

A receptor of chaperone-mediated autophagy regulates glioblastoma stem cells and may serve as a potential biomarker for advanced tumor grade and poor survival in this disease.
Abstract
Chaperone-mediated autophagy (CMA) is a homeostatic process essential for the lysosomal degradation of a selected subset of the proteome. CMA activity directly depends on the levels of LAMP2A, a critical receptor for CMA substrate proteins at the lysosomal membrane. In glioblastoma (GBM), the most common and aggressive brain cancer in adulthood, high levels of LAMP2A in the tumor and tumor-associated pericytes have been linked to temozolomide resistance and tumor progression. However, the role of LAMP2A, and hence CMA, in any cancer stem cell type or in glioblastoma stem cells (GSC) remains unknown. In this work, we show that LAMP2A expression is enriched in patient-derived GSCs, and its depletion diminishes GSC-mediated tumorigenic activities. Conversely, overexpression of LAMP2A facilitates the acquisition of GSC properties. Proteomic and transcriptomic analysis of LAMP2A-depleted GSCs revealed reduced extracellular matrix interaction effectors in both analyses. Moreover, pathways related to mitochondrial metabolism and the immune system were differentially deregulated at the proteome level. Furthermore, clinical samples of GBM tissue presented overexpression of LAMP2, which correlated with advanced glioma grade and poor overall survival. In conclusion, we identified a novel role of CMA in directly regulating GSCs activity via multiple pathways at the proteome and transcriptome levels.
Significance:
A receptor of chaperone-mediated autophagy regulates glioblastoma stem cells and may serve as a potential biomarker for advanced tumor grade and poor survival in this disease.
Introduction
Glioblastoma (GBM) represents the most common and aggressive primary brain tumor, it is classified as grade IV glioma by the WHO, and it presents an average patient survival lower than 15 months (1). Fast growing GBM cells contain damaged DNA and numerous misfolded proteins, causing a significantly imbalanced proteostasis. Thus, processes such as unfolded protein response, ubiquitin–proteasome system or autophagy seem to be important for GBM cells to prevent proteotoxic crisis (2).
Autophagy is a key process to maintain cellular homeostasis in response to various stimuli, based on the recycling of cytoplasmic content and elimination of damaged molecules via lysosomal degradation. To date, three main types of autophagy have been described in mammals, which include macroautophagy (MA), microautophagy, and chaperone-mediated autophagy (CMA). They differ on how cargo is delivered to lysosomes for degradation. MA is specialized in the removal of abnormal proteins, damaged organelles or invading pathogens by activating the formation of double-membrane vesicles, whereas microautophagy delivers cargo to the lysosome through direct invagination of the lysosomal membrane (3). CMA is a unique selective type of autophagy specialized in the targeting and translocation of individual proteins for their degradation within the lysosome (4). In this latest type, heat shock cognate 71 kDa protein (HSC70) chaperone recognizes cytosolic proteins containing a “KFERQ”-like motif and targets them to the lysosome-associated membrane protein type 2A (LAMP2A), the key limiting mediator of CMA (4). Herein, the cargo protein is directly translocated across the lysosomal membrane for its degradation (4).
During homeostasis, CMA can be activated in the presence of stress such as starvation (5), oxidative stress (6), or genotoxic insults (7), and both, MA and CMA, protect cells against malignant transformation, by the preservation of cellular integrity or inactivating neoplastic precursors, among others (8, 9). However, once cellular damage gets accumulated, cancer cells deregulate MA and CMA, which facilitate their survival or cell death promotion depending on the context and stage of the tumor (8, 9). In GBM, MA activity has been associated with the metabolism and phenotype of glioma stem cells (GSC), a subpopulation of cells responsible for tumor progression, therapy resistance, and relapse (10, 11). However, a dual role of MA suppressing or promoting tumor progression has been described in GBM (2). In regard to CMA, very little is known about its impact in GBM. Clinical samples of patients treated with temozolomide (TMZ) revealed increased expression of LAMP2A, as well as the MA and endolysosomal markers LC3B and LAMP1, compared with pretreated condition (12). Moreover, recent evidence showed that GBM-induced upregulated expression of LAMP2A and CMA activity in pericytes promotes GBM tumor progression (13). However, no evidence has been described linking CMA to any cancer stem cell type, and especially, to GSCs population. In this work, we addressed this unresolved issue.
Materials and Methods
Patient databases and association studies
Control and glioma patient clinical and transcriptomic data have been collected from GlioVis database (http://gliovis.bioinfo.cnio.es/; ref. 14). Microarray results from The Cancer Genome Atlas (TCGA; 10 control, 226 grade II, 244 grade III, 528 grade IV GB) and Rembrandt (28 control, 98 grade II, 85 grade III, 219 grade IV GB) cohorts for LAMP2 have been represented. For survival studies, cutoffs for Kaplan–Maier have been designated as optimal by GlioVis database. Pairwise comparisons with corrections for multiple testing (P values with Bonferroni correction) have been performed for statistical analysis. Association studies have been developed on the basis of transcriptomic data from TCGA cohort using cBioPortal database (https://www.cbioportal.org/; ref. 15). ROC curves of TMZ treated patients with GBM were performed by ROC plotter online tool (nonresponders n = 154, responders n = 165; http://www.rocplot.org/; ref. 16). Single-cell RNA sequencing (RNA-seq) analysis was performed on the basis of UCSC Cell Browser (https://cells.ucsc.edu/?ds=gbm; ref. 17). The study also included a total of 20 biopsies for mRNA study and 98 biopsies for tissue microarray IHC analysis from patients seen at Donostia University Hospital (San Sebastian, Spain), and diagnosed as primary GBM according to the WHO criteria. For mRNA expression analysis, human brain total RNA (Invitrogen AM7962) was used as healthy control tissue. All study participants signed an informed consent form approved by the Institutional Ethical Committee.
Cell lines, cultures, and reagents
Patient-derived GNS166, GNS179, and GNS144 glioma stem cell lines, kindly provided by Dr. Steven Pollard (18), were cultured in 2 μg/mL laminin (Sigma-Aldrich)-coated culture plates, and GSC medium consisting of DMEM/F-12 (Sigma-Aldrich) supplemented by N2, B27 (Thermo Fisher Scientific), glucose (Gibco), 100 U/mL penicillin, 100 μg/mL streptomycin, and growth factors (20 ng/mL basic FGF and 20 ng/mL EGF; Sigma-Aldrich). GB2 was generated in the laboratory (19). Glioma cell lines U87MG (U87), U251MG (U251), U373MG (U373), T98G (T98), and A172 were purchased from the ATCC and cultured in DMEM (Gibco), supplemented with 10% FBS (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin for monolayer cultures and in GSC medium for oncosphere cultures. Cells were maintained at standard conditions of 37°C and 5% CO2 in humidified atmosphere. TMZ (Sigma) and QX77 (MedChemExpress) were dissolved in DMSO.
CMA activity measurement assay
Cells were transduced with lentivirus carrying the KFERQ-PS-Dendra reporter (20). Cells were photoactivated by exposure to a 3.5 mA (current constant) light-emitting diode (LED: Norlux, 405 nm) for 3 minutes and then plated in glass-bottom 96-well plates. After 16 hours, cells were fixed with 4% paraformaldehyde, nuclei stained with Hoechst, and imaged using high-content microscopy (Operetta system, Perkin Elmer). Images were quantified using the manufacturer's software in a minimum of 1,500 cells per well. In all cases, focal plane thickness was set at 0.17 μm and sections with maximal nucleus diameter were selected for quantification. Values are presented as number of puncta per cell section that in our acquisition conditions represents 10% to 20% of the total puncta per cell. Where indicated, CMA was induced by replacing the media by serum-free media after extensively washing or by addition of 50 μmol/L paraquat to induce mild oxidative stress.
Lentivirus-mediated shRNA and overexpression studies
For stable downregulation of LAMP2A, lentiviral plasmid pGK shL2A was used, together with the empty vector pGK as described previously (21). Due to LAMP2A protein long half-life, infected cells were maintained in culture for 5 days post-infection before seeding of experiments, and efficiency of the infection was tested by GFP+ signal. For overexpression studies, pAMC1 lentiviral plasmid was used, which was generated from the insertion of human LAMP2A transcript in pCR3.1 plasmid.
Colony assay
Five hundred pGK and shL2A U251 cells/well were seeded in triplicate into six-well plates and were cultured for 9 days. After that, colonies were fixed with 37% paraformaldehyde and stained with 5% Giemsa.
Oncosphere formation assay
A total of 1 × 104pGK and shL2A U251 or U373 cells/well were seeded in nontreated six-well flat-bottom plates in GSC medium and primary oncospheres (1ry CSCs) were grown after 10 days of culture. After quantification, spheres were disaggregated with accutase (Gibco) and seeded for secondary oncospheres (2ry CSCs) to maintain them for other 10 days. Fresh media was added every 2 to 3 days to the plate. When indicated, TMZ or QX77 was added into the media.
Proliferation assay
For cell count experiments, 2.5 × 104 cells/well were seeded in duplicate in treated six-well flat-bottom plates. Total number of cells was determined at day 1, 3, and 5 after cell seeding, using Neubauer counting chamber and a light microscope.
Immunofluorescence
It was performed as described in previous studies (22). Cells were incubated overnight at 4°C with primary antibody for p-H3 (ab14955 Abcam, 1/2000) and active caspase-3 (AF835 R&D Systems, 1/500). Then, cells were incubated for 1 hour with secondary Alexa Fluor 555 IgG anti-mouse (A21422 Invitrogen, 1/500) or anti-rabbit (A31572 Invitrogen, 1/500) antibodies at room temperature in darkness. For nuclear DNA staining, Hoechst 33342 (Sigma) was used. Pictures were taken with an Eclipse 80i microscope and processed with the NIS Elements Advances Research software (Nikon).
Cell viability assay
Cells were seeded at a density of 8 × 103 cells/well for GNS166 and 3 × 103 cells/well for U251 in 96-well plates, with six replicates per condition. After overnight incubation for cell adhesion, 2 mmol/L (GNS166 cells) or 250 μmol/L (U251 cells) TMZ was applied into each well. Cell viability was determined by MTT assay at 72 hours after treatment. For this, MTT reagent was added, and after 3.5 hours incubation at 37°C and 5% CO2, media was removed. Formed crystals were resuspended in DMSO and absorbance was measured at 570 nm using a spectrophotometer. Results were analyzed by GraphPad Prism software.
In vivo intracranial injection carcinogenesis assay
For xenotransplantation by intracranial injection of cells, infected GNS166 pGK and shL2A cells were harvested with accutase (Gibco) and resuspended in PBS before injection. A total of 2 × 105 cells were injected in 6- to 8-week-old NOD.CB17-Prkdcscid/NCrHsd mice brains, at 2.5 mm lateral and 1 mm anterior to bregma, by an infusion pump (PHD ULTRA Harvard Apparatus). Mice weight and welfare was monitored during the experiment and they were culled when cognitive and motor failure. Survival distributions were determined using the long-rank test and GraphPad Prism 8 software.
IHC studies
Subcutaneous tumors generated in mice were dissected, fixed in 4% formalin for 48 hours, and embedded in paraffin. Heat-induced antigen retrieval was performed. Tumor sections were incubated overnight at 4°C with primary antibody for LAMP2A (ab18528, Abcam, 1 μg/mL). After that, sections were incubated for 1 hour at room temperature with goat Anti-Rabbit IgG H&L (HRP; ab6721, Abcam, 1/1000). Same antibody for LAMP2A was used for tissue microarrays of GBM biopsies.
Proteomic study
Samples were incubated and digested following the filter-aided FASP protocol with minor modifications (23). Trypsin was added to a trypsin:protein ratio of 1:50, and the mixture was incubated overnight at 37°C, dried out in a RVC2 25 speedvac concentrator (Christ), and resuspended in 0.1% FA. Peptides were desalted and resuspended in 0.1% FA using C18 stage tips (Millipore).
Samples (200 ng) were next analyzed in a hybrid trapped ion mobility spectrometry—quadrupole time of flight mass spectrometer (timsTOF Pro with PASEF; Bruker Daltonics) coupled online to either a nanoElute (Bruker) or a Evosep ONE (Evosep) liquid chromatograph.
Protein identification and quantification were performed using MaxQuant software under default settings. Searches were carried out against a database consisting of human protein entries (Uniprot/Swissprot), with precursor and fragment tolerances of 20 ppm and 0.05 Da. Only proteins identified with at least two peptides at FDR <1% were considered for further analysis. Data were loaded onto Perseus platform (24) and further processed (log2 transformation, imputation). Significantly differential proteins (P < 0.05), with a ratio >1.5 in either direction, were analyzed using DAVID (https://david.ncifcrf.gov/) to characterize the biological pathways they are involved in. Processes enriched with a P ≤ 0.05 were considered as significantly correlated to the set of differential proteins. Venn diagram for the comparison of proteomic results from GNS166 and U251 was generated by an online tool from Bioinformatics and Evolutionary Genomics (https://bioinformatics.psb.ugent.be/webtools/Venn/). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD027069 for GNS166 and PXD029393 for U251 cells.
RNA-seq study and target validation
RNA-seq was performed in CD Genomics. Briefly, for library construction, RNA was extracted by TRIzol (Life Technologies). After performing quality control, 200 ng of high quality total RNA was proceeded to library construction. Magnetic beads with Oligo (dT) were used to isolate mRNA. The mRNA was fragmented randomly by adding fragmentation buffer, then the cDNA was synthesized. Short fragments were purified and resolved with EB elution buffer for end repair and single nucleotide A (adenine) addition. After that, the short fragments were connected with sequencing adapters. The double-stranded cDNA library was completed through size selection and PCR enrichment. During the quality control steps, Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System were used in quantification and qualification of the sample library. Finally, the qualified RNA-seq libraries were sequenced using Illumina NovaSeq6000 in CD Genomics. The sequencing was paired-end 150 bp. The FastQC tool was used to perform basic statistics on the quality of the raw reads. Then, sequencing adapters and low-quality data of the sequencing data were removed by Trim Galore. For the alignment, HISAT2 software was used, having human GRCh38 as the reference genome. HTseq software and DESeq were used to quantify transcripts and differential gene expression levels using mapped reads' positional information on the gene. During the process, fold change ≥2 and FDR <0.01 are set as screening criteria. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis results have been represented.
For validation of target genes, reverse transcription was performed using random priming and Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). qRT-PCR was performed by Absolute SYBR Green mix (Thermo Fisher Scientific) in a CFX384 real-time thermal cycler (Bio-Rad). Variations in input RNA were corrected by subtracting PCR threshold cycle values obtained for 18s rRNA. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible in GSE181556.
Western blot analysis
Immunoblots were performed following standard procedures (25). Specific antibodies against LAMP2A (ab18528, Abcam, 1/500), SOX2 (AB5603, Millipore, 1/250), SOX9 (AB5535, Millipore, 1/1,000), TIM23 (11123–1-AP, Proteintech, 1/1,000), MRSP23 (SAB2701383, Sigma, 1/500), p-STAT1 (9167S, Cell Signaling, 1/1,000), STAT1 (14994S, Cell Signaling, 1/1,000), MX1 (ab95926, Abcam, 1/500), p-STAT3 (9145S, Cell Signaling, 1/1,000), STAT3 (9139, Cell Signaling, 1/1,000), ITGA6 (3750S, Cell Signaling, 1/250), ITGB4 (HPA036348, Sigma, 1/500), p-AKT (9271, Cell Signaling, 1/1,000), AKT (sc-8312, Santa Cruz Biotechnology, 1/200), and β-actin (A5441, Sigma-Aldrich, 1/100,000) were used in the study, performing overnight incubations at 4°C in orbital agitation. For secondary antibodies, horseradish peroxidase (HRP)-linked anti-rabbit (7074S, Cell Signaling, 1/2,000), or anti-mouse (7076S, Cell Signaling, 1/2,000) were used, performing incubations of 1 hour at room temperature in orbital agitation. Detection was performed by chemiluminiscence using NOVEX ECL Chemi Substrate and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). Same β-actin were represented when analyzed samples were the same and have been treated in same conditions.
Cytokine detection assay
Human Cytokine Antibody Array C5 Kit (Raybiotech) was performed, following the manufacturer's guidance. Before sample incubation, GNS166 pGK and shL2A supernatants were centrifuged at 1 × 104 rpm for 4 minutes. Array membranes were incubated overnight with 1 mL of infected undiluted supernatant. After that, both biotin-conjugated antibodies and HRP-linked secondary antibody were incubated overnight. Detection was performed by chemiluminescence using Novex ECL Chemi Substrate (Thermo Fisher Scientific).
Metabolic analysis
A total of 1.5 × 104 GNS166 cells were seeded in 10 μg/mL laminin (Sigma-Aldrich) coated XF 96-well plates (Seahorse/Agilent) in six replicates, in 100 μL of GSC medium. For U251 cells assay, experiment was adapted to their culture conditions. All measurements of OCR and ECAR were performed by XF Extracellular Flux Analyzer (Seahorse Bioscience). Mitochondrial activity was evaluated by Seahorse XF Cell Mito Stress Test Kit (Agilent), following manufacturer's guidelines. Oligomycin (Sigma-Aldrich), FCCP (Sigma-Aldrich), and rotenone/antimycin A (Sigma-Aldrich) were used at 1 μmol/L. For the study of glycolytic activity, XF Glycolysis Stress Test (Agilent) was performed. 10 mmol/L glucose (Sigma-Aldrich), 1 μmol/L oligomycin and 50 mmol/L 2-D-deoxy-glucose (Sigma-Aldrich) were used. Results were normalized on the basis of cell content by crystal violet.
Mitochondrial depolarization and reactive oxygen species production measurement
Mitochondrial depolarization and reactive oxygen species (ROS) production were studied by flow cytometry (SH800, Sony). For that, MitoTracker Red FM dye (M22425, Invitrogen), which stains active and polarized mitochondria, and MitoSox Red dye (M36008, Invitrogen), whose mitochondrial oxidation by superoxide produces red fluorescence, were employed. Both dyes were diluted in Hank's Balanced Salt Solution (HBSS; Gibco) and studied separately to avoid spectral overlapping. Briefly, 2 × 105 GNS166, pGK, and shL2A cells were incubated with control HBSS, 5 μmol/L MitoSox or 0.2 μmol/L MitoTracker for 25 minutes at 37°C. After this time, cells were centrifuged, resuspended in HBSS, and analyzed. Thus, we first gated the population by BSC-A versus FSC-A, and excluded doublets by FSC-H versus FSC-W. As both pGK and shL2A conditions were GFP+, manual compensation was applied, where, besides MitoSox and MitoTracker unstained controls, wild-type GNS166 was used as a negative control for GFP. Thus, gates were set for both dyes and analysis was performed reaching at least 104 final events per condition. Fluorescence intensity (%) was represented, which comprises the number of positive events multiplied by the mean fluorescence intensity of them. For ROS scavenging experiment, 100 μmol/L butylated hydroxyanisole (BHA, Sigma, 24 hours) antioxidant agent was employed and 10 μmol/L antimycin A (Sigma, 24 hours) was used as positive control of ROS induction.
Invasion/migration assay
GNS166 pGK and shL2A cell migration was evaluated using 6.5 mm transwell with 8 μm pore polycarbonate membrane inserts (Corning), in a density of 2.5 × 104 cells/well. For invasion assay, 8 μm pore collagen cell invasion assay (Merck-Millipore) was performed in the same density of cells. Both transwell and invasion inserts were previously coated with 2 μg/mL laminin (Sigma-Aldrich), for 3 hours at 37°C. Quantification of migrating and invading cells was performed 72 hours after seeding, by unstaining the inserts and measuring the absorbance at 560 nm in a MultiSkan Ascent microplate reader (Thermo Fisher Scientific).
Data analysis
Data are represented as mean values ± SEM, with the number of experiments (n) carried out for each assay. Unless otherwise indicated, statistical significance was calculated by Student t test (#, P ≤ 0.1; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001). One-way ANOVA with Tukey multiple comparison ad hoc test was used in CMA studies. In Gliovis, pairwise comparisons between group levels with corrections for multiple testing (P values with Bonferroni correction) were used.
Ethics approval
This study was approved by the Clinical Research Ethics Committee of the Donostia University Hospital (protocol AMF-EGM-2016–01) and adhered to the tenets of the Declaration of Helsinki. All processes involving animals were subjected to approval by the Research Animal Care of Biodonostia Institute.
Results
LAMP2 is overexpressed in human GBM and GSCs
We first characterized the expression of LAMP2A in a set of conventional glioma cell lines and patient-derived GSCs. LAMP2A levels varied between cell lines, being patient-derived GSCs the cells with higher LAMP2A expression (Fig. 1A). Moreover, glioma cells cultured as oncospheres in GSC media as well as tumors derived from them (19), presented consistently higher expression of LAMP2A compared with controls (Fig. 1B and C; Supplementary Fig. S1A). In these conditions, SOX2 and SOX9 GSC markers were also elevated (Fig. 1A; Supplementary Fig. S1B; ref. 19). These results suggest that LAMP2A is enriched in GSC population. This finding was extended into clinical data where single-cell transcriptomic in GBM samples showed high expression of LAMP2 in immature astrocytes, glycolytic progenitors, and radial glia GSC cell subtypes (Fig. 1D). In line with this, the levels of LAMP2 positively correlated with several GSCs markers in GBM samples from TCGA cohort (Supplementary Fig. S1C).
Figure 1.

LAMP2 is overexpressed in human GBM and GSCs. A, Representative immunoblot of LAMP2A, SOX2, SOX9, and β-actin in a set of glioma (U87, U373, U251, T98, A172) and patient-derived glioma stem (GNS166, GNS179, GNS144, GB2) cell lines (n = 4). B, Representative image of Western blot analysis for LAMP2A and β-actin in parental and 2ry oncospheres of glioma cell lines (n = 3). C, Representative images of the IHC staining of LAMP2A in subcutaneous tumors from parental U373 and respective oncospheres (n = 2). Scale bar, 100 μm. D, Single-cell RNA-seq results for the expression of LAMP2 among different cell types in glioblastoma samples (https://cells.ucsc.edu/?ds=gbm). E,LAMP2 mRNA expression in control and GBM samples in TCGA (P = 2.4e−13) and Rembrandt (P = 4.5E−03) cohorts. F, mRNA expression of LAMP2 in classical (cla), mesenchymal (me), and proneural (pro) subtypes of GBM from TCGA cohort (Pcla-me = 2.9e−09, Pcla-pro = 2.9e–08, Pme-pro = 4.5e−27) and Rembrandt cohort (Pcla-me = 4.8E−02, Pcla-pro = n.s., Pme-pro = 6.3E−04). G,LAMP2 mRNA expression in grade II–IV of glioma in TCGA (PII–III = n.s., PII–IV = 1.3e−02, PIII–IV = 7.5e−03) and Rembrandt (PII–III = n.s, PII–IV = 3.5e−05, PIII–IV = 3.1e−08) cohorts. H,LAMP2 expression in TMZ treatment of nonresponder (n = 154) and responder (n = 165) patients with GBM (http://www.rocplot.org/). Mann–Whitney test P value: 0.036. I, ROC curve of LAMP2 gene expression in patients with GBM treated with TMZ (n = 319; http://www.rocplot.org/). J, mRNA expression of LAMP2A, LAMP2B, and LAMP2C in GBM samples (n = 20) compared with a mix of healthy brain tissues. K, Representative tissue microarray and overall quantification for LAMP2A protein expression in GBM samples (n = 98). L and M, Representative image and quantification of CMA activity as the average number of puncta per cell (n > 1,500 cells per well in 4–6 independent wells) after transducing control (NIH3T3, N2A), GSCs (GNS166 and 179), and indicated GBM cell lines with photo switchable KFERQ-Dendra CMA reporter. Scale bar, 10 mm. Differences with NIH3T3 cells are compared. *, P ≤ 0.05; ***, P ≤ 0.001; ****, P < 0.0001.
We next studied LAMP2 gene expression in control and GBM human tissues from publicly available TCGA and Rembrandt cohorts, which include a total of 538 and 247 samples, respectively (Gliovis website; ref. 14). Herein, we found that LAMP2 was significantly upregulated in human GBM samples compared with control tissue in both cohorts (Fig. 1E). Within GBM samples, we next wondered whether LAMP2 expression could be differential in any specific molecular subtype. This analysis showed that classical and mesenchymal subtypes presented higher expression of LAMP2 compared with proneural subtype (Fig. 1F). However, no statistical significances were detected in patient survival in each subtype (Supplementary Fig. S2A). Notably, when comparing its expression among different glioma grade samples, LAMP2 expression was higher in grade IV cases in TCGA cohort, linking its high levels to tumor malignancy (Fig. 1G). Consistent with this, TMZ treatment nonresponder patients displayed a significantly higher expression of LAMP2 (Fig. 1H), and these high levels presented a significant AUC sensitivity (Fig. 1I). Moreover, we analyzed the expression of the three isoforms of LAMP2 in a cohort established in Donostia Hospital (19), finding that LAMP2A and LAMP2B were elevated in human GBM samples when compared with healthy brain tissue (Fig. 1J). In line with this, tissue microarray analysis revealed that LAMP2A staining was high in 60% of GBM samples with only 13% expressing low or no levels (Fig. 1K).
To directly test that elevated LAMP2A levels resulted in higher CMA activity in GBM, we monitored CMA activity in the different glioma cell lines using a photoconvertible reporter that behaves as a CMA substrate and highlight lysosomes as fluorescent puncta upon its delivery to this organelle via CMA (20). Quantification of the fluorescent puncta revealed that basal CMA activity was significantly higher in all glioma cells analyzed when compared with nontumor cells, being the highest in patient-derived GSCs and U373 cells (Fig. 1L and M). Similar results were obtained in conditions where CMA activity is induced such as starvation by serum removal or treatment with the oxidative stress inductor paraquat (Supplementary Figs. S2B and S2C). Combined, these data reveal an overexpression of LAMP2A and elevated CMA activity in GSCs and GBM biopsies.
LAMP2A knockdown suppresses patient-derived GSCs activity
Given that LAMP2A expression is enriched in GSC subpopulation, we characterized its function on them. First, we developed lentiviral infections for the knockdown of LAMP2A (shL2A) in U251 and U373 cells (Fig. 2A). The shL2A cells presented a reduction of an 80% in colony formation ability (Fig. 2B) and a nearly 60% diminishment in the ability of 1ry and 2ry oncosphere generation (Fig. 2C; Supplementary Fig. S3A). These results were accompanied by a significant decrease in the expression of SOX2 and SOX9 stem cell markers in shL2A cells (Supplementary Fig. S3B). On the contrary, LAMP2A overexpression significantly increased the ability of U251 glioma cells to form oncospheres (Fig. 2D). In line with this, treatment with QX77, a pharmacologic activator of CMA, also enhanced the number of oncospheres (Fig. 2E). Moreover, LAMP2A overexpression rescued the impaired oncosphere and colony formation capacity of shL2A cells (Fig. 2D; Supplementary Fig. S3C). These data suggest that LAMP2A could modulate the activity of glioma cells with stem cell properties.
Figure 2.

LAMP2A silencing reduces GSC proliferation and tumorigenicity. A, Representative Western blot analysis for LAMP2A and β-actin in pGK and shL2A U251 cells (n = 3). B, Relative number of colonies formed by pGK and shL2A U251 cells (n = 3). C, Relative number of primary and secondary oncospheres generated by pGK and shL2A U251 cells at 10 and 20 days, respectively (n = 3). D, Relative number of primary oncospheres generated by control and LAMP2A overexpressing cells in pGK or shL2A U251 (n = 3). E, Relative number of oncospheres generated by control or after 20 μmol/L QX77 treatment (n = 3). F, Immunoblot of LAMP2A in pGK and shL2A GNS166 and GNS179 cells (n = 3). G, Number of proliferating cells at day 5 relative to day 1 in pGK and shL2A indicated cells (n ≥ 3). H, Quantification of immunofluorescence of p-H3 in pGK and shL2A GNS166 (n = 3), GNS179 (n = 4), U251 (n = 2), and U373 (n = 3) cells. I, Number of proliferating cells at day 5 relative to day 1 after LAMP2A expression rescue (n = 3). J, Quantification of immunofluorescence of caspase-3 in pGK and shL2A GNS166 and GNS179 cells (n = 4). K, Relative mRNA expression of GSC markers SOX2, SOX9, and NESTIN in pGK and shL2A GNS166 (n = 3) and GNS179 cells (n = 2). L, Relative cell viability of pGK and shL2A GNS166 and U251 cells after 72 hours with control and 2 mmol/L or 250 μmol/LTMZ treatment, respectively (n = 3). M, Relative primary oncospheres generated by pGK and shL2A U251 cells, after 50 μmol/L TMZ treatment (n = 3). #, P ≤ 0.1; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
To directly unravel the impact of LAMP2A in GSC population, we carried out the same strategy of silencing of LAMP2A in two patient-derived glioma stem cell lines (GNS166 and GNS179 cells; Fig. 2F). Interestingly, shL2A cells displayed a significant reduction in proliferative capacity, measured by cell counting (Fig. 2G; Supplementary Fig. S3D), and number of positive cells for the mitosis marker phospho-histone3 (p-H3) by immunofluorescence (Fig. 2H; Supplementary Fig. S3E). Similar results were also observed in U251 and U373 glioma cells in these experiments (Fig. 2G and H; Supplementary Figs. S3D and S3E). Of note, impaired proliferation phenotype was also rescued by LAMP2A overexpression (Fig. 2I; Supplementary Fig. S3F). Finally, shL2A GNS 166 and 179 cells were more prone to apoptosis as the number of positive cells for caspase-3 were significantly increased in shL2A GNS cells compared with controls (Fig. 2J). Besides, they displayed reduced levels of several stem cell markers (Fig. 2K). Consistent with these results, GNS166 and U251 shL2A cells were more sensitive to TMZ treatment in cell viability and oncosphere formation assays (Fig. 2L and M; Supplementary Fig. S3G). Together, these results reveal a role of LAMP2A in GSC activity.
Proteome and transcriptome studies reveal multiple pathways altered in LAMP2A knockdown GSCs
To get a deep view regarding molecular changes associated to LAMP2A knockdown in GSCs, we performed high throughput proteome and transcriptome-wide studies in control and shL2A GNS166 cells. The proteome study showed a total number of 101 upregulated and 172 downregulated proteins in shL2A cells (Fig. 3A). Among them, previously described CMA substrates such as Annexin A6 (26) and HSC70 (27) proteins were elevated (Fig. 3B; Supplementary Data S1), further validating the inhibition of CMA function in those cells. Moreover, shL2A cells showed diminished protein expression of CD44, CD109, and EGFR (Fig. 3B; Supplementary Data S1), whose implication in GBM and GSC proliferation and maintenance has been reported previously (28–30). When analyzing all altered proteins by integrating them into biological pathways and processes, significant deregulation in chaperonins and other protein translation regulators, consistent with CMA role in protein quality control, as well as IFN signaling and antigen presentation, mitochondrial metabolism, and extracellular matrix (ECM)-related pathways were detected (Fig. 3C; Supplementary Fig. S4A). A similar proteome study in U251 shL2A cells showed over 400 altered proteins (Fig. 3D), with only 60 deregulated proteins shared in bulk tumor cells and glioma stem cells (Fig. 3D and E; Supplementary Data S2), revealing differences among bulk tumor and glioma stem cells. However, among biological pathways and processes, chaperone function and other protein-related activities were also significantly altered, as well as ECM interaction, metabolism, and mitochondria, among others (Supplementary Fig. S4B).
Figure 3.

LAMP2A silencing in GSCs alters crucial proteomic and transcriptomic pathways. A, Volcano plot of upregulated and downregulated proteins in proteome study of GNS166 pGK versus shL2A cells (n = 4). B, Representative heat maps of upregulated and downregulated proteins in the proteome study. C, Ontology analysis based on Reactome database of altered proteome. D, Volcano plot of upregulated and downregulated proteins in U251 pGK versus shL2A proteomics (n = 4). E, Venn diagram representing the comparison of proteins altered in GNS166 and U251 proteomic studies. F, Representative heatmap of upregulated and downregulated transcripts in RNA-seq study of pGK vs. shL2A GNS166 cells (n = 3). G, Dot plot representation of the main altered biological processes after gene ontology analysis of RNA-seq study in F.
Among altered pathways in the proteomic study, processes related to mRNA processing were also highlighted (Fig. 3C; Supplementary Fig. S4B), suggesting that LAMP2A downregulation may also alter the transcriptome of GSCs. Interestingly, RNA-seq study revealed a total number of 1299 transcripts significantly downregulated, whereas 1673 appeared upregulated (Fig. 3F; Supplementary Data S3). KEGG pathway analysis highlighted PI3K–AKT and p53 signaling and pathways associated with ECM interactions, actin cytoskeleton, and focal adhesion as the main altered pathways (Fig. 3G). These results indicate that CMA regulates multiple pathways at proteome and transcriptome level in GSCs.
Knockdown of LAMP2A impairs metabolism and mitochondrial function of GSCs
CMA can selectively promote the degradation of enzymes operating in glucose or lipid metabolism pathways and regulate energy homeostasis (31). A main feature altered in the proteome was mitochondrial activity (Fig. 3C; Supplementary Figs. S4A and S4B). Blockage of CMA in hepatocytes has been shown to result in higher levels of mitochondrial proteins, mostly enzymes implicated in the TCA cycle and lipid metabolism (32). In contrast, the proteome study of LAMP2A knockdown in GNS166 cells revealed a significant decrease in levels of mitochondrial ribosomal proteins such as MRP-S23, MRP-S11, or MRP-L3, and proteins regulating mitochondrial functionality such as TIM23, COX VIb-1, NDUFAF7, and AKB ligase (Fig. 3B, Supplementary Data S1), suggesting a distinct molecular mitochondrial pattern in cancer stem cells. Of note, immunoblot studies validated the decrease of TIM23 and MRP-S23 protein levels in shL2A cells (Fig. 4A; Supplementary Fig. S5A), and there was a positive correlation between LAMP2 and all of them in clinical samples (Fig. 4B). To functionally validate these results, we determined the effects of LAMP2A knockdown on the cellular metabolism of glioma cells using Agilent Seahorse XF technology. We first measured the oxygen consumption rate (OCR) in both control and LAMP2A knockdown GNS166 and U251 cells (Fig. 4C; Supplementary Fig. S5B). Notably, shL2A cells presented a reduction of nearly 40% and 60% of the basal and carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) induced maximal respiration, respectively (Fig. 4D and E), which lead to a reduction of approximately 75% in the spare respiratory capacity of these cells (Fig. 4F). These results were accompanied by a reduction in ATP production via mitochondrial oxidative phosphorylation system (OXPHOS) activity (Fig. 4G), indicating that LAMP2A knockdown impairs mitochondrial activity and thus, the production of ATP in GSCs. Moreover, this assay also demonstrated that the shL2A cells presented reduced nonmitochondrial O2 consumption at approximately 50% (Fig. 4H). In addition, flow cytometry assays in GNS166 confirmed that shL2A cells showed more depolarized mitochondria (Fig. 4I) and demonstrated that they accumulated more mitochondrial ROS (Fig. 4J), which was partially rescued with BHA hydrophilic antioxidant agent (Supplementary Figs. S5C and S5D).
Figure 4.

LAMP2A silencing diminishes mitochondrial metabolism and induces glycolysis in GSCs. A, Immunoblot of mitochondrial markers TIM23 and MRP-S23, and β-actin in pGK and shL2A GNS166, GNS179, and U251 (n ≥ 3) cells; β-actin blots in U251 and GNS179 were shared with Fig. 5A. B, Association study of LAMP2 with metabolism genes MRPS23 (P = 1.336e−05), MRPS11 (P = 2.972e−05), MRPL3 (P = 5.80e−10), COX6B1 (P = 2.086e−04), NDUFAF7 (P = 3.970e−04), and GCAT (AKB ligase; P = 0.0430) in TCGA cohort (cBioPortal for cancer genomics, https://www.cbioportal.org/). C, Normalized OCR response in pGK (black) and shL2A (green) GNS166 in basal conditions and after consecutive addition of oligomycin 1 μmol/L, FCCP 1 μmol/L, and rotenone/antimycin A 1 μmol/L. A representative experiment is shown from a total of n = 3. D–H, Quantification of mitochondrial respiratory functions of pGK and shL2A GNS166 and U251 (n = 3). I, Quantification of mitochondrial polarization measured by fluorescent intensity of MitoTracker Red FM dye via flow cytometry (n = 3). J, Quantification of mitochondrial ROS analyzed by MitoSox Red via flow cytometry (n = 3). K–M, Quantification of glycolytic functions based on kinetic normalized ECAR response of pGK and shL2A GNS166 in basal conditions and after consecutive addition of glucose 10 mmol/L, oligomycin 1 μmol/L, and 2-D-deoxy-glucose 50 mmol/L (n = 3). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Next, we studied glycolysis, a main metabolic pathway for cancer cells, by measuring the extracellular acidification (ECAR) of GNS166 cells. Intriguingly, and contrary to what has been described before in bulk cancer cells (33), shL2A GNS166 cells displayed an approximately 50% increase on the levels of glycolysis in glucose saturating conditions (Fig. 4K), indicating that these cells induce this metabolic pathway to compensate the diminished ATP production occurred by mitochondrial dysfunction. Indeed, when mitochondrial OXPHOS is inhibited, both control and LAMP2A silenced cells present the same glycolytic capacity (Fig. 4L), which highlights the basal dysfunction of the mitochondria in shL2A cells. Thus, these cells presented a smaller difference between the basal and the OXPHOS inhibited glycolysis levels, which is represented by a reduction in the glycolytic reserve (Fig. 4M). All these data indicate that LAMP2A knockdown in GSCs impairs mitochondrial function and increases glycolysis as a potential compensatory mechanism. Importantly, these results confirm the impact of CMA in GSC metabolism and mitochondrial function.
LAMP2A knockdown impairs inflammatory response and immune system in GSCs
In relation to inflammatory response and immune system, a total number of 10 proteins of IFN signaling were upregulated in shL2A cells (Supplementary Data S1), which are implicated in both α/β and γ IFN signaling (Fig. 3B). Of note, these proteins included critical mediators such as STAT1, which also exerts an important function in growth arrest and apoptosis (34), as well as MX1, an IFN-induced dynamin-like GTPase (Fig. 3C; ref. 35). Immunoblot studies validated the higher levels of STAT1 and MX1 in shL2A cells (Fig. 5A; Supplementary Fig. S6A). These cells also presented elevation of the phosphorylated form of STAT1 (Fig. 5A; Supplementary Fig. S6A), indicating an activation of the protein. In line, these data were accompanied by elevation of IFNγ mRNA expression (Fig. 5B). In addition to the increase of those proteins directly degraded by CMA upon LAMP2A knockdown, we also observed subsets of proteins found at lower levels in these cells. Among them, the proteomic study revealed MHC class II antigen presentation alterations including reduction of key effectors such as AP2M1, UKHC, DPP1, and cathepsin D (Fig. 3B and C; Supplementary Data S1).
Figure 5.

LAMP2A downregulation reduces cytokine secretion of GSCs. A, Representative immunoblot of indicated proteins in pGK versus shL2A GNS166, GNS179, and U251 (n ≥ 3). β-Actin blots in this figure and Fig. 4A in GNS179 and U251 are the same as they derive from the same samples. B, Relative mRNA expression of IFNγ in pGK and shL2A GNS166, GNS179, and U251 cells (n = 4). C, Representative heatmap of the altered secretion of cytokines from supernatants of GNS166 pGK and shL2A (P < 0.05, n = 4). D, Relative mRNA expression of LAMP2A, IGFBP2, IGFBP4, ANG, Eotaxin-3, SDF-1, and IP10 in pGK and shL2A GNS166 (n = 8). E, Association study of LAMP2 with IGFBP4 (P = 1.904e−03), ANG (P = 2.40e−13), Eotaxin-3 (P = 3.56e−07), and SDF1 (P = 1.701e−03) in the TCGA cohort (cBioPortal for cancer genomics, https://www.cbioportal.org/). F, Dot plot of KEGG enrichment analysis of LAMP2 high versus low in the TCGA cohort (P < 0.05). #, P ≤ 0.1; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
To further characterize the impact of CMA blockage in the immune response, we studied the comprehensive profile of cytokines secreted by shL2A GSCs. For this, we performed a cytokine expression array from supernatants of control and shL2A GNS166 cells. Interestingly, this study showed 50 significantly reduced cytokines including among the most reduced IGFBP4, ANG, VEGF, IL6, IGFBP2, TGFβ, IFNγ, Eotaxin-3, and SDF1 (Fig. 5C; Supplementary Fig. S6B). On the contrary, only 1 cytokine, IP10, also known as CXCL10, was elevated on shL2A supernatants (Fig. 5C; Supplementary Fig. S6B). In line with this, cytokines such as IGFBP4, ANG, IGFBP2, Eotaxin-3, and SDF1 were reduced at mRNA level, whereas IP10 was elevated in shL2A GNS 166, and 179 cells (Fig. 5D; Supplementary Fig. S6C). However, this analysis also revealed elevated mRNA levels of VEGF, IGFBP4, IL6, and IL1-α (Supplementary Fig. S6D). Moreover, the significant reduction of cytokines in shL2A GSCs was accompanied by a reduction of the phosphorylated form of STAT3 (Fig. 5A; Supplementary Fig. S6A), a major modulator of inflammation and immune modulation and well-established downstream effector of cytokines and growth factors in GBM (36). To further characterize the link between LAMP2A and immune modulation, we moved to clinical samples. Data of GBM samples from TCGA cohort showed positive correlation between LAMP2 mRNA and IGFBP4, ANG, Eotaxin-3, and SDF1 as well as STAT3, STAT1, and MX1 expression and MHC class II antigen presentation related genes (Fig. 5E; Supplementary Fig. S6E). Moreover, KEGG analysis of pathways differentially expressed in high versus low LAMP2 expressing brain GBM tissues from TCGA cohort presented inflammation-associated pathways (Fig. 5F). All these data indicate that CMA controls GSCs inflammatory response and immune system.
LAMP2A knockdown alters ECM-related pathways in GSCs
ECM proteins have been described to establish a physical and biochemical niche for GSCs (37), as well as regulating GSCs migratory response (38). Interestingly, pathways related to ECM interactions were both deregulated in proteome and transcriptome analysis of shL2A GSCs (Supplementary Data S1 and S3). The reduction in the expression of markers such as ITGA3, ITGA6, ITGAV, ITGB3, ITGB4, LAMC1, COL4A5, and FN1 was validated in both GNS and U251 cells at mRNA and protein levels (Fig. 3B; Fig. 6A–C; Supplementary Fig. S7A). Of note, association studies in GBM samples from TCGA cohort supported the role of CMA in integrin-mediated GSC–ECM interaction since all the studied ECM interaction markers positively correlated with LAMP2 gene expression (Fig. 6D; Supplementary Fig. S7B). To test the impact of these molecular changes in GSC phenotype, we next performed transwell migration and collagen invasion assays in control and shL2A GNS166 cells. Herein, shL2A cells presented ∼ 60% reduced migration capacity (Fig. 6E), and approximately 40% reduction in the invasion ability (Fig. 6F).
Figure 6.

LAMP2A silencing in GSCs diminishes ECM interaction and content. A and B, Representative immunoblot of ITGA6, ITGB4, and β-actin in GNS166 (n = 4) and U251 (n = 3) cells (A), and respective quantification (B). C, Relative mRNA expression of ECM interaction markers ITGA3, ITGA6, ITGAV, ITGB3, ITGB4, LAMC1, COL4A5, and FN1 in pGK and shL2A GNS166 and U251 cells (n = 3/4). D, Association study of LAMP2 with ITGAV (P = 1.31e−20), ITGA6 (P = 8.90e−10), ITGA3 (P = 8.92e−09), and ITGB4 (P = 1.98e−08) in the TCGA cohort (cBioPortal for cancer genomics, https://www.cbioportal.org/). E and F, Representative images (left; E) and quantification (right) of migration and invasion of pGK and shL2A GNS166 cells (n = 4; F). #, P ≤ 0.1; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
RNA-seq study revealed within PI3K–AKT pathway elevated level of PI3CA and mTOR in shL2A GSCs (Supplementary Data S2), which was further validated in both GNS166 and U251 cell lines (Supplementary Fig. S8A). Accordingly, phosphorylated form of AKT was more abundant in LAMP2A-silenced GSCs (Supplementary Fig. S8B), Moreover, transcriptomic analysis and subsequent validation also showed a downregulation of the receptor PDGFRA and an upregulation of the negative regulator PTEN (Supplementary Fig. S8C), suggesting a role of this pathway in the effects on metabolism and proliferation. Consistent with the decreased proliferation of shL2A cells, p53 pathway was activated and Cyclin A was downregulated (Supplementary Data S2), being the latter one further validated by qPCR in shL2A GNS166 and U251 cells (Supplementary Fig. S8D).
LAMP2A levels modulate GBM survival
To extend the in vitro studies, we injected control and LAMP2A knockdown GNS166 cells intracranially into the caudate nucleus of immunodeficient mice and checked tumor formation and survival rates. Notably, shL2A cells presented delayed and less tumors than controls and led to significantly extended mice survival (Fig. 7A). In particular, although control cells formed tumors from day 64 onwards, shL2A did not until day 114 post-inoculation. Moreover, 75% of control mice bared detectable tumors at day 130, whereas only 22% did in shL2A conditions.
Figure 7.
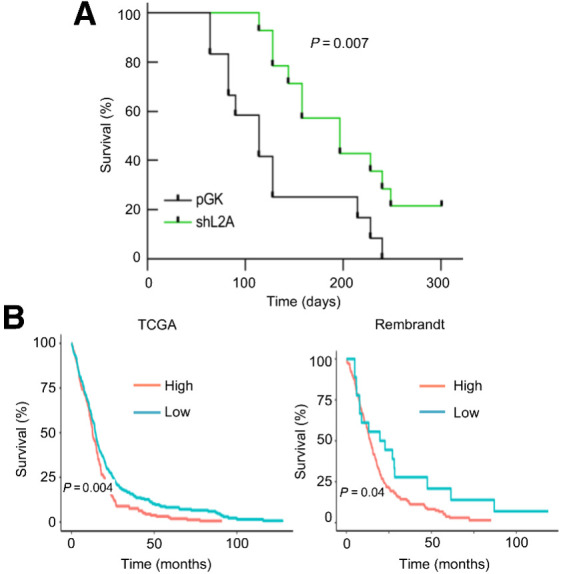
LAMP2A correlates with poor overall survival in GBM. A, Kaplan–Meier survival curve of orthotopic intracranial xenograft of GNS166 pGK and shL2A cells in NOD.SCID immunodeficient mice (n = 12 per condition). B, Kaplan–Meier curves representing survival of patients with low versus high expression of LAMP2 in TCGA (left, n = 340 vs. n = 185, respectively) and Rembrandt (right, n = 18 vs. n = 185, respectively) cohorts. Optimal cutoff points for Kaplan–Meier curves were designated by GlioVis database.
Finally, we studied whether LAMP2 levels could correlate with patient survival in the whole set of patients from TCGA and Rembrandt cohorts. Notably, high levels of LAMP2 correlated with decreased overall patient survival in both cohorts (Fig. 7B). Combined, these data reveal a role of LAMP2A in GSC tumorigenicity and demonstrate its potential as prognosis marker in GBM.
Discussion
Although CMA and LAMP2A overexpression has been detected in several types of cancer (4), only two recent studies determined its implication in GBM, one particularly in tumor-associated pericytes (13) and another one in glioma bulk cells (39). Importantly, the role of CMA in cancer stem cells has remained unknown so far. Our data revealed an intrinsic role of CMA in maintaining GSC activity through the modulation of multiple pathways and processes.
The role of autophagy in GBM is complex and seems to parallel the functions that it exerts during physiology. Several studies showed that MA is activated and promotes apoptosis in response to several stressors in glioma cells (40–42). In this line, clinical trials with inhibitors of lysosomal proteolysis that will block MA but also any other type of autophagy such as chloroquine have been performed with promising results (43, 44). Under unfavorable conditions during advanced stages, the activation of MA plays a crucial role in the survival of GBM as it provides metabolic support and prevents senescence (45), supports the maintenance and invasion of GSCs (46, 47), and confers resistance to chemotherapy. In relation to CMA, LAMP2A levels were elevated in small cohorts of glioblastoma clinical samples, paralleling high levels of MA markers (39, 48). Our results validated and extended these results as we detected increased LAMP2 in advanced glioma grade and in GBM samples in TCGA and Rembrandt large cohorts as well as a specific upregulation of LAMP2A in an additional one established in Donostia Hospital. Importantly, LAMP2 higher levels were associated to poor overall survival in the two GBM large and public cohorts.
Our results show LAMP2A upregulation in GSC population using different approaches, whereas its knockdown significantly decreases GSC activities. Although a possible limitation of our study is that we used only a short hairpin RNA-targeting LAMP2A, the fact that this shRNA was extensively compared with two additional ones in early studies and the three shRNA led to similar changes in cell proliferation and cell death in response to stressors, minimize concerns about unintended off-target effects of this shRNA (21). In this line, an additional study silenced LAMP2A in conventional U87MG glioma cell line showing also enhancement of apoptosis and reduction of tumor growth (39). Moreover, we performed experiments with LAMP2A overexpression detecting enhancement of GSC activities. Consistent with this and the idea that GSCs play a relevant role in therapy resistance and recurrence in GBM (11), we detected that LAMP2A knockdown sensitizes GSCs to TMZ treatment, whereas TMZ nonresponder patients with GBM present higher expression of LAMP2 gene compared with responders. In line with our data, a previous study showed that tumor samples of patients of GBM treated with TMZ displayed increased expression of LAMP2A compared with pretreated conditions (12). Of note, our study not only focused on LAMP2A expression but also used a reporter that behaves as a CMA substrate and confirmed functional upregulation of CMA. A recent publication revealed that CMA is involved in sustaining of hematopoietic stem cell (HSC) function in adult mice (49). Thus, our study, together with this, suggests that CMA activity regulates stem cell maintenance not only under physiologic state, but also in pathologic conditions.
Proteomic and transcriptomic analysis revealed a variety of pathways and processes by which CMA could control GSC activity. They seem to be modulated at distinct regulatory levels, as anticipated by the variety of functional protein groups normally degraded by this pathway (4). Among them, ECM effectors were decreased both at the transcriptional and protein level, whereas immune system and mitochondrial metabolism-related pathways were deregulated at proteome level, and PI3K–AKT and p53 pathways were altered in RNA-seq study. In the same way, previous analysis of CMA targetome in ovarian, breast, fibrosarcoma, and lung cancer cells demonstrated alterations in processes such as translation and RNA regulation or intercellular transport (50, 51), which are only partly observed in our model. These differences could suggest a cell-type–dependent regulation of CMA, with some common processes in normal tumor bulk and cancer stem cells, but for the most part, very distinctive mechanism in these last ones. Further extending the differences between stem and differentiated cells, the recent study in HSCs of LAMP2A knockout mice revealed changes in cell growth, survival, movement, cell-to-cell signaling, and metabolic alterations in proteomic and transcriptomic studies (49). In contrast, neurons from LAMP2A knockout mice presented alterations in protein trafficking, calcium homeostasis, and metabolism (52).
Cellular energetics have been associated with CMA activity during the last decade (31). In our work, we reveal that LAMP2A downregulation reduces mitochondrial activity in GSCs, leading to a diminished production of ATP. Previous studies reported that CMA blockage compromises mitochondrial respiration in lung and melanoma cancer cell lines (33), whereas increased CMA activity alleviated mitochondrial dysfunction under oxidative stress in mouse dopaminergic neurons (53). LAMP2A knockdown GSCs displayed upregulation of glycolysis along with mitochondrial dysfunction, suggesting a potential compensatory mechanism for the obtention of ATP. Previous studies revealed reduced glycolysis and lipid metabolism in LAMP2A knockout HSCs (49) and neurons (52), indicating a cell-type and context-dependent modulation of metabolic pathways upon CMA downregulation. GSCs are a metabolically heterogeneous and adaptive cell population able to shift between glycolytic and oxidative phenotypes (54, 55).Thus, although blockage of CMA in other cancer cells led to reduced mitochondrial function, as the one observed here in GSCs, in that case glycolysis was significantly downregulated and restoration of glycolytic function was enough to restore tumorigenic ability (33). In contrast, in the case of GSCs, the observed changes in glycolysis are instead secondary to the mitochondrial function that appears to be the primary defect.
A clinical hallmark of GBM is the extensive infiltrative capacity of tumor cells (56), where remodeling of ECM is crucial (38). Indeed, although gliomas very rarely metastasize out of the brain, their invasive capacity let them colonize diverse areas, triggering relapse in distant areas from the primary tumor (56). Our results show that LAMP2A downregulation alters the expression of key effectors in ECM interaction of GSCs, both at protein and mRNA level, suggesting that CMA controls ECM content and GSC interaction with ECM.
Our proteomic study also revealed an elevation of IFN signaling and reduction of MHC class II antigen-presentation and a set of pro-inflammatory cytokines, suggesting a role of CMA in immune response in GBM via STAT family members. Future work should elicit which cytokines are critical, as some of the factors were not validated, and whether the effect is specific of GSCs. Among the identified factors, insulin growth factor binding proteins (IGFBP), ILs, and angiogenic factors were the most diminished ones and have been previously associated with GBM progression (13, 57, 58). The alteration in the GSCs secretion of angiogenic factors by LAMP2A downregulation was of special interest, as a previous study has demonstrated the implication of CMA in the crosstalk between glioma cells and pericytes (13), functional and critical contributors of tumor angiogenesis. Indeed, there are evidences that demonstrate that GSCs can transdifferentiate into pericyte-like cells, supporting the interaction between these cell types (59). Thus, our results might suggest that high levels of CMA in GSCs could contribute to the secretion of angiogenic factors, inducing the interactions with tumor-associated pericytes, and thus, possibly contributing to tumor survival.
Overall, our work demonstrates that LAMP2A expression and CMA is enriched in GSC population, it is associated with poor patient survival and it is necessary for the maintenance of GSC tumorigenic activities. At molecular level, LAMP2A controls distinct biological and regulatory pathways including mitochondrial activity, immune response, and interactions with ECM in GSCs.
Supplementary Material
Acknowledgments
J. Auzmendi-Iriarte, M. Otaegi-Ugartemendia, A. Saenz-Antoñanzas, and M. Garcia-Puga received predoctoral fellowships from Department of Education, University and Research of the Basque Government (PRE_2016_1_0375), Spanish Ministry of Universities (FPU18/04540), Instituto de Salud Carlos III (FI17/00250), and University of the Basque Country (PIF 15/245), respectively. E. Carrasco-Garcia received a Stop Fuga de Cerebros fellowship and a Miguel Servet contract (CP19/00085) from Instituto de Salud Carlos III. The authors thank the Neurooncology Committee of the Donostia Hospital and the Pathology Service of Biodonostia for their help, the Basque Biobank for Research O+EHUN (http://www.biobancovasco.org) for providing the human glioblastoma samples and CD Genomics (USA) company for performing RNA-seq study. The proteostasis of aging core (autophagy platform) was supported by NIH grant no. P30AG038072. A. Matheu lab was supported by grants from Instituto de Salud Carlos III and FEDER Funds (DTS16/00184, PI16/01580, DTS18/00181, PI19/01355) and Industry and Health Departments of the Basque Country.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
A. Cuervo reports grants from NIH during the conduct of the study; personal fees from Generian Therapeutics, Cognition Pharmaceutical, and Life Biosciences outside the submitted work; also has a patent for US9512092 issued. No disclosures were reported by the other authors.
Authors' Contributions
J. Auzmendi-Iriarte: Formal analysis, investigation, methodology, writing–original draft. M. Otaegi-Ugartemendia: Investigation, writing–original draft. E. Carrasco-Garcia: Investigation, writing–review and editing. M. Azkargorta: Investigation, writing–review and editing. A. Diaz: Investigation, writing–review and editing. A. Saenz-Antoñanzas: Investigation, writing–original draft. J.A. Andermatten: Resources, investigation, writing–review and editing. M. Garcia-Puga: Formal analysis, investigation, writing–review and editing. I. Garcia: Investigation, writing–review and editing. A. Elua-Pinin: Resources, investigation, writing–review and editing. I. Ruiz: Resources, investigation, writing–review and editing. N. Sampron: Resources, investigation, writing–review and editing. F. Elortza: Formal analysis, investigation, writing–review and editing. A.M. Cuervo: Resources, funding acquisition, investigation, writing–review and editing. A. Matheu: Conceptualization, formal analysis, supervision, funding acquisition, investigation, writing–review and editing.
References
- 1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 2. Yang K, Niu L, Bai Y, Le W. Glioblastoma: targeting the autophagy in tumorigenesis. Brain Res Bull 2019;153:334–40. [DOI] [PubMed] [Google Scholar]
- 3. Abdrakhmanov A, Gogvadze V, Zhivotovsky B. To eat or to die: deciphering selective forms of autophagy. Trends Biochem Sci 2020;45:347–64. [DOI] [PubMed] [Google Scholar]
- 4. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 2018;19:365–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol 1995;269:C1200–8. [DOI] [PubMed] [Google Scholar]
- 6. Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 2004;15:4829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun 2015;6:6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rybstein MD, Bravo-San Pedro JM, Kroemer G, Galluzzi L. The autophagic network and cancer. Nat Cell Biol 2018;20:243–51. [DOI] [PubMed] [Google Scholar]
- 9. Arias E, Cuervo AM. Pros and Cons of chaperone-mediated autophagy in cancer biology. Trends Endocrinol Metab 2020;31:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryskalin L, Gaglione A, Limanaqi F, Biagioni F, Familiari P, Frati A, et al. The autophagy status of cancer stem cells in gliobastoma multiforme: from cancer promotion to therapeutic strategies. Int J Mol Sci 2019;20:3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parada LF, Dirks PB, Wechsler-Reya RJ. Brain tumor stem cells remain in play. J Clin Oncol 2017;35:2428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Natsumeda M, Aoki H, Miyahara H, Yajima N, Uzuka T, Toyoshima Y, et al. Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 2011;31:486–93. [DOI] [PubMed] [Google Scholar]
- 13. Valdor R, Garcia-Bernal D, Riquelme D, Martinez CM, Moraleda JM, Cuervo AM, et al. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 2019;116:20655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol 2017;19:139–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Menyhart O, Fekete JT, Gyorffy B. Gene expression-based biomarkers designating glioblastomas resistant to multiple treatment strategies. Carcinogenesis 2021;42:804–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sören Müller EDL, Bhaduri A, Alvarado B, Yagnik G, Kohanbash G, Aghi M, Aaron Diaz. A single-cell atlas of human glioblastoma reveals a single axis of phenotype in tumor-propagating cells. bioRxiv 2018. doi: 10.1101/377606. [DOI]
- 18. Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009;4:568–80. [DOI] [PubMed] [Google Scholar]
- 19. Garros-Regulez L, Aldaz P, Arrizabalaga O, Moncho-Amor V, Carrasco-Garcia E, Manterola L, et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin Ther Targets 2016;20:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun 2011;2:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 2006;103:5805–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arrizabalaga O, Moreno-Cugnon L, Auzmendi-Iriarte J, Aldaz P, Ibanez de Caceres I, Garros-Regulez L, et al. High expression of MKP1/DUSP1 counteracts glioma stem cell activity and mediates HDAC inhibitor response. Oncogenesis 2017;6:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009;6:359–62. [DOI] [PubMed] [Google Scholar]
- 24. Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 2016;13:731–40. [DOI] [PubMed] [Google Scholar]
- 25. Matheu A, Klatt P, Serrano M. Regulation of the INK4a/ARF locus by histone deacetylase inhibitors. J Biol Chem 2005;280:42433–41. [DOI] [PubMed] [Google Scholar]
- 26. Cuervo AM, Gomes AV, Barnes JA, Dice JF. Selective degradation of annexins by chaperone-mediated autophagy. J Biol Chem 2000;275:33329–35. [DOI] [PubMed] [Google Scholar]
- 27. Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, et al. Chaperone-mediated autophagy at a glance. J Cell Sci 2011;124:495–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown DV, Filiz G, Daniel PM, Hollande F, Dworkin S, Amiridis S, et al. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS One 2017;12:e0172791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Silver DJ, Lathia JD. Revealing the glioma cancer stem cell interactome, one niche at a time. J Pathol 2018;244:260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Talukdar S, Emdad L, Das SK, Fisher PB. EGFR: An essential receptor tyrosine kinase-regulator of cancer stem cells. Adv Cancer Res 2020;147:161–88. [DOI] [PubMed] [Google Scholar]
- 31. Tasset I, Cuervo AM. Role of chaperone-mediated autophagy in metabolism. FEBS J 2016;283:2403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 2014;20:417–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 2011;3:109ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swiatek-Machado K, Kaminska B. STAT signaling in glioma cells. Adv Exp Med Biol 2013;986:189–208. [DOI] [PubMed] [Google Scholar]
- 35. Aljohani AI, Joseph C, Kurozumi S, Mohammed OJ, Miligy IM, Green AR, et al. Myxovirus resistance 1 (MX1) is an independent predictor of poor outcome in invasive breast cancer. Breast Cancer Res Treat 2020;181:541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Piperi C, Papavassiliou KA, Papavassiliou AG. Pivotal role of STAT3 in shaping glioblastoma immune microenvironment. Cells 2019;8:1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nallanthighal S, Heiserman JP, Cheon DJ. The role of the extracellular matrix in cancer stemness. Front Cell Dev Biol 2019;7:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Herrera-Perez M, Voytik-Harbin SL, Rickus JL. Extracellular matrix properties regulate the migratory response of glioblastoma stem cells in three-dimensional culture. Tissue Eng Part A 2015;21:2572–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y, Zhang B, Wang J, Wu H, Xu S, Zhang J, et al. Discovery of LAMP-2A as potential biomarkers for glioblastoma development by modulating apoptosis through N-CoR degradation. Cell Commun Signal 2021;19:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu WT, Huang CY, Lu IC, Gean PW. Inhibition of glioma growth by minocycline is mediated through endoplasmic reticulum stress-induced apoptosis and autophagic cell death. Neuro Oncol 2013;15:1127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pallichankandy S, Rahman A, Thayyullathil F, Galadari S. ROS-dependent activation of autophagy is a critical mechanism for the induction of anti-glioma effect of sanguinarine. Free Radic Biol Med 2015;89:708–20. [DOI] [PubMed] [Google Scholar]
- 42. Abdul Rahim SA, Dirkse A, Oudin A, Schuster A, Bohler J, Barthelemy V, et al. Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br J Cancer 2017;117:813–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weyerhauser P, Kantelhardt SR, Kim EL. Re-purposing chloroquine for glioblastoma: potential merits and confounding variables. Front Oncol 2018;8:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Compter I, Eekers DBP, Hoeben A, Rouschop KMA, Reymen B, Ackermans L, et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. Autophagy 2021;17:2604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gammoh N, Fraser J, Puente C, Syred HM, Kang H, Ozawa T, et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016;12:1431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dolma S, Selvadurai HJ, Lan X, Lee L, Kushida M, Voisin V, et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell 2016;29:859–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang C, Zhang X, Xu R, Huang B, Chen AJ, Li C, et al. TGF-beta2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells' invasion. J Exp Clin Cancer Res 2017;36:162. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Giatromanolaki A, Sivridis E, Mitrakas A, Kalamida D, Zois CE, Haider S, et al. Autophagy and lysosomal related protein expression patterns in human glioblastoma. Cancer Biol Ther 2014;15:1468–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dong S, Wang Q, Kao YR, Diaz A, Tasset I, Kaushik S, et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021;591:117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hao Y, Kacal M, Ouchida AT, Zhang B, Norberg E, Vakifahmetoglu-Norberg H. Targetome analysis of chaperone-mediated autophagy in cancer cells. Autophagy 2019;15:1558–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kacal M, Zhang B, Hao Y, Norberg E, Vakifahmetoglu-Norberg H. Quantitative proteomic analysis of temporal lysosomal proteome and the impact of the KFERQ-like motif and LAMP2A in lysosomal targeting. Autophagy 2021;17:3865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bourdenx M, Martín-Segura A, Scrivo A, Rodriguez-Navarro JA, Kaushik S, Tasset I, et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021;184:2696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nie T, Tao K, Zhu L, Huang L, Hu S, Yang R, et al. Chaperone-mediated autophagy controls the turnover of E3 ubiquitin ligase MARCHF5 and regulates mitochondrial dynamics. Autophagy 2020;17:2923–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saga I, Shibao S, Okubo J, Osuka S, Kobayashi Y, Yamada S, et al. Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro Oncol 2014;16:1048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shibao S, Minami N, Koike N, Fukui N, Yoshida K, Saya H, et al. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro Oncol 2018;20:343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vollmann-Zwerenz A, Leidgens V, Feliciello G, Klein CA, Hau P. Tumor cell invasion in glioblastoma. Int J Mol Sci 2020;21:1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kumar Praveen VR, Sehgal P, Thota B, Patil S, Santosh V, Kondaiah P. Insulin like growth factor binding protein 4 promotes GBM progression and regulates key factors involved in EMT and invasion. J Neurooncol 2014;116:455–64. [DOI] [PubMed] [Google Scholar]
- 58. Yeung YT, McDonald KL, Grewal T, Munoz L. Interleukins in glioblastoma pathophysiology: implications for therapy. Br J Pharmacol 2013;168:591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guelfi S, Duffau H, Bauchet L, Rothhut B, Hugnot JP. Vascular transdifferentiation in the CNS: a focus on neural and glioblastoma stem-like cells. Stem Cells Int 2016;2016:2759403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.