K4me3) and decreases in repressive H3K27me3 at its promoter (68). Overexpression of TXNIP increased oxidative stress, mitochondrial and endothelial dysfunction and impaired glucose uptake, inflammation, fibrosis, and apoptosis in various target cells/tissues affected by complications, including retinal cells (69), kidney (70), nerves (71), and vascular cells (72). In podocytes from kidneys of diabetic mice, HG induced sustained elevation of H3K9/14ac and H3K4me1 at the promoter of SHP-1, encoding a cytosolic tyrosine phosphatase, causing persistent gene upregulation, subsequently leading to persistent insulin resistance, podocyte dysfunction, and nephropathy even after glucose re-normalization (73).
Overall, in vitro (cells) and in vivo (animal) studies demonstrated persistent changes in histone PTMs even after reversal from HG to NG. This resulted in sustained changes in the expression of genes associated with organ dysfunction, which in turn facilitated development of persistent diabetes-related complications (Fig. 3).
ncRNAs in Experimental Models of Metabolic Memory
Persistence of changes in noncoding RNAs like miRNAs and lncRNAs after glucose normalization has also been implicated in metabolic memory in a few studies (Fig. 3). Human aortic ECs challenged with HG followed by NG showed continued expression of miR-125b and miR-146a-5p, sustained NF-κB signaling, and endothelial dysfunction (74). Genome-wide analysis on hearts from diabetic mice identified 268 miRNAs displaying persistent changes after animals achieved glucose balance, potentially related to cardiomyopathy (75). In ECs challenged with HG to NG, a signaling pathway involving miR-27a-3p induced by NF-κB remained persistently activated, causing subsequent downregulated NRF2 expression, reduced nitric oxide (NO), increased ROS, and increased TGF-β, leading to endothelial to mesenchymal transition and ultimately causing perivascular fibrosis and cardiac dysfunction (76). In murine retinas or human retinal ECs, HG facilitated the recruitment of NF-κB p65 and promoted transcription of the miR-23b-1 gene, which in turn induced acetylation at NF-κB via targeting SIRT1, suggesting that the miR-23b-3p/SIRT1/NF-κB feedback loop may initiate and maintain metabolic memory (77).
LncRNAs act via epigenetic mechanisms and a number of lncRNAs have been implicated in the development of diabetic complications (21). However, the involvement of lncRNAs in metabolic memory is relatively less explored. Lnc-Ang362, a lncRNA induced by AngII and regulating VSMC proliferation (78), is the host gene of miR-221/222 that are known to mediate VSMC growth, and myocyte hypertrophy, were also persistently upregulated in the diabetic heart (75), suggesting potential role in metabolic memory. Another lncRNA, lnc-MGC, which is a host gene for 40 miRNAs in the miR-379 cluster, was upregulated in renal glomeruli of diabetic mice and in mesangial cells treated with HG or TGF-β. Notably, inhibition of this lncRNA could reduce glomerular matrix accumulation, fibrosis and hypertrophy associated with early DKD (79). Expression of lnc-MGC, and several of its hosted miRNAs, was induced by the endoplasmic reticulum (ER) stress-related CHOP transcription factor, which in turn caused more ER stress due to upregulation of hosted miRNAs like miR-379. The feed-forward amplifying circuits could cause long-lasting downstream changes associated with diabetic nephropathy, potentially contributing to metabolic memory. However, further studies needed to establish the links between key lncRNAs and metabolic memory.
Interplay Between Multiple Epigenetic Marks in Metabolic Memory
Metabolic memory can also be a consequence of the dynamic interplay and cooperation between multiple epigenetic factors, including DNAme, histone PTMs, chromatin accessibility, and ncRNAs. For example, analysis of the promoter of angiotensinogen (associated with kidney injury and DKD) in the kidney, especially proximal tubules, of diabetic versus nondiabetic mice revealed epigenetic changes beginning with H3K9ac and gradually extending to H3K4me3 followed by DNA demethylation at later time points, leading to increased gene expression. Alterations in DNAme and gene expression persisted even after restoration of glucose balance in the mice suggesting cooperative epigenetic mechanisms in metabolic memory of DKD (51). Upregulation of the redox gene p66Shc under diabetic conditions was associated with continued ROS production, reduced NO, inflammation, and apoptosis in various cell type/tissues including peripheral blood mononuclear cells (PBMCs), ECs, kidney, heart, and blood vessels and was implicated in both micro- and macrovascular complications (80). Notably, there was sustained expression of p66Shc with HG in human ECs despite return to normal glucose, and in diabetic mice following restoration of normoglycemia (53). This persistent upregulation was found to be epigenetically regulated by DNA hypomethylation and histone H3 hyperacetylation at the p66Shc promoter in ECs further supporting the concept of epigenetic interplay in metabolic memory (53). In addition, the H3 hyperacetylation was also regulated by alteration in SIRT1 (81). Similar changes were found in diabetic mice hearts secondary to downregulation of Dnmt3b and deacetylase SIRT1 (82). Interestingly, miR-218 and miR-34a were also implicated (82), illustrating the cooperation between DNAme, histone modifications, and miRNAs in processes regulating metabolic memory. In another study with human ECs, transient HG could increase key active histone marks and DNA hypomethylation at promoters of several hyperglycemia responsive genes, which might be potentially associated with memory of EC dysfunction, although their persistency after return to normoglycemia was not investigated (83).miRNAs, besides targeting genes related to diabetic complications, may participate in metabolic memory by targeting histone modifying enzymes. For example, in VSMC from diabetic db/db mice, upregulation of miR-125b (even after culture under nondiabetic conditions) promoted sustained increases expression of inflammatory genes by reducing their promoter H3K9me3 repressive signals through downregulating its target, the KMT Suv39h1 (84).
Relative to cells from healthy nondiabetic controls, when kidney proximal tubule epithelial cells derived from subjects with T2D were cultured for few passages under nondiabetic conditions, they depicted persistent promoter hyper-DNAme, decreased chromatin accessibility, and reduced H3K27ac at key downregulated transport-associated genes including claudin (CLDN)10, CLDN14, CLDN16, solute carrier family members SLC16A2, and SLC16A5 associated with tubular function. In addition, the tubular cells from T2D subjects were more sensitive to TGF-β, a major profibrotic growth factor associated with DKD, illustrating the crosstalk between epigenetic marks and chromatin remodeling in metabolic memory (52).
ROLE OF EPIGENETICS IN METABOLIC MEMORY IN HUMANS
Although studies in cell systems and rodents support the role of epigenetics in metabolic memory, investigations with biosamples directly collected from diabetic subjects, and particularly those experiencing metabolic memory of complications, have provided firmer evidence. These studies in humans have been primarily performed with archived whole blood (WB) genomic DNA or DNA from isolated white blood cells (WBC; which can be accessed noninvasively, unlike target tissues), which play an important role in inflammation.
Profiling of DNAme at ∼27 K CpGs in WBCs obtained from adult nondiabetic children of mothers with gestational T1D versus nondiabetic offspring of fathers with T1D revealed differentially methylated CpGs where the DNAme was associated with kidney function, indicating that T1D and exposure to hyperglycemia in utero might alter kidney function in offspring via DNAme (85). Blood monocytes from individuals with poorly controlled T2D before and after a 6-mo interval of glucose control showed persistent overexpression of p66Shc secondary to persistent promoter DNA hypomethylation and H3 acetylation (86). However, these studies did not examine clinical cohorts experiencing metabolic memory. To address this, we performed three epigenome-wide studies using blood cell DNA collected from participants of DCCC/EDIC clinical trial, in which the phenomenon of metabolic memory was first documented.
To test the role of PTMs (H3K9ac, H3K4me3, and H3K9me2) in metabolic memory, their enrichment levels in blood monocytes and lymphocytes collected at EDIC years 16/17 were studied. Genome-wide profiles were compared in the T1D participants of the DCCT/EDIC study between 30 cases (who received CONV therapy, had high HbA1c during DCCT, and developed vascular complications during EDIC) and 30 controls (received INT therapy with relatively normal HbA1c during DCCT and no complication development during EDIC) (87). Results showed significant increases in H3K9ac in cases versus controls at the promoters of key genes associated with inflammation and vascular complications, especially NF-κB pathway. Moreover, monocyte H3K9ac levels were positively associated with glycemic history (HbA1c). Since histone H3K9ac is associated with active chromatin and gene expression, these data provided support for the potential role of epigenetics in human metabolic memory.
To next examine the persistence of epigenetic marks over time, DNAme profiles were examined in WB and leukocyte samples from the same cohort (cases and controls) of DCCT/EDIC study subjects obtained at two time points 16/17 years apart (88). DNAme was profiled using the HumanMethylation450K BeadChip arrays. Impressively, several differentially methylated loci between cases and controls were found to persist even after 16/17 years. Among these changes, persistent hypomethylation at CpG cg19693031 (and two other CpGs) located in 3'-UTR of TXNIP was noted. Monocytes exposed to HG in culture likewise showed hypomethylation at 3’UTR of TXNIP along with upregulation of TXNIP expression. This was noteworthy because TXNIP is known to play a major role in the pathogenesis of multiple diabetic complications as mentioned earlier. The study provided clear support for connections between sustained DNAme changes over time and metabolic memory in humans with TXNIP being a top affected candidate. The next study was performed in a bigger DCCT cohort to not only verify the connections between DNAme and prior glycemic history but also determine a mediatory role for DNAme between the glycemic history and future complications development over an 18-yr period (89). We performed an EWAS using a subset of DCCT cohort T1D subjects who were regularly followed up for multiple complications during the EDIC study. DNAme profiles (generated with Illumina Infinium DNAmethylation EPIC BeadChip arrays) of archived WB DNAs from 499 DCCT/EDIC participants identified 186 CpGs whose DNAme was associated with mean DCCT HbA1c. Many of these were in genes related to complications and located in regulatory regions in hematopoietic stem cells (HSCs) and myeloid cells (as revealed by active chromatin states). Interestingly, CpG cg19693031 in TXNIP 3'-UTR region was again the most significant HbA1c-associated CpG. Remarkably, several HbA1c associated CpGs together were found to explain 68%–97% of the association between mean DCCT HbA1c and the risk of complications (retinopathy and nephropathy phenotypes) during EDIC (89).
Notably, since these data indicated DNAme changes can occur via modifying enhancer activity in HSCs and myeloid cells, metabolic memory of human diabetic complications may arise from hyperglycemia-induced epigenetic marks in stem cells that may persist in differentiated cells long after reinstitution of glycemic control, contributing to continued complications progression. These results further suggest that immune/inflammatory cells actively participate in metabolic memory (89). This intriguing concept that hyperglycemia-induced epigenetic change on HSCs could be transferred to myeloid cells, leading to diabetic complications, was also recently investigated in trained immunity.
TRAINED IMMUNITY AND METABOLIC MEMORY
Trained immunity refers to the long-term functional reprogramming of innate immune cells when challenged with exogenous or endogenous stimuli, which leads to altered function of these cells (especially myeloid cells such as monocyte/macrophages) on rechallenge (90). The induction and maintenance of trained immunity were reported to result from an interplay between metabolic reprogramming and epigenetic modifications. Many epigenetic enzymes utilize energy-related metabolites as essential cofactors. For example, acetyl-CoA is required for HAT activity, a-ketoglutarate is a cofactor of both TET and the demethylase JMJD3, and NAD+ is a cofactor of SIRT deacetylases. Alterations in these substrates during metabolic shifts can change the epigenetic landscape, including DNAme, PTMs, open chromatin, TADs, and ncRNAs (91).
Importantly, trained immunity can function in undifferentiated HSCs, which can further skew hematopoiesis toward myeloid cells, providing an explanation for why circulating trained monocytes may be maintained for periods much longer than their normal lifespan in circulation (92). It was shown that hyperglycemia increased proinflammatory gene expression and/or proatherogenic functional characteristics through glycolysis-dependent mechanisms in both macrophages and bone marrow–derived macrophages (BMDM) isolated from diabetic mice even after culturing in physiological glucose (93). Interestingly, atherosclerosis was markedly increased in normoglycemic mice transplanted with bone marrow from diabetic mice. This trained immunity was driven by persistent H3K4me3 related to chromatin accessibility in HSCs and macrophages (93). These data further support our previous data in humans (89) that metabolic memory can result from the propagation of epigenetics-driven chromatin remodeling and resultant proinflammatory states from HSCs to their progenies.
Other epigenetic changes identified in trained immunity may also contribute to metabolic memory. Restimulation with interferon (IFN)β resulted in faster and higher induction of some IFNβ-stimulated genes (memory ISGs) in murine fibroblasts and BMDM (94). This was associated with accelerated PolII recruitment on memory ISGs and correlated with accumulation of histone variant H3.3 and H3K36me3 modification. Because we previously found IFN signaling was activated by HG in human THP1 monocytes (95), IFNβ-stimulated transcriptional memory in monocytes may also be involved in metabolic memory. Besides promoters, the sustained residual H3K4me1 at latent enhancers in response to a stimulus also mediated a faster and stronger response on restimulation in differentiated macrophages (96), providing another level of epigenetic regulation.
The molecular mechanisms underlying epigenetics changes in trained immunity are not yet fully understood. Immune-gene priming lncRNAs located within the same TADs of several TNF-responsive cytokines could act in cis to prime these genes by epigenetic mechanisms (97), suggesting lncRNAs may also participate in trained immunity. But additional studies are needed to examine these connections in metabolic memory.
TARGETING EPIGENETICS TO LIMIT AND ERASE METABOLIC MEMORY
Since metabolic memory remains a challenge in the effective prevention and treatment of diabetic complications, it is imperative to identify therapeutic targets. Unlike genetics, epigenetic marks can be reversed, and hence in theory, epigenetics can be intervened on to limit metabolic memory. One approach would be to target epigenetic machineries. In experimental studies described earlier, the reversal of epigenetic changes resulted in corresponding changes in gene expression, pathway, or phenotypes after mutation of specific epigenetic writers/erasers or treatment with their specific inhibitors. Such results verified the functional involvement of both DNAme and key PTMs in metabolic memory. HDAC inhibitors have shown some benefits against complications in mice and rats, but their specificity is unclear (98, 99).
Drugs specifically targeting epigenetic factors, including DMNT, HDACs, KMTs, and KDMs, have been used for clinical treatment primarily in cancer, and other pathologies (100). But there are only limited reports in diabetic complications. Apabetalone, a selective inhibitor of the PTM reader BET protein, could reduce the incidence of major adverse cardiovascular events in individuals with T2D and chronic kidney disease (101) and decrease hospitalizations rates for heart failure in T2D (102). Interestingly, both curcumin and metformin, agents used in treatment of inflammation, diabetes and its complications, have been found to exert epigenetic effects (103, 104).
Targeting ncRNAs is another potential approach to interrupt metabolic memory. In this regard, locked nucleic acid (LNA)-modified oligonucleotides are commonly employed to target miRNAs and lncRNAs. LNA-modified anti-miRNA or antagomirs (miRNA inhibitors) or miRNA mimics can modulate miRNA function. For example, the LNA-modified inhibitor of miR-192 limited miR-192 and its downstream miRNAs and ameliorated renal fibrosis and related features of DKD in diabetic mice (105). In addition, a LNA-modified antisense oligonucleotide, the GapmeR designed to target DKD-associated lncRNA megacluster lnc-MGC, which effectively reduced features of early DKD in mice, was also effective in human kidney cells (79). This GapmeR could be a modality to interrupt the ER-stress-mediated amplifying circuits and persistent adverse events in the diabetic kidney. Such ncRNA targeting strategies could be expanded to include other lncRNAs for treatment of metabolic memory.
However, cell-specific and site-specific targeting is critical to limit off-target effects since epigenetic changes can be cell-type and locus specific. Site-directed CRISPR–Cas9 editing (106) may provide this specificity by enabling direct transcriptional regulation, locus-specific alterations in DNAme or PTMs using modified fusion constructs (107).
Nonetheless, the science behind these various options remains emergent and there is ample room for discovery and clinical translation. The field of nucleotide-specific gene editing to reverse disease-related genetic abnormalities is already showing promise in the clinic. Epigenetic editing as well as epigenetic drugs are likely to confer translational benefits for metabolic memory in the future.
CONCLUSIONS
As a genetically and environmentally complex disease, diabetes appears to modify the epigenetic landscape via alterations in DNAme, histone PTMs, and ncRNAs in target cells to promote the expression of factors associated with complications in multiple organs. Some epigenetic changes during diabetes persist despite normalization of glucose to drive uncontrolled oxidative stress, inflammation, fibrosis, and other pathological phenotypes (Fig. 1). Interestingly, trained immunity in HSC and peripheral immune cells may also participate in metabolic memory with epigenetic changes primed after initial exposure to HG. However, additional investigation is needed to fully understand the upstream and downstream mechanisms driving these persistent epigenetic variations in target cells. Furthermore, other factors affecting epigenetics such as circular RNAs (108) and RNA methylation involving N6-methyladenosine modification (109) may also contribute to metabolic memory and continue to be defined. Advances in single-cell RNA sequencing and single-cell epigenomics coupled with sophisticated bioinformatics tools can reveal cell-specific changes that are not evident from heterogeneous blood cells or tissues. EWAS in diabetic complications are expected to expand rapidly thanks to these unprecedented technological and computational advances coupled with publicly available data sets. Importantly, research biopsies of target tissues (e.g., kidney) from diabetic subjects at different time points would be a valuable resource to determine organ-/cell-specific epigenetic changes involved in metabolic memory that that can further aid in identifying much-needed new drug targets for clinical development.
GRANTS
The study was supported by National Institutes of Health Grants R01 DK065073, R01 HL106089, R01 DK081705, and DP3 DK106917; the Schaeffer Foundation; the Wanek Family Project for Cure of Type 1 Diabetes at City of Hope; and a DiaComp Pilot and Feasibility grant.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Z.C. and R.N. drafted manuscript; Z.C. and R.N. edited and revised manuscript; R.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Jeffrey Isenberg for valuable assistance with the manuscript.
REFERENCES
- 1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, Stein C, Basit A, Chan JCN, Mbanya JC, Pavkov ME, Ramachandaran A, Wild SH, James S, Herman WHH, Zhang P, Bommer C, Kuo SH, Boyko EJJ, Magliano DJ. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract 183: 109119, 2022. doi: 10.1016/j.diabres.2021.109119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev 93: 137–188, 2013. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- 3. Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J 34: 2436–2443, 2013. doi: 10.1093/eurheartj/eht149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 58: 443–455, 2015. doi: 10.1007/s00125-014-3462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol 15: 327–345, 2019. doi: 10.1038/s41581-019-0135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 107: 1058–1070, 2010. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol 79: 1527–1534, 2008. doi: 10.1902/jop.2008.080246. [DOI] [PubMed] [Google Scholar]
- 8. Nguyen DV, Shaw LC, Grant MB. Inflammation in the pathogenesis of microvascular complications in diabetes. Front Endocrinol (Lausanne) 3: 170, 2012. doi: 10.3389/fendo.2012.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ban CR, Twigg SM. Fibrosis in diabetes complications: pathogenic mechanisms and circulating and urinary markers. Vasc Health Risk Manag 4: 575–596, 2008. doi: 10.2147/vhrm.s1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kowluru RA, Santos JM, Mishra M. Epigenetic modifications and diabetic retinopathy. Biomed Res Int 2013: 635284, 2013. doi: 10.1155/2013/635284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Keating ST, Plutzky J, El-Osta A. Epigenetic changes in diabetes and cardiovascular risk. Circ Res 118: 1706–1722, 2016. doi: 10.1161/CIRCRESAHA.116.306819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Russo V. Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbor: Cold Spring harbor laboratory Press, 1996. [Google Scholar]
- 13. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet 17: 487–500, 2016. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 14. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492, 2012. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 15. Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18: 517–534, 2017. doi: 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- 16. Kouzarides T. Chromatin modifications and their function. Cell 128: 693–705, 2007. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 17. Fujisawa T, Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol 18: 246–262, 2017. doi: 10.1038/nrm.2016.143. [DOI] [PubMed] [Google Scholar]
- 18.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74, 2012. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roadmap Epigenomics Consortium; Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330, 2015. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bartel DP. Metazoan microRNAs. Cell 173: 20–51, 2018. doi: 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanwar VS, Reddy MA, Natarajan R. Emerging role of long non-coding RNAs in diabetic vascular complications. Front Endocrinol (Lausanne) 12: 665811, 2021. doi: 10.3389/fendo.2021.665811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Statello L, Guo C-J, Chen L-L, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22: 96–118, 2021. [Erratum in Nat Rev Mol Cell Biol 2021]. doi: 10.1038/s41580-020-00315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morrison O, Thakur J. Molecular complexes at euchromatin, heterochromatin and centromeric chromatin. Int J Mol Sci 22: 6922, 2021. doi: 10.3390/ijms22136922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380, 2012. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen C-A, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503: 290–294, 2013. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature 571: 489–499, 2019. doi: 10.1038/s41586-019-1411-0. [DOI] [PubMed] [Google Scholar]
- 27. Ling C, Bacos K, Rönn T. Epigenetics of type 2 diabetes mellitus and weight change—a tool for precision medicine? Nat Rev Endocrinol 18: 433–448, 2022. doi: 10.1038/s41574-022-00671-w. [DOI] [PubMed] [Google Scholar]
- 28. Cagliero E, Maiello M, Boeri D, Roy S, Lorenzi M. Increased expression of basement membrane components in human endothelial cells cultured in high glucose. J Clin Invest 82: 735–738, 1988. doi: 10.1172/JCI113655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nathan DM. Long-term complications of diabetes mellitus. N Engl J Med 328: 1676–1685, 1993. doi: 10.1056/NEJM199306103282306. [DOI] [PubMed] [Google Scholar]
- 30. Nathan DM; DCCT/EDIC Research Group. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care 37: 9–16, 2014. doi: 10.2337/dc13-2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DCCT/EDIC Research Group. Effect of intensive diabetes treatment on albuminuria in type 1 diabetes: long-term follow-up of the Diabetes Control and Complications Trial and Epidemiology of Diabetes Interventions and Complications study. Lancet Diabetes Endocrinol 2: 793–800, 2014. doi: 10.1016/S2213-8587(14)70155-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nathan DM. Realising the long-term promise of insulin therapy: the DCCT/EDIC study. Diabetologia 64: 1049–1058, 2021. doi: 10.1007/s00125-021-05397-4. [DOI] [PubMed] [Google Scholar]
- 33. Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359: 1577–1589, 2008. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- 34. Gæde P, Lund-Andersen H, Parving H-H, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med 358: 580–591, 2008. doi: 10.1056/NEJMoa0706245. [DOI] [PubMed] [Google Scholar]
- 35.Control Group; Turnbull FM, Abraira C, Anderson RJ, Byington RP, Chalmers JP, Duckworth WC, Evans GW, Gerstein HC, Holman RR, Moritz TE, Neal BC, Ninomiya T, Patel AA, Paul SK, Travert F, Woodward M. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia 52: 2288–2298, 2009. [Erratum in Diabetologia 52: 2470, 2009]. doi: 10.1007/s00125-009-1470-0. [DOI] [PubMed] [Google Scholar]
- 36. Miller RG, Orchard TJ. Understanding metabolic memory: a tale of two studies. Diabetes 69: 291–299, 2020. doi: 10.2337/db19-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 36: 808–812, 1987. doi: 10.2337/diab.36.7.808. [DOI] [PubMed] [Google Scholar]
- 38. Intine RV, Olsen AS, Sarras MP Jr.. A zebrafish model of diabetes mellitus and metabolic memory. J Vis Exp 72: e50232, 2013. doi: 10.3791/50232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roy S, Sala R, Cagliero E, Lorenzi M. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci USA 87: 404–408, 1990. doi: 10.1073/pnas.87.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kowluru RA. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes 52: 818–823, 2003. doi: 10.2337/diabetes.52.3.818. [DOI] [PubMed] [Google Scholar]
- 41. Kowluru RA, Zhong Q, Kanwar M. Metabolic memory and diabetic retinopathy: role of inflammatory mediators in retinal pericytes. Exp Eye Res 90: 617–623, 2010. doi: 10.1016/j.exer.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kowluru RA, Abbas SN, Odenbach S. Reversal of hyperglycemia and diabetic nephropathy: effect of reinstitution of good metabolic control on oxidative stress in the kidney of diabetic rats. J Diabetes Complications 18: 282–288, 2004. doi: 10.1016/j.jdiacomp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 43. Olsen AS, Sarras MP Jr, Leontovich A, Intine RV. Heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes 61: 485–491, 2012. doi: 10.2337/db11-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tewari S, Zhong Q, Santos JM, Kowluru RA. Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Invest Ophthalmol Vis Sci 53: 4881–4888, 2012. doi: 10.1167/iovs.12-9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mishra M, Kowluru RA. Epigenetic modification of mitochondrial DNA in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci 56: 5133–5142, 2015. doi: 10.1167/iovs.15-16937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mishra M, Kowluru RA. DNA methylation—a potential source of mitochondria DNA base mismatch in the development of diabetic retinopathy. Mol Neurobiol 56: 88–101, 2019. doi: 10.1007/s12035-018-1086-9. [DOI] [PubMed] [Google Scholar]
- 47. Kowluru RA, Shan Y, Mishra M. Dynamic DNA methylation of matrix metalloproteinase-9 in the development of diabetic retinopathy. Lab Invest 96: 1040–1049, 2016. doi: 10.1038/labinvest.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kowluru RA, Mohammad G. Epigenetics and mitochondrial stability in the metabolic memory phenomenon associated with continued progression of diabetic retinopathy. Sci Rep 10: 6655, 2020. doi: 10.1038/s41598-020-63527-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mishra M, Kowluru RA. The role of DNA methylation in the metabolic memory phenomenon associated with the continued progression of diabetic retinopathy. Invest Ophthalmol Vis Sci 57: 5748–5757, 2016. doi: 10.1167/iovs.16-19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park LK, Maione AG, Smith A, Gerami-Naini B, Iyer LK, Mooney DJ, Veves A, Garlick JA. Genome-wide DNA methylation analysis identifies a metabolic memory profile in patient-derived diabetic foot ulcer fibroblasts. Epigenetics 9: 1339–1349, 2014. doi: 10.4161/15592294.2014.967584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marumo T, Yagi S, Kawarazaki W, Nishimoto M, Ayuzawa N, Watanabe A, Ueda K, Hirahashi J, Hishikawa K, Sakurai H, Shiota K, Fujita T. Diabetes induces aberrant DNA methylation in the proximal tubules of the kidney. J Am Soc Nephrol 26: 2388–2397, 2015. doi: 10.1681/ASN.2014070665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bansal A, Balasubramanian S, Dhawan S, Leung A, Chen Z, Natarajan R. Integrative omics analyses reveal epigenetic memory in diabetic renal cells regulating genes associated with kidney dysfunction. Diabetes 69: 2490–2502, 2020. doi: 10.2337/db20-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, Lüscher TF, Cosentino F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res 111: 278–289, 2012. doi: 10.1161/CIRCRESAHA.112.266593. [DOI] [PubMed] [Google Scholar]
- 54. Zhong Q, Kowluru RA. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J Cell Biochem 110: 1306–1313, 2010. doi: 10.1002/jcb.22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci USA 105: 9047–9052, 2008. doi: 10.1073/pnas.0803623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yu XY, Geng YJ, Liang JL, Zhang S, Lei HP, Zhong SL, Lin QX, Shan ZX, Lin SG, Li Y. High levels of glucose induce “metabolic memory” in cardiomyocyte via epigenetic histone H3 lysine 9 methylation. Mol Biol Rep 39: 8891–8898, 2012. doi: 10.1007/s11033-012-1756-z. [DOI] [PubMed] [Google Scholar]
- 57. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 205: 2409–2417, 2008. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, Calkin AC, Brownlee M, Cooper ME, El-Osta A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58: 1229–1236, 2009. doi: 10.2337/db08-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cooper ME, El-Osta A, Allen TJ, Watson AMD, Thomas MC, Jandeleit-Dahm KAM. Metabolic karma—the atherogenic legacy of diabetes: the 2017 Edwin Bierman Award Lecture. Diabetes 67: 785–790, 2018. doi: 10.2337/dbi18-0010. [DOI] [PubMed] [Google Scholar]
- 60. Okabe J, Orlowski C, Balcerczyk A, Tikellis C, Thomas MC, Cooper ME, El-Osta A. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ Res 110: 1067–1076, 2012. doi: 10.1161/CIRCRESAHA.112.266171. [DOI] [PubMed] [Google Scholar]
- 61. Takizawa F, Mizutani S, Ogawa Y, Sawada N. Glucose-independent persistence of PAI-1 gene expression and H3K4 tri-methylation in type 1 diabetic mouse endothelium: implication in metabolic memory. Biochem Biophys Res Commun 433: 66–72, 2013. doi: 10.1016/j.bbrc.2013.02.064. [DOI] [PubMed] [Google Scholar]
- 62. Calandrelli R, Xu L, Luo Y, Wu W, Fan X, Nguyen T, Chen C-J, Sriram K, Tang X, Burns AB, Natarajan R, Chen ZB, Zhong S. Stress-induced RNA–chromatin interactions promote endothelial dysfunction. Nat Commun 11: 5211, 2020. doi: 10.1038/s41467-020-18957-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhong Q, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes 60: 1304–1313, 2011. doi: 10.2337/db10-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhong Q, Kowluru RA. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: role of histone methylation. Invest Ophthalmol Vis Sci 54: 244–250, 2013. doi: 10.1167/iovs.12-10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mishra M, Zhong Q, Kowluru RA. Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic Biol Med 75: 129–139, 2014. doi: 10.1016/j.freeradbiomed.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mishra M, Zhong Q, Kowluru RA. Epigenetic modifications of Keap1 regulate its interaction with the protective factor Nrf2 in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci 55: 7256–7265, 2014. doi: 10.1167/iovs.14-15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Perrone L, Devi TS, Hosoya K, Terasaki T, Singh LP. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol 221: 262–272, 2009. doi: 10.1002/jcp.21852. [DOI] [PubMed] [Google Scholar]
- 68. De Marinis Y, Cai M, Bompada P, Atac D, Kotova O, Johansson ME, Garcia-Vaz E, Gomez MF, Laakso M, Groop L. Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int 89: 342–353, 2016. doi: 10.1016/j.kint.2015.12.018. [DOI] [PubMed] [Google Scholar]
- 69. Singh LP. Thioredoxin interacting protein (TXNIP) and pathogenesis of diabetic retinopathy. J Clin Exp Ophthalmol 4: 10.4172/2155-9570.1000287, 2013. doi: 10.4172/2155-9570.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shah A, Xia L, Masson EA, Gui C, Momen A, Shikatani EA, Husain M, Quaggin S, John R, Fantus IG. Thioredoxin-interacting protein deficiency protects against diabetic nephropathy. J Am Soc Nephrol 26: 2963–2977, 2015. doi: 10.1681/ASN.2014050528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang X, Zhao S, Yuan Q, Zhu L, Li F, Wang H, Kong D, Hao J. TXNIP, a novel key factor to cause Schwann cell dysfunction in diabetic peripheral neuropathy, under the regulation of PI3K/Akt pathway inhibition-induced DNMT1 and DNMT3a overexpression. Cell Death Dis 12: 642, 2021. doi: 10.1038/s41419-021-03930-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Myers RB, Fomovsky GM, Lee S, Tan M, Wang BF, Patwari P, Yoshioka J. Deletion of thioredoxin-interacting protein improves cardiac inotropic reserve in the streptozotocin-induced diabetic heart. Am J Physiol Heart Circ Physiol 310: H1748–H1759, 2016. doi: 10.1152/ajpheart.00051.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lizotte F, Denhez B, Guay A, Gévry N, Côté AM, Geraldes P. Persistent insulin resistance in podocytes caused by epigenetic changes of SHP-1 in diabetes. Diabetes 65: 3705–3717, 2016. doi: 10.2337/db16-0254. [DOI] [PubMed] [Google Scholar]
- 74. Zhong X, Liao Y, Chen L, Liu G, Feng Y, Zeng T, Zhang J. The microRNAs in the pathogenesis of metabolic memory. Endocrinology 156: 3157–3168, 2015. doi: 10.1210/en.2015-1063. [DOI] [PubMed] [Google Scholar]
- 75. Costantino S, Paneni F, Lüscher TF, Cosentino F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur Heart J 37: 572–576, 2016. doi: 10.1093/eurheartj/ehv599. [DOI] [PubMed] [Google Scholar]
- 76. Yao Y, Song Q, Hu C, Da X, Yu Y, He Z, Xu C, Chen Q, Wang QK. Endothelial cell metabolic memory causes cardiovascular dysfunction in diabetes. Cardiovasc Res 118: 196–211, 2022. doi: 10.1093/cvr/cvab013. [DOI] [PubMed] [Google Scholar]
- 77. Zhao S, Li T, Li J, Lu Q, Han C, Wang N, Qiu Q, Cao H, Xu X, Chen H, Zheng Z. miR-23b-3p induces the cellular metabolic memory of high glucose in diabetic retinopathy through a SIRT1-dependent signalling pathway. Diabetologia 59: 644–654, 2016. doi: 10.1007/s00125-015-3832-0. [DOI] [PubMed] [Google Scholar]
- 78. Leung A, Trac C, Jin W, Lanting L, Akbany A, Sætrom P, Schones DE, Natarajan R. Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ Res 113: 266–278, 2013. doi: 10.1161/CIRCRESAHA.112.300849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kato M, Wang M, Chen Z, Bhatt K, Oh HJ, Lanting L, Deshpande S, Jia Y, Lai JY, O'Connor CL, Wu Y, Hodgin JB, Nelson RG, Bitzer M, Natarajan R. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat Commun 7: 12864, 2016. doi: 10.1038/ncomms12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Francia P, Cosentino F, Schiavoni M, Huang Y, Perna E, Camici GG, Lüscher TF, Volpe M. p66(Shc) protein, oxidative stress, and cardiovascular complications of diabetes: the missing link. J Mol Med (Berl) 87: 885–891, 2009. doi: 10.1007/s00109-009-0499-3. [DOI] [PubMed] [Google Scholar]
- 81. Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, Huang S, Liu JJ, Lu YB, Zhang ZQ, Yang RF, Zhang R, Cai H, Liu DP, Liang CC. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res 109: 639–648, 2011. doi: 10.1161/CIRCRESAHA.111.243592. [DOI] [PubMed] [Google Scholar]
- 82. Costantino S, Paneni F, Mitchell K, Mohammed SA, Hussain S, Gkolfos C, Berrino L, Volpe M, Schwarzwald C, Lüscher TF, Cosentino F. Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66(Shc). Int J Cardiol 268: 179–186, 2018. doi: 10.1016/j.ijcard.2018.04.082. [DOI] [PubMed] [Google Scholar]
- 83. Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, Ziemann M, Karagiannis T, Tonna S, Kowalczyk A, Beresford-Smith B, Macintyre G, Kelong M, Hongyu Z, Zhu J, El-Osta A. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 21: 1601–1615, 2011. doi: 10.1101/gr.116095.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Villeneuve LM, Kato M, Reddy MA, Wang M, Lanting L, Natarajan R. Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes 59: 2904–2915, 2010. doi: 10.2337/db10-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gautier JF, Porcher R, Abi Khalil C, Bellili-Munoz N, Fetita LS, Travert F, Choukem SP, Riveline JP, Hadjadj S, Larger E, Boudou P, Blondeau B, Roussel R, Ferré P, Ravussin E, Rouzet F, Marre M. Kidney dysfunction in adult offspring exposed in utero to type 1 diabetes is associated with alterations in genome-wide DNA methylation. PLoS One 10: e0134654, 2015. doi: 10.1371/journal.pone.0134654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Costantino S, Paneni F, Battista R, Castello L, Capretti G, Chiandotto S, Tanese L, Russo G, Pitocco D, Lanza GA, Volpe M, Lüscher TF, Cosentino F. Impact of glycemic variability on chromatin remodeling, oxidative stress, and endothelial dysfunction in patients with type 2 diabetes and with target hba1c levels. Diabetes 66: 2472–2482, 2017. doi: 10.2337/db17-0294. [DOI] [PubMed] [Google Scholar]
- 87. Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X, Li SM, Cleary P, Riggs A, Harlan DM, Lorenzi G, Kolterman O, Sun W, Lachin JM, Natarajan R; DCCT/EDIC Research Group. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 63: 1748–1762, 2014. doi: 10.2337/db13-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chen Z, Miao F, Paterson AD, Lachin JM, Zhang L, Schones DE, Wu X, Wang J, Tompkins JD, Genuth S, Braffett BH, Riggs AD, Natarajan R; DCCT/EDIC Research Group. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci USA 113: E3002–E3011, 2016. doi: 10.1073/pnas.1603712113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chen Z, Miao F, Braffett BH, Lachin JM, Zhang L, Wu X, Roshandel D, Carless M, Li XA, Tompkins JD, Kaddis JS, Riggs AD, Paterson AD, Braffet BH, Lachin JM, Chen Z, Miao F, Zhang L, Natarajan R, Paterson AD, Natarajan R; DCCT/EDIC Study Group. DNA methylation mediates development of HbA1c-associated complications in type 1 diabetes. Nat Metab 2: 744–762, 2020. doi: 10.1038/s42255-020-0231-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, Riksen NP, Schlitzer A, Schultze JL, Stabell Benn C, Sun JC, Xavier RJ, Latz E. Defining trained immunity and its role in health and disease. Nat Rev Immunol 20: 375–388, 2020. doi: 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Fanucchi S, Domínguez-Andrés J, Joosten LAB, Netea MG, Mhlanga MM. The intersection of epigenetics and metabolism in trained immunity. Immunity 54: 32–43, 2021. doi: 10.1016/j.immuni.2020.10.011. [DOI] [PubMed] [Google Scholar]
- 92. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, Chatzigeorgiou A, Schlitzer A, Beyer M, Joosten LAB, Isermann B, Lesche M, Petzold A, Simons K, Henry I, Dahl A, Schultze JL, Wielockx B, Zamboni N, Mirtschink P, Coskun U, Hajishengallis G, Netea MG, Chavakis T. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172: 147–161.e12, 2018. doi: 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Edgar L, Akbar N, Braithwaite AT, Krausgruber T, Gallart-Ayala H, Bailey J, Corbin AL, Khoyratty TE, Chai JT, Alkhalil M, Rendeiro AF, Ziberna K, Arya R, Cahill TJ, Bock C, Laurencikiene J, Crabtree MJ, Lemieux ME, Riksen NP, Netea MG, Wheelock CE, Channon KM, Rydén M, Udalova IA, Carnicer R, Choudhury RP. Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis. Circulation 144: 961–982, 2021. doi: 10.1161/CIRCULATIONAHA.120.046464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kamada R, Yang W, Zhang Y, Patel MC, Yang Y, Ouda R, Dey A, Wakabayashi Y, Sakaguchi K, Fujita T, Tamura T, Zhu J, Ozato K. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc Natl Acad Sci USA 115: E9162–E9171, 2018. doi: 10.1073/pnas.1720930115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Miao F, Chen Z, Zhang L, Wang J, Gao H, Wu X, Natarajan R. RNA-sequencing analysis of high glucose-treated monocytes reveals novel transcriptome signatures and associated epigenetic profiles. Physiol Genomics 45: 287–299, 2013. doi: 10.1152/physiolgenomics.00001.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell 152: 157–171, 2013. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 97. Fanucchi S, Fok ET, Dalla E, Shibayama Y, Börner K, Chang EY, Stoychev S, Imakaev M, Grimm D, Wang KC, Li G, Sung WK, Mhlanga MM. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat Genet 51: 138–150, 2019. doi: 10.1038/s41588-018-0298-2. [DOI] [PubMed] [Google Scholar]
- 98. Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, Yuen DA, Connelly KA, Marsden PA, Gilbert RE. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol 178: 2205–2214, 2011. doi: 10.1016/j.ajpath.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang AJ, Wang S, Wang BJ, Xiao M, Guo Y, Tang Y, Zhang J, Gu J. Epigenetic regulation associated with Sirtuin 1 in complications of diabetes mellitus. Front Endocrinol (Lausanne) 11: 598012, 2020. doi: 10.3389/fendo.2020.598012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenetics 11: 174, 2019. doi: 10.1186/s13148-019-0776-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kalantar-Zadeh K, Schwartz GG, Nicholls SJ, Buhr KA, Ginsberg HN, Johansson JO, Kulikowski E, Lebioda K, Toth PP, Wong N, Sweeney M, Ray KK; BETonMACE Investigators. Effect of apabetalone on cardiovascular events in diabetes, CKD, and recent acute coronary syndrome: results from the BETonMACE randomized controlled trial. Clin J Am Soc Nephrol 16: 705–716, 2021. doi: 10.2215/CJN.16751020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nicholls SJ, Schwartz GG, Buhr KA, Ginsberg HN, Johansson JO, Kalantar-Zadeh K, Kulikowski E, Toth PP, Wong N, Sweeney M, Ray KK; BETonMACE Investigators . Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: a prespecified analysis of the BETonMACE study. Cardiovasc Diabetol 20: 13, 2021. doi: 10.1186/s12933-020-01199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tang C, Liu Y, Liu S, Yang C, Chen L, Tang F, Wang F, Zhan L, Deng H, Zhou W, Lin Y, Yuan X. Curcumin and its analogs as potential epigenetic modulators: prevention of diabetes and its complications. Pharmacology 107: 1–13, 2022. doi: 10.1159/000520311. [DOI] [PubMed] [Google Scholar]
- 104. Bridgeman SC, Ellison GC, Melton PE, Newsholme P, Mamotte CDS. Epigenetic effects of metformin: from molecular mechanisms to clinical implications. Diabetes Obes Metab 20: 1553–1562, 2018. doi: 10.1111/dom.13262. [DOI] [PubMed] [Google Scholar]
- 105. Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol 23: 458–469, 2012. doi: 10.1681/ASN.2011050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096, 2014. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 107. Pulecio J, Verma N, Mejía-Ramírez E, Huangfu D, Raya A. CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell 21: 431–447, 2017. doi: 10.1016/j.stem.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20: 675–691, 2019. doi: 10.1038/s41576-019-0158-7. [DOI] [PubMed] [Google Scholar]
- 109. Yang C, Hu Y, Zhou B, Bao Y, Li Z, Gong C, Yang H, Wang S, Xiao Y. The role of m6A modification in physiology and disease. Cell Death Dis 11: 960, 2020. doi: 10.1038/s41419-020-03143-z. [DOI] [PMC free article] [PubMed] [Google Scholar]