

Keywords: calcium channel, calcium release, mouse, skeletal muscle, sodium channel
Abstract
Hypokalemic periodic paralysis (HypoPP) is a channelopathy of skeletal muscle caused by missense mutations in the voltage sensor domains (usually at an arginine of the S4 segment) of the CaV1.1 calcium channel or of the NaV1.4 sodium channel. The primary clinical manifestation is recurrent attacks of weakness, resulting from impaired excitability of anomalously depolarized fibers containing leaky mutant channels. Although the ictal loss of fiber excitability is sufficient to explain the acute episodes of weakness, a deleterious change in voltage sensor function for CaV1.1 mutant channels may also compromise excitation-contraction coupling (EC-coupling). We used the low-affinity Ca2+ indicator Oregon Green 488 BAPTA-5N (OGB-5N) to assess voltage-dependent Ca2+-release as a measure of EC-coupling for our knock-in mutant mouse models of HypoPP. The peak ΔF/F0 in fibers isolated from CaV1.1-R528H mice was about two-thirds of the amplitude observed in WT mice; whereas in HypoPP fibers from NaV1.4-R669H mice the ΔF/F0 was indistinguishable from WT. No difference in the voltage dependence of ΔF/F0 from WT was observed for fibers from either HypoPP mouse model. Because late-onset permanent muscle weakness is more severe for CaV1.1-associated HypoPP than for NaV1.4, we propose that the reduced Ca2+-release for CaV1.1-R528H mutant channels may increase the susceptibility to fixed myopathic weakness. In contrast, the episodes of transient weakness are similar for CaV1.1- and NaV1.4-associated HypoPP, consistent with the notion that acute attacks of weakness are primarily caused by leaky channels and are not a consequence of reduced Ca2+-release.
INTRODUCTION
Hypokalemic periodic paralysis (HypoPP) is a dominantly inherited disorder of skeletal muscle that presents with recurrent episodes of weakness lasting for hours to a day or even longer (1, 2). Recovery of strength is initially complete, with normal muscle function between attacks, but mild to moderate permanent weakness and vacuolar changes in muscle may occur with aging. HypoPP is an ion channelopathy caused by missense mutations of CACNA1S encoding the CaV1.1 subunit of the L-type calcium channel (3), or less frequently by missense mutations of SCN4A encoding the NaV1.4 subunit of the sodium voltage-gated channel (4). Remarkably, 24 of the 25 reported HypoPP mutations are missense substitutions at arginine residues in S4 transmembrane segments of the voltage-sensor domains of these channels (5, 6).
The transient episodes of weakness are caused by sustained depolarization of the fiber resting potential (Vrest) that inactivates sodium channels and thereby impairs fiber excitability (1). This depolarized shift of Vrest paradoxically occurs in low extracellular [K+] (<3.5 mM) because of the anomalous “gating pore” leakage current created by the missense mutations of S4 in either CaV1.1 or NaV1.4 (7–9). Because the S4 segment is a critical component of the CaV1.1 voltage sensor that couples transverse tubule depolarization to activation of the ryanodine receptor and Ca2+ release from the sarcoplasmic reticulum (SR), the possibility of a defect in excitation-contraction coupling (EC-coupling) has been proposed for the pathogenesis of HypoPP.
The integrity of EC-coupling in CaV1.1-associated HypoPP has been assessed in only two prior studies (9, 10), which had conflicting results, and has never been examined for NaV1.4-associated HypoPP. Jurkat-Rott et al. (10) reported Ca2+-release “was not grossly altered” between wild-type (WT) and CaV1.1-R528H muscle, using fura-2 microfluorimetry in voltage-clamped human HypoPP myotubes (WT/R528H heterozygous) or in a mouse muscle cell line that is null for CaV1.1 (GLT cells) with transiently expressed R528H mutant channels. We created a knock-in mutant mouse model of CaV1.1-R528H with a robust HypoPP phenotype and measured voltage-dependent Ca2+ transients with fluo-4 in dissociated fibers (9). Ca2+-release was comparable for WT and heterozygous CaV1.1-R528H fibers, but the average response from a pool of 14 homozygous CaV1.1-R528H fibers was markedly reduced to 30% of WT levels.
In the present study, we use a low-affinity fast Ca2+ dye, Oregon Green 488 BAPTA-5N (OGB-5N), to address the prior conflicting results for CaV1.1-R528H (9, 10), and also assess Ca2+-release for NaV1.4-R669H fibers. We confirm a reduced peak ΔF/F0 for CaV1.1-R528H fibers but at a more modest degree of about two-thirds of WT levels. We propose this milder impairment of Ca2+-release compared with our prior report is because more stringent criteria were used to select healthy fibers that presumably do not have the triad disruption that occurs with vacuolar myopathy in HypoPP. No defect of Ca2+-release was detected for NaV1.4-R669H fibers.
MATERIALS AND METHODS
Mouse Models of HypoPP
The generation of knock-in mutant mouse models for hypokalemic periodic paralysis (HypoPP), screening progeny for mutant alleles, confirmation of mutant allele expression, and characterization of the periodic paralysis phenotype with paradoxical depolarization of Vrest and loss of force in a low-K+ challenge has been previously described (9, 11). Two murine models of HypoPP were studied here: a missense mutation of Cacna1s coding for CaV1.1 (mR528H) and a missense mutation of Scn4a coding for NaV1.4 (mR663H). These homologous mutant alleles in mice correspond to the human disease-causing mutations CaV1.1-R528H and NaV1.4-R669H, and this nomenclature is used herein to facilitate the comparison to the clinical literature. Mouse lines have been propagated in the 129/sv strain for more than 20 generations. All procedures on mice were in accordance with animal protocols approved by the Institutional Animal Care and Use Committee at the David Geffen School of Medicine, University of California, Los Angeles, CA.
Muscle Fiber Preparation
All recordings were performed on dissociated single fibers from the flexor digitorum brevis muscle (FDB) as previously described (12). Animals aged 2–4 mo were euthanized by isoflurane inhalation followed by cervical dislocation. Both male and female mice were used, and the data were pooled. The FDB was rapidly dissected free and pinned to the bottom of a sylgard-coated dish containing collagenase type II (6 mg/mL, Gibco) in standard Tyrode’s solution. The dish was gently agitated for 45 min in an incubator at 35°C. The enzymatically digested muscle was then triturated in a series of fire-polished glass pipettes, with progressively smaller tip diameters. Dissociated single fibers were rinsed four times with collagenase-free Tyrode’s solution and then transferred to a glass-bottomed recording chamber constructed from a 35-mm plastic dish fitted with a glass coverslip (FisherFinest No. 12-548-A).
Electrophysiology
Single FDB fibers of length 400–600 µm (mean = 537 µm) were impaled using an approach angled 15° from the vertical perpendicular with two sharp microelectrodes (10–12 MΩ), near the midpoint and longitudinally separated by 10–15 µm. Recordings were initiated in two-electrode current-clamp (TEV-200, Dagan Corporation), and a holding current was applied to polarize the fiber to −80 mV. We intentionally selected “healthy” fibers, as defined: 1) optically by sharp edges, clear sharply contrasted striations, and no intracellular inclusions and 2) electrophysiologically by a spontaneous membrane potential of −40 mV to −50 mV upon initial impalement, and full polarization to −80 mV with a holding current in the range of −5 to −25 nA in Tyrode’s solution.
After the fiber equilibrated for 10 min at −80 mV, action potentials were elicited by application of a brief depolarizing current of 0.2–0.3 µA for 0.5 ms. Fiber contraction was prevented by using an internal solution for the microelectrodes that contained a high concentration of EGTA (see Solutions).
To improve the voltage control of the clamped fiber (speed, spatial uniformity, steady-state error), we exchanged the Tyrode bath with a tetraethylammonium (TEA)-Cl solution that also contained blockers for sodium, calcium, and chloride channels (see Solutions). The holding current was manually adjusted to keep Vm near −80 mV, and then the TEV-200 amplifier was switched to voltage-clamp mode at a clamp potential of −80 mV. The voltage protocol was a sequence of 10-ms step depolarizations, over a range from −80 mV to +80 mV, from a holding potential of −80 mV. The sequence progressed from −80 mV to more positive values in 10 mV increments, and the fiber was returned to −80 mV for 20 s between pulses. Current (low-pass filtered at 5 kHz) and voltage signals were sampled at 25 kHz using a 16-bit A/D and D/A converter, controlled with LabVIEW software (National Instruments).
Detection of Ca2+ Transients
A fluorescent, low affinity, impermeant Ca2+-sensitive dye, Oregon Green 488 BAPTA-5N (OGB-5N), was used to follow fast changes of myoplasmic [Ca2+], in response to action potentials or voltage pulses. The dye was excited at 480 ± 20 nm, and its emission was restricted to 535 ± 22 nm. Excitation and emission bandwidths were separated by a 510 nm dichroic mirror. All optical components were from Semrock. The light source was an LED (SP-05-B6, Luxeon StarLeds) driven by a custom-made power source under computer control. The experimental chamber was placed on the stage of an inverted microscope (Olympus XI-171) equipped with a homemade epifluorescence attachment. Fibers were imaged with a ×100, 1.4 numerical aperture (NA) oil immersion objective. The illumination spot diameter was adjusted to ∼90% of the fiber diameter. The light detector consisted of a low capacitance PIN photodiode (UV-001, OSI Optoelectronics) connected to the head stage of a patch-clamp amplifier (Axopatch 2B, Molecular Devices Inc.) with no bias current applied. The dark current was set to zero. The photocurrent was filtered at 2 kHz. Changes in myoplasmic [Ca2+] are presented as Ca2+-dependent fluorescence transients, F, expressed as fractional changes above the base line, ΔF/F0 = (F − F0)/F0, where F0 is the prestimulus photocurrent.
Solutions
The standard Tyrode’s solution contained (mM): 150 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 MOPS, pH 7.4 with NaOH. The extracellular solution for voltage-clamp studies contained (mM): 140 TEA-Cl, 10 CsOH, 2 CaCl2, 1 MgCl2, 10 glucose, 10 MOPS, pH 7.4 with HCl. Channel blockers were added from stock solutions for a working concentration of 200 nM TTX, 20 µM nifedipine, and 200 µM 9-anthracene carboxylic acid. The internal solution contained (mM): 140 K-aspartate, 20 K-MOPS, 5 MgCl2, 5 Na2 creatine phosphate, 5 ATP-K2, 5 glucose, 5 reduced glutathione, 30 EGTA, and 15 mM Ca(OH)2 for a 30:15 EGTA:Ca2+ ratio equivalent to 97 nM free Ca2+ (see Ref. 13). The membrane impermeant Ca2+ indicator OGB-5N (Thermo Fisher) was added to the internal solution for a final concentration of 125 µM. All experiments were performed at room temperature (22°C).
Statistical Analysis
Data are presented as means ± SE in Tables or error bars in Figures. ANOVA was used to test for statistically different mean values (P < 0.05), followed by a post hoc Bonferroni correction to perform pairwise t test among the four genotypes (WT, heterozygous CaV1.1-R528H, homozygous CaV1.1-R528H, and homozygous NaV1.4-R669H).
RESULTS
Ca2+ Release by an Action Potential Is Reduced for CaV1.1-R528H but Not for NaV1.4-R669H HypoPP Fibers
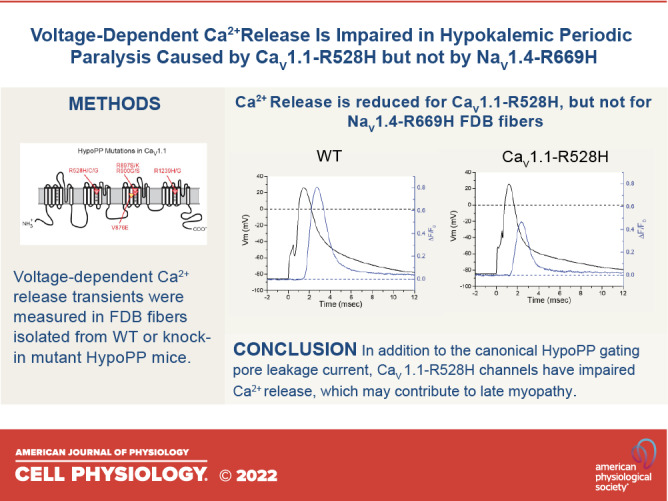
The resting potential of acutely dissociated FDB fibers is typically −40 mV to −50 mV (12), a range that is substantially depolarized from the value of −80 mV to −90 mV measured from intact whole muscle using sharp microelectrodes (14). Therefore, we applied a holding current to set Vm = −80 mV after fiber impalement with two microelectrodes. This paradigm also compensates for the mild depolarization of Vrest in HypoPP fibers (9, 11), so that action potentials were elicited from the same initial conditions for fibers from WT and mutant mice. Representative action potentials triggered by a 0.5 ms current pulse of 0.2–0.3 µA, and the corresponding ΔF/F0 for the Ca2+ transient, are shown for WT and homozygous CaV1.1-R528H fibers in Fig. 1A. The ΔF/F0 waveforms were similar, but the peak amplitude was consistently smaller for homozygous CaV1.1-R528H fibers compared with WT. A superposition of amplitude-normalized ΔF/F0 responses for WT, CaV1.4-R528H, and NaV1.4-R669H fibers (Fig. 1B) shows no perceptible difference in kinetics, which was confirmed quantitatively by measuring the rise times and decay time constants. The distribution of peak ΔF/F0 values recorded in FDB fibers from each of the mouse lines is shown in the box plot of Fig. 2. On average, the peak ΔF/F0 for homozygous CaV1.1-R528H was 66% of WT (0.48 ± 0.038, n = 12 fibers and 0.73 ± 0.028, n = 18 fibers; respectively, P < 0.0001 ANOVA). Heterozygous WT/CaV1.1-R528H fibers had an intermediate peak ΔF/F0 = 0.62 ± 0.047, n = 10 fibers that was not statistically different from WT. The peak ΔF/F0 for homozygous NaV1.4-R669H fibers (0.72 ± 0.074, n = 8 fibers) was identical to that of WT.
Figure 1.

Ca2+ release elicited by an action potential. A: each record shows the ΔF/F0 response for a single trial, in which an action potential was triggered in an isolated flexor digitorum brevis muscle (FDB) fiber by a 0.5 ms current pulse. These representative traces show a larger ΔF/F0 for wild type (WT) (left) compared with a fiber from a homozygous CaV1.1-R528H mouse (right). B: the kinetics of Ca2+ release was not altered by either hypokalemic periodic paralysis (HypoPP) mutation. Traces show the mean of amplitude-normalized ΔF/F0 responses that were aligned by the time of the peak (WT n = 8 fibers, CaV1.1 R528H n = 7 fibers, NaV1.4 R669H n = 5 fibers). Each trace has shaded edges, barely perceptible in this figure, that show the SE.
Figure 2.

Ca2+ release by an action potential is smaller for CaV1.1-R528H fibers than for wild type (WT). Data points in the box plots show the peak ΔF/F0 recorded for individual flexor digitorum brevis muscle (FDB) fibers. The sample mean (large circle and horizontal line) was lower for homozygous CaV1.1-R528H than WT (P < 0.001) or for heterozygous CaV1.1-R528H (P < 0.02), whereas the peak ΔF/F0 for homozygous NaV1.4-R669H fibers was not different from WT. Box dimensions show 25th and 75th percentiles, and the smooth curve is a Gaussian best fit.
To determine whether the smaller peak ΔF/F0 for homozygous CaV1.1-R528H fibers was possibly a consequence of a smaller amplitude action potential in HypoPP fibers, we generated a scatter plot of ΔF/F0 as a function of the ΔVm for the action potential for each fiber (Fig. 3). In the overlapping range of action potential amplitudes (100–115 mV), the peak ΔF/F0 was consistently smaller for homozygous CaV1.1-R528H fibers than for WT ones. A linear fit to those data shows that Ca2+ release has a comparable voltage sensitivity for WT and homozygous CaV1.1-R528H (despite a partial loss of charge in the DII voltage sensor), smaller ΔF/F0 with smaller ΔV, but the response for the latter is shifted downward to lower values.
Figure 3.

Reduced Ca2+ release for CaV1.1-R528H fibers is not because the action potential has a smaller amplitude. The ΔF/F0 is plotted as a function of ΔV for the corresponding action potential. Each point represents the response of a single trial from a different fiber. Lines show the least-squares fit for all the data from wild type (WT, black squared) or homozygous CaV1.1-R528H (red circles).
We previously reported a modest disruption of the coupling between voltage-sensor movement and channel opening for NaV1.4-R669H (15). This gating defect may result in a reduction of the peak Na+ current, and consequently smaller amplitude action potentials. Consistent with this prediction, ΔVm for action potentials elicited in NaV1.4-R669H fibers were smaller than for WT fiber (102 ± 2.3 mV n = 6 fibers; 108 ± 1.2 mV n = 13 fibers; respectively P < 0.05). However, this small reduction in ΔVm for the action potential was not associated with a reduction of the peak ΔF/F0 for NaV1.4-R669H (Fig. 2). Amplitudes for action potentials in CaV1.1-R528H fibers (homozygous and heterozygous) were not different from those in WT.
A Larger Holding Current Is Required for HypoPP Fibers, Consistent with a Gating Pore Leakage Current
The primary functional defect in HypoPP is the anomalous gating pore leak current, resulting from missense mutations for the S4 segment in voltage sensor domains of CaV1.1 (9, 16) or NaV1.4 (7, 8). The gating pore leak, although pathogenic, is only ∼2% of the resting fiber conductance (9, 11, 16, 17), and so the detection of this anomalous current by voltage-clamp is technically challenging. Consequently, the voltage-clamp strategy requires either blocking all of the ion channels (conventional pores) in a HypoPP muscle fiber or using the oocyte system to achieve high expression of the mutant channel. The current-clamp studies herein, provide a new opportunity to detect the presence and magnitude of an anomalous inward current in HypoPP fibers, consistent with the gating pore current. Importantly, this measurement was performed in a standard mammalian bath solution without ion channel blockers. The box plot in Fig. 4 shows the distribution of holding currents that were required to set Vm = −80 mV. On average, larger negative holding currents were required to hyperpolarize HypoPP fibers, homozygous for either CaV1.1-R528H or NaV1.4-R669H. As expected for heterozygous CaV1.1-R528H with half the gating pore current density, an intermediate holding current was required, between WT and homozygous HypoPP. Moreover, the magnitude of the additional negative holding current (about −10 µA/cm2) is consistent with the reported conductance (10–20 µS/cm2) and reversal potential (−20 mV) for the anomalous gating pore current in murine HypoPP fibers (9, 11).
Figure 4.

The holding current required to maintain the fiber Vm = −80 mV is larger (more negative) for hypokalemic periodic paralysis (HypoPP) fibers than for wild type (WT). Each point in the box plot represents the original holding current required to set Vm to −80 mV for an individual fiber. The response from a fiber was included in the dataset if the measured voltage several minutes later was in the range −88 to −78 mV. Large circles and horizontal lines show the mean, the box indicates the 25% and 75% range, and the smoother curves are the Gaussian fit.
Voltage Dependence of Ca2+ Release Is Not Altered by the HypoPP Mutations
The voltage dependence of Ca2+ release was assessed by measuring ΔF/F0 in voltage-clamped FDB fibers. Figure 5 shows a set of responses observed for series of voltage steps from −40 mV to +80 mV, from a holding potential of −80 mV. The first detectable change in ΔF/F0 was at −30 mV (blue traces) for both WT and homozygous R528H fibers. The ΔF/F0 saturated for voltage steps > +30 mV, and this level was larger for WT than for homozygous CaV1.1-R529H as shown by the representative traces in Fig. 5. The peak ΔF/F0 is shown as a function of voltage in Fig. 6A. The maximum ΔF/F0, determined by fitting a Boltzmann function to the responses for each fiber over the voltage range from −40 mV to +40 mV, was 76% of WT for homozygous CaV1.1-R528H fibers (0.71 ± 0.026, n = 16 fibers and 0.53 ± 0.028, n = 7 fibers respectively; P = 0.01 ANOVA). The maximum ΔF/F0 for heterozygous CaV1.1-R528H was 0.60 ± 0.062, n = 9 fibers, a value that lies between WT and homozygous CaV1.1-R528H but is not statistically different from either (P > 0.05 ANOVA).
Figure 5.

Ca2+ release transients in response to a series of step depolarizations for voltage-clamped flexor digitorum brevis muscle (FDB) fibers. Each sweep is for a single trial (no averaging), and the responses are superimposed for voltage steps over the range −40 to +80 mV. Representative responses show the peak ΔF/F0 was larger for wild type (WT; A) than for homozygous CaV1.1-R528H (B). The traces in the lower panels are the measured Vm, not the digital command pulses.
Figure 6.

Voltage-dependent Ca2+ release is reduced for homozygous CaV1.1-R528H fibers compared with wild type (WT) fibers. A: the peak ΔF/F0, averaged from measurements in several fibers, is shown as a function of step potential. B: amplitude-normalized ΔF/F0 shows a comparable voltage-dependence of Ca2+ release in fibers from WT and either hypokalemic periodic paralysis (HypoPP) mutant line. Symbols show means ± SE.
A comparison for the voltage dependence of ΔF/F0 is illustrated with the amplitude-normalized data in Fig. 6B. Although some variability was apparent for the voltage of the midpoint, none of the differences in V1/2 among the four genotypes were statistically distinguishable. The steepness of the voltage dependence of ΔF/F0 was also identical, despite the partial loss of gating charge for CaV1.1-R528H mutant channels. Parameter values for the Boltzmann fits (smooth lines, Fig. 6B) are shown in Table 1.
Table 1.
Parameters for Boltzmann fit of ΔF(V)/F0
| Genotype | (ΔF/F0)max | V1/2 (mV) | k (mV) | n (fibers) |
|---|---|---|---|---|
| WT | 0.71 ± 0.026 | −13 ± 2.7 | 8.1 ± 0.43 | 16 |
| het CaV1.1-R528H | 0.60 ± 0.061 | −8.2 ± 1.4 | 8.7 ± 0.49 | 9 |
| homo CaV1.1-R528H | 0.53 ± 0.028† | −18 ± 2.3 | 8.8 ± 0.56 | 7 |
| homo NaV1.4-R669H | 0.79 ± 0.098 | −11 ± 1.7 | 8.1 ± 0.92 | 5 |
P = 0.01. WT, wild type.
DISCUSSION
The prevailing view for the pathogenesis of recurrent episodes of weakness in HypoPP is that an aberrant sustained depolarization of Vrest inactivates Na+ channels, resulting in severe attenuation or even failure of action potential propagation (1). Although this reduction of excitability that drives EC-coupling is sufficient to explain the impaired generation of contractile force, a possible contribution from altered voltage sensing or disrupted coupling to activation of ryanodine receptor 1 (RyR1) and Ca2+-release remained an open question (10, 18). The data herein show impaired Ca2+ release for CaV1.1-R528H but not for the NaV1.4 HypoPP mutation R669H. The S4 segment in the domain II voltage sensor, containing R528H, was recently shown to translocate rapidly in response to membrane depolarization (τ = 3 ms) consistent with a role in EC-coupling, as opposed to the slow translocation of the domain I S4 segment (τ = 100 ms) that occurs with the same time course as channel opening (19). Indeed, the HypoPP mutations of CACNA1S are all located in voltage sensor domains (II–IV), and nine out of 10 are missense substitutions at positively charged arginine residues in S4 segments (20). This canonical pattern of HypoPP mutation sites has a common functional correlate. An anomalous “gating pore” leakage current, that can account for the paradoxical depolarization of Vrest in low K+, has been reported for seven HypoPP-CaV1.1 mutations (17, 21–23). On the other hand, no gating pore current was detected for two mutations (R897K and R900S in IIIS4), which raises the possibility of other mechanisms for the intermittent weakness (22, 24).
The assessment of Ca2+-release integrity in HypoPP has previously been inadequate because of the challenges in obtaining muscle biopsies for this very rare disorder or for limited availability of suitable model systems. Only a single study of Ca2+-release in human HypoPP muscle has been reported (10), and these fura-2 measurements in voltage-clamped CaV1.1-R528H myotubes showed no difference in the midpoint or the steepness for the voltage-dependence of Ca2+-release. Amplitude-normalized responses were shown and no comment was made about the relative amplitude for HypoPP versus WT. In this same study, expression of CaV1.1-R528H in CaV1.1-null myotubes (GLT cells immortalized from the mdg mouse) also failed to reveal any differences in fura-2 transients compared with WT.
The Reduced Ca2+-Release in CaV1.1-R528H Fibers Is Likely Because of Lower Functional Channel Density
Our measurements of Ca2+ transients using the low-affinity fast dye OGB-5N show a 24% to 34% reduction in the peak ΔF/F0 elicited by a voltage pulse or by an action potential, respectively, for homozygous CaV1.1-R528H fibers. This value is comparable with the 36% reduction in maximal gating charge displacement, Qmax, that we previously reported (9). Recordings from heterozygous CaV1.1-R528H fibers show a milder reduction of peak ΔF/F0 (Figs. 2 and 6) and of Qmax (9), consistent with a dosage effect of the mutant allele. The difference in Ca2+ release is not likely to be caused by changes in the SR Ca2+ gradient because we previously showed SR Ca2+ content is not altered in CaV1.1-R528H fibers [no difference in 4-chloro-m-cresol (4-CMC) release (9)] and in the present experiments the myoplasmic-free Ca2+ is strongly buffered at ∼100 nM. Taken together, these data suggest that a reduced density of functional channels at the plasma membrane produces the reduction of peak Ca2+-release in CaV1.1-R528H fibers. This interpretation also supports the notion that CaV1.1-R528H channels are functionally equivalent to WT channels with regard to effectiveness for EC-coupling.
We previously reported a more pronounced reduction by 67% for the peak ΔF/F0 measured with Fluo-4 in homozygous CaV1.1-R528H fibers in whole cell recording mode (9). That study did not use the stringent criteria for fiber quality that were applied herein (see materials and methods). It is very likely that the population of fibers in our prior study included ones with structurally disrupted triads, as shown histologically by transverse tubular aggregates and vacuoles with dilated SR demonstrated on ultrastructural studies with transmission EM (9). We applied more stringent optical and electrical criteria for fiber integrity herein, in an effort to specifically assess the intrinsic capability of CaV1.1-R528H channels for EC-coupling; rather than detecting the apparent effectiveness of EC-coupling in HypoPP muscle that may be compromised by a combination of altered CaV1.1 function and structurally disrupted triads. The absence of a detectable impairment of Ca2+-release for NaV1.4-R669H also supports the notion that a primary disruption of EC-coupling is not a mechanism shared in common for HypoPP mutations in the pathogenesis of episodic weakness.
In contrast to our findings earlier, Jurkat-Rott et al. (10) concluded Ca2+-release “was not grossly altered” as reported by fura-2 measurements in human fibers heterozygous for CaV1.1-R528H or for GLT cells expressing a uniform population of R528H HypoPP mutant channels. There are several plausible explanations for this discrepancy: 1) the reduction detected with the fast dyes (OGB-5N, Fluo-4) may not be apparent with the slower high-affinity fura-2; 2) the signal-to-noise was much less favorable in the fura-2 study, which was further exacerbated by the heterozygous state for the human myotubes; 3) the relative amplitude of ΔF/F for R528H mutant compared with WT was not reported in the fura-2 study, whereas both studies are concordant for no differences in V1/2 or k for the voltage dependence; and 4) the lower membrane expression of CaV1.1-R528H that we detected in our HypoPP mouse model may not be a feature of human HypoPP muscle or of heterologous expression in GLT cells. Nevertheless, both studies agree that CaV1.1-R528H does not disrupt the ability of the channel to participate in EC-coupling.
Clinical Implications of Reduced Ca2+-Release in CaV1.1-R528H HypoPP Muscle Fibers
The confluence of HypoPP mutations, almost all of which are in S4 segments of voltage sensors, raised questions about whether disrupted EC-coupling contributes to the acute attacks of weakness or to the late-onset permanent weakness. Although a reduction of Ca2+-release is clearly present in our CaV1.1-R528H mouse model of HypoPP, our overall impression is that a primary defect of EC-coupling is not a substantive contributing factor to the transient attacks of weakness. First, a “static” impairment of EC-coupling is more likely to act as a modifying effect for the severity of an episode of weakness rather than contributing to the occurrence of an ictal event. Second, the decrease in peak Ca2+-release after an action potential is only 24% in heterozygous CaV1.1-R528H fibers, which is modest in comparison to the safety-factor of Ca2+ release for muscle contraction (25, 26). This interpretation is consistent with the clinical observation that muscle strength is normal between episodic attacks of HypoPP (1, 2). Third, the clinical severity of acute episodes of weakness is equivalent for HypoPP from NaV1.4 mutations that are not expected to affect Ca2+-release, as now confirmed herein experimentally. Fourth, the electromyogram consistently shows a severe impairment of muscle excitability during an acute attack in HypoPP (27, 28), which is sufficient to produce the severe loss of force. The available data therefore support the view that the transient episodes of weakness in HypoPP are not a consequence of an intrinsic defect of EC-coupling.
The reduced Ca2+-release for CaV1.1-associated HypoPP may contribute to the susceptibility to permanent muscle weakness with aging. In a cohort of 36 patients with HypoPP, permanent weakness was more pronounced for patients with CaV1.1 mutations than with NaV1.4 mutations (16), consistent with the notion that impaired Ca2+-release contributes to permanent weakness. Moreover, studies in WT mice show an age-dependent decline in voltage-dependent Ca2+-release (50%) with an associated 35% decrease in maximal specific force of a tetanic contraction (29). These changes in WT muscle are caused by an age-dependent decrease of functional CaV1.1 channels in the plasma membrane (30). We propose the reduced Ca2+-release for CaV1.1-associated HypoPP and the decrease of functional CaV1.1 channels with normal aging will synergistically increase the risk of permanent muscle weakness in HypoPP. This hypothesis also explains why patients with NaV1.4 mutations are less susceptible to permanent muscle weakness, because the channel defect does not reduce Ca2+-release. Thus far, studies of Ca2+-release in CaV1.1-associated HypoPP are limited to R528H. Permanent muscle weakness is more severe and has an earlier age of onset for patients with the CaV1.1-R1239H mutation (16). The prediction is Ca2+-release may be more severely impaired for R1239H than for R528H, which warrants additional studies of this and other CaV1.1-associated HypoPP mutations.
GRANTS
This work was supported by Grants AR063182 and AR078198 from the National Institute of Arthritis, Musculoskeletal, and Skin Diseases of the National Institutes of Health (NIH).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.D. and S.C.C. conceived and designed research; M.D. performed experiments; S.C.C. analyzed data; M.D. and S.C.C. interpreted results of experiments; S.C.C. prepared figures; S.C.C. drafted manuscript; M.D. and S.C.C. edited and revised manuscript; M.D. and S.C.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Marbella Quiñonez for maintaining the mouse colonies.
This article is part of the special collection “Musculoskeletal Biology and Bioengineering.” Dr. Frank Zaucke, Dr. Thomas Hawke, and Dr. Liliana Schaefer, served as Guest Editors of this collection.
REFERENCES
- 1.Cannon SC. Channelopathies of skeletal muscle excitability. Compr Physiol 5: 761–790, 2015. doi: 10.1002/cphy.c140062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehmann-Horn F, Rüdel R, Jurkat-Rott K. Nondystrophic myotonias and periodic paralyses. In: Myology, edited by Engel AG, Franzini-Armstrong C.. New York: McGraw-Hill, 2004, p. 1257–1300. [Google Scholar]
- 3.Ptacek LJ, Tawil R, Griggs RC, Engel AG, Layzer RB, Kwiecinski H, McManis PG, Santiago L, Moore M, Fouad G, Bradley P, Leppert MF. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 77: 863–868, 1994. doi: 10.1016/0092-8674(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 4.Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, Tollar LL, Ebers GC. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 53: 1932–1936, 1999. doi: 10.1212/wnl.53.9.1932. [DOI] [PubMed] [Google Scholar]
- 5.Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, Meola G, Schorge S, Kullmann DM, Davis MB, Hanna MG. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 72: 1544–1547, 2009. doi: 10.1212/01.wnl.0000342387.65477.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cannon SC. Sodium channelopathies of skeletal muscle. Handb Exp Pharmacol 246: 309–330, 2018. doi: 10.1007/164_2017_52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature 446: 76–78, 2007. doi: 10.1038/nature05598. [DOI] [PubMed] [Google Scholar]
- 8.Struyk AF, Cannon SC. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol 130: 11–20, 2007. doi: 10.1085/jgp.200709755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu F, Mi W, Hernandez-Ochoa EO, Burns DK, Fu Y, Gray HF, Struyk AF, Schneider MF, Cannon SC. A calcium channel mutant mouse model of hypokalemic periodic paralysis. J Clin Invest 122: 4580–4591, 2012. doi: 10.1172/JCI66091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurkat-Rott K, Uetz U, Pika-Hartlaub U, Powell J, Fontaine B, Melzer W, Lehmann-Horn F. Calcium currents and transients of native and heterologously expressed mutant skeletal muscle DHP receptor alpha1 subunits (R528H). FEBS Lett 423: 198–204, 1998. doi: 10.1016/s0014-5793(98)00090-8. [DOI] [PubMed] [Google Scholar]
- 11.Wu F, Mi W, Burns DK, Fu Y, Gray HF, Struyk AF, Cannon SC. A sodium channel knockin mutant (NaV1.4-R669H) mouse model of hypokalemic periodic paralysis. J Clin Invest 121: 4082–4094, 2011. doi: 10.1172/JCI57398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woods CE, Novo D, DiFranco M, Vergara JL. The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J Physiol 557: 59–75, 2004. doi: 10.1113/jphysiol.2004.061291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol 99: 1–26, 2010. doi: 10.1016/B978-0-12-374841-6.00001-3. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Nawaz M, DuPont C, Myers JH, Burke SR, Bannister RA, Foy BD, Voss AA, Rich MM. The role of action potential changes in depolarization-induced failure of excitation contraction coupling in mouse skeletal muscle. eLife 11: e71588, 2022. doi: 10.7554/eLife.71588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mi W, Rybalchenko V, Cannon SC. Disrupted coupling of gating charge displacement to Na+ current activation for DIIS4 mutations in hypokalemic periodic paralysis. J Gen Physiol 144: 137–145, 2014. doi: 10.1085/jgp.201411199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jurkat-Rott K, Weber MA, Fauler M, Guo XH, Holzherr BD, Paczulla A, Nordsborg N, Joechle W, Lehmann-Horn F. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci USA 106: 4036–4041, 2009. doi: 10.1073/pnas.0811277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuster C, Perrot J, Berthier C, Jacquemond V, Charnet P, Allard B. Na leak with gating pore properties in hypokalemic periodic paralysis V876E mutant muscle Ca channel. J Gen Physiol 149: 1139–1148, 2017. doi: 10.1085/jgp.201711834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev 79: 1317–1372, 1999. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- 19.Savalli N, Angelini M, Steccanella F, Wier J, Wu F, Quinonez M, DiFranco M, Neely A, Cannon SC, Olcese R. The distinct role of the four voltage sensors of the skeletal CaV1.1 channel in voltage-dependent activation. J Gen Physiol 153: e202112915, 2021. doi: 10.1085/jgp.202112915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannon SC. Voltage-sensor mutations in channelopathies of skeletal muscle. J Physiol 588: 1887–1895, 2010. doi: 10.1113/jphysiol.2010.186874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu F, Quinonez M, DiFranco M, Cannon SC. Stac3 enhances expression of human CaV1.1 in Xenopus oocytes and reveals gating pore currents in HypoPP mutant channels. J Gen Physiol 150: 475–489, 2018. doi: 10.1085/jgp.201711962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu F, Quinonez M, Sc. C. Gating pore currents occur in CaV1.1 domain III mutants associated with HypoPP. J Gen Physiol 153: e202112946, 2021. doi: 10.1085/jgp.202112946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuster C, Perrot J, Berthier C, Jacquemond V, Allard B. Elevated resting H+ current in the R1239H type 1 hypokalaemic periodic paralysis mutated Ca2+ channel. J Physiol 595: 6417–6428, 2017. doi: 10.1113/JP274638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kubota T, Wu F, Vicart S, Nakaza M, Sternberg D, Watanabe D, Furuta M, Kokunai Y, Abe T, Kokubun N, Fontaine B, Cannon SC, Takahashi MP. Hypokalaemic periodic paralysis with a charge-retaining substitution in the voltage sensor. Brain Commun 2: fcaa103, 2020. doi: 10.1093/braincomms/fcaa103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westerblad H, Allen DG. Changes of myoplasmic calcium concentration during fatigue in single mouse muscle fibers. J Gen Physiol 98: 615–635, 1991. doi: 10.1085/jgp.98.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashley CC, Mulligan IP, Lea TJ. Ca2+ and activation mechanisms in skeletal muscle. Q Rev Biophys 24: 1–73, 1991. doi: 10.1017/s0033583500003267. [DOI] [PubMed] [Google Scholar]
- 27.Engel AG, Lambert EH, Rosevear JW, Tauxe WN. Clinical and electromyographic studies in a patient with primary hypokalemic periodic paralysis. Am J Med 38: 626–640, 1965. doi: 10.1016/0002-9343(65)90138-5. [DOI] [PubMed] [Google Scholar]
- 28.Links TP, van der Hoeven JH, Zwarts MJ. Surface EMG and muscle fibre conduction during attacks of hypokalaemic periodic paralysis. J Neurol Neurosurg Psychiatry 57: 632–634, 1994. doi: 10.1136/jnnp.57.5.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jimenez-Moreno R, Wang ZM, Gerring RC, Delbono O. Sarcoplasmic reticulum Ca2+ release declines in muscle fibers from aging mice. Biophys J 94: 3178–3188, 2008. doi: 10.1529/biophysj.107.118786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang ZM, Messi ML, Delbono O. L-Type Ca2+ channel charge movement and intracellular Ca2+ in skeletal muscle fibers from aging mice. Biophys J 78: 1947–1954, 2000. doi: 10.1016/S0006-3495(00)76742-7. [DOI] [PMC free article] [PubMed] [Google Scholar]