

Abstract
Human embryonic stem cells demonstrate a unique ability to respond to morphogens in vitro by self-organizing patterns of cell fate specification that correspond to primary germ layer formation during embryogenesis. Thus, these cells represent a powerful tool with which to examine the mechanisms that drive early human development. We have developed a method to culture human embryonic stem cells in confined colonies on compliant substrates that provides control over both the geometry of the colonies and their mechanical environment in order to recapitulate the physical parameters that underlie embryogenesis. The key feature of this method is the ability to generate polyacrylamide hydrogels with defined patterns of extracellular matrix ligand at the surface to promote cell attachment. This is achieved by fabricating stencils with the desired geometric patterns, using these stencils to create patterns of extracellular matrix ligand on glass coverslips, and transferring these patterns to polyacrylamide hydrogels during polymerization. This method is also compatible with traction force microscopy, allowing the user to measure and map the distribution of cell-generated forces within the confined colonies. In combination with standard biochemical assays, these measurements can be used to examine the role mechanical cues play in fate specification and morphogenesis during early human development.
Keywords: Bioengineering, Issue 151, human embryonic stem cells (hESCs), polyacrylamide hydrogels, traction force microscopy (TFM), photolithography, cell patterning, extracellular matrix (ECM), tissue engineering, regenerative medicine
Introduction
Human embryonic stem cells (hESCs) hold great promise for use in regenerative medicine and tissue engineering applications. The pluripotent nature of these cells gives them the ability to differentiate into any adult cell type. While great strides have been made in directing the fate of hESCs to particular cell types, it has remained very difficult to generate whole tissues or organs de novo1,2,3,4,5. This is due, in large part, to a limited understanding of the mechanisms that drive the formation of these tissues during human development. In order to fill this gap in knowledge, a number of methods have emerged in recent years to model the early embryo and subsequent stages of development with embryonic stem cells6,7,8,9,10,11,12,13.
Shortly after the derivation of the first hESC lines14, it was demonstrated that embryoid bodies formed from hESCs were capable of spontaneously producing cells of the three primary germ layers6. However, due to the inherent lack of control over the size and morphology of embryoid bodies, the organization of germ layers varied significantly and failed to match the organization of the early embryo. More recently, Warmflash et al. developed a method to confine colonies of hESCs on glass substrates via micropatterning, providing control and consistency over the size and geometry of the colonies8. In the presence of BMP4, an important morphogen in early development, these confined colonies were capable of self-organizing reproducible patterns of specification to fates representing the primary germ layers. Although this provided a useful model for studying the mechanisms by which primary germ layers are established, the patterns of fate specification did not precisely match the organization and morphogenesis observed during embryogenesis15. A more faithful recapitulation of early embryonic development was achieved by embedding hESCs in a three-dimensional extracellular matrix (ECM) of matrigel11, providing the strongest evidence to date for the ability of hESCs to self-organize and model the early stages of embryogenesis ex vivo. However, this method yields inconsistent results and is thus incompatible with a number of assays that could be used to reveal the underlying mechanisms of self-organization and fate specification.
Given these existing methods and their respective limitations, we sought to develop a method for reproducibly culturing hESC colonies of defined geometries in conditions that model the extracellular environment of the early embryo. To achieve this, we used polyacrylamide hydrogels of tunable elasticity to control the mechanical properties of the substrate. Using atomic force microscopy on gastrulation-stage chicken embryos, we found that the elasticity of the epiblast ranged from hundreds of pascals to a few kilopascals. Thus, we focused on generating polyacrylamide hydrogels with elasticity in this range to serve as the substrate for hESC colonies. We modified our previous methods for culturing hESCs on polyacrylamide hydrogels7,9 to provide robust control over the geometry of the colonies. We achieved this by first patterning ECM ligands, namely matrigel, onto glass coverslips through microfabricated stencils, as previously reported16. We then designed a novel technique to transfer the patterned ligand to the surface of polyacrylamide hydrogels during polymerization. The method we describe here involves using photolithography to fabricate a silicon wafer with the desired geometric patterns, creating stamps of theses geometric features with polydimethylsiloxane (PDMS), and using these stamps to generate the stencils that ultimately allow patterning of ligand onto the surface of glass coverslips and transfer to polyacrylamide.
In addition to recapitulating the mechanical environment of the early embryo, confining hESC colonies on polyacrylamide enables the measurement of cell-generated forces with traction force microscopy (TFM), as reported in our previous method9. In brief, fluorescent beads can be embedded in the polyacrylamide and used as fiducial markers. Cell-generated forces are calculated by imaging the displacement of these beads after seeding hESCs onto the patterned substrate. Furthermore, the resulting traction force maps can be combined with traditional assays, such as immunostaining, to examine how the distribution of cell-generated forces in confined hESC colonies may regulate or modulate downstream signaling. We expect these methods will reveal that mechanical forces play a critical role in the patterning of cell fate specification during early embryonic development that is currently overlooked.
Protocol
All methods described here pertaining to the use of hESCs have been approved by the Human Gamete, Embryo and Stem Cell Research (GESCR) Committee at the University of California San Francisco.
1. Preparation of silicon wafer with geometric features
-
Create a photomask with desired geometric features. Use computer-aided drafting software to design the features. For use with negative photoresist, make features opaque and the remainder of the mask transparent.
NOTE: For patterning onto 18 mm diameter coverslips, group features for each experimental condition into 10 × 10 mm areas to ensure the stencils generated in Step 3 fit onto the coverslips.
-
Spin coat negative photoresist onto a 100 mm silicon wafer. Use the photoresist data sheet to determine the speed and length of spin to generate a film thickness of 100–250 μm.
NOTE: The film thickness should be adjusted such that the aspect ratio of the width of the patterns to the height of the stencil is as close to 1:1 as possible. However, we recommend a minimum thickness of 100 μm, as anything less produces delicate stencils that easily tear.
-
Process the photoresist according to the product data sheet. This typically involves a soft bake, UV exposure with photomask in a mask aligner, post exposure bake, development, and hard bake. As an example, the procedure used to process the wafers used in this protocol is outlined in Table 1.
NOTE: Be cautious when handling silicon wafers as they are fragile. Once generated, wafers can be used repeatedly as long as they are not damaged.
Table 1: Example SU8 wafer fabrication protocol.
Silicon wafers used to generate PDMS stamps and subsequent stencils were fabricated using the steps outlined in this table. This protocol was generated using the SU8 3000 data sheet with the aim of creating a film thickness of approximately 100–250 μm.
| Example SU8 Wafer Fabrication | |
|---|---|
| 1. Spin coat wafer with SU8–3050 | Spin at 1,000 RPM for 30 s |
| 2. Soft bake wafer on hot plate | i. 3 min at 65 °C |
| ii. 45 min at 95 °C | |
| iii. 3 min at 65 °C | |
| iv. Cool to room temperature | |
| 3. Expose in mask aligner | i.Align wafer and photomask in mask aligner |
| ii.Expose with energy of 250 mJ/cm2 | |
| • E.g. for lamp with intensity of 11 mW/cm2, expose for 23 s | |
| 4. Post exposure bake on hot plate | i. 1 min at 65 °C |
| ii.15 min at 95 °C | |
| iii. 1 min at 65 °C | |
| iv. Cool to room temperature | |
| 5. Develop | i. Agitate in SU8 Developer, approx. 5–10 min |
| ii.Check development with isopropyl alcohol (IPA) | |
| • If under-developed, IPA rinse will produce a white residue | |
| 6. Hard bake on hot plate (optional) | 1–2 h at 150 °C |
2. Preparation of coverslips
1. Preparation of acid-washed “top” coverslips
Place 18 mm diameter #1 thickness coverslips into a glass Petri dish. The appropriate number of coverslips depends on the size of the dish. For a 100 mm dish, prepare 50–100 coverslips at a time. Volume amounts given through the remainder of this section correspond to a 100 mm dish.
-
Add 20 mL of 1 M HCl to the Petri dish and gently shake the dish to disperse the coverslips. Ensure all the coverslips are submerged and release air bubbles from between the coverslips. Incubate overnight at room temperature with gentle shaking.
CAUTION: HCl is acidic and corrosive. Use caution and wear appropriate PPE while working with HCl.
Decant HCl from the dish. Add 20 mL of ultrapure water and wash with gentle shaking for 10 min. Discard water and repeat for a total of five washes.
After discarding the final wash, add 20 mL of 100% ethanol to the dish. Using forceps, remove the coverslips individually and arrange between two pieces of filter paper to dry.
Store dried coverslips in a sealed container. Acid-washed coverslips can be prepared in advance and stored for a month or longer in dust-free conditions.
2. Preparation of glutaraldehyde-activated “bottom” coverslips
NOTE: Refer to previous methods for additional details7,9.
Place 18 mm diameter #1 thickness coverslips into a Petri dish.
Add 20 mL of 0.2 M HCl to the petri dish and gently shake the dish to disperse the coverslips. Ensure all the coverslips are submerged and release air bubbles from between the coverslips. Incubate overnight at room temperature with gentle shaking.
Decant HCl from the dish. Add 20 mL of ultrapure water and wash with gentle shaking for 10 min. Discard water and repeat for a total of five washes.
After discarding the final wash, add 20 mL of 0.1 M NaOH and shake gently to disperse and submerge coverslips. Incubate at room temperature with gentle shaking for 1 h.
Decant NaOH from the dish. Add 20 mL of ultrapure water and wash with gentle shaking for 10 min. Discard water and repeat for a total of five washes.
-
After discarding the final wash, add 20 mL of a 1:200 dilution of 3-aminopropyltrimethoxysilane in ultrapure water and shake gently to disperse and submerge coverslips. Incubate at room temperature with gentle shaking for 1 h or overnight.
CAUTION: 3-aminopropyltrimethoxysilane is flammable and can cause skin irritation. Handle with care, wear appropriate PPE, and discard waste according to local disposal regulations.
Decant the diluted 3-aminopropyltrimethoxysilane solution from the dish. Add 20 mL of ultrapure water and wash with gentle shaking for 10 min. Discard water and repeat for a total of at least five washes. It is critical to remove all 3-aminopropyltrimethoxysilane before proceeding.
-
After discarding the final wash, add 20 mL of a 1:140 dilution of 70% glutaraldehyde in phosphate buffered saline (PBS) and shake gently to disperse and submerge coverslips. Incubate at room temperature with gentle shaking for 1 h or overnight.
CAUTION: 70% glutaraldehyde is toxic and can cause skin irritation. Handle with care, wear appropriate PPE, and discard waste according to local disposal regulations.
Decant the diluted glutaraldehyde solution from the dish. Add 20 mL of ultrapure water and wash with gentle shaking for 10 min. Discard water and repeat for a total of five washes.
After discarding the final wash, add 20 mL of 100% ethanol. Using forceps, remove the coverslips individually and arrange between two pieces of filter paper to dry.
Store dried coverslips in a sealed container. Glutaraldehyde-activated coverslips can be prepared in advance and stored for up to six months.
3. Generation of stencils for patterning ECM ligand
1. Generating polydimethylsiloxane (PDMS) intermediate stamps
-
Mix PDMS base with PDMS curing agent at a 10:1 ratio, by weight. Mix thoroughly.
NOTE: For the initial application of PDMS, prepare approximately 100 g of PDMS to fill a 100 mm dish. For subsequent applications, remove PDMS from only the center of the dish where the silicon wafer features are located and prepare 20–30 g of new PDMS.
Degas the PDMS mixture in a desiccator for 30–60 min or until all air bubbles are removed.
Place the modified silicon wafer from Step 1 into a plastic 100 mm dish. Slowly and evenly pour the PDMS mixture over the surface of the wafer. Tap the dish on the work surface to release any air bubbles from the surface of the wafer.
Degas the PDMS mixture poured over the wafer for 10 min or until all air bubbles are removed or have come to the surface of the PDMS.
Bake the PDMS at 70 °C for 2 h. Allow to cool to room temperature.
Using a scalpel or box cutter, cut out the section of PDMS from the center of the dish that contains the geometric features.
Cut the PDMS into roughly 10 × 10 mm squares, each containing the features for a single experimental condition. These are referred to as “stamps” in subsequent steps of the protocol.
2. Generating flat slabs of PDMS
Mix PDMS base with PDMS curing agent at an 8:1 ratio, by weight. Prepare approximately 20 g for each 100 mm dish to be used. Mix thoroughly.
Degas the PDMS mixture in a desiccator for 30–60 min or until all air bubbles are removed.
Slowly and evenly pour the PDMS into a clean 100 mm dish. Tap the dish on the work surface to release any air bubbles.
Bake the PDMS at 70 °C for 2 h. Allow to cool to room temperature.
Remove the PDMS from the dish. Invert such that the surface of the PDMS that was in contact with the bottom of the dish is facing up.
For each stamp generated in Step 3.1, cut a 15 × 15 mm square of PDMS. These are referred to as “flat slabs” in subsequent steps of the protocol.
3. Generating stencils
Arrange flat slabs of PDMS on the lid of a 150 mm dish, or other flat surface for easier handling. Ensure sufficient spacing between slabs (e.g., for a 150 mm dish, arrange nine flat slabs with even spacing).
Invert each stamp generated in Step 3.1 onto a flat slab of PDMS, such that the features on the stamp are in contact with the flat slab of PDMS. Gently press on the top of the stamp with forceps to ensure even contact.
Carefully prop the lid holding PDMS stamp/slab pairs up on the base of the dish, such that the lid rests at an angle. This assists the wicking of the UV-curable polymer in the next step.
-
Place a small drop of UV-curable polymer at the top interface of each stamp/slab pair. The polymer will be wicked between the two by surface tension, assisted by gravity.
NOTE: Many different UV-curable polymers may work in this protocol. We selected Norland Optical Adhesive 74 for the following properties: i) Fast curing upon exposure to UV light, ii) Easy removal from PDMS stamps upon curing, iii) Robust adhesion to glass coverslips with manual pressure.
Once the polymer has been completely wicked through, place the lid with stamp/slab pairs flat on the work surface. Place a small drop of UV-curable polymer at each of the remaining three sides of each stamp/slab interface.
-
Using a 200 μL pipette tip, connect the drops of polymer around the edges of the stamp/slab interface. This creates a border that will hold ligand solution in Step 4.
NOTE: Be careful when working the polymer around the edges of the stamp. If the stamp/slab interface is disrupted, the polymer will wick underneath the features and the stencil will not form properly.
Carefully place the stamp/slab pairs into a UV-sterilization box. Use the sterile “Str” power setting and expose for 10 min.
Remove the stamp/slab pairs from the UV-sterilization box. Using two pairs of forceps, gently remove the PDMS stamp while holding down the flat slab and stencil that formed from the UV-curable polymer.
-
Using forceps, carefully remove the stencil and invert it such that the surface that was in contact with the flat slab of PDMS is facing up.
NOTE: The stencils are delicate. Be careful while handing them, especially while initially removing them from the PDMS, so that they do not tear.
Place the inverted stencils back into a UV-sterilization box. Use the sterile “Str” power setting and expose for 3 min.
4. Patterning ECM ligand on coverslips
1. Pressing stencils onto acid-washed coverslips
Place one acid-washed coverslip for each stencil onto a clean piece of laboratory film.
Using forceps, carefully place each stencil, flat-side-down, onto an acid-washed coverslip. Place such that features are centered on the coverslip.
-
Place a small piece of laboratory film on top of each stencil. Firmly and evenly press down on the stencil to create strong contact between the stencil and the coverslip.
NOTE: This is a critical step! Press firmly and across the entire surface of the stencil. If sufficient contact is not created, the ligand solution will leak between the stencil and the coverslips and the patterning will fail. OPTIONAL: To confirm sufficient contact between the stencil and the coverslip, pipette 100 μL of ultrapure sterile water onto the surface of each stencil and incubate at room temperature. After 1 h, check for leaking. Aspirate water from successfully bonded stencils and proceed.
2. Plasma cleaning the surface of the stenciled coverslips to increase hydrophilicity
Place the coverslips with stencils into a plasma cleaner. Apply high power plasma for 30 s.
3. Preparation of ECM ligand solution
NOTE: All subsequent steps should be completed in sterile conditions, if possible.
- Place sterile 100 mM HEPES, 100 mM NaCl, pH 8.0 solution on ice. Once ice cold, add matrigel and collagen to achieve concentrations of 225 μg/mL and 25 μg/mL, respectively. OPTIONAL: For troubleshooting or for ligand visualization, include a fluorescently tagged ligand in the ligand solution.
- NOTE: Different ECM ligands can be used with this method. For different ligands, additional optimization may be required to determine ideal concentration, incubation time, and incubation temperature. See discussion section for more information.
Pipette the ligand solution onto the surface of each stencil. For stencils made from 10 × 10 mm square stamps, apply 100 μL per stencil.
-
Check for air bubbles in stencil features. If necessary, use fine-tipped forceps or a 2 μL pipette tip to remove air bubbles from features.
NOTE: This is a critical step! If air bubbles remain trapped at the surface of the coverslip, the ligand will not adsorb to the surface and the resulting pattern will be incomplete.
Place the stenciled coverslips with ligand into a dish, wrap with laboratory film, and incubate at 4 °C overnight.
5. Transfer of ligand to polyacrylamide gel
1. Removing ligand solution and stencil from each coverslip
Aspirate the ligand solution from the surface of a stencil. Ligand solution can be collected and stored at 4 °C and reused for up to a month.
Using forceps, briefly submerge the coverslip with stencil into a dish containing sterile PBS to wash.
Briefly submerge the coverslip with stencil into a second dish of sterile PBS to wash.
Remove the stencil from the surface of the coverslip. Be careful to not break the coverslip.
Briefly submerge the coverslip into a dish containing ultrapure sterile water to remove salts from the PBS washes. Tap the edge of the coverslip to a delicate task wipe to wick away excess water.
Dry the coverslip under an inert gas, such as nitrogen. Mark the underside of the coverslip to keep track of orientation from this point forward. Be careful to keep the patterned side facing up.
Repeat steps 5.1.1 through 5.1.6 for each stenciled coverslip.
2. Making polyacrylamide hydrogels with patterned coverslips
NOTE: Refer to previous methods for additional details7,9 Sterilize all cap holders, tubes, and spacers used to make polyacrylamide gels by washing with 10% bleach overnight, rinsing with water at least five times to remove bleach, and washing briefly in 70% ethanol. Place all pieces on delicate task wipes in a sterile biosafety cabinet to dry before use.
-
Prepare polyacrylamide solution to obtain desired hydrogel elasticity (Table 2). Mix together all components except the fluorescent microspheres and the potassium persulfate (PPS).
NOTE: Prepare 1% potassium persulfate solution fresh each time.
Degas the polyacrylamide solution, microspheres, and PPS in separate tubes under vacuum for 30 min.
For each patterned coverslip, prepare a glutaraldehyde-activated “bottom” coverslip with a spacer (18 mm outer diameter, 14 mm inner diameter) placed on top.
After degassing, briefly sonicate microspheres and add appropriate volume to polyacrylamide solution. Mix by pipetting up and down, being careful not to introduce air bubbles. Briefly sonicate.
-
Add appropriate volume of PPS to the polyacrylamide solution. Pipette up and down to mix the solution, being careful not to introduce air bubbles.
NOTE: After adding PPS, work rapidly to complete steps 5.2.6 through 5.2.11 before polyacrylamide polymerizes.
Pipette 75–150 μL (depending on thickness of spacer) to the center of each glutaraldehyde-activated coverslip.
Using forceps, place a patterned coverslip onto each glutaraldehyde-activated coverslip with polyacrylamide and spacer, such that the patterned ligand faces the polyacrylamide solution. If sufficient polyacrylamide solution was added, surface tension will wick the polyacrylamide between the two coverslips and no air bubbles will be present.
Pick up each polyacrylamide “sandwich” and carefully touch the side to a delicate task wipe to wick away excess polyacrylamide solution.
Carefully place each polyacrylamide “sandwich” into a cap holder with threads that are compatible with 15 mL conical-bottom tubes. The orientation should be such that the bottom of the glutaraldehyde coverslip is in contact with the bottom of the cap holder, and the patterned coverslip is on top.
Screw a 15 mL conical-bottom tube into each cap holder to hold the polyacrylamide “sandwiches” in place. The tubes should be tight to prevent leaking, but be careful to not overtighten and crack the coverslips.
-
Centrifuge the polyacrylamide “sandwiches” in the tubes in swing-buckets at 200 × g for 10 min at room temperature. Remove the tubes from the centrifuge and place in tube racks to maintain orientation for an additional 50 min to ensure full polymerization. Keep covered with foil to prevent bleaching of fluorescent microspheres.
NOTE: Centrifugation is critical when using this method to perform TFM. The centrifugal force moves all the microspheres to a single plane, which will ultimately be just below the surface of the hydrogel. This is ideal for imaging microsphere displacements that are used to calculate cell-generated traction stresses. If not performing TFM, microspheres can be left out of the polyacrylamide solution and polymerization can occur at the benchtop without centrifugation.
Remove polymerized polyacrylamide “sandwiches” from the tubes and submerge in PBS in a 100 mm dish. Wrap in laboratory film and incubate for 3 h at room temperature or overnight at 4 °C.
Table 2: Polyacrylamide gel formulations.
Volumes of components for generating polyacrylamide hydrogels of various elastic moduli with fluorescent microspheres for TFM. Volumes can be scaled up or down based on amount of polyacrylamide solution needed. All components except the fluorescent microspheres and PPS are mixed together prior to degassing. PBS = phosphate-buffered saline, TEMED = tetramethylethylenediamine, PPS = potassium persulfate.
| Polyacrylamide Gel Formulations | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Volumes for 1 mL complete solution | |||||||||
| Elastic Modulus (Pa) | Final % acrylamide | Final % Bis-acrylamide | ddH2O (μL) | 40% acrylamide (μL) | 2% Bis-acrylamide (μL) | 10× PBS (μL) | 1% TEMED in ddH2O (μL) | Microspheres 0.5% solids in ddH2O (μL) | 1% PPS in ddH2O (μL) |
| 1050 | 3 | 0.1 | 565 | 75 | 50 | 100 | 75 | 60 | 75 |
| 2700 | 7.5 | 0.035 | 485 | 187.5 | 17.5 | 100 | 75 | 60 | 75 |
| 4000 | 7.5 | 0.05 | 477.5 | 187.5 | 25 | 100 | 75 | 60 | 75 |
| 6000 | 7.5 | 0.07 | 467.5 | 187.5 | 35 | 100 | 75 | 60 | 75 |
3. Preparation of patterned gels for seeding cells
-
Use a scalpel and forceps to carefully remove the patterned coverslip and the spacer from the polyacrylamide “sandwich” while it remains submerged in PBS. The patterned ligand will have transferred to the polyacrylamide during polymerization and will remain after removing the coverslip. The polyacrylamide gel will remain attached to the glutaraldehyde-activated coverslip.
NOTE: This is a critical step! The polyacrylamide must be submerged in PBS while removing the coverslip or the ligand patterns will be destroyed.
Place each coverslip with patterned polyacrylamide into a cap holder. Place a gasket (18 mm outer diameter, 14 mm inner diameter) on top of the coverslip and around the polyacrylamide, where the spacer was located. Screw a sawed-off 15 mm conical-bottom tube into the cap holder, forming a well with the patterned polyacrylamide at the bottom. These assemblies fit into the wells of a standard 12-well plate for easy handling.
Wash each gel by adding 500 μL of PBS to the well assembly. Incubate for 10 min at room temperature with gentle shaking. Remove PBS and repeat twice for a total of three washes.
After the final PBS wash, add 500 μL of knockout-DMEM media to gels and incubate at 37 °C overnight. Gels can be kept at 37 °C with media for up to 5 days before seeding cells.
The day before seeding cells, replace knockout-DMEM for complete KSR media and incubate at 37 °C overnight.
6. Culturing hESCs on patterned gels
NOTE: If planning to fix samples for immunostaining after TFM, take images of unstressed microsphere positions prior to seeding cells. In this case, fluorescently tagged ligand should be used in step 4.3.1 so that the eventual locations of cells will be known before seeding and microspheres can be imaged in those regions.
-
Maintain stock hESC cultures and secondary feeder-free cultures prepared on matrigel-coated plates in conditioned KSR media prior to seeding on patterned polyacrylamide gels as previously described9.
NOTE: Generate conditioned KSR media by culturing irradiated mouse embryonic fibroblasts in KSR media supplemented with 4 ng/mL basic fibroblast growth factor (bFGF). Collect and replace media every 24 h.
Aspirate the media from hESCs. Briefly wash with knockout-DMEM media. Add 0.05% trypsin-EDTA supplemented with 10 μM Y27632 (Rho kinase inhibitor) and incubate at 37 °C for 5–10 min.
Add media with serum to inhibit trypsin. Gently aspirate media and pipette over the dish to remove cells. Continue pipetting gently to remove all cells and dissociate cell clusters to single cells.
Collect the cell suspension and centrifuge at 200 × g for 3 min.
Aspirate the supernatant and resuspend the pellet in an equivalent volume of KSR media to wash. Centrifuge at 200 × g for 3 min.
Aspirate the supernatant and resuspend the pellet in conditioned KSR media supplemented with 10 ng/mL bFGF and 10 μM Y27632.
Count the cells with a hemocytometer and adjust the cell suspension to a concentration of 300,000 cells per mL. Pipette 500 μL of the cell suspension onto each patterned gel (150,000 cells per gel).
-
3 h after seeding, use a pipette to carefully aspirate the media from each gel and replace with fresh conditioned KSR media supplemented with 10 ng/mL bFGF and 10 μM Y27632. This removes excess cells that did not adhere to patterned regions of the polyacrylamide.
NOTE: This is a critical step! At this stage cells will be loosely adhered to the patterned ligand. Be very careful when swapping media to not detach the cells. Equal care should be taken with each of the media swaps in the subsequent steps.
24 h after seeding, use a pipette to carefully aspirate the media and exchange for conditioned KSR media supplemented with 10 ng/mL bFGF and 5 μM Y27632.
48 h after seeding, use a pipette to carefully aspirate the media and exchange for conditioned KSR media supplemented with 10 ng/mL bFGF. This removes the Rho kinase inhibitor from the media and experiments can begin the next day, 72 h after seeding.
7. Performing TFM
When desired, perform TFM as previously described9 under desired experimental conditions.
If unstressed microsphere positions were imaged before seeding cells, fix the cells after taking stressed microsphere positions and perform immunostaining to determine localization of proteins of interest relative to regions of high and low traction forces.
Representative Results
The main challenge to overcome in attempting to culture hESCs in colonies of controlled geometry on compliant substrates is to generate a homogenous pattern of ECM-ligand on the surface of the substrate. The strategy presented in this method involves first generating the desired pattern on the surface of a glass coverslip and then subsequently transferring that pattern to the surface of a polyacrylamide hydrogel during polymerization of the gel (Figure 1A). Thus, it is important to ensure the desired pattern is created successfully on the surface of the glass coverslip prior to proceeding to polymerization of the hydrogel and transfer of the pattern (Figure 1B). Based on imaging of fluorescent ligand patterns transferred to polyacrylamide, the ligand appears to be present only in a single plane at the surface of the polyacrylamide, though we did not precisely characterize the thickness of this layer. There are two common types of defects observed following pattern generation on the glass coverslip, each with its own source of error. The first is the appearance of fluorescent ligand extending beyond the margins of the desired pattern (Figure 1C, top), which results from leaking of the fluorescent ligand solution due to a small tear in the stencil or insufficient sealing of the stencil to the glass coverslip. The second is the appearance of an incomplete pattern (Figure 1C, bottom), which is typically due to an air bubble trapped at the coverslip interface that prevents adsorption of the fluorescent ligand.
Figure 1: Patterning of ECM ligand onto acid-washed coverslips and transfer to polyacrylamide hydrogels.
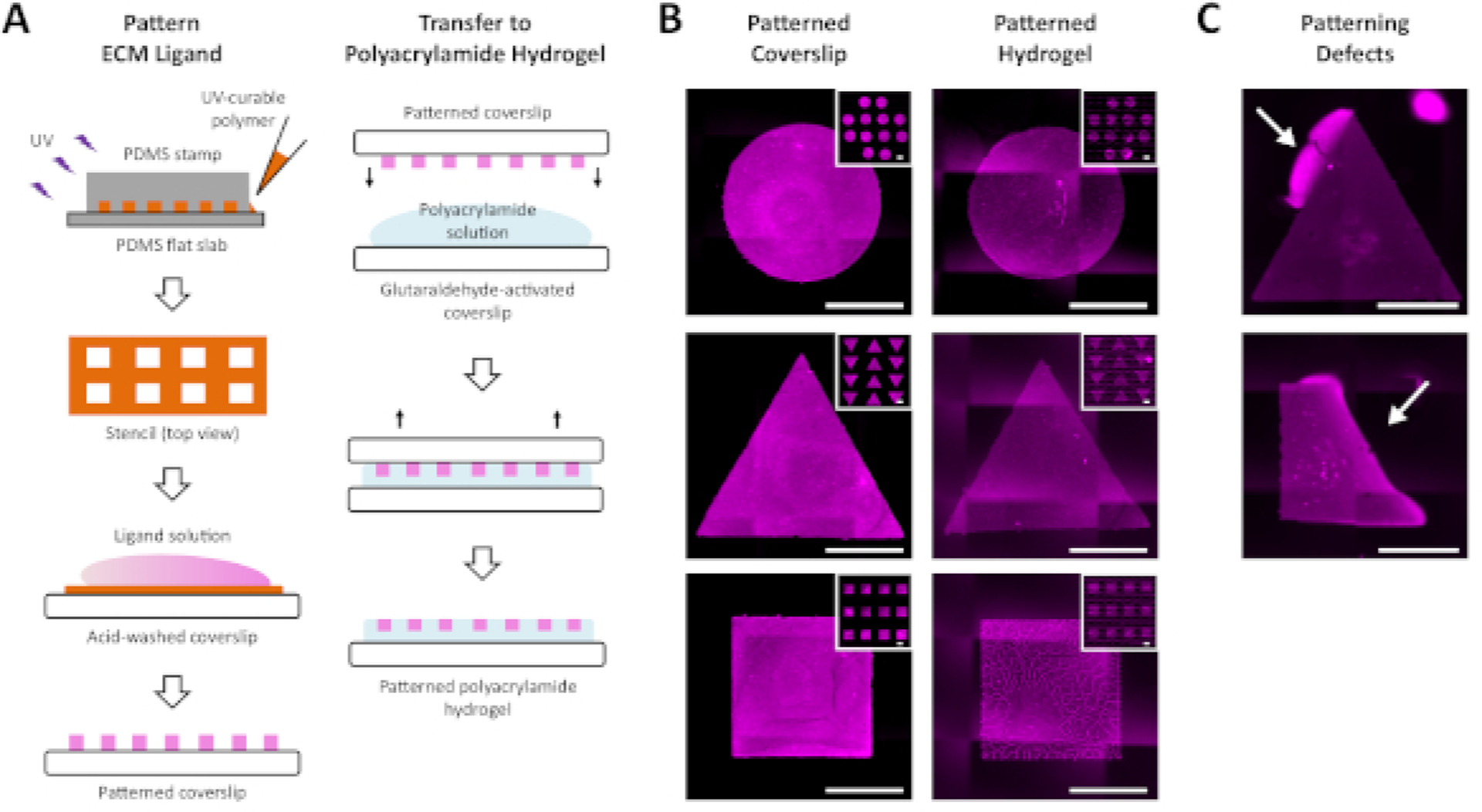
(A) Schematic representation of the protocol for patterning ECM ligand on acid-washed coverslips and transferring the patterned ligand to polyacrylamide hydrogels. (B) Immunofluorescent images of patterned ECM ligand on acid-washed coverslips (left) and following transfer to polyacrylamide hydrogel (right). Biotin-tagged matrigel was patterned onto the coverslip and labeled with Alexa Fluor 555 streptavidin prior to transfer to polyacrylamide. Insets show zoomed-out view of the full patterns generated. (C) Representative fluorescent images demonstrating patterning defects of ligand on the glass coverslip. These result from common errors in the protocol, such as leaking of the ligand solution outside the patterned geometry (top, arrow indicates site of leak), and trapping of air bubbles inside the patterned geometries of the stencil upon adding the ligand solution (bottom, arrow indicates site of air bubble). All scale bars = 500 μm.
The ultimate measure of success for this method is the ability to culture hESCs in desired geometries on the patterned hydrogels (Figure 2). In order to achieve this, hESCs are seeded at a relatively high density (300,000 cells/mL) in the presence of a Rho kinase inhibitor (Y27632) and incubated for 3 h to facilitate adhesion to the patterns of ligand. Media is then replaced to remove non-adhered cells. Over the course of 72 h, the Y27632 is gradually diluted out of the media by a series of media exchanges at 24 and 48 h post-seeding. Typically, the hESCs proliferate to complete the patterned geometries by 48–72 h, such that experiments can begin at 72 h post-seeding, once the Y27632 is completely removed from the media.
Figure 2: Seeding of hESCs onto patterned polyacrylamide hydrogels.

Representative brightfield images demonstrating successful seeding of hESCs onto polyacrylamide hydrogels with patterned ligand. The timeline at the top indicates the series of media changes used to remove unattached cells and dilute out the Y27632. Note that after initial seeding, cells adhere stochastically to various regions within the patterned ligand and then proliferate to fill the patterned regions over the course of 72 h. Scale bar = 500 μm.
Culturing hESC colonies in confined geometries on polyacrylamide hydrogels permits the measurement of cell-generated traction forces using TFM. These measurements are made by embedding fluorescent microspheres in the hydrogel and imaging the positions of these beads before and after seeding hESCs (Figure 3A). The displacement of the beads following cell seeding is a function of cell-generated forces and the elasticity of the hydrogel, thus the images of the bead positions can be used to generate maps of bead displacements and subsequently used to calculate the underlying traction stresses. In circular colonies of hESCs, the largest traction stresses are found near the peripheral edge of the colonies, while the center of the colonies display uniformly low traction stresses (Figure 3B). Interestingly, the highest traction stresses are found in clusters near the edge of the colonies, rather than forming a continuous ring of maximal stress. This implies that although colony geometry plays a key role in determining the distribution of traction stresses, more localized regulation and feedback determines the precise locations of maximal stress. Additionally, so long as the image of microsphere positions without adhered cells is taken prior to cell seeding, hESC colonies can be fixed for immunostaining of proteins of interest following traction force measurements. Despite the observed non-uniform distributions of traction stresses, hESCs cultured as patterned circles in maintenance conditions display uniform expression of the pluripotency marker Oct3/4 and cell adhesion molecule E-cadherin (Figure 3C).
Figure 3: Regional localization of traction stresses and immunostaining of confined hESC colonies.
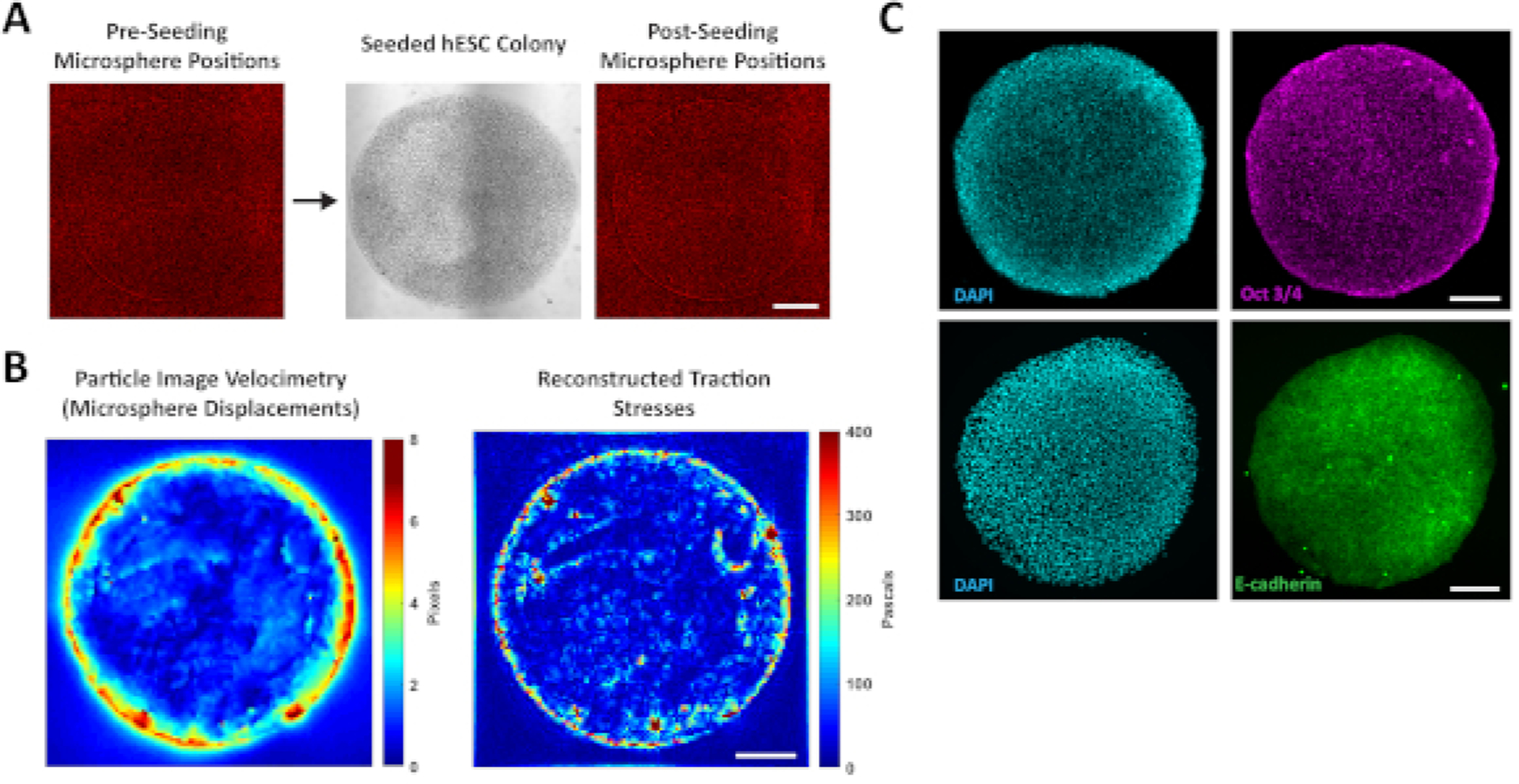
(A) Fluorescent images of microspheres within the polyacrylamide hydrogel before and after seeding hESCs. (B) Representative particle image velocimetry (PIV) plot depicting the displacement of microspheres due to traction stresses (left) and corresponding reconstructed traction stresses (right). (C) Immunostaining of confined hESC colonies on patterned polyacrylamide hydrogels, demonstrating the ability to compare localization of proteins of interest to traction stresses. All scale bars = 200 μm.
The presented method for using stencils to generate patterns of ligand is demonstrably superior to the common technique of microcontact printing for the relatively large geometries used in this method (i.e., for circular colonies 1 mm in diameter as well as triangles and squares of equivalent area). Microcontact printing patterns of ligand at this length scale results in heterogeneous transfer at the edges of patterns, with very little ligand deposited in the central regions of the patterns (Figure 4A). This is clearly insufficient for consistently producing hESC colonies of specific geometries. Reducing the cell seeding density also leads to inconsistency in achieving completed colonies. Cells seeded at 200,000 cells/mL, rather than 300,000 cells/mL, are not able to generate sufficient cell-cell contacts to survive the reduced concentration of Y27632 at 24 h post-seeding (Figure 4B). It may be possible to generate complete patterns with lower cell densities by extending the period of Y27632 dilution; however, overall it is more efficient to seed at the higher 300,000 cells/mL density. Occasionally, errors in pattern generation that are not detected earlier in the protocol become apparent upon seeding of hESCs. One such error is the leaking of ligand solution underneath the stencil due to poor contact with the coverslip. This ultimately results in ligand being transferred to regions of the polyacrylamide outside the desired geometries, and unconfined growth of hESCs upon seeding (Figure 4C).
Figure 4: Non-ideal results yielded by alternative methods and errors.
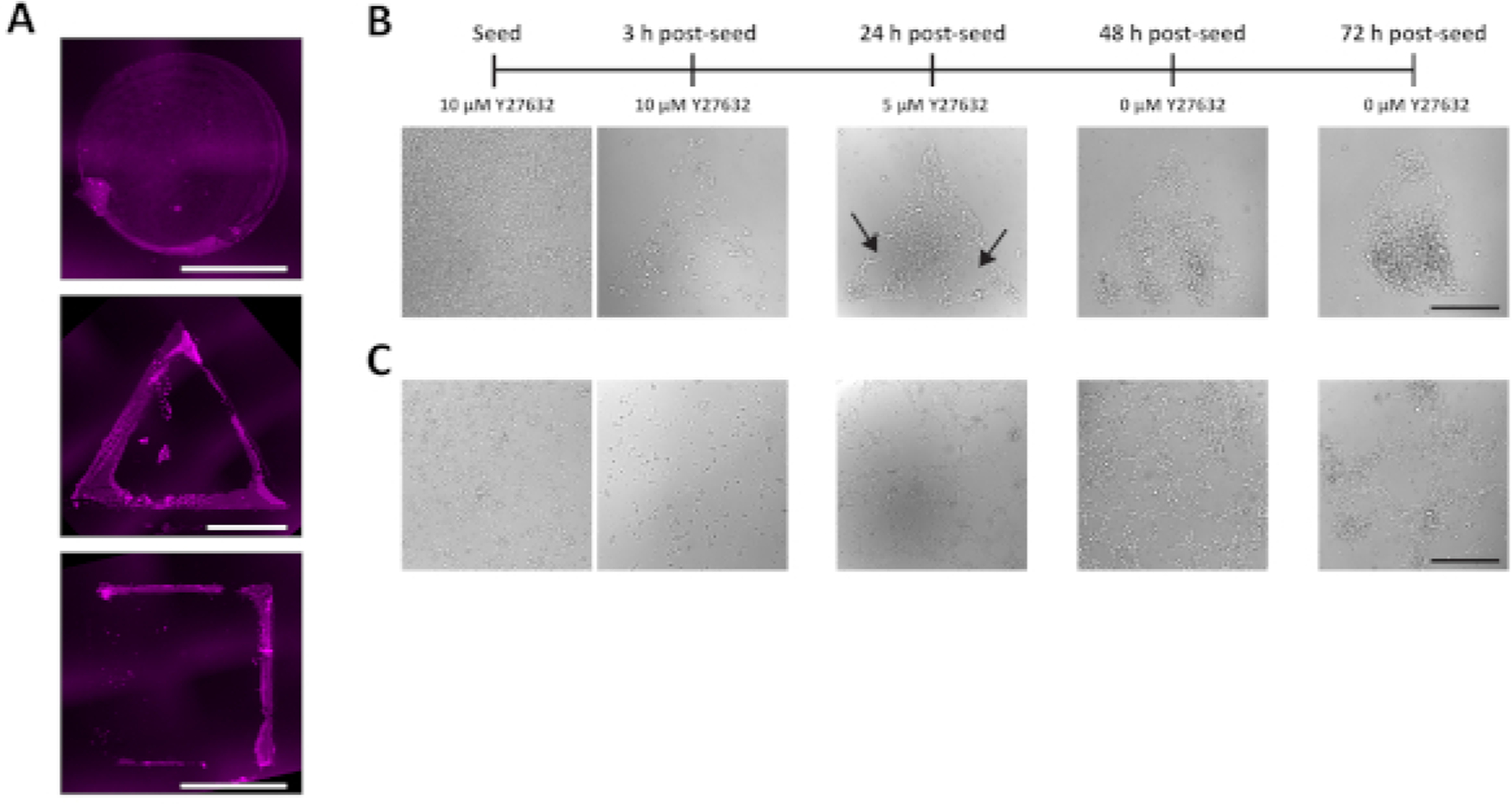
(A) Representative fluorescent images of ECM ligand patterned onto acid-washed coverslips via microcontact printing. (B) Brightfield images of hESCs that fail to form completed colonies due to insufficient adherence of cells. Large gaps remaining in the colonies at 24 h post-seeding (arrows) are an early indicator of this issue. (C) Brightfield images of hESCs that fail to form confined colonies due to errors in patterning that led to presence of ligand outside the desired geometries. All scale bars = 500 μm.
Discussion
To simplify a long and detailed protocol, this method consists of three critical stages: 1) generating patterns of ECM ligand on glass coverslips, 2) transferring the patterns to polyacrylamide hydrogels during polymerization of the gel, and 3) seeding hESCs on the patterned hydrogel. There are critical steps that must be considered at each of these three stages. In order to generate high-fidelity patterns on the glass coverslips, the stencil must be firmly pressed onto the coverslip to prevent leaking of the ligand solution and all air bubbles must be removed after adding ligand solution to the surface of the stencil (Figure 1C). If ligand solution does leak through the stencil due to poor contact with the coverslip, ligand will be transferred to the entire surface of the polyacrylamide hydrogel and hESCs will not be confined to the desired colony geometry (Figure 4C). The most important step in transferring the patterned ligand to polyacrylamide is gently separating the top coverslip from the hydrogel while all components remain submerged in PBS. If the hydrogel does not remain submerged during separation, the patterns may be completely destroyed. Furthermore, if the separation occurs too rapidly, the surface of the hydrogel may tear or be otherwise damaged. The softer the hydrogel, the more it is at risk for damage during separation. Finally, the user must pipette very carefully when exchanging media during the seeding and culture of hESCs on the patterned hydrogels. The hESCs remain loosely adhered throughout the protocol and the patterned colonies can be easily disrupted by careless or rushed pipetting.
In addition to the critical steps discussed above, there are a number of other steps that may require modification and troubleshooting when adapting this protocol for different applications. While the patterns demonstrated here are on the length scale of hundreds of microns to a millimeter, generating patterned features on silicon wafers with transparency photomasks and negative photoresist allows for feature sizes all the way down to 7–10 μm. Thus, this protocol could be adapted for confining the geometry of single cells or smaller colonies of a few cells on compliant substrates.
Two parameters that will likely require optimization when adapting this protocol for different cell types are the type of ECM ligand used and the concentration of the ligand in solution during adsorption to the glass coverslip through the stencils. A total ligand concentration of 250 μg/mL was sufficient for producing homogenous patterns of matrigel and facilitating attachment of hESCs (Figure 1B and Figure 2), though it may be possible to achieve similar results with lower concentrations. On the other hand, it may be necessary to further increase the concentration of ligand for cell types that are less adherent or for applications that require shorter incubation times. Increased concentration of ligand may also be required for different ECM ligands, such as fibronectin or collagen, which are less hydrophobic than matrigel and therefore may not adsorb to the hydrogel as strongly. Because the transfer of ligand from coverslip to hydrogel occurs during polymerization of the hydrogel, commonly used techniques for robustly cross-linking the ECM ligand to the hydrogel surface (such as sulfo-SANPAH treatment) are impossible. Using fluorescently-labelled ECM ligands to enable visualization of the patterns at each step of the protocol is extremely helpful when optimizing and troubleshooting these parameters.
Additionally, the protocol for seeding cells may require optimization depending on the cell type and media conditions used. For cells that adhere more rapidly or efficiently, a lower seeding density may be required to prevent cell-cell adhesions that span between patterns and result in aggregates rather than patterned monolayer colonies. For cells that display very poor attachment, a larger seeding density or longer length of time before the initial media swap may be required to facilitate complete formation of confined colonies (Figure 4B).
The key limitation of this method is its technical complexity, which results in relatively low-throughput results compared to similar methods that involve culturing cells on patterned glass substrates8. However, this drawback is far outweighed by the physiological relevance achieved by culturing confined hESC colonies on compliant substrates. By effectively recapitulating the mechanical properties of the early embryo, we are able to better model and understand the processes that lead to self-organization of the primary germ layers.
An additional benefit of confining hESC colonies on polyacrylamide hydrogels is that it enables the use of TFM to examine the link between the organization of an hESC colony, as a model of the early embryo, and the distribution of cell-generated forces that may underlie morphogenesis and cell fate specification. Confining hESC colonies results in distributions of traction stresses that are dependent on colony geometry (Figure 3B). Despite the non-uniformity of these traction stresses, cells throughout the colonies remain pluripotent in maintenance conditions (Figure 3C). However, we hypothesize that the traction stress distributions may be involved in regulating patterns of cell fate specification by tuning the response to induction cues, such as soluble morphogens. We anticipate that this method will allow us and other groups to better model the early human embryo with hESCs, leading to a more complete understanding of the fundamental processes that underlie human embryogenesis.
Acknowledgments
We would like to acknowledge funding from CIRM grant RB5-07409. J.M.M. would like to thank FuiBoon Kai, Dhruv Thakar, and Roger Oria for various discussions that guided the generation and troubleshooting of this method. J.M.M. also thanks the UCSF Discovery Fellowship for the ongoing support of his work.
Footnotes
Video Link
The video component of this article can be found at https://www.jove.com/video/60334/
Disclosures
The authors have nothing to disclose.
References
- 1.Tabar V, Studer L Pluripotent stem cells in regenerative medicine: Challenges and recent progress. Nature Reviews Genetics. 15, 82–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avior Y, Sagi I, Benvenisty N Pluripotent stem cells in disease modelling and drug discovery. Nature Reviews Molecular Cell Biology. 17, 170–182 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Trounson A, DeWitt ND Pluripotent stem cells progressing to the clinic. Nature Reviews Molecular Cell Biology. 17, 194–200 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Li L, Chen ZN, Gao G, Yao R, Sun W Engineering-derived approaches for iPSC preparation, expansion, differentiation and applications. Biofabrication. 9 (3), 032001 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Stevens KR, Murry CE Human Pluripotent Stem Cell-Derived Engineered Tissues: Clinical Considerations. Cell Stem Cell. 22 (3), 294–297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itskovitz-Eldor J, et al. Differentiation of human embryonic stem cells into embryoid bodies comprising the three embryonic germ layers. Molecular Medicine. 6 (2), 88–95 (2000). [PMC free article] [PubMed] [Google Scholar]
- 7.Lakins JN, Chin AR, Weaver VM Exploring the link between human embryonic stem cell organization and fate using tension-calibrated extracellular matrix functionalized polyacrylamide gels. In: Mace K, Braun K (eds) Progenitor Cells. Methods in Molecular Biology (Methods and Protocols). Humana Press, Totowa, NJ. 916, 317–350 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Warmflash A, Sorre B, Etoc F, Siggia ED, Brivanlou AH A method to recapitulate early embryonic spatial patterning in human embryonic stem cells. Nature Methods. 11 (8), 847–854 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Przybyla L, Lakins JN, Sunyer R, Trepat X, Weaver VM Monitoring developmental force distributions in reconstituted embryonic epithelia. Methods. 94, 101–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Przybyla L, Lakins JN, Weaver VM Tissue Mechanics Orchestrate Wnt-Dependent Human Embryonic Stem Cell Differentiation. Cell Stem Cell. 19 (4), 462–475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shahbazi MN, et al. Self-organization of the human embryo in the absence of maternal tissues. Nature Cell Biology. 18, 700–708 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shao Y, Taniguchi K, Townshend RF, Miki T, Gumucio DL, Fu J A pluripotent stem cell-based model for post-implantation human amniotic sac development. Nature Communications. 8 (208), 1–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simunovic M, Brivanlou AH Embryoids, organoids and gastruloids: new approaches to understanding embryogenesis. Development. 144, 976–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 282 (5391), 1145–1147 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Shahbazi MN, Zernicka-Goetz M Deconstructing and reconstructing the mouse and human early embryo. Nature Cell Biology. 20, 878–887 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Li Q, et al. Extracellular matrix scaffolding guides lumen elongation by inducing anisotropic intercellular mechanical tension. Nature Cell Biology. 18 (3), 311–318 (2016). [DOI] [PubMed] [Google Scholar]